
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long- and short-range structure studies of KBT-KBZ solid-solutions using synchrotron radiation
Samuel
Liu
a,
Peter E. R.
Blanchard
bc,
Zhaoming
Zhang
d,
Brendan J.
Kennedy
a and
Chris D.
Ling
*a
aSchool of Chemistry, University of Sydney, Sydney 2006, Australia. E-mail: chris.ling@sydney.edu.au; Fax: +61293513329; Tel: +61293514504
bCanadian Light Source, Saskatoon, SK, Canada S7N 2V3
cDepartment of Chemistry, University of Saskatchewan, Saskatoon, SK, Canada S7N 5C9
dAustralian Nuclear Science and Technology Organisation, Lucas Heights, New South Wales 2234, Australia
First published on 14th January 2015
Abstract
The relaxor ferroelectric K0.5Bi0.5TiO3 has been synthesised in a solid-solution series with K0.5Bi0.5ZrO3, as K0.5Bi0.5Ti1−xZrxO3. High-resolution synchrotron X-ray powder diffraction and X-ray absorption near edge structure spectroscopy were used to characterise the long-range average and local structural behaviour. Rietveld refinements against diffraction data show that a pseudocubic tetragonal region exists across the whole solid-solution series, with truly cubic symmetry only observed at x = 1 (pure KBZ). Variable-temperature diffraction data for x = 0 (pure KBT) showed a broad ferroelectric transition from tetragonal to cubic symmetry at approximately 683 K with a coexistence of both phases close to that temperature, accompanied by a marked volume contraction. Ti K-edge data showed that Zr doping has a minimal effect on Ti off-centering, and revealed no evidence for local clustering. Metal L-edges showed that Ti4+ cations remain off-centered with increasing Zr content, while Zr4+ cations approach a higher-symmetry coordination environment, most likely due to the increased size of the Zr atoms. Although there is a minimal effect on actual Ti-offsets, an effective dilution of these environments by Zr doping leads to a reduction in polar domains and a diminished ferroelectric response.
1. Introduction
A thorough understanding of structure–property relationships underlies any rational search for new functional materials. In the case of ferroelectric materials, the most fundamental relationship is between the role of second-order Jahn–Teller distortions of metal cations with a d0 electronic state,1 and local (or long-range) ferroelectric order. In metal oxide perovskite (ABO3) systems, the empty d orbitals of the B-site cation can hybridise with oxygen 2p orbitals causing a favourable offset of the B-site cation itself.2 For a non-centrosymmetric crystal structure, this offset causes a net dipole.3 An increase in temperature can remove the B-site cation offset, rendering the material paraelectric at the Curie temperature TC.Ferroelectricity is widely found in perovskites and perovskite-related materials. The most important commercially is PbZrxTi1−xO3 (PZT), which has a number of particular features, chief among which is the lead cation (Pb2+) which enhances the ferroelectric effect via additional distortions from its stereochemically active 6s2 electron lone pair. It has been found that the ferroelectric properties of PZT are further enhanced near a structural transition known as the morphotropic phase boundary (MPB).4 A great deal of research effort has been dedicated to finding lead-free alternatives to PZT in order to minimise the use of the toxic element, and encouraging the growth of MPBs appears to be a promising route.5 Bismuth-based materials have been shown to be feasible candidates, noting that Bi3+ is isoelectronic to Pb2+ and thus preserves the enhanced distortive effects. Charge balance upon replacing Pb2+ with Bi3+ can be achieved by an alkali metal cation of similar size, such as Na+ or K+. There has been extensive research into bismuth titanates, including Na0.5Bi0.5TiO3 (NBT) and K0.5Bi0.5TiO3 (KBT), solid-solutions systems such as NBT-KBT,6,7 combinations with SrTiO3,8,9 BaTiO3,10 and the multiferroic BiFeO3.11
Relatively little research has been done into the effects of replacing the B-site cation, compared to the vast amounts of literature in A-site modifications. The 4d0 cation Zr4+ is only slightly larger than the 3d0 Ti4+ (ref. 12), and complete B-site substitution is possible in BaTiO313 and NBT14. The present study concerns the solid-solution between KBT and K0.5Bi0.5ZrO3 (KBZ), first reported by Yamada et al.,15 and more recently the subject of a physical property study by Bengagi et al.16 A smooth structural transition was reported on the basis of sealed-tube X-ray powder diffraction (XRD) data, from the relaxor ferroelectric tetragonal KBT (Ti end member) to the paraelectric cubic KBZ (Zr end member). A pseudocubic region was reported for x = 0.1 to 0.4, in which a dramatic decrease in the ferroelectric moment was observed.16
This work is specifically concerned with investigating chemical changes to the B-site and their effects on structural distortions in the KBT-KBZ system. Due to the presence of very heavy metals such as Bi, conventional XRD is inadequate to resolve the details of oxygen atomic displacements. Our approach is therefore to use a combination of high-resolution synchrotron XRD (SXRD) to investigate long-range average structure and X-ray absorption near edge structure (XANES) spectroscopy to investigate the local coordination environment of Ti4+ and Zr4+ cations. Our recent study using these techniques on the NBT-NBZ solid-solution concluded that Zr doping had little effect on Ti-offset;14 noting the slightly different sizes of Na+ and K+, slightly different Zr doping effects can be expected in the KBT-KBZ series.
2. Experimental
∼5 g samples of polycrystalline K0.5Bi0.5Ti1−xZrxO3, where x = 0 to 1 (in 0.1 steps), were prepared by first weighing stoichiometric amounts of Bi2O3 (Aithaca, 99.999%, dried at 1023 K for 20 h before use), K2CO3, TiO2, and ZrO2. The reagents were finely ground with a mortar and pestle with the addition of acetone to improve homogeneity. The mixture was then reacted as a powder in alumina crucibles initially at 1023 K for 8 h, then reground and heated to 1223 K for 8 h twice. At high temperatures, significant potassium loss was noted by Bengagi et al.16 An additional 10–15% K2CO3 was therefore added to each sample to help minimise impurities. Long heating times improve the stability of the tetragonal phase of KBT, therefore the x = 0 and 0.1 samples were additionally heated to 1223 K for 72 h.16 Progress was monitored by X-ray powder diffraction (XRD) using a PANalytical X'pert PRO diffractometer in Bragg–Brentano geometry with a sealed-tube source of unmonochromated Cu-Kα radiation (λα1 = 1.5405 Å, λα2 = 1.5443 Å).Synchrotron X-ray powder diffraction (SXRD) data were obtained at the Australian Synchrotron, Melbourne, on the Powder Diffraction beamline.17 Samples were ground into a very fine powder and placed in 0.3 mm quartz capillaries for high-temperature measurements of KBT, or 0.3 mm glass capillaries for room-temperature measurements. Data were collected in Debye–Sherrer geometry at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 eV. The corresponding refined wavelength against a LaB6 NIST standard was λ = 0.953296 (1) Å. Data collection under non-ambient conditions were achieved using a hot air blower. The temperature range was controlled between room temperature and 973 K (10 K steps, 5 K min−1 ramp) with 2 × 150 s (2 detector positions) acquisition time at each step from 10° ≤ 2θ ≤ 90°. Room-temperature samples were collected over 2 × 120 s acquisition times.
