
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The titanium tris-anilide cation [Ti(N[tBu]Ar)3]+ stabilized as its perfluoro-tetra-phenylborate salt: structural characterization and synthesis in connection with redox activity of 4,4′-bipyridine dititanium complexes†
Heather A.
Spinney‡
,
Christopher R.
Clough§
and
Christopher C.
Cummins
*
6-435 Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139-4307, USA. E-mail: ccummins@mit.edu; Tel: +(617) 253-5332
First published on 16th February 2015
Abstract
This work explores the reduction of 4,4′-bipyridine using two equivalents of the titanium(III) complex Ti(N[tBu]Ar)3 resulting in formation of a black, crystalline complex, (4,4′-bipy){Ti(N[tBu]Ar)3}2, for which an X-ray structure determination is reported. The neutral, black, 4,4′-bipyridine-bridged bimetallic was found to be redox active, with mono- and di-anions being accessible electrochemically, and with the mono- and di-cations also being accessible chemically, and isolable, at least when using the weakly coordinating anion [B(C6F5)4]− as the counter-ion. It proved possible to crystallize the salt [(4,4′-bipy){Ti(N[tBu]Ar)3}2][B(C6F5)4]2 for a single-crystal X-ray structure investigation; in this instance it was revealed that the aromaticity of the 4,4′-bipyridine ligand, that had been disrupted upon reduction, had been regained. A rare cationic d0 metal tris-amide complex, shown by X-ray crystallography to contain an intriguing pyramidal TiN3 core geometry, namely {Ti(N[tBu]Ar)3}+, could also be isolated when using [B(C6F5)4] as the essentially non-interacting counter-ion. This highly reactive cation should be considered as a potential intermediate in the plethora of reactions wherein Ti(N[tBu]Ar)3 has been shown to effect the reduction of substrates including halogenated organic molecules, carbonyl compounds, organic nitriles, and metal complexes.
1 Introduction
This investigation took shape as an inquiry into the potential ability of the tris-anilide titanium(III) complex Ti(N[tBu]Ar)3 (Ar = 3,5-Me2C6H3) to reductively bind or couple pyridine. Pyridine coupling as a route to bipyridines is a reaction of significant interest, and it has been shown previously by Rothwell et al. that pyridine reductive coupling could be effected by a titanium(II) synthon resulting in a dititanium(III) system containing a bridging {–NC4H4C(H)–C(H)C4H4N–} moiety.1 In addition, we showed previously that Ti(N[tBu]Ar)3 was capable of promoting pyridine reductive cross-coupling with benzonitrile, with the molybdenum(III) tris-anilide complex Mo(N[tBu]Ar)3 in this case responsible for reductive complexation of the organic nitrile prior to coupling with titanium-complexed pyridine.2 Related to Rothwell's para-coupled pyridine dimer by dehydrogenation is a system in which 4,4′-bipyridine bridges two titanium centers, and accordingly, this article treats the ability of Ti(N[tBu]Ar)3 (Fig. 1) to reductively complex that organic molecule in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 fashion. A study of the redox behavior of the (4,4′-bipy){Ti(N[tBu]Ar)3}2 bimetallic system led to the significant discovery that the highly electrophilic cation {Ti(N[tBu]Ar)3}+ could be isolated when generated in particular as its {[B(C6F5)4]}− salt. The synthesis and structural characterization of cation {Ti(N[tBu]Ar)3}+ is a key finding delivered in the present work, as this species is a plausible reactive intermediate in a variety of reductive processes effected by the special one-electron reducing agent that is embodied by Ti(N[tBu]Ar)3.
:1 fashion. A study of the redox behavior of the (4,4′-bipy){Ti(N[tBu]Ar)3}2 bimetallic system led to the significant discovery that the highly electrophilic cation {Ti(N[tBu]Ar)3}+ could be isolated when generated in particular as its {[B(C6F5)4]}− salt. The synthesis and structural characterization of cation {Ti(N[tBu]Ar)3}+ is a key finding delivered in the present work, as this species is a plausible reactive intermediate in a variety of reductive processes effected by the special one-electron reducing agent that is embodied by Ti(N[tBu]Ar)3.
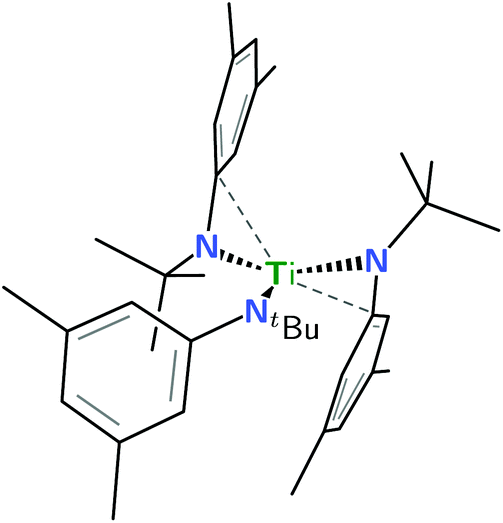 | ||
| Fig. 1 Line drawing of neutral Ti(N[tBu]Ar)3 based upon coordinates from an X-ray crystal structure3 and emphasizing (dashed lines) the two aryl groups that make close contact with the electrophilic d1 metal center. | ||
1.1 Reductions effected by titanium tris-anilide
As a one-electron reducing agent, Ti(N[tBu]Ar)3 may be compared to SmI2,4 but in general Ti(N[tBu]Ar)3 is “hotter” in being able to effect halogen atom abstraction from less-activated halogen-containing organic molecules than SmI2 is capable of; an example of this is X abstraction from stoichiometric bromobenzene or iodobenzene to produce the corresponding titanium(IV) complex XTi(N[tBu]Ar)3 rapidly and quantitatively at 25 °C in a nonpolar solvent such as benzene.5 While 7-chloronorbornadiene similarly gave a rapid reaction,6 neat chlorobenzene proved to be less reactive, requiring ca. 8 h to produce ClTi(N[tBu]Ar)3 (also in essentially quantitative yield), while fluorobenzene was observed to be effectively inert to Ti(N[tBu]Ar)3.5 Since Ti(N[tBu]Ar)3 has little propensity to capture organic radicals, its halogen abstraction propensity could be harnessed in a radical synthesis of PR3 molecules utilizing white phosphorus.7 In general, carbonyl compounds are attacked rapidly by Ti(N[tBu]Ar)3 leading to ketyl radical chemistry;5 this behavior is similar to that reported by Covert and Wolczanski for the related three-coordinate titanium(III) complex Ti(OSitBu3)3.8tert-Butyl esters react giving substitution of tert-butyl radical by Ti(N[tBu]Ar)3, a facile pathway for the synthesis of formate derivatives.9 Titanium tris-anilide has also been shown to reduce inorganic substrates, for example in the synthesis of WCl3(THF)3 from WCl4(DME),10 in the reductive complexation of metal oxo and nitrido systems,3,11 and in the synergistic (together with Mo(N[tBu]Ar)3) binding of N2.12 Additionally, there is evidence that Ti(N[tBu]Ar)3 engages in (reversible) reductive complexation of CO2 and pyridine.22 Results and discussion
2.1 Reaction of Ti(N[tBu]Ar)3 with 4,4′-bipyridine
The dinuclear titanium complex (4,4′-bipy){Ti(N[tBu]Ar)3}2 was synthesized in excellent yield (89%) via treatment of a bright green solution of Ti(N[tBu]Ar)3 in thawing Et2O (−110 °C) with 0.5 equiv. of 4,4′-bipyridine. Addition of the latter reactant elicited a color change from green to black and then the product (4,4′-bipy){Ti(N[tBu]Ar)3}2 precipitated as a fine black powder. The bimetallic complex is diamagnetic, and its structure (a single anilide ligand environment and D2h symmetry of the bridging 4,4′-bipyridine ligand) was readily assigned using 1H and 13C NMR spectroscopies. Integration of 1H NMR signals indicated that there were six anilide ligands for every one molecule of 4,4′-bipyridine. The complex is diamagnetic by virtue of two electron reduction of the 4,4′-bipyridine bridge by the two equivalents of titanium(III) tris-anilide; the 1H NMR chemical shifts observed for the bipyridine protons (δ = 5.43 and 6.97 ppm) are typical of those observed in other doubly reduced 4,4′-bipyridine species.13,142.2 Molecular structure of (4,4′-bipy){Ti(N[tBu]Ar)3}2
Crystals of (4,4′-bipy){Ti(N[tBu]Ar)3}2 that were grown from a saturated toluene solution were found to be suitable for analysis by X-ray crystallography; the space group was P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with a crystallographic inversion center located at the midpoint of the central C–C bond. The observed molecular structure of (4,4′-bipy){Ti(N[tBu]Ar)3}2 confirms the presence of a reduced 4,4′-bipyridine bridge in the molecule (Fig. 2). The central C–C bond is markedly contracted (1.394(2) Å) relative to the single C–C bond in free 4,4′-bipyridine (1.484(2) Å), and is essentially equal in length to the double C–C bond observed in bis(trimethylsilyl)dihydro-4,4′-bipyridine (1.382(3) Å), another doubly-reduced derivative of 4,4′-bipyridine.13,14 The shortest C–C distance observed in the pyridyl rings of the complex (C41–C42 = 1.358(2) Å) also points to the existence of localized C–C double bonds in the molecule. The Ti1–N4 distance (1.9913(13) Å) is similar to the average Ti1–N distance for the anilide ligands (1.97 Å), indicating that N4 may be regarded similarly as an amide nitrogen atom, as would be expected in a system containing a [4,4′-bipyridine]2− moiety. A search of the Cambridge Structural Database returned one other system containing two titanium atoms bridged by a 4,4′-bipyridine ligand. In 2005, Kraft et al. reported a molecular square formed from four molecules of 4,4′-bipyridine connected by four titanocene fragments at the corners.14 In this case the 4,4′-bipyridine ligands were only partially reduced, with interatomic distances intermediate between those of neutral 4,4′-bipyridine and bis(trimethylsilyl)dihydro-4,4′-bipyridine.
with a crystallographic inversion center located at the midpoint of the central C–C bond. The observed molecular structure of (4,4′-bipy){Ti(N[tBu]Ar)3}2 confirms the presence of a reduced 4,4′-bipyridine bridge in the molecule (Fig. 2). The central C–C bond is markedly contracted (1.394(2) Å) relative to the single C–C bond in free 4,4′-bipyridine (1.484(2) Å), and is essentially equal in length to the double C–C bond observed in bis(trimethylsilyl)dihydro-4,4′-bipyridine (1.382(3) Å), another doubly-reduced derivative of 4,4′-bipyridine.13,14 The shortest C–C distance observed in the pyridyl rings of the complex (C41–C42 = 1.358(2) Å) also points to the existence of localized C–C double bonds in the molecule. The Ti1–N4 distance (1.9913(13) Å) is similar to the average Ti1–N distance for the anilide ligands (1.97 Å), indicating that N4 may be regarded similarly as an amide nitrogen atom, as would be expected in a system containing a [4,4′-bipyridine]2− moiety. A search of the Cambridge Structural Database returned one other system containing two titanium atoms bridged by a 4,4′-bipyridine ligand. In 2005, Kraft et al. reported a molecular square formed from four molecules of 4,4′-bipyridine connected by four titanocene fragments at the corners.14 In this case the 4,4′-bipyridine ligands were only partially reduced, with interatomic distances intermediate between those of neutral 4,4′-bipyridine and bis(trimethylsilyl)dihydro-4,4′-bipyridine.
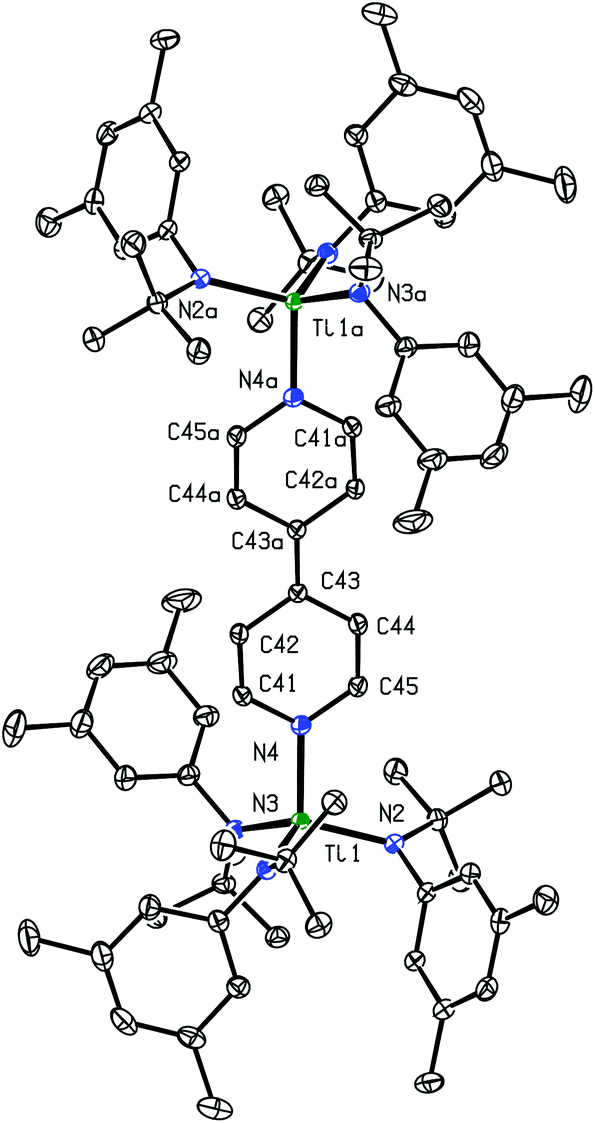 | ||
| Fig. 2 Diagram of (4,4′-bipy){Ti(N[tBu]Ar)3}2 with thermal ellipsoids at the 50% probability level.15 | ||
2.3 Electronic structure of (4,4′-bipy){Ti(N[tBu]Ar)3}2
To gain insight into the electronic structure of the neutral dititanium complex of 4,4′-bipyridine, density functional theory (DFT) calculations were performed on a truncated version of the molecule: (4,4′-bipy){Ti(N[Me]Ph)3}2; the model was built based upon the crystallographic atomic coordinates for (4,4′-bipy){Ti(N[tBu]Ar)3}2 and then geometry-optimized. The calculated HOMO (Fig. 3) contains both π-bonding and π-antibonding character, with the most significant contribution coming from the central C–C π-bond; the pattern of π bonding in the HOMO corresponds nicely to what is likely the dominant Lewis resonance structure for this system. Notably, the HOMO also contains a contribution from Ti–N π-bonding. The LUMO is composed almost exclusively of metal-centered empty d orbital character. A significant HOMO–LUMO energy gap is predicted by the calculation (0.83 eV) and this is supported by the presence of a strong ligand-to-metal charge transfer band at 910 nm (ε = 32000 M−1 cm−1) in the UV/Vis spectrum of black (4,4′-bipy){Ti(N[tBu]Ar)3}2 (Fig. 4).
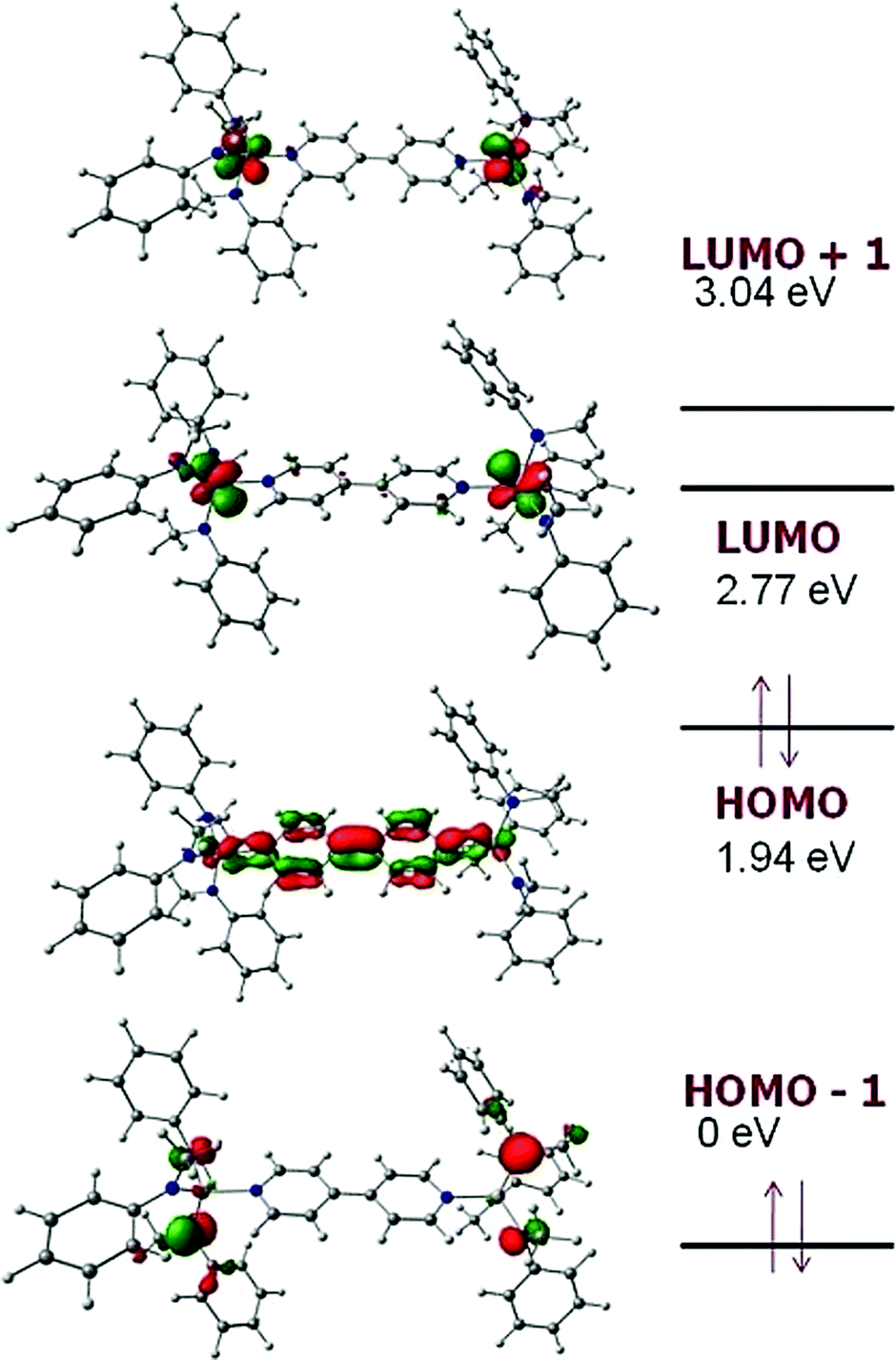 | ||
| Fig. 3 Frontier MO energy levels calculated for model system (4,4′-bipy){Ti(N[Me]Ph)3}2.16 | ||
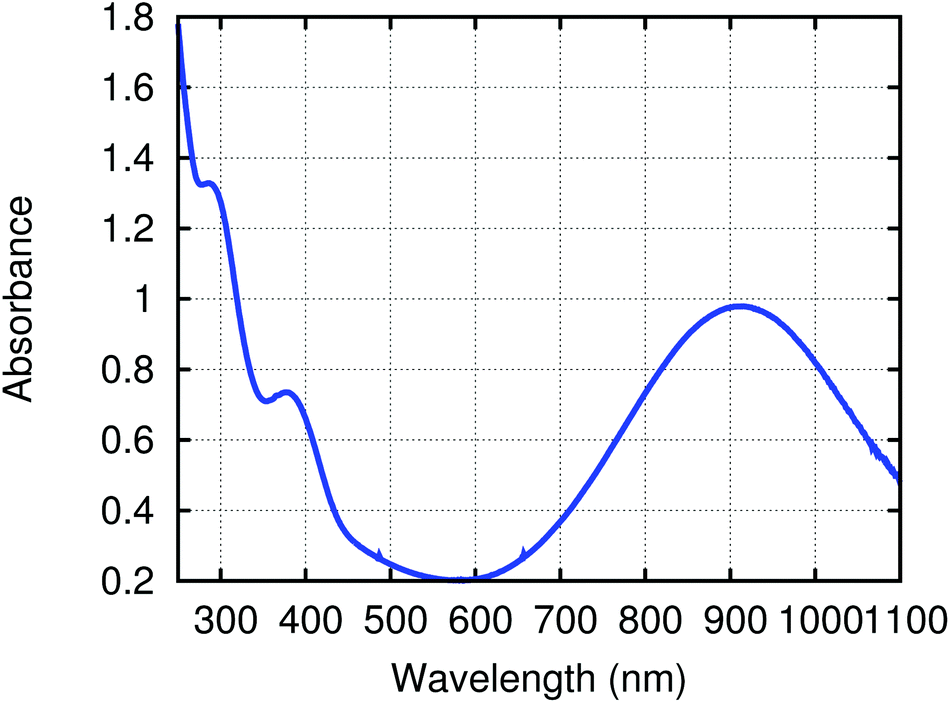 | ||
| Fig. 4 Electronic absorption spectrum of (4,4′-bipy){Ti(N[tBu]Ar)3}2 as a solution in THF. | ||
2.4 Electrochemistry and chemical redox of (4,4′-bipy){Ti(N[tBu]Ar)3}2
The electronic structure of (4,4′-bipy){Ti(N[tBu]Ar)3}2 leads to interesting electrochemical behavior in solution. Cyclic voltammetry performed on THF solutions of (4,4′-bipy){Ti(N[tBu]Ar)3}2 (Fig. 5) indicates two reversible oxidation events and two quasi reversible reduction events and the electrochemical data are summarized in Table 1. The observed redox features can be explained by considering the different charged states of the molecule (illustrated in Fig. 6 for the isolable neutral, cationic, and di-cationic species). Starting from the neutral species, removal of an electron from the [4,4′-bipy]2− ligand results in formation of a radical cation without any change to titanium oxidation state. Nevertheless, it is possible to draw a resonance structure for this species wherein the unpaired electron resides on one of the titanium centers, such that the radical cation could also be formulated as a mixed-valent titanium(III)/titanium(IV) species. Removal of an additional electron from the radical cation leads to dicationic [(4,4′-bipy){Ti(N[tBu]Ar)3}2]2+, in which aromaticity is restored to the bridging 4,4′-bipy ligand.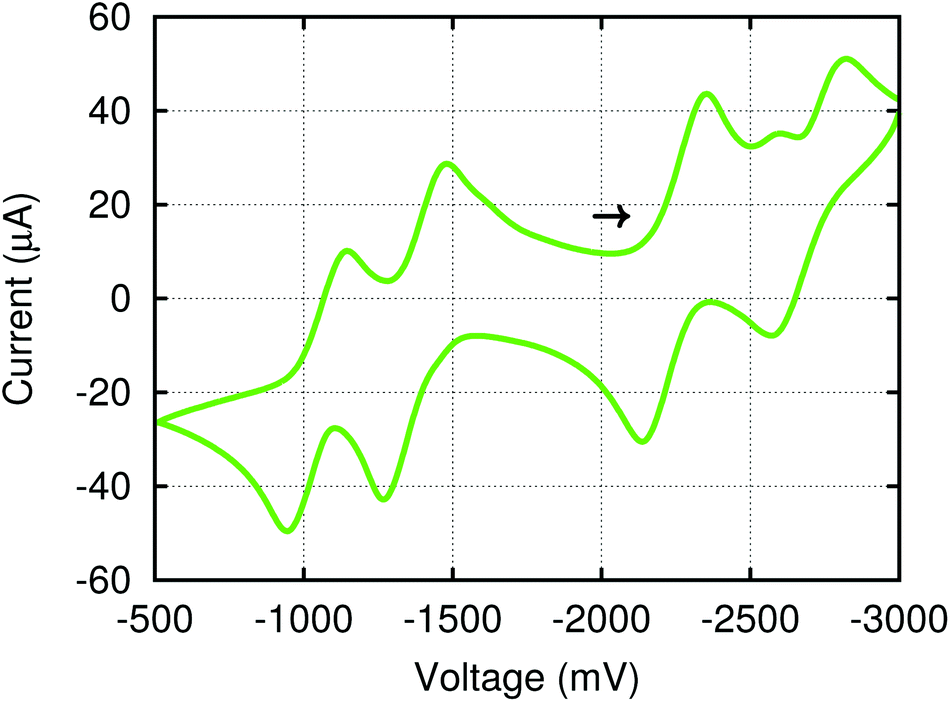 | ||
| Fig. 5 Cyclic voltammogram for (4,4′-bipy){Ti(N[tBu]Ar)3}2 in THF with a sweep rate of 800 mV s−1 referenced vs. Fc/Fc+ with [NnBu4][B(C6F5)4] (0.2 M) as the supporting electrolyte. The arrow signifies the rest potential and the direction of the sweep. | ||
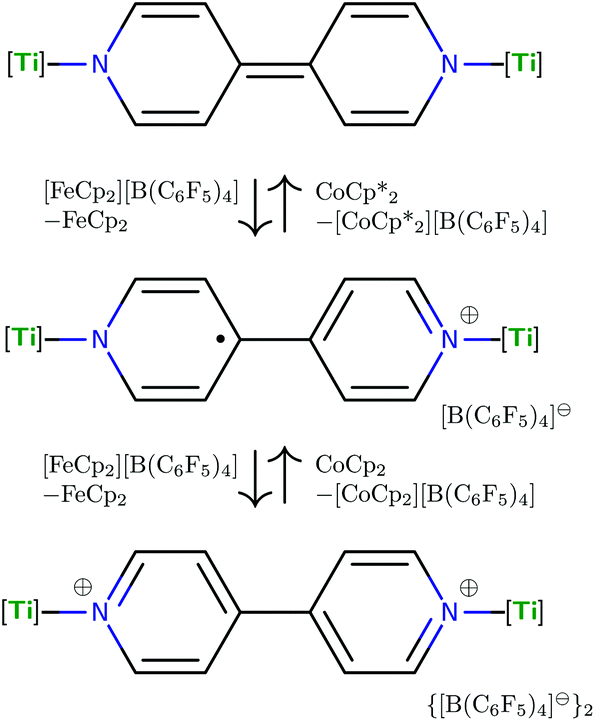 | ||
| Fig. 6 Isolable in three states of charge, the figure depicts schematically (4,4′-bipy){Ti(N[tBu]Ar)3}2, its mono-cation, and di-cation, the latter cations both having been isolated as perfluoro-tetraphenyl borate salts. | ||
| Couple | E 1/2/Va | ΔE1/2/Vb |
K
cc |
|---|---|---|---|
|
a
E
1/2 values are referenced versus ferrocene/ferrocenium.
b Separation between two adjacent half-wave potentials representing the range wherein the respective ion exists.
c Calculated according to lnKc = −nFΔE1/2/RT.17
|
|||
| 2−/1− | −2.65 | 0.43 | 1.9 × 107 |
| 1−/0 | −2.22 | ||
| 0/1+ | −1.36 | 0.32 | 2.6 × 105 |
| 1+/2+ | −1.04 | ||
Sweeping negative, addition of an electron to (4,4′-bipy){Ti(N[tBu]Ar)3}2 converts one of the titanium(IV) centers in the molecule to titanium(III) and generates an anionic mixed-valent complex. Addition of a second electron reduces the remaining titanium(IV) center to titanium(III). It is indicated that all five charge states of (4,4′-bipy){Ti(N[tBu]Ar)3}2 should be accessible through chemical oxidation and reduction reactions, with the exception of the second reduction event. At −2.7 V vs. the Fc/Fc+ couple, the latter is too near the solvent window. Here we note that the cyclic voltammogram of bis(trimethylsilyl)dihydro-4,4′-bipyridine exhibits two reversible oxidation waves at potentials (vs. Fc/Fc+) nearly identical to those observed here;13 however, for that molecule the two reversible reduction waves are absent as there are present at silicon vis-à-vis titanium no low-lying empty d orbitals.
Differential pulse voltammetry (DPV)18 was used to obtain accurate E1/2 values for each of the one-electron couples observed in the cyclic voltammogram. The comproportionation constants Kc were calculated for the proposed mixed-valence species [(4,4′-bipy){Ti(N[tBu]Ar)3}2]+ in order to evaluate the extent of electronic communication between the titanium centers. The Kc values were derived from the potential separation between ΔE1/2 between couples (Table 1).17 For the cation [(4,4′-bipy){Ti(N[tBu]Ar)3}2]+, the ΔE1/2 value of 0.32 V corresponds to Kc = 2.6 × 105, which is in the range usually associated with Class II mixed-valent compounds.19 Therefore, some degree of electronic communication is occurring between the two metal centers. This communication is more pronounced in the anion, [(4,4′-bipy){Ti(N[tBu]Ar)3}2]−, where the larger ΔE1/2 value of 0.43 V corresponds to Kc = 1.9 × 107. A greater degree of electronic delocalization is therefore observed in the mixed-valent anion than in the mixed-valent cation.
Chemical oxidation reactions of (4,4′-bipy){Ti(N[tBu]Ar)3}2 have shown that it is possible to release an equivalent of 4,4′-bipyridine from the molecule. For example, treatment of (4,4′-bipy){Ti(N[tBu]Ar)3}2 with an equivalent of iodine resulted in its quantitative conversion to two equivalents of ITi(N[tBu]Ar)35,11 with concomitant release of an equivalent of 4,4′-bipyridine. A similar transformation was also achieved with two equivalents of the outer-sphere oxidant ferrocenium triflate (Fc[OTf]), and demonstrates that the reduction of 4,4′-bipyridine by titanium(III) trisanilide Ti(N[tBu]Ar)3 is chemically reversible. Treatment of (4,4′-bipy){Ti(N[tBu]Ar)3}2 with a single equivalent of Fc[OTf] did not result in conversion to a one-electron oxidation product, but instead, half an equivalent of the dinuclear complex was consumed in being oxidized by two electrons, leaving half an equivalent unreacted. This is not entirely surprising considering how closely spaced the two oxidation events are in the cyclic voltammogram and that only mild oxidizing agents are required for both transformations.
The one-electron oxidation product [(4,4′-bipy){Ti(N[tBu]Ar)3}2]+ is available via oxidation with a ferrocenium salt containing a weakly coordinating anion. For example, treatment of (4,4′-bipy){Ti(N[tBu]Ar)3}2 with one equivalent of Fc[B(C6F5)4]20,21 at room temperature in THF resulted upon mixing in a color change from black to brown. After separation from ferrocene, the [B(C6F5)4]− salt of [(4,4′-bipy){Ti(N[tBu]Ar)3}2]+ was isolated as a brown powder (84% yield). The latter cation proved to be completely NMR silent. It also was found to be sensitive thermally, appearing gradually to decompose in solution at room temperature over a period of a few days. Efforts to grow crystals of this salt at low temperature did not succeed.
The radical nature of [(4,4′-bipy){Ti(N[tBu]Ar)3}2]+ was confirmed by EPR spectroscopy. A room-temperature EPR spectrum of a toluene solution containing the cation displayed only one signal (g = 2.0004), with no additional hyperfine coupling. The g-value of the signal is typical of an organic π-radical; however, this does not rule out a resonance contributor to the radical cation in which the unpaired electron is located on one of the titanium ions. As the HOMO of the neutral precursor is largely π-bonding in nature, removal of an electron from this orbital should result in π bonds of reduced order. Consistent with this, the IR spectrum of [(4,4′-bipy){Ti(N[tBu]Ar)3}2]+ (as its [B(C6F5)4]− salt) displays a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch at 1614 cm−1, compared to the CC stretch at 1622 cm−1 that is observed for the neutral precursor. Additionally, the UV/Vis spectrum of the radical cation in THF shows a band at 965 nm (ε = 10000 M−1 cm−1), this being less intense than the corresponding band observed at 910 nm (ε = 32000 M−1 cm−1) in the unoxidized species. This can be interpreted as a diminution of the LMCT interaction in the system upon oxidation.
C stretch at 1614 cm−1, compared to the CC stretch at 1622 cm−1 that is observed for the neutral precursor. Additionally, the UV/Vis spectrum of the radical cation in THF shows a band at 965 nm (ε = 10000 M−1 cm−1), this being less intense than the corresponding band observed at 910 nm (ε = 32000 M−1 cm−1) in the unoxidized species. This can be interpreted as a diminution of the LMCT interaction in the system upon oxidation.
In line with the inference that one-electron oxidation of (4,4′-bipy){Ti(N[tBu]Ar)3}2 is not accompanied by any major structural change, we found that the cation [(4,4′-bipy){Ti(N[tBu]Ar)3}2]+ can be reduced back to its neutral and diamagnetic precursor by treatment with decamethyl cobaltocene. The cation can also be further oxidized by treatment with an additional equivalent of Fc[B(C6F5)4] to yield the new dicationic complex [(4,4′-bipy){Ti(N[tBu]Ar)3}2]2+ as its ([B(C6F5)4]−)2 salt. The latter red salt, as expected, could also be accessed (in 84% yield) upon treatment of (4,4′-bipy){Ti(N[tBu]Ar)3}2 with two equivalents of Fc[B(C6F5)4]. Additionally, two equiv. of the radical cation [(4,4′-bipy){Ti(N[tBu]Ar)3}2]+ are generated quantitatively upon mixing of one equivalent each of neutral (4,4′-bipy){Ti(N[tBu]Ar)3}2 and [(4,4′-bipy){Ti(N[tBu]Ar)3}2]2+, demonstrating the relationship among this series of complexes whose compositions differ only in terms of their state of charge.
Crystals of the ([B(C6F5)4]−)2 salt of dication [(4,4′-bipy){Ti(N[tBu]Ar)3}2]2+ suitable for an X-ray diffraction study were grown by liquid–liquid diffusion of hexane into an o-difluorobenzene solution. o-Difluorobenzene is polar and has a higher dielectric constant (Ds = 13.8) than THF (Ds = 7.4), but is a weaker donor.22 The ([B(C6F5)4]−)2 salt of dication [(4,4′-bipy){Ti(N[tBu]Ar)3}2]2+ is insoluble in alkane and arene solvents, but dissolves in coordinating solvents including THF and pyridine. However, dissolution in pyridine-d5 results in a ligand displacement reaction, liberating 4,4′-bipyridine and resulting in coordination of pyridine-d5 to the liberated {Ti(N[tBu]Ar)3}+ moieties (vide infra). When the dication salt is dissolved in THF-d8, the THF-d8 solvent molecules compete with 4,4′-bipyridine for binding to the {Ti(N[tBu]Ar)3}+ cations, resulting in an equilibrium between free and bound 4,4′-bipyridine being observed in the solution 1H NMR (benzene-d6, 20 °C, 500 MHz) spectrum. Thus, the use of o-difluorobenzene as a crystallization solvent was key to determining the solid-state structure of the dication's ([B(C6F5)4]−)2 salt. The molecular structure of dication [(4,4′-bipy){Ti(N[tBu]Ar)3}2]2+ is shown in Fig. 7.
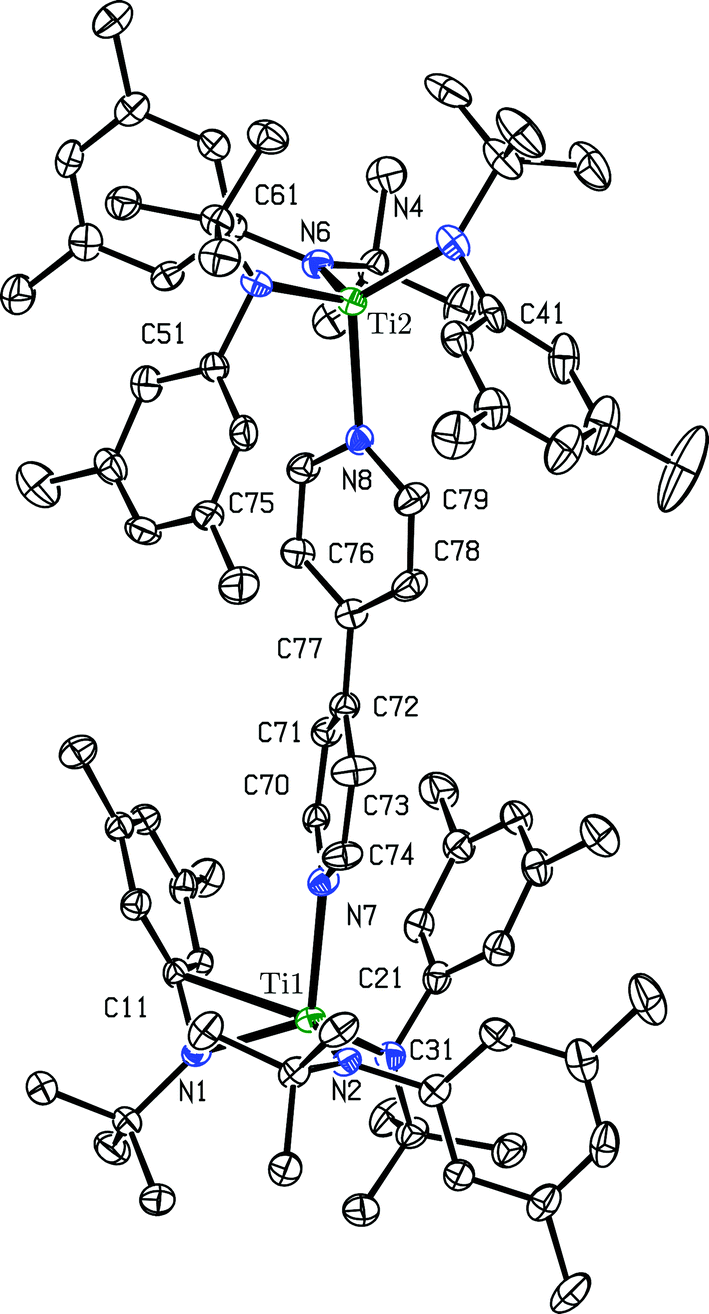 | ||
| Fig. 7 Diagram (50% probability ellipsoids) of dication [(4,4′-bipy){Ti(N[tBu]Ar)3}2]2+ with hydrogen atoms, [B(C6F5)4]− ions, and an o-difluorobenzene solvent molecule omitted.15 | ||
The most striking structural feature of dication [(4,4′-bipy){Ti(N[tBu]Ar)3}2]2+ is the twist about the central C–C bond of the bipyridine (torsion angle = 37.16°). A twist of similar magnitude is observed in free 4,4′-bipyridine (torsion angle = 34.20°).14 Additionally, the central C–C bond is 1.484(4) Å in length, coincident with the corresponding distance in free 4,4′-bipyridine (1.484(2) Å) and 0.09 Å longer than in neutral (4,4′-bipy){Ti(N[tBu]Ar)3}2 (1.394(2) A). This demonstrates that the removal of two electrons from (4,4′-bipy){Ti(N[tBu]Ar)3}2 results in rearomatization of the 4,4′-bipyridine unit. In further support of the latter interpretation, the Ti–N(bipyridine) distances in [(4,4′-bipy){Ti(N[tBu]Ar)3}2]2+ (2.209(2) Å and 2.213(2) Å) are substantially longer than they are in neutral (4,4′-bipy){Ti(N[tBu]Ar)3}2 (1.9913(13) Å), and are characteristic of the interaction between a neutral N-donor and titanium(IV).23 Compensating for the weaker Ti–N(bipyridine) bonds, the Ti–N(anilide) distances in the dication (avg. = 1.92 Å) are substantially shorter than in the neutral precursor (avg. = 1.97 Å), consistent with increased π-donation to the cationic titanium centers from the anilide nitrogens. As well, there appears to be an additional bonding interaction between C11 and Ti1 (C11⋯Ti1 = 2.500(3) Å), and C41 also closely approaches Ti2 (C41⋯Ti2 = 2.554(3) Å). It is not unprecedented for N-tert-butylanilide ligands to bind in an η2 or even in an η3 fashion in the process of stabilizing an electrophilic metal center.3
2.5 Synthesis and molecular structure of cation {Ti(N[tBu]Ar)3}+ ([B(C6F5)4]− salt)
The dication of the previous section can reasonably be viewed as two {Ti(N[tBu]Ar)3}+ cations linked by a neutral 4,4′-bipyridine ligand. As the {Ti(N[tBu]Ar)3}+ cation had never been independently synthesized in our laboratory, we set out to accomplish this via oxidation of Ti(N[tBu]Ar)3 using [FeCp2][B(C6F5)4]. Treatment of a green, ethereal solution of Ti(N[tBu]Ar)3 with an Et2O slurry of [FeCp2][B(C6F5)4] at room temperature resulted upon mixing in a color change to red. After removal of solvent under reduced pressure, what remained was a red oil that could be converted to an orange powder upon repeated trituration using n-hexane. The orange powder was determined to be the salt {Ti(N[tBu]Ar)3}[B(C6F5)4], obtained (90% yield) in a form that is entirely free of any coordinating solvent molecules. In this form, salt {Ti(N[tBu]Ar)3}[B(C6F5)4] is insoluble in Et2O, benzene and toluene, but does dissolve in these solvents upon addition of a few drops of THF. The relative inertness of the [B(C6F5)4]− counter-ion is key to the isolation of the {Ti(N[tBu]Ar)3}+ cation. Prior experiments aimed at similarly generating the cation {Ti(N[tBu]Ar)3}+via ferrocenium oxidation using [FeCp2][B[3,5-C6H3(CF3)2]4]24 instead resulted in isolation of the fluoride, FTi(N[tBu]Ar)3.5 It is likely that the reactive {Ti(N[tBu]Ar)3}+ cation is formed under the latter conditions but that it abstracts fluoride from the insufficiently inert [B[3,5-C6H3(CF3)2]4]− anion.Crystals of {Ti(N[tBu]Ar)3}[B(C6F5)4] were grown by liquid–liquid diffusion of hexane into an o-difluorobenzene solution of the salt, conducive to an X-ray diffraction study (Fig. 8). The titanium(IV) ion of cation [Ti(N[tBu]Ar)3]+ pulls the N-tert-butylanilide ligands in very close (Ti1–N = 1.8788(15), 1.8830(16), and 1.8937(16) Å). Additionally, all three anilide ligands in this otherwise unsolvated cation appear to engage titanium in an η2-N,C fashion, with the Ti1–C distances (2.3870(18), 2.3984(18), and 2.4048(19) Å) being shorter than is typically observed for η2 or η3 anilide aryl ring ligation to titanium; for example, see the structure of {(THF)Ti(N[tBu]Ar)3}+, described below (Fig. 9).
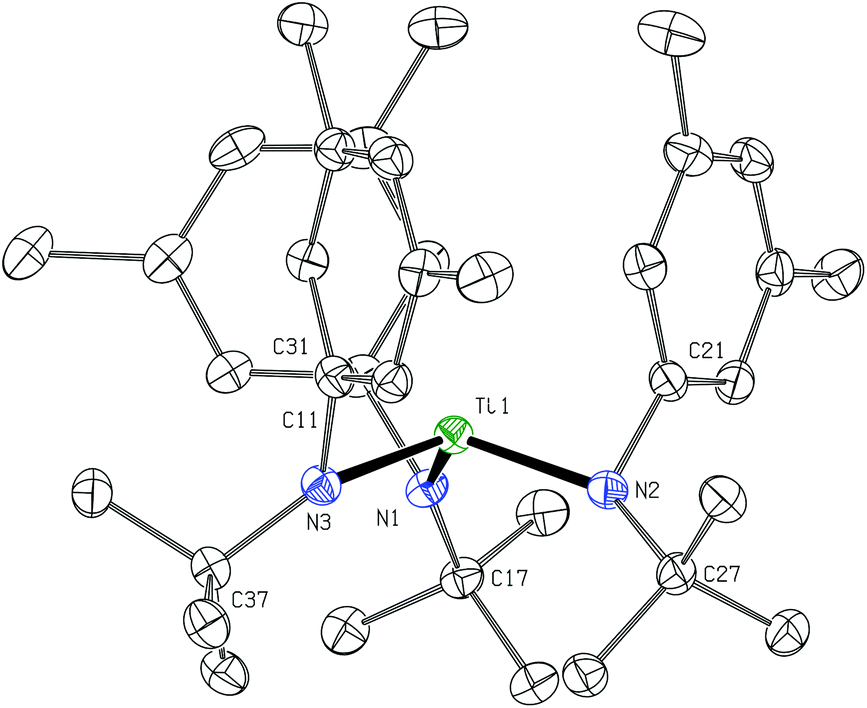 | ||
| Fig. 8 Diagram of cation {Ti(N[tBu]Ar)3}+ with 50% probability ellipsoids.15 The [B(C6F5)4]− counter-anion and hydrogen atoms are omitted for clarity (no close anion/cation contacts were observed). | ||
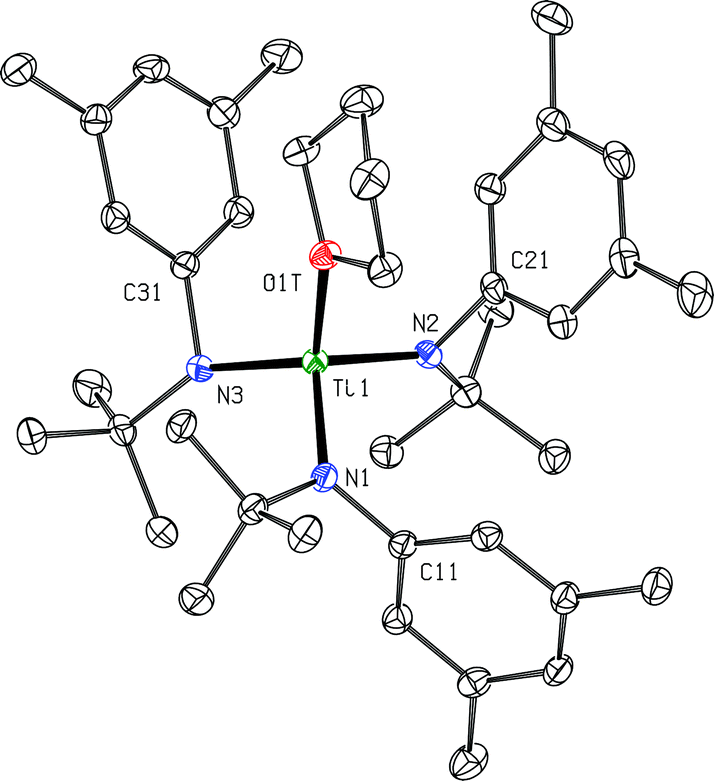 | ||
| Fig. 9 Diagram of cation {(THF)Ti(N[tBu]Ar)3}+ with 50% probability ellipsoids.15 The [B(C6F5)4]− counter-anion, ether molecule of crystallization and hydrogen atoms are omitted for clarity (no close anion/cation contacts were observed). | ||
A most unusual feature of the structure (Fig. 8) is the trigonal pyramidal nature of the TiN3 core unit (the average N–Ti–N angle is ca. 111.3°); despite the presence of two η3 anilide interactions in the structure of neutral Ti(N[tBu]Ar)3,3 the TiN3 moiety in that case is rigorously trigonal planar, a structural feature common to all of the neutral M(N[R]Ar)3 complexes that have been characterized to date (M = Ti,3 V,25 Cr,26 Mo12,27–29). Trigonal pyramidal MN3 units are, however, a characteristic structural feature for homoleptic rare earth metal phenyl(trimethylsilyl) amide complexes.30 In addition, Andersen has remarked on the trigonal pyramidal coordination environment at uranium in U[N(SiMe3)2]3.31 Finally, Hursthouse et al. showed that the coordination environment at scandium in Sc[N(SiMe3)2]3 is similarly trigonal pyramidal with N–Sc–N angles of ca. 117°.32
The N-tert-butylanilide ligands in the otherwise unsolvated [Ti(N[tBu]Ar)3]+ cation can certainly be viewed as providing a protective steric sheath about the electrophilic titanium(IV) center that is essential, together with the inert counter anion, for its isolation. Inspection of the packing arrangement in this crystal structure reveals no significant cation–anion interactions.
2.6 Electronic structure of cation {Ti(N[tBu]Ar)3}+
The trigonal pyramidal nature of {Ti(N[tBu]Ar)3}+ is not the only unusual feature of the observed structure. The Ti–N–CMe3 angles are quite obtuse (average value, 147.08°) while the Ti–N–C(aryl) angles are correspondingly acute (avg.: 91.23°). The latter acute Ti–N–C(aryl) angles bring the three aryl ipso carbon atoms in suggestively close to the titanium center, such that structure drawing programs (based on their default element radii) draw lines between Ti1 and each of C11, C21, and C31. The average Ti–C(aryl ipso) distance is 2.3967 Å. Because of these relatively close Ti–C(aryl ipso) contacts, it is tempting to conclude that there is an associated chemical bonding interaction. In order to probe for this, an Atoms in Molecules (AIM) analysis33 was carried out on a wavefunction34 that was calculated at the observed structure (only the hydrogen positions were optimized) with the result that no bond critical points were located between titanium and the aryl ipso carbon atoms; by this electron density criterion, no chemical bonds exist between titanium and the aryl ipso carbon atoms and we must conclude that the relatively close contacts occur incidentally as a consequence of the titanium–nitrogen multiple bonding together with unusual hybridization at nitrogen.Regarding the Ti–N bonding being multiple in {Ti(N[tBu]Ar)3}+, natural bond orbital (NBO)35,36 and natural resonance theory (NRT)37–39 analysis of the same wavefunction34 used for AIM leads to the conclusion that each nitrogen is tetravalent (engages in four electron-pair bonds), that the titanium–nitrogen natural bond order is ca. 1.96, and that the natural valence at titanium is six (titanium engages in six electron-pair bonds).40 This analysis also gives Ti–C(aryl ipso) bond orders of near zero, consistent with the lack of associated bond critical points from the AIM analysis. The indicated TiN double bonding is associated with short titanium–nitrogen inter-atomic distances (avg.: 1.885 Å; for reference, an average value of 1.928 Å was reported for the corresponding chloride derivative, ClTi(N[tBu]Ar)3).41
2.7 Known metal tris-amide cations
The metal tris-amide cations [M{N(SiMe3)2}3]+, where M is zirconium or hafnium, have been generated by methide abstraction using B(C6F5)3.42 The latter cations were studied by X-ray crystallography and found to exhibit short M ← (Si–C) contacts interpreted as weak bonding interactions that serve to stabilize the positive charge in these systems.42 In view of the AIM and NBO results discussed above for the bonding in similarly trigonal pyramidal {Ti(N[tBu]Ar)3}, the invocation of M ← (Si–C) contacts as the reason for the pyramidal nature of the MN3 (M = Zr, Hf) unit in these cationic amides may warrant reconsideration. Donor-free, cationic transition-metal tris-amide complexes are otherwise unknown. An example of a donor-stabilized titanium tris-amide cation that has been characterized crystallographically is the simple homoleptic dimethylamide-derived system [(Me2NH)Ti(NMe2)3]+ of importance in alkyne carboamination catalysis.43 A cationic titanium ferrocene-diamide system has been generated by reaction of a dimethyl precursor with the trityl borate [CPh3][B(C6F5)4] reagent.442.8 Binding and ring-opening of tetrahydrofuran
We hypothesized that the highly electrophilic titanium(IV) center in the {Ti(N[tBu]Ar)3}+ cation might allow us to access some previously inaccessible species, one target of interest being the neutral titanium(IV) hydride, HTi(N[tBu]Ar)3. If obtained, the latter would be a member of the small set of documented titanium(IV) hydride complexes that includes HTi(ODipp)3(PMe3),45 where Dipp represents 2,6-C6H3-iPr2, as well as {HTiCp*(NPtBu3)(THF)}+.46 Addition of stoichiometric [Li][HBEt3] (as a 1.0 M solution in THF) to a thawing red solution of {Ti(N[tBu]Ar)3}[B(C6F5)4] in o-difluorobenzene did not result in any immediate sign of reaction, but the color of the reaction mixture gradually turned from red to orange to yellow as the solution warmed to room temperature. After workup, a major product could be obtained in 58% yield as a free-flowing yellow powder, characterization of which revealed it to be the n-butoxy complex nBuOTi(N[tBu]Ar)3 that ostensibly arose by way of hydride attack on a coordinated THF ligand. Complexation by strongly Lewis-acidic metal cations is known to activate THF to nucleophilic attack;47 In one example, addition of KCp* to the cation [Cp*2Sm(THF)2]+ (as its tetraphenylborate salt) led to THF ring-opening and C–C bond formation, rather than to the formation of the desired product, SmCp*3.482.9 Structure of cation {(THF)Ti(N[tBu]Ar)3}+
Both the n-butoxy complex and the THF adduct {(THF)Ti(N[tBu]Ar)3}+ (the latter as its [B(C6F5)4]+ salt) have been isolated and characterized by single-crystal X-ray diffraction studies (Fig. 9 and 10). In the case of the cationic THF adduct {(THF)Ti(N[tBu]Ar)3}+, we find that binding of the THF molecule displaces two of the three Ti⋯Carylipso close contacts involving the anilide ligands with respect to the structure observed in the solid state for the solvent-free cation, {Ti(N[tBu]Ar)3}+ (Fig. 8). The relevant distance that characterizes the remaining Ti1⋯C31 close contact, 2.5250(16) Å, is longer by greater than 0.1 Å than any of the three such distances observed for {Ti(N[tBu]Ar)3}+ (Fig. 8). The Ti1–O1 T distance of 2.0962(12) Å characterizing the titanium–THF interaction is not remarkable, but it should be noted that the crystallographic data reported here (Fig. 9) represent rare structural characterization of a group 4 metal cation having the general composition {(THF)M(NR1R2)3}+.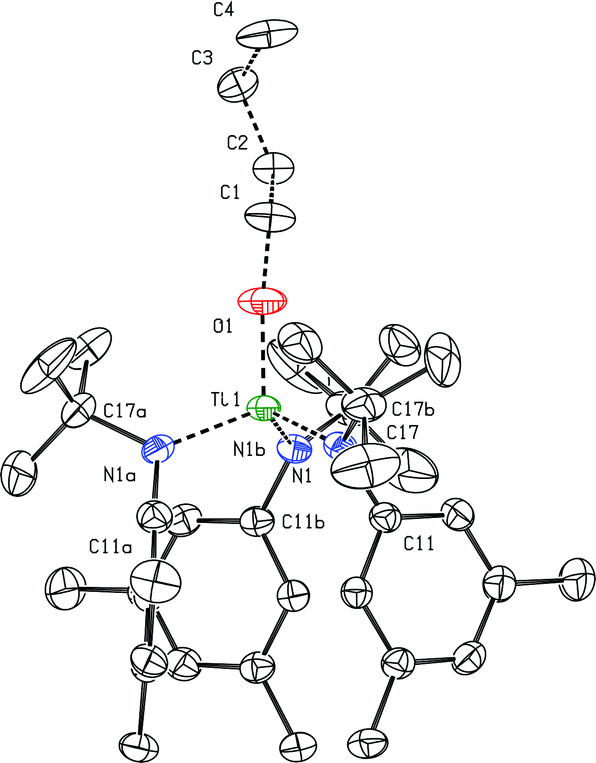 | ||
| Fig. 10 Diagram of n-butoxy complex nBuOTi(N[tBu]Ar)3 with 50% probability ellipsoids.15 The molecule has a crystallographic C3 axis coincident with the Ti–O vector and the n-butyl group occupies two positions (one shown). | ||
3 Experimental details
3.1 General procedures
All manipulations were performed under an atmosphere of purified N2 in a vacuum atmospheres model MO-40M glovebox or in standard Schlenk glassware in the hood. Solvents were obtained anhydrous and oxygen-free from a Contour Glass Solvent Purification System, built by SG Water recently rebranded as Pure Process Technology (http://www.pureprocesstechnology.com/) with the exception of pyridine which was distilled from sodium. All anhydrous solvents were stored at room temperature over 4 Å molecular sieves. Deuterated solvents were purchased from Cambridge Isotope Labs. Benzene-d6 was degassed and stored over molecular sieves for at least two days prior to use. THF-d8 was distilled from sodium/benzophenone and stored over molecular sieves at −35 °C. Pyridine-d5 was distilled from calcium hydride and stored over molecular sieves. Celite 435 (EM Science), 4 Å molecular sieves (Aldrich), and alumina (EM Science) were dried by heating at 200 °C under a dynamic vacuum for at least 24 hours prior to use. All glassware was oven-dried at temperatures greater than 170 °C prior to use. Ti(N[tBu]Ar)3 was synthesized following a reported literature procedure11 that has also been the subject of a video (see http://web.mit.edu/ccclab/videos/). Ferrocenium triflate was prepared by oxidation of ferrocene with silver triflate in dichloromethane.49 All other chemicals were purchased from Aldrich and used without further purification unless otherwise stated. NMR spectra were obtained on Bruker Avance 400, Varian Mercury 300 or Varian Inova 500 spectrometers. 1H NMR spectra were referenced internally to residual solvent resonances (benzene-d6, 7.16 ppm; THF-d8, 3.58 ppm; pyridine-d5, 8.74 ppm). 13C NMR spectra were referenced internally to solvent signals (benzene-d6, 128.39 ppm; THF-d8, 67.57 ppm). 19F NMR spectra were referenced externally to CFCl3. EPR spectra were collected on a Bruker EMX spectrometer outfitted with 13-inch magnets, an ER 4102ST cavity and a Gunn diode microwave source producing X-band (8–10 GHz) radiation. Samples for IR spectroscopy were prepared as Nujol mulls on KBr windows and IR spectra were collected on a Perkin-Elmer 2000 FT-IR spectrophotometer. UV/Vis spectra were collected on a HP8453 spectrophotometer using 1 cm quartz cells manufactured by Starna. Elemental analyses were performed by Midwest Microlab, LLC, Indianapolis, Indiana.3.2 Preparation of (4,4′-bipy){Ti(N[tBu]Ar)3}2
Ti(N[tBu]Ar)3 (3.01 g, 5.22 mmol) was dissolved in 60 mL of Et2O to create an emerald green solution. This solution was frozen in the dry box cold well. The 4,4′-bipyridine (0.405 g, 2.59 mmol) was dissolved in 15 mL of Et2O to create a colorless solution. This solution was also frozen in the dry box cold well. The thawing bipyridine slurry (4,4′-bipyridine is insoluble in Et2O at low temperatures) was added dropwise with stirring to the thawing Ti(N[tBu]Ar)3 solution. There was an immediate color change from green to black upon addition. The reaction mixture was allowed to stir for one hour while it slowly warmed to room temperature. The Et2O was removed under vacuum, and the resulting black residue was slurried in 50 mL of n-hexane. The black solid was isolated by filtration and washed with n-hexane until the washings became colorless. The powder was further dried under dynamic vacuum for two hours. Total yield: 3.12 g, 2.38 mmol, 89% (based on C82H116N8Ti2, 1309.58 g mol−1). Crystals suitable for X-ray diffraction were grown from a saturated solution of the material in toluene, which was left in the freezer at −35 °C for one week. Anal. Calcd: C 75.21; H 8.93; N 8.56. Found: C 74.98; H 8.78; N 8.67. 1H NMR (benzene-d6, 20 °C, 400 MHz): δ = 7.12 (broad s, 4H, CC(H)–N), 6.97 (s, 12H, HAr), 6.77 (s, 6H, HAr), 5.43 (d, JHH = 8 Hz, 4H, (H)CC–N), 2.34 (s, 36H, Ar–CH3), 1.23 (s, 54H, C(CH3)3) ppm. 13C NMR (benzene-d6, 20 °C, 100.6 MHz): δ = 150.4 (s, aryl ipso), 137.3 (s, m-Ar), 137.0 (s, central CC), 129.6 (s, p-Ar), 127.3 (s, o-Ar), 118.4 (s, ring CC), 105.7 (s, ring CC), 63.8 (s, NC(CH3)3), 31.0 (s, ArCH3), 22.1 (s, NC(CH3)3) ppm. UV/Vis Data (THF, 3.05 × 10−5 M, 20 °C): λmax = 287 nm (ε = 32000 M−1 cm−1), λmax = 379 nm (ε = 19000 M−1 cm−1), λmax = 910 nm (ε = 32000 M−1 cm−1). IR Data (nujol mull, KBr plates): 1623 (s), 1586 (vs), 1354 (s), 1287 (vs), 1182 (vs), 1148 (vs), 1043 (vs), 982 (vs), 937 (vs), 885 (vs), 717 (vs), 686 (s), 638 (vs), 581 (s), 418 (s) cm−1.
3.3 Iodine oxidation of (4,4′-bipy){Ti(N[tBu]Ar)3}2
Complex (4,4′-bipy){Ti(N[tBu]Ar)3}2 was dissolved in THF (5 mL) to create a black solution (0.100g, 0.0764 mmol). Iodine (0.020 g, 0.079 mmol) was dissolved in THF (2 mL) to create a red-brown solution. The I2 solution was added to the titanium solution dropwise with stirring. After 30 seconds of stirring, the reaction mixture turned from black to orange. The reaction mixture was allowed to stir for half an hour at which point the solvent was removed under vacuum. The resulting crude material (orange powder) was dissolved in benzene-d6 for analysis by 1H NMR spectroscopy. This demonstrated that titanium(IV) iodide complex ITi(N[tBu]Ar)35 was cleanly formed in quantitative yield, together with free 4,4′-bipyridine. 1H NMR (benzene-d6, 20 °C, 400 MHz): δ = 8.57 (dd, JHH = 4 Hz and 2 Hz, 4H, CC(H)–N, 4,4′-bipyridine), 6.92 (broad s, 12H, HAr), 6.83 (dd, JHH = 8 Hz, 4H, (H)CC–N, 4,4′-bipyridine), 6.81 (broad s, 6H, HAr), 2.25 (s, 36H, Ar–CH3), 1.38 (s, 54H, C(CH3)3) ppm.
3.4 Fc[OTf] reaction with (4,4′-bipy){Ti(N[tBu]Ar)3}2
Complex (4,4′-bipy){Ti(N[tBu]Ar)3}2 (0.100 g, 0.0764 mmol) was dissolved in THF (5 mL) to create a black solution. Ferrocenium triflate (Fc[OTf],49 0.026 g, 0.078 mmol) was slurried in THF (3 mL) to create a dark blue suspension. The Fc[OTf] suspension was added to the titanium solution dropwise, with stirring. After 30 seconds of stirring, the reaction mixture turned from black to dark red-black. The reaction mixture was allowed to stir for half an hour at which time the solvent was removed under reduced pressure. The resulting crude material (dark red powder) was dissolved in benzene-d6 for analysis by 1H NMR spectroscopy. It is clear from this analysis that no one-electron oxidation product was formed. Half of the starting material (4,4′-bipy){Ti(N[tBu]Ar)3}2 was oxidized to produce one equivalent of the triflate complex TfOTi(N[tBu]Ar)3,11 one equivalent of ferrocene, and half an equivalent of 4,4′-bipyridine. 1H NMR (benzene-d6, 20 °C, 400 MHz): δ = 8.57 (dd, JHH = 4 Hz and 2 Hz, 2H, CC(H)–N, 4,4′-bipyridine), 7.12 (broad s, 4H, CC(H)–N, (4,4′-bipy){Ti(N[tBu]Ar)3}2), 6.97 (s, 6H, HAr, (4,4′-bipy){Ti(N[tBu]Ar)3}2), 6.83 (dd, JHH = 8 Hz, 2H, (H)CC–N, 4,4′-bipyridine), 6.77 (s, 3H, HAr, (4,4′-bipy){Ti(N[tBu]Ar)3}2), 6.74 (s, 3H, HAr, TfOTi(N[tBu]Ar)3), 6.56 (broad s, 6H, HAr, TfOTi(N[tBu]Ar)3), 5.43 (d, JHH = 8 Hz, 2H, (H)CC–N, (4,4′-bipy){Ti(N[tBu]Ar)3}2), 4.00 (s, 10H, ferrocene), 2.34 (s, 18H, Ar–CH3, (4,4′-bipy){Ti(N[tBu]Ar)3}2), 2.23 s, 18H, Ar–CH3, TfOTi(N[tBu]Ar)3), 1.24 (s, 27H, C(CH3)3, TfOTi(N[tBu]Ar)3), 1.23 (s, 27H, C(CH3)3, (4,4′-bipy){Ti(N[tBu]Ar)3}2) ppm.
3.5 Preparation of [FeCp2][B(C6F5)4]
This salt was prepared using Geiger's reported procedure for Fc[PF6],21 using [Li(OEt2)3][B(C6F5)4]50 in place of Na[PF6]. In a 100 mL round bottom flask, ferrocene (1.00 g, 5.38 mmol) was suspended in a mixture of water (20 mL) and acetone (8 mL). To this suspension, FeCl3 (1.13 g, 6.95 mmol) was added as a solid, immediately creating a dark green solution and evolving heat. The solution gradually changed from dark green to dark blue as it was stirred for 20 minutes. At this point, the dark blue solution was filtered through Celite and a small amount of additional acetone was used to wash the filter cake. The [Li(OEt2)3][B(C6F5)4] (5.02 g, 5.52 mmol) was added with stirring to the dark blue solution. There was no color change, but a dark blue oil began to form on the sides of the flask. After stirring for 15 minutes, ethanol (10 mL) was added to the reaction mixture, causing precipitation of a dark blue solid. The solid was collected by filtration and washed with water (20 mL) and hexane (20 mL). A second crop of solid was obtained by concentrating the volume of the filtrate to about 15–20 mL on a rotary evaporator and then adding additional ethanol (10 mL). The blue solid thus obtained was collected and washed in a similar manner. The two crops of solid were combined and dried under vacuum for 48 hours. Yield: 3.28 g, 3.79 mmol, 70%. 19F{1H} NMR (THF, 20 °C, 282 MHz): 133 (s), 162 (t, JFF = 21 Hz), 166 (t, JFF = 18 Hz) ppm. Note: the 19F NMR signals show some broadening due to the presence of the paramagnetic Fc+ cation in solution.3.6 Preparation of [(4,4′-bipy){Ti(N[tBu]Ar)3}2][B(C6F5)4]
(4,4′-bipy){Ti(N[tBu]Ar)3}2 (0.500 g, 0.382 mmol) was dissolved in THF (20 mL) to create a black solution. The ferrocenium salt, Fc[B(C6F5)4] (0.330 g, 0.381 mmol), was dissolved in THF (5 mL) to create a dark blue solution. The ferrocenium solution was added dropwise with stirring to the solution of (4,4′-bipy){Ti(N[tBu]Ar)3}2, the color of which gradually changed from black to brown during the course of the addition. The reaction mixture was allowed to stir for half an hour, at which time all volatile materials were removed under vacuum. The brown oil so obtained was dissolved in Et2O (5 mL) and to the solution was added n-hexane (20 mL). All volatile materials were again removed under vacuum, yielding a brown, oily solid. The oily solid was slurried in n-hexane (20 mL) and then, again, all volatile materials were removed under reduced pressure, leaving a brown powder. The brown powder was in turn slurried in n-hexane (20 mL), and then isolated by filtration. The solids were washed with n-hexane until the washings became colorless. (Note: the orange filtrate was concentrated to dryness under vacuum and found to contain ferrocene.) After removal of all volatile materials under reduced pressure, salt [(4,4′-bipy){Ti(N[tBu]Ar)3}2][B(C6F5)4] was isolated as a brown powder. Yield: 0.6382 g, mmol, 84% (based on C106H116BN8F20Ti2, 1988.62 g mol−1). Anal. Calcd: C 64.02; H 5.88; N 5.63. Found (Try 1): C 59.76; H 5.67; N 4.85. Found (Try 2): C 59.18; H 5.24; N 5.45. Poor analytical results for [(4,4′-bipy){Ti(N[tBu]Ar)3}2][B(C6F5)4] are likely a result of partial decomposition of the radical cation at room temperature. Attempts to further purify the compound failed. The complex gradually decomposes in solution, even at −35 °C. This is noted by a change in color from brown to green or violet. The salt dissolves in benzene-d6 to form a dark brown solution, but was found to be NMR silent by both 1H and 19F NMR spectroscopy. EPR (toluene, 20 °C): singlet at g = 2.0004. UV/Vis Data (THF, 2.85 × 10−5 M, 20 °C): λmax = 372 nm (ε = 20000 M−1 cm−1), λmax = 965 nm (ε = 10000 M−1 cm−1). IR Data (nujol mull, KBr plates): 1614 (vs), 1587 (s), 1356 (vs), 1279 (vs), 1209 (m), 1167 (vs), 1128 (vs), 1045 (s), 1022 (s), 974 (vs), 935 (s), 884 (s), 838 (m), 789 (m), 713 (s), 682 (s), 667 (s), 638 (s), 566 (w), 423 (w) cm−1.
3.7 Preparation of [(4,4′-bipy){Ti(N[tBu]Ar)3}2][B(C6F5)4]2
(4,4′-bipy){Ti(N[tBu]Ar)3}2 (0.200 g, 0.153 mmol) was dissolved in THF (10 mL) to create a black solution. The ferrocenium salt, Fc[B(C6F5)4] (0.264 g, 0.305 mmol), was dissolved in THF (5 mL) to create a dark blue solution. The ferrocenium solution was added dropwise with stirring to the (4,4′-bipy){Ti(N[tBu]Ar)3}2 solution, the color of which gradually changed from black to light red during the course of the addition. The reaction mixture was allowed to stir for half an hour at which time all volatile materials were removed under vacuum. The red oil so obtained was dissolved in Et2O (5 mL) and to the solution was added n-hexane (20 mL). Solvent was removed from this mixture under vacuum, yielding a red, oily solid. The oily solid was slurried in n-hexane (20 mL) and then the mixture was subjected again to solvent removal under reduced pressure, leaving behind a light red-orange powder. The powder was in turn slurried in n-hexane (20 mL), and then isolated by filtration. The solids were washed with n-hexane until the washings became colorless. (Note: upon solvent removal under reduced pressure, the orange filtrate was found to contain ferrocene.) After complete solvent removal under reduced pressure, salt [(4,4′-bipy){Ti(N[tBu]Ar)3}2]-[B(C6F5)4]2 was isolated as a red powder. Yield: 0.341 g, 0.128 mmol, 84% (based on C130H116B2N8F40Ti2, 2667.66 g mol−1). Crystals suitable for X-ray diffraction were grown by dissolving 0.15 g in 1.5 mL of o-difluorobenzene. The red solution was transferred to an NMR tube, and n-hexane was layered on top. The tube was left undisturbed at room temperature for two weeks, during which time large, red, shard-like crystals grew on the sides of the tube (crystals began to appear after one day). Anal. Calcd: C 58.50; H 4.38; N 4.20. Found: C 58.38; H 4.34; N 4.19. 1H NMR (pyridine-d5, 20 °C, 400 MHz): δ = 8.86 (d, JHH = 6 Hz, 2H, free 4,4′-bipy), 7.81 (d, JHH = 6 Hz, 2H, free 4,4′-bipy), 6.99 (s, 3H, HAr, [(pyridine-d5)Ti(N[tBu]Ar)3]+), 6.91 (s, 6H, HAr, [(pyridine-d5)Ti(N[tBu]Ar)3]+), 2.31 (s, 18H, Ar-CH3, [(pyridine-d5)Ti(N[tBu]Ar)3]+), 1.29 (s, 27H, C(CH3)3, [(pyridine-d5)Ti(N[tBu]Ar)3]+) ppm.3.8 [(4,4′-bipy){Ti(N[tBu]Ar)3}2][B(C6F5)4]: alternative preparation
[(4,4′-bipy){Ti(N[tBu]Ar)3}2][B(C6F5)4]2 (0.050 g, 0.038 mmol), was dissolved in THF (5 mL) to create a red solution. In a separate vial, (4,4′-bipy){Ti(N[tBu]Ar)3}2 (0.102 g, 0.0382 mmol) was dissolved in THF (5 mL) to create a black solution. The black solution was added to the red solution dropwise, with stirring. Upon completion of the addition, the reaction mixture was a dark brown color. The reaction mixture was allowed to stir for one hour at which time all volatile materials were removed under reduced pressure to yield a brown oil. The oil was dissolved in Et2O (3 mL) and n-hexane (15 mL) was added to the solution. After removing all volatile materials from the mixture under vacuum, an oily, brown solid was obtained. The solid was slurried in n-hexane (15 mL) and once again subjected to the removal of all volatile materials, yielding a light brown powder. The brown powder was slurried in n-hexane (10 mL) and the solids were isolated by filtration. The material was characterized as [(4,4′-bipy){Ti(N[tBu]Ar)3}2][B(C6F5)4] by its color and by the fact that it was observed to be NMR silent (paramagnetic). Yield: 0.124 g, 0.0616 mmol, 81% (based on C106H116BN8F20Ti2, 1988.62 g mol−1).3.9 Preparation of [Ti(N[tBu]Ar)3][B(C6F5)4]
Titanium trisanilide, Ti(N[tBu]Ar)3 (0.500 g, 0.867 mmol), was dissolved in Et2O (30 mL) to create a bright green solution. In a separate flask, the ferrocenium salt, Fc[B(C6F5)4] (0.750 g, 0.867 mmol), was slurried in Et2O (10 mL). The slurry was added dropwise, with stirring, to the Ti(N[tBu]Ar)3 solution. Additional Et2O (10 mL) was used to transfer the Fc[B(C6F5)4] that remained behind in the original flask. During the course of the addition, the color of the reaction mixture turned from bright green to bright red-orange. The reaction mixture was allowed to stir for an additional 30 minutes and then were removed all volatile materials. The resulting sticky, red oil was dissolved in Et2O (5 mL), n-hexane (30 mL) was added, and then from the new mixture were removed all volatile materials. This step was repeated, and the resulting orange powder was slurried in n-hexane (30 mL) before removing once again all volatile materials. It is essential to repeatedly triturate the material with n-hexane in order to remove all the coordinated solvent molecules. Finally, the orange powder was slurried in n-hexane (30 mL), collected by filtration using a frit, and washed with n-hexane until the washings became colorless. The orange filtrate was concentrated under vacuum and ferrocene (0.1284 g) was recovered (80% of expected). The orange powder in the frit was further kept under vacuum for 1 hour. Yield: 1.057 g, 0.842 mmol, 97% (based on C60H54BN3F20Ti, 1255.74 g mol−1). Crystals suitable for X-ray diffraction were grown by dissolving [Ti(N[tBu]Ar)3][B(C6F5)4] (0.20 g) in o-difluorobenzene. The red solution was transferred to an NMR tube and n-hexane was layered on top. The tube was left undisturbed at room temperature for five days, during which time large, red, block-like crystals grew on the sides of the tube. Anal. Calcd: C 57.39; H 4.33; N 3.35. Found: C 57.13; H 4.61; N 3.07. 1H NMR (pyridine-d5, 20 °C, 400 MHz): δ = 6.99 (s, 3H, HAr), 6.91 (s, 6H, HAr), 2.31 (s, 18H, Ar–CH3), 1.29 (s, 27H, C(CH3)3) ppm. 1H NMR (THF-d8, 20 °C, 400 MHz): δ = 7.13 (s, 3H, HAr), 6.84 (s, 6H, HAr), 2.40 (s, 18H, Ar–CH3), 1.28 (s, 27H, C(CH3)3) ppm. 13C NMR (pyridine-d5, 20 °C, 125.7 MHz): δ = 149 (d, half of doublet hidden under pyridine-d5 resonance, C–F), 142.5 (s, aryl ipso), 139.6 (s, m-Ar), 139.3 (d, JCF = 246 Hz, C–F), 137.4 (d, JCF = 232 Hz, C–F), 132.0 (s, p-Ar), 130.2 (s, o-Ar), 125.8 (m, C–F), 66.9 (s, NC(CH3)3), 30.7 (s, ArCH3), 21.6 (s, NC(CH3)3) ppm. 13C NMR (THF-d8, 20 °C, 125.7 MHz): δ = 149.3 (d, JCF = 238 Hz, C–F), 144.4 (s, aryl ipso), 140.3 (d, JCF = 246 Hz, C–F) 139.8 (s, m-Ar), 137.3 d, JCF = 250 Hz, C–F), 131.6 (s, p-Ar), 129.4 (s, o-Ar), 125.5 (broad, C–F), 67.3 (s, NC(CH3)3), 30.9 (s, ArCH3), 21.4 (s, NC(CH3)3) ppm.3.10 Crystallization of [(THF)Ti(N[tBu]Ar)3][B(C6F5)4]
[Ti(N[tBu]Ar)3][B(C6F5)4] (ca. 100 mg) was slurried in Et2O (10 mL). THF was added (three to four drops) to completely dissolve the material. The orange solution was placed in the freezer at −35 °C. After two days, large, orange crystals were observed in the vial. The material continued to crystallize over the next two days, with large, orange crystals covering the base of the vial. A crystal was selected for the X-ray structure determination after about a week had passed. Attempts to grow crystals in a similar manner from toluene resulted in the material oiling out of solution. The material also did not crystallize out of concentrated THF solutions stored in the freezer.3.11 Preparation of (nBuO)Ti(N[tBu]Ar)3
[Ti(N[tBu]Ar)3][B(C6F5)4] (0.192 g, 0.152 mmol) was dissolved in o-difluorobenzene (5 mL) to form a red solution, which was subsequently frozen solid in the dry box cold well. A solution of [Li][HBEt3] (1.0 M in THF, 152 μL, 0.152 mmol) was added to the thawing red solution using a microsyringe. There was no immediate color change; however, the color of the reaction mixture gradually changed from red to orange to yellow upon warming to room temperature. The reaction mixture was allowed to stir for a total of one hour and then pumped down to dryness. The resulting yellow oil was triturated twice with n-hexane (10 mL) to give a yellow powder. This material was slurried in n-hexane and filtered to remove [Li][B(C6F5)4]. The white solids present in the fritted funnel were washed with n-hexane until the washings became colorless. The yellow filtrate was pumped down to dryness to yield a yellow powder. Yield: 0.058 g, 0.089 mmol, 59%. Crystals suitable for X-ray diffraction were grown by first creating a slurry of the yellow powder in n-pentane and then adding Et2O dropwise until all the material dissolved. The yellow solution so obtained was placed in the dry box freezer at −35 °C for five days, during which time yellow cubic crystals deposited. Anal. Calcd: C 73.92; H 9.78; N 6.47. Found: C 71.05; H 8.91; N 6.09. 1H NMR (benzene-d6, 20 °C, 400 MHz): δ 6.68 (s, 3H, p-ArH), 5.83 (br s, 6H, o-ArH), 4.53 (t, 3JHH = 7.6 Hz, 2H, OCH2CH2CH2CH3), 2.16 (s, 18H, Ar–CH3), 1.83 (apparent pentet, J = 7.6 Hz, 2H, OCH2CH2CH2CH3), 1.41 (apparent sextet, J = 7.6 Hz, 2H, OCH2CH2CH2CH3), 1.10 (s, 27H, C(CH3)3), 0.99 (t, 3JHH = 7.6 Hz, OCH2CH2CH2CH3); 13C NMR (benzene-d6, 20 °C, 100.6 MHz): δ 152.7 (s, NAr ipso), 136.0 (s, m-Ar), 128.5 (s, o-Ar), 125.5 (s, p-Ar), 77.0 (s, OCH2CH2CH2CH3), 60.3 (s, C(CH3)3), 35.7 (s, OCH2CH2CH2CH3), 31.2 (s, C(CH3)3), 21.7 (s, Ar–CH3), 19.3 (s, OCH2CH2CH2CH3), 14.3 (OCH2CH2CH2CH3).3.12 X-Ray crystallography
Crystals were mounted in hydrocarbon oil on a nylon loop or a glass fiber. Low-temperature (100 K) data were collected on a Siemens Platform three-circle diffractometer coupled to a Bruker-AXS Smart Apex CCD detector with graphite-monochromated Mo-Kα radiation (λ = 0.71073) performing ψ- and ω-scans. A semi-empirical absorption correction was applied to the diffraction data using SADABS.51 All structures were solved by direct or Patterson methods using SHELXS52,53 and refined against F2 on all data by full-matrix least squares with SHELXL-97.53 All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were included in the model at geometrically calculated positions and refined using a riding model. The isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the U value of the atoms to which they are linked (1.5 times for methyl groups). In structures where disorders were present, the disorders were refined within SHELXL with the help of rigid bond restraints as well as similarity restraints on the anisotropic displacement parameters for neighboring atoms and on 1,2- and 1,3-distances throughout the disordered components. The relative occupancies of disordered components were refined freely within SHELXL.54 The unit cell of [(4,4′-bipy){Ti(N[tBu]Ar)3}2][B(C6F5)4]2 contains one o-difluorobenzene molecule residing atop the inversion center which has been treated as a diffuse contribution to the overall scattering without specific atom positions by SQUEEZE/PLATON.55 Further details on all structures are provided in Tables S1 and S2.†3.13 Electrochemistry
Electrochemical measurements were made using a BAS CV50W potentiostat with a platinum disk working electrode, platinum wire counter electrode, and a silver wire pseudoreference electrode. Potentials were internally referenced to the ferrocene/ferrocenium redox couple at 0 mV. Measurements were made under an atmosphere of purified nitrogen using THF solutions of [NnBu4][B(C6F5)4] (0.2 M), which was synthesized using modified literature methods.56 The cyclic voltammograms of the complexes were collected at the sweep rates specified in the figure captions.4 Conclusions
The exploration recounted herein, irrespective of initial motivation, led to an important outcome in the form of the synthesis, isolation, and thorough characterization of an exceedingly rare metal tris-amide cation, {Ti(N[tBu]Ar)3}+, in a form that is unstabilized by coordination of donor solvent molecules. This accomplishment was made possible by the judicious choice of a robust counter-anion, namely the perfluorinated tetraphenylborate anion, [B(C6F5)4]−. The latter has been reported previously to permit isolation of extremely electrophilic cations, such as in the salt originally formulated as triethylsilyl perfluoro-tetraphenylborate, [Et3Si][B(C6F5)4],57,58 but recently shown actually to have the composition [Et3Si–H–SiEt3][B(C6F5)4],59 and thus still able to serve as a source of triethylsilylium ion via release of an equivalent of triethylsilane. The latter silylium ion source has exhibited impressive stoichiometric reactivity that has spawned applications in catalysis.60 The titanium tris-amide cation {Ti(N[tBu]Ar)3}+ can now be explored in analogous fashion as a unique and powerful Lewis acid: based upon the observation that neutral Ti(N[tBu]Ar)3 readily abstracts a halogen atom from substrates including ISiMe3, bromobenzene, and triphenyltin chloride to form XTi(N[tBu]Ar)3 (X = I, Br, or Cl),5–7 the titanium(IV)–halogen bond energy in this system exceeds those found in the corresponding group 14 element–halogen compounds.Acknowledgements
The authors are grateful to Syngenta for support and for helpful discussions with Dr David A. Jackson and Prof. Clark R. Landis. Nicholas Piro is thanked for his help in solving the structure of (4,4′-bipy){Ti(N[tBu]Ar3}2 and Matthew Rankin is thanked for aiding with NMR data collection.References
- L. D. Durfee, P. E. Fanwick, I. P. Rothwell, K. Folting and J. C. Huffman, J. Am. Chem. Soc., 1987, 109, 4720–4722 CrossRef CAS.
- A. Mendiratta and C. C. Cummins, Inorg. Chem., 2005, 44, 7319–7321 CrossRef CAS PubMed.
- P. W. Wanandi, W. M. Davis, C. C. Cummins, M. A. Russell and D. E. Wilcox, J. Am. Chem. Soc., 1995, 117, 2110–2111 CrossRef CAS.
- (a) P. Girard, J. L. Namy and H. B. Kagan, J. Am. Chem. Soc., 1980, 102, 2693–2698 CrossRef CAS; (b) A. Krief and A.-M. Laval, Chem. Rev., 1999, 99, 745–778 CrossRef CAS PubMed.
- T. Agapie, P. L. Diaconescu, D. J. Mindiola and C. C. Cummins, Organometallics, 2002, 21, 1329–1340 CrossRef CAS.
- T. Agapie, P. L. Diaconescu and C. C. Cummins, J. Am. Chem. Soc., 2002, 124, 2412–2413 CrossRef CAS PubMed.
- B. M. Cossairt and C. C. Cummins, New J. Chem., 2010, 34, 1533–1536 RSC.
- (a) K. J. Covert and P. T. Wolczanski, Inorg. Chem., 1989, 28, 4565–4567 CrossRef CAS; (b) K. J. Covert, P. T. Wolczanski, S. A. Hill and P. J. Krusic, Inorg. Chem., 1992, 31, 66–78 CrossRef CAS.
- A. Mendiratta, J. S. Figueroa and C. C. Cummins, Chem. Commun., 2005, 3403–3405 RSC.
- E. Kim, A. L. Odom and C. C. Cummins, Inorg. Chim. Acta, 1998, 278, 103–107 CrossRef CAS.
- J. C. Peters, A. R. Johnson, A. L. Odom, P. W. Wanandi, W. M. Davis and C. C. Cummins, J. Am. Chem. Soc., 1996, 118, 10175–10188 CrossRef CAS.
- J. C. Peters, J.-P. F. Cherry, J. C. Thomas, L. Baraldo, D. J. Mindiola, W. M. Davis and C. C. Cummins, J. Am. Chem. Soc., 1999, 121, 10053–10067 CrossRef CAS.
- A. Lichtblau, H. D. Hausen, W. Schwarz and W. Kaim, Inorg. Chem., 1993, 32, 73–78 CrossRef CAS.
- S. Kraft, E. Hanuschek, R. Beckhaus, D. Haase and W. Saak, Chem. – Eur. J., 2005, 11, 969–978 CrossRef CAS PubMed.
- PLATON55 was used to generate all ORTEP61 diagrams in this work.
- This Figure is based upon a model system that was geometry optimized using ADF 2006.0162 using the LDA functional of Vosko, Wilk, and Nusair63 together with the GGA functional known as OLYP64 which is a combination of Handy and Cohen's OPTX functional65 for exchange and Lee, Yang, and Parr's nonlocal correlation functional.66 The zero-order regular approximation67 (ZORA) was used for inclusion of relativistic effects. The basis sets were QZ4P for Ti and N, and TZ2P for C and H as supplied with ADF. Orbital contours were analyzed using the DGrid package68 of Kohout.
- D. E. Richardson and H. Taube, Inorg. Chem., 1981, 20, 1278–1285 CrossRef CAS.
- A. Bard and L. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2001 Search PubMed.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247–466 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- A. Nafady, T. T. Chin and W. E. Geiger, Organometallics, 2006, 25, 1654–1663 CrossRef CAS.
- T. R. O'Toole, J. N. Younathan, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1989, 28, 3923–3926 CrossRef.
- C. Boisson, J. C. Berthet, M. Ephritikhine, M. Lance and M. Nierlich, J. Organomet. Chem., 1997, 531, 115–119 CrossRef CAS.
- I. Chávez, A. Alvarez-Carena, E. Molins, A. Roig, W. Maniukiewicz, A. Arancibia, V. Arancibia, H. Brand and J. Manuel Manríquez, J. Organomet. Chem., 2000, 601, 126–132 CrossRef.
- K. B. P. Ruppa, N. Desmangles, S. Gambarotta, G. Yap and A. L. Rheingold, Inorg. Chem., 1997, 36, 1194–1197 CrossRef CAS PubMed.
- D. Reardon, I. Kovacs, K. B. P. Ruppa, K. Feghali, S. Gambarotta and J. Petersen, Chem. – Eur. J., 1997, 3, 1482–1488 CrossRef CAS.
- C. E. Laplaza, A. L. Odom, W. M. Davis, C. C. Cummins and J. D. Protasiewicz, J. Am. Chem. Soc., 1995, 117, 4999–5000 CrossRef CAS.
- C. E. Laplaza, M. J. A. Johnson, J. C. Peters, A. L. Odom, E. Kim, C. C. Cummins, G. N. George and I. J. Pickering, J. Am. Chem. Soc., 1996, 118, 8623–8638 CrossRef CAS.
- J.-P. F. Cherry, A. R. Johnson, L. M. Baraldo, Y.-C. Tsai, C. C. Cummins, S. V. Kryatov, E. V. Rybak-Akimova, K. B. Capps, C. D. Hoff, C. M. Haar and S. P. Nolan, J. Am. Chem. Soc., 2001, 123, 7271–7286 CrossRef CAS PubMed.
- C. Schädle, C. Meermann, K. W. Törnroos and R. Anwander, Eur. J. Inorg. Chem., 2010, 2010, 2841–2852 CrossRef.
- R. A. Andersen, Inorg. Chem., 1979, 18, 1507–1509 CrossRef CAS.
- J. S. Ghotra, M. B. Hursthouse and A. J. Welch, J. Chem. Soc., Chem. Commun., 1973, 669–670 RSC.
- R. F. W. Bader, Atoms in molecules: a quantum theory, Clarendon Press, Oxford, New York, 1990 Search PubMed.
- The calculations used the M06 functional69 and the def2-TZVPP basis set of Alrichs70 as implemented in ORCA71 (Default-Basis-4).
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, WIREs Comput. Mol. Sci., 2012, 2, 1–42 CrossRef CAS.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 593–609 CrossRef CAS.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 610–627 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop and F. Weinhold, J. Comput. Chem., 1998, 19, 628–646 CrossRef CAS.
- The natural resonance theory (NRT) routine provided by NBO employs an alternative definition of valence as compared to the traditional one described by Sidgwick72 and more recently by Parkin.73 For example, NRT gives boron a natural valence of four for the boron atom in trigonal-planar BF3 rather than a traditional valence of three due to equating “natural valence” to the total number of electron-pair bonds formed, rather than to the number of central-atom valence electrons used in bonding.
- A. R. Johnson, P. W. Wanandi, C. C. Cummins and W. M. Davis, Organometallics, 1994, 13, 2907–2909 CrossRef CAS.
- J. R. Galsworthy, M. L. H. Green, N. Maxted and M. Müller, J. Chem. Soc., Dalton Trans., 1998, 387–392 RSC.
- H. Aneetha, F. Basuli, J. Bollinger, J. C. Huffman and D. J. Mindiola, Organometallics, 2006, 25, 2402–2404 CrossRef CAS.
- A. Shafir and J. Arnold, J. Am. Chem. Soc., 2001, 123, 9212–9213 CrossRef CAS PubMed.
- H. Nöth and M. Schmidt, Organometallics, 1995, 14, 4601–4610 CrossRef.
- K. Ma, W. E. Piers, Y. Gao and M. Parvez, J. Am. Chem. Soc., 2004, 126, 5668–5669 CrossRef CAS PubMed.
- Z. Y. Guo, P. K. Bradley and R. F. Jordan, Organometallics, 1992, 11, 2690–2693 CrossRef CAS.
- W. J. Evans, T. A. Ulibarri, L. R. Chamberlain, J. W. Ziller and D. Alvarez, Organometallics, 1990, 9, 2124–2130 CrossRef CAS.
- R. R. Schrock, L. G. Sturgeoff and P. R. Sharp, Inorg. Chem., 1983, 22, 2801–2806 CrossRef CAS.
- A. Massey and A. Park, J. Organomet. Chem., 1964, 2, 245–250 CrossRef CAS.
- G. M. Sheldrick, SADABS, Bruker AXS, Inc., 2005 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. Müller, R. Herbst-Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, Crystal structure refinement: a crystallographers guide to SHELXL, Oxford University Press, Oxford, New York, 2006 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 7–13 CrossRef CAS.
- R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395–6401 CrossRef CAS PubMed.
- J. B. Lambert and S. Zhang, J. Chem. Soc., Chem. Commun., 1993, 383–384 RSC.
- J. B. Lambert, S. Zhang and S. M. Ciro, Organometallics, 1994, 13, 2430–2443 CrossRef CAS.
- M. Nava and C. A. Reed, Organometallics, 2011, 30, 4798–4800 CrossRef CAS PubMed.
- H. F. T. Klare and M. Oestreich, Dalton Trans., 2010, 39, 9176–9184 RSC.
- M. N. Burnett and C. K. Johnson, ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, Oak Ridge National Laboratory Report ORNL-6895, 1996.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- J. Baker and P. Pulay, J. Comput. Chem., 2003, 24, 1184–1191 CrossRef CAS PubMed.
- W.-M. Hoe, A. J. Cohen and N. C. Handy, Chem. Phys. Lett., 2001, 341, 319–328 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- (a) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS; (b) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS; (c) E. van Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- A. I. Baranov and M. Kohout, J. Phys. Chem. Solids, 2010, 71, 1350–1356 CrossRef CAS.
- Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- N. V. Sidgwick, The electronic theory of valency, Clarendon Press, Oxford, 1927 Search PubMed.
- G. Parkin, J. Chem. Educ., 2006, 83, 791–799 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectra, tables of crystallographic data, DFT-optimized coordinates. CCDC 893531–893535. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/C5DT00105F |
| ‡ Present address: The Dow Chemical Company, 1776 Building, Midland, MI 48674, USA. |
| § Present address: SAFC Hitech, 5485 County Road V, Sheboygan Falls, WI 53085, USA. |
| This journal is © The Royal Society of Chemistry 2015 |