
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Binuclear platinum–iridium complexes: synthesis, reactivity and luminescence†‡
Brian T.
Sterenberg
ab,
Christian T.
Wrigley
a and
Richard J.
Puddephatt
*a
aDepartment of Chemistry, University of Western Ontario, London, Ontario, Canada N6A 5B7. E-mail: pudd@uwo.ca
bDepartment of Chemistry and Biochemistry, University of Regina, Regina, Saskatchewan, Canada
First published on 9th February 2015
Abstract
The chemistry of the heterobinuclear platinum–iridium complex [PtIr(CO)3(μ-dppm)2][PF6], 1, dppm = Ph2PCH2PPh2, is described. The reaction of a hydride with 1 gave [HPtIr(CO)2(μ-dppm)2], by displacement of the carbonyl ligand from platinum, while reaction of 1 with dihydrogen, hydrogen chloride or Ph2MeSiH gave the fluxional complex [PtIrH4(CO)(μ-dppm)2][PF6], [PtIrH2Cl2(CO)(μ-dppm)2][PF6], or [PtIrH(SiMePh2)(CO)2(μ-dppm)2][PF6], respectively, by oxidative addition at iridium. Complex 1 reacted, often regioselectively, with several alkynes to give the μ–η1,η1 bridging alkyne complexes [PtIr(μ-RCCR′)(CO)2(μ-dppm)2][PF6], R = H, R′ = Ph, 4-C6H4Me, CO2Me; R = Ph, R′ = CO2Me; R = R′ = CO2Me. The complex [PtIr(μ-HCC-4-C6H4Me)(CO)2(μ-dppm)2][PF6] reacted reversibly with CO to give [PtIr(μ-HCC-4-C6H4Me)(CO)3(μ-dppm)2][PF6] and [PtIr(CO)3(μ-dppm)2][PF6], 1. With HCl, [PtIr(μ-HCC-4-C6H4Me)(CO)2(μ-dppm)2][PF6] reacted to give [PtIrHCl(μ-HCC-4-C6H4Me)(CO)2(μ-dppm)2][PF6], by oxidative addition at iridium, and then the alkenylplatinum derivative [PtIrCl{HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(4-C6H4Me)}(CO)2(μ-dppm)2][PF6]. [PtIr(μ-HCC-4-C6H4Me)(CO)2(μ-dppm)2][PF6] reacted slowly with dihydrogen to give 4-MeC6H4CHCH2 and [PtIrH4(CO)(μ-dppm)2][PF6]. The complex [PtIr(μ-HCCPh)(CO)2(μ-dppm)2][PF6] is intensely luminescent in solution at room temperature, with features characteristic of a d8–d8 face-to-face complex.
CH(4-C6H4Me)}(CO)2(μ-dppm)2][PF6]. [PtIr(μ-HCC-4-C6H4Me)(CO)2(μ-dppm)2][PF6] reacted slowly with dihydrogen to give 4-MeC6H4CHCH2 and [PtIrH4(CO)(μ-dppm)2][PF6]. The complex [PtIr(μ-HCCPh)(CO)2(μ-dppm)2][PF6] is intensely luminescent in solution at room temperature, with features characteristic of a d8–d8 face-to-face complex.
Introduction
The study of heterobimetallic or cluster complexes has relevance in the testing of current bonding concepts,1,2 in modelling the reactions proposed to occur during catalysis using bimetallic catalysts3,4 and, in some cases, in developing photonic devices.5 The concepts that are implicit in Wade's rules and explicit in the isolobal analogy have been crucially important in providing a framework for understanding complex chemistry and in predicting future developments.1,2 Bimetallic Pt–Ir, Pt–Re and Pt–Sn catalysts are universally used in reforming petroleum, to increase the octane number by converting linear alkanes to branched or cyclic alkanes, alkenes and aromatics, and they can also be used for catalytic oxidation in fuel cells and for liquid phase catalytic isotope exchange.3,4 The ability of a second metal complex to interact with a square planar d8 metal centre, such as a platinum(II) centre, is proving to be important in the development of brightly phosphorescent complexes.5In this context, we and others have been interested in the synthesis of heterobinuclear complexes with platinum–metal bonds and in studies of their reactivity and photophysical properties.3,5,6–11 In particular, during the synthesis of PtIr2 cluster complexes, two binuclear complexes containing Pt–Ir bonds bridged by bis(diphenylphosphino)methane (dppm) ligands were prepared as shown in Scheme 1. In the cationic complex [PtIr(CO)3(μ-dppm)2]+, 1, which was isolated as the hexafluorophosphate salt, the Pt–Ir distance is 2.7674(4) Å, and the square planar platinum and trigonal bipyramidal iridium centres have 16 and 18-electron configurations respectively.6 This article reports a study of the chemistry of complex 1.
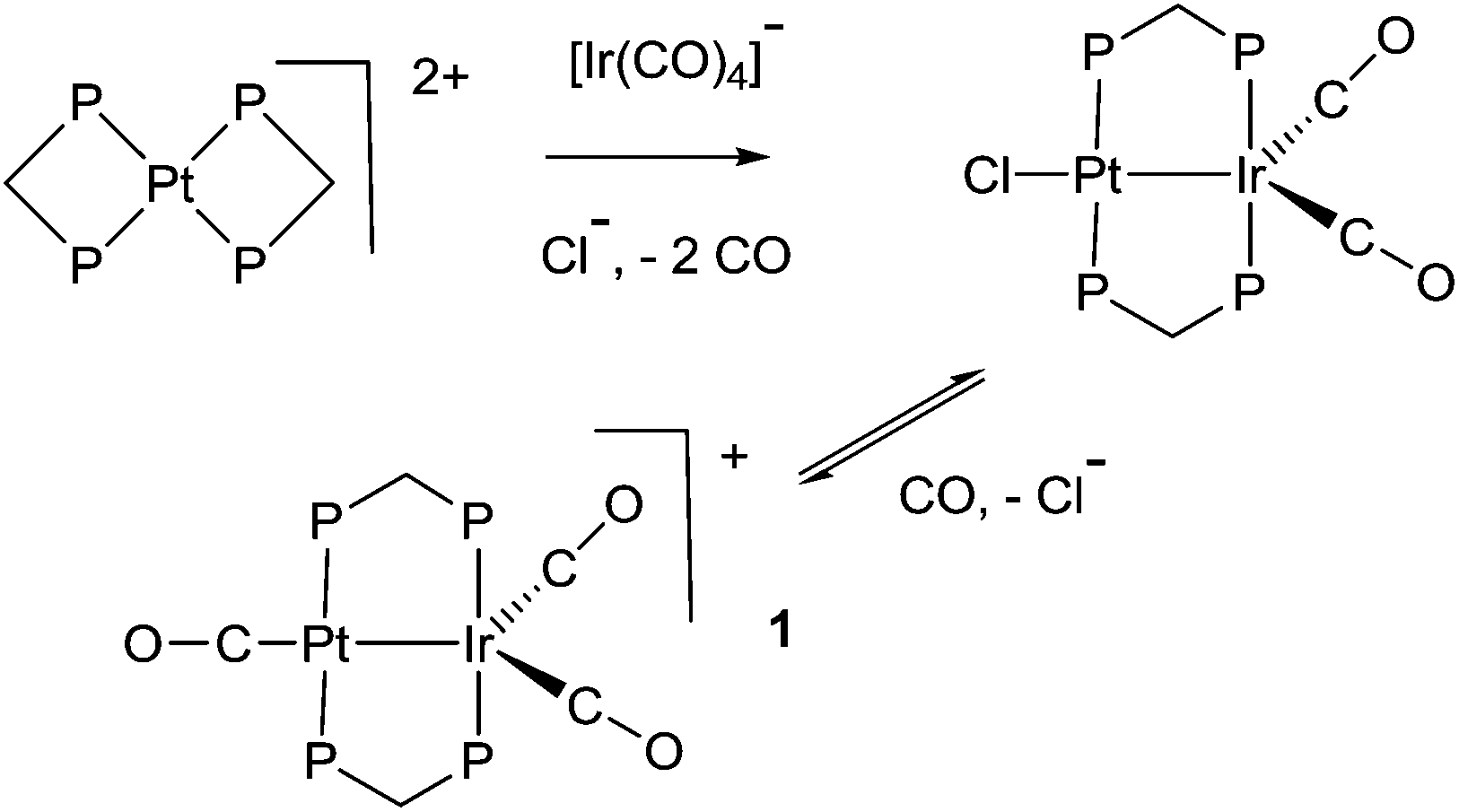 | ||
| Scheme 1 Synthesis of [PtIr(CO)3(μ-dppm)2][PF6], 1 (P = PPh2). | ||
Results and discussion
Hydride complexes derived from complex 1
Some hydrido derivatives derived from complex 1 are shown in Scheme 2. We were not able to grow crystals of any of the hydrides suitable for structure determination, but the main features of the complexes could be determined spectroscopically.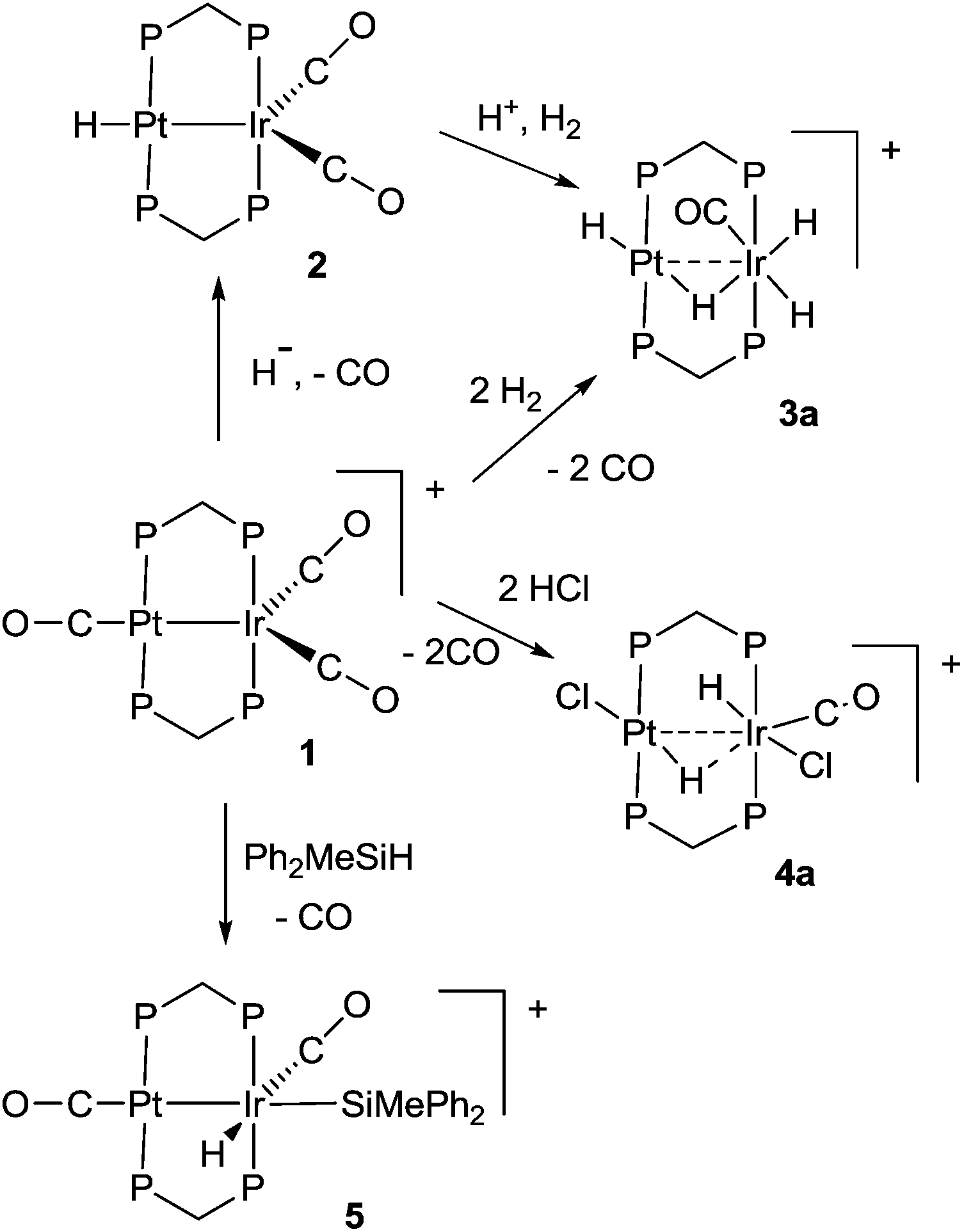 | ||
| Scheme 2 Synthesis and possible structures of hydride derivatives 2–5 (P = PPh2). | ||
Complex 2 was most readily prepared by reaction of complex 1 with sodium triethylborohydride. It is characterized by a hydride resonance at δ −3.3 with a coupling constant 1J(PtH) of 1123 Hz, showing that the hydride is bound as a terminal ligand to platinum. Homobinuclear complexes [HPtPt(L)(μ-dppm)2]+, with the hydride trans to a Pt–Pt bond, give somewhat smaller values of 1J(PtH), such as 1J(PtH) 990 Hz when L = CO, but this increases to 1326 Hz in [HPtPt(CO)2(μ-dppm)2]+, which is isoelectronic to complex 1.12 The CH2 protons of the dppm ligands in 2 appeared as a single multiplet, showing that there is an effective plane of symmetry containing the PtIrP4C2 atoms of the PtIr(μ-dppm)2 unit.8,9,12–14 The 31P NMR spectrum contained two dppm resonances at δ 16.1, 1J(PtP) 2873 Hz, and −16.4, 2J(PtP) 69 Hz, for the PtP and IrP groups respectively.
The reaction of complex 1 with dihydrogen gave the product of double oxidative addition [PtIrH4(CO)(μ-dppm)2][PF6], 3 (Scheme 2). Complex 3 was also formed during attempted synthesis of 2 by the reaction of 1 with Na[BH4] using an aqueous workup procedure, and this reaction was later shown to involve reaction of 2 with dihydrogen in the presence of a proton source (Scheme 2). The presence of four hydride ligands in complex 3 was readily shown by the 1H NMR spectrum, which contained four equal intensity resonances in the hydride region (Fig. 1). At room temperature, the spectrum contained two well resolved hydride resonances and two very broad ones, which sharpened on cooling to −30 °C (Fig. 1). There were two resonances for the CH2P2 protons of the dppm ligands, which were broad at room temperature but which also sharpened at −30 °C. These data suggest that complex 3 is fluxional in such a way that two of the hydride ligands and the CHxHyP2 protons can become equivalent at higher temperatures, while two of the hydrides do not exchange. The activation energies estimated using the Eyring equation from coalescence of the CHxHyP2 protons [coalescence temperature, Tc = 323 K, Δν = 405 Hz] and the hydride Hc,Hd protons [coalescence temperature, Tc = 333 K, Δν = 750 Hz] were 61.1 and 61.3 kJ mol−1 respectively in C2D2Cl4 solution. These values are equal within experimental error [61(1) kJ mol−1] and indicate that a common step is rate determining. The two hydrides which do not exchange are identified as a terminal PtH group [δ −3.90, 1J(PtH) 1225 Hz, Ha] and a terminal IrH group [δ −8.17, no PtH coupling resolved, Hb], while the two that do exchange are identified as a bridging hydride [δ −9.52, 1J(PtH) 540 Hz, Hc] and a terminal IrH group [δ −12.02, 3J(PtH) ca. 90 Hz, Hd]. There are four potential isomers of complex 3 labelled as 3a–3d in Scheme 3, of which 3a, 3b and 3d contain a single bridging hydride ligand and 3c contains two bridging hydride ligands. The ground state structure is likely to be 3a or 3b, in each of which the bridging hydride is trans to a terminal hydride ligand on iridium, and so more nucleophilic than the hydride trans to carbonyl on iridium. In order to give the observed spectra, the slow step in the fluxionality should exchange positions of Hc and Hd and also generate a mirror plane containing the PtIrP4C2 atoms of the PtIr(μ-dppm)2 unit. We suggest that the motion involves mostly rotation of the IrH3(CO) unit about the PIrP axis, in a windscreen wiper fashion (Scheme 3). From 3a, only anticlockwise rotation is possible because the carbonyl group cannot pass through the Pt–Ir bond. Conversion of 3a to 3b involves inversion of Hc through the Pt–Ir bond, 3b to 3c involves Hb also moving into a bridging position, and 3c to 3d involves moving Hc out of the bridging position (3c could be a transition state rather than an intermediate, but the transition state is likely to have a roughly linear PtHIr group). This completes the first half of the motion, and is followed by inversion of Hb in 3d through the Pt–Ir bond to give 3d* and then further anticlockwise rotation gives in turn 3c*, 3b* and 3a*. No further anticlockwise rotation is possible, and clockwise rotation from 3a* simply reverses the sequence. It is the 3d to 3d* step which leads to effective equivalence of the Hc and Hd hydrides and creates the effective mirror plane needed to give equivalence of the CHxHyP2 protons. The hydride Ha remains bonded to platinum and Hb remains on iridium in the position trans to CO throughout the rotation and, although it is possible to envisage ways in which exchange with Hc or Hd might occur,12,14 it is evident that any such exchange must have a significantly higher activation energy.
 | ||
| Fig. 1 1H NMR spectra (300 MHz) of complex 3: above at −30 °C; below at 20 °C. The asterisks * indicate 195Pt satellite spectra. | ||
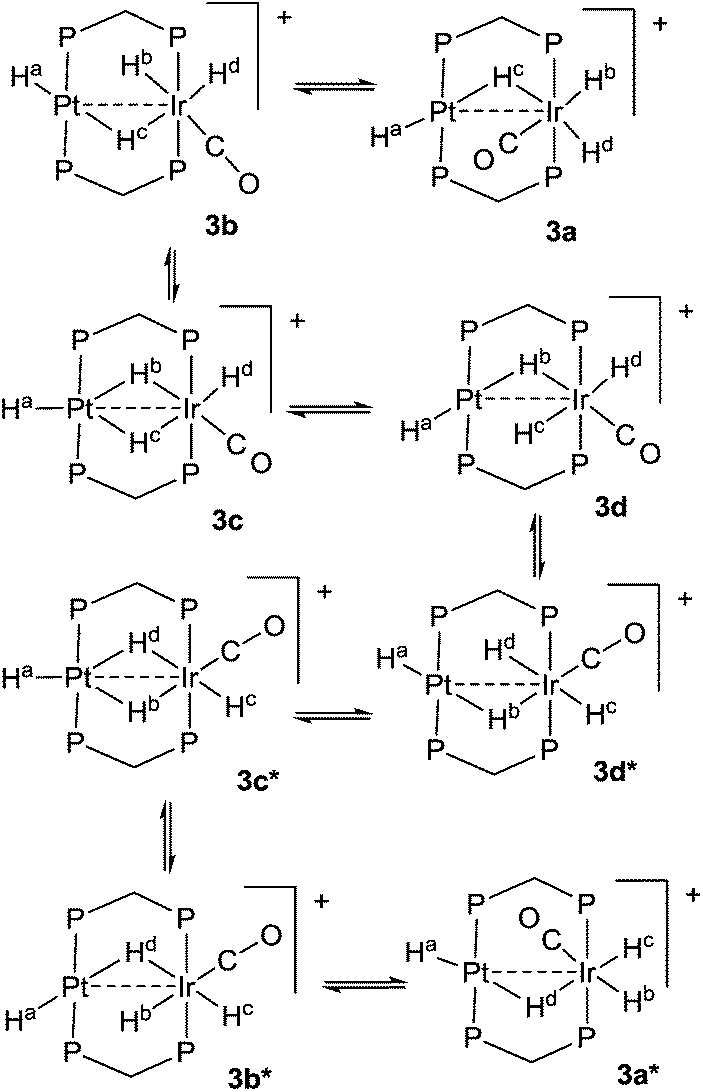 | ||
| Scheme 3 Proposed fluxionality of complex 3. | ||
The reaction of complex 1 with excess HCl gave the complex [PtIrH2Cl2(CO)(μ-dppm)2][PF6], 4, with loss of CO (Scheme 2).
Complex 1 also reacted with one equivalent of HCl but a mixture of products was obtained which could not be characterized. Complex 4 gave two hydride resonances in the 1H NMR spectrum at δ −15.0 [t, 2J(PH) = 10 Hz, no resolved coupling to platinum] and −15.5 [s, 1J(PtH) = 858 Hz] which can be assigned to IrH and PtH groups, respectively. There were two resonances for the CHxHyP2 protons of the dppm ligands, indicating the absence of a mirror plane containing the PtIrP4C2 atoms. The 13CO NMR spectrum contained a single resonance with a coupling constant 2J(PtC) of 180 Hz, which is too small for a direct Pt–CO bond, indicating that the carbonyl group is bound to iridium.6,7 The 31P NMR spectrum contained three resonances, which were readily assigned to the PtP groups [δ −7.8, 1J(PtP) 2344 Hz], the IrP groups [δ −6.3] and the [PF6]− anions [δ −143.3, 1J(PF) 711 Hz]. There was no evidence of fluxionality of complex 4. Several isomers of 4 are possible but only one was observed and the structure 4a (Scheme 2) is considered most consistent with the NMR data. At the platinum centre, the coupling constant 1J(PtH) = 858 Hz is lower than expected for a simple terminal hydride and higher than for a symmetrical bridging hydride, but it is consistent with an unsymmetrical bridging hydride or a hydride bound to a 5-coordinate platinum(II) centre.14,15 For example, the T-frame Pt–Pt bonded complex [HClPt(μ-dppm)2PtH]+ gives 1J(PtH) = 1360 and 962 Hz for the 4- and 5-coordinate platinum(II) centres respectively, with a long range coupling constant 2J(PtH) = 212 Hz for the hydride at the 4-coordinate centre trans to the Pt–Pt bond.14 At the iridium centre of 4, the hydride shows no resolved long range coupling to platinum while the carbonyl does, suggesting that the carbonyl group is trans to the Pt⋯Ir bond.
The reactions of 1 to give 3 or 4 occur by double oxidative addition of H2 or HCl respectively, and may be considered to convert Pt(I)Ir(0) in 1 to Pt(II)Ir(III) in 3 or 4. In each case, there must be an intermediate formed by a single oxidative addition step, but it has not been possible to characterize it. We therefore studied reactions of complex 1 with silane derivatives, hoping that, after the first oxidative addition, the bulky silyl group might prevent a second addition. The reagents Ph3SiH or (PhCH2)3SiH failed to react with 1, while Ph2SiH2 and PhSiH3 reacted but gave products which could not be characterized. However, excess Ph2MeSiH did react with complex 1 to give [PtIrH(SiMePh2)(CO)2(μ-dppm)2][PF6], 5, Scheme 2. The reaction was reversible and 5 reacted with excess CO to regenerate complex 1. In the 1H NMR spectrum of complex 5, a single hydride resonance was observed at δ −8.29, with coupling constant 2J(PtH) = 33 Hz, showing that the hydride is bound to iridium and cis to the Pt–Ir bond. The 13C{1H} NMR spectrum of a 13CO enriched sample contained two carbonyl resonances, a triplet at δ = 186.8, 2J(PC) = 10 Hz, with no resolved coupling to platinum, and a broad singlet at δ = 170.1, 1J(PtC) = 1130 Hz, which are therefore assigned as IrCO and PtCO groups respectively. Important structural information is obtained from a 13C (1H coupled) NMR experiment, in which the IrCO resonance shows additional doublet splitting due to the coupling 2J(HC) = 32 Hz. The magnitude of the 2J(HC) coupling in 5 can be compared with the values of 43 Hz and 5 Hz found in the isomers of [IrHBr(CO){Si(OEt)3}(dppe)], in which the hydride and carbonyl ligands are mutually trans or cis respectively, indicating that the trans-IrH(CO) group is present in 5.16 These data define the stereochemistry of 5 unambiguously. An unusual feature in the 31P NMR spectrum of 5 is that the phosphorus atoms of the dppm ligands are all inequivalent. The PtP resonances were well separated and occurred as an “AB” multiplet at δ = −3.6 and −6.2, with 2J(PP) = 350 Hz typical of trans P–Pt–P groups,12,14,17 and with 1J(PtP) = 2946 Hz and 3032 Hz respectively. The Ir–P resonances overlapped at δ = −20.8 in CD2Cl2 solution, but were resolved in CD3CN solution. The inequivalence of the phosphorus centres is no doubt due to the bulky SiMePh2 group being locked into an unsymmetrical conformation. Complex 5 is formed by cis oxidative addition of the Si–H bond at the iridium centre of complex 1, and so is a likely model for the first step in the oxidative addition of dihydrogen to 1. The iridium centre in 1 has an 18-electron configuration so the oxidative addition should be preceded by an effective dissociative step at iridium, which might be loss of CO, heterolytic cleavage of the Pt–Ir bond or migration of a CO ligand from iridium to platinum, but loss of CO must occur at some stage during the reaction.14 The oxidative addition of the Si–H bond to complex 1 may also provide a model for the first step in more complex reactions of silanes with dppm bridged complexes of rhodium and iridium.18
Reactions of alkynes with complex 1
Some reactions of complex 1 with alkynes are shown in Scheme 4. The products were characterized spectroscopically and, in three cases, by structure determination. During each reaction, one carbonyl ligand is displaced and the alkyne coordinates in the μ2–η1–η1 bonding mode, which is common in dppm bridged complexes.19 The alkynes RCCH (R = Ph, 4-C6H4Me, CO2Me) react selectively to give 6a–6c (Scheme 4), in which the CH and CR groups are bound to platinum and iridium, respectively. The symmetrical alkyne RCCR (R = CO2Me) gave only complex 7, but the unsymmetrical alkyne PhCCCO2Me gave an equal mixture of the two possible isomers 8a and 8b (Scheme 4). Diphenyl acetylene failed to react with complex 1. No rearrangement of the complexes 6 to give the μ2–η2–η2 bonded isomers, alkynyl-hydride complexes containing PtIrH(CCR) groups, or bridging vinylidene complexes containing PtIr(μ-CCHR) groups, was observed though related reactions are known in palladium, rhodium and iridium complexes with bridging dppm ligands.20
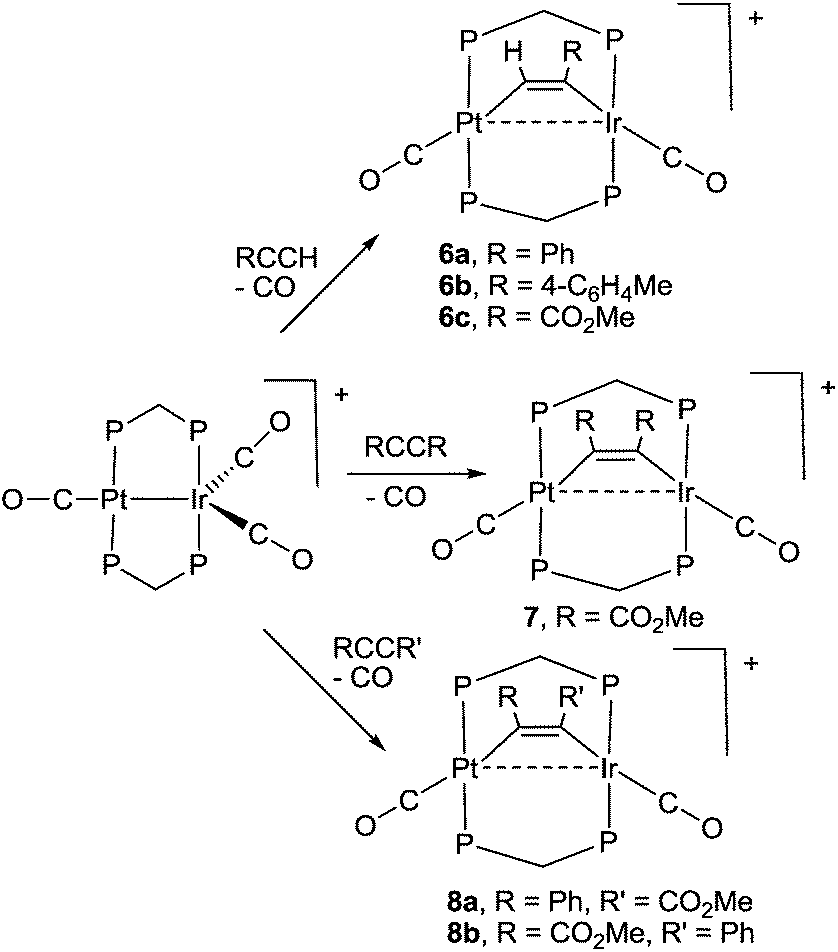 | ||
| Scheme 4 Synthesis of alkyne complexes 6–8. | ||
The structures of complexes 6a, 6b and 6c are similar and are shown in Fig. 2–4. In each case, the cation contains the expected trans,trans-PtIr(μ-dppm)2 group, with a bridging alkyne and a terminal carbonyl group on each square planar metal centre. The Pt⋯Ir distance of 2.9180(4) Å for 6b is longer than the sum of the covalent radii (ca. 2.75 Å) but shorter than the sum of the van der Waals radii (ca. 3.77 Å) of platinum and iridium.21 In addition, the Pt⋯Ir distance for 6b is somewhat shorter than the non-bonding distances P(1)P(4) and P(2)P(3) of 3.041(2) and 3.053(2) Å, and the angles C(4)–C(3)–Pt = 111.4(6) and C(3)–C(4)–Ir = 113.9(6)° are less than the natural sp2 bond angle of 120°. The parameters can be compared with those for [Pt2Cl2(μ-PhCCH)(μ-dppm)2] in which the Pt⋯Pt distance of 3.480(4) Å is longer and the angles CC–Pt of 121(1) and 124(1)° are greater than 120°, indicative of no metal–metal bonding.19 Thus, the data for 6b indicate that there is a weak bonding interaction between the platinum and iridium atoms, which could be of the donor–acceptor or secondary metallophilic bonding type.5–7 It should be noted that the platinum and iridium atoms could not be distinguished in the structure determination, and the assignments in Fig. 2–4 are based on the structure determination by NMR analysis described below. For example, the 31P NMR spectrum of complex 6a contained dppm resonances at δ 16.5 (IrP) and at δ 3.8 (1J(PtP) = 3260 Hz, PtP). The 1H NMR spectrum contained two resonances for the dppm methylene groups at δ 3.79 and 4.31, as expected for an A-frame structure,22 and a resonance for the HCC proton of the bridging alkyne at δ 7.04 [tt, 3J(PH) 14 Hz, 3J(PH) 1 Hz], with no resolved coupling to platinum. The 13C–1H HSQC NMR spectrum was used to identify the HC = carbon resonance at δ 119.2 [1J(PtC) 820 Hz] and the magnitude of the 195Pt13C coupling constant clearly shows that this carbon atom is directly bonded to platinum. The 13C–1H HMBC NMR spectrum was used to identify the HCCPh carbon resonance at δ 129.9 [t, 2J(PC) 25 Hz ]. The carbonyl resonances appeared at δ 177 [t, 2J(PC) = 10 Hz, IrCO] and 184 [t, 2J(PC) = 8 Hz, 1J(PtC) = 1105 Hz, PtCO] and a correlation between the PtCO and HCC resonances was also seen in the 13C–1H HMBC NMR spectra. The infrared spectrum of 6a shows two terminal carbonyl bands at 2067 and 1964 cm−1, as well as the CC stretch of the bridging alkyne at 1606 cm−1. Thus, the structure determination by a combination of X-ray and NMR techniques leaves no doubt that the assigned structure (Scheme 4, Fig. 2) is correct. The structure obtained for complex 6a was of low quality, and only the connectivity is established with confidence.
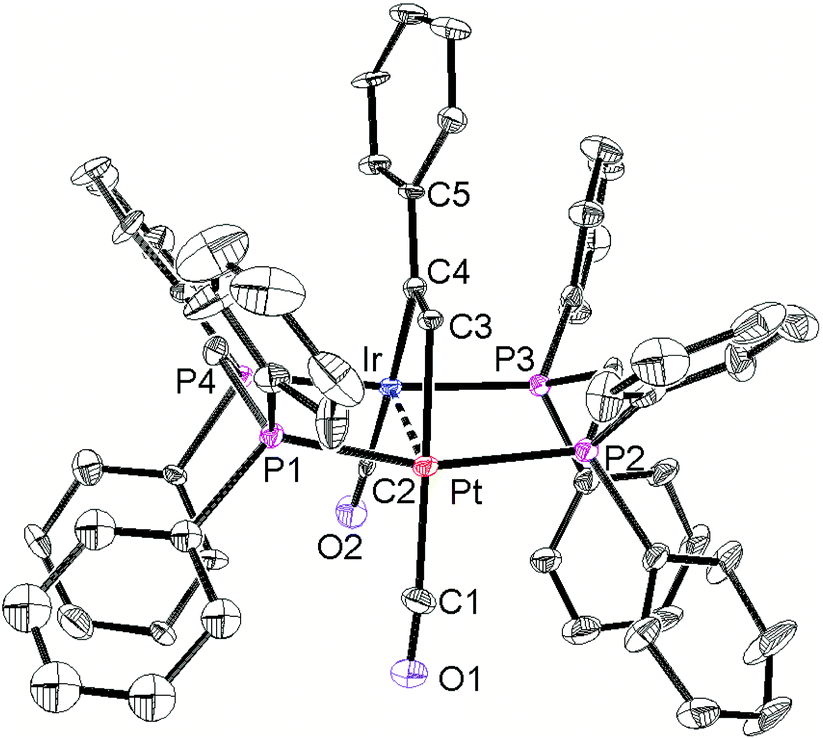 | ||
| Fig. 2 The structure of the cationic complex 6a. | ||
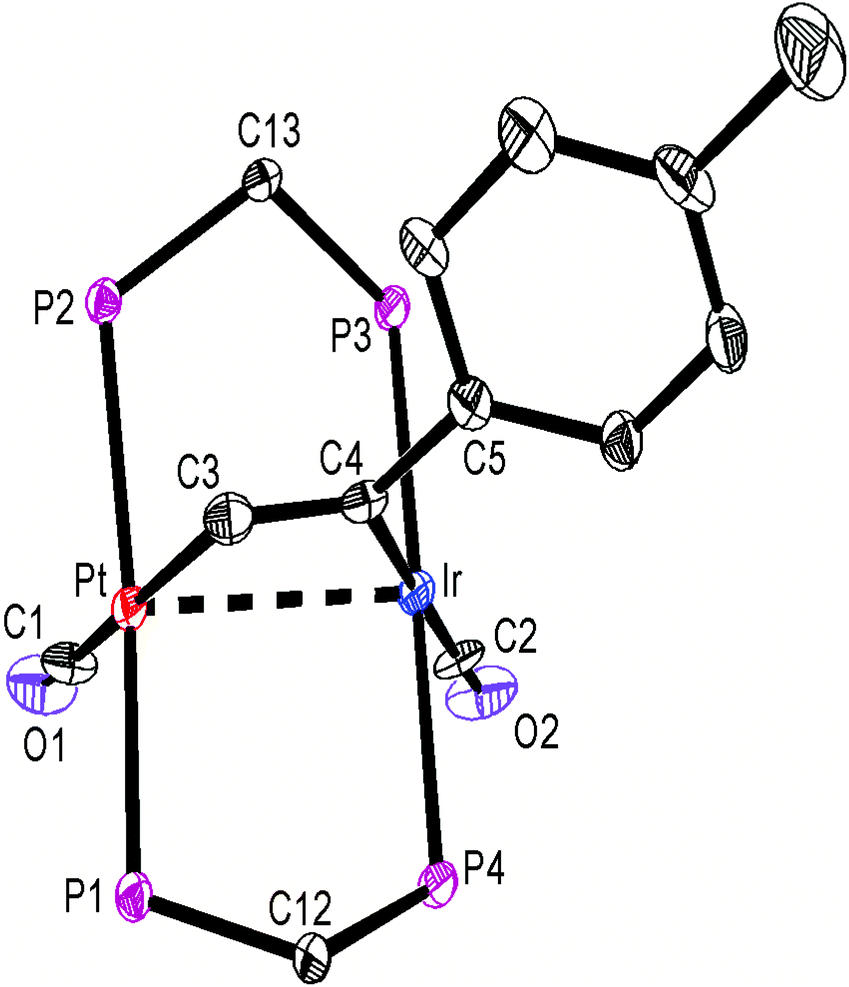 | ||
| Fig. 3 The structure of the cationic complex 6b, with phenyl groups omitted for clarity. | ||
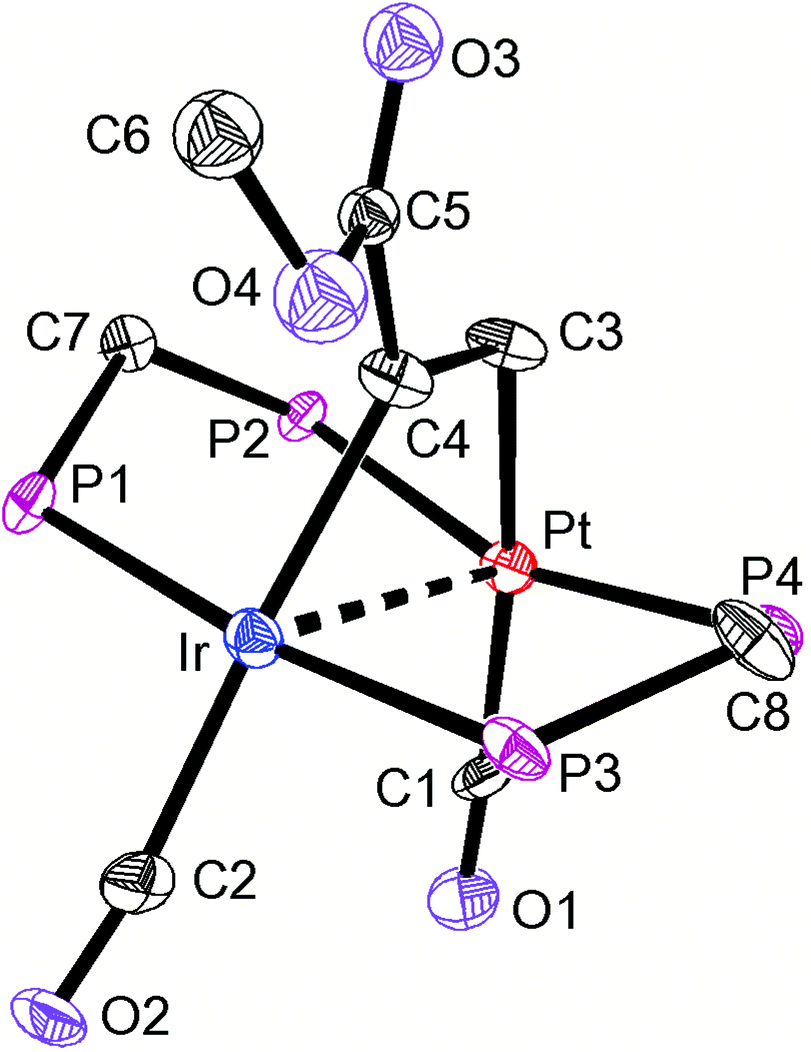 | ||
| Fig. 4 The structure of the cationic complex 6c, with phenyl groups omitted for clarity. | ||
A comparison of some bond parameters for 6b and 6c is given in Table 1. One feature is that the P–Pt–P angles are more distorted from linearity (19–23°) than the P–IrP angles (5–7°). This is consistent with a donor–acceptor metal–metal interaction with iridium as the donor. In all cases, the methylene linkages of the dppm groups are folded toward the coordinated alkyne in order to minimize steric interactions between the axial phenyl rings of the dppm ligands and the alkyne.8,12,14
| 6b | 6c | |
|---|---|---|
| Pt–Ir | 2.9180(4) | 3.0047(4) |
| Pt–C(1) | 1.918(9) | 1.95(1) |
| Pt–C(3) | 2.075(8) | 2.069(7) |
| Ir–C(2) | 1.96(2) | 1.89(1) |
| Ir–C(4) | 2.109(8) | 2.088(8) |
| Pt–P(1) | 2.323(2) | 2.352(7) |
| Pt–P(2) | 2.327(2) | 2.292(9) |
| Ir–P(3) | 2.298(2) | 2.315(8) |
| Ir–P(4) | 2.299(2) | 2.278(9) |
| P(1)–Pt–P(2) | 156.71(7) | 161.07(8) |
| P(3)–Ir–P(4) | 174.58(8) | 173.03(8) |
| C(4)–C(3)–Pt | 111.4(6) | 108.8(6) |
| C(3)–C(4)–Ir | 113.9(6) | 118.5(6) |
The reactivity of selected alkyne complexes has been studied. Complexes 6a and 6b reacted reversibly with CO to form the adducts 9a and 9b (Scheme 5), but 6c, 7 and 8 did not react. The complexes 9 could not be isolated because the reactions were reversed to give 6 on evaporation of the solvents. They were characterized by reaction of 6a or 6b with 13CO in an NMR tube. For example, the reaction of 6b with excess 13CO in CD2Cl2 solution at low temperature gave 9b essentially quantitatively, with a change in colour from pink/orange to yellow. At −30 °C, two dppm resonances were observed in the 31P NMR spectrum at δ(31P) = 5.78 [1J(PtP) = 3164 Hz, PtP] and −4.42 [IrP], with the iridium–phosphorus shifted from δ(31P) = 16.19 [IrP] in 6b. In the 13C NMR spectrum, three carbonyl resonances were observed at δ(13C) = 175.67 [m, IrCO], 178.35 [m, IrCO] and 184.80 [s, 1J(PtC) = 1184 Hz, PtCO]. At room temperature, the IrP resonance was broad in the 31P NMR spectrum and a single broad IrCO resonance was observed in the 13C NMR spectrum, while resonances for 1 and 4-MeC6H4CCH were also observed. These data are interpreted in terms of rapid exchange between 6a and 9a at room temperature and with slower, partial displacement of the alkyne to give complex 1. No CO insertion into the Ir–C or Pt–C bond of the coordinated alkyne was observed.
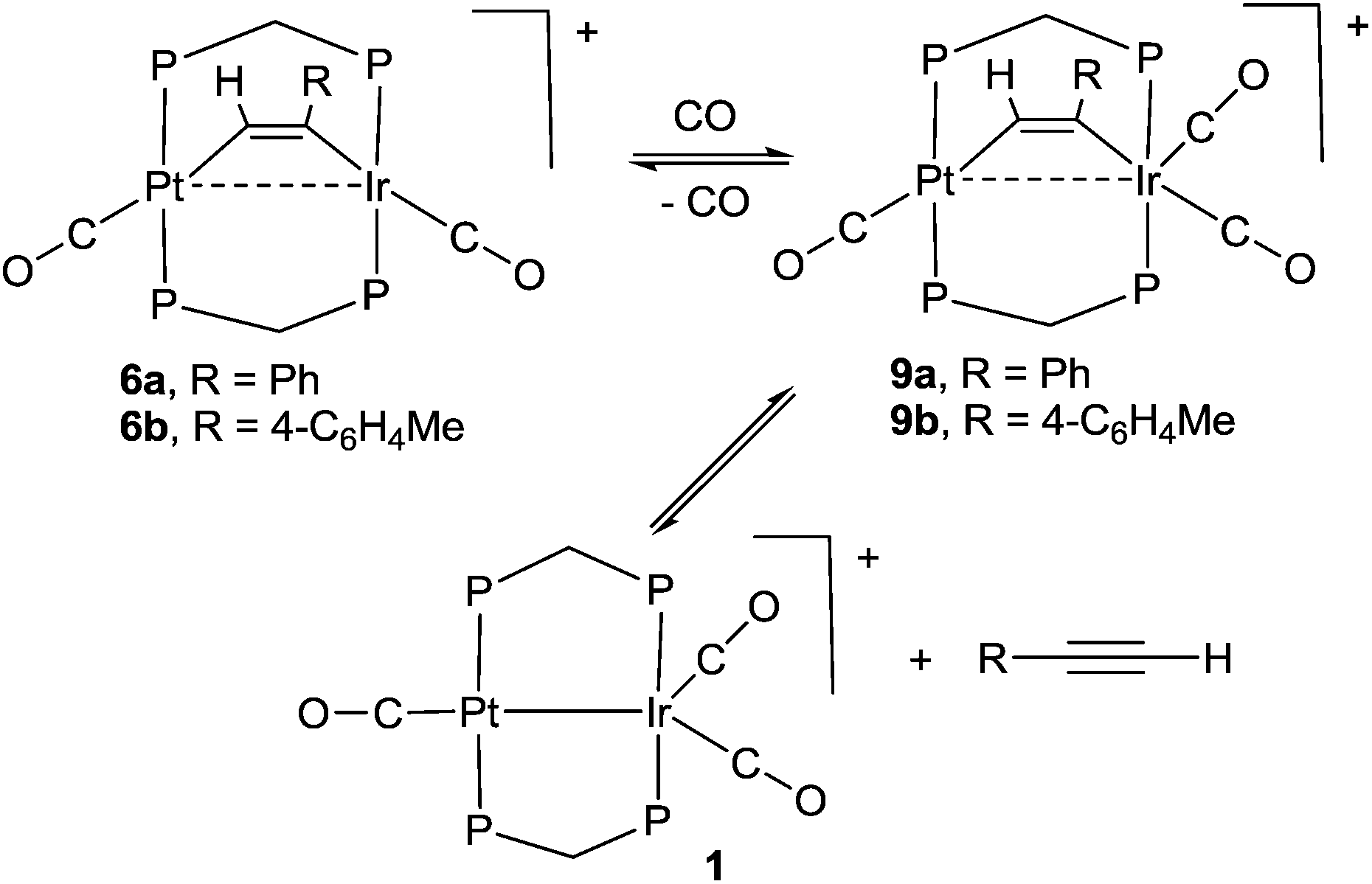 | ||
| Scheme 5 Reactions of 6a and 6b with CO. | ||
The reactions of complex 6b with dihydrogen and with hydrogen chloride are shown in Scheme 6. With dihydrogen a slow reaction occurred to give the same hydride complex [PtIrH4(CO)(μ-dppm)2][PF6], 3, which had previously been isolated by reaction of dihydrogen with complex 1 (Scheme 2). The alkyne group was hydrogenated to 4-methylstyrene, which was characterized by comparison of its 1H NMR spectrum with that of an authentic sample. The reaction must involve several steps, but no intermediates were detected in significant concentration. It is therefore likely that an initial oxidative addition of dihydrogen, probably to the iridium centre, is the slow step in the sequence. The hydrogenation of phenylacetylene to styrene has been observed previously with the homobinuclear complexes [Ir2(μ-S)(CO)2(μ-dppm)2] and [Rh2Cl2(μ-CO)(μ-dppm)2], but the alkyne was not coordinated prior to the introduction of H2.23 In contrast to the dihydrogen reaction, the initial reaction of 6b with HCl involved rapid oxidative addition to iridium(I) to give complex 10 and this was followed by slow reductive elimination to give the alkenylplatinum complex 11. The 1H NMR spectrum of 10 contained a hydride resonance at δ −19.34 [t, 1H, 2J(PH) = 14 Hz, 4J(PtH) = 139 Hz], assigned as an iridium hydride, and the 13C NMR spectrum of a 13CO enriched sample contained resonances for both iridium carbonyl and platinum carbonyl groups at δ(13C) = 171.2 [s, IrCO] and 178.3 [s, 1J(PtC) = 1110 Hz, PtCO], respectively. In the 1H NMR spectrum of 11, the vinyl protons appeared at δ 5.41 and 5.68, in the range expected for a terminal alkenyl group, and there was a coupling 3J(HH) = 17 Hz between the two vinyl protons, showing that they are mutually trans.12 The β-hydrogen at δ 5.68 couples to platinum with 3J(PtH) = 63 Hz, showing that the alkenyl group is bound to platinum. In the 13C NMR spectrum, there was only one carbonyl resonance, assigned as an iridium carbonyl because there was no resolved coupling to platinum. The carbonyl group is terminal, so a bridging chloride ligand is suggested to give a stable structure.12,14,23,24
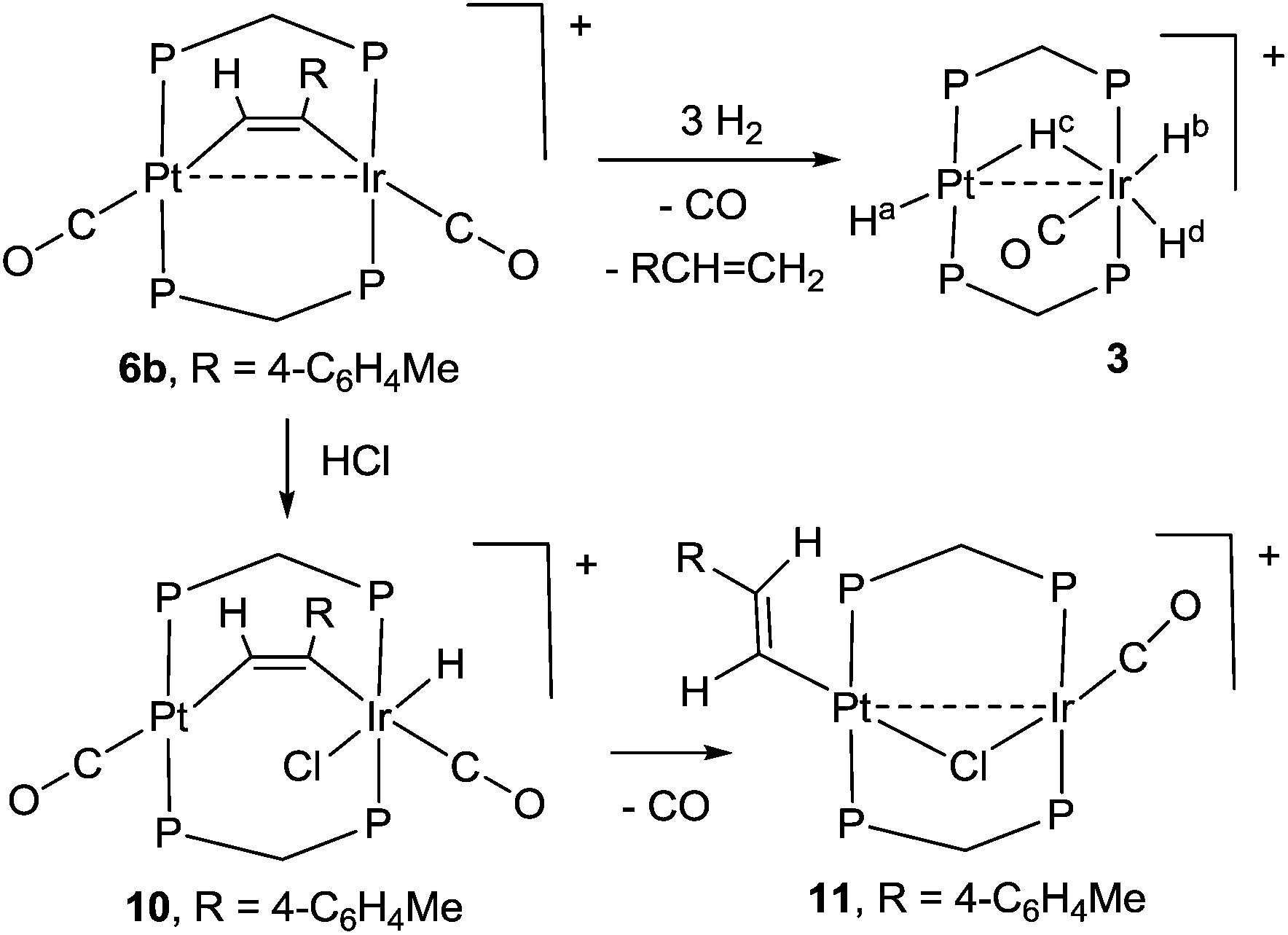 | ||
| Scheme 6 Reactions of 6b with H2 and HCl. | ||
Absorption and emission spectra of complex 6a
The complex [PtIr(CO)2(μ-PhCCH)(μ-dppm)2][PF6], 6a, which is pink in the solid state, forms dichroic solutions which may appear orange (concentrated solution) or bright pink (dilute solution), depending on the concentration and whether viewed by transmitted or reflected light. This unusual colour led us to investigate its absorption and emission spectra (Fig. 5). The UV-visible spectrum of 6a contains a very weak absorption at 650 nm (ε = 100 M−1 cm−1, barely visible in Fig. 5) and a strong absorption at 522 nm (ε = 4.8 × 103 M−1 cm−1). There are also partially resolved, higher energy, absorptions at ca. 420 and 365 nm.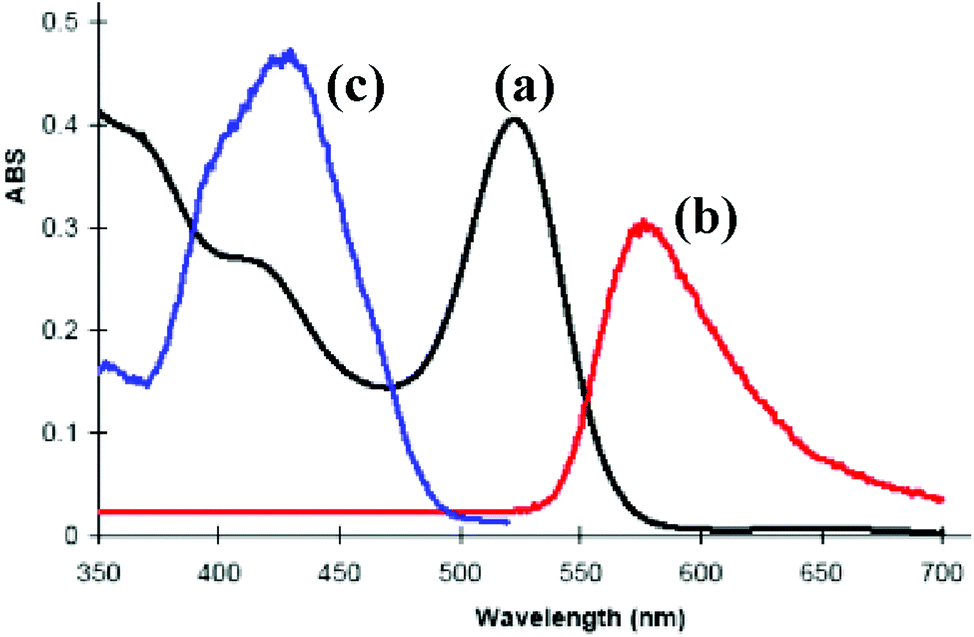 | ||
| Fig. 5 The photophysical properties of complex 6a in solution in CH2Cl2: (a) absorption spectrum (8.5 × 10−5 M); (b) emission spectrum (λex = 520 nm); (c) excitation spectrum (λem = 570 nm). | ||
There have been several detailed studies of the photophysical properties of d8–d8 face-to-face complexes, for which the two lower energy bands have been assigned, for third row transition metal complexes, as primarily due to the spin forbidden singlet–triplet and spin-allowed singlet–singlet 5dσ*→6pσ transitions.5,25 Complex 6a can be considered as a distorted face-to-face complex, because of the constraints of the bridging alkyne ligand, and it has strong π-acceptor carbonyl ligands, so the transitions are likely to be primarily 5dσ*→6pσ/COπ* transitions (Fig. 6). The Pt⋯Ir bonding should be stronger in the excited state.25 In the heterobinuclear PtIr complex 6a, the HOMO will have more iridium 5dz2 character and the LUMO will have more platinum 6pz and CO π* character (Fig. 6), so the lowest energy transitions will involve some iridium to platinum charge transfer.25 The absorption spectrum is very similar to that of the face-to-face Pt(II)Rh(I) complex [Pt(CN)2Rh(tBuNC)2(μ-dppm)2]+, A, [λmax 547 nm (triplet) and 469 (singlet)] except that the bands in 6a are shifted to considerably lower energy [λmax 650 nm (triplet) and 522 (singlet)].5,25 This shift can be understood in terms of the neutral iridium(I) centre in 6a being more electron rich than the cationic rhodium(I) centre in A and the cationic platinum(II) centre in 6a being more electron deficient than the neutral platinum(II) centre in A.
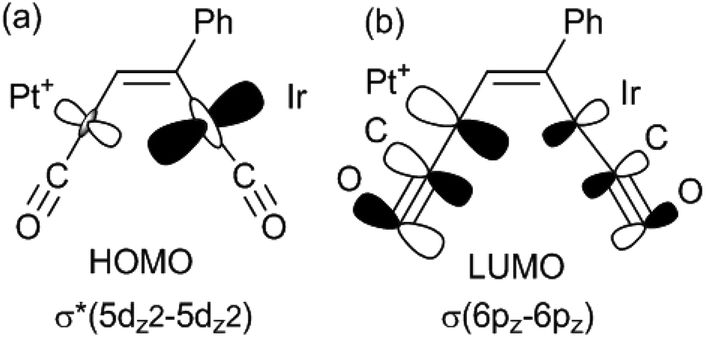 | ||
| Fig. 6 Schematic diagram of the likely HOMO and LUMO in complexes such as 6a (dppm ligands omitted for clarity). The different orbital sizes are intended only to depict the likely differences in orbital character in the HOMO and LUMO. | ||
Complex 6a is strongly emissive at room temperature in a dichloromethane solution, giving an emission band at 575 nm which is assigned to the 6pσ→5dσz2* fluorescence, with a shoulder at ca. 670 nm which might arise from the corresponding phosphorescence. The Stokes shift of 53 nm for the main fluorescence band is similar to that observed in related complexes.5,25 The addition of carbon monoxide to this solution results in the complete suppression of the room temperature luminescence as complex 9a is formed (Scheme 5).
Computational studies
In order to gain further insight into the above chemistry, DFT calculations were carried out on some of the complexes, using the ligand CH2(PMe2)2, dmpm, in place of dppm in order to make the computation times reasonable (see experimental for details).26 The calculated structures of [PtIr(CO)3(μ-dmpm)2]+, 1*, [Pt–CO 1.96, Ir–CO 1.91, Pt–Ir 2.78 Å, P–Ir–P 175°] and [PtIrH(CO)2(μ-dmpm)2], 2*, [Pt–H 1.64, Ir–CO 1.91, Pt–Ir 2.85 Å, P–Ir–P 156°] are shown in Fig. 7. The structure of complex 1 has been determined [Pt–CO 1.91, Ir–CO 1.90, Pt–Ir 2.77 Å, P–Ir–P 169°] but that of 2 has not. The calculation predicts a greater twist of the diphosphine ligand, a longer Pt–Ir distance and a greater distortion of the P–Ir–P bond angle from linearity in 2 when compared to 1. These calculated features can be understood in terms of the greater trans-influence of hydride in 2 and 2* compared to carbonyl in 1 and 1*, leading to a higher degree of Pt(II)–Ir(−I) character in 2 and 2*. Note that the complexes can be formulated as Pt(0)–Ir(I)+, Pt(I)+–Ir(0) or Pt(II)2+–Ir(−I)−, depending on how the electrons in the PtIr bond are assigned.27 The HOMO in both 1 and 2 is expected to be Pt–Ir bonding, with a high degree of iridium 6p/5d character, Fig. 7, and the calculation for 1* and 2* predicts polarity Ptδ+–Irδ− [calculated Hirshfeld charges: 1*, Pt 0.03e, Ir −0.17e; 2*, Pt −0.08e, Ir −0.22e]. Oxidation of both 1 and 2 is expected to occur at the more electron-rich iridium centre, provided there is a low energy pathway. | ||
| Fig. 7 The calculated structure of (a) [PtIr(CO)3(μ-dmpm)2]+, 1*, [Pt–CO 1.96, Ir–CO 1.91, Pt–Ir 2.78 Å, P–Ir–P 175°] and (b) PtIrH(CO)2(μ-dmpm)2], 2*, [Pt–H 1.64, Ir–CO 1.91, Pt–Ir 2.85 Å, P–Ir–P 156°] with (c) and (d) the corresponding HOMO. | ||
Calculations were carried out on the isomers of the model complex cation [PtIrH4(CO)(μ-dmpm)2]+, 3*, which is a model for the complex 3 formed by reaction of dihydrogen with complex 1 (Schemes 2 and 3). Good minima were found for isomers 3a* and 3b* (Fig. 8), but attempts to optimize the geometry of isomers 3c* or 3d* (or isomers with only terminal hydrides) led to spontaneous isomerisation to 3b*. A plausible reaction coordinate diagram for the fluxionality of complex 3 based on these calculations and on the experimental observations (Fig. 1, Scheme 3) is shown in Fig. 8. The high point is the transition state associated with inversion of the PtHIr group in 3d*, and this is the step that leads to Hc–Hd exchange.
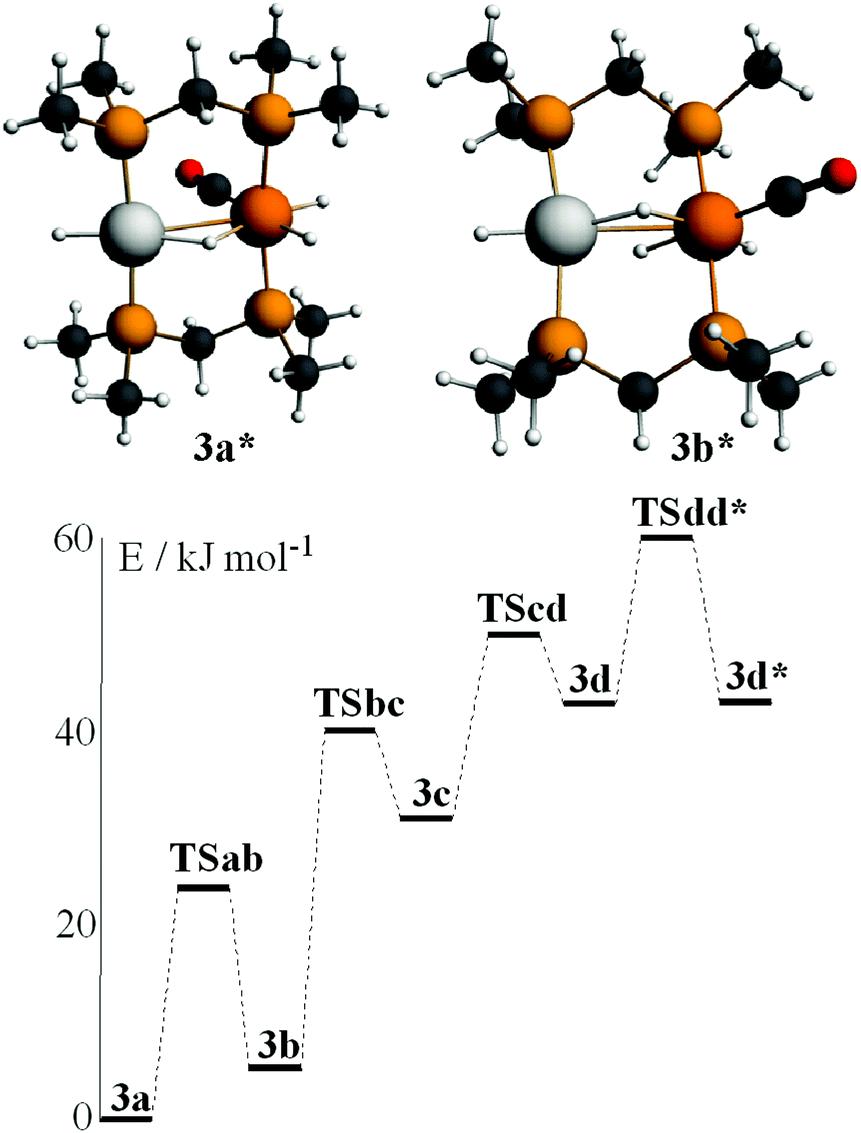 | ||
| Fig. 8 The calculated structures of isomers 3a* and 3b* of [PtIrH4(CO)(μ-dmpm)2]+ and a plausible reaction coordinate diagram for the observed fluxionality of the dppm analogue (Scheme 3). | ||
The calculated structure of the complex [PtIrH(SiMePh2)(CO)2(μ-dmpm)2]+, 5*, as a model for the dppm analogue 5 (Scheme 2), is shown in Fig. 9. The structure is rigid with a highly twisted PtIr(μ-dmpm)2 unit, as a result of the steric effects of the silyl group. The corresponding complex [PtIrH2(CO)2(μ-dmpm)2]+, 12*, was also studied as a model for the first step in the oxidative addition of dihydrogen to complex 1. In this case, the isomer 12a*, which is analogous to 5*, was predicted to be the most stable isomer but the complex is much more flexible than 5* and isomers with a bridging hydride, such as 12b* (ΔE +63 kJ mol−1 from 12a*) or with one hydride transferred to platinum, such as 12c* (ΔE +76 kJ mol−1 from 12a*), are predicted to be kinetically accessible (Fig. 9).
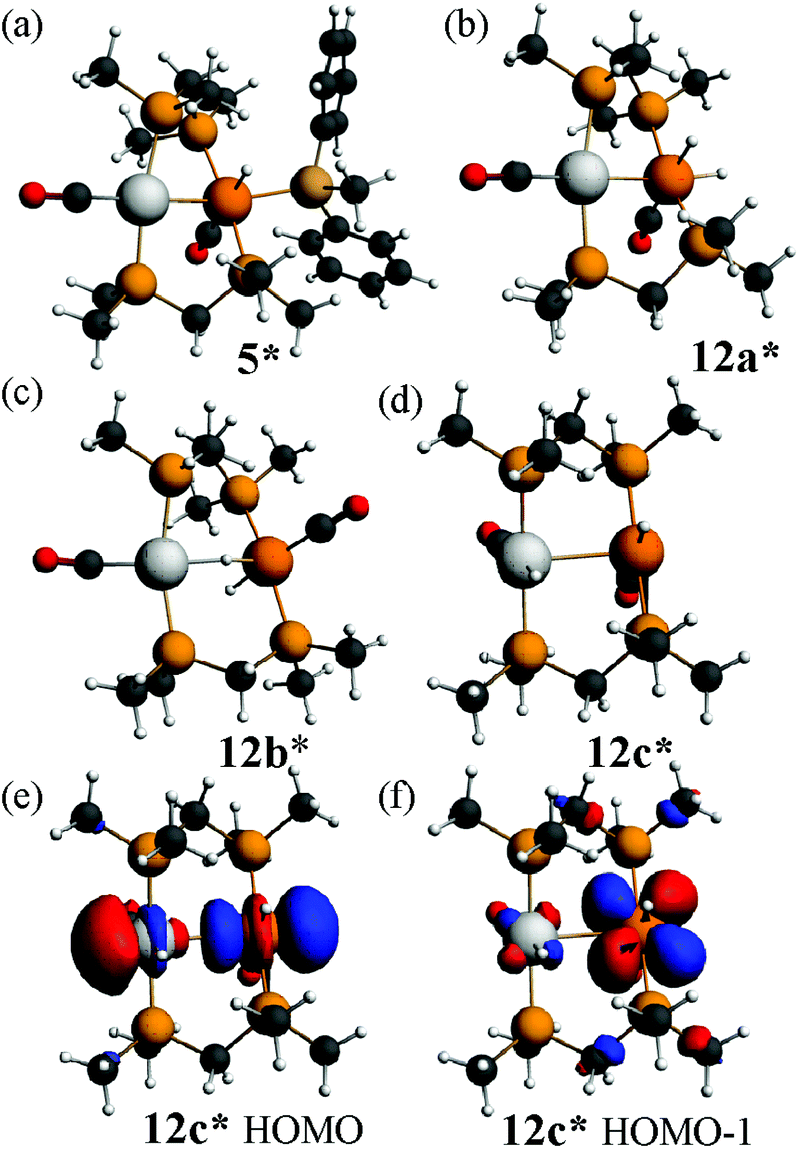 | ||
| Fig. 9 The calculated structures of (a) the complex [PtIrH(SiMePh2)(CO)2(μ-dmpm)2]+, 5*, (b), (c), (d) possible isomers of [PtIrH2(CO)2(μ-dmpm)2]+, 12*, and (e), (f) the highest energy 5dσ* and 5dπ* occupied MOs of the face-to-face isomer 12c*. | ||
Several mechanisms can therefore be considered possible for a second oxidative addition of dihydrogen to [PtIrH2(CO)2(μ-dppm)2]+, 12, to give complex 3 (Scheme 2). In isomer 12a the iridium centre has an 18-electron configuration, so concerted oxidative addition would occur either at platinum or across the Pt–Ir bond. However, the face-to-face isomer 12c contains a 16-electron iridium(I) centre, and the highest occupied molecular orbitals have mostly iridium 5d character, so oxidative addition might occur at iridium after isomerisation of 12a to 12c. The carbonyl dissociation from platinum might occur during or after the oxidative addition of dihydrogen.
Calculated structures of some isomers of [PtIrH2Cl2(CO)(μ-dmpm)2]+, 4*, are shown in Fig. 10. The most stable isomer is 4c*, followed by 4d*, 4a* and 4b*, with isomers having the iridium chloride ligand trans to the Pt–Ir bond at higher energy. Complexes 4a* and 4b*, and 4c* and 4d*, can interconvert by inversion of the PtHIr group, but there is no easy way for 4a* to isomerise to 4c*. The NMR spectra of the complex [PtIrH2Cl2(CO)(μ-dppm)2]+, 4, were considered to favour isomer 4a, but the evidence is not definitive and a structure analogous to 4c* cannot be ruled out. The calculations support the presence of a very unsymmetrical bridging hydride (Fig. 10), with a short Pt–H and a long Ir⋯H distance, as suggested by the hydride NMR data.
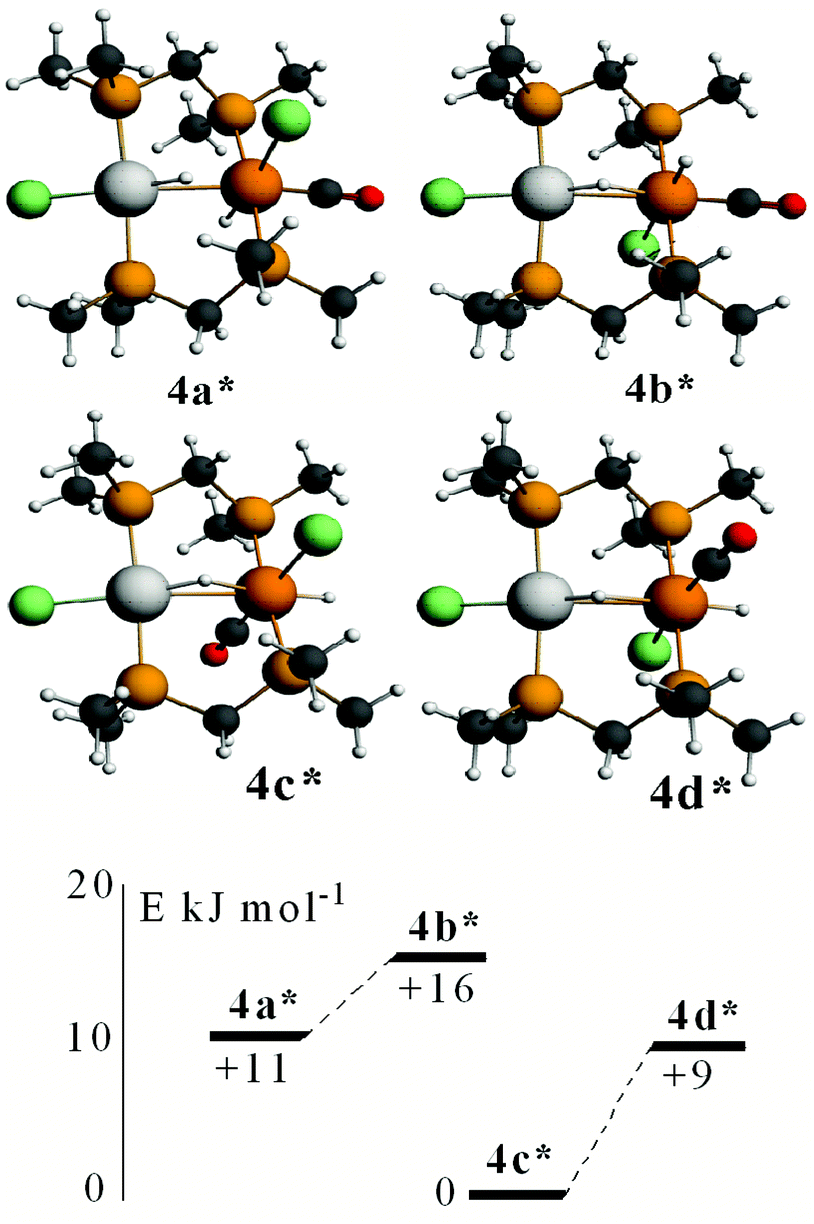 | ||
| Fig. 10 The calculated structures of some isomers of [PtIrH2Cl2(CO)(μ-dmpm)2]+, 4*, and their relative energies. Selected calculated distances: 4a*, Pt–H 1.66, Ir⋯μ-H 1.92, Ir–H 1.62, Ir–CO 1.88, Pt⋯Ir 2.85 Å; 4b*, Pt–H 1.67, Ir⋯μ-H 1.89, Ir–H 1.62, Ir–CO 1.88, Pt⋯Ir 2.86 Å; 4c*, Pt–H 1.65, Ir⋯μ-H 1.95, Ir–H 1.61, Ir–CO 1.87, Pt⋯Ir 2.93 Å; 4d*, Pt–H 1.64, Ir⋯μ-H 2.00, Ir–H 1.61, Ir–CO 1.87, Pt⋯Ir 2.91 Å. | ||
Some calculated structures for the dmpm analogues of alkyne complexes 6 and 8 (Scheme 3) are shown in Fig. 11 and 12.
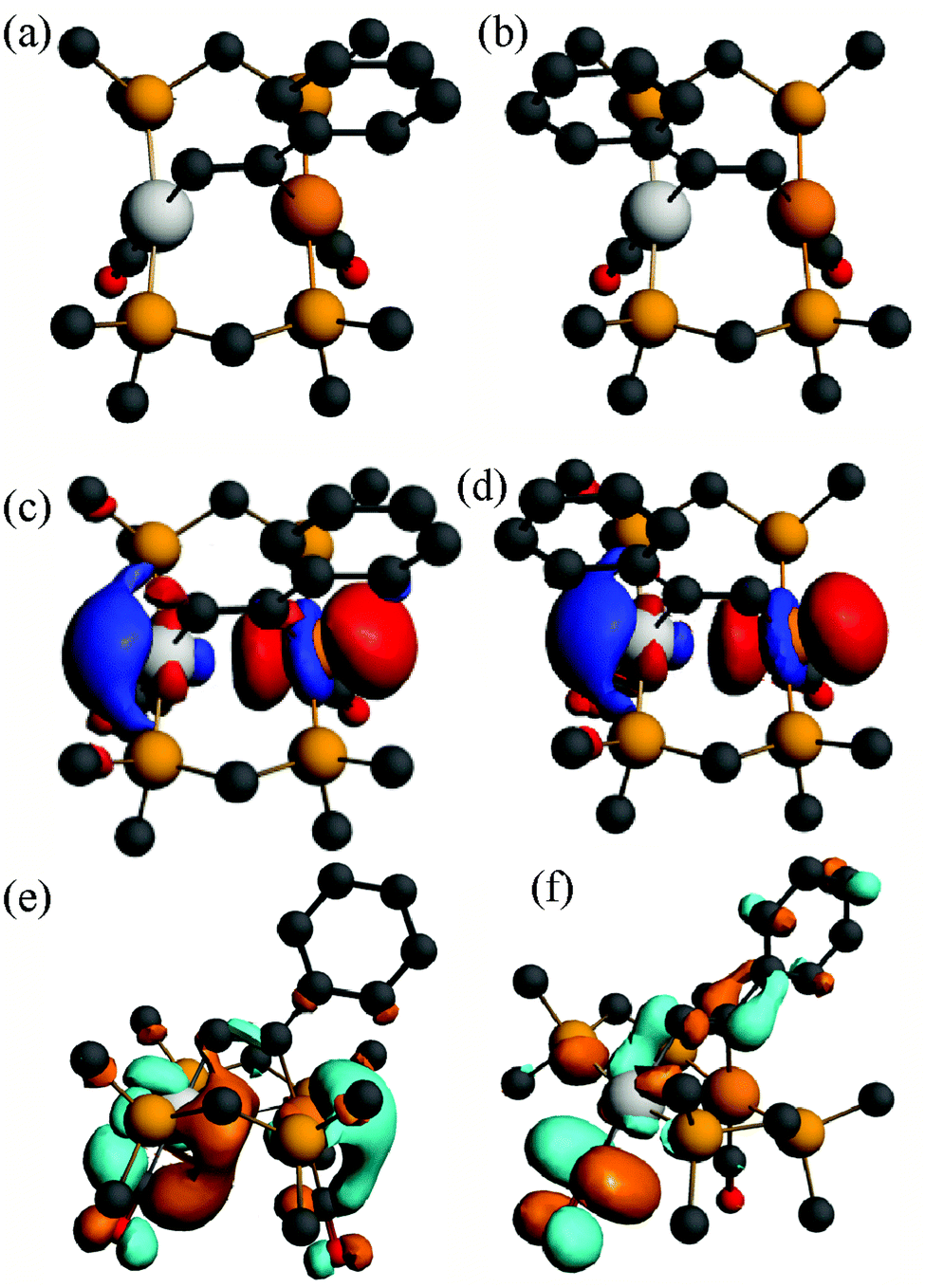 | ||
| Fig. 11 (a), (b) The calculated structures of the alkyne complex [PtIr(HCCPh)(CO)2(μ-dmpm)2]+, 6a*, and its isomer 6a*′, (c), (d) the HOMO for 6a* and 6a*′, (e), (f) the LUMO and LUMO+1 for 6a*. | ||
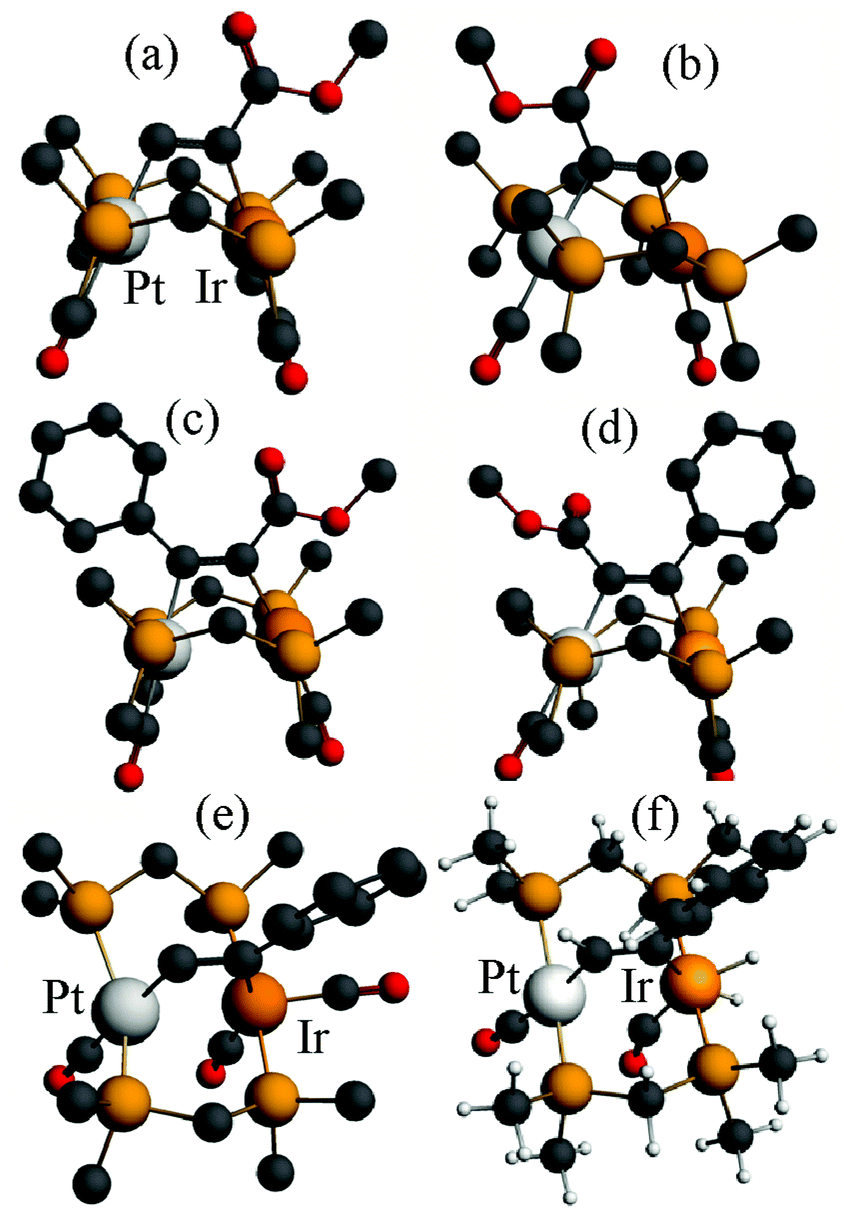 | ||
| Fig. 12 (a), (b) The calculated structures of the alkyne complex [PtIr(HCCCO2Me)(CO)2(μ-dmpm)2]+, 6c* and its isomer 6c*′, (c), (d) complex 8a* and its isomer 8b*, (e) complex 9a, (f) the possible intermediate dihydride complex 13*. | ||
The calculated energies of reaction to form the alkyne complexes 6a*, 6c*, 7* and 8a* from complex 1*, with displacement of one carbonyl ligand are −22, −85, −99 and −72 kJ mol−1 respectively, predicting that more electronegative substituents on the alkyne, and especially the –CO2Me groups, favour the reaction. For the complex 6a* or 6c* the conformation of the phenyl or –CO2Me group respectively is close to coplanar with the Pt–CC–Ir unit, which allows maximum π-conjugation, but in the disubstituted alkyne complex 8a* or 8b* the substituents are twisted out of the Pt–CC–Ir plane to reduce steric effects (Fig. 12). The reduction in π-bonding because of this twisting effect can explain the lack of reactivity of diphenylacetylene with complex 1 (the calculated energy of reaction is −13 kJ mol−1). The calculation predicts that 8a is more stable than 8b, but by only 4 kJ mol−1, consistent with the experimental observation that both isomers are formed. However, the calculations predict that, based on the ground state energies, there might also be an equilibrium between the isomers 6a* and 6a*′ [6a*′ favoured by 4 kJ mol−1] and between 6c* and 6c*′ [6c*′ favoured by 3 kJ mol−1] (Scheme 7) whereas, experimentally, only isomers 6a and 6c were observed (Scheme 4). No significant differences between steric effects in the isomers are expected. Unless the calculations give a wrong prediction, it is likely that the observed selectivity is based on kinetic rather than thermodynamic control. Perhaps the alkyne first coordinates to iridium with the bulky substituent oriented outwards, then slides over to the bridging position. Fig. 12e shows the calculated structure of [PtIr(μ-HCCPh)(CO)3(μ-dmpm)2]+, 9a*, which is a model for the complex 9a, observed on the initial reaction of 6a with CO (Scheme 5). Complex 9a might also be an intermediate in the reaction of phenylacetylene with complex 1. Dihydrogen is also expected to react with 6a at the iridium centre, and the structure of a potential dihydride complex [PtIrH2(μ-HCCPh)(CO)2(μ-dmpm)2]+, 13a*, is shown in Fig. 12f. Initial C–H reductive elimination from an analogous intermediate [PtIrH2(μ-HCCPh)(CO)2(μ-dmpm)2]+, 13a, would give a styrenyl complex, related to the observed complex 11 (Scheme 6), and a further oxidative addition of hydrogen and C–H reductive elimination would give styrene. However, given the ease with which hydride and carbonyl ligands can migrate between metal centres, there are several mechanisms that might apply.
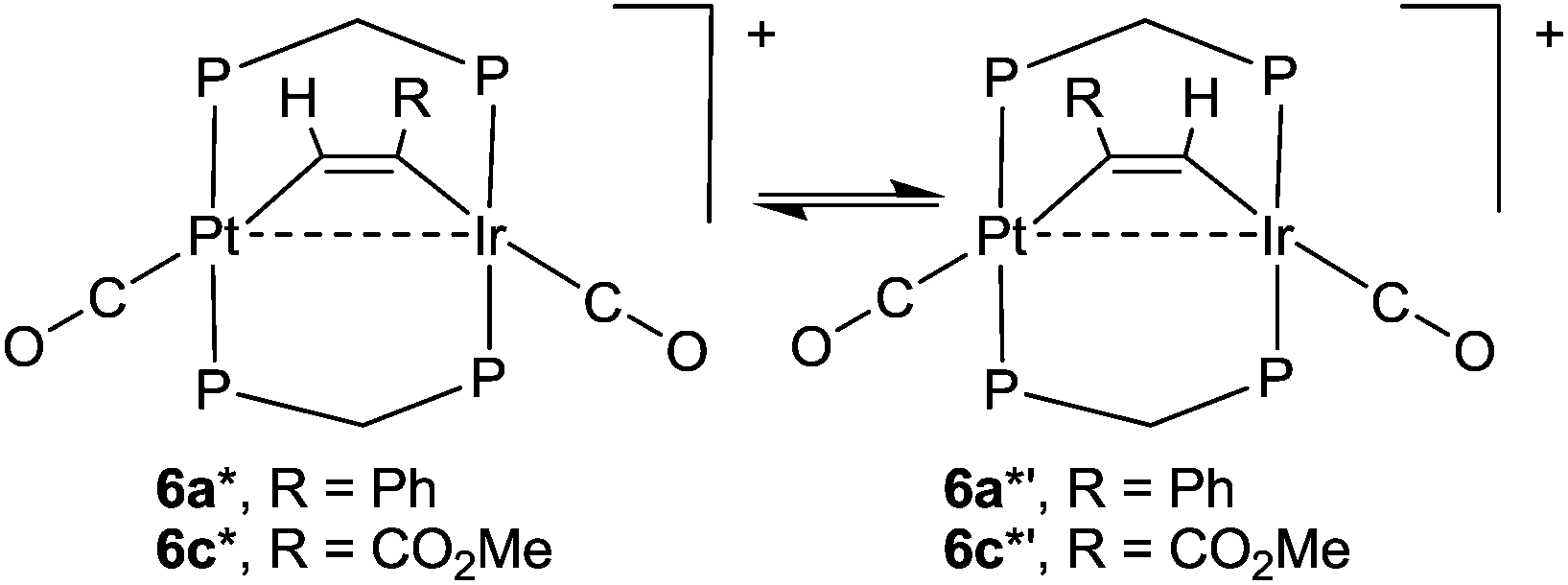 | ||
| Scheme 7 Predicted equilibrium between isomers of the alkyne complexes 6 (PP = dmpm). | ||
The absorption and emission spectra of complex 6a can be understood in terms of the frontier orbitals for the model complex 6a* shown in Fig. 11. The HOMO (Fig. 11c) is primarily the Pt–Ir 5dσ* molecular orbital, which is similar to that in the face-to-face complex 12c* (Fig. 9e), though the planes of the platinum(II) and iridium(I) are at an angle from the ideal face-to-face orientation. This HOMO has a greater character of the more electron rich iridium(I) centre, and it is very similar to that calculated for the isomeric 6a*′ (Fig. 11d). The LUMO has mostly Pt–Ir 6pz bonding character, with significant contribution of the pz–π* character of the carbonyl ligands, and is mostly centred on the PtCO group. The lowest energy singlet–singlet absorption and emission bands for 6a (Fig. 5) are associated with the transition between these molecular orbitals, in agreement with literature assignments for related compounds.5,25 The first singlet–singlet absorption band for complex 6a* is calculated to have a maximum at 537 nm, compared to the observed band for 6a at 522 nm.
Conclusions
The metal oxidation states in complex 1 can be considered as Pt(0)–Ir(I)+, Pt(I)+–Ir(0) or Pt(II)2+–Ir(−I)−, depending on how the electrons in the PtIr bond are assigned, but the reactivity is most easily interpreted in terms of the Pt(II)2+–Ir(−I)− formalism, in which the metal–metal bond can be considered as a donor–acceptor bond, formed by donation of electrons from Ir(−I) to Pt(II).27 This is also consistent with the nature of the HOMO (Fig. 7) and with the calculated charges on platinum and iridium in 1*. Thus the nucleophilic substitution of hydride for carbonyl in 1 occurs at the platinum centre, while oxidative addition reactions occur, at least initially, at the iridium centre (Scheme 2). Alkynes react with 1 at the metal–metal bond and the products 6–8 (Scheme 4) are considered as distorted face-to-face Pt(II)⋯Ir(I) complexes, and complex 6a exhibits strong room temperature emission, which is a characteristic property of such complexes.5The unusual chemistry of the polar Pt–Ir bond in complex 1 may provide insight into the mechanisms of reaction of the important bimetallic PtIr catalysts.3,4
Experimental
The syntheses were carried out using standard Schlenk techniques under an atmosphere of nitrogen. The complex [PtIr(CO)3(μ-dppm)2][PF6], 1, was prepared by the literature method, from [Pt(η2-dppm)2][PF6]2 and [PPN][Ir(CO)4], and 13CO enriched samples were prepared by stirring under an atmosphere of 13CO.6,28 The 1H, 31P{1H}, and 13C{1H} NMR spectra were recorded using a Varian Gemini 300, Varian Inova 400 or Inova 600 spectrometer. The gCOSY, gHSQC, and gHMBC spectra were recorded using the Varian Inova 400 or Inova 600 spectrometer. Chemical shifts are cited with respect to TMS or 85% phosphoric acid (31P). IR spectra were recorded with Nujol mulls or solutions using a Perkin Elmer 2000 FTIR spectrometer. Emission spectra were recorded by using a Fluorolog-3 spectrofluorimeter (ISA Jobin Yvon Spex), using a solution in CH2Cl2 at room temperature in a quartz cuvette. DFT calculations (gas phase only) were carried out by using the Amsterdam Density Functional (ADF) program based on the BP functional, with double-zeta basis set and first-order scalar relativistic corrections.26X-ray crystallography29
Crystals of compounds 6a, 6b, and 6c were mounted on glass fibers. Programs for diffractometer operation, data collection, cell indexing, data reduction and absorption correction were those supplied by Nonius. Diffraction measurements were made using a Nonius Kappa-CCD diffractometer using graphite-monochromated Mo-Kα radiation at 200 K (6a and 6c) or 150 K (6b). Structure solution and refinement was carried out using the SHELX97 or the SHELXT suite of programs, using the WinGX graphical interface. The initial solutions were obtained by direct methods and refined by successive least-squares cycles. Compound 6a co-crystallized with two solvent acetone molecules, one of which was disordered over two positions, as was the PF6− counterion. All non-hydrogen atoms in the main residue were refined anisotropically. Disordered C, O, and F atoms in the disordered solvent and anion were refined isotropically. The agreement factors were poor and so this is considered as a partial structure determination only. Compound 6b co-crystallized with a small amount of a chloro analog, presumably formed from the CH2Cl2 solvent during crystallization. The resulting CO–Cl (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25) disorder was successfully modeled and all non-H atoms were refined anisotropically. Compound 6c was refined as a racemic twin, and also contains disorder in the CO2CH3 group of the main residue, as well as in two phenyl rings of the main residue. In each case, the disorder was modelled over two positions using isotropic thermal parameters for the disordered C and O positions. All other non-H atoms in the main residue were refined anisotropically. Disorder was also present in the PF6− counterion and in two co-crystallized CH2Cl2 molecules. Details of the data collection and refinement can be found in the cif files (CCDC 1040457–1040458).
:25) disorder was successfully modeled and all non-H atoms were refined anisotropically. Compound 6c was refined as a racemic twin, and also contains disorder in the CO2CH3 group of the main residue, as well as in two phenyl rings of the main residue. In each case, the disorder was modelled over two positions using isotropic thermal parameters for the disordered C and O positions. All other non-H atoms in the main residue were refined anisotropically. Disorder was also present in the PF6− counterion and in two co-crystallized CH2Cl2 molecules. Details of the data collection and refinement can be found in the cif files (CCDC 1040457–1040458).
[HPt(μ-dppm)2Ir(CO)2], 2
To a stirred solution of [(CO)Pt(μ-dppm)2Ir(CO)2][PF6] (35 mg) in thf (10 mL) was added a solution of NaBHEt3 in thf (1.0 mL, 2 M). The solution was vigorously stirred for 3 hours, over which time the orange colour of the solution changed to yellow colour. The solvent was removed under vacuum, the solid was extracted with CH2Cl2 (1 mL) and the product was precipitated as a yellow solid by addition of pentane (5 mL). Yield: 17 mg, 55%. Anal. Calc. for C52H45IrO2P4Pt: C, 51.48; H, 3.74. Found: C, 50.97; H, 3.53%. NMR in CD2Cl2: δ(1H) = −3.33 [m, 1H, 1J(PtH) = 1123 Hz, PtH], 4.85 [br t, 4H, 2J(PH) = 4 Hz, CH2], 7.0–7.8 [m, 40H, Ph]; δ(31P) = 16.08 [t, J(PP) = 30 Hz, 1J(PtP) = 2873 Hz, PtP]; −16.40 [t, J(PP) = 30 Hz, 2J(PtP) = 69 Hz, IrP].[HPt(μ-dppm)2(μ-H)Ir(H)2(CO)][PF6], 3
To a stirred, degassed solution of [PtIr(CO)3(μ-dppm)2][PF6] (30 mg) in CH2Cl2 (10 mL) was added dihydrogen (1 atm) and the flask was sealed. The colour slowly changed from orange to yellow. After 3 h, the solvent was removed under vacuum, the residue was redissolved in a minimum amount of CH2Cl2 (ca. 1 mL) and the product was precipitated as a yellow solid by addition of ether (5 mL). Yield: 25 mg, 82%. Anal. Calc. for C51H48F6IrOP5Pt: C, 45.95; H, 3.63. Found: C, 45.49; H, 3.55%. IR (Nujol): ν(CO) = 2080 cm−1 (s). NMR in CD2Cl2 at −30 °C: δ(1H) = −12.02 [br s, 1H, 3J(PtH) 90 Hz, IrHd], −9.52 [br s, 1H, 1J(PtH) 540 Hz, PtHbIr], −8.17 [m, 1H, 2J(PH) 15 Hz, IrHc], −3.90 [m, 1H, 2J(PH) 15 Hz, 1J(PtH) 1225 Hz, PtHa], 3.88 [m, 2H, CH2P2], 5.23 [m, 2H, 3J(PtH) 55 Hz, CH2P2], 7.0–8.0 [m, 40H, Ph]; δ(31P) = 19.61 [m, J(PP) = 36 Hz, 1J(PtP) = 2817 Hz, PtP], −1.60 [t, J(PP) = 36 Hz, IrP], −143.42 [septet, 1J(PF) = 711 Hz, PF6].[HClPt(μ-dppm)2IrHCl(CO)][PF6], 4
To a solution of [(CO)Pt(μ-dppm)2Ir(CO)2][PF6] (127.5 mg, 0.0919 mmol) in CH2Cl2 (10 mL) was added a solution of HCl in CH2Cl2 (10 mL, 0.09 M). Immediately upon addition, effervescence was noted and the colour of the solution changed from orange to yellow. The solvent was removed in vacuo and the residue recrystallized from CH2Cl2–ether to give a pale yellow solid. Yield: 88 mg, 78%. Anal. Calc. for C51H46Cl2F6IrOP5Pt: C, 43.69; H, 3.31. Found: C, 43.25; H, 3.08%. IR: ν(CO) = 2048 cm−1 (s). NMR in CD2Cl2: δ(1H) = −15.53 [s, 1H, 1J(PtH) = 858 Hz, PtH]; −15.02 [t, 1H, 2J(PH) = 10 Hz, IrH]; 4.52 [m, 2H, CH2P2]; 4.78 [m, 2H, CH2P2]; 7.2–7.8 [m, 40H, Ph]; δ(31P) = 7.84 [t, J(PP) = 34 Hz, 1J(PtP) = 2344 Hz, PtP]; −6.28 [m, J(PP) = 34 Hz, IrP]; −143.3 [septet, 1J(PF) = 711 Hz, PF6]; δ(13C) = 159.99 [m, 2J(PtC) = 180 Hz, IrCO].[(CO)Pt(μ-dppm)2IrH(CO)(SiPh2Me)][PF6], 5
To a stirred solution of [(CO)Pt(μ-dppm)2Ir(CO)2][PF6] (61.1 mg, 0.441 mmol) in CH2Cl2 (10 mL) was added diphenylmethylsilane (0.2 g, 1 mmol). The colour of the solution slowly changed from orange to yellow. After 24 h, the solvent was removed in vacuo and the residue was recrystallized from CH2Cl2–ether. Yield: 46 mg, 67%. Anal. Calc. for C65H58F6IrO2P5PtSi: C, 51.12; H, 3.83. Found: C, 50.63; H, 3.77%. IR: ν(CO) = 2046 cm−1 (s), 1945 cm−1 (m). NMR in CD2Cl2: δ(1H) = −8.29 [t, 1H, 2J(PH) = 14 Hz, 2J(PtH) = 33 Hz, IrH]; 0.42 [s, 3H, 2J(SiH) = 40 Hz, SiMe]; 5.07 [m, 2H, CH2P2]; 6.12 [m, 2H, CH2P2]; 6.4–8.2 [m, 52H, Ph]; δ(13C) = 186.83 [m, 2J(PC) = 10 Hz, 2J(HC) = 32 Hz, IrCO]; 170.08 [m, 1J(PtC) = 1130 Hz, PtCO]; δ(29Si) = −10.0; δ(31P) = −3.64 [m, J(PP) = 40 Hz, 2J(PPtP) = 350 Hz, 1J(PtP) = 2946 Hz, PtP]; −6.16 [m, J(PP) = 40 Hz, 2J(PPtP) = 350 Hz, 1J(PtP) = 3032 Hz, PtP]; −20.84, −20.86 [m, IrP]; −143.39 [septet, 1J(PF) = 710 Hz, PF6].[PtIr(CO)2(μ-HCCPh)(μ-dppm)2][PF6], 6a
To a solution of [PtIr(CO)3(μ-dppm)2][PF6], 1, (50 mg, 0.036 mmol) in CH2Cl2 (10 mL) was added phenylacetylene (3.7 mg, 3.9 μL, 0.072 mmol) and the mixture was stirred under nitrogen for 16 h. Over the reaction time, a change from an orange solution to a bright pink/orange dichroic solution was observed. The solvent was removed in vacuo and the pink residue was recrystallized from CH2Cl2–Et2O to yield the product as a dark pink solid. Yield: 35 mg, 68%. Anal. Calc. for C60H50F6IrO2P5Pt: C, 49.39; H, 3.45. Found: C, 49.47; H, 3.45%. IR(Nujol): ν(CO) = 2067 (m), 1964 (s); ν(CC) = 1606 (s). NMR in CD2Cl2: δ(1H) = 3.79 [m, 2H, 2J(HH) = 14 Hz, 3J(PH) = 7 Hz, CH2P2], 4.31 [m, 2H, 3J(PtH) = 60 Hz, CH2P2], 6.42 [d, 2H, 3J(HH) = 7 Hz, Ph Ho], 6.59 [t, 2H, 3J(HH) = 7 Hz, Ph Hm], 6.72 [t, 1H, 3J(HH) = 7 Hz, Ph Hp], 7.04 [tt, 1H, 3J(PH) = 14 Hz, 4J(PH) = 1 Hz, PhCCH], 7.2–7.8 [m, 40H, dppm Ph]; δ(31P) = 16.5 [m, IrP], 3.8 [m, 1J(PtP) = 1630 Hz, PtP]; δ(13C) = 17.7 [m, CH2P2]; 119.2 [s, 1J(PtC) = 820, PtCCIr], 129.9 [t, 2J(PC) = 25 Hz, PtCCIr], 129.0–134.3 [Ph], 177 [t, 2J(PC) = 10 Hz, IrCO], 184 [t, 2J(PC) = 8 Hz, 1J(PtC) = 1105 Hz, PtCO].[PtIr(CO)2(μ-HCC-4-C6H4Me)(μ-dppm)2][PF6], 6b
This was prepared in a similar way from complex 1 (29.8 mg, 0.022 mmol) and 4-ethynyltoluene (4 μL, 0.032 mmol). Yield: 24.7 mg, 84%. Anal. Calc. for C61H52F6IrO2P5Pt: C, 49.73; H, 3.56. Found: C, 49.38; H, 3.47%. IR (Nujol): ν(CO) = 2065 m, 1954 m. NMR in CD2Cl2: δ(1H) = 2.00 [s, 3H, Me], 3.25 [m, CH2P2], 3.75 [m, CH2P2], 6.28 [d, 2H, 3J(HH) = 8 Hz, C6H4–Ho], 6.37 [d, 2H, 3J(HH) = 8 Hz, C6H4–Hm], 6.97 [tt, 1H, 3J(PH) = 14 Hz, 4J(PH) = 2 Hz, CCH], 7.10–7.70 [m, 40H, Ph]; δ(31P) = 16.19 [m, IrP]; 2.83 [m, 1J(PtP) = 3236 Hz, PtP]; −142.2 [septet, 1J(PF) = 710 Hz, PF6]; δ(13C) = 18.0 [m, CH2], 20.9 [s, Me], 119.2 [m, CCH], 128–134 [m, Ph], 183.88 [t, 2J(PC) = 8 Hz, 1J(PtC) = 1117 Hz, PtCO]; 188.65 [t, 2J(PC) = 10 Hz, IrCO].
[PtIr(CO)2(μ-HCCCO2Me)(μ-dppm)2][PF6], 6c
This was prepared in a similar way from complex 1 (71.9 mg, 0.0519 mmol) and methyl propiolate (4.7 μL, 0.0528 mmol). Yield: 51.1 mg, 68%. Anal. Calc. for C56H48F6IrO4P5Pt: C, 46.67; H, 3.36. Found: C, 46.65; H, 3.01%. IR(Nujol): ν(CO) = 2064 (m), 1959 (m); ν(CO) of CO2CH3 = 1687 (m). NMR in CD2Cl2: δ(1H) = 2.63 [s, 3H, Me], 3.60 [m, 2H, CH2P2], 3.84 [m, 2H, CH2P2], 7.0–8.1 [m, 40H, Ph]; δ(31P) = 12.95 [m, IrP]; 2.45 [m, 1J(PtP) = 3105 Hz, PtP]; −143.36 [septet, 1J(PF) = 710 Hz, PF6].[PtIr(CO)2(μ-MeO2CCCCO2Me)(μ-dppm)2][PF6], 7
This was prepared in a similar way from complex 1 (176 mg, 0.1271 mmol) and dimethyl acetylenedicarboxylate (16 μL, 0.130 mmol). Yield: 140 mg, 73%. Anal. Calc. for C58H50F6IrO6P5Pt: C, 46.47; H, 3.36. Found: C, 45.98; H, 3.22%. IR(Nujol): ν(CO) = 2043 (m), 1952 (m); ν(CO) of CO2CH3 = 1702 (m). NMR in CD2Cl2: δ(1H) = 2.20 [s, 3H, OMe], 2.47 [s, 3H, OMe], 3.75 [m, 2H, CH2P2], 3.88 [m, 2H, CH2P2], 7.1–8.3 [m, 40H, Ph]; δ(31P) = 8.30 [m, IrP]; 0.12 [m, 1J(PtP) = 2862 Hz, PtP]; −143.39 [septet, 1J(PF) = 710 Hz, PF6]; δ(13C) = 178.86 [t, 2J(PC) = 10 Hz, 1J(PtC) = 1096 Hz, PtCO]; 187.72 [t, 2J(PC) = 9 Hz, IrCO].[PtIr(CO)2(μ-PhCCCO2Me)(μ-dppm)2][PF6], 8a and 8b
This was prepared in a similar way from complex 1 (71.7 mg, 0.0518 mmol) and methyl phenylpropiolate (8.3 μL). Yield: 47 mg, 60%. Anal. Calc. for C62H52F6IrO4P5Pt: C, 49.08; H, 3.45. Found: C, 49.26; H, 3.41%. IR(Nujol): ν(CO) = 2059 (m), 1948 (m); ν(CO) of CO2CH3 = 1698 (m). NMR in CD2Cl2: δ(1H) = 2.26 [s, OMe], 2.35 [s, OMe], 3.80 [m, CH2P2], 3.80 [m, CH2P2], 3.92 [m, CH2P2], 3.97 [m, CH2P2], 4.07 [m, CH2P2], 5.95–8.00 [m, Ph]; δ(31P) = 9.02 [m, IrP]; 7.97 [m, IrP]; −0.28 [m, 1J(PtP) = 2995 Hz, PtP]; −1.54 [m, 1J(PtP) = 2887 Hz, PtP]; −143.37 [septet, 1J(PF) = 710 Hz, PF6]. It was not possible to assign resonances to specific isomers because they were formed in equal amounts.Reaction of 6a and 6b with CO to give [PtIr(CO)3(μ-RCCH)(μ-dppm)2][PF6], 9a and 9b
An NMR tube containing a solution of complex 6b (29.5 mg, 0.020 mmol) in CD2Cl2 (1 mL) was cooled to −80 °C and then evacuated and refilled with 13CO. On shaking the tube, the colour of the solution changed from pink/orange to yellow, and spectra were recorded at −30 °C and at 20 °C. NMR in CD2Cl2 for 9b: δ(1H) = 2.18 [s, 3H, Me], 3.73 [m, 2H, CH2P2], 3.87 [m, 2H, CH2P2], 6.19 [d, 2H, 3J(HH) = 8 Hz, C6H4–Ho], 6.73 [d, 2H 3J(HH) = 8 Hz, C6H4–Hm], 7.1–7.7 [m, 41H, Ph andCH]; δ(13C) = 20.1 [s, Me], 126.0 [C6H4–Co], 128.8 [C6H4–Cm], 128–135 [Ph], 175.67 [m, IrCO], 178. 35 [m, IrCO], 184.80 [s, 1J(PtC) = 1184 Hz, PtCO]; δ(31P) = 5.78 [m, 1J(PtP) = 3164 Hz, PtP]; −4.42 [s, IrP]; −143.31 [septet, 1J(PF) = 711 Hz, PF6]. At 20 °C, resonances for 9b were still observed, but there were also resonances for complex 1 and MeC6H4CCH. The IrP (δ −3.96) and IrCO (δ 176) resonances were broad. When the CO was removed, the resonances for 6b returned.
The reaction of CO with 6a was carried out in a similar way to give reversible formation of 9a. NMR in CD2Cl2: δ(1H) = 3.81 [m, 2H, CH2P2], 3.93 [m, 2H, CH2P2], 6.32 [m, 2H, C6H5–Ho], 6.91[m, 2H, C6H5–Hm], 7.04 [m, 1H, C6H5–Hp], 7.2–7.8 [m, 41H, Ph and =CH]; δ(13C) = 182 [br, IrCO], 184.7 [s, 1J(PtC) = 1176 Hz, PtCO]; δ(31P) = 5.5 [m, 1J(PtP) = 3170 Hz, PtP], −4 [br, IrP].
Reaction of 6b with dihydrogen
A solution of complex 6b (74 mg, 0.050 mmol) in CD2Cl2 (1 mL) in an NMR tube was treated with hydrogen (1 atmos.). The colour of the solution changed from pink-orange to orange over a period of 48 h., and NMR spectra were recorded during this period. Resonances for 6b decayed to zero after 48 h and were replaced by those for complex 3 (data as above) and 4-MeC6H4CHCH2, which were identical to those of an authentic sample. No resonances for 4-MeC6H4CCH were observed. The product 3 was precipitated by addition of pentane. Yield: 45 mg, 67%.
[PtIrHCl(CO)2(μ-dppm)2(μ-HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C-4-C6H4Me][PF6], 10, and [PtIr(μ-Cl)(CHCH-4-C6H4Me)(CO)(μ-dppm)2][PF6], 11
C-4-C6H4Me][PF6], 10, and [PtIr(μ-Cl)(CHCH-4-C6H4Me)(CO)(μ-dppm)2][PF6], 11
To a solution of complex 6b (72.2 mg, 0.049 mmol) in CD2Cl2 (0.6 mL) in an NMR tube was added a solution of HCl in CD2Cl2 (0.42 mL, 0.12 M, 0.0504 mmol). There was an immediate colour change from pink to orange. The initial product 10 was characterized spectroscopically. NMR in CD2Cl2: δ(1H) = −19.34 [t, 1H, 2J(PH) = 14 Hz, 4J(PtH) = 139 Hz, IrH], 2.00 [s, 3H, Me], 3.33 [m, 2H, CH2P2], 3.62 [m, 2H, CH2P2], 6.09 [d, 2H, 3J(HH) = 8 Hz, C6H4–Ho], 6.23 [d, 2H, 3J(HH) = 8 Hz, C6H4–Hm], 6.70 [m, 1H, CCH], 7.0–7.9 [m, 40H, Ph]; δ(13C) = 171.2 [s, IrCO], 178.3 [s, 1J(PtC) = 1110 Hz, PtCO]; δ(31P) = 12.89 [t, 2J(PP) = 22 Hz, 1J(PtP) = 2398 Hz, PtP]; −9.09 [m, IrP]; −143.31 [septet, 1J(PF) = 711 Hz, PF6]. After 17 h., the conversion to complex 11 was complete, and the product was isolated by evaporation of the solvent and precipitation from a solution in CH2Cl2 by addition of pentane. Yield: 54%. Anal. Calc. for C60H53ClF6IrOP5Pt: C, 48.64; H, 3.61. Found: C, 48.22; H, 3.46%. IR(Nujol) ν(CO) = 1996 cm−1. NMR in CD2Cl2: δ(1H) = 2.06 [s, 3H, Me], 4.24 [m, 2H, CH2P2], 4.55 [m, 2H, CH2P2], 5.41 [dt, 1H, 3J(HH) = 17 Hz, 3J(PH) = 7 Hz, Pt–CHC], 5.68 [d, 1H, 3J(HH) = 17 Hz, 3J(PtH) = 63 Hz, PtCCH], 5.88 [d, 3J(HH) = 8 Hz, C6H4–Ho], 6.63 [d, 3J(HH) = 8 Hz, C6H4–Hm], 6.8–8.2 [m, 40H, Ph]; δ(13C) = 171.13 [s, IrCO]; δ(31P) = 17.50 [m, 1J(PtP) = 2858 Hz, PtP]; −5.00 [s, br, IrP]; −143.34 [septet, 1J(PF) = 711 Hz, PF6].
Acknowledgements
We thank the NSERC (Canada) for financial support.Notes and references
- K. Wade, J. Chem. Soc., Chem. Commun., 1971, 792 RSC; K. Wade, Inorg. Nucl. Chem. Lett., 1972, 8, 559 CrossRef CAS; K. Wade, Chem. Br., 1975, 11, 177 Search PubMed.
- R. Hoffman, Angew. Chem., Int. Ed. Engl., 1982, 21, 711 CrossRef CAS; D. M. P. Mingos, Acc. Chem. Res., 1984, 17, 311 CrossRef.
- P. Braunstein and J. Rose, Comprehensive Organometallic Chemistry, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon, Oxford, 1995, ch. 7, vol. 10 CrossRef CAS; R. D. Adams, Polyhedron, 1988, 7, 2251 CrossRef CAS; J. Xiao and R. J. Puddephatt, Coord. Chem. Rev., 1995, 143, 457 CrossRef; M. Cowie, Can. J. Chem., 2005, 83, 1043 CrossRef; V. Ritleng and M. J. Chetcuti, Chem. Rev., 2007, 107, 797 CrossRef PubMed.
- L. N. Lewis, Chem. Rev., 1993, 93, 2693 CrossRef CAS; J. H. Sinfelt, Bimetallic Catalysts: Discoveries, Concepts, and Applications, Wiley, New York, 1983 CrossRef; L. D. Menard, Q. Wang, J. H. Kang, A. J. Sealey, G. S. Girolami, X. Teng, A. I. Frenkel and R. G. Nuzzo, Phys. Rev. B: Condens. Matter, 2009, 80, 064111 CrossRef; Y. J. Huang, S. C. Fung, W. E. Gates and G. B. McVicker, J. Catal., 1989, 118, 192 CrossRef; G. B. McVicker, M. Daage, M. S. Touvelle, C. W. Hudson, D. P. Klein, W. C. Baird, B. R. Cook, J. G. Chen, S. Hantzer and D. E. W. Vaughan, J. Catal., 2002, 210, 137 CrossRef; D. W. Flaherty and E. Iglesia, J. Am. Chem. Soc., 2013, 135, 18586 CrossRef PubMed; A. Djeddi, I. Fachete and F. Garin, Appl. Catal., A, 2012, 413, 340 CrossRef PubMed; S. G. Ramos, A. Calafiore, A. R. Bonesi, W. E. Triaca, A. M. C. Luna, M. S. Moreno, G. Zampieri and S. Bengio, Int. J. Hydrogen Energy, 2012, 37, 14849 CrossRef PubMed; F.-S. Ke, B. C. Solomon, S.-G. Ma and X.-D. Zhou, Electrochim. Acta, 2012, 85, 444 CrossRef PubMed; F.-D. Kong, S. Zhang, G.-P. Yin, J. Liu and A.-X. Ling, Catal. Lett., 2014, 144, 242 CrossRef; S. Hu, L. Xiong, X. Ren, C. Wang and Y. Luo, Int. J. Hydrogen Energy, 2009, 34, 8723 CrossRef PubMed.
- M.-N. Betzile, X. Wang, Z. M. Hudson and S. Wang, Dalton Trans., 2014, 43, 13696 RSC; L. Zhou, C.-L. Kwong, C.-C. Kwok, G. Cheng, H. Zhang and C.-M. Che, Chem. – Asian J., 2014, 9, 2984 CrossRef CAS PubMed; Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Chem. Sci., 2012, 3, 1185 RSC; D. R. Striplin and G. A. Crosby, J. Phys. Chem., 1995, 99, 7977 CrossRef; H.-K. Yip, H.-M. Lin, Y. Wang and C.-M. Che, Inorg. Chem., 1993, 32, 3402 CrossRef; A. L. Balch and V. J. Catalano, Inorg. Chem., 1992, 31, 2569 CrossRef; A. Kobayashi and M. Kato, Eur. J. Inorg. Chem., 2014, 4469 CrossRef; A. Diez, E. Lalinde and M. T. Moreno, Coord. Chem. Rev., 2011, 255, 2426 CrossRef PubMed.
- B. T. Sterenberg, H. A. Jenkins and R. J. Puddephatt, Organometallics, 1999, 18, 219 CrossRef CAS.
- B. T. Sterenberg, M. C. Jennings and R. J. Puddephatt, Organometallics, 1999, 18, 2162 CrossRef CAS; B. T. Sterenberg, M. C. Jennings and R. J. Puddephatt, Organometallics, 1999, 18, 3737 CrossRef; B. T. Sterenberg, G. J. Spivak, G. P. A. Yap and R. J. Puddephatt, Organometallics, 1998, 17, 2433 CrossRef.
- R. W. Hilts, O. Oke, M. J. Ferguson, R. McDonald and M. Cowie, Organometallics, 2005, 24, 4393 CrossRef CAS; M. E. Slaney, D. J. Anderson, M. J. Ferguson, R. McDonald and M. Cowie, Organometallics, 2012, 31, 2286 CrossRef; D. S. A. George, R. McDonald and M. Cowie, Organometallics, 1998, 17, 2553 CrossRef.
- D. M. McEwan, D. P. Markham, P. G. Pringle and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1986, 1809 RSC; F. S. M. Hassan, D. P. Markham, P. G. Pringle and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1985, 279 RSC; S. W. Carr, P. G. Pringle and B. L. Shaw, J. Organomet. Chem., 1988, 341, 543 CrossRef CAS.
- W. A. Schenk and G. H. J. Hilpert, Chem. Ber., 1991, 124, 433 CrossRef CAS; P. Braunstein, C. D. Debellefon, B. Oswald, M. Ries, M. Lanfranchi and A. Tiripicchio, Inorg. Chem., 1993, 32, 1638 CrossRef; T. Tanase, R. A. Begum, H. Toda and Y. Yamamoto, Organometallics, 2001, 20, 968 CrossRef.
- R. D. Adams, B. Captain, M. B. Hall, J. L. Sith Jr. and C. E. Webster, J. Am. Chem. Soc., 2005, 127, 1007 CrossRef CAS PubMed; M. J. Freeman, A. D. Miles, M. Murray, A. G. Orpen and F. G. A. Stone, Polyhedron, 1984, 3, 1093 CrossRef; S. Bhaduri, K. R. Sharma, W. Clegg, G. M. Sheldrick and D. Stalke, J. Chem. Soc., Dalton Trans., 1984, 2851 RSC; A. Fumagalli, R. D. Pergola, F. Bonacina, L. Garlaschelli, M. Moret and A. Sironi, J. Am. Chem. Soc., 1989, 111, 165 CrossRef; D. H. Cao, P. J. Stang and A. M. Arif, Organometallics, 1995, 14, 2733 CrossRef; M. H. Araujo, A. A. Avent, P. B. Hitchcock, J. F. Nixon and M. D. Vargas, Organometallics, 1998, 17, 5460 CrossRef; M. V. Jimenez, E. Sola, A. P. Martinez, F. J. Lahoz and L. A. Oro, Organometallics, 1999, 18, 1125 CrossRef; I. Jourdain, M. Knorr, C. Strohmann, C. Unkelbach, S. Rojo, P. Gomez-Iglesias and F. Villafane, Organometallics, 2013, 32, 5343 CrossRef.
- M. P. Brown, J. R. Fisher, L. Manojlovic-Muir, K. W. Muir, R. J. Puddephatt, M. A. Thomson and K. R. Seddon, J. Chem. Soc., Chem. Commun., 1979, 931 RSC; M. P. Brown, J. R. Fisher, R. H. Hill, R. J. Puddephatt and K. R. Seddon, Inorg. Chem., 1981, 20, 3516 CrossRef CAS; M. P. Brown, J. R. Fisher, A. J. Mills, R. J. Puddephatt and M. A. Thomson, Inorg. Chim. Acta, 1980, 44, L271 CrossRef; H. C. Foley, R. H. Morris, T. S. Targos and G. L. Geoffroy, J. Am. Chem. Soc., 1981, 103, 7337 CrossRef; L. Manojlovic-Muir, K. W. Muir, A. A. Frew, S. S. M. Ling, M. A. Thomson and R. J. Puddephatt, Inorg. Chem., 1981, 20, 1500 CrossRef; R. H. Hill, P. de Mayo and R. J. Puddephatt, Inorg. Chem., 1982, 21, 3642 CrossRef; J. R. Fisher, A. J. Mills, S. Sumner, M. P. Brown, M. A. Thomson, R. J. Puddephatt, A. A. Frew, Lj. Manojlovic-Muir and K. W. Muir, Organometallics, 1982, 1, 1421 CrossRef; R. H. Hill and R. J. Puddephatt, J. Am. Chem. Soc., 1983, 105, 5797 CrossRef; A. J. McLennan and R. J. Puddephatt, Organometallics, 1986, 5, 811 CrossRef; M. P. Brown, J. R. Fisher, S. J. Franklin, R. J. Puddephatt and K. R. Seddon, J. Organomet. Chem., 1978, 161, C46 CrossRef.
- Z. S. Wilson, G. G. Stanley and D. A. Vicic, Inorg. Chem., 2010, 49, 5385 CrossRef CAS PubMed; W. H. Lam and V. W.-W. Yam, Inorg. Chem., 2010, 49, 10930 CrossRef PubMed; B. J. Burkhart and R. L. DeKock, Comput. Theor. Chem., 2012, 994, 1 CrossRef PubMed.
- B. Chaudret, B. Delavaux and R. Poilblanc, Coord. Chem. Rev., 1988, 86, 191 CrossRef CAS; S. M. Oldham, J. F. Houlis, C. J. Leigh, S. B. Duckett and R. Eisenberg, Organometallics, 2000, 19, 2985 CrossRef; R. A. Stockland, G. K. Anderson and N. P. Rath, J. Am. Chem. Soc., 1999, 121, 7945 CrossRef; B. A. Vaartstra and M. Cowie, Inorg. Chem., 1989, 28, 3138 CrossRef; B. A. Vaartstra, K. N. O'Brien, R. Eisenberg and M. Cowie, Inorg. Chem., 1988, 27, 3668 CrossRef; R. McDonald, B. R. Sutherland and M. Cowie, Inorg. Chem., 1987, 26, 3333 CrossRef; M. C. Grossel, J. R. Batson, R. P. Moulding and K. R. Seddon, J. Organomet. Chem., 1986, 304, 391 CrossRef; K. A. Azam, A. A. Frew, B. R. Lloyd, L. Manojlovic-Muir, K. W. Muir and R. J. Puddephatt, Organometallics, 1985, 4, 1400 CrossRef; R. J. Puddephatt, K. A. Azam, R. H. Hill, M. P. Brown, C. D. Nelson, R. P. Moulding, K. R. Seddon and M. C. Grossel, J. Am. Chem. Soc., 1983, 105, 5642 CrossRef.
- E. A. V. Ebsworth, D. W. H. Rankin, I. M. Blacklaws and H. E. Robertson, J. Chem. Soc., Dalton Trans., 1978, 753 Search PubMed; R. J. Puddephatt, Coord. Chem. Rev., 2001, 219, 157 CrossRef.
- C. E. Johnson and R. Eisenberg, J. Am. Chem. Soc., 1985, 107, 6531 CrossRef CAS.
- P. S. Pregosin and R. W. Kunz, 31P and 13C NMR of Transition Metal Complexes, Springer-Verlag, Berlin, 1979 Search PubMed.
- W. D. Wang and R. Eisenberg, Organometallics, 1992, 11, 908 CrossRef CAS; M. H. Mobarok, O. Oke, M. J. Ferguson, R. McDonald and M. Cowie, Inorg. Chem., 2010, 49, 11556 CrossRef PubMed.
- C.-L. Lee, C. T. Hunt and A. L. Balch, Inorg. Chem., 1981, 20, 2498 CrossRef CAS; R. J. Puddephatt and M. A. Thomson, Inorg. Chem., 1982, 21, 725 CrossRef; M. Cowie and T. G. Southern, Inorg. Chem., 1982, 21, 246 CrossRef; K. A. Azam and R. J. Puddephatt, Organometallics, 1983, 2, 1396 CrossRef; J. A. Davies, K. Kirschbaum and C. Kluwe, Organometallics, 1994, 13, 3664 CrossRef; M. J. Irwin, G. Jia, J. J. Vittal and R. J. Puddephatt, Organometallics, 1996, 15, 5321 CrossRef.
- D. S. A. George, R. McDonald and M. Cowie, Can. J. Chem., 1998, 74, 2289 CrossRef CAS; D. S. A. George, R. McDonald and M. Cowie, Organometallics, 1998, 17, 2553 CrossRef; H. C. Clark and R. J. Puddephatt, Inorg. Chem., 1971, 10, 18 CrossRef.
- A. Albinati, T. J. Emge, T. F. Koetzle, S. V. Meille, A. Musco and L. M. Venanzi, Inorg. Chem., 1986, 25, 4821 CrossRef CAS.
- C. P. Kubiak and R. Eisenberg, J. Am. Chem. Soc., 1977, 99, 6129 CrossRef CAS.
- C. P. Kubiak, C. Woodcock and R. Eisenberg, Inorg. Chem., 1980, 19, 2733 CrossRef CAS; M. Cowie and T. G. Southern, Inorg. Chem., 1982, 21, 246 CrossRef.
- S. J. Cooper, M. P. Brown and R. J. Puddephatt, Inorg. Chem., 1981, 20, 1374 CrossRef CAS.
- C.-M. Che, L. G. Butler and H. B. Gray, J. Am. Chem. Soc., 1981, 103, 1593 CrossRef CAS; W. A. Fordyce, J. G. Brummer and G. A. Crosby, J. Am. Chem. Soc., 1981, 103, 7061 CrossRef; D. M. Roundhill, H. B. Gray and C.-M. Che, Acc. Chem. Res., 1989, 22, 55 CrossRef; M. I. S. Kenney, J. W. Kenney III and G. A. Crosby, Organometallics, 1986, 5, 230 CrossRef; C.-M. Che, V. V.-W. Yam, W.-T. Wong and T.-F. Lai, Inorg. Chem., 1989, 28, 2908 CrossRef; A. L. Balch and V. J. Catalano, Inorg. Chem., 1992, 31, 3934 CrossRef; V. W.-W. Yam, P. K.-Y. Yeung, L.-P. Chan, W.-K. Kwok, D. L. Phillips, K.-L. Yu, R. W.-K. Wong, H. Yan and Q.-J. Meng, Organometallics, 1998, 17, 2590 CrossRef; W. H. Lam and V. W.-W. Yam, Inorg. Chem., 2010, 49, 10930 CrossRef PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, S. van Gisbergen, C. F. Guerra, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS; A. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef.
- C. Mealli, F. Pichierri, L. Randaccio, E. Zangrando, M. Krumm, D. Holtenrich and B. Lippert, Inorg. Chem., 1995, 34, 3418 CrossRef CAS; M. P. Brown, S. J. Cooper, A. A. Frew, L. Manojlovic-Muir, K. W. Muir, R. J. Puddephatt, K. R. Seddon and M. A. Thomson, Inorg. Chem., 1981, 20, 1500 CrossRef; M. Rashidi, M. C. Jennings and R. J. Puddephatt, Organometallics, 2003, 22, 2612 CrossRef; B. Shafaatian, A. Akbari, S. M. Nabavizadeh, F. W. Heinemann and M. Rashidi, Dalton Trans., 2007, 4715 RSC.
- L. Garlaschelli, R. Della Pergola and S. Martinengo, Inorg. Synth., 1990, 28, 211 CrossRef CAS.
- (a) Computer programs (a) COLLECT, DENZO, SCALEPACK, Nonius B. V., Amsterdam, 1998 Search PubMed; (b) G. M. Sheldrick, SHELXTL, Madison, Wisconsin, Release 97-2, 1998, Release 2014-1, 2014 Search PubMed; (c) L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
Footnotes |
| † This article is dedicated to the memory of Professor Ken Wade, whose work has been so important in understanding not only the structure but also the reactivity of complexes with metal–metal bonds. |
| ‡ CCDC 1040456–1040458 for 6a–6c. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03966a |
| This journal is © The Royal Society of Chemistry 2015 |