500 eV. The corresponding refined wavelength against a LaB6 NIST standard was λ = 0.953296 (1) Å. Data collection under non-ambient conditions were achieved using a hot air blower. The temperature range was controlled between room temperature and 973 K (10 K steps, 5 K min−1 ramp) with 2 × 150 s (2 detector positions) acquisition time at each step from 10° ≤ 2θ ≤ 90°. Room-temperature samples were collected over 2 × 120 s acquisition times.
Initial structural models were adapted from the already reported structure of KBT.18 Rietveld refinements against diffraction data were carried out using the GSAS19 program with the EXPGUI20 interface and extracted atomic positions were visualised using the VESTA software package.21 Cell parameters, anisotropic atomic displacement parameters (ADPs) as well as instrumental parameters and a zero offset were refined.
XANES measurements at the Ti L-edge for K0.5Bi0.5Ti1−xZrxO3, where x = 0 to 0.9 were collected on the Soft X-ray beamline at the Australian Synchrotron.22 Powder samples were thinly dusted on double-sided carbon tape (SPI Supplies) and inserted into the vacuum chamber via a load lock. The pressure inside the analysis chamber was maintained at better than 10−9 Torr. All spectra were taken simultaneously with a total electron yield (TEY) signal measured from a Ti metal reference foil in the beamline. Approximately 10% of the beam intensity is removed by the reference foil. The Ti L3-edge of metallic Ti is well known, with a distinct feature located at 453.8 eV.23 This allowed for a precise energy calibration of the spectra from the various samples. The Ti K- and Zr L3-edge XANES spectra were collected on beamline 16A1 at the National Synchrotron Radiation Research Center (NSRRC) in Hsinchu, Taiwan.24 Samples were finely ground and dispersed onto Kapton tape and placed at a 45° angle to the incident X-ray beam. Spectra were collected in total fluorescence yield (TFY) mode using a Lytle detector with a step size of 0.2 eV near the absorption edge. The Ti K-edge spectra were calibrated against metallic Ti foil with the maximum in the first derivative of the K-edge set to 4966.4 eV. The Zr L3-edge spectra were calibrated against elemental Zr with the maximum in the first derivative of the L3-edge set to 2222.3 eV.25 All XANES spectra were normalised using the Athena26 software.
3. Results and discussion
The solid-solution K0.5Bi0.5Ti1−xZrxO3 (x = 0 to 1 with 0.1 steps) was successfully synthesised and confirmed by Rietveld refinement against laboratory XRD. Refined occupancies showed that the Ti/Zr ratio were within their nominal composition ranges and hence fixed for refinement models. Interestingly, the refinement for KBT (x = 0) by Bengagi et al. shows the (111) reflection to be significantly undercalculated.16 A better fit to the peak profile was obtained here by refining for strain factors. The improvement is shown in Fig. 1 with the relevant statistics. This suggests that closer to the Ti-rich end member of the solid-solution series, there is a significant contribution from microstrain in the material, which explains the increased synthesis time required to stabilise this phase. For x = 0.1–1, the strain parameter did not significantly improve the fit and was therefore not used. This suggests that the introduction of a slightly larger cation rapidly removes the microstrain by granting additional flexibility to the crystal lattice. The best statistics in Rietveld refinements using laboratory XRD data were achieved using a non-centrosymmetric tetragonal P4mm (#99) model from x = 0–0.8 and a cubic Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (#221) model for x = 0.9–1. This preliminary result suggested that the tetragonal pseudocubic region extends to x > 0.4, above the previously reported limit.16
m (#221) model for x = 0.9–1. This preliminary result suggested that the tetragonal pseudocubic region extends to x > 0.4, above the previously reported limit.16
 | ||
| Fig. 1 The room-temperature laboratory XRD data of KBT. Black crosses represent data points with the calculated pattern shown as a red line. Bragg reflection markers are shown in black below the observed peaks. Diff. Curve 1 refers to the difference curve of the Rietveld refinement including strain parameters shown above (χ2 = 1.673, Rp = 0.0264). Diff. Curve 2 refers to a Rietveld refinement not including strain (χ2 = 1.948, Rp = 0.0279). | ||
3.1. Synchrotron XRD
Higher-resolution SXRD data were used to refine models to greater precision. The room-temperature data confirmed that the samples were pure apart from a very small (<2%) fraction of ZrO2 in some of the samples, which could not be entirely eliminated despite the use of excess K to compensate for loss by evaporation.In the conventional XRD data of Bengagi et al.16 diffraction peaks were generally observed to broaden with increasing Ti content through the pseudocubic region (x = 0.1–0.4). Visually, the same behaviour was observed in our SXRD data for x = 0.1–0.4, with only the pure KBT compound found to possess a clear tetragonal distortion (i.e., split peaks) (Fig. 2). In order to test this more rigorously, Rietveld refinements were carried out against SXRD data for all samples using both the non-centrosymmetric tetragonal P4mm (#99) and cubic Pmm (#221) space groups. A comparison of the fits for x = 0.8 is shown in Fig. 3. Refining equivalent parameters, the tetragonal model provides a significantly improved fit. Refined crystal parameters also were more plausible using a tetragonal model, as shown in Table 1, where the ADPs for the B-site cations and oxygen atoms are unreasonably large using the cubic model. Using the tetragonal model, large ADPs are only found for the K/Bi mixed occupancy A-site, as expected due to static disorder. The tetragonal model was superior for all samples except pure KBZ, where the best fit was obtained using the cubic model. The evolution of unit cell parameters and cell volume across the KBT-KBZ solid-solution is shown in Fig. 4. The unit cell volume is consistent with Vegard's law. The difference between the a and c parameters drops rapidly as x (Zr content) increases, but remains statistically significant until x = 1.
