DOI:
10.1039/C4DT04043K
(Paper)
Dalton Trans., 2015,
44, 5771-5776
Mono(boratabenzene) rare-earth metal dialkyl complexes: synthesis, structure and catalytic behaviors for styrene polymerization†
Received
31st December 2014
, Accepted 9th February 2015
First published on 10th February 2015
Abstract
Four mono(boratabenzene) rare-earth metal dialkyl complexes, [(3,5-Me2-C5H3BR)Ln(CH2SiMe3)2(THF)] (1: R = NEt2, Ln = Sc; 2: R = NEt2, Ln = Lu; 3: R = Ph, Ln = Sc; 4: R = Ph, Ln = Lu), were synthesized efficiently via a one-pot strategy with Li[3,5-Me2-C5H3BR] (R = NEt2, Ph), LnCl3(THF)x (Ln = Sc, x = 3; Ln = Lu, x = 0), and LiCH2SiMe3. The solid-state structures of 1 and 2 were determined by single-crystal X-ray diffraction. Variable-temperature NMR studies indicated that the energy barrier for the rotation of aminoboratabenzene in 1 (ΔG‡ ≈ 71 kJ mol−1) is higher than that of phenylboratabenzene in 3 (ΔG‡ ≈ 59 kJ mol−1). These mono(boratabenzene) rare-earth metal dialkyl complexes’ catalytic behaviors for styrene polymerization were investigated, and found that mono(boratabenzene) scandium dialkyl complexes show high catalytic activities for syndiotactic polymerization upon activation with cocatalysts.
Introduction
Organometallic complexes of the rare-earth metals are of importance. These complexes not only have rich coordination properties but also promote a variety of useful transformations,1 and have been widely used in organic2 and polymer synthesis.3 The most widely investigated organometallic complexes of rare-earth metals are those bearing Cp-type ligands. To further explore the chemistry of rare-earth metal complexes, ancillary ligands other than Cp and Cp derivatives have been introduced recently.4
Boratabenzene is a heterocyclic, 6π-electron aromatic anion that has been introduced into organometallic chemistry as an isoelectronic analogue of the well-known cyclopentadienide anion (Cp).5 A large number of transition-metal complexes bearing boratabenzenes, in particular the derivatives of Group 4, 6, and 8 metals, have been reported.6 On the other hand, only a handful examples of boratabenzene derivatives of rare-earth metals have been synthesized before 2007, and most of them are chloride derivatives.7 Recently, our group carried out a study on the chemistry of boratabenzene rare-earth metal complexes, and were able to synthesize a series of bis(boratabenzene) trivalent rare-earth metal chlorides, amides and alkyl complexes,8 bis(boratabenzene) divalent rare-earth metal complexes,9ansa-heteroborabenzene divalent ytterbium,10 and amidino-boratabenzene trivalent rare-earth metal complexes,11 some of which show interesting activities.8c,d,9 Herein we report the synthesis and structural characterization of mono(boratabenzene) rare-earth metal alkyl complexes. The catalytic activity of these complexes for styrene polymerizations is also presented.
Results and discussion
Our choice of the specific boratabenzene ligands to use in our studies was based on stability considerations, which are crucial for isolation of the targeted complexes and catalytic behavior studies. One challenge in the synthesis of mono-Cp rare-earth metal complexes is ligand redistribution, which results in the formation of bis- and/or tris-Cp rare-earth metal complexes, and the sterically demanding Cp ligands are generally required to prevent ligand redistribution. Thus, we selected the multi-substituted boratabenzene ligands [3,5-Me2-C5H3BR]− (R = NEt2, Ph). Li[3,5-Me2-C5H3BNEt2] (LiL1) and Li[3,5-Me2-C5H3BPh] (LiL2) were prepared by a modified procedure (Scheme 1) reported by Herberich and co-workers.12 A salt elimination reaction between dipotassium salt of 2,4-dimethyl-1,3-pentadiene and diethylaminoboron dichloride or dichlorophenylborane gave 1-(diethylamino)-5-methyl-3-methylene-1,2,3,6-tetrahydro-borinine or 1-phenyl-3,5-dimethyl-1,6-dihydroborinine, which was subsequently deprotonated by LDA to produce LiL1 or LiL2.
 |
| | Scheme 1 Synthesis of LiL1 and LiL2. | |
The 1H NMR spectral monitoring of the reaction of one equivalent of LiL1 with ScCl3(THF)3 in THF-d8 at ambient temperature showed that the corresponding mono(boratabenzene) scandium dichloride was produced in nearly quantitative yield after 15 min. Followed by the salt elimination reaction with two equivalents of LiCH2SiMe3, the desired mono(boratabenzene) scandium dialkyl complex [(3,5-Me2-C5H3BNEt2)Sc(CH2SiMe3)2(THF)] (1) was formed in 10 min with high yield. The reaction was subsequently scaled up in THF, and complex 1 was obtained in 99% yield (Scheme 2). Accordingly, [(3,5-Me2-C5H3BR)Ln(CH2SiMe3)2(THF)] (2: R = NEt2, Ln = Lu; 3: R = Ph, Ln = Sc; 4: R = Ph, Ln = Lu) was synthesized by reactions between LiL1 (or LiL2), LnCl3(THF)x (Ln = Sc, x = 3; Ln = Lu, x = 0) and LiCH2SiMe3 in THF at ambient temperature in 86–94% yields (Scheme 2). Attempts to prepare the yttrium analogues by reactions of LiCH2SiMe3 with in situ generated mono(boratabenzene) yttrium dichlorides failed as the formed mono(boratabenzene) yttrium dialkyls readily decomposed at room temperature.
 |
| | Scheme 2 Synthesis of mono(boratabenzene) rare-earth metal dialkyl complexes 1–4. | |
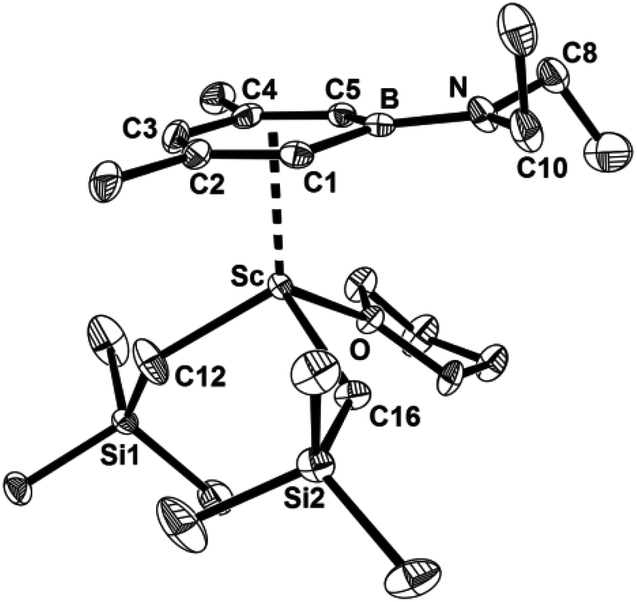
Single crystals of 1 and 2 were obtained from hexane solutions at −30 °C and characterized by X-ray diffraction. An ORTEP diagram of 1 is shown in Fig. 1, while that of 2 is presented in the ESI.† The structural features of 1 and 2 are very similar and 1 was taken as an example to analyse the structural features. A scandium ion is four-coordinated by an anionic boratabenzene, two –CH2SiMe3 groups and one THF molecule. The geometry at the metal center can be described as a pseudotetrahedral with the boratabenzene taking an apical position. Inspection of the Sc–C(boratabenzene) bond lengths in 1 shows that the Sc–C(boratabenzene) distances are significantly longer for C5 (2.626(3) Å) and C4 (2.615(3) Å) vs. C1 (2.569(3) Å), C2 (2.547(3) Å), and C3 (2.539(3) Å), which minimizes steric interactions between the boratabenzene ring and the –CH2SiMe3 groups. The average Sc–C (boratabenzene) bond length in 1 is 2.579(3) Å, which is significantly longer than that in its Cp analogue [C5Me5Sc(CH2SiMe3)2(THF)] (2.493(3) Å).13 It is also noteworthy that the bonding situation in 1 is rather different from that in the amidino-boratabenzene scandium dialkyl complex [{C5H5BN(iPr)C(H)N(iPr)}Sc(CH2SiMe3)2(THF)],11 where the scandium ion is far from the para and meta carbon atoms (2.679(2), 2.674(2) and 2.676(2) Å) and closer to the ortho carbon atoms (2.606(2) and 2.604(2) Å) and the boron atom (2.573(2) Å). The B–N bond length (1.428(4) Å) in 1 is much shorter than that in [{C5H5BN(iPr)C(H)N(iPr)}Sc(CH2SiMe3)2(THF)] (1.512(3) Å). The trigonal-planar geometry around the nitrogen atom (∑N = 395.5°) and the rather shorter B–N bond length in 1 indicate a fairly strong π-interaction between boron and nitrogen.
 |
| | Fig. 1 Molecular structure of 1 with thermal ellipsoids set at 30% probability. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Sc–B 2.741(3), Sc–C1 2.569(3), Sc–C2 2.547(3), Sc–C3 2.539(3), Sc–C4 2.615(3), Sc–C5 2.626(3), Sc–C12 2.202(3), Sc–C16 2.221(3), Sc–O 2.157(2), B–N 1.428(4), B–N–C8 122.8(2), B–N–C10 122.1(2), C8–N–C10 114.6(2). | |
The rotation barriers of boratabenzene in the mono(boratabenzene) rare-earth metal dialkyls are influenced by the substituents on the boron atom. For example, the 1H NMR spectra of the scandium complexes 1 and 3 in toluene-d8 at 25 °C showed an AB system (−0.02 and −0.15 ppm) and a singlet (−0.10 ppm) for the methylene protons in Sc-CH2SiMe3, respectively. Increasing the temperature of a solution of 1 from 25 to 85 °C results in broadening and coalescence of the Sc-CH2SiMe3 resonances followed by sharpening of the resulting coalesced peak (Fig. 2). The coalescence temperature (ca. 348 K) and the Δδ for the individual 1H NMR resonances due to Sc-CH2SiMe3 groups (ca. 69 Hz) have been used to estimate a ΔG‡ value of ca. 71 kJ mol−1 for the rotation of the diethylamino-substituted boratabenzene.14 For 3, the Sc-CH2SiMe3 resonance splits into an AB system upon lowering the temperature from 25 to −50 °C (Fig. 3). The coalescence temperature (ca. 288 K) and the Δδ (ca. 42 Hz) gives a ΔG‡ value of ca. 59 kJ mol−1 for the rotation of the phenyl-substituted boratabenzene. The observed rotation barrier difference is consistent with the fact that the aminoboratabenzene is a better electron donor than the phenyl-substituted one.
 |
| | Fig. 2 Variable-temperature 1H NMR spectra of 1 (toluene-d8, 600 MHz). The complex slowly decomposes at elevated temperature. | |
 |
| | Fig. 3 Variable-temperature 1H NMR spectra of 3 (toluene-d8, 600 MHz). | |
Recently, mono-Cp rare-earth metal dialkyl complexes were found to be the efficient catalysts for styrene polymerization upon activation with [Ph3C][B(C6F5)4].15,16 The above synthesized mono(boratabenzene) rare-earth metal dialkyl complexes, in combination with [Ph3C][B(C6F5)4] and iBu3Al, were investigated for styrene polymerization. The two scandium complexes 1 and 3 show high catalytic activities for styrene polymerization (Table 1), while the two lutetium alkyls 2 and 4 only give a trace of polymers under the same conditions. The styrene polymerization activity of the combined system of mono(boratabenzene) scandium dialkyl complexes with [Ph3C][B(C6F5)4] was low, if iBu3Al was not added (Table 1, entries 1 and 4). However, when five molar ratios of iBu3Al were used, activities up to 1944 and 2061 kg PS mol−1 Sc h−1 were achieved for 1 and 3, respectively (Table 1, entries 2 and 5). Although the rare-earth metal dialkyl-[Ph3C][B(C6F5)4]-AlR3 ternary cationic catalytic systems have been extensively used for olefin coordination polymerization, the elucidation of the effects of AlR3 in the reaction remains a challenge.15b,17 The molecular weights (Mw) of the resulting polymers are 4.29 × 105 (Mw/Mn = 1.89) and 1.65 × 105 (Mw/Mn = 1.74), respectively. The molecular weights of the polymers decrease as the aluminate-to-scandium dialkyl ratio increase, while the Mw/Mn values are almost the same (Table 2, entries 3 and 6). All the resulting polymers are highly syndiotactic and have high melting points (ca. 267 °C).
Table 1 Styrene polymerization catalyzed by monoboratabenzene-Sc-dialkyl/[Ph3C][(C6F5)4]/iBu3Al systems.a
| Entry |
Complex |
iBu3Al (eq.) |
t (min) |
Yield (%) |
Activityb |
sPSc (%) |
aPSd (%) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e (103) e (103) |
M
we (103) |
M
w/Mn |
T
mf (°C) |
|
Polymerization conditions: Sc complex (18 μmol), 26 °C, Sc complex–[Ph3C][(C6F5)4] (1:1 mol/mol), monomer–Sc complex (1000:1 mol/mol), toluene–monomer (7:1 v/v).
kg PS mol−1 Sc h−1.
Percentage of polymers that are insoluble in 2-butanone under reflux.
Percentage of polymers that are soluble in 2-butanone under reflux.
Determined by gel permeation chromatography (GPC) relative to the polystyrene standards.
Differential scanning calorimetry (DSC) analysis of the polymers that are insoluble in 2-butanone under reflux.
Monomer–Sc complex (500:1 mol/mol).
|
| 1g |
1
|
0 |
30 |
Trace |
— |
— |
— |
— |
— |
— |
— |
| 2 |
1
|
5 |
3 |
95 |
1944 |
96 |
4 |
227.2 |
429.4 |
1.89 |
267 |
| 3 |
1
|
10 |
3 |
68 |
1409 |
95 |
5 |
172.6 |
347.6 |
2.01 |
268 |
| 4 |
3
|
0 |
8 |
85 |
657 |
99 |
1 |
99.1 |
157.5 |
1.59 |
267 |
| 5 |
3
|
5 |
3 |
97 |
2061 |
99 |
1 |
95.0 |
164.8 |
1.74 |
265 |
| 6 |
3
|
10 |
3 |
85 |
1737 |
98 |
2 |
74.4 |
129.8 |
1.74 |
267 |
Table 2 Crystallographic data and refinement for complexes 1 and 2
| Complex |
1
|
2
|
| Empirical formula |
C23H49BNOScSi2 |
C23H49BLuNOSi2 |
| Formula weight |
467.58 |
597.59 |
| Colour |
Yellow |
Yellow |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P2(1)/n |
P2(1)/n |
|
a/Å |
10.945(2) |
10.9985(9) |
|
b/Å |
13.877(3) |
14.0655(11) |
|
c/Å |
19.505(4) |
19.6993(15) |
|
α/° |
90.00 |
90.00 |
|
β/° |
91.922(4) |
91.8000(10) |
|
γ/° |
90.00 |
90.00 |
| Volume/Å3 |
2960.9(11) |
3046.0(4) |
|
Z
|
4 |
4 |
| Density (calculated)/g cm−3 |
1.049 |
1.303 |
|
F(000) |
1024 |
1224 |
|
Θ range/° |
1.80 to 27.56 |
1.78 to 27.00 |
| Reflections collected |
19868 |
21338 |
| Unique reflections |
6792 |
6608 |
| Unique reflections (I > 2σ(I)) |
4446 |
5621 |
| Parameters |
272 |
272 |
| Goodness of fit |
1.024 |
1.028 |
| Final R indices [I > 2σ(I)] |
0.0573, 0.1214 |
0.0190, 0.0420 |
| Δρmax, min/e Å−3 |
0.701, −0.337 |
0.918, −0.565 |
Conclusions
In summary, the synthesis of mono(boratabenzene) rare-earth metal dialkyl complexes was achieved by using multi-substituted boratabenzene ligands. One-pot reactions of boratabenzene lithiums, rare-earth metal chlorides, and LiCH2SiMe3 provided [(3,5-Me2-C5H3BR)Ln(CH2SiMe3)2 (THF)] (1: R = NEt2, Ln = Sc; 2: R = NEt2, Ln = Lu; 3: R = Ph, Ln = Sc; 4: R = Ph, Ln = Lu) in high yields. Single-crystal X-ray diffraction analysis of 1 and 2 showed that the complexes have a three-legged piano-stool geometry, which is similar to their Cp analogues; however, the Ln–boratabenzene distances are significantly longer than the Ln–Cp ones as the boratabenzene is a poorer electron donor in comparison with Cp. The rotation barriers of boratabenzene in the mono(boratabenzene) rare-earth metal alkyls are influenced by the substituents on the boron atom. Complex 1 displays a higher rotation barrier than complex 3, which is consistent with the fact that the aminoboratabenzene is a better electron donor than the phenyl-substituted one due to the strong π-interaction between boron and nitrogen. The mono(boratabenzene) scandium dialkyl complexes, activated with [Ph3C][B(C6F5)4] and iBu3Al, act as good catalyst systems for syndiotactic styrene polymerization.
Experimental
General procedures
All operations were carried out under an atmosphere of argon using Schlenk techniques or in a nitrogen filled glovebox. Toluene, hexane, THF, C6D6, THF-d8 and toluene-d8 were dried over the Na/K alloy, transferred under vacuum, and stored in the glovebox. Dichlorophenylborane was purchased from Aldrich and used as received. Diethylaminoboron dichloride18 and LiCH2SiMe319 were synthesized according to the literature procedures. 1-(Diethylamino)-5-methyl-3-methylene-1,2,3,6-tetrahydro-borinine and 1-phenyl-3,5-dimethyl-1,6-dihydroborinine were prepared by using the procedure reported by Herberich and co-workers with the dipotassium salt of 2,4-dimethyl-1,3-pentadiene and diethylaminoboron dichloride (or dichlorophenylborane) as the reactants.121H and 13C NMR spectra were recorded on a Varian Mercury 400 MHz spectrometer at 400 MHz and 100 MHz, respectively. 11B NMR spectra were recorded on a Bruker DXP 400 MHz spectrometer at 128 MHz. Variable-temperature 1H NMR spectra were recorded on an Agilent 600 MHz spectrometer. All chemical shifts were reported in δ units with references to the residual solvent resonance of the deuterated solvents for proton and carbon chemical shifts, and to external BF3·OEt2 for boron chemical shifts. Gel permeation chromatographic (GPC) analysis was performed on a Waters high temperature GPC 2000 at 135 °C. 1,2,4-Trichlorobenzene was used as the eluent, and the system was calibrated using polystyrene standards. Differential scanning calorimetry (DSC) measurements were performed on a Perkin-Elmer Pyris 1 instrument under a nitrogen atmosphere. For the elimination of the thermal history, the samples were heated to 300 °C at 20 °C min−1, and then cooled to 40 °C at 20 °C min−1. In the second runs, the samples were heated to 300 °C at 10 °C min−1, and the melting points recorded were reported. Elemental analysis was performed at the Analytical Laboratory of Shanghai Institute of Organic Chemistry.
Li[3,5-Me2-C5H3BNEt2].
2.5 M BuLi solution in hexane (3.1 mL, 7.75 mmol) was added to diisopropylamine (0.95 g, 9.34 mmol) in 10 mL of THF at 0 °C. After stirring at 0 °C for 30 min, the reaction mixture was warmed to room temperature and stirred for 1 h. This formed LDA solution was cooled to −70 °C, and 1-(diethylamino)-5-methyl-3-methylene-1,2,3,6-tetrahydro-borinine (1.38 g, 7.79 mmol) in 10 mL of THF was added dropwise. After stirring at −70 °C for 2 h, the reaction mixture was warmed to room temperature and stirred overnight to give a red solution. The volatiles of the solution were removed under vacuum, the residue was washed with 4 × 7 mL of hexane and dried under vacuum to give Li[3,5-Me2-C5H3BNEt2] as a pale yellow solid (530 mg, 38% yield). 1H NMR (400 MHz, THF-d8, 25 °C): δ (ppm) 5.14 (s, 2H, 2-/6-H), 5.12 (s, 1H, 4-H), 3.05 (q, 3JH–H = 6.8 Hz, 4H, NCH2CH3), 2.03 (s, 6H, 3-/5-CH3), 1.00 (t, 3JH–H = 6.8 Hz, 6H, NCH2CH3). 13C NMR (100 MHz, THF-d8, 25 °C): δ (ppm) 142.2, 107.3 (br), 101.6 (boratabenzene-C), 43.6 (NCH2CH3), 25.7 (3-/5-CH3), 16.3 (NCH2CH3). 11B NMR (128 MHz, THF-d8, 25 °C): δ (ppm) 30.4.
Li[3,5-Me2-C5H3BPh].
Following the procedure described for Li[3,5-Me2-C5H3BNEt2], reaction of 2.5 M BuLi solution in hexane (1.7 mL, 4.25 mmol) with diisopropylamine (0.42 g, 4.15 mmol) in 10 mL of THF gave a LDA solution, which subsequently reacted with 1-phenyl-3,5-dimethyl-1,6-dihydroborinine (0.63 g, 3.46 mmol) to give Li[3,5-Me2-C5H3BPh] as a pale yellow solid (525 mg, 81% yield). 1H NMR (400 MHz, THF-d8, 25 °C): δ (ppm) 7.72 (d, 3JH–H = 7.6 Hz, 2H, Ph-H), 7.08 (t, 3JH–H = 7.4 Hz, 2H, Ph-H), 6.93 (t, 3JH–H = 8.0 Hz, 1H, Ph-H), 6.21 (s, 2H, 2-/6-H), 5.64 (s, 1H, 4-H), 2.08 (s, 6H, 3-/5-CH3). 13C NMR (100 MHz, THF-d8, 25 °C): δ (ppm) 150.8 (br), 141.3, 132.8, 126.8, 124.4, 121.4 (br), 113.0 (Ph-C and boratabenzene-C), 25.5 (3-/5-CH3). 11B NMR (128 MHz, THF-d8, 25 °C): δ (ppm) 33.0.
[(3,5-Me2-C5H3BNEt2)Sc(CH2SiMe3)2(THF)] (1).
A THF solution of Li[3,5-Me2-C5H3BNEt2] (100 mg, 0.55 mmol in 4 mL of THF) was added to ScCl3(THF)3 (201 mg, 0.55 mmol) in 5 mL of THF. After stirring for 15 min at room temperature, the solution of LiCH(SiMe3)2 (103 mg, 1.09 mmol in 2 mL of THF) was added, and the reaction mixture was stirred for 10 min at room temperature. The volatiles were removed under vacuum, and the residue was extracted with 4 × 2 mL of hexane. Evaporation of the yellow extract under vacuum gave 1 as a yellow solid (255 mg, 99% yield). 1H NMR (400 MHz, C6D6, 25 °C): δ (ppm) 6.13 (s, 1H, 4-H), 5.61 (s, 2H, 2-/6-H), 3.68 (m, 4H, THF), 3.23 (br, 2H, NCH2CH3), 3.02 (br, 2H, NCH2CH3), 2.16 (s, 6H, 3-/5-CH3), 1.23 (m, 4H, THF), 1.11 (t, 3JH–H = 6.8 Hz, 6H, NCH2CH3), 0.30 (s, 18H, SiMe3), −0.02 (d, 2JH–H = 10.4 Hz, 2H, CH2SiMe3), −0.15 (d, 2JH–H = 10.4 Hz, 2H, CH2SiMe3). 13C NMR (100 MHz, C6D6, 25 °C): δ (ppm) 150.5, 114.7 (br), 106.3 (boratabenzene-C), 72.0 (THF), 44.4 (br), 43.0 (CH2SiMe3 and NCH2CH3), 26.5, 25.0, 16.0 (3-/5-CH3, THF and NCH2CH3), 4.2 (SiMe3). 11B NMR (128 MHz, C6D6, 25 °C): δ (ppm) 32.7. Anal. calcd (%) for C23H49BNOScSi2: C, 59.08; H, 10.56; N, 3.00. Found: C, 58.58; H, 10.57; N, 2.91.
[(3,5-Me2-C5H3BNEt2)Lu(CH2SiMe3)2(THF)] (2).
Following the procedure described for 1, reaction of Li[3,5-Me2-C5H3BNEt2] (100 mg, 0.55 mmol in 4 mL of THF), LuCl3 (154 mg, 0.55 mmol in 5 mL of THF), and LiCH2SiMe3 (105 mg, 1.10 mmol in 2 mL of THF) gave 2 as a yellow solid (307 mg, 94% yield). 1H NMR (400 MHz, C6D6, 25 °C): δ (ppm) 6.08 (t, 4JH–H = 2.0 Hz, 1H, 4-H), 5.61 (d, 4JH–H = 2.0 Hz, 2H, 2-/6-H), 3.57 (br, 4H, THF), 3.21 (br, 2H, NCH2CH3), 3.06 (br, 2H, NCH2CH3), 2.20 (s, 6H, 3-/5-CH3), 1.15 (br, 4H, THF), 1.12 (t, 3JH–H = 7.2 Hz, 6H, NCH2CH3), 0.31 (s, 18H, SiMe3), −0.73 (s, 4H, CH2SiMe3). 13C NMR (100 MHz, C6D6, 25 °C): δ (ppm) 151.4, 113.1 (br), 104.8 (boratabenzene-C), 71.6 (THF), 43.0, 40.8 (CH2SiMe3 and NCH2CH3), 26.1, 24.9, 16.1 (3-/5-CH3, THF and NCH2CH3), 4.6 (SiMe3). 11B NMR (128 MHz, C6D6, 25 °C): δ (ppm) 32.9. Anal. calcd (%) for C23H49BLuNOSi2: C, 46.23; H, 8.26; N, 2.34. Found: C, 46.53; H, 8.06; N, 2.27.
[(3,5-Me2-C5H3BPh)Sc(CH2SiMe3)2(THF)] (3).
Following the procedure described for 1, reaction of Li[3,5-Me2-C5H3BPh] (79 mg, 0.42 mmol in 4 mL of THF), ScCl3(THF)3 (154 mg, 0.42 mmol in 5 mL of THF) and LiCH2SiMe3 (79 mg, 0.84 mmol in 2 mL of THF) gave 3 as a yellow solid (170 mg, 86% yield). 1H NMR (400 MHz, C6D6, 25 °C): δ (ppm) 7.96 (dd, 3JH–H = 8.0 Hz, 4JH–H = 1.6 Hz, 2H, Ph-H), 7.32 (t, 3JH–H = 6.8 Hz, 2H, Ph-H), 7.23 (tt, 3JH–H = 7.2 Hz, 4JH–H = 1.6 Hz, 1H, Ph-H), 6.83 (s, 2H, 2-/6-H), 6.78 (s, 1H, 4-H), 3.36 (br, 4H, THF), 2.36 (s, 6H, 3-/5-CH3), 0.88 (br, 4H, THF), 0.25 (s, 18H, SiMe3), −0.10 (s, 4H, CH2SiMe3). 13C NMR (100 MHz, C6D6, 25 °C): δ (ppm) 150.5, 132.9, 128.3, 128.2, 127.9, 127.5 (br), 117.5 (Ph-C and boratabenzene-C), 72.0 (THF), 47.9 (br, CH2SiMe3), 26.4 (3-/5-CH3), 24.5 (THF), 4.1 (SiMe3). 11B NMR (128 MHz, C6D6, 25 °C): δ (ppm) 36.7. Anal. calcd (%) for C25H44BOScSi2: C, 63.54; H, 9.39. Found: C, 62.87; H, 9.13.
[(3,5-Me2-C5H3BPh)Lu(CH2SiMe3)2(THF)] (4).
Following the procedure described for 1, reaction of Li[3,5-Me2-C5H3BPh] (80 mg, 0.43 mmol in 4 mL of THF), LuCl3 (120 mg, 0.43 mmol in 5 mL of THF) and LiCH2SiMe3 (80 mg, 0.85 mmol in 2 mL of THF) gave 4 as a yellow solid (220 mg, 86% yield). 1H NMR (400 MHz, C6D6, 25 °C): δ (ppm) 8.00 (dd, 3JH–H = 8.0 Hz, 4JH–H = 1.2 Hz, 2H, Ph-H), 7.33 (t, 3JH–H = 6.8 Hz, 2H, Ph-H), 7.23 (tt, 3JH–H = 7.2 Hz, 4JH–H = 1.2 Hz, 1H, Ph-H), 6.86 (s, 2H, 2-/6-H), 6.71 (s, 1H, 4-H), 3.24 (br, 4H, THF), 2.38 (s, 6H, 3-/5-CH3), 0.91 (br, 4H, THF), 0.28 (s, 18H, SiMe3), −0.78 (br, 4H, CH2SiMe3). 13C NMR (100 MHz, C6D6, 25 °C): δ (ppm) 150.8, 133.0, 128.2, 128.0, 127.0, 116.5 (Ph-C and boratabenzene-C), 71.0 (THF), 43.1 (CH2SiMe3), 26.0, 24.6 (3-/5-CH3 and THF), 4.5 (SiMe3). 11B NMR (128 MHz, C6D6, 25 °C): δ (ppm) 37.0. Anal. calcd (%) for C25H44BLuOSi2: C, 49.83; H, 7.36. Found: C, 49.32; H, 7.39.
A typical procedure for styrene polymerization.
The mono(boratabenzene) scandium dialkyl complex (18 μmol) and [Ph3C][B(C6F5)4] (18 μmol) were mixed in toluene. The above reaction mixture was stirred at room temperature for one minute, and then iBu3Al (90 μmol) and styrene (1.87 g, 18 mmol) were added under vigorous stirring. The reaction solution became very viscous after several minutes, and the polymerization was quenched with 2 mL of acidic methanol. The reaction mixture was poured into 50 mL of methanol to precipitate the polymer. The resulting polymer was isolated, washed with methanol, and dried under vacuum for one day to a constant weight.
X-ray crystallography
Single crystals of 1 and 2 were grown from hexane solutions. Suitable single crystals were mounted under a nitrogen atmosphere on a glass fiber, and data collection was performed on a Bruker APEX2 diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 133(2) K. The SMART program package was used to determine the unit cell parameters. The absorption correction was applied using SADABS. The structures were solved by direct methods and refined on F2 by full-matrix least-squares techniques with anisotropic thermal parameters for non-hydrogen atoms. Hydrogen atoms were placed at calculated positions and included in the structure calculations. All calculations were carried out using the SHELXL-97 and SHELXL-2014 programs. The software used is given in ref. 20. Crystallographic data and refinement for 1 and 2 are listed in Table 2.
Acknowledgements
This work was supported by the State Key Basic Research & Development Program (grant no. 2011CB808705), the National Natural Science Foundation of China (grant no. 21272256, 21132002, 21325210 and 21421091), and the CAS/SAFEA International Partnership Program for Creative Research Teams.
Notes and references
-
(a) H. Schumann, J. A. Meese-Marktscheffel and L. Esser, Chem. Rev., 1995, 95, 865 CrossRef CAS;
(b) S. Arndt and J. Okuda, Chem. Rev., 2002, 102, 1953 CrossRef CAS PubMed;
(c) W. J. Evans and B. L. Davis, Chem. Rev., 2002, 102, 2119 CrossRef CAS PubMed;
(d) W. J. Evans, Inorg. Chem., 2007, 46, 3435 CrossRef CAS PubMed.
-
(a) G. A. Molander and J. A. C. Romero, Chem. Rev., 2002, 102, 2161 CrossRef CAS PubMed;
(b) S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef CAS PubMed.
-
(a) H. Yasuda, J. Organomet. Chem., 2002, 647, 128 CrossRef CAS;
(b) Z. M. Hou and Y. Wakatsuki, Coord. Chem. Rev., 2002, 231, 1 CrossRef CAS;
(c) J. Gromada, J. F. Carpentier and A. Mortreux, Coord. Chem. Rev., 2004, 248, 397 CrossRef CAS;
(d) M. Nishiura and Z. M. Hou, Nat. Chem., 2010, 2, 257 CrossRef CAS PubMed.
-
(a) F. T. Edelmann, D. M. M. Freckmann and H. Schumann, Chem. Rev., 2002, 102, 1851 CrossRef CAS PubMed;
(b) W. E. Piers and D. J. H. Emslie, Coord. Chem. Rev., 2002, 233–234, 131 CrossRef CAS;
(c) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283 CrossRef CAS PubMed.
-
(a) G. E. Herberich, G. Greiss and H. F. Heil, Angew. Chem., Int. Ed. Engl., 1970, 9, 805 CrossRef CAS;
(b) A. J. Ashe III and P. Shu, J. Am. Chem. Soc., 1971, 93, 1804 CrossRef.
- For reviews and some representative research papers of transition metal boratabenzene complexes, see:
(a) G. E. Herberich and O. Holger, Adv. Org. Chem., 1986, 25, 199 CAS;
(b) A. J. Ashe III, S. Al-Ahmad and X. D. Fang, J. Organomet. Chem., 1999, 581, 92 CrossRef;
(c) G. C. Fu, Adv. Organomet. Chem., 2001, 47, 101 CrossRef CAS;
(d) G. C. Bazan, G. Rodriguez, A. J. Ashe III, S. Al-Ahmad and C. Müller, J. Am. Chem. Soc., 1996, 118, 2291 CrossRef CAS;
(e) G. E. Herberich, U. Englert, A. Fischer, J. H. Ni and A. Schmitz, Organometallics, 1999, 18, 5496 CrossRef CAS;
(f) M. A. Putzer, J. S. Rogers and G. C. Bazan, J. Am. Chem. Soc., 1999, 121, 8112 CrossRef CAS;
(g) J. S. Rogers, X. H. Bu and G. C. Bazan, J. Am. Chem. Soc., 2000, 122, 730 CrossRef CAS;
(h) A. J. Ashe III, S. Al-Ahmad, X. D. Fang and J. W. Kampf, Organometallics, 2001, 20, 468 CrossRef;
(i) B. Wang, X. L. Zheng and G. E. Herberich, Eur. J. Inorg. Chem., 2002, 2002, 31 CrossRef;
(j) N. Auvray, T. S. BasuBaul, P. Braunstein, P. Croizat, U. Englert, G. E. Herberich and R. Welter, Dalton Trans., 2006, 2950 RSC;
(k) A. Languérand, S. S. Barnes, G. Bélanger-Chabot, L. Maron, P. Berrouard, P. Audet and F. G. Fontaine, Angew. Chem., Int. Ed., 2009, 48, 6695 CrossRef PubMed;
(l) G. Bélanger-Chabot, P. Rioux, L. Maron and F. G. Fontaine, Chem. Commun., 2010, 46, 6816 RSC;
(m) P. Frank, R. A. Lalancette and F. Jäkle, Chem. – Eur. J., 2011, 17, 11280 CrossRef PubMed;
(n) A. Glöckner, P. Cui, Y. F. Chen, C. G. Daniliuc, P. G. Jones and M. Tamm, New J. Chem., 2012, 36, 1392 RSC;
(o) B. B. Macha, J. Boudreau, L. Maron, T. Maris and F. G. Fontaine, Organometallics, 2012, 31, 6428 CrossRef CAS.
-
(a) G. E. Herberich, U. Englert, A. Fischer, J. H. Ni and A. Schmitz, Organometallics, 1999, 18, 5496 CrossRef CAS;
(b) M. A. Putzer, J. S. Rogers and G. C. Bazan, J. Am. Chem. Soc., 1999, 121, 8112 CrossRef CAS;
(c) X. L. Zheng, B. Wang, U. Englert and G. E. Herberich, Inorg. Chem., 2001, 40, 3117 CrossRef CAS PubMed;
(d) B. Wang, X. L. Zheng and G. E. Herberich, Eur. J. Inorg. Chem., 2002, 31 CrossRef.
-
(a) Y. Y. Yuan, Y. F. Chen, G. Y. Li and W. Xia, Organometallics, 2008, 27, 6307 CrossRef CAS;
(b) Y. Y. Yuan, Y. F. Chen, G. Y. Li and W. Xia, Organometallics, 2010, 29, 3722 CrossRef CAS;
(c) Y. Y. Yuan, X. F. Wang, Y. X. Li, L. Y. Fan, X. Xu, Y. F. Chen, G. Y. Li and W. Xia, Organometallics, 2011, 30, 4330 CrossRef CAS;
(d) E. L. Lu, Y. Y. Yuan, Y. F. Chen and W. Xia, ACS Catal., 2013, 3, 521 CrossRef CAS.
-
(a) P. Cui, Y. F. Chen, X. H. Zeng, J. Sun, G. Y. Li and W. Xia, Organometallics, 2007, 26, 6519 CrossRef CAS;
(b) P. Cui, Y. F. Chen, G. P. Wang, G. Y. Li and W. Xia, Organometallics, 2008, 27, 4013 CrossRef CAS.
-
(a) P. Cui, Y. F. Chen, G. Y. Li and W. Xia, Angew. Chem., Int. Ed., 2008, 47, 9944 CrossRef CAS PubMed;
(b) P. Cui, Y. F. Chen, Q. Zhang, G. Y. Li and W. Xia, J. Organomet. Chem., 2010, 695, 2713 CrossRef CAS (Special Issue for Organo-f-Element Chemistry at Pacifichem 2010);
(c) P. Cui, Y. F. Chen, G. Y. Li and W. Xia, Organometallics, 2011, 30, 2012 CrossRef CAS.
- X. F. Wang, W. J. Peng, P. Cui, X. B. Leng, W. Xia and Y. F. Chen, Organometallics, 2013, 32, 6166 CrossRef CAS.
- G. E. Herberich, U. Englert, M. U. Schmidt and R. Standt, Organometallics, 1996, 15, 2707 CrossRef CAS.
- X. F. Li, M. Nishiura, L. H. Hu, K. Mori and Z. M. Hou, J. Am. Chem. Soc., 2009, 131, 13870 CrossRef CAS PubMed.
- ΔG‡ = αTc(9.972 + log(Tc/Δδ)), α is a constant of 1.914 × 10−2 kJ mol−1, Tc(K) is the coalescence temperature, and Δδ (Hz) is the frequency difference of the coalescing signals.
J. Sandström, Dynamic NMR Spectroscopy, Academic Press Inc., London, 1982, Chapter 7 Search PubMed.
-
(a) Y. J. Luo, J. Baldamus and Z. M. Hou, J. Am. Chem. Soc., 2004, 126, 13910 CrossRef CAS PubMed;
(b) J. Hitzbleck, K. Beckerle, J. Okuda, T. Halbach and R. Mülhaupt, Macromol. Symp., 2006, 236, 23 CrossRef CAS;
(c) X. F. Li, M. Nishiura, K. Mori, T. Mashiko and Z. M. Hou, Chem. Commun., 2007, 4137 RSC;
(d) X. Xu, Y. F. Chen and J. Sun, Chem. – Eur. J., 2009, 15, 846 CrossRef CAS PubMed;
(e) Y. P. Pan, W. F. Rong, Z. B. Jian and D. M. Cui, Macromolecules, 2012, 45, 1248 CrossRef CAS.
-
K. Beckerle and J. Okuda, Rare-earth Metal Complexes as Catalysts for Syndiotactic Styrene Polymerization, in Syndiotactic Polystyrene: Synthesis, Characterization, Processing, and Applications, ed. J. Schellenberg, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2010 Search PubMed.
-
(a) L. Zhang, M. Nishiura, M. Yuki, Y. Luo and Z. M. Hou, Angew. Chem., Int. Ed., 2008, 47, 2642 CrossRef CAS PubMed;
(b) X. F. Li, X. Y. Wang, X. Tong, H. X. Zhang, Y. Y. Chen, Y. Liu, H. Liu, X. J. Wang, M. Nishiura, H. He, Z. G. Lin, S. W. Zhang and Z. M. Hou, Organometallics, 2013, 32, 1445 CrossRef CAS.
- J. F. Brown Jr., J. Am. Chem. Soc., 1952, 74, 1219 CrossRef.
- P. T. Davidson, D. H. Harris and M. F. Lappert, J. Chem. Soc., Dalton Trans., 1976, 2268 RSC.
-
(a)
G. M. Sheldrick, SADABS, An Empirical Absorption Correction Program for Area Detector Data, University of Göttingen, Göttingen, Germany, 1996 Search PubMed;
(b)
G. M. Sheldrick, SHELXS-97 and SHELXL-97, University of Göttingen, Göttingen, Germany, 1997 Search PubMed;
(c)
G. M. Sheldrick, SHELXL-2014, University of Göttingen, Göttingen, Germany, 2014 Search PubMed;
(d)
SMART Version 5.628, Bruker AXS Inc., Madison, WI, 2002 Search PubMed;
(e)
SAINT+ Version 6.22a, Bruker AXS Inc., Madison, WI, 2002 Search PubMed;
(f)
SHELXTL NT/2000, Version 6.1, Bruker AXS Inc., Madison, WI, 2002 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Molecular structure of complex 2, 1H (13C,11B) NMR spectra of complexes 1–4. CCDC 1041234 (1) and 1041235 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt04043k |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence