
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Binding of an organo–osmium(II) anticancer complex to guanine and cytosine on DNA revealed by electron-based dissociations in high resolution Top–Down FT-ICR mass spectrometry†
Christopher A.
Wootton
a,
Carlos
Sanchez-Cano
a,
Hong-Ke
Liu
ab,
Mark P.
Barrow
 a,
Peter J.
Sadler
*a and
Peter B.
O'Connor
*a
a,
Peter J.
Sadler
*a and
Peter B.
O'Connor
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: p.oconnor@warwick.ac.uk; p.j.sadler@warwick.ac.uk; Fax: +44 (0)24 761 51009, +44 (0)24 765 23819; Tel: +44 (0)24 761 51008, +44 (0)24 765 23818
bJiangsu Collaborative Innovation Center of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Wenyuan Road 1, Nanjing 210023, PR China
First published on 4th February 2015
Abstract
The OsII arene anticancer complex [(η6-bip)Os(en)Cl]+ (Os1-Cl; where bip = biphenyl, and en = ethylenediamine) binds strongly to DNA. Here we investigate reactions between Os1-Cl and the self-complementary 12-mer oligonucleotide 5′-TAGTAATTACTA-3′ (DNA12) using ultra high resolution Fourier Transform-Ion Cyclotron Resonance Mass Spectrometry (FT-ICR MS). Identification of the specific sites of DNA osmiation with {(η6-bip)Os(en)}2+ was made possible by the use of Electron Detachment Dissociation (EDD) which produced a wide range of assignable osmiated MS/MS fragments. In contrast, the more commonly used CAD and IRMPD techniques produced fragments which lose the bound osmium. These studies reveal that not only is guanine G3 a strong binding site for {(η6-bip)Os(en)}2+ but, unexpectedly, so too is cytosine C10. Interestingly, the G3/C10 di-osmiated adduct of DNA12 also formed readily but did not undergo such facile fragmentation by EDD, perhaps due to folding induced by van der Waal's interactions of the bound osmium arene species. These new insights into osmium arene DNA adducts should prove valuable for the design of new organometallic drugs and contribute to understanding the lack of cross resistance of this organometallic anticancer complex with cisplatin.
Introduction
Metal based therapeutics are currently at the forefront of anticancer therapy, with compounds such as cisplatin being used extensively in chemotherapy treatments.1 Potent cytotoxic and anti-proliferative compounds have also been developed based on a range of transition metals, including ruthenium,2 iridium,3 vanadium,4 cobalt,5 osmium,6 as well as platinum.7–9 Though many of these compounds have been shown to exhibit different and interesting mechanisms of action (MoA), many metal-based drugs are believed to achieve their cytotoxic/anti-proliferative effect via binding to the strands of DNA within the nuclei of cancer cells.10 This binding of metallodrugs to DNA residues often causes a change in the conformation/structure of the DNA strands, and the deformation of tertiary structure can then trigger the cytotoxic and/or anti-proliferative effects, e.g. via apoptosis.10,11It is therefore of much interest to study how these new metallodrugs can bind to DNA, which nucleotide residues are preferred binding partners, whether certain nucleotide sequences are targeted selectively, and to characterise the coordination sphere of the bound metal. Many metallodrugs change their composition during their journey to DNA, often multiple times, depending on various environmental factors (e.g. pH changes, chloride concentration, small/large biomolecule binding) and so the metallodrug injected into a system may not be the active species eventually causing the desired anticancer effects.12 Due to the multiple activation pathways in which injected pro-drugs become active metallodrugs, and many pathways of deactivation of these compounds, it has often been observed that a variety of reaction products are observed when metal-based compounds encounter biomolecules.13
Many analytical techniques have been utilised in order to study metal complex–DNA interactions, often 1H-NMR or X-ray crystallography, with both techniques providing powerful structural information and sometimes identifying the location of DNA modification.14,15 However, the mixture of products produced pose problems for both NMR and X-ray crystallography, and although separation of reaction products, e.g. via High Pressure Liquid Chromatography (HPLC), can assist in the acquisition of purer samples for analysis, this invariably ends in further dilution of samples and can often affect equilibria in dynamic systems, which may render separations ineffective via decomposition, hydrolysis etc.
To address these issues, other analytical techniques are being explored which offer superior sensitivity (and reduced sample requirements) and can handle mixtures of adducts more effectively. Mass spectrometry is increasingly being utilised to study biological and complex chemical systems, due to its inherent high sensitivity and ability to cope with extremely complex samples (over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 species have been detected in a single mass spectrum16). With the advent of tandem mass spectrometry (MS/MS), the ability to fragment and interrogate ionised species in the gas phase has paved the way for in-depth chemical and structural analysis of complex systems,17 with biomolecules being the main focus for contemporary tandem mass spectrometry studies. An extensive array of ion dissociation techniques has been developed including Collisionally Activated Dissociation (CAD),18 Electron Capture Dissociation (ECD),19 Electron Transfer Dissociation (ETD),20 Infra-Red Multi Photon Dissociation (IRMPD),21 Electron Induced Dissociation (EID),22 Ultra-Violet Photo Dissociation (UVPD),23 and Electron Detachment Dissociation (EDD).24 There are now many established methods for interrogating and studying biomolecules and their modifications via MS/MS analysis, each suited to a variety of different analytes, and many able to offer complementary data to provide increased confidence in analysis.
000 species have been detected in a single mass spectrum16). With the advent of tandem mass spectrometry (MS/MS), the ability to fragment and interrogate ionised species in the gas phase has paved the way for in-depth chemical and structural analysis of complex systems,17 with biomolecules being the main focus for contemporary tandem mass spectrometry studies. An extensive array of ion dissociation techniques has been developed including Collisionally Activated Dissociation (CAD),18 Electron Capture Dissociation (ECD),19 Electron Transfer Dissociation (ETD),20 Infra-Red Multi Photon Dissociation (IRMPD),21 Electron Induced Dissociation (EID),22 Ultra-Violet Photo Dissociation (UVPD),23 and Electron Detachment Dissociation (EDD).24 There are now many established methods for interrogating and studying biomolecules and their modifications via MS/MS analysis, each suited to a variety of different analytes, and many able to offer complementary data to provide increased confidence in analysis.
Fourier Transform Ion Cyclotron Resonance Mass Spectrometry (FT-ICR MS) uses electric and magnetic fields to trap ions of interest within a Penning trap during MS/MS experiments and detection,25,26 allowing ultra-high resolving powers of 500000–10000000+ with the latest commercial setups.27 Combining the flexibility of the ICR cell with quadrupole isolation and collisional activation, FT-ICR MS offers the largest array of fragmentation techniques and the highest mass accuracy of any mass spectrometer, providing the highest possible confidence in spectral assignment, with mass errors into the sub part per million (ppm) and even ppb (part per billion) ranges. Importantly, FT-ICR MS is uniquely and perfectly suited to Top Down fragmentation studies.28,29
Here we study the metallation of a DNA 12-mer oligonucleotide by the organometallic OsII arene anticancer complex, Os1-Cl·PF6 (Scheme 1). We use Ultra High Resolution Mass Spectrometry (UHR-MS) together with MS/MS analysis to locate the osmium binding sites and to identify the coordinated ligands. In particular we use a range of fragmentation techniques to gain new insights into the nature of the osmium binding sites on DNA.
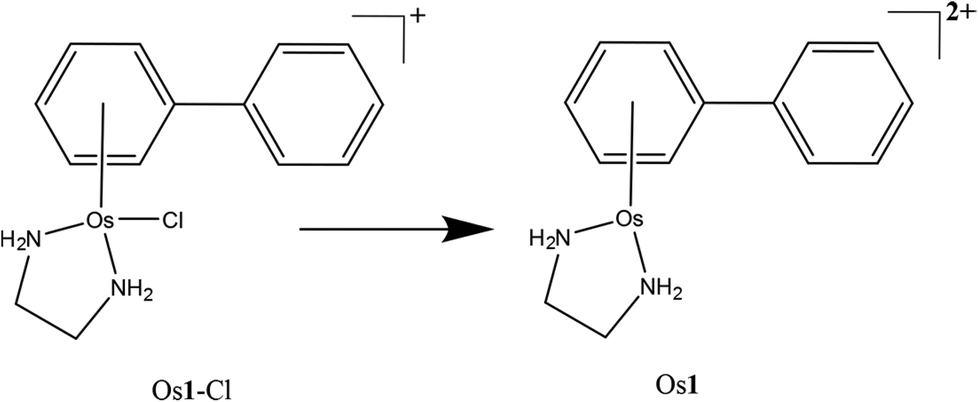 | ||
| Scheme 1 The OsII arene complex Os1-Cl studied in this work and the detected species/modification observed via nESI-MS (Os1). | ||
Experimental
[(η6-bip)Os(en)Cl]PF6 (Os1-Cl·PF6) was synthesised and characterised as described previously.6The 12-mer DNA oligonucleotide DNA12 is defined here for MS purposes as fully protonated, i.e. each of the 11 phosphates is protonated:
| 5′-TAGTAATTACTA-3′ (DNA12) |
DNA12 (Tm 28 °C) was purchased desalted from DNA Technology (Denmark). Further purification was performed by HPLC, using an Agilent 1200 series liquid chromatography system. The oligonucleotide was separated from shorter oligonucleotides (present as minor impurities in the commercial sample) using a PL-SAX ion exchange column (1000 Å pore size; 8 μM particle size; 150 mm length, 4.6 mm diameter; Polymer Laboratories, Amherst, USA); buffer A: 7% acetonitrile + 0.1 M triethylammonium acetate (TEAA), pH 7; buffer B: 7% acetonitrile + 0.1 M TEAA + 1 M sodium acetate, pH 7; temp 40 °C; flow rate 1.5 ml min−1; gradient: 0–5 min 0% B, 5–45 min 0–40% B, 45–55 min 40–100% B, 55–65 min 100% B, 65–75 min 0% B (see ESI Fig. S1:† HPLC traces of commercial sample and inset; the resulting purified fraction). The solvent was removed on a freeze-drier, and a reverse phase chromatography PLRP-S column (100 Å pore size; 8 μM particle size; 300 mm length, 7.5 mm diameter; Polymer Laboratories, USA) was used to remove sodium acetate; buffer A: 50 mM TEAA, pH 7; buffer B: 70% acetonitrile + 50 mM TEAA, pH 7; temp 40 °C; flow rate 2 ml min−1; gradient: 0–5 min 5% B, 5–35 min 5–100% B, 35–45 min 100% B, 45–45 min 0% B. The solvent was removed again on a freeze-drier.
Aqueous solutions of the oligonucleotide (250 μM) and osmium complex Os1-Cl·PF6 (250 μM) were mixed in 0.5:1 and 1:1 Os:oligonucleotide ratios. Under these conditions of low ionic strength, DNA12 would be present largely as a single strand. Osmium-oligonucleotide samples were then incubated in the dark at 37 °C for 3 hours before dilution with purified (Milli-Q) water to mass spec concentrations (ca. 0.5–1 μM total concentration) for FT-ICR MS analysis.
FT-ICR mass spectrometry analysis
All samples were analysed via nano-electrospray ionisation (nESI) for increased sensitivity and lower sample consumption compared to traditional electrospray ionisation (ESI).30 All experiments were carried out on a solariX FT-ICR Mass Spectrometer, fitted with a 12 tesla actively-shielded magnet (Bruker Daltonik GmbH, Bremen, Germany), and samples were sprayed in negative-ion mode unless otherwise stated. For MS experiments, ions were accumulated for 0.01 s in the hexapole-based collision cell before transfer to the infinity cell31 for detection.For Collisionally Activated Dissociation (CAD) MS/MS experiments, ions of interest were isolated in the front end quadrupole using an isolation window of 3–10 m/z to isolate deprotonated species only. Ions were then accelerated into argon collision gas at 8 V (for unmodified DNA species) and 10–12 V (for Os-modified species). Ions were continuously accumulated for 0.1–2 s before transmission and detection.
For Infra-Red Multi Photon Dissociation (IRMPD) experiments, ions of interest were isolated, accumulated for 0.1–2 s, then transferred and trapped in the infinity cell. Trapped ions were then subjected to infra-red photons prior to detection. IR photons were produced from a continuous wave, 25 W, CO2 laser (Synrad Inc., Washington, USA) held at 60% power output and pulsed into the infinity cell for 30–100 ms.
For Electron Detachment Dissociation MS/MS experiments, ions of interest were isolated in the front end quadrupole, accumulated in the hexapole for 0.7–3 s, then transferred and trapped in the infinity cell. Trapped ions were then irradiated for 0.8 s with 20.2 eV electrons produced from a 1.5 A indirectly-heated hollow cathode dispenser via an extraction lens held at 18 V.
All spectra were internally calibrated using a quadratic calibration function32 and then manually interpreted and assigned via Data Analysis v4.2 (Bruker Daltonik GmbH, Bremen, Germany). All fragments in the low to sub-ppm range were assigned; tables of assignments can be found in the electronic supplementary information† for each MS/MS spectrum assigned here.
Results and discussion
Electrospray ionisation (including nESI) forms ions via the addition or removal of protons during the desolvation process occurring after the emission of sample droplets from the electrospray needle tip and while passing through the electric fields in the source region of the mass spectrometer.33 Due to the high density of phosphate groups in oligonucleotides; negative mode electrospray produces abundant [M − nH]n− ions in the resulting mass spectrum, with the phosphate groups stabilising the negative charges. This stabilisation is much greater than that seen in most peptide and protein mass spectra. As a result, DNA and RNA ESI/nESI spectra show charge states higher (per monomer) than commonly observed for poly(amino acid) species, even without the addition of additives such as bases to aid deprotonation.Fig. 1A shows the full mass spectrum of the 12-mer DNA produced by nESI. Isotopic simulations (Fig. 1A inset) match the expected pattern for the 12-mer DNA (sequence inset) and show no modification of the DNA before reaction. CAD MS/MS of the 7-charge-state species is shown in Fig. 2a; although very stable in solution, the high charge states (per nucleotide) of the DNA precursor make the gas phase species more fragile, causing it to dissociate under lower-than-average collision energies. The CAD MS/MS spectrum showed many abundant sequence fragments, most notably w ions and a-base ions.34 The nomenclature devised by McLuckey et al.34 based on the McCloskey et al. system35 for assignment of oligonucleotide MS/MS is summarised in Fig. 2a (inset, with assignments listed in Table S1 in ESI†) and was used for all assignments. Nucleotide base loss is a common fragmentation channel for CAD MS/MS, especially for a ions, which was also observed during these experiments. The overall sequence coverage was excellent, with 100% cleavage coverage for the unmodified species produced during one experimental run (Fig. 2A). It is worth noting that, due to the fragility of DNA ions in the gas phase, careful tuning of transmission and extraction voltages was essential to preserve ions of interest as they move through the mass spectrometer to avoid unwanted fragmentation of precursor ions before and after MS and MS/MS experiments. Due to the large extent of fragmentation (even at low voltages) numerous internal fragments are often observed in DNA CAD MS/MS spectra.36 These species are most often produced via fragmentation in two separate areas of the molecule, creating peaks not easily identified using a “linear” fragmentation model, though are still often predictable to some extent.37
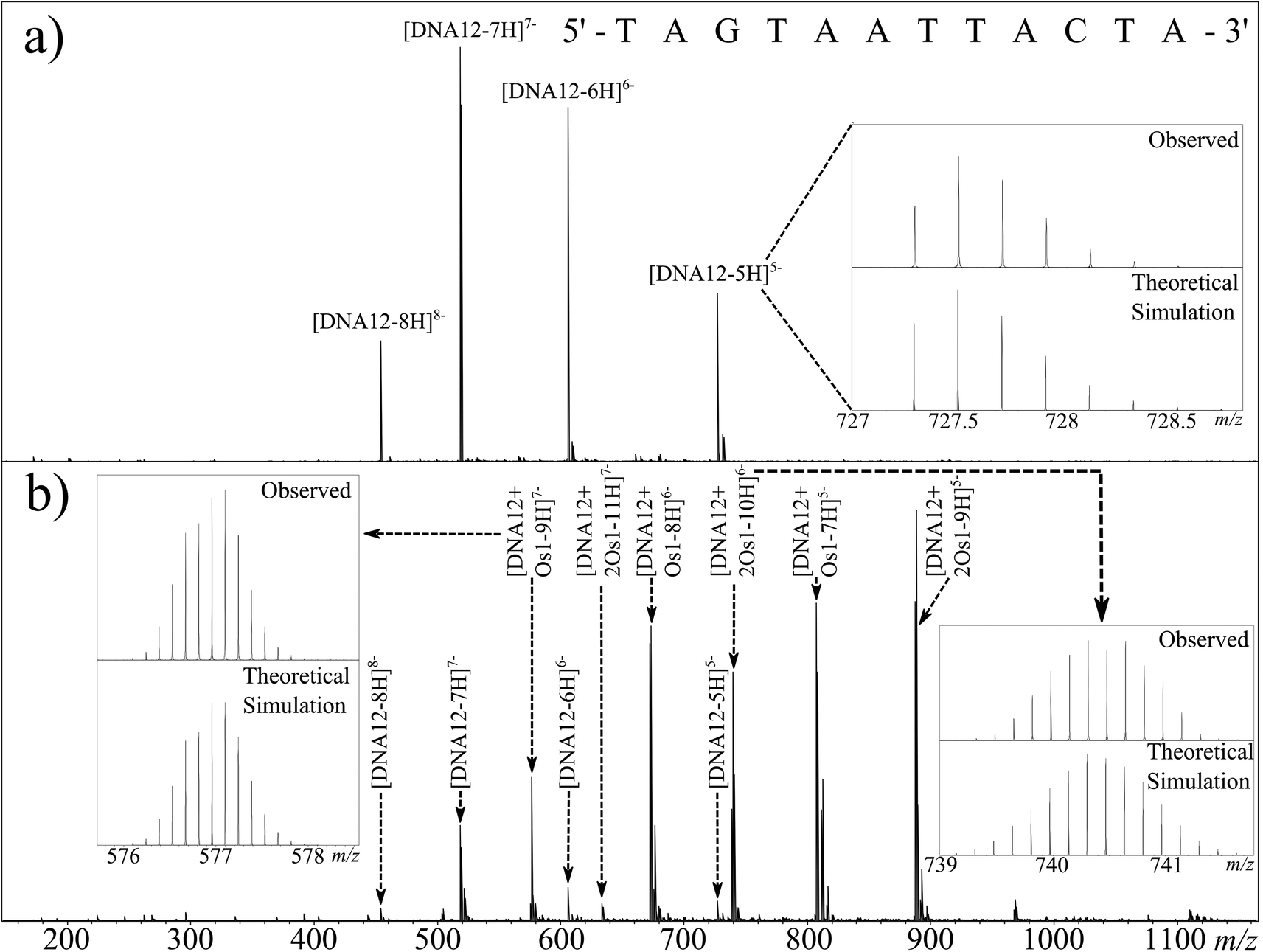 | ||
| Fig. 1 nESI Mass spectrum of (a) DNA12 and (b) DNA12 + Os1 reaction mixture after 3 h incubation at 37 °C (b). Inset: nucleotide sequence of the oligonucleotide 12-mer and observed vs. simulated isotope patterns for unreacted DNA12 (top right), DNA12 + Os1 (bottom left), and DNA12 + 2Os1 (bottom right). | ||
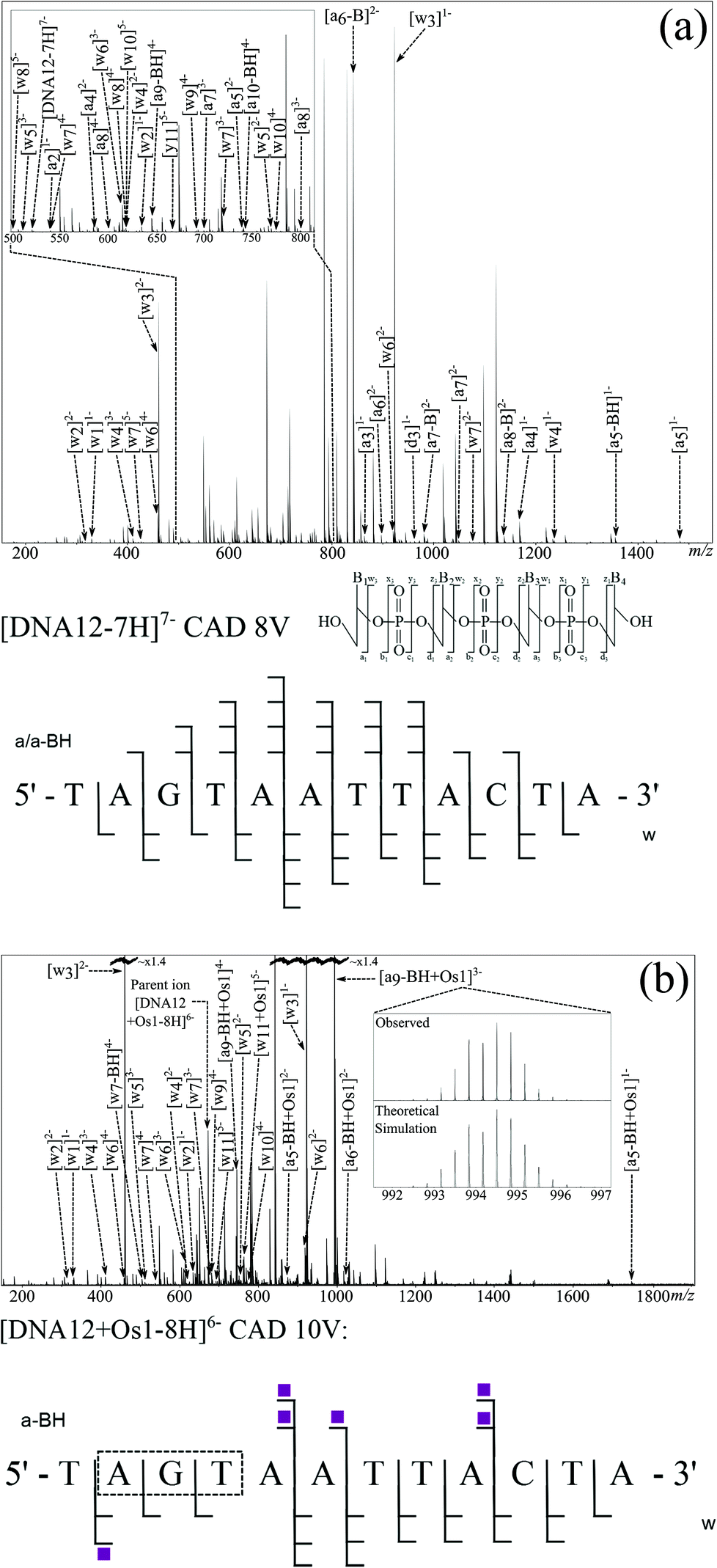 | ||
| Fig. 2 FT-ICR CAD MS/MS spectra for [DNA12-7H]7− (a) and [DNA12 + Os1-9H]7− (b) species, along with corresponding fragmentation maps; osmiated fragments are indicated with shaded squares. Inset: theoretical simulation of the [a9-BH + Os1-5H]3− fragment and the observed isotopic pattern during CAD MS/MS. | ||
Fig. 1b shows the full mass spectrum of a 1:1 solution of Os1-Cl·PF6:DNA12 (1 μM) after 12 h of reaction. The unreacted DNA peaks were readily observable as before, as were a series of peaks of osmiated DNA. Osmium-containing species are easily identifiable by UHR-MS due to the characteristic isotope pattern for osmium isotopes (Fig. 1b inset). It was immediately apparent from the full MS spectrum that the osmium complex had lost the monodentate chloride ligand, and was bound as {(η6-bip)Os(en)}2+. Since the oligonucleotides were analysed in negative ion mode and the Os1 modification is doubly charged; two additional protons have to be removed from the precursor to achieve the same charge state as the corresponding unmodified species, as detected by comparison with the theoretical isotope simulation shown in Fig. 1b (inset) depicting the simulation and observed spectrum for [DNA12 + Os1-9H]7−. Additionally two distinct Os containing oligonucleotide isotopic distributions were observed, one for the mono-osmiated DNA 12 mer ([DNA12 + Os1-(n + 2)H]n−) and one for the di-osmiated DNA 12 mer ([DNA12 + 2Os1-(n + 4)H]n−).
The usual binding site for RuII and OsII organometallic complexes within DNA is N7 of guanine, due to its high basicity and availability, even in the DNA duplex.38 However, the DNA 12 mer used during this study contained only one guanine residue. This suggested that in the present case a nucleotide other than guanine could also be a target for this metallodrug.
The CAD MS/MS spectrum for the [DNA12 + Os1-8H]6− ion is shown in Fig. 2b. Again, as with the corresponding unmodified DNA, CAD produced a spectrum with a large number of fragments, some osmiated, some not, and a range of assignable fragments (Fig. 2b and Table S2 in ESI†). Analysis of the MS/MS data showed Os-containing fragments could be used to assign the binding location to the A2–G3 region of the oligonucleotide. CAD is based upon multiple collisions of the species of interest with background gas (in this case Ar), producing a “slow heating effect” of ions until the weakest bonds within the molecule break, causing the dissociation.18 Unfortunately, due to the slow heating caused by CAD, biomolecule modifications are often dissociated along with the biomolecule backbone;39 this effect was also observed for the CAD spectra of osmiated DNA in this study, as shown by the observation of [DNA12-xH]x− species in the CAD spectra and further fragmentation. There are clear peaks showing the loss of the en ligand, followed by the loss of the whole complex during CAD, producing the unmodified species ([DNA12-xH]x−), which then fragments again under the multiple collisions in CAD and produces a string of unmodified fragments. These fragments from the unmodified species can lead to problems during data analysis, such as creating fragments which contradict the location of the modification sites, and can add extra fragment peaks to already dense MS/MS spectra. Unfortunately the loss of the DNA modification under CAD meant that unmodified fragments could not be used to locate the modification, instead only the Os-containing fragments were used during this analysis. However, despite this limitation the modification was narrowed down to the A2–G3–T4 region for the DNA 12-mer from the CAD MS/MS spectrum.
In order to identify the osmium binding sites unequivocally, further fragmentation techniques were investigated to explore their ability to fragment the backbone, while maintaining the osmium modification. It has been recently shown by Xu et al.40 that although different slow heating MS/MS techniques produce very similar MS/MS spectra of poly(amino acids) (CAD vs. IRMPD), if tuned correctly, these dissociations show a much greater difference for oligonucleotides; with changes in the proportion of fragments exhibiting base loss, internal fragmentation, and proportions of different types of ions (e.g.a & w). IRMPD was conducted on the same isolated osmiated DNA species as the CAD shown in Fig. 2b. Unfortunately, although the laser-based fragmentation produced many fragments, IRMPD dissociated the osmium modification to an even greater extent than CAD, despite varying the laser power and pulse length. Due to the extensive secondary fragmentation and modification loss; IRMPD was shown to be unsuitable for studying the osmiated DNA species here.
In view of the problems associated with dissociations caused by slow heating when studying biomolecule modifications, many reported studies now utilise electron-based fragmentations, which can cleave backbone bonds while preserving modifications/adducts. By transferring or creating radical sites on/at the backbone, fragmentation is achieved via radical reactions/rearrangements.19,41 These electron-based fragmentations have been effective in studies of modifications in positive ion mode via electron capture, both for natural modifications42 and for metal/metallodrug binding to peptides/proteins.13,43 However for negative ions, capture of electrons, such as those from Electron Capture Dissociation (ECD), is less likely due to the negative nature of the analyte, causing electrostatic repulsion. Instead other electron-based techniques such as Electron Detachment Dissociation (EDD) have been developed to allow electron-based fragmentation of negative ions.24 EDD is a dissociative technique unique to FT-ICR MS, which causes backbone cleavage by irradiating oligonucleotides or other biomolecules, with medium energy electrons (ca. 14–24 eV).44,45 The incoming electrons interact with the negative ions and cause emission/release of an electron from the analyte, creating a radical site, which, like other electron based fragmentations, can then cause dissociations along the backbone. A general pathway for this process is shown in Scheme 2.
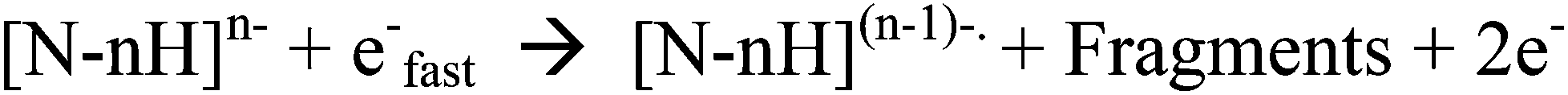 | ||
| Scheme 2 A general EDD reaction pathway, adapted from Zubarev et al.24 | ||
EDD requires very fine tuning of the electron energy and other cathode parameters to cause effective dissociation, but once tuned can result in extremely high quality fragmentation patterns. Fig. 3a shows the EDD MS/MS spectrum created by irradiation of the [DNA12-7H]7− species with 20.2 eV electrons. Due to the removal of electrons from the original ion, so called “Charge Reduced Species” (CRS) for the [DNA12-7H]6− and [DNA12-7H]5− ions are observed as high intensity peaks in the spectrum. Along with the CRS peaks, an extensive series of w and d ions can be observed and assigned, summarised in the oligonucleotide fragmentation map in Fig. 3a (and Table S3 in ESI†).
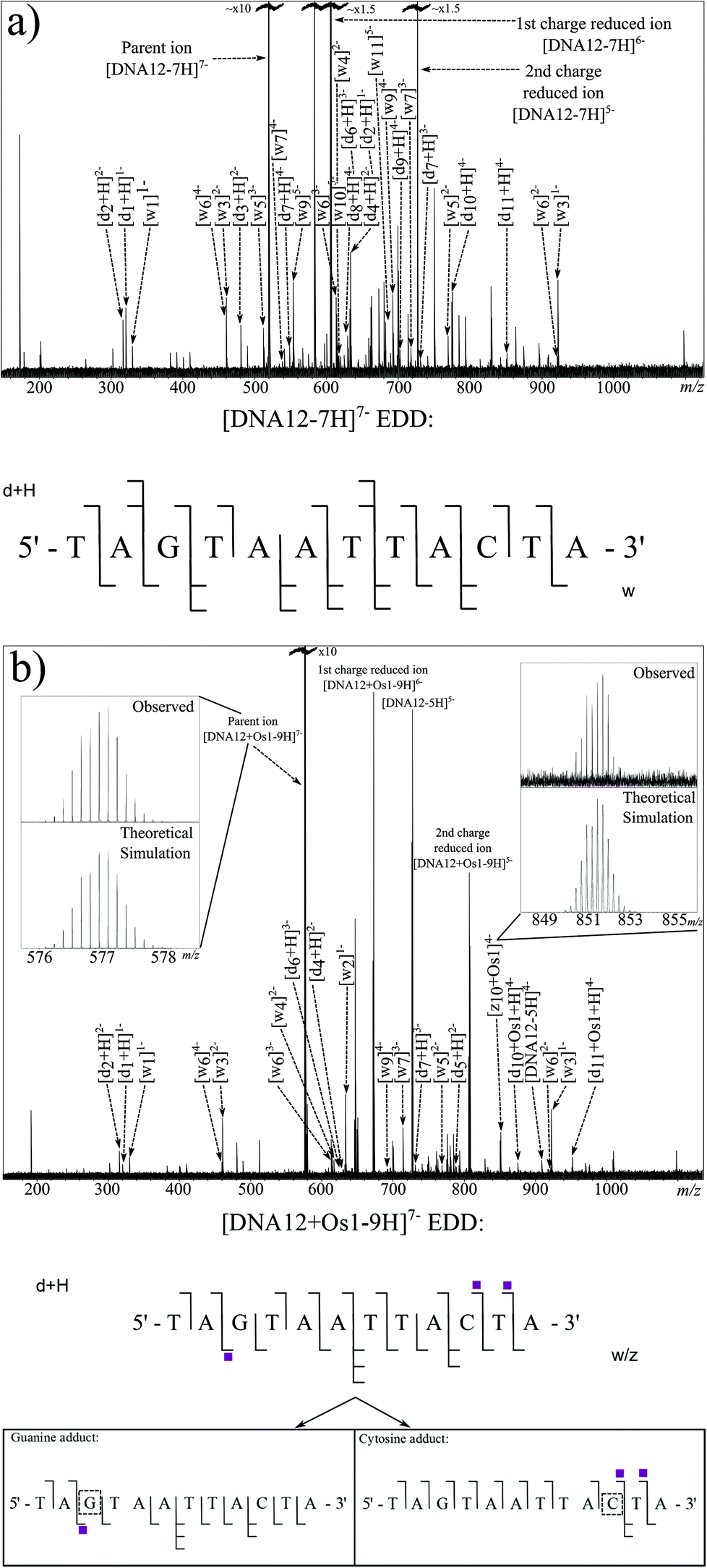 | ||
| Fig. 3 Electron detachment dissociation FT-ICR MS/MS spectra of (a) [DNA12-7H]7− and (b) [DNA12 + Os1-9H]7− ions, along with corresponding fragmentation maps. Shaded squares indicate the presence and number of osmium complex modifications bound to observed fragments. Inset: comparisons of selected theoretical simulations and observed species. | ||
EDD was then carried out on the [DNA12 + Os1-9H]− species using the same parameters as for the unmodified species. The resulting MS/MS spectrum is shown in Fig. 3b. Again, as with the previous EDD MS/MS spectrum, very intense peaks for the CRS were produced, as were a string of lower intensity fragments. Assigned fragments are summarised in the fragmentation map in Fig. 3b (and Table S4 in ESI†). EDD produced 100% cleavage coverage of the isolated species. The resulting spectrum also contained far fewer internal