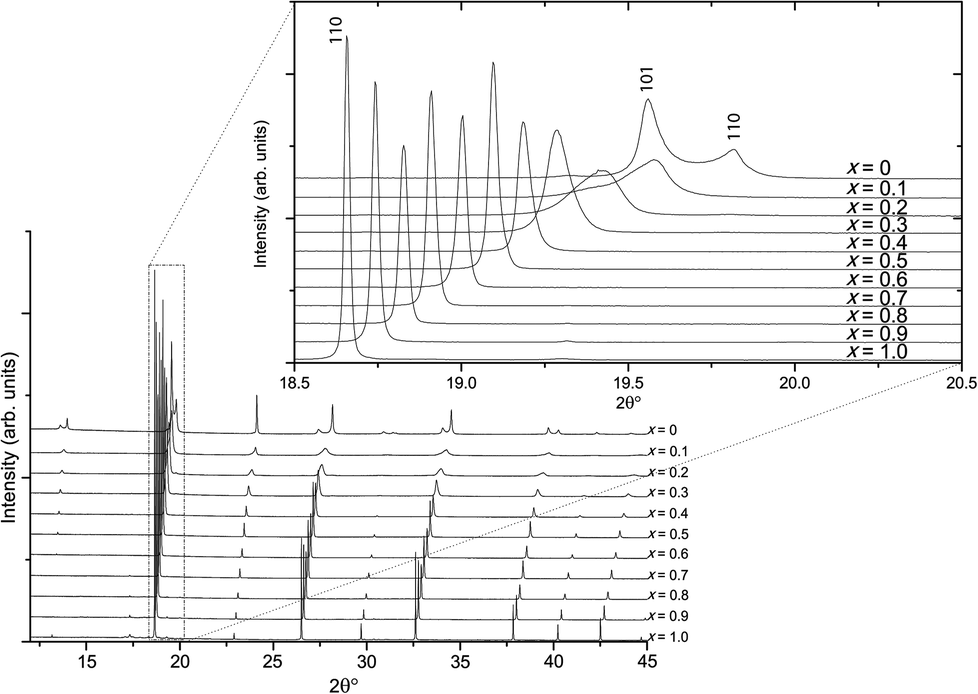 | ||
| Fig. 2 Room-temperature SXRD patterns of KBT-KBZ solid-solution. | ||
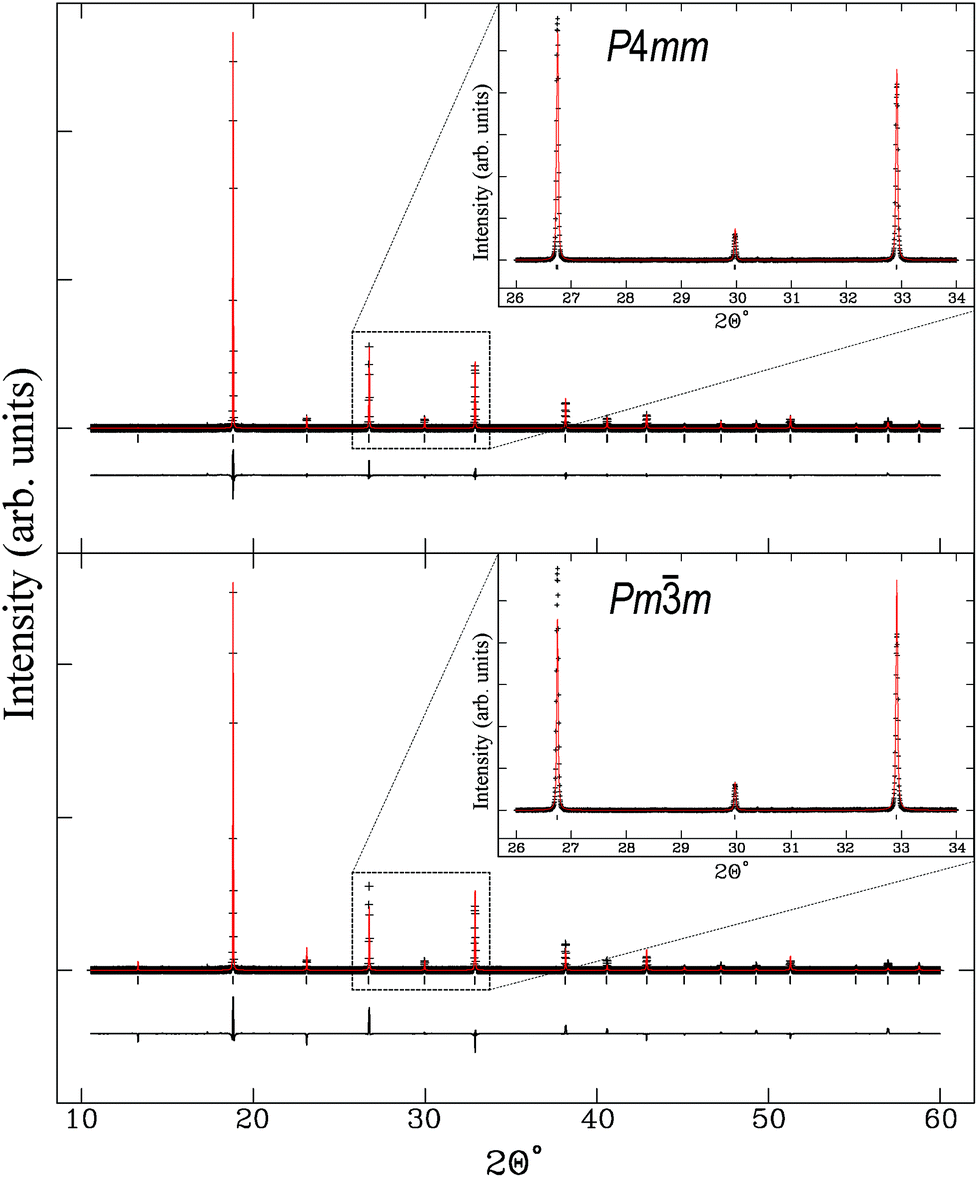 | ||
| Fig. 3 K0.5Bi0.5Ti0.2Zr0.8O3 fit to tetragonal P4mm (#99) Rp = 0.0347 and χ2 = 9.294 above and fit to cubic Pmm (#221) Rp = 0.0520 and χ2 = 29.98 below. | ||
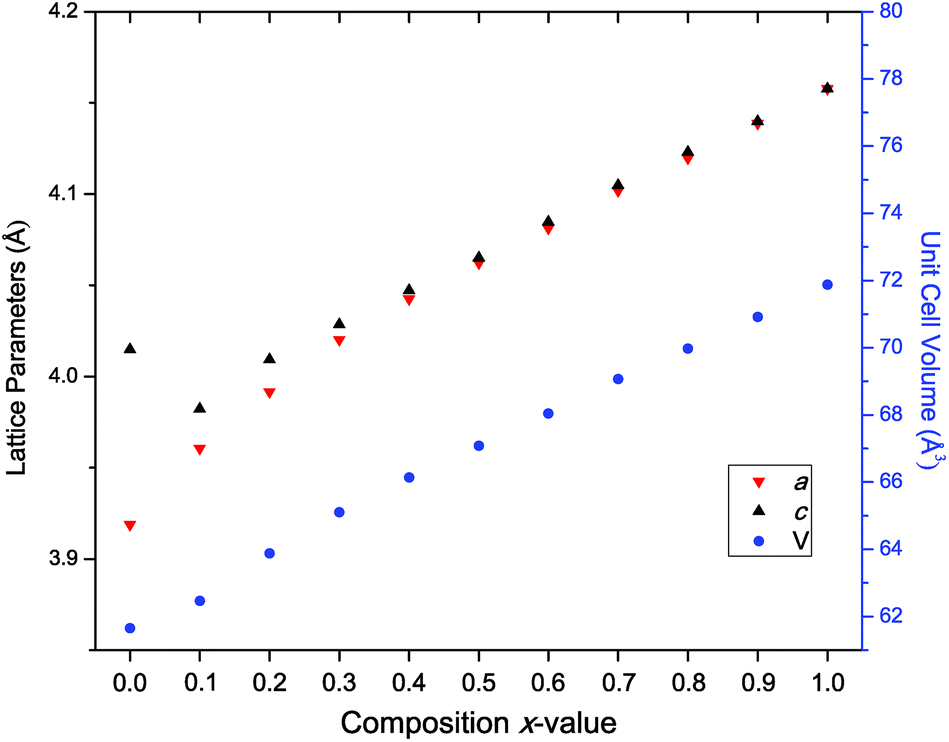 | ||
| Fig. 4 The lattice parameters extracted from Rietveld refinement across the solid-solution with a (red ∇ triangles), c (black Δ triangles) and unit cell volume V (blue circles). For x = 1 only, the data were modelled in cubic Pmm hence a and c are equal. Error bars are smaller than symbols. | ||
| 100 × Uiso | P4mm (#99) |
Pmm (#221) |
|---|---|---|
| A-site (Å2) | 14.47(5) | 3.48(4) |
| B-site (Å2) | 0.89(2) | 7.74(8) |
| Oxygen (Å2) | 2.91(12) | 21.7(4) |
The observation that the KBT-KBZ solid-solution retains tetragonal symmetry – i.e., is pseudocubic rather than truly cubic – up to at least x = 0.9 is consistent with its relaxor ferroelectric behaviour for x = 0–0.9 (albeit very weak at higher x).16
Interestingly, the sodium analogue Na0.5Bi0.5ZrO3 is reported to be rhombohedral/orthorhombic instead of cubic.14 This structural difference between K- and Na-versions can be explained by the tolerance factor:
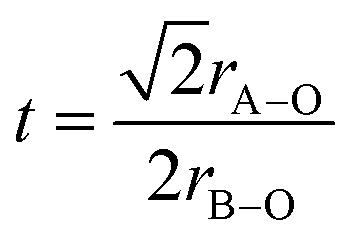 | (1) |
3.2. In situ synchrotron XRD
The temperature-dependent behaviour through the ferroelectric-paraelectric phase transition was studied by in situ variable-temperature SXRD. Data were collected for pure KBT in order to track the evolution of lattice parameters. This builds on previous work using conventional XRD.7,27 The evolution of the diffraction patterns (Fig. 5) clearly shows the transition from tetragonal to cubic symmetry at TC ∼ 683 K where the lower symmetry split peaks converge to higher symmetry single peaks. Slightly below this temperature, at approximately 663 K, there is a region of phase coexistence. This was not observed in earlier studies, but is a known behaviour of relaxor ferroelectric compounds, in which the presence of polar nano-regions creates a distribution of ferroelectric domains and hence a range of transition temperatures;27–29 and is in agreement with the broad peak in dielectric measurements reported by Bengagi et al.16 Around TC, from 633 to 693 K, a two-phase model in Rietveld refinement was used to track the behaviour of the coexisting phases. This region is highlighted by the shaded area in Fig. 6.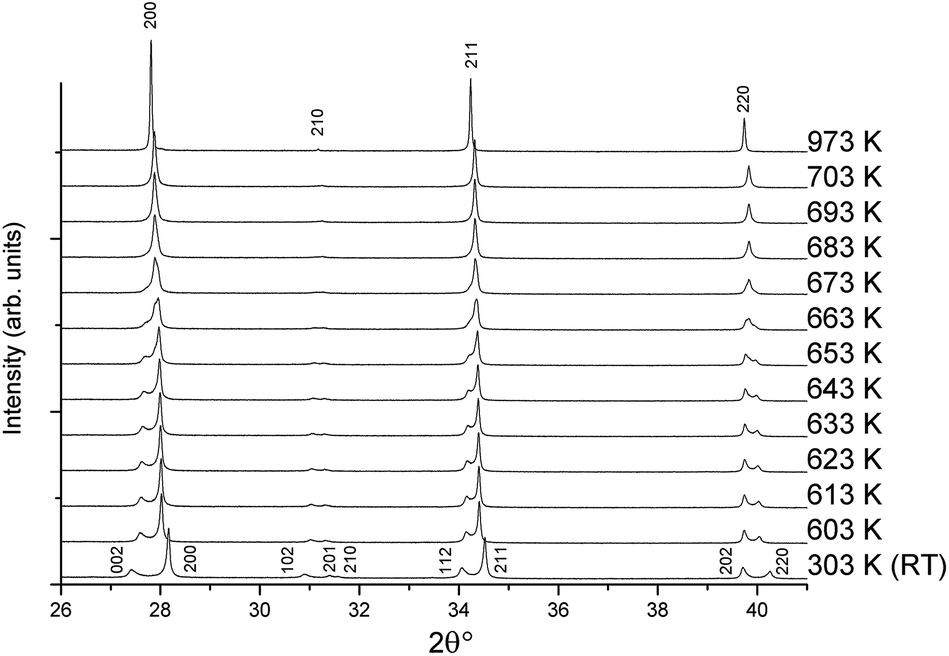 | ||
| Fig. 5 Variable-temperature SXRD patterns from 303 to 973 K showing the tetragonal to cubic transition with indexed peaks. | ||
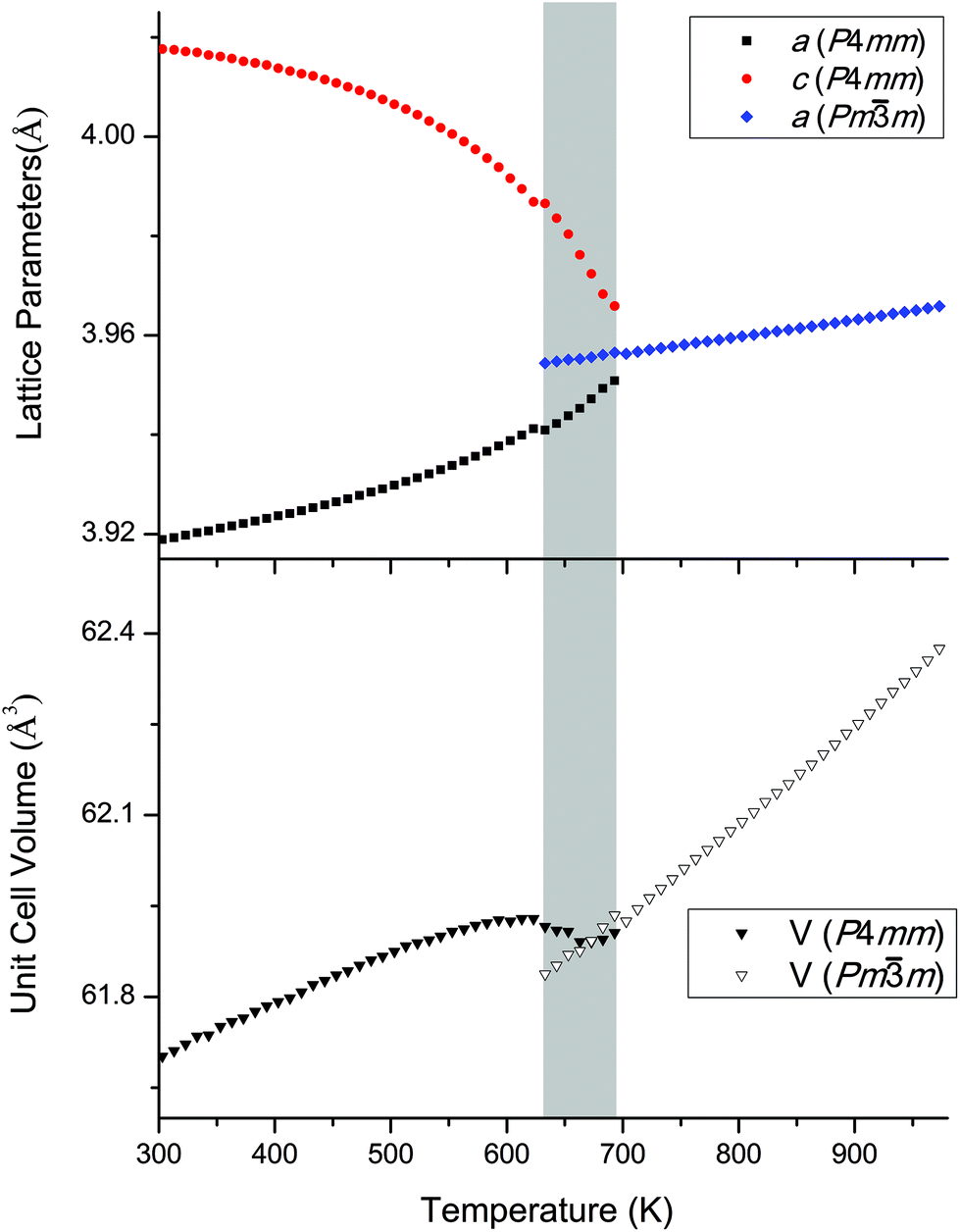 | ||
| Fig. 6 Evolution of lattice parameters and cell volume for KBT obtained from Rietveld refinements against SXRD data. The grey region indicates the temperature range in which the tetragonal and cubic phases coexist. | ||
Fig. 6 shows the Rietveld-refined lattice parameters and cell volume as the sample is heated from room temperature. The a and c parameters of the tetragonal phase converge to the common a parameter of the cubic phase in an essentially continuous second order manner, but with the unusual addition of a narrow coexistence region close to TC, the origin of which is discussed above. Another unusual feature is the lattice volume contraction from the tetragonal to the cubic phase. We can rationalise this by considering the relationship between bond length and bond strength, which in solid-state chemistry is commonly considered in terms of effective bond valence sum (BVS) (eqn (2)):
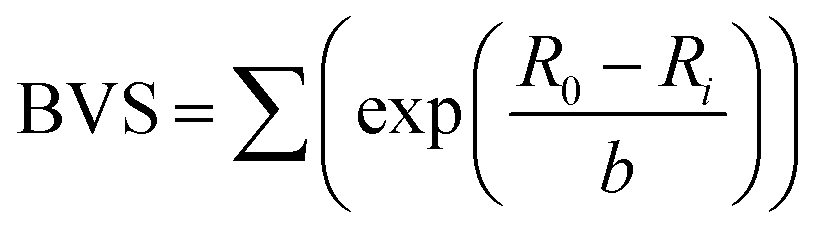 | (2) |
3.3. XANES
Relaxor ferroelectrics have complex behaviour due to their locally mixed compositions, sizes and charges across the crystal lattice.29 While diffraction provides a good measure for the average crystal structure, knowledge of the local structure is crucial to understanding the behaviour of solid-solution ferroelectrics. We have used XANES as a probe of the local coordination environment. Changes in local cation environments cause changes in orbital hybridisation, which manifest as peak shifts and intensity changes at the metal absorption edges. The Ti K-edge is particularly sensitive to the oxidation state and coordination environment of Ti in systems such as these.14Normalised Ti K-edge spectra across the KBT-KBZ solid-solution are shown in Fig. 7 (note that for x = 0.9, statistics were too poor to obtain usable data). There are two major features: the main edge feature (labelled A); and a pre-edge feature (labelled B1–B4). Feature A corresponds to the dipole-allowed transition of a 1s electron into unoccupied 4p states.14 The line shape of A implies minor changes to the hybridisation of Ti 4p states with the bonding states of nearest- and next-nearest neighbouring atoms, largely due to the introduction of Zr content. Although small intensity changes are observed in the main peak A, likely due to replacing Ti 4p states with Zr 5p states, the lack of a shift in the main edge energy confirms that Ti remains tetravalent – i.e., there is no evidence for oxygen vacancies.14
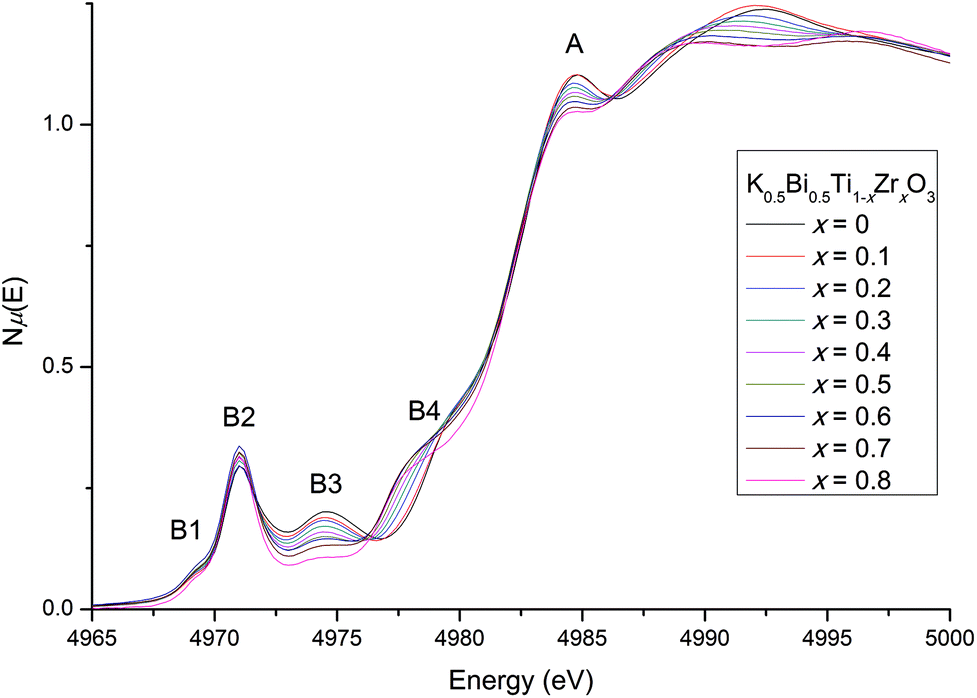 | ||
| Fig. 7 The Ti K-edge (A) including the distinct pre-edge features (B1-B4). No energy shifts are observed at the main edge, but pre-edge changes indicate an increasingly centrosymmetric environment for the B-site with increasing Zr content, while the Ti4+ cation distortion remains largely unchanged. | ||
More subtle information about the coordination environment can be obtained by analysing the forbidden transitions in the pre-edge region. The pre-edge features arise from a dipole-forbidden transition of a 1s electron into unoccupied 3d states.31 The first pre-edge feature, B1, corresponds to the dipole-forbidden transition of a 1s electron into the 3d t2g states for octahedrally coordinated Ti and remains largely the same with increasing Zr content. The higher-energy feature B2 arises from 1s → 3d eg transitions as well as non-local inter-site hybridisation, and B3 originates from the non-local 1s → p transitions (where the empty p states of the absorbing Ti atom is hybridised with the empty d states of the Ti second neighbours via O 2p states).31,32 Similarly to previously reported Zr doping effects on NBT, a small increase is observed in B2 suggesting that the Ti-offset increases slightly with increasing Zr content.13,14 Together, B3 and B4 are sensitive to the local ratio of Ti/Zr around the absorbing atom, and therefore to local clustering.13 In the present case, B3 gradually decreases as Ti content decreases; while B4 increases as Zr content increases. These systematic trends suggest that Ti/Zr atoms are randomly distributed in the lattice,13,14 with no significant clustering. The local crystal structure from x = 0.5 to 0.9 (previously reported as cubic16) does not appear to ever become completely centrosymmetric, with Ti remaining off-centered. This is consistent with our high-resolution SXRD diffraction results, and the observation of weak ferroelectricity up to x = 0.9 by Bengagi et al.16
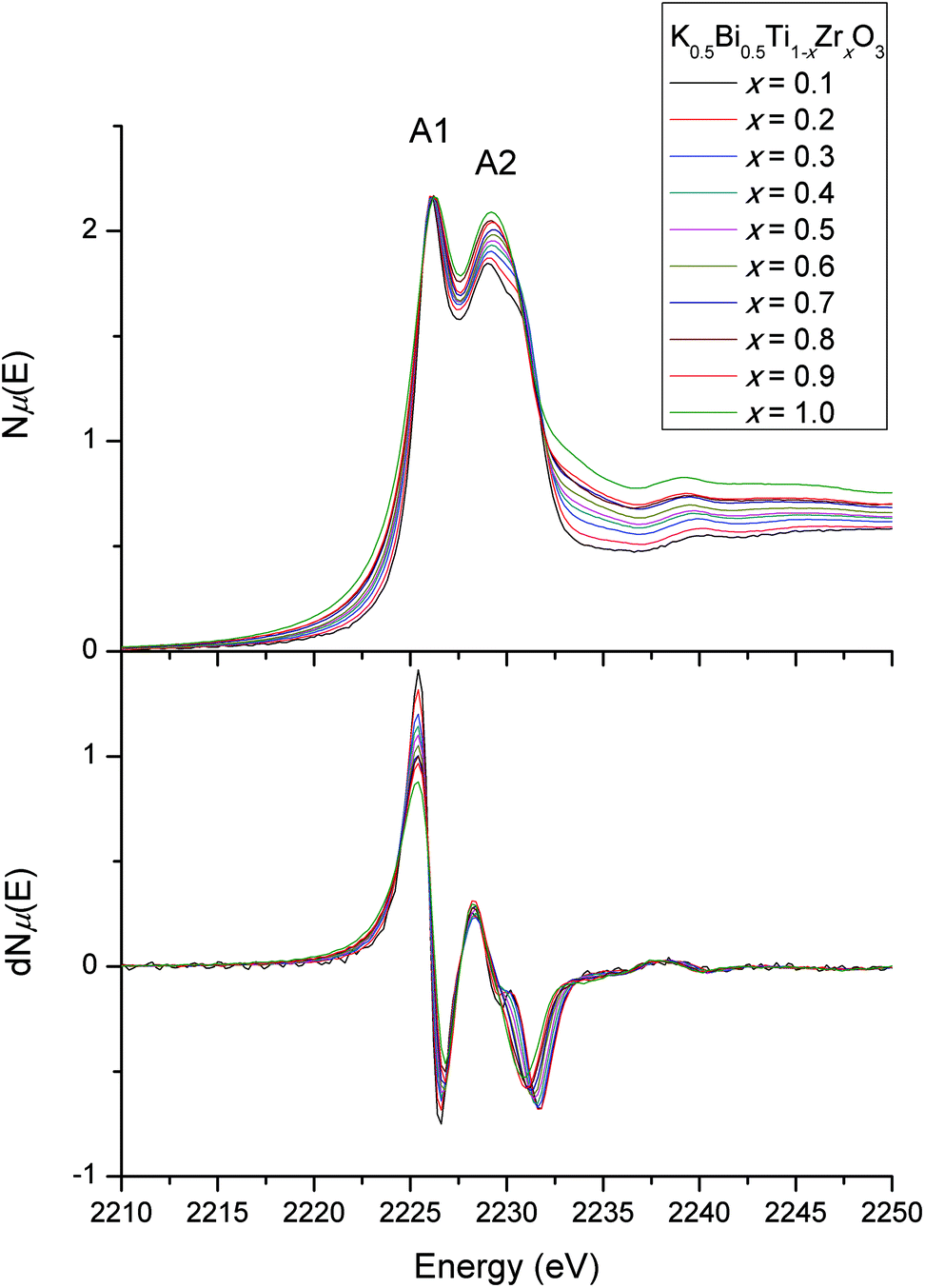
The Ti L-edge and Zr L-edge transitions correspond to the dipole-allowed transition of a Ti or Zr 2p electron into the unoccupied d states and hence probes the B-site distortion.23,33 Changes in line shape with composition for the L-edges of Ti and Zr are shown in Fig. 8 and 9 respectively. In Fig. 8, the four strong features correspond to the Ti L3-edge (two lower-energy transitions labelled A1 and A2) and the L2-edge (two higher-energy transitions B1 and B2). The separation between the L3- and L2-edges comes from spin–orbit splitting of Ti 2p3/2 (L3) and 2p1/2 (L2). In an octahedral environment, the main edge splits into the 3d subbands, corresponding to excitations to the 3d t2g (A1 and B1), and eg (A2 and B2) orbitals.34 The L3-edge is usually more resolved due to the lifetime-related broadening of the L2-edge. Consequently, after normalising the data for the Ti L3-edge to A1, one can observe any line shape changes across the solid-solution series measured. Our interest lies in the A2 feature, the eg states, which consist of dz2 and dx2−y2 orbitals. These orbitals are directed toward the ligand and are therefore sensitive to deviations in octahedral symmetry by Ti off-centering. In our normalised Ti L-edge data, peak asymmetry in A2 (less apparent in B2) is observed. This indicates a splitting of the eg states due to a deviation from octahedral symmetry (i.e., off-centered Ti4+). However, no obvious line shape changes are observed across the solid-solution series. This is consistent with the observed feature B2 in our normalised Ti K-edge data, where there were minimal changes in line shape with changes in composition, suggesting that Zr doping does not affect off-centering of the Ti4+ cations.
 | ||
| Fig. 8 Ti L3- and L2-edges. Asymmetry is observed in peak A2, with no line shape changes across the solid-solution series. | ||
 | ||
| Fig. 9 Normalised Zr L3-edge (upper) showing distinct line shape changes in A2 across the solid-solution series, indicative of increased symmetry with Zr doping. Line shape changes are emphasised by the plot of the first derivative (lower). | ||
The Zr L3-edge (Fig. 9) corresponds to the transition of a 2p3/2 electron into 4d states, which are split into t2g (A1) and eg (A2) states. Unlike the Ti L-edge, the Zr L3-edge line shape changes with Zr doping, particularly with respect to the eg state. In general, the asymmetry of the eg peak (A2), which is due to the splitting of the 4d eg states into 4dx2−y2 and 4dz2 states, decreases with increasing Zr content. This indicates a decrease in Zr4+ cation off-centering with doping.35–37 The decrease in peak splitting is more obvious in the first derivative plot of the Zr L3-edge shown in Fig. 9. Such changes in the Zr environment have been observed for BaTi1−xZrxO3 (BTO-BZO).14 The similar behaviour can be explained by similarities in the effective ionic radii (IR) of Ba2+ and K+/Bi3+ cations. In twelve-coordinate environments, IR(Ba2+) = 1.61 Å, mean IR(K+, Bi3+) = 1.51 Å and mean IR(Na+, Bi3+) = 1.385 Å. This gives ranges for t(BTO-BZO) = 1.06–1.00, t(KBT-KBZ) = 1.03–0.97 and t(NBT-NBZ) = 0.98–0.93. However, in practice, IR(K+, Bi3+) is larger due to the asymmetry of the Bi3+ cation, which brings t(KBT-KBZ) closer to t(BTO-BZO), but still different to t(NBT-NBZ).
In contrast, Zr cations did not approach higher symmetry environments in the NBT-NBZ series.14 This is probably due to the fact that the end-members were already stabilised in much lower-symmetry environments (rhombohedral, monoclinic and orthorhombic) as discussed above on the basis of calculated tolerance factors. For the KBT-KBZ series under investigation here, the combined results from the normalised metal L3-edges suggest that B-site off-centering is reduced with Zr doping. However, it appears that this is due to the Zr4+ cations moving to more symmetric coordination environments, rather than a reduction in Ti off-centering (as shown by the lack of line shape changes in our observed Ti L3-edge). This can be rationalised by a cation size effect, where the larger Zr4+ cation environment “pushes” neighbouring Ti4+ environments at low Zr content compositions. At high Zr content, larger cations are more likely to remain centered in the octahedra rather than offset. The dominating presence of larger Zr4+ cations over fewer and smaller Ti4+ cations brings the average B-site closer to centrosymmetric, as shown by diffraction data. However, on a local scale, Ti4+ cations remain offset in the octahedra. This effect was also observed for BTO-BZO.13,14
In summary, analysis of XANES spectra shows that Zr doping in this series of compounds effectively dilutes the number of non-centrosymmetric environments rather than affecting the Ti-offset directly. This in turn reduces the effective number of polar nano-regions and hence weakens the ferroelectric properties in the material.
4. Conclusions
Synchrotron X-ray techniques have provided new insights into the evolution of long- and short-range structure across the solid-solution series K0.5Bi0.5Ti1−xZrxO3, where x = 0 to 1 with 0.1 steps. Rietveld refinements against high-resolution SXRD data showed that the pseudocubic tetragonal region exists across the whole solid-solution, rather than 0 < x < 0.4 as previously observed. Only the pure KBT end member was clearly tetragonal, and only the pure KBZ end member was clearly cubic. On heating through the tetragonal-cubic transition at TC, a narrow region of tetragonal and cubic phase coexistence was observed despite the overall continuous second-order nature of the transition. This was explained by the presence of polar nano-regions in these relaxor ferroelectric perovskites. A marked volume contraction from the tetragonal to the cubic phase was explained in terms of the bonding (BVS) requirements of the cations as they move from an off-centered to a centered location. A future transmission electron microscopy study, such as has been carried out on the KBT end-member,27 might be able to reveal direct evidence of these polar nanoregions.Analysis of metal L-edge XANES data across the solid-solution series revealed that Ti4+ cations remain off-centered with increasing Zr content, while Zr4+ cations approach a higher symmetry coordination environment. Ti K-edge data showed a similar lack of change in the Ti off-centering. We can therefore conclude that Zr doping has a minimal effect on Ti off-centering, but rather, increases the centrosymmetry of the Zr local coordination environment with its size leading to a reduction in polar domains and hence diminished ferroelectric response.
Acknowledgements
Work at the Australian Synchrotron was performed with the assistance of Dr Helen Brand at the powder diffraction beamline, and Dr Bruce Cowie and Dr Anton Tadich at the Soft X-ray Spectroscopy beamline. Work at the National Synchrotron Radiation Research Center (NSRRC) was performed with the assistance of Dr Ling-Yun Jang and supported by the Australian Synchrotron International Access Program. CDL and BJK acknowledge the Australian Research Council and AINSE for financial support.References
- P. Halasyamani, Chem. Mater., 2004, 16, 3586–3592 CrossRef CAS.
- R. Cohen, Nature, 1992, 358, 136–138 CrossRef CAS.
- P. S. Halasyamani and K. R. Poeppelmeier, Chem. Mater., 1998, 10, 2753–2769 CrossRef CAS.
- B. Jaffe, R. Roth and S. Marzullo, J. Res. Natl. Bur. Stand., 1955, 55, 239–254 CrossRef CAS.
- K. S. Wallwork, B. J. Kennedy and D. Wang, Synchrotron Radiation Instrumentation, Pts 1 and 2, 2 Huntington Quadrangle, STE 1NO1, Melville, NY 11747-4501, USA, 2007, pp. 879–882.
- V. Isupov, Ferroelectrics, 2005, 315, 123–147 CrossRef CAS.
- O. Elkechai, M. Manier and J. Mercurio, Phys. Status Solidi A, 1996, 157, 499–506 CrossRef.
- V. Bogatko, Y. Venevtsev and A. Abramova, Inorg. Mater., 1980, 16, 72–74 Search PubMed.
- J. Gomah-Pettry, A. Salak, P. Marchet, V. Ferreira and J. Mercurio, Phys. Status Solidi B, 2004, 241, 1949–1956 CrossRef CAS.
- J. Gomah-Pettry, P. Marchet, A. Simon, R. Von der Muhll, M. Maglione and J. Mercurio, Integr. Ferroelectr., 2004, 61, 155–158 CrossRef CAS.
- V. Dorcet, P. Marchet, O. Pena and G. Trolliard, J. Magn. Magn. Mater., 2009, 321, 1762–1766 CrossRef CAS PubMed.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767 CrossRef.
- I. Levin, E. Cockayne, V. Krayzman, J. Woicik, S. Lee and C. Randall, Phys. Rev. B: Condens. Matter, 2011, 83, 094122 CrossRef.
- P. E. R. Blanchard, S. Liu, B. J. Kennedy, C. D. Ling, Z. Zhang, M. Avdeev, L.-Y. Jang, J.-F. Lee, C.-W. Pao and J.-L. Chen, Dalton Trans., 2014, 43, 17358–17365 RSC.
- Y. Yamada, T. Akutsu, H. Asada, K. Nozawa, S. Hachiga, T. Kurosaki, O. Ikagawa, H. Fujiki, K. Hozumi, T. Kawamura, T. Amakawa, K. Hirota and T. Ikeda, Jpn. J. Appl. Phys., Part 1, 1995, 34, 5462–5466 CrossRef CAS.
- M. Bengagi, F. Morini, M. El Maaoui and P. Marchet, Phys, Status Solidi A, 2012, 209, 2063–2072 CrossRef CAS.
- K. S. Wallwork, B. J. Kennedy and D. Wang, Synchrotron Radiation Instrumentation, Pts 1 and 2, 2 Huntington Quadrangle, STE 1NO1, Melville, NY 11747-4501 USA, 2007, pp. 879–882.
- G. Jones, J. Kreisel and P. Thomas, Powder Diffr., 2002, 17, 301–319 CrossRef CAS.
- A. Larson and R. V. Dreele, Gen. Struct. Anal. Syst. (GSAS), 2000 Search PubMed , LAUR 86-748.
- B. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- B. C. C. Cowie, A. Tadich and L. Thomsen, SRI 2009: The 10th International Conference on Synchrotron Radiation Instrumentation, 2 Huntington Quadrangle, STE 1NO1, Melville, NY 11747-4501, USA, 2010, pp. 307–310.
- G. Henderson, X. Liu and M. Fleet, Phys. Chem. Miner., 2002, 29, 32–42 CrossRef CAS.
- T. Dann, S. Chung, L. Huang, J. Juang, C. Chen and K. Tsang, J. Synchrotron Radiat., 1998, 5, 664–666 CrossRef CAS PubMed.
- T. Schneller, H. Kohlstedt, A. Petraru, R. Waser, J. Guo, J. Denlinger, T. Learmonth, P.-A. Glans and K. E. Smith, Journal of Sol-gel Sci. Technol., 2008, 48, 239–252 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- M. Otonicar, S. D. Skapin, B. Jancar, R. Ubic and D. Suvorov, J. Am. Ceram. Soc., 2010, 93, 4168–4173 CrossRef CAS PubMed.
- A. Bokov and Z. Ye, J. Mater. Sci., 2006, 41, 31–52 CrossRef CAS PubMed.
- L. Cross, Ferroelectrics, 1987, 76, 241–267 CrossRef CAS.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- E. R. Aluri, J. R. Hayes, J. D. S. Walker and A. P. Grosvenor, J. Phys. Chem. C, 2014, 118, 7910–7922 CAS.
- D. Cabaret, A. Bordage, A. Juhin, M. Arfaoui and E. Gaudry, Phys. Chem. Chem. Phys., 2010, 12, 5619–5633 RSC.
- P. Nachimuthu, S. Thevuthasan, E. Adams, W. Weber, B. Begg, B. Mun, D. Shuh, D. Lindle, E. Gullikson and R. Perera, J. Phys. Chem. B, 2005, 109, 1337–1339 CrossRef CAS PubMed.
- V. Mastelaro, P. Neves, S. de Lazaro, E. Longo, A. Michalowicz and J. Eiras, J. Appl. Phys., 2006, 99, 044104 CrossRef PubMed.
- H. Ikeno, M. Krause, T. Hoeche, C. Patzig, Y. Hu, A. Gawronski, I. Tanaka and C. Ruessel, J. Phys.: Condens. Matter, 2013, 25, 165505 CrossRef PubMed.
- P. E. R. Blanchard, S. Liu, B. J. Kennedy, C. D. Ling, Z. Zhang, M. Avdeev, B. C. C. Cowie, L. Thomsen and L.-Y. Jang, Dalton Trans., 2013, 42, 14875–14882 RSC.
- E. Reynolds, P. E. R. Blanchard, B. J. Kennedy, C. D. Ling, S. Liu, M. Avdeev, Z. Zhang, G. J. Cuello, A. Tadich and L.-Y. Jang, Inorg. Chem., 2013, 52, 8409–8415 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |