
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural and dielectric studies of the phase behaviour of the topological ferroelectric La1−xNdxTaO4†
Keith J.
Cordrey
a,
Magda
Stanczyk
a,
Charlotte A. L.
Dixon
a,
Kevin S.
Knight
b,
Jonathan
Gardner
a,
Finlay D.
Morrison
a and
Philip
Lightfoot
*a
aEaStCHEM and School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK. E-mail: pl@st-and.ac.uk
bISIS Facility, STFC Rutherford Appleton Laboratory, Harwell, Oxfordshire OX11 0QX, UK
First published on 19th January 2015
Abstract
The layered perovskite LaTaO4 has been prepared in its polar orthorhombic polymorphic form at ambient temperature. Although no structural phase transition is observed in the temperature interval 25° C < T < 500 °C, a very large axial thermal contraction effect is seen, which can be ascribed to an anomalous buckling of the perovskite octahedral layer. The non-polar monoclinic polymorph can be stabilised at ambient temperature by Nd-doping. A composition La0.90Nd0.10TaO4 shows a first-order monoclinic-orthorhombic (non-polar to polar) transition in the region 250° C < T < 350 °C. Dielectric responses are observed at both the above structural events but, despite the ‘topological ferroelectric’ nature of orthorhombic LaTaO4, we have not succeeded in obtaining ferroelectric P–E hysteresis behaviour. Structural relationships in the wider family of AnBnX3n+2 layered perovskites are discussed.
Introduction
The mechanism of the paraelectric-ferroelectric phase transition in most well-studied oxide ferroelectrics lies in the condensation of a polar soft-phonon mode, typically driven by off-centring of an octahedral ‘d0 cation’ or a ‘lone-pair cation’ via the 2nd-order Jahn–Teller effect.1 This is called proper ferroelectricity, and examples are the classic perovskite ferroelectrics BaTiO3, PbTiO3 and BiFeO3. However, in recent years several new mechanisms that drive ferroelectricity have been discovered or postulated. These include so-called ‘geometric ferroelectricity’, first proposed theoretically for hexagonal YMnO32,3 and later confirmed experimentally,4 and ‘hybrid improper ferroelectricity’ (HIF),5 which requires coupling of two distinct non-polar modes to produce a net polar symmetry. These latter two mechanisms are both types of improper ferroelectricity, where the polar modes arise as a natural ‘side-effect’ from strictly non-polar lattice modes. In turn these non-polar modes are generally driven by purely geometric (e.g., ionic size) effects: tilting of MnO5 polyhedra in the case of YMnO3, and octahedral rotations and/or cation ordering in the more general case of HIF. Significantly, the non-polar modes are the primary order parameters driving the phase transition, which then permit the polar modes as secondary order parameters. More recently,6 it has been observed that a purely ‘geometric’ effect can also operate to produce a proper ferroelectric; i.e., the lattice distortion is not driven by one of the two common ‘electronic’ factors above, but simply by a structural (size) mismatch of some sort. In this case, however, this geometric effect directly gives rise not only to a polar space group, but to the polarisation itself, without the need for a ‘slave’ polar mode. It has now been suggested that the name ‘topological ferroelectricity’ rather than geometric ferroelectricity be applied to this type of mechanism.7 Note that use of the term ‘ferroelectric’ in these definitions refers to an intrinsic structural polarity, which is in principle switchable, and does not necessarily imply experimental observation of switchability. The mechanism has been proposed within the class of layered perovskites of generic formula AnBnX3n+2,8 specifically for the cases BaMnF4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 (n = 2) and La2Ti2O77 (n = 4). The key point is that the layered nature of the perovskite topology means that rigid octahedral tilting, ubiquitous in perovskite crystallography, can naturally give rise to a polarisation since the number of ‘oxygen planes’ is odd (Fig. 1); this contrasts with the case in three-dimensionally connected perovskites, where octahedral tilt modes are intrinsically centrosymmetric.
6 (n = 2) and La2Ti2O77 (n = 4). The key point is that the layered nature of the perovskite topology means that rigid octahedral tilting, ubiquitous in perovskite crystallography, can naturally give rise to a polarisation since the number of ‘oxygen planes’ is odd (Fig. 1); this contrasts with the case in three-dimensionally connected perovskites, where octahedral tilt modes are intrinsically centrosymmetric.
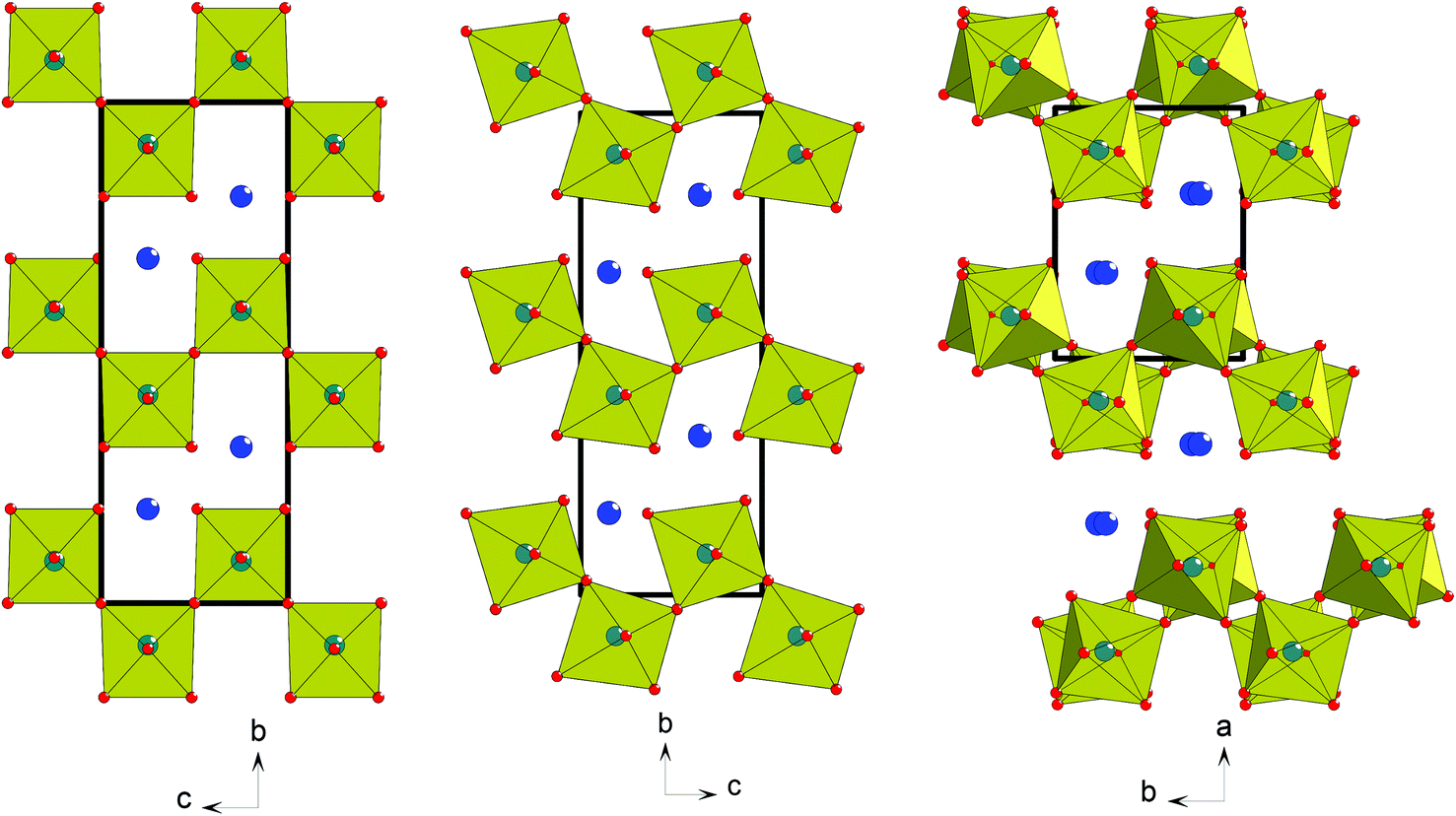 | ||
| Fig. 1 (a) Aristotype ABX4 structure type, space group Cmcm, with no octahedral tilting. (b) O-LaTaO4 structure, space group Cmc21: note that the c-axis polarity arises as a natural consequence of the ‘in-phase’ tilting around the a-axis. (c) M-LaTaO4 structure, space group P21/c: in this case the ‘anti-phase’ tilting around c permits retention of centrosymmetricity. | ||
In this paper we explore some of the crystal chemistry of another member of the AnBnX3n+2 family, viz. LaTaO4. This compound can exist in two polymorphic forms when prepared by ‘traditional’ high-temperature solid-state routes9–12 and a third polymorph can also be prepared via decomposition of a hydrated precursor.13 Of the former two phases, one is orthorhombic and polar and the other monoclinic and centrosymmetric (hereafter designated O-LaTaO4 and M-LaTaO4, respectively, Fig. 1). Earlier studies9–12 suggest M-LaTaO4 is the most stable form at ambient temperature, and that this phase undergoes a phase transition to O-LaTaO4 at ∼175 °C9 or 240 °C.10 In our study we have successfully prepared a near-pure sample of O-LaTaO4 and we study the structural behaviour of this system versus temperature using high-resolution powder neutron diffraction. We also show that the M-LaTaO4 phase can be stabilised further at ambient temperature by Nd-doping, and study the thermal evolution of this phase, and its transformation into the polar O-LaTaO4 polymorph. Finally, we carry out some general structural comparisons of these phases and related ones, which may be of relevance to their physical properties.
Experimental
Synthesis
Several different procedures were trialled in order to produce phase-pure samples. Typically, stoichiometric ratios of dried component oxides (La2O3, Ta2O5 and Nd2O3; all >99%) were mixed, either by ball-milling at 600 rpm for 1 hour or manually in a pestle and mortar, before being pressed into a pellet and annealed at temperatures between 1200 °C and 1400 °C for 12 hours. For LaTaO4 all these procedures produced phase mixtures of either orthorhombic and monoclinic polymorphs of LaTaO4, or orthorhombic plus a small amount of an unidentified impurity. The highest phase fraction of LaTaO4 (as the O-LaTaO4 polymorph) was produced in a two-stage process: annealing at 1250 °C for 6 hours, followed by ball-milling, re-pelleting and annealing for a further 6 hours at 1450 °C. For the Nd-doped samples, La1−xNdxTaO4 0.05 < x < 0.60, starting mixtures were hand-ground, pelleted and annealed at 1200 °C for 6 hours, then re-ground, pelleted and annealed at 1350 °C for 12 hours. In each case, the best samples were reacted on Pt foil in open alumina crucibles. For both the O-LaTaO4 and La0.9Nd0.1TaO4 samples used for electrical characterisation a heating rate of 10 °C min−1 was employed and at the end of the heating stage, the furnace was switched off and samples allowed to cool inside the furnace.Powder diffraction
Preliminary phase purity and structure analysis for each material was confirmed by Rietveld refinement of X-ray powder diffraction (XPD) data collected on a PANalytical Empyrean X-ray diffractometer using Cu-Kα1 radiation.Variable temperature neutron powder diffraction (NPD) data on LaTaO4 and La0.9Nd0.1TaO4 were collected on the high-resolution instrument (HRPD) at ISIS. For LaTaO4 data were collected at ambient temperature and between 100 °C and 500 °C at 50 °C intervals; for La0.9Nd0.1TaO4 date were collected between 100 °C and 600 °C at 50 °C intervals. In each case, data collection times were approximately 40 minutes for ∼5 g samples. For LaTaO4, a preliminary dataset was collected at 25 °C on the GEM diffractometer.
All quantitative data analysis was carried out by Rietveld refinement using the GSAS program14 and its EXPGUI user interface. For each of the HRPD runs, detector banks at 2θ ∼ 168° and 90° were analysed simultaneously.
Electrical properties
Electrodes were fabricated on opposing faces of pelleted samples using either Ag paint or sputtered gold. Dielectric properties were measured from room temperature to 600 °C using either an Agilent 4294A or Wayne Kerr 6500B impedance analyser with 500 mV excitation and heating/cooling rates of 2 K min−1. Polarisation-field data were obtained using an aixACCT TF2000 analyzer with a TREK 4 kV voltage amplifier.Results and discussion
LaTaO4
All attempted preparations resulted in phase mixtures of either O-LaTaO4 and M-LaTaO4 or O-LaTaO4 plus a small amount of unidentified impurity, as evidenced by XPD (see ESI† for standard XPD patterns). Comparing to previous studies, it is not entirely clear why previous authors have isolated predominantly the M-LaTaO4 phase from apparently similar reactions; for example Cava9 used a much higher reaction temperature (1750 °C), whereas Siqueira12 used a lower temperature (1300 °C). It is possible that the present ball-milling method (not used in previous studies) is critical, although sample cooling rate is another parameter that has not been quoted or studied in detail in any of the studies so far. The sample with the highest phase fraction of O-LaTaO4 (and negligible M-LaTaO4) was chosen for further study by NPD. A typical Rietveld fit is shown in Fig. 2. This refinement is based on a single phase fit to O-LaTaO4, space group Cmc21, with a ∼ 3.94, b ∼ 14.7, c ∼ 5.6 Å (Fig. 1(b)). Further details of the refinement model and outcomes are given in ESI.† No phase changes were detected throughout the temperature range studied, and the quality of fit to the O-LaTaO4 model remained similar from 100 °C to 500 °C. However, an unusual trend in lattice parameters versus temperature was observed. Fig. 3 shows the thermal evolution of unit cell a, b, c, and V parameters. Of particular note is the very large negative expansivity of the b-axis (αb ∼ − 40 × 10−6 K−1) in the temperature range 100–300 °C, leading to a near-zero volume expansivity around 200–250 °C.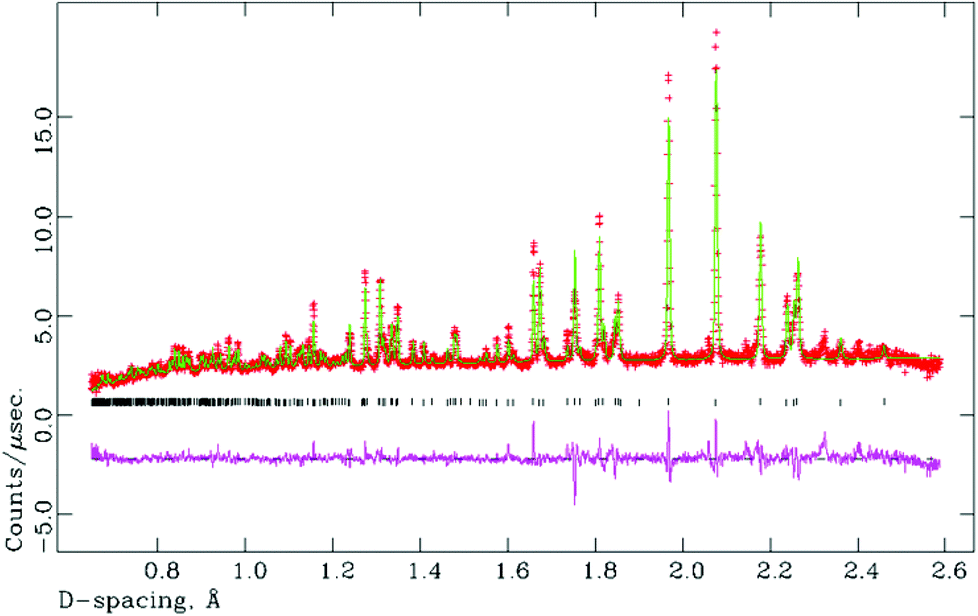 | ||
| Fig. 2 Rietveld fit (NPD) for single phase O-LaTaO4 model at 100 °C. Overall χ2 = 6.67 for 37 refined variables. | ||
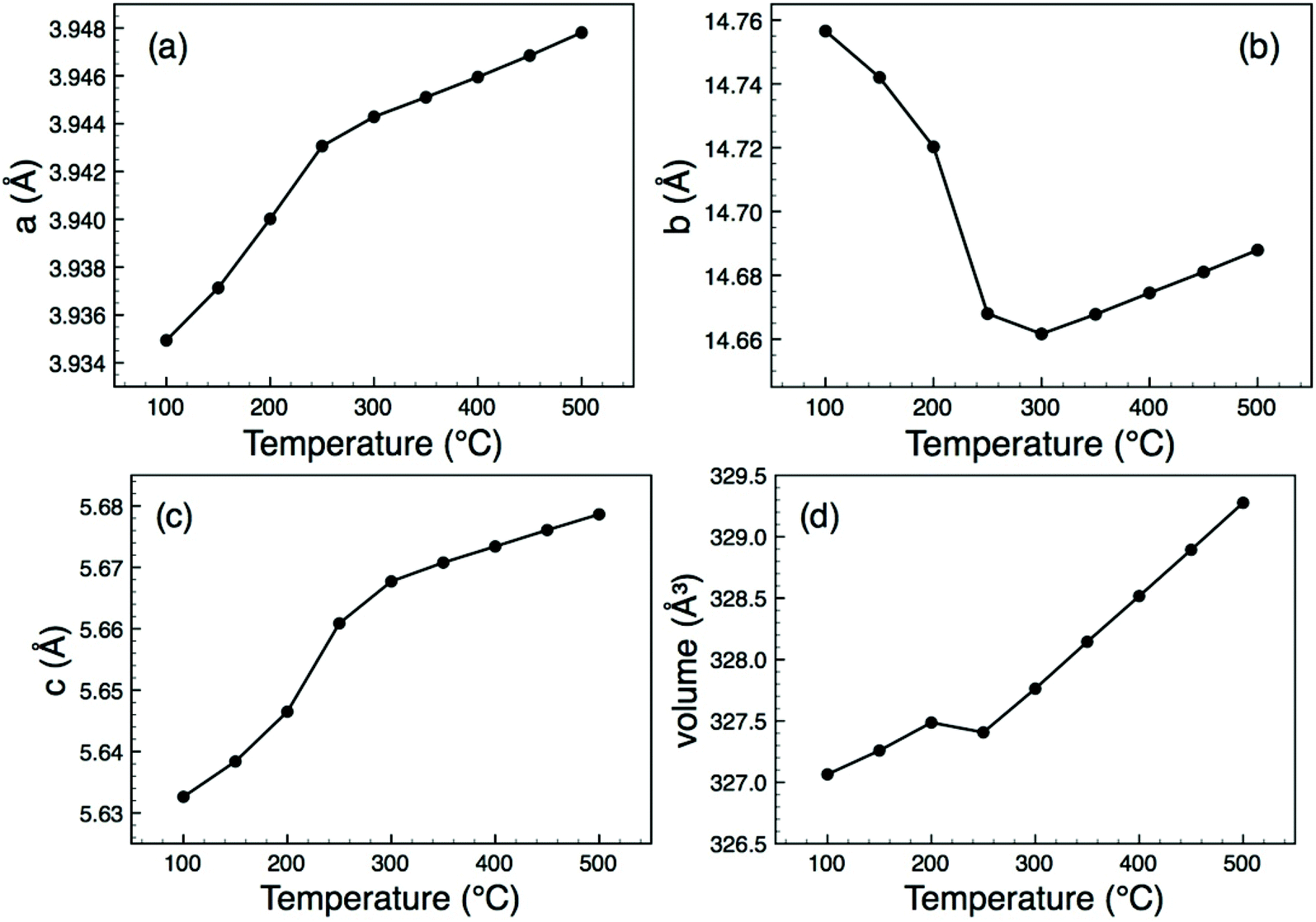 | ||
| Fig. 3 Thermal evolution of lattice parameters and unit cell volume for O-LaTaO4, derived from Rietveld refinement of NPD data. | ||
The crystal structure (Fig. 1(b), 4) consists of perovskite-like blocks of corner-shared octahedra extending in the ac plane, separated by the La3+ cations, which may be regarded as nine-coordinated to oxide. In order to pinpoint the structural features leading to the anomalous expansivity behaviour, we can define three parameters within these two distinct blocks: the thickness of the inter-layer block, d1, the thickness of the perovskite block, d2, and the angle of tilt within the corrugated octahedral layers, ω. From the geometry of these definitions it can be seen that
| b = 2(d1 + d2) |
 | ||
| Fig. 4 View of adjacent octahedral blocks in the O-LaTaO4 structure, with the definitions of the parameters, d1, d2 and ω shown. | ||
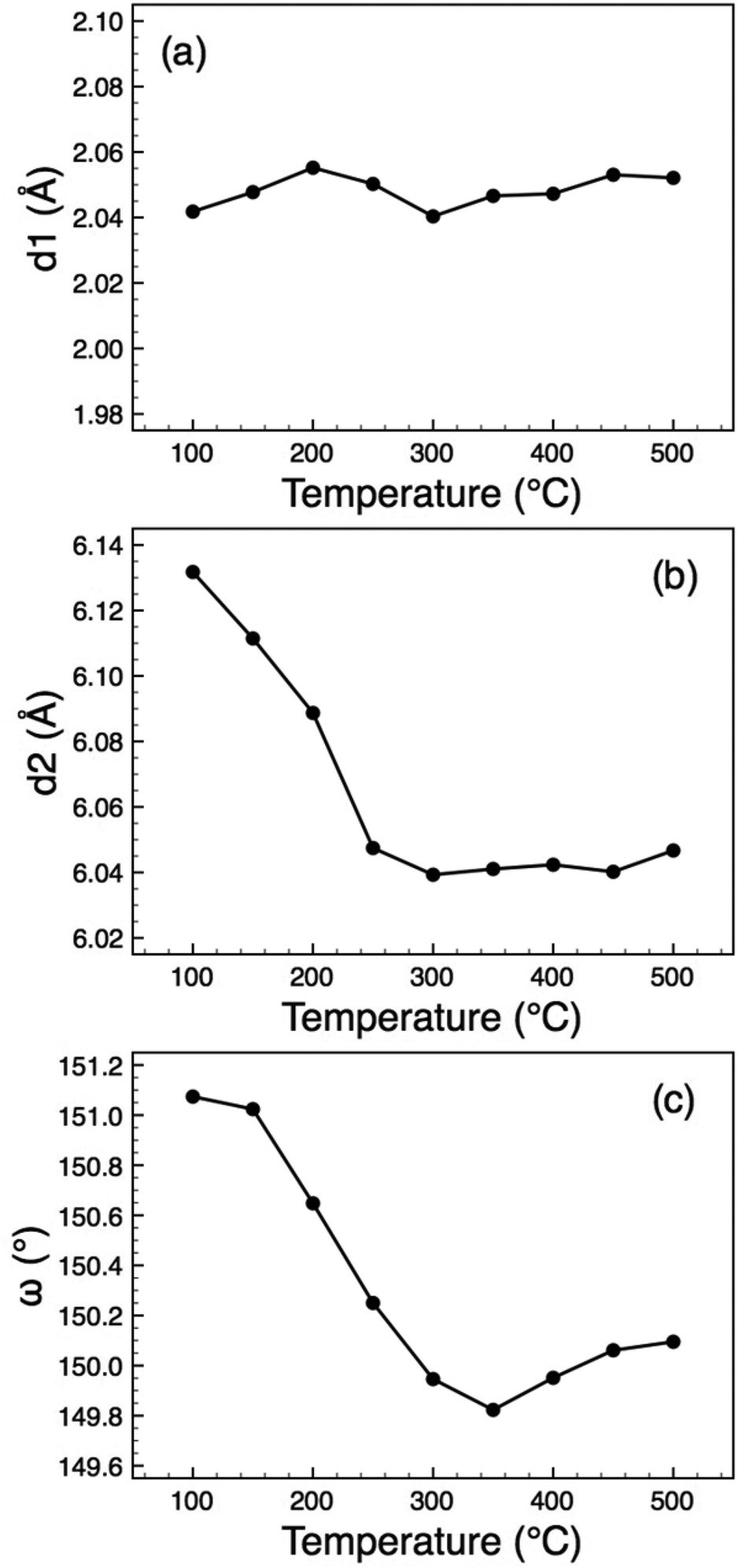 | ||
| Fig. 5 Thermal evolution of the distortion parameters (defined in Fig. 4) for O-LaTaO4, (a) d1, (b) d2, (c) ω. | ||
A recent variable temperature (90–350 K) structural study of some higher n members of this family, Lan(Ti,Fe)nO3n+2 (n = 5 and 6)15 showed that in those cases the thermal variation in the interlayer distance parameter is also insignificant, whereas the thickness of the perovskite block showed a marked increase, with no observable anomalies.
La1−xNdxTaO4 (0.05 < x < 0.60)
The motivation for preparing this series was to stabilise the M-LaTaO4 structure rather than O-LaTaO4 phase at ambient temperature. This postulate was based on the previous observation by Cava9 that the M-LnTaO4 to O-LnTaO4 phase transition temperature increases rapidly with decreasing ionic size of Ln3+: La (175 °C) < Ce (818 °C) < Pr (1300 °C). These are the only three lanthanides that adopt either of these structure types under ambient pressure, although we note that pure M-NdTaO4 can be prepared from the fergusonite polymorph under extreme conditions (8 MPa, 1500 °C).16 As the change in stability of the M- versus O-polymorphs appears to be so dramatically influenced by small changes in ionic size, it was anticipated that a small amount of Nd3+-doping into O-LaTaO4 would trigger a transformation to the M-phase; this was indeed found to be the case.For values of 0.05 ≤ x ≤ 0.40, preliminary analysis of XPD data suggested that the M-LaTaO4 could be prepared almost phase-pure. For x = 0.50 and 0.60 a significant additional phase (fergusonite type LnTaO4) was observed. Rietveld analysis of this series confirmed the extent of the M-LaTaO4 solid solution in La1−xNdxTaO4 as 0.05 ≤ x ≤ 0.40. Lattice parameters across the solid solution vary monotonically with x; these were determined by using a model with fixed atomic coordinates. An example Rietveld plot is given in Fig. 6, and further details are given in the ESI.†
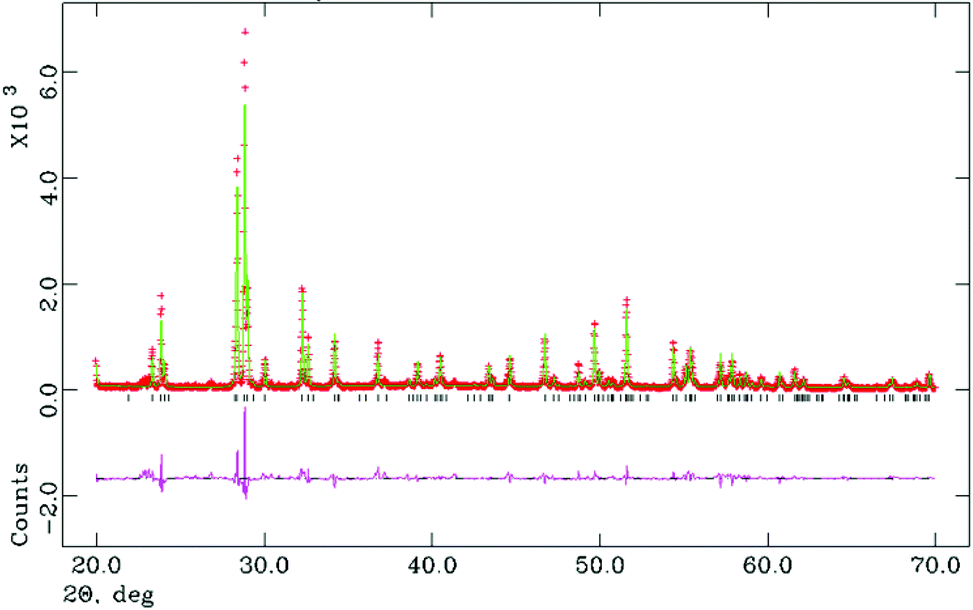 | ||
| Fig. 6 Rietveld fit (XPD) for La0.80Nd0.20TaO4 (M-LaTaO4) model at 25 °C. χ2 = 4.91. | ||
A sample of M-La0.9Nd0.1TaO4 was studied versus temperature by NPD, up to 600 °C. As anticipated, a phase transition from the M- to the O-phase was observed within the temperature range studied. This transition is of a first-order nature, as shown by the co-existence of both phases with the region 250 < T < 350 °C (M/O phase fractions (%) at 250 °C, 300 °C and 350 °C are 90/10, 59/41 and 24/76, respectively). In fact, there is no simple group-subgroup relationship between the two crystal structures, so the transition is expected to be 1st order according to Landau theory. Rietveld fits are shown in Fig. 7(a) and (b) for single-phase fits to the M- and O-models at 100 and 500 °C, respectively. Attempts to refine the high-temperature phase in the centrosymmetric parent phase, space group Cmcm (Fig. 1(a)), led to significantly poorer fits and unrealistic atomic displacement parameters (Cmc21 model: 37 variables, χ2 = 3.26, Cmcm model: 30 variables, χ2 = 13.5). A similar contraction of the b-axis, as observed for the O-LaTaO4 phase, is seen here in the region 250 < T < 450 °C. Further details are given in ESI.†
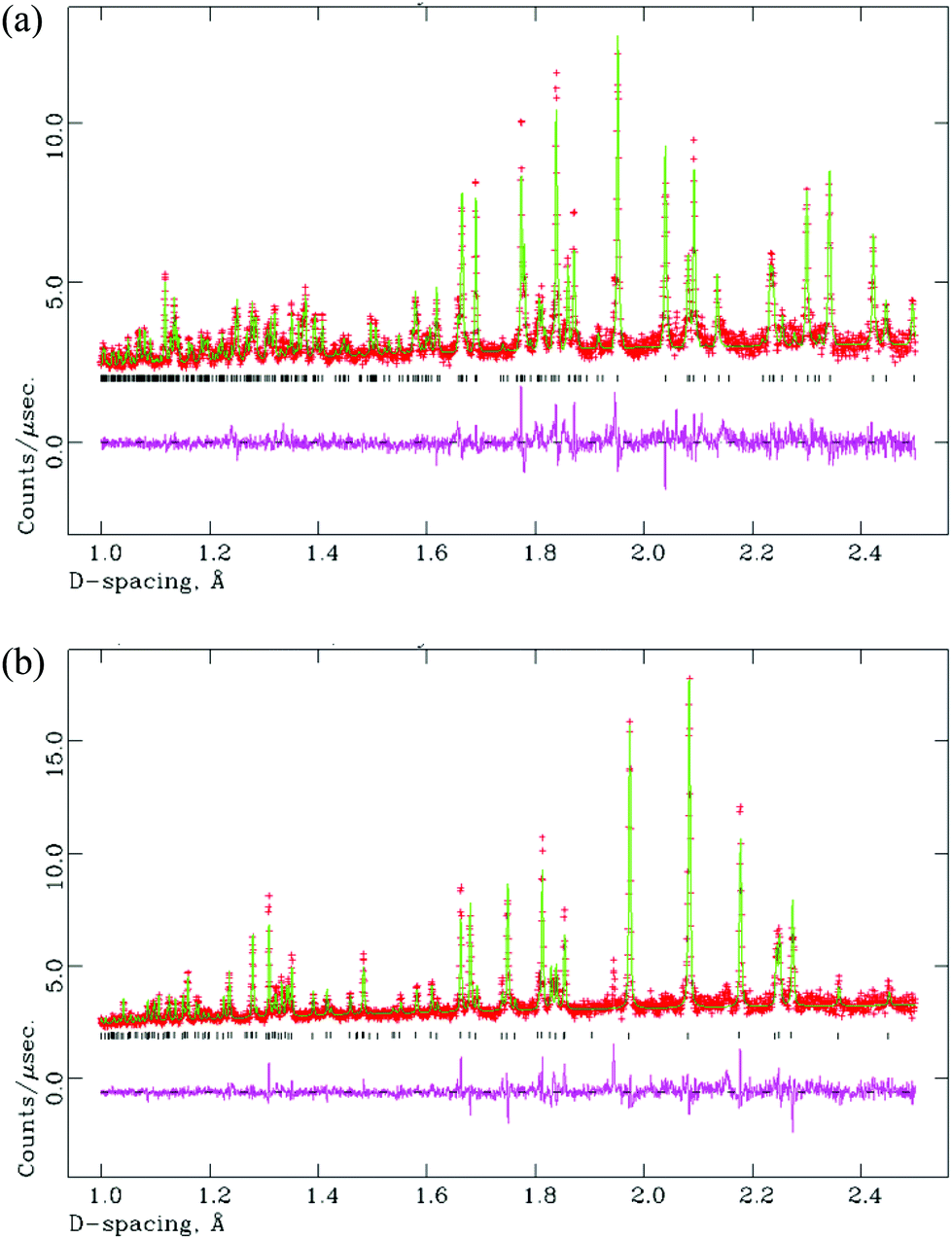 | ||
| Fig. 7 Rietveld fits (NPD) for La0.90Nd0.10TaO4 at (a) 100 °C (M-LaTaO4 model); χ2 = 2.58 for 45 variables, and (b) 500 °C (O-LaTaO4 model); χ2 = 3.26 for 37 variables. Note that the prominent peak at d ∼ 1.95 Å is instrumental (it does not appear in the room-temperature XPD data). | ||
Electrical properties
Dielectric data for O-LaTaO4 and M-La0.9Nd0.1TaO4 are shown in Fig. 8 (additional plots at a range of frequencies are provided in ESI†). It should be noted that the presence of other phase(s) in these samples are at level unlikely to contribute significantly to the overall dielectric response given the low volume fraction and similarity in relative permittivity. O-LaTaO4 exhibits a broad peak in permittivity with a maximum at ca. 200 °C, Fig. 8(a), which corresponds to the structural change observed in the diffraction data. The peak is observed on both heating and cooling, indicating this structural relaxation is reversible and shows no thermal hysteresis. Data for M-La0.9Nd0.1TaO4 show a sharp increase in permittivity beginning at ca. 250 °C on heating, corresponding to the M–O phase transition, Fig. 8(b). This feature shows a large thermal hysteresis consistent with the first order nature of the phase transition. A second feature in the permittivity in the form of a small peak at ca. 400 °C is evident on both heating and cooling (and with no temperature hysteresis). The overall dielectric response for this composition is reminiscent of that observed for the ferroelectric tetragonal tungsten bronze compound GdK2Nb5O15 recently reported by Gagou et al.;17 based on diffraction data the authors of that study assigned the small peak in permittivity to an antiferroelectric-paraelectric phase transition. Unfortunately we have insufficient high temperature diffraction data for our La0.9Nd0.1TaO4 sample in this region to be able to make a similar comparison, but it is worthy to note that short range antiferroelectric ordering has been reported in isostructural BaMnF4,18 which arises from an alternate rotation direction of perovskite blocks along the a-axis giving an incommensurate modulation. Also, if the “anti-polar” alternate octahedral rotations have no long-range order but are present as nano-domains or interspersed in a manner akin to stacking faults, these are unlikely to be readily detectable by diffraction.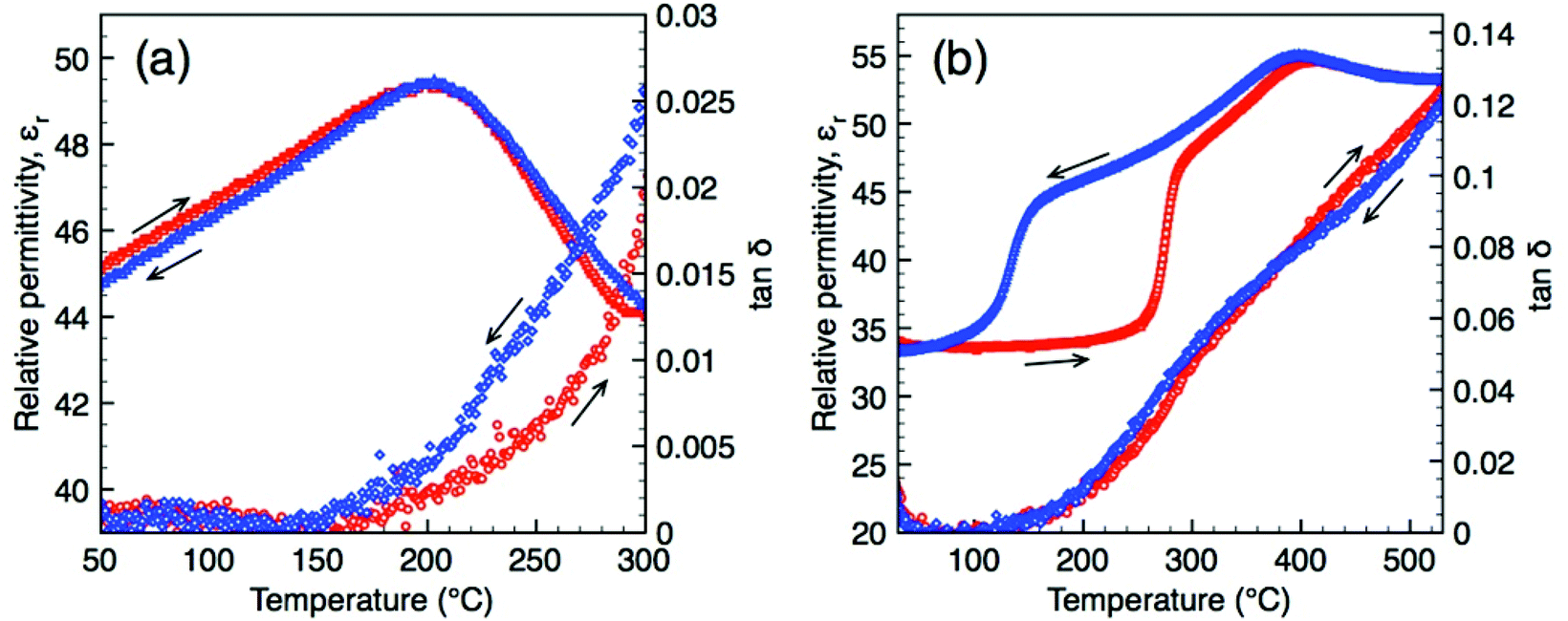 | ||
| Fig. 8 Dielectric data (obtained at 1 MHz) as a function of temperature for LaTaO4 (a) and La0.90Nd0.10TaO4 (b). | ||
Polarisation-field (P–E) measurements were carried out on O-LaTaO4 in an attempt to confirm the ferroelectric nature of this polymorph. P–E data obtained at room temperature under an applied field of 100 kV cm−1 at 1 kHz showed a linear dielectric response and no evidence of ferroelectric switching (see ESI†). The coercive field for isostructural BaMgF4 is known to increase dramatically with frequency,19,20 suggesting a high nucleation energy for switching. In order to investigate this, measurements were repeated at lower frequencies (down to 0.1 Hz). Unfortunately, however, under these conditions the samples became increasingly leaky (ESI†) and so no conclusive evidence for ferroelectric switching was obtained. The polar (orthorhombic) phase of La0.9Nd0.1TaO4 is only stable at temperatures above our measuring capabilities.
Structural comparisons
Both structure types, M- and O-, can be regarded as derived from an aristotype phase having no octahedral tilting, which may be described in space group Cmcm (a ∼ 4 Å, b ∼ 14.8 Å, c ∼ 5.65 Å (∼a√2), as shown in Fig. 1(a). Naturally, there are many possible ways to lower the symmetry of the aristotype phase through structural distortions; those based on ‘octahedral tilting’ have been classified for the wider family of AnBnX3n+2 phases by Levin.21 In Levin's notation, the O-LaTaO4 structure corresponds to the a+ tilt system and the M-LaTaO4 structure corresponds to a−b (note that this notation is similar but distinct from that used in the Glazer system for conventional perovskites). The key difference is that in the O-phase tilts of successive octahedra along the a-axis are in-phase, whereas in the M-phase the corresponding tilts (c-axis) are out-of-phase, and tilting relative to the other aristotype axes is also permitted (Fig. 1). This aspect of the tilting, coupled with the layered nature of these structures, leads to the topological ferroelectric nature of the a+ tilt system, but allows retention of centrosymmetricity in the a− tilt system.Using first-principles calculations, Ederer and Spaldin6 suggested that the topological ferroelectricity observed in the BaMF4 family was driven by a single polar soft-mode, essentially a ‘rigid’ octahedral tilt. However, employing similar methods, López-Pérez and Íñiguez7 found that in the n = 4 member of this layered perovskite family, La2Ti2O7, the cooperative interaction of two distinct soft-modes is fundamentally important. They suggest that although the tilt mode is the driver for polarity, coupling with an octahedral distortion mode is significant, and is actually the majority contributor to the overall calculated polarisation. This may suggest that the conventional ‘d0-ness’ criterion of ferroelectric activity does actually play a part in the oxide members of this family but not so much in the fluorides. In order to probe these effects further we can compare the degree of distortion within the octahedral units of several of these phases. Firstly, from the present work we can analyse the distortions present in the O-phase of LaTaO4 over a range of temperatures, and compare this to the O- and M-phases in La0.9Nd0.1TaO4. We can define simple ‘distortion indices’ for the octahedra, based on deviations of the six bond lengths from the mean, and the twelve bond angles from 90°:
| Δ1 = 1/6∑|Rav − Ri| |
| Δ2 = 1/12∑|90 − ϕi| |
These are presented in Table 1, and compared to selected examples from the BaMF4 series, based on well-determined single crystal X-ray studies.22,23 Also included for comparison are values for the related n = 4 phases La2Ti2O7 (theoretical study7) and Sr2Nb2O7 (single crystal X-ray study24), and for BiReO4 and NaAlF4, which both adopt the parent Cmcm structure for the n = 2 series.25,26 In the case of the n = 4 phases there are two distinct B sites (Fig. 9), representing ‘inner two’ and ‘outer two’ octahedra of each perovskite block. It has previously been noted8 that the distortion of the outer octahedra are greater than those of the inner ones; this is a natural consequence of the weaker inter-block versus intra-block bonding (i.e., the inner octahedra are directly linked to six neighbouring octahedra, whereas the outer ones are linked to four).
 | ||
| Fig. 9 Nature of the octahedral distortions in selected members of the AnBnX3n+2 family: (a) O-LaTaO4 at 100 °C (present study) (b) M-NaCrF4 at 25 °C (ref. 23) (c) O-Sr2Nb2O7 at 25 °C (ref. 24). Note that the latter contains two distinct octahedral sites. | ||
| Phase | Δ1 (Å) | Δ2 (°) | Distortion type |
|---|---|---|---|
| O-LaTaO4 (100 °C) | 0.044 | 5.9 | Edge |
| O-LaTaO4 (500 °C) | 0.073 | 5.0 | Edge |
| M-La0.9Nd0.1TaO4 (100 °C) | 0.039 | 4.8 | Indistinct |
| O-La0.9Nd0.1TaO4 (500 °C) | 0.053 | 5.1 | Edge |
| O-BaMnF4 (25 °C)22 | 0.024 | 4.3 | Indistinct |
| O-BaCoF4 (25 °C)22 | 0.047 | 3.3 | Axial |
| O-BaNiF4 (25 °C)22 | 0.055 | 3.1 | Axial |
| M-NaCrF4 (25 °C)23 | 0.031 | 2.0 | Edge |
| NaAlF4 (25 °C)25 | 0.018 | 1.3 | Edge |
| BiReO4 (25 °C)26 | 0.056 | 3.2 | Edge |
| O-La2Ti2O7 (calc)7 | 0.088/0.071 | 6.2/5.6 | Apex |
| O-Sr2Nb2O7 (25 °C)24 | 0.125/0.084 | 7.3/5.6 | Apex |
As a further comparison to these values, the corresponding Δ1 and Δ2 values for the tetragonal ferroelectric phase of BaTiO3 at room temperature are 0.067 Å and 2.5°, respectively.27
It can be immediately seen that all the examples given do have significant octahedral distortions. Moreover, these distortions have a significant component along the polar c-axis in the O-phases, and hence will contribute to any net polarisation. From the relatively limited data available it can also be suggested that (i) the oxide members of the n = 2 series generally have larger distortions than the fluorides, (ii) the n = 4 members of the series have enhanced octahedral distortions relative to the n = 2 members, even in the case of the ‘inner’ octahedral layers. However, the representation of the octahedral distortions in terms of these two numerical parameters does not necessarily tell the full story; the nature of the distortion differs, as represented in Fig. 9. In general, the B cation in a perovskite can be considered to be displaced towards an octahedral apex, edge or face (compare the tetragonal, orthorhombic and rhombohedral ferroelectric phases of BaTiO3, for example). Also shown in Table 1, and illustrated for selected examples in Fig. 9, is a rough classification of the different distortion types present in this family. As can be seen, for the fluorides there is no clear trend to the distortion types, whereas for the oxides there are two distinct types of displacement: edge-oriented for the n = 2 O-phases and apex-oriented for the n = 4 O-phases. Due to the limited data available, it is dangerous to draw any global conclusion from this, but it is clear that the d0 cations do display the types of distortion commonly seen in oxides; whether there is an inherent preference for a particular cation to show a particular type of distortion is unclear. Such effects have been discussed by Halasyamani and co-workers,28 who suggest that Ti4+, Nb5+ and Ta5+ show roughly similar preferences for edge- and apex-distortions. Therefore it is likely that the specific details of inter-block versus intra-block bonding and the nature of the A cation in these complex, highly distorted structures is also significant.
We suggest that, although rigid octahedral tilting in the O-phases intrinsically provides a polarisation, in reality the octahedra are far from rigid, and the complex nature and differences in these deviations from ideality are not necessarily straightforward to predict, depending on a subtle difference in compatibility of the perovskite blocks and separating layers, together with the size and electronic nature of the B cations.
Conclusions and further work
We have studied various aspects of the phase behaviour of LaTaO4 and related members of the AnBnX3n+2 series of layered perovskites. This work demonstrates that, under appropriate reaction conditions, the polar O-LaTaO4 phase can be stabilised at room temperature, whereas previous work had suggested that the centrosymmetric M-LaTaO4 polymorph is dominant. Variable temperature powder diffraction studies reveal an unusual anisotropic thermal expansion effect, driven by anomalous behaviour of inter-octahedral bond angles. This structural anomaly correlates with a dielectric maximum. Despite the polar nature of this polymorph it has not been possible to demonstrate ferroelectric switching under the conditions employed here.Further, we have shown that the M-LaTaO4 polymorph can be stabilised to room temperature by a small amount of Nd doping at the La site: a consequence of the smaller A-cation size. In the series La0.9Nd0.1TaO4 we have shown that the stability range of the M-phase at ambient temperature extends to x ∼ 0.50. For the x = 0.1 case, we have shown that a first-order phase transition from the M- to the O-phase occurs in the region 250–350 °C. It is anticipated that the temperature of this phase transition will increase markedly for higher x-values, and further work on this is prompted. The dielectric data for x = 0.1 also suggests a further structural feature to be present at ca. 400 °C, and the intriguing possibility of antiferroelectric ordering; this also requires further study.
By comparing the present crystallographic data with selected previous examples of this structural family, we conclude that significant octahedral distortions are prevalent in all cases. This supports the previous theoretical studies, which suggest that although rigid octahedral tilting drives the transition into the polar O-phase, octahedral distortions are key in enhancing and stabilising the polarisation in this topological ferroelectric family. Although some broad general comments can be made on the nature and magnitude of these distortions, there is insufficient high quality structural data available to clarify specific ‘design’ principles, for the optimisation of net polarisation. The contributions of octahedral tilting versus octahedral distortion should therefore be considered on a case-by-case basis, and further systematic structural studies, on a more diverse range of compositions and structural variants, need to be carried out.
The transformation of an ambient temperature centrosymmetric phase into a higher temperature polar phase is highly unusual; indeed we are aware of no other examples of this amongst oxide ferroelectrics. It is anticipated that both the M- and O-polymorphs within this family will transform to an aristotype centrosymmetric phase (space group Cmcm) at high temperature. Although examples of the aristotype phase have been structurally characterised (e.g., BiReO4 and Sr2Ta2O7, which transforms to the polar O-phase at −107 °C), the nature of this transition has not been studied in detail previously. This would provide a very interesting study, in order to probe the relative importance of the octahedral tilt and distortion modes near TC.
Note added in proof
We note the recent observation of a uniaxial NTE in another family of layered perovskites (Senn et al., Phys. Rev. Lett., 2015, 114, 035701).Acknowledgements
We thank the University of St Andrews and EPSRC (via DTG studentships to CALD and JG) for funding, STFC for providing neutron facilities and Cameron Black and Irene Munaò for experimental assistance. MS would like to acknowledge the International Association for the Exchange of Students for Technical Experience (IAESTE) for the opportunity to study at St Andrews.Notes and references
- R. E. Cohen, Nature, 1992, 358, 136 CrossRef CAS.
- B. B. van Aken, T. T. M. Palstra, A. Filippetti and N. A. Spaldin, Nat. Mater., 2004, 3, 164 CrossRef CAS PubMed.
- C. J. Fennie and K. M. Rabe, Phys. Rev. B: Condens. Matter, 2005, 72, 100103 CrossRef.
- A. S. Gibbs, K. S. Knight and P. Lightfoot, Phys. Rev. B: Condens. Matter, 2011, 83, 094111 CrossRef.
- N. A. Benedek and C. J. Fennie, Phys. Rev. Lett., 2011, 106, 107204 CrossRef PubMed.
- C. Ederer and N. A. Spaldin, Phys. Rev. B: Condens. Matter, 2006, 74, 024102 CrossRef.
- J. López-Pérez and J. Íñiguez, Phys. Rev. B: Condens. Matter, 2011, 84, 075121 CrossRef.
- F. Lichtenberg, A. Herrnberger and K. Wiedenmann, Prog. Solid State Chem., 2008, 36, 253 CrossRef CAS.
- R. J. Cava and R. S. Roth, J. Solid State Chem., 1981, 36, 139 CrossRef CAS.
- F. Vullum, F. Nitsche, S. M. Selbach and T. Grande, J. Solid State Chem., 2008, 181, 2580 CrossRef CAS.
- I. Hartenbach, F. Lissner, T. Nikelski, S. F. Meier, H. Müller-Bunz and T. Schleid, Z. Anorg. Allg. Chem., 2005, 631, 2377 CrossRef CAS.
- K. P. F. Siqueira and A. Dias, Mater. Res., 2014, 17, 167 CrossRef CAS.
- M. Nyman, M. A. Rodriguez, L. E. S. Rohwer, J. E. Martin, M. Waller and F. E. Osterloh, Chem. Mater., 2009, 21, 4731 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, Los Alamos National Laboratory Report No. LA-UR-86-748, 2000 (unpublished).
- A. Wölfel, P. Dorscht, F. Lichtenberg and S. van Smaalen, Acta Crystallogr., Sect. B: Struct. Sci., 2013, 69, 137 Search PubMed.
- Yu. A. Titov, A. M. Sych, A. N. Sokolov, A. A. Kapshuk, V. Ya. Markiv and N. M. Belyavina, J. Alloys Compd., 2000, 311, 252 CrossRef CAS.
- Y. Gagou, Y. Amira, I. Lukyanchuk, D. Mezzane, M. Courty, C. Masquelier, Yu. I. Yuzyuk and M. El Marssi, J. Appl. Phys., 2014, 115, 064104 CrossRef.
- M. Hidaka, T. Nakayama, J. F. Scott and J. S. Storey, Physica B and C, 1987, 144, 310 CrossRef CAS.
- K. Shimamura, E. G. Víllora, H. Zeng, M. Nakamura, S. Takekawa and K. Kitamura, Appl. Phys. Lett., 2006, 89, 232911 CrossRef.
- E. G. Víllora, K. Shimamura, F. Jing, A. Medvedev, S. Takekawa and K. Kitamura, Appl. Phys. Lett., 2007, 90, 192909 CrossRef.
- I. Levin and L. A. Bendersky, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 853 CrossRef.
- S. W. Kim, H. Y. Chang and P. S. Halasyamani, J. Am. Chem. Soc., 2010, 132, 17684 CrossRef CAS PubMed.
- G. Knoke, W. Verscharen and D. Babel, J. Chem. Res.Synop, 1979, 7, 213 Search PubMed.
- N. Ishizawa, F. Marumo, T. Kawamura and M. Kimura, Acta Crystallogr., Sect. B: Struct. Sci., 1975, 31, 1912 CrossRef.
- A. Le Bail, Powder Diffr., 2009, 24, 301 CrossRef CAS.
- A. R. Rae-Smith and A. K. Cheetham, J. Solid State Chem., 1979, 30, 345 CrossRef.
- G. H. Kwei, A. C. Lawson Jr., S. J. L. Billinge and S.-W. Cheong, J. Phys. Chem., 1993, 97, 2368 CrossRef CAS.
- K. M. Ok, P. S. Halasyamani, D. Casanova, M. Llunell, P. Alemany and S. Alvarez, Chem. Mater., 2006, 18, 3176 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt03721a |
| This journal is © The Royal Society of Chemistry 2015 |