
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal evolution of the crystal structure of proton conducting BaCe0.8Y0.2O3−δ from high-resolution neutron diffraction in dry and humid atmosphere
Annika K.
Eriksson Andersson
a,
Sverre M.
Selbach
a,
Tor
Grande
a and
Christopher S.
Knee
*b
aDepartment of Materials Science and Engineering, Norwegian University of Science and Technology, NO-7491 Trondheim, Norway
bEnvironmental Inorganic Chemistry, Department of Chemical and Biological Engineering, Chalmers University of Technology, SE-412 96 Göteborg, Sweden. E-mail: knee@chalmers.se
First published on 16th March 2015
Abstract
The crystal structure of the proton conducting perovskite BaCe0.8Y0.2O3−δ (BCY20) has been studied via high-resolution in situ neutron diffraction performed in controlled dry and humid (heavy water) oxygen flow. Two phase transitions, cubic Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m→Rc (775 °C)→Imma (250 °C) were observed on cooling from 1000 °C in dry O2. A significant shift of the phase stability fields was observed on cooling in wet oxygen (pD2O ≈ 0.2 atm) with the Rc structure stabilised at 900 °C, and the Rc→Imma transition occurring at 675 °C. On cooling below 400 °C a monoclinic, I2/m, phase started to appear. The structural dependence on hydration level is primarily due to the de-stabilisation of the correlated, octahedra tilts as a consequence of structural relaxation around the oxygen vacancies present in the non-hydrated phase. The tendency of hydrated BaCe0.8Y0.2O3−δ to show octahedral tilting is also found to be enhanced, indicating that the deuteronic (protonic) defects influence the crystal structure, possibly via hydrogen bonding. Stabilisation of the monoclinic I2/m phase is attributed to the structural effect of deuterons that is inferred to increase on cooling as deuterons localise to a greater extent. Changing from wet oxidising (O2 + D2O(g)) to wet reducing (5% H2 in Ar + D2O(g)) atmosphere did not influence the structure or the phase stability, indicating that Ce4+ was not reduced under the present conditions. Based on the observed cell volume expansion protonic defects are present in the material at 900 °C at a D2O partial pressure of ∼0.2 atm. The origin of the chemical expansion is explained by the effective size of the oxygen vacancy being significantly smaller than the [OD] defect. Rietveld analysis has been used to locate possible sites for the deuterons in the high temperature, Rc and Imma, phases that are most relevant for proton transport.
m→Rc (775 °C)→Imma (250 °C) were observed on cooling from 1000 °C in dry O2. A significant shift of the phase stability fields was observed on cooling in wet oxygen (pD2O ≈ 0.2 atm) with the Rc structure stabilised at 900 °C, and the Rc→Imma transition occurring at 675 °C. On cooling below 400 °C a monoclinic, I2/m, phase started to appear. The structural dependence on hydration level is primarily due to the de-stabilisation of the correlated, octahedra tilts as a consequence of structural relaxation around the oxygen vacancies present in the non-hydrated phase. The tendency of hydrated BaCe0.8Y0.2O3−δ to show octahedral tilting is also found to be enhanced, indicating that the deuteronic (protonic) defects influence the crystal structure, possibly via hydrogen bonding. Stabilisation of the monoclinic I2/m phase is attributed to the structural effect of deuterons that is inferred to increase on cooling as deuterons localise to a greater extent. Changing from wet oxidising (O2 + D2O(g)) to wet reducing (5% H2 in Ar + D2O(g)) atmosphere did not influence the structure or the phase stability, indicating that Ce4+ was not reduced under the present conditions. Based on the observed cell volume expansion protonic defects are present in the material at 900 °C at a D2O partial pressure of ∼0.2 atm. The origin of the chemical expansion is explained by the effective size of the oxygen vacancy being significantly smaller than the [OD] defect. Rietveld analysis has been used to locate possible sites for the deuterons in the high temperature, Rc and Imma, phases that are most relevant for proton transport.
1. Introduction
Solid state proton conductors have applications as electrolytes in fuel cells, solid oxide electrolysers, gas sensors and mixed proton and electron conducting membranes for hydrogen separation.1–4 Electrochemical devices based on these materials are therefore of wide relevance for environmentally cleaner and more efficient electricity generation technologies; spanning direct energy conversion using protonic ceramic fuel cells, the production of hydrogen from steam or from fossil fuels, and pre-combustion separation of hydrogen and simultaneous CO2 capture for gas or coal burning power plants.1,4 The leading solid state proton conducting materials were summarised by Norby,5 and in the intermediate temperature region, 300–700 °C, oxides with the perovskite crystal structure are presently the outstanding candidates.Protonic defects are typically introduced into perovskite oxides via the hydration of oxygen vacancies generated due to charge compensation whereby the B-cation is replaced by a cation, B′, with a lower oxidation state in a process commonly referred to as acceptor doping, i.e., AB1−xB′xO3−y. In dry atmospheres the oxygen vacancies facilitate oxygen mobility often leading to high oxide ion conductivity particularly at elevated temperatures. However, in humid environments some perovskites hydrate as their oxygen vacancies are filled with hydroxyl ions according to reaction (1):
| H2O(g) + vO˙˙ + OOx = 2OHO˙ | (1) |
In common with many A2+B4+O3 perovskites with a Goldschmidit tolerance factor,10t < 1.0 BaCeO3 adopts an orthorhombic, Pnma, symmetry at room temperature as the cubo-octahedral void formed by the octahedral BO6 units is too large for the A cation and cooperative tilts of the octahedra reduce this volume. The high proton conductivity of acceptor doped BaCeO3 has motivated many studies of the system's crystal structure as a function of temperature and dopant, utilising a range of techniques including Raman spectroscopy, dilatometery and neutron diffraction.11–13 High-resolution neutron diffraction data eventually provided the definitive description of the thermal dependence of the structure of BaCeO3 on heating from RT as being, Pnma→Imma→Rc→Pmm.14,15 More recently the structural evolution of acceptor doped BaCe1−xYxO3−δ has been examined as a function of temperature,16 and humidity,17 and a noteworthy deviation to monoclinic I2/m symmetry is reported at RT for hydrated samples with x ≥ 0.2.17–19
Proton conduction in perovskites is mediated via the hopping of a proton from one oxygen ion to another, a process commonly referred to as the Grotthuss mechanism.2,3 By probing the location of the protons in perovskite structure with density functional theory (DFT) and atomistic or molecular dynamics simulation calculations the conduction mechanism can be predicted, see for example ref. 20–22. However, only a few articles have been published reporting the proton position in perovskites by practical experiments. In relation to BaCeO3, two studies were found by the present authors: An early neutron powder diffraction (NPD) study by Knight23 suggested a potential proton position in BaCe0.9Y0.1O3−δ at 4.2 K bonded to the O(2) oxygen of the orthorhombic Pnma structure. Azad et al.24 recently suggested the proton to be close to the apical O(1) site based on analysis of neutron data recorded at RT on the same composition.
The focus of the present article is the detailed examination of the thermal and atmospheric dependence of the crystal structure of BaCe1−xYxO3−δ. In our previous in situ powder X-ray diffraction work focused on the chemical expansion of BaCe1−xYxO3−δ and BaZr1−xYxO3−δ a considerable volume expansion associated with the protonation reaction (eqn (1)) was observed.19 It was also found that the pre-hydrated material, BaCe0.8Y0.2O2.8(OH)0.2, demonstrates three structural phase transitions on heating in air; I2/m→Imma→Rc→Pmm but only two phase transitions on cooling in low vacuum: Pmm→Rc→Imma. To investigate the structural dependence of this leading proton conductor further we report here high-resolution neutron diffraction measurements as a function of temperature and atmosphere. As neutron scattering lengths do not scale with atomic number in the same way as X-ray scattering form factors, neutron diffraction is much better suited to the investigation of lighter elements such as oxygen and hydrogen. To avoid the large incoherent scattering cross-section of protons to thermal neutrons, it is generally preferable to use heavy water (D2O) to prepare samples containing deuterons. This approach also increases the chance of locating D ions within the oxide matrix, as the magnitude of the neutron scattering length of D is approx. double that of H.25 The findings show a remarkable structural dependence on the level of hydration; the presence or absence of oxygen vacancies strongly influences the structural transition temperatures related to the cooperative octahedral titling. Significant hydration induced chemical expansion is also observed. Possible positions for the deuterons are presented for the high temperature Rc and Imma crystal structures most relevant for proton conduction in the material.
2. Experimental
Synthesis of polycrystalline powder of BaCe0.8Y0.2O3−δ was performed by spray pyrolysis of aqueous nitric solutions (Cerpotech AS, Trondheim, Norway). As-pyrolysed powder was calcined at 1200 °C for 6 h in air, pressed into dense pellets (∼14 g, Ø 10 mm, with 2 tons load) and re-heated at 1200 °C for a further 6 h.In situ high-resolution diffraction data were collected on the time-of-flight HRPD instrument at the ISIS neutron and muon source, Rutherford Appleton Labs, UK. Loosely compacted pellets of the sintered samples were crushed into small pieces and loaded into the silica gas flow sample cell where they rested upon a gas permeable glass frit. The sample was exposed to oxidising (dry O2 followed by wet O2 + D2O vapour) and then wet reducing (5% H2 in Ar + D2O vapour) conditions produced in an external gas mixing system with an average gas flow of 23 ml min−1 (1.4 L h−1). For more details of the experimental set-up see ref. 26. The D2O was heated to 60 °C and the gas tubing was heated to 50 °C to avoid condensation giving an estimated pD2O ≈ 0.2 atm at the sample. As-prepared BaCe0.8Y0.2O3−δ was found to be hygroscopic on the basis of TGA measurements and PXRD scans that revealed a monoclinic phase indicative of partial hydration, i.e., the as-prepared material's composition should more accurately be written as BaCe0.8Y0.2O3−x(OH2)x. The sample was therefore pre-heated in situ to 1000 °C and was left for 1 h to dehydrate fully before commencing the neutron data collection on cooling in the O2 flow. Data were recorded in steps from 1000, 950, 900, 800, 700, 650, 600, 575, 550, 525, 500, 475, 450, 425, 400, 350, 300, 250, 200 and 150 °C employing 2 hours scans and equilibration dwells of 15 minutes. For the following runs in wet gas data were recorded from 900 °C using the same temperature interval and dwells as for the dry oxygen temperature scan.
The data sets were analysed by the Rietveld method27 using the GSAS program.28 The majority of the refinements utilised data from both the high-resolution backscattering detector bank (data analysed in region 0.8 ≤ d ≤ 2.6 Å) and the 90° detector bank (0.9 ≤ d ≤ 3.5 Å). Additional refinements using only the high-resolution data were employed to help determine the phase behaviour at temperatures close to the Rc→Imma and Imma→Imma + I2/m phase transitions. To aid the assignment of the phase transitions analysis of the FWHM of diagnostic peaks obtained from the high-resolution data was also used. A feature of the refinements was the presence of a modulated background originating from the silica sample container which was calculated with the use of approx. 20 background parameters of the background function type 1 for each histogram. In addition it was necessary to use anisotropic atomic displacement parameters (ADPs) to model the oxygen ion sites in order to obtain high quality fits.
3. Results
3.1. Structural analysis – temperature and atmospheric dependence of phase transitions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14,15 and BaPrO3 and BaPr0.9Y0.1O3−δ,29 the growth of these superlattice reflections reflect a cubic Pmm to trigonal Rc structure transition that occurs due to the stabilisation of antiphase tilting of the (Ce/Y)O6−δ units, along all three pseudo-cubic directions by the same magnitude, a process described as a0a0a0→a−a−a− in the Glazer notation.30 The second phase transition is not immediately apparent from Fig. 1. Instead, the transformation from a rhombohedral to an orthorhombic cell metric is revealed by considering the line widths of certain diffraction peaks. Fig. 2 plots the evolving FWHM of the intensity found in the region of the diagnostic (022) reflection of the Rc phase, as estimated using a Gaussian function. The resolution of our neutron data (backscattering detector bank) is sufficient to reveal a significant increase in the FWHM of this reflection (see inset) that occurs at T < 300 °C. This broadening is attributed to splitting of the (022) peak into the (202) and (040) reflections in orthorhombic Imma symmetry as previously observed for BaCeO313,14 and BaPrO3.29 Comparative refinements of the high-resolution data revealed a small but meaningful decrease in the Rwp agreement parameter with the Imma model for the data sets collected at T < 300 °C. For example, for the data set collected at 150 °C a Rwp = 2.11% was obtained with 43 variables using the Imma model vs. a Rwp = 2.35% for the Rc structure with 41 variables. This transition Rc (a−a−a−)→Imma (a0b−b−) reflects the loss of one antiphase tilt.
14,15 and BaPrO3 and BaPr0.9Y0.1O3−δ,29 the growth of these superlattice reflections reflect a cubic Pmm to trigonal Rc structure transition that occurs due to the stabilisation of antiphase tilting of the (Ce/Y)O6−δ units, along all three pseudo-cubic directions by the same magnitude, a process described as a0a0a0→a−a−a− in the Glazer notation.30 The second phase transition is not immediately apparent from Fig. 1. Instead, the transformation from a rhombohedral to an orthorhombic cell metric is revealed by considering the line widths of certain diffraction peaks. Fig. 2 plots the evolving FWHM of the intensity found in the region of the diagnostic (022) reflection of the Rc phase, as estimated using a Gaussian function. The resolution of our neutron data (backscattering detector bank) is sufficient to reveal a significant increase in the FWHM of this reflection (see inset) that occurs at T < 300 °C. This broadening is attributed to splitting of the (022) peak into the (202) and (040) reflections in orthorhombic Imma symmetry as previously observed for BaCeO313,14 and BaPrO3.29 Comparative refinements of the high-resolution data revealed a small but meaningful decrease in the Rwp agreement parameter with the Imma model for the data sets collected at T < 300 °C. For example, for the data set collected at 150 °C a Rwp = 2.11% was obtained with 43 variables using the Imma model vs. a Rwp = 2.35% for the Rc structure with 41 variables. This transition Rc (a−a−a−)→Imma (a0b−b−) reflects the loss of one antiphase tilt.
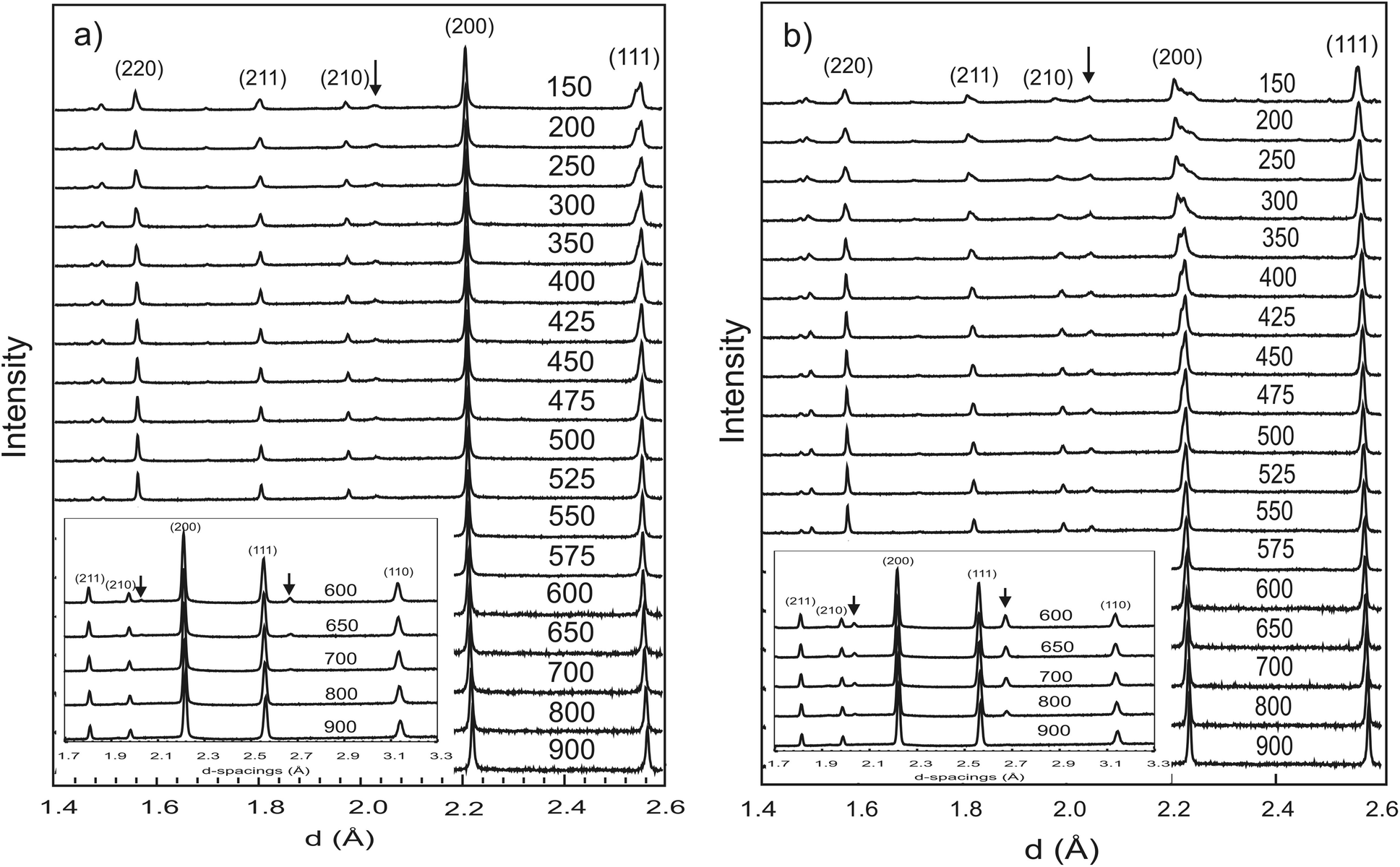 | ||
| Fig. 1 Neutron diffraction patterns from the high-resolution backscattering detector of HRPD collected in dry O2 flow (a) and in O2 + D2O(g) flow (b) from 900 to 150 °C. Pseudo-cubic Miller indices are labelled. The insets show patterns from the 90° detector bank where new peaks marked with arrows appear linked to the Rc structure. | ||
 | ||
| Fig. 2 Analysis of the FWHM of the intensity in the region of the cubic (200) reflection in O2 and O2 + D2O(g) atmospheres. Inset shows a close-up of the dry O2 behaviour revealing broadening linked to the appearance of the Imma phase at T < 300 °C. | ||
In the latter stages of the Rietveld analyses the oxygen site occupancy was refined, giving values of ∼0.96(1) for the Rc phase, consistent with the expected level of oxygen vacancies in BaCe0.8Y0.2O2.9. The Imma structure has two non-equivalent oxygen sites and the occupancy of the sites were probed for the 150 °C data. These refinements revealed a minor improvement in fit on reducing the oxygen site occupancies, and suggested a slight preference for the location of the oxygen vacancy at the O(1) 4e position. The refined structure models at 900 °C (cubic), 600 °C and 300 °C (rhombohedral) and 150 °C (orthorhombic) are given in Table 1. In Fig. 3a Rietveld fit to the 90° detector bank data in the Rc symmetry at 700 °C is shown.
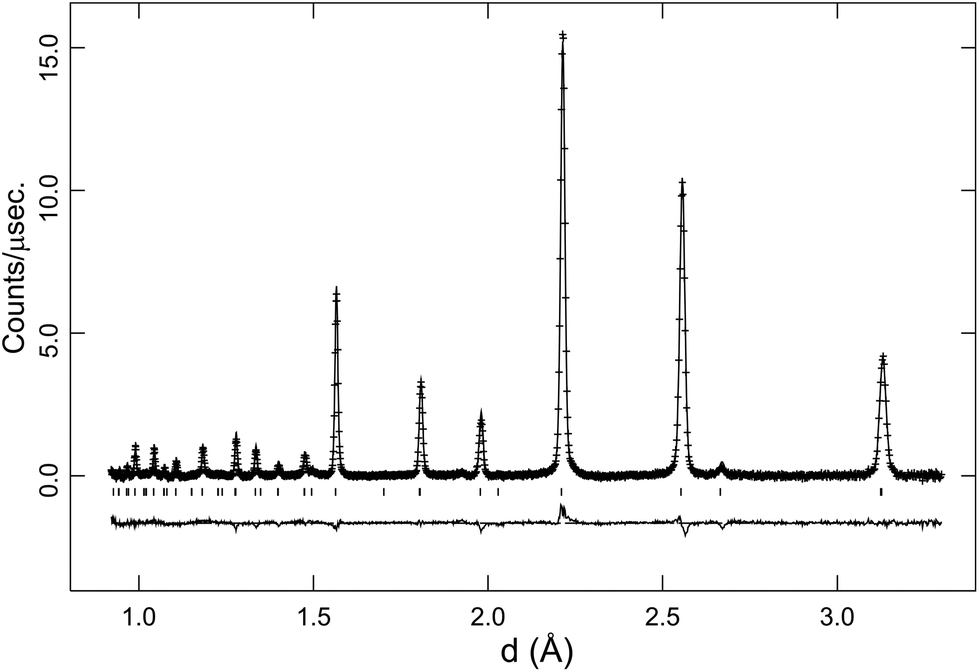 | ||
| Fig. 3 Rietveld fit to the 90° angle detector bank data for BaCe0.8Y0.2O3−δ at 700 °C in O2. Crosses are observed data, lines are the calculated profile and difference plot. Tick marks indicate reflection positions for the Rc structural model. | ||
|
Pmm, 900 °C |
Rc, 600 °C |
Rc, 300 °C |
Imma, 150 °C | |
|---|---|---|---|---|
| a (Å) | 4.4359(3) | 6.2538(2) | 6.2467(6) | 6.2192(2) |
| b (Å) | 4.4359(3) | 6.2538(2) | 6.2467(6) | 8.8038(2) |
| c (Å) | 4.4359(3) | 15.3075(8) | 15.2401(3) | 6.2522(2) |
| Vol. (Å3) | 87.29(1) | 518.47(4) | 515.00(1) | 342.32(2) |
| Ba | 1b | 6b | 6b | 4e |
| x | 0.5 | 0 | 0 | 0 |
| y | 0.5 | 0 | 0 | 0.25 |
| z | 0.5 | 0.25 | 0.25 | 1.0010(7) |
| Uiso (×100 Å2) | 4.88(5) | 3.86(4) | 2.86(4) | 2.41(4) |
| Ce/Y | 1a | 6a | 6a | 4b |
| x | 0 | 0 | 0 | 0 |
| y | 0 | 0 | 0 | 0 |
| z | 0 | 0 | 0 | 0.5 |
| U iso (x 100 Å2) | 3.03(4) | 2.51(4) | 1.99(4) | 1.95(4) |
| O(1) | 3d | 18e | 18e | 4e |
| x | 0 | 0.4714(3) | 0.4509(2) | 0 |
| y | 0.5 | 0 | 0 | 0.25 |
| z | 0 | 0.25 | 0.25 | 0.4328(4) |
| U iso (×100 Å2) | 9.78a | 8.42a | 6.00a | 3.07a |
| Occup. n | 0.957(3) | 0.960(3) | 0.966(3) | 0.90(1) |
| O(2) | 8g | |||
| x | 0.25 | |||
| y | 0.5296(3) | |||
| z | 0.25 | |||
| U iso (×100 Å2) | 6.70b | |||
| Occup. n | 1.0 | |||
| R wp (%) | 2.81 | 3.07 | 3.19 | 2.79 |
| R p (%) | 2.79 | 3.02 | 2.65 | 2.50 |
| χ 2 | 1.553 | 1.835 | 7.780 | 11.25 |
| U 11 | 12.8(7)a | 9.0(3)a | 6.0(6)a | 6.8(4)a, 7.2(2)b |
| U 22 | 3.8(7)a | 8.3(4)a | 5.7(2)a | 1.7(3)a, 3.9(2)b |
| U 33 | 12.8(7)a | 7.9(4)a | 6.2(1)a | 0.7(1)a, 8.9(2)b |
| U 13 | 0a, 6.1(2)b | |||
c structure at d ∼ 2.7 Å is just visible in the first data set collected at 900 °C. The intensity of the superlattice peaks grows rapidly in the pattern collected at 800 °C (see inset). Refinements with either cubic Pmm or Rc models produced satisfactory fits, of comparable quality, to the 900 °C data. However, the presence of the super-lattice peak that was fitted via the stable refinement of a displaced oxygen site (0.489(1), 0, 0.25), confirm that the structure is most accurately described using the Rc structural model. In fact, based on the temperature dependence of the antiphase octahedral tilt angle extracted from the refined oxygen position following31 (see Fig. 4), the Pmm to Rc transition is expected to occur at approx. 925 °C.
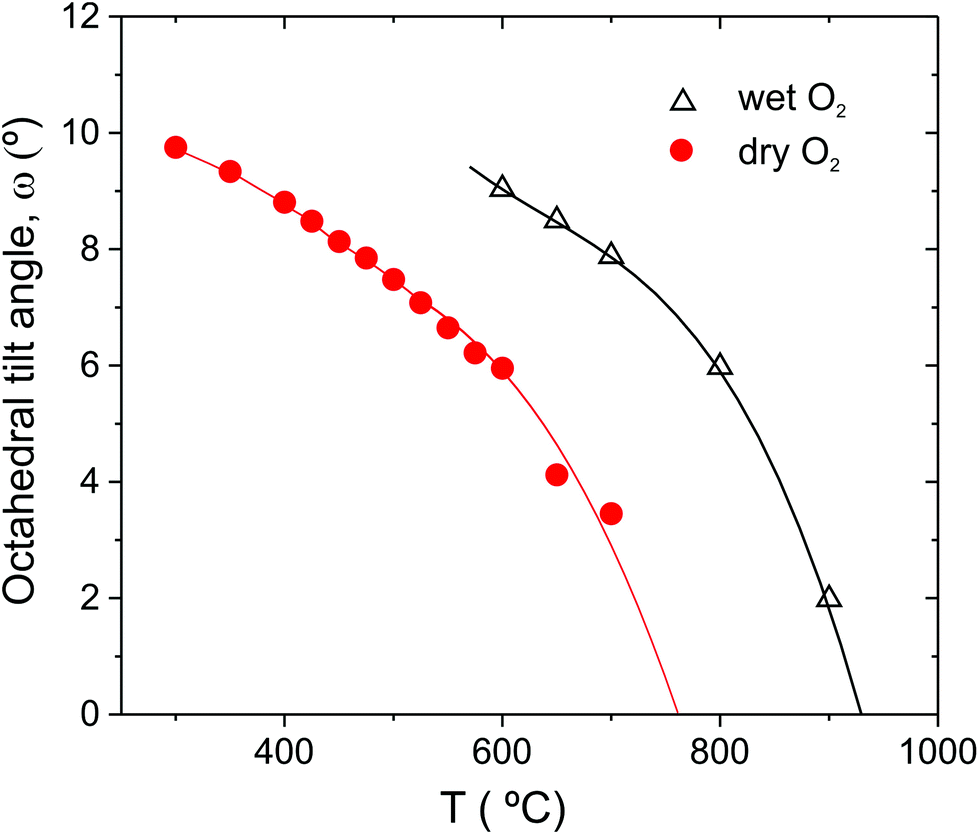 | ||
| Fig. 4 Antiphase octahedral tilt angle of BCY20 extracted from Rc refinements of the data collected in dry O2 and O2 + D2O(g). Lines are included as guides to the eye only. | ||
The behaviour of the FWHM of the pseudo-cubic (200) peak in O2 + D2O(g) is also shown in Fig. 2, and it allows two structural phase transitions to be discerned. The FWHM started to increase more rapidly at T ≈ 650 °C indicating the onset of the Rc to Imma transition. The broadening of this intensity was more pronounced in the wet oxygen data in comparison to the dry oxygen scans (Fig. 2) and a clear shoulder was distinguishable in the high-resolution data for T ≤ 500 °C. On further cooling, the profile broadening increased more sharply and the changing peak structure is clearly apparent in Fig. 1b for T < 400 °C. This marked the second phase transition with new intensity emerging consistent with the growth of a monoclinic I2/m phase. In order to successfully fit the data recorded at T ≤ 400 °C a two phase approach was required. As expected the amount of the I2/m phase was found to increase with decreasing T. The best fit achieved to the 150 °C data set (backscattering data) is shown in Fig. 5, obtained with refined weight percentage of 38.8(2)% and 61.2(2)% for the Imma and I2/m phases, respectively. These refinements were often unstable, displaying a high level of correlation between the peak shape parameters, phase fractions and atomic parameters, reflecting the high degree of peak overlap at this ferroelastic transition. Due to the two phase nature of the sample attempts to include deuteron sites were not successful. The failure to model the intensities shown in Fig. 5 to a greater extent is believed to originate, at least partly, from the absence of deuteron sites in the structural models as discussed further below. The I2/m structure corresponds to a further lowering of symmetry as the magnitude of the tilts along the y and z directions become dissimilar, a transition denoted a0b−b− (Imma)→a0b−c− (I2/m).
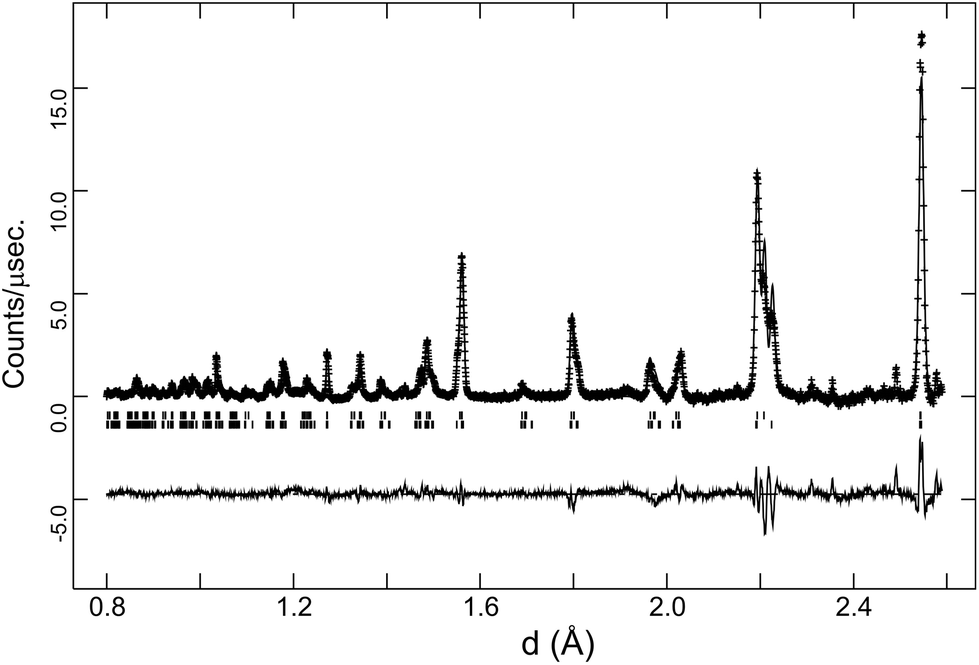 | ||
| Fig. 5 Rietveld fit to the high-resolution data collected at 150 °C in O2 + D2O(g) for BaCe0.8Y0.2O3−δ. Crosses are observed data, lines are the calculated profile and difference plot. Upper and lower tick marks indicate reflection positions for the Imma and I2/m structural models respectively. | ||
In addition to the growth of the monoclinic phase, the data sets collected at T < 300 °C revealed a number of minor reflections (<5%) attributed to the presence of impurity phases. These phases vanished upon reheating. The analysis of the data collected on cooling from 900 °C in 5% H2/95% Ar + D2O(g), revealed a basically identical thermal evolution of the structures as observed for the wet O2 scans. Therefore we do not present details of the refined structural parameters as no significant differences were apparent. The refined pseudo-cubic cell parameters obtained in O2 and O2 + D2O gas flows are plotted in Fig. 6 and refined structural parameters at selected temperatures are found in Table 2. The normalised cell volume dependence in the three different atmospheres is shown in Fig. 7.
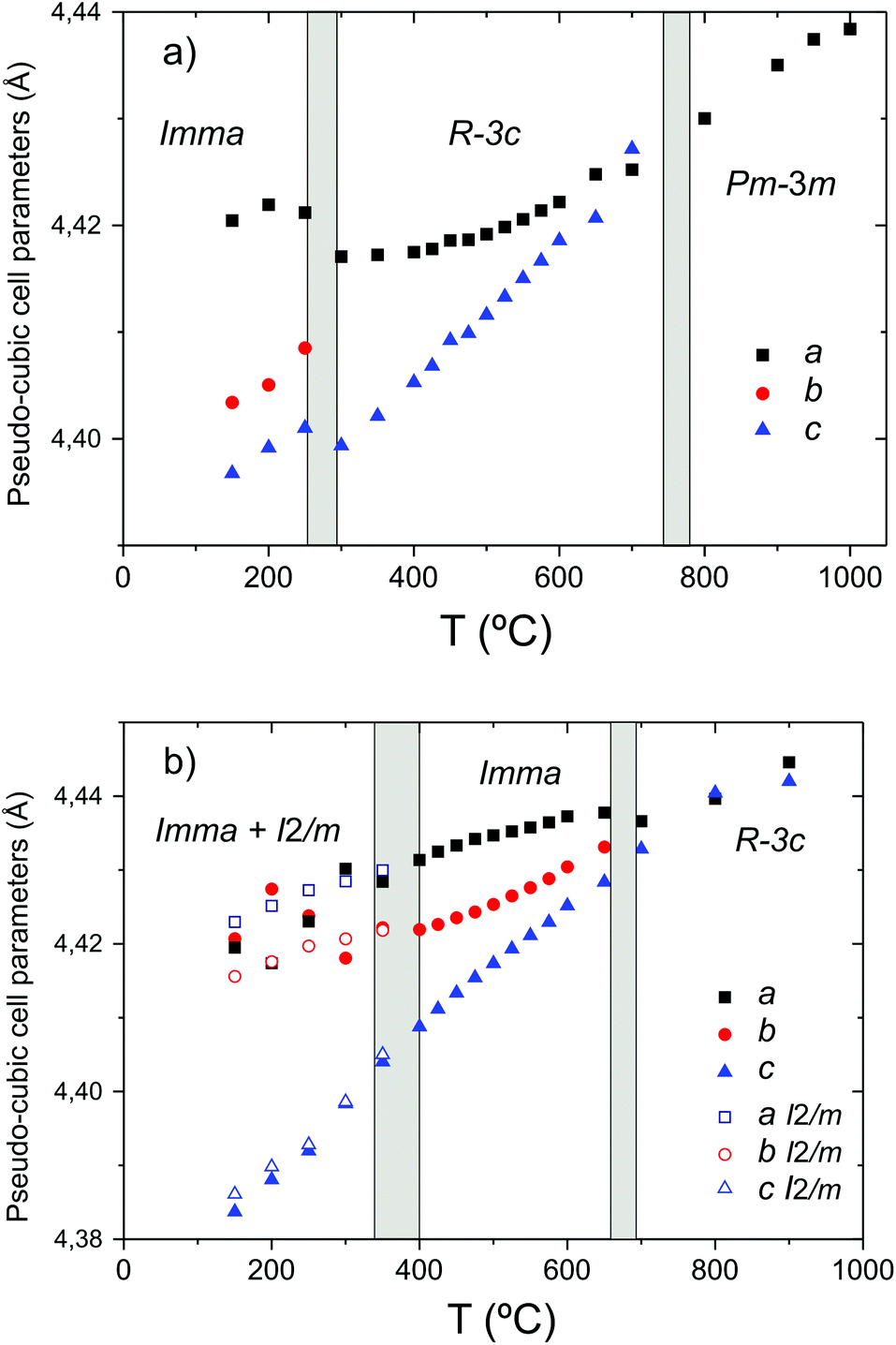 | ||
| Fig. 6 Phase stability fields and pseudo-cubic lattice parameters of BaCe0.8Y0.2O3−δ in (a) dry O2 and (b) O2 + D2O(g). | ||
 | ||
| Fig. 7 The pseudo-cubic cell volume of BaCe0.8Y0.2O3−δ in O2, O2 + D2O(g) and 5% H2/95% Ar + D2O(g). | ||
|
Rc, 900 °C |
Rc, 700 °C |
Imma, 450 °C | |
|---|---|---|---|
| a D site occupancy set to give 0.2 moles of D consistent with full hydration. b U iso not refined. | |||
| a (Å) | 6.2860(2) | 6.2735(1) | 6.2559(1) |
| b (Å) | 6.2860(2) | 6.2735(1) | 8.8265(1) |
| c (Å) | 15.386(1) | 15.3597(8) | 6.2692(1) |
| Vol./Å3 | 526.51(4) | 523.52(3) | 346.17(1) |
| Ba | 6b | 6b | 4e |
| x | 0.0 | 0 | 0 |
| y | 0.0 | 0 | 0.25 |
| z | 0.25 | 0.25 | 0.003(8) |
| U iso (×100 Å2) | 5.03(5) | 4.24(5) | 2.87(3) |
| Ce/Y | 6a | 6a | 4b |
| x | 0 | 0 | 0 |
| y | 0 | 0 | 0 |
| z | 0 | 0 | 0.5 |
| U iso (×100 Å2) | 3.63(4) | 3.10(5) | 2.65(3) |
| O(1) | 18e | 18e | 4e |
| x | 0.489(1) | 0.4590(3) | 0 |
| y | 0 | 0 | 0.25 |
| z | 0.25 | 0.25 | 0.4339(4) |
| U iso (×100 Å2) | 8.66c | 7.35c | 4.68c |
| Occup. n | 1 | 1 | 1 |
| O(2) | — | — | 8g |
| x | — | — | 0.25 |
| y | — | — | 0.5339(2) |
| z | — | — | 0.25 |
| U iso (×100 Å2) | 6.06d | ||
| Occup. n | 1 | ||
| D | 36f | 16j | |
| x | — | 0.606(4) | 0.235(9) |
| y | — | 0.147(4) | 0.054(2) |
| z | — | 0.244(2) | 0.292(7) |
| U iso (×100 Å2) | 2.34(2) | 4.0b | |
| Occup. n | 0.034b | 0.05a | |
| R wp (%) | 2.82 | 2.86 | 2.51 |
| R p (%) | 2.78 | 2.95 | 2.34 |
| χ 2 | 1.57 | 1.76 | 4.83 |
| U 11 | 13.1(4)c | 7.2(1)c | 9.4(3)c, 9.2(3)d |
| U 22 | 2.6(6)c | 2.2(2)c | 1.9(2)c, 4.3(1)d |
| U 33 | 10.2(7)c | 12.6(4)c | 2.8(2)c, 4.7(2)d |
| U 12 | 1.3(1)c | 1.1(1)c | 0c,0d |
| U 13 | −0.9(1)c | −1.64(6)c | 0c, 4.1(2)d |
| U 23 | −1.8(3)c | −3.2(1)c | 0c, 0d |
3.2. Structural analysis – location of deuteron sites
A feature of the Rietveld analyses of the data collected in D2O containing atmosphere at elevated T was the fact that a structural model based on the same polymorph, Rc or Imma, obtained from the dry gas oxygen runs did not adequately reproduce the data. This is illustrated in Fig. 8a, that shows significant discrepancies between the observed diffraction profile collected in wet O2 at 700 °C, and the calculated intensities based on a non-hydrated, Rc structure. In the hydrated state, the filling of oxygen vacancies alters the chemical composition from BaCe0.8Y0.2O3−δ to BaCe0.8Y0.2O2.8(OD)0.2 assuming that all vacancies are filled, and both the presence of additional oxygen ions, and the deuterons, can now contribute to the structure factors determining the intensity of the diffraction peaks. Initial refinement of the oxygen site occupancy factor, n, revealed a tendency for the site to favour full occupancy, (in some cases n > 1.0 was obtained) leading to improvements in the fit. However, the simulated intensities were still unsatisfactory and therefore Fourier difference maps were calculated in order to locate missing nuclear scattering density. This is an approach widely used to locate D+/H+ ions in proton conductors.32–34Fig. 9 shows a slice of the ab-plane of the Fourier difference map calculated based on the Rc structure for the 700 °C data. Note that many peaks are present in the plot and most can be attributed to spurious artefacts originating from either noise or mis-fitting of the major intensities. The missing positive nuclear scattering density at the site (0.62, 0.15, 0.25) close to the oxygen position, which corresponds to the 36f Wyckoff site in Rc, was found to give a sensible O–D distance of ∼1 Å. A deuteron atom was therefore introduced with its occupancy set to give the expected stoichiometry of a fully hydrated phase. This site refined stably producing a decrease in the Rwp agreement parameter from 3.02 to 2.86%. In the final stages the isotropic ADP of the D site was refined independently. The fit achieved to the 90° detector bank data with the complete structural model, including deuterons, is shown in Fig. 8b. The improvement in agreement between calculated and observed intensity, particularly for the high d-spacing peaks, is clearly visible.
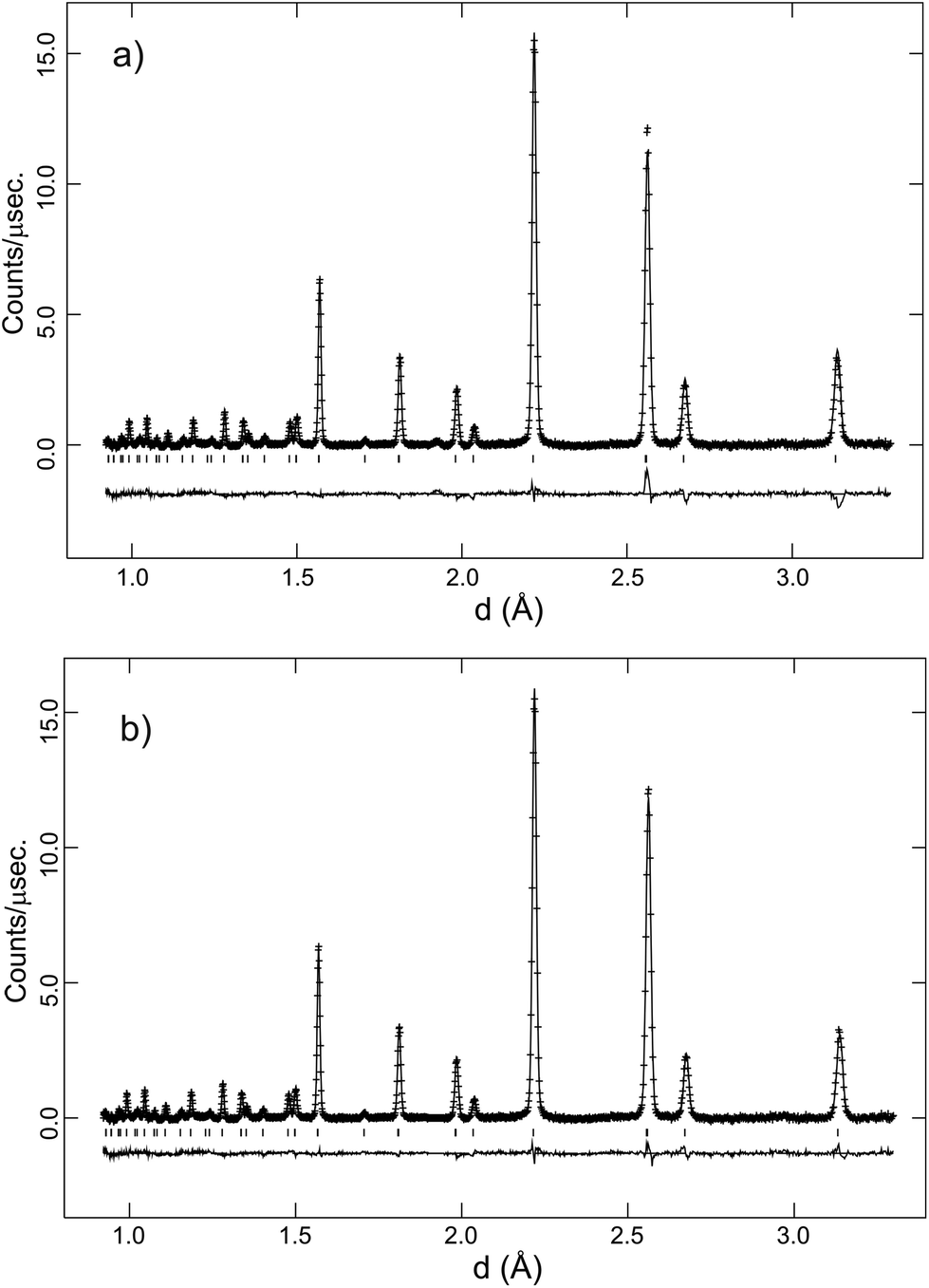 | ||
| Fig. 8 (a) Rietveld fit achieved, with no deuteron position defined, to the 700 °C, O2 + D2O(g) data for BaCe0.8Y0.2O3−δ. (b) Rietveld fit achieved with the deuteron position defined at the 36f site to the same data. Crosses are observed data, lines are the calculated profile and difference plot. Tick marks indicate reflection positions for the Rc structural model. | ||
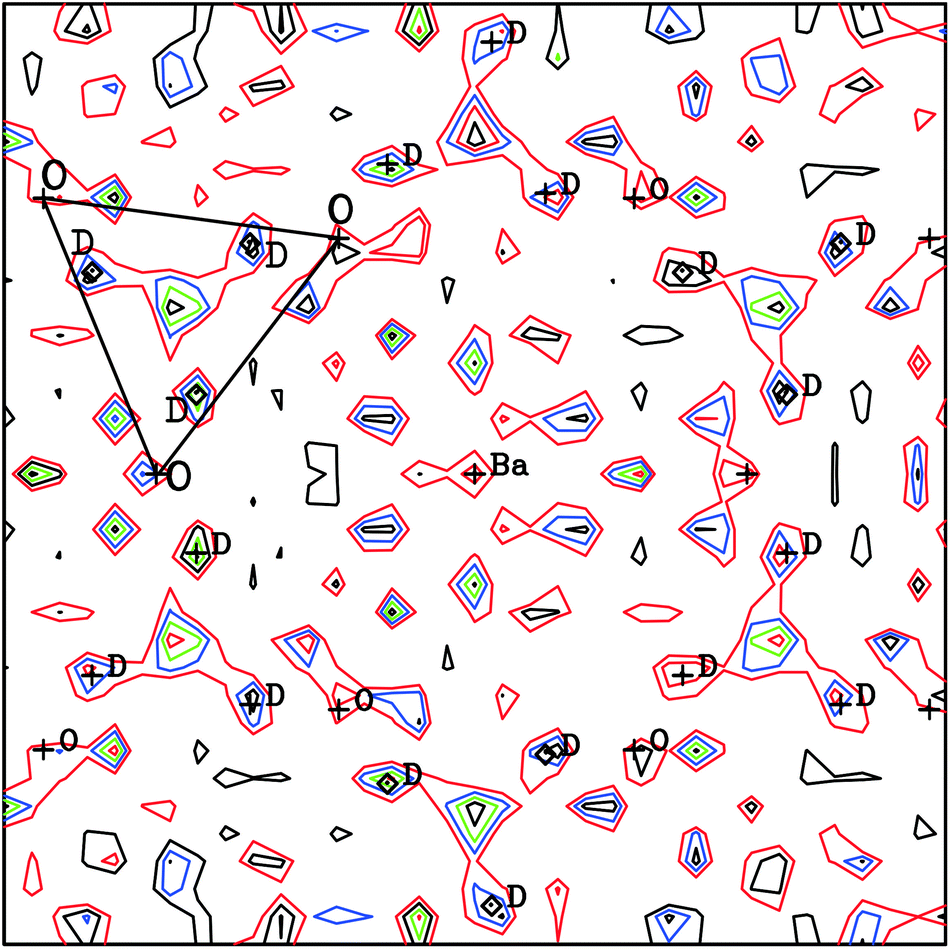 | ||
| Fig. 9 Fourier difference map in the ab-plane calculated for a Rc model without defining a deuteron site for the 700 °C in O2 + D2O(g). Missing positive scattering density attributed to D close to O is labelled. One face of a oxygen octahedra is also drawn. Contours are drawn at 0.0273 (red), 0.0547 (blue), 0.0820 (green) and 0.124 (black) fm Å−3. | ||
A similar analysis was performed for the data set at 450 °C in which the Imma structure is favoured. The Fourier difference maps suggested missing scattering density positioned at a 16j site (0.23, 0.05, 0.25) close to the O(2) oxygen. Introduction of a D site at the position refined stably and produced a reduction in the χ2 goodness of fit parameter from 5.6 to 4.8. The details of the refined structural parameters at 450 °C, including the deuteron site, are presented in Table 2.
As noted above, independent refinement of the oxygen site(s) occupancy factor for the analyses of the wet gas data favoured complete filling, indicative of full hydration of the vacancies for all temperatures studied. For all analyses (Rc or Imma) free refinements of the D site occupancy, with the isotropic ADP fixed, also gave values consistent with the expected stoichiometry of 0.2 moles of D. These observations may be serendipitous, given the high degree of coupling between ADP and site occupancy. However, taken together and considering the marked lattice expansion observed for the 900 °C data sets collected in wet gas (Fig. 7), it seems likely that the material was full hydrated throughout the temperature window of the wet gas runs.
4. Discussion
Table 3 below provides a complete summary of the available data with respect to BCY20 samples collected in situ as a function T, and in some cases under controlled pH2O (pD2O). Also, included is the accepted thermal behaviour of BaCeO3 for which little or no functional dependence based on the atmosphere is expected due to the absence of acceptor doped oxygen vacancies. The data reveal a level of consistency with respect to crystal structures but a wide spread of phase transition temperatures, reflecting the particular balance between the radiation (X-ray or neutron), instrument resolution and atmospheric conditions. Some important general points of consensus though are clear:(i) In the dry state, acceptor doping with 20% of yttrium suppresses the temperature of the Pmm to Rc phase transition considerably in comparison to BaCeO3.
(ii) Measuring in enhanced pH2O atmospheres conversely leads to large shifts of the Pmm to Rc phase transition and of the Rc to Imma (orthorhombic) transition to higher T.
(iii) The presence of the monoclinic I2/m structure at T < 400 °C is unequivocally linked to hydration of the material.
| Material | Method | Experimental conditions | Phase behaviour | Ref. |
|---|---|---|---|---|
|
a The presence of the I2/m monoclinic phase at RT indicates partial hydration.
b
Imma phase also present at RT.
c
Rc phase also present at 800 °C.
d Authors suggest Imma as symmetry of possible orthorhombic intermediate phase.
|
||||
| BaCeO3 | High-resolution NPD (HRPD @ ISIS) | Heating in vacuum (5 × 10−4 mbar) in vanadium can | Pnma→Imma at 290 °C | Knight14 |
|
Imma→Rc at 400 °C |
||||
|
Rc→Pmm at 900 °C |
||||
| BaCe0.8Y0.2O3−δ | High-resolution NPD (HRPD @ ISIS) | O2 flow in silica tube (cooling from 1000 °C) |
Pmm→Rc at ∼775 °C |
This study |
|
Rc→Imma at ∼275 °C |
||||
| O2 or 5% H2 in Ar + pD2O ∼ 0.2 atm flow (cooling from 900 °C) |
Rc→Imma at ∼675 °C |
|||
| Imma→Imma + I2/m at ∼375 °C | ||||
| BaCe0.8Y0.2O3−δ | Low resolution NPD (QENS at IPNS) | Air + pH2O ∼0.02 atm flow in silica tube (heating) |
Rc→Pmm at ∼700 °C |
Malikova et al.35 |
| Air flow (cooling after dehydration) |
Pmm→Rc ∼540 °C |
|||
| BaCe0.8Y0.2O3−δ | Medium resolution NPD (D1A at ILL) | Static air in closed silica tube (heating) | I2/m→Imma at 500 °Ca | Malavasi et al.16 |
|
Imma→Rc at 600 °Cb |
||||
|
Rc→Pmm at 800 °Cc |
||||
| BaCe0.8Y0.2O3−δ | PXRD (Cu Kα) | O2 or Ar (cooling from 1000 °C) |
Pmm→Rc at 500–450 °C |
Han et al.18 |
| O2 + pH2O ∼ 0.031 atm (cooling from 1000 °C) |
Pmm→orthorhombicd at 600 °C |
|||
| Orthorhombic→I2/m at 450–400 °C | ||||
| BaCe0.8Y0.2O3−δ | PXRD (Cu Kα) | Heating pre-hydrated sample in synthetic air | I2/m→Imma at ∼360 °C | Andersson et al.19 |
|
Imma→Rc at ∼480 °C |
||||
|
Rc→Pmm at ∼600 °C |
||||
| Cooling under low vacuum (1 × 10−1 mbar) |
Pmm→Rc at ∼475 °C |
|||
|
Rc→Imma at ∼370 °C |
||||
Based on the comparison in Table 3 a clear tendency for results based on PXRD to underestimate the temperature for the Pmm to Rc transition is also clear, and this is trivially assigned to the relative insensitivity of X-rays to the oxygen positions. In what follows below we discuss the three main observations, with a particular emphasis on our present high-resolution neutron diffraction data that provides the most accurate description of the material's thermal and atmospheric dependence so far.
4.1. Role of yttrium dopant and oxygen vacancies on the crystal structure in the dry state
As a precursor to understanding the complex thermal phase stability dependence of BCY20 it is valuable to establish the origin of the octahedral tilting in perovskites, such as BaCeO3, with Goldschmidt tolerance factors, t = (RA + RO)/√2(RB + RO),10 less than 1, is competition between the bonding preferences of the B–O and A–O sub-lattices. In particular, for t < 1, the B–O bonding is under compressive and the A–O environment under tensile stress if the cubic structure is retained, and tilting of the BO6 octahedra, typically as semi-rigid units, is a way to satisfy the competing bonding requirements. Due to the 3-dimensional connectivity of the structure this produces long range ordered, cooperative tilts propagating either in-phase or out of phase with each other. The exact ordering scheme is dependent on the interplay of several factors, such as the relative size of the cations and degree of covalency within the bonding. In addition external parameters, e.g. temperature and pressure, often exert a strong influence.It is also important to realise that the structural impact of replacing Ce4+ (0.87 Å36) by 20% Y3+ (0.90 Å36) and creating oxygen deficiency, is reflected in the RT structures of the materials. The undoped parent phase adopts a Pnma structure at T < 390 °C, with a 3-tilt, a+b−b− scheme, whereas we expect BCY20 to adopt the Imma a0b−b− (2-tilt system) structure in dry ambient conditions. As noted our sample was found to be hygroscopic and this complicated the RT comparison, however our high-resolution neutron data under oxygen show the Imma structure is stable at 150 °C. We expect no further transition on cooling to RT based on the PXRD results that show Imma symmetry is obtained at RT after cooling a pre-hydrated sample from 900 °C in low vacuum.19 A conclusion further supported by the results of Takeuchi et al.17 who report the Rc structure at RT for samples cooled from 800 °C in dry oxygen based on moderate resolution NPD. Given that our high-resolution neutron data was used to discriminate between the Rc and Imma structures we judge that the sample studied by Takeuchi et al. was, in all likelihood, Imma.
The assignment of the Imma structure for our BCY20 sample in the dry state was supported by the peak broadening of the pseudo-cubic (200) intensity. In the original high-resolution neutron study of BaCeO3 the splitting of this peak is easily resolved using the HRPD instrument.14 The greater peak widths observed in the present study are ascribed to the presence of 20% yttrium at the B-site contributing to high levels of strain in the crystallites. An increase in peak widths with dopant concentration was observed previously for the BaCe1−xNdxO3−δ series37 using HRPD, and this factor may also contribute to the common use of the Rc structure for the dry state of BCY20 (Table 3). Of further relevance is that substitution of the larger Y3+ ion at the B-site does not produce the expected expansion of the cell volume for BaCe1−xYxO3−δ.17 Similar behaviour has been reported for BaCe1−xNdxO3−δ (Nd3+ = 0.983 Å) with the cell volume decreasing for x ≤ 0.12 before increasing and surpassing that of the undoped phase at x ≥ 0.16.37 An unexpected contraction of the cell volume was also observed for BaPr0.9Y0.1O3−δ in comparison to BaPrO3.29 Structural relaxation around the oxygen vacancy was suggested to account for this behaviour29 and this seems likely to be the main cause for the dependence of the cell volume of acceptor doped BaCeO3.
On heating to 290 °C BaCeO3 transforms to Imma symmetry,14 and BaCeO3 and BCY20 both then follow the same crystallographic sequence, Imma to Rc to Pmm, on further heating but the stability fields of the Imma and Rc structures are strongly suppressed to lower T by the yttrium substitution (Table 3). Based on the present, high-resolution analysis the temperature for the Rc→Imma transition decreases from 400 °C to 300 °C and that of the Pmm→Rc conversion drops from 900 °C to 775 °C. In summary, doping and the accompanying vacancies, act to strongly suppress the tendency for tilting of the octahedral (Ce/Y)O6−δ units as reflected in both the RT crystal structures of BaCeO3 and BCY20, and the relative phase stability fields observed at higher temperatures. We therefore propose that significant structural relaxation around the oxygen vacancy functions as an alternative mechanism to alleviate the competing bond strain, in effect reducing the driving force for octahedral tilting in comparison to BaCeO3. This is the key factor responsible for extending the stability of the cubic aristotype to lower temperature in BaCe0.8Y0.2O3−δ.
The propensity for oxygen vacancies to lower the tilt induced transition temperatures from cubic to rhombohderal (Rc) was recently observed for La1−xSrxCoO3−δ (x = 0.3 and 0.4) and La1−xSrxFeO3−δ (x = 0.3, 0.4 and 0.5) perovskites.38 For example a suppression of the phase transition by ∼125 °C for a more reduced form of La0.7Sr0.3CoO3−δ was noted. While there is no indication of change in oxygen content in BCY20, with concomitant reduction of Ce4+ to Ce3+, Co4+ is easily reduced to Co3+ in La1−xSrxCoO3−δ. The reduction of Co4+ to Co3+ reduces the average tolerance factor, which in itself should favour the tilted Rc structure over the Pmm structure. This serves to reinforce the conclusion that the structural impact of oxygen vacancies on octahedral tilting in perovskites is more important than arguments based on the tolerance factor.
Finally, in the above we have focused mainly on the role of the oxygen vacancy. It may, however, be more accurate to consider the structural relaxation around dopant-vacancy pairs, or even extended Y–VO–Y defects, remembering that two dopants ions are required per oxygen vacancy and that a strong associative attraction between the positively charged vacancies and the acceptor dopants is expected from computational studies, see for example refs.39,40 Experimental evidence for the vacancy being close to the dopant ion is available for yttrium doped BaZrO341 and for BaZr1−xScxO3−δ.42
4.2. Comparison of phase stability in hydrated BaCe0.8Y0.2O3−δ and BaCeO3
Having identified structural relaxation around oxygen vacancies as being critical for rationalising the dry state behaviour we now turn our attention to the structural dependence of BCY20 in hydrated form, bearing in mind the hydration reaction, eqn (1), replaces a vacancy and a lattice oxide ion with two [OH] defects. For the hydrated system, the phase transitions revert to much higher temperatures than seen for dry BCY20, and in fact our present data, collected at a relatively high p(D2O), indicate both the Pmm to Rc and, particularly the Rc to Imma phase transitions now occur at temperatures above those of BaCeO3 (Table 3). The transition to Imma for BCY20 is at ∼675 °C in wet gas (Fig. 6b) and at 400 °C for BaCeO3. Based mainly on the cell volume data (Fig. 7) that shows a significant expansion of the cell for the 900 °C data in wet condition we infer that BCY20 is hydrated at 900 °C when such high water partial pressures are used. Furthermore, the smooth temperature dependence of the cell volume indicates that the hydration level is constant on cooling and we conclude the sample is basically fully hydrated from 900 to 150 °C. Therefore, to a first approximation, the differing phase stability fields of the hydrated material can be explained by the filling of all, or at least the vast majority, of oxygen vacancies. The concomitant removal of the vacancy induced relaxation leads to a restoration of the phase transition to temperatures more in line with those observed for the undoped material.
The present observation that BCY20 is hydrated at 900 °C is a noteworthy finding attributed to the high p(D2O) used in the study. Most proton conducting perovskites dehydrate at T ≤ 600 °C under the usual experimental conditions of ∼0.02 pH2O atm. In a rare study the behaviour of BCY10 in the pH2O range 0.02 to 0.3 atm was recently reported.43 Based on thermodynamic parameters extracted from modelling of conductivity data it was demonstrated that increasing pH2O increases proton conductivity and suppresses hole/oxide ion conductivity. Further, it was estimated that at a pH2O of 0.3 atm. approx. 70% of the vacancies are occupied by [OH] at 800 °C, and that protons become the dominant charge carriers at T = 700 °C in a pH2O of 0.1 atm. It is worth noting that BaCe1−xYxO3−δ systems in general have high hydration enthalpies, and the higher hydration enthalpy of BCY20 over BCY10,44 will favour retention of protons to higher temperatures, i.e., the equilibrium of the hydration reaction eqn (1), will be shifted further to the right. This lends support to our conclusion that full hydration of the material at 900 °C is possible. Further studies of the (de-)hydration characteristics of BCY20 at elevated pH2O are needed, but we note that in terms of practical applications these conditions are of most relevance for solid oxide electrolysers that operate at much higher pH2O than used for protonic ceramic fuel cells.
The second point to consider further is why the phase stability of the Imma structure is so sensitive to the hydration state of the material, and in particular why the transition temperature of the hydrated BCY20 phase is so much higher (∼275 °C) than that observed for BaCeO3. We note that Cordero et al. have previously observed a significant but smaller shift to higher T of approx. 75 °C for the Imma transition of hydrated BaCe0.9Y0.1O3−δ in comparison to BaCeO3 based on anaelastic spectroscopy measurements.45 This provides support for the current findings and suggests a dependence on the dopant level and, more importantly we will argue, the associated concentration of protonic defects. The sequence of transitions Pmm→Rc (a−a−a−)→Imma (a0b−b−)→Pnma (a+b−b) is rare among perovskites, only BaCeO3 and BaPrO3 display this route, which involves the loss of a tilt on cooling at the Rc→Imma transition. The appearance of the Imma phase becomes more understandable by considering it as an intermediate in the conversion of anti-phase (a−a−a−) to an in-phase tilt (a+b−b−). We hypothesise that within the Rc (a−a−a−) structure increased anti-phase tilting driven by the reduction of temperature, is only stable up to a certain threshold level. Increasing the tilt angle, ω, means that the oxygen ions from neighbouring octahedra get closer and closer, which becomes increasingly unfavourable. In the limit ω = 30°, the octahedra must change from corner to edge sharing.46 In such a scenario, the threshold tilt value is reached at a much higher temperature in the hydrated material as shown by the tilt angle temperature dependence in Fig. 4, and the Imma structure emerges.
Further, one can speculate as to whether the presence of protons, although highly mobile, can be acting to enhance the tendency of tilting either via hydrogen bonding interactions or changes in the local electrostatic environment. An additional interaction favouring tilting would explain the increased temperature of the Imma transition and also explain the marginally higher Pmm to Rc transition of hydrated BCY20 over BaCeO3. A tendency for hydration to increase the level of tilting in the Pnma structure of La0.6Ba0.4ScO2.8 has previously been noted based on data collected at 4.2 K,34 and several computational studies provide evidence that protonic defects induce local relaxations of the oxide lattice in perovskites.47,48
4.3. The monoclinic I2/m phase and the impact of proton mobility on crystal structure
We now consider the appearance of the monoclinic phase for hydrated BCY20 samples. Given the rectification of the Pmm to Rc phase transition temperature coupled with the appearance of the Imma phase at high T, a further transition to a Pnma structure (a+b−b− tilting) as observed for BaCeO3 may have been expected as the natural next step for hydrated BCY20. Instead, the hydrated material favours a transition to monoclinic I2/m symmetry as witnessed by the gradual growth of this phase below 400 °C apparent in the high-resolution data collected in D2O bearing gases. Takeuchi et al.17 were the first to identify this monoclinic phase for BaCe1−xYxO3−δ with x ≥ 0.2 and link it explicitly to the hydration level based on neutron data collected at RT. Han et al.18 and Andersson et al.19 also recently confirmed its presence for hydrated forms of BCY20 from in situ PXRD data. The material therefore stays as a two tilt system, but becomes more anisotropic as the magnitude of the antiphase tilts are different, a change that this is reflected by the presence of three crystallographically distinct oxygen sites. The fact that the I2/m (a0b−c−) structure is an extremely unusual tilt system provides further evidence for a strong structural directing effect of the presence of protons. PrAlO3 is the only example of I2/m symmetry known to the authors.49 The material undergoes a Imma to I2/m transition at 150 K, due to a Jahn–Teller distortion of the Pr3+ ion.49 Based on symmetry arguments the Imma to I2/m transition is expected to be continuous in nature.50 Under equilibrium conditions two phases separated by a second order phase transition cannot coexist. The very wide temperature range of this phase transition shows that chemical inhomogenities due to the slower kinetics of proton diffusion are most likely involved.
In summary then comparison between the structural dependence of BCY20 in dry and hydrated states leads us to conclude that the structural influence of protonic defects becomes more and more important on cooling, i.e., as the proton mobility decreases. This interpretation is strengthened from the study of BCY20 by Loong et al.51 that used a combined NPD and Quasielastic neutron scatting (QENS) approach. In their investigation a possible link between an increase in the hydrogen mean square displacement, indicative of greater proton mobility, and the onset of the disappearance of the monoclinic phase at approx. 360 °C based on data collected in moist air was reported. Such a connection between the mobility of the protons and the crystal structure is consistent with the present analysis in relation to the growing structural impact of protons on cooling. Given that proton dynamics are usually considered to be intimately linked to the oxygen ion dynamics2,52 the present study, that provides an unambiguous link between the presence of protonic defects and the global crystal symmetry at temperatures where peak proton conductivity occurs, is profound and highlights the importance of studying materials in situ and under realistic operating conditions.
Based on molecular dynamics simulations on BaCeO3 at elevated temperatures Münch et al.52 proposed the Ce–O bending mode as being critical in reducing the oxygen to oxygen separation and allowing the proton transfer to occur via the formation of a transient hydrogen bond. Recent in situ Raman spectroscopy data collected under wet air (0.03 atm H2O) for BaCe0.9Y0.1O3−δ42 shows the growth of O–H stretch bands in the region 2500–2900 cm−1 on cooling below 700 °C. These results were interpreted as evidence of transient, strongly hydrogen bonded protons and they strengthen the view that H-bonding interactions are primarily responsible for influencing the oxide lattice. For further insight in relation to the structural influence of hydration it is natural to consider the position of the deuterons in more detail.
4.4. Deuteron sites in BaCe0.8O2.8(OD)0.2
We begin by noting that our analysis is, to the best of our knowledge, the first time deuteron sites have been reported at such high temperatures for any perovskite proton conducting system. The only exception we are aware of is the study by Ito et al. of the D site at temperatures up to 200 °C (473 K), for a heavily substituted, cubic BaSn0.5In0.5O3−δ perovskite phase,32 and no great difference between the D site at 77 K and 473 K was observed. It is therefore worthwhile to assess the reliability of the sites we propose. Firstly, it is clear that the low D/H concentrations in proton conducting oxides make it challenging to confidently locate deuteron/proton positions and in general, the majority of previous studies have used cryogenic temperatures to freeze out the motion of the oxygen sub-lattice and help localise the protons. At elevated temperatures, the protons can be expected to access a wider range of stable sites. Nonetheless, assuming that the deuterons still spend the vast majority of their time in the lowest energy site(s), i.e., dwelling between either the proton transfer step (from one oxygen to another) or the rotation step (around a given oxygen), it is plausible that the scattering from the D will contribute to the time averaged diffraction intensity. Indeed, differences in the peak intensities recorded in dry oxygen and D2O containing atmospheres were apparent that reflect, primarily, the changed sample stoichiometry, and improvements to the Rietveld fits were obtained through refinement of the proposed D sites. However, the interpretation of powder diffraction data is not straightforward and other factors may also subtlety influence the fitting of observed intensities, ranging from the atomic thermal motion to microstructural factors such as strain broadening effects, both of which are affected, to some degree, by the hydration reaction. Further studies based on exploiting the contrast in scattering length of H and D are therefore suggested as a viable route to confirm or refute the proposed deuteron positions.The Rc and Imma structures, along with the possible deuteron sites obtained from the Rietveld analyses at 700 °C and 450 °C, are shown in Fig. 10 and 11. A common feature is that the D site is located along or close to the edge of the (Ce/Y)O6 octahedra. For the 36f site in the Rc structure, the deuterons occupy two, opposed faces, of the octahedral units and the refined O–D distance of 0.93(2) Å is close to that expected for a O–H bond. This site gives shortest cation to deuterons separations of ∼1.75 Å and 2.65 Å for the Ce/Y and Ba sites respectively. The former, in particular, is shorter than may have been expected based on repulsion between the positively charged deuteron and the B-site. For the Imma structure the refined O–D distance is 0.82(2) Å and the shortest (Ce/Y)–D and Ba–D interactions are 2.02 Å and 2.73 Å, respectively. A possible explanation for the shorter than expected O(2)–D distance lies with the anisotropic atomic displacement parameters (ADP) associated with the oxygen ions (Fig. 11). It is conceivable that the oxygen ion is located at the farthest extreme of the ADP on those occasions where the 16j position is simultaneously occupied. It should be recalled that we obtain the time averaged structure from our diffraction experiments and that the fraction of anion sites occupied by [OD] groups is small compared to the more commonly occurring oxide ion. In fact the oxygen displacement parameters for both structures are highly anisotropic, and as is usual for perovskites, display greater amplitude in the directions perpendicular to the B–O–B bonds than parallel to them.
 | ||
| Fig. 10 Possible deuteron positions (purple) for the Rc structure of BCY20 at 700 °C. | ||
 | ||
| Fig. 11 Possible deuteron site (purple) for the Imma structure of BCY20 at 450 °C. | ||
Previous neutron diffraction studies of BCY10 with the orthorhombic Pnma structure have suggested differing locations for possible H+ sites. Knight,23 suggested a site bonded to the basal O(2) oxygen for data recorded at 4.2 K whilst, Azad et al.24 report a position close to the apical O(1) from analysis of data collected in a wet 5% H2/Ar condition at RT. Our results for the Imma structure of hydrated BCY20 that is stable between ∼650–400 °C, indicate that the deuterons are bonded preferably to the O(2) sites. In a recent density functional theory (DFT) investigation of BaCe0.875Gd0.125O2.9375 the stability of four proton sites around the two distinct oxygen ions in Pnma symmetry was examined.40 The findings revealed a reasonably strong tendency for the proton to be found in the first coordination shell of the Gd dopant, and a smaller preference for the proton to bond to the apical O(1) rather than the basal O(2) oxygen, regardless of whether the B-site was occupied by Gd or Ce. Further calculations are needed to explore the energy landscape of the proton in the Imma and Rc phases identified here as being most relevant for proton transport in BCY20.
Based on our analysis there appears to be a reduction in the dimensionality of the proton environment at the Rc to Imma transition, with the deuterons localising in the ac-plane of the orthorhombic Imma structure. The next nearest oxygen is the O(1), that has a separation of 2.44 Å whilst the closest O(2) is found at 2.94 Å. The larger interatomic distances found to oxygen ions on neighbouring octahedra (>3.5 Å) indicate that inter-octahedral transfer is unlikely. Further cooling of the deuterated material stabilises the monoclinic phase and, as discussed above, proton localisation appears to play a role in this structural transition.
4.5. Hydration induced chemical expansion
Chemical expansion upon hydration can be detrimental to the mechanical stability of proton conducting ceramics. In our recent work we presented a defect model for BCY20 where the chemical expansion was modelled from the different effective ionic radii of O2−, OH− and VO.19 With increasing hydration the average anion radius increases, thus causing a unit cell expansion. An empirical approximation of this model was to set the VO to 1.18 Å in order to fit the experimentally observed unit cell volumes in dry and wet atmospheres. This implies that the effective radius of VO is smaller than the radius of OH− (1.35 Å), which in turn is smaller than O2− (1.38 Å).36 As hydration also converts O2− to OH−, OH− must be significantly larger than the effective radius of VO to give a net volume expansion. Attributing such an ionic “size” for the oxygen vacancy is consistent with the significant structural relaxation around the vacancy site discussed above. However, in reality neither the VO nor the OH− defect are spherical and assigning one single effective radius is thus a simplification. The effects should be more accurately described in terms of the individual strain tensors associated with the point defects that will produce anisotropic lattice distortions at a local level.An interesting observation from Fig. 7 is that the chemical expansion is strongly dependent on the crystal symmetry. At high temperature and higher symmetry the volume difference of the dry and wet BCY20 is fairly independent on temperature, while at low temperature and low symmetry the two volumes approach each other. The reason for this behaviour is the strongly non-linear temperature dependence of dry BCY20, while the volume of BCY20 wet is roughly linear with temperature. One final point to note is that the cell volume, and the overall structural dependence, i.e., sequence of phase transitions, obtained on cooling in wet 5% hydrogen containing gas flow was found to be very similar to that observed on cooling in wet oxygen. We therefore find no evidence of a structural response indicative of reduction of the Ce4+ ion under the present conditions. A minor reduction of Ce4+ has been reported to occur for samples of BaCe1−xYxO3−δ (0.5 ≤ x ≤ 0.15) heated in 5% H2/Ar + 0.03 atm H2O at T ≥ 800 °C based on TGA data.53
5. Conclusions
The powerful combination of high-resolution neutron diffraction data and in situ sample cells allowing the study of materials at realistic temperatures and atmospheres have here been used to study the structural dependence of the leading proton conducting perovskite BaCe0.8Y0.2O3−δ. Remarkable differences between the phase stability fields of the crystal structures are observed dependent on the degree of hydration of the material. The symmetry and magnitude of the octahedral tilting is strongly influenced by the degree of hydration; the phase transitions being depressed due to the presence of oxygen vacancies and the tendency for octahedral tilting is enhanced by protonic defects. This coupling between the proton and oxygen sub-lattice, most likely mediated via strong hydrogen bonding interactions, helps to shed new light on the factors governing the proton dynamics in the material. Finally, the neutron measurements performed at a relatively high pD2O indicate that protonic defects are stable in the material at 900 °C, suggesting future investigations in relation to the dominant charge carriers of BCY20 at elevated water partial pressures are justified.Acknowledgements
The authors thank the Science and Technology Facilities Council (STFC) for provision of neutron beam time at the ISIS neutron and muon facility. We are also grateful for the assistance of Kevin Knight and the ISIS technical staff during the experiments on the HRPD diffractometer. Financial support from the Climit (grant 17796/I30) at the research council of Norway and NTNU is also acknowledged.References
- H. Iwahara, Solid State Ionics, 1995, 77, 289–298 CrossRef CAS.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- T. Norby and Y. Larring, Current Opinion in Solid State and Materials Science, 1997, 2, 593–599 CrossRef CAS.
- T. Norby and R. Haugsrud, Nonporous Inorganic Membranes; Dense ceramic membranes for hydrogen separation, ed. E. b. A. F. S. a. M. V. Mundschau, WILEY-VCH Verlag, Weinheim, Germany, 2006 Search PubMed.
- T. Norby, Solid State Ionics, 1999, 125, 1 CrossRef CAS.
- K. D. Kreuer, Solid State Ionics, 1999, 125, 285–302 CrossRef CAS.
- K. H. Ryu and S. M. Haile, Solid State Ionics, 1999, 125, 355–367 CrossRef CAS.
- H. Iwahara, H. Uchida, K. Ono and K. Ogaki, J. Electrochem. Soc., 1988, 135, 529–533 CrossRef CAS PubMed.
- J. Guan, S. E. Dorris, U. Balachandran and M. Liu, J. Electrochem. Soc., 1998, 145, 1780–1786 CrossRef CAS PubMed.
- V. M. Goldschmidt, Str. Nor. Vidensk-Akad. Oslo, 1926, 1, 1 Search PubMed.
- A. V. Kuzmin, V. P. Gorelov, B. T. Melekh, M. Glerup and F. W. Poulsen, Solid State Ionics, 2003, 162–163, 13 CrossRef CAS.
- S. Loridant, L. Abello, E. Siebert and G. Lucazeau, Solid State Ionics, 1995, 78, 249 CrossRef CAS.
- F. Genet, S. Loridant, C. Ritter and G. Lucazeau, J. Phys. Chem. Solids, 1999, 60, 2009–2021 CrossRef CAS.
- K. S. Knight, Solid State Ionics, 1994, 74, 109–117 CrossRef CAS.
- K. S. Knight, Solid State Ionics, 2001, 145, 275–294 CrossRef CAS.
- L. Malavasi, C. Ritter and G. Chiodelli, Chem. Mater., 2008, 20, 2343–2351 CrossRef CAS.
- K. Takeuchi, C.-K. Loong, J. W. Richardson Jr., J. Guan, S. E. Dorris and U. Balachandran, Solid State Ionics, 2000, 138, 69–77 CrossRef.
- D. Han, M. Majimab and T. Uda, J. Solid State Chem., 2013, 205, 122–128 CrossRef CAS PubMed.
- A. K. E. Andersson, S. M. Selbach, C. S. Knee and T. Grande, J. Am. Ceram. Soc., 2014, 97, 2654–2661 CrossRef CAS PubMed.
- P. G. Sundell, M. E. Bjorketun and G. Wahnström, Phys. Rev. B: Condens. Matter, 2007, 76, 094301 CrossRef.
- M. S. Islam, R. A. Davies and J. D. Gale, Chem. Mater., 2001, 13, 2049–2055 CrossRef CAS.
- F. Shimojo, K. Hoshino and H. Okazaki, J. Phys. Soc. Jpn., 1998, 67, 2008–2011 CrossRef CAS.
- K. S. Knight, Solid State Ionics, 2000, 127, 43–48 CrossRef CAS.
- A. K. Azad, A. Kruth and J. T. S. Irvine, Int. J. Hydrogen Energy, 2014, 39, 12804–12811 CrossRef CAS PubMed.
- V. F. Sears, Neutron News, 1992, 3, 26–37 CrossRef.
- R. Haynes, S. T. Norberg, S. G. Eriksson, M. A. H. Chowdbury, C. M. Goodway, G. D. Howells, O. Kirichek and S. Hull, J. Phys.: Conf. Ser., 2010, 251, 012090 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS.
- A. C. Larson and R. B. von Dreele, General Structure Analysis System (GSAS) Los Alamos National Laboratory Report, LAUR, 1994, pp. 86–748.
- C. S. Knee, A. Magraso, T. Norby and R. I. Smith, J. Mater. Chem., 2009, 19, 3238–3247 RSC.
- A. M. Glazer, Acta Crystallogr., Sect. B: Struct. Sci., 1972, 28, 3384–3392 CrossRef CAS.
- C. J. Howard and B. J. Kennedy, J. Phys.: Condens. Matter, 1999, 11, 3229–3236 CrossRef CAS.
- T. Ito, T. Nagasaki, K. Iwasaki, M. Yoshino, T. Matsui, H. Fukazawa, N. Igawa and Y. Ishii, Solid State Ionics, 2007, 178, 607–613 CrossRef CAS PubMed.
- I. Ahmed, C. S. Knee, M. Karlsson, S. G. Eriksson, P. F. Henry, A. Matic, D. Engberg and L. Börjesson, J. Alloys Compd., 2008, 450, 103–110 CrossRef CAS PubMed.
- E. Kendrick, K. S. Knight, M. S. Islam and P. R. Slater, Solid State Ionics, 2007, 178, 943–949 CrossRef CAS PubMed.
- N. Malikova, C. K. Loong, J. M. Zanotti and F. Fernandez-Alonso, J. Phys. Chem. C, 2007, 111, 6574–6580 CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32, 751 CrossRef.
- K. S. Knight and N. Bonanos, Solid State Ionics, 1995, 77, 189–194 CrossRef CAS.
- (a) X. Chen and T. Grande, Chem. Mater., 2013, 25, 3296–3306 CrossRef CAS; (b) X. Chen and T. Grande, Chem. Mater., 2013, 25, 927–934 CrossRef CAS.
- S. M. Islam, J. Mater. Chem., 2000, 10, 1027–1038 RSC.
- J. Hermet, F. O. Bottin, G. Dezanneau and G. Geneste, Phys. Rev. B: Condens. Matter, 2012, 85, 205137 CrossRef.
- P. Haro-González, M. Karlsson, S. M. Gaita, C. S. Knee and M. Bettinelli, Solid State Ionics, 2013, 247–248, 94–97, DOI:10.1016/j.ssi.2013.06.008.
- L. Buannic, F. Blanc, D. S. Middlemiss and C. P. Grey, J. Am. Chem. Soc., 2012, 134, 14483–14498 CrossRef CAS PubMed.
- A. Grimaud, J. M. Bassat, F. Mauvy, P. Simon, A. Canizares, B. Rousseau, M. Marrony and J. C. Grenier, Solid State Ionics, 2011, 191, 24–31 CrossRef CAS PubMed.
- K. D. Kreuer, W. Münch, M. Ise, T. He, A. Fuchs, U. Traub and J. Maier, Ber. Bunsenges. Phys. Chem., 1997, 101, 1344 CAS.
- F. Cordero, F. Trequattrini, F. Deganello, V. La Parola, E. Roncari and A. Sanson, Phys. Rev. B: Condens. Matter, 2010, 82, 104102 CrossRef.
- M. O'Keeffe and B. G. Hyde, Acta Crystallogr., Sect. B: Struct. Sci., 1977, 33, 3802–3381 CrossRef.
- C. Shi, M. Yoshino and M. Morinaga, Solid State Ionics, 2005, 176, 1091–1096 CrossRef CAS PubMed.
- S. J. Stokes and M. S. Islam, J. Mater. Chem., 2010, 20, 6258–6264 RSC.
- S. M. Moussa, B. J. Kennedy, B. A. Hunter, C. J. Howard and T. Vogt, J. Phys.: Condens. Matter, 2001, 13, L203–L209 CrossRef CAS.
- C. J. Howard and H. T. Stokes, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 782–789 CrossRef.
- C. K. Loong, M. Ozawa, K. Takeuchi, K. Ui and N. Koura, J. Alloys Compd., 2006, 408, 1065–1070 CrossRef PubMed.
- W. Münch, G. Seifert, K. D. Kreuer and J. Maier, Solid State Ionics, 1996, 86–88, 647–652 CrossRef.
- A. Kruth and J. T. S. Irvine, Solid State Ionics, 2003, 162, 83–91 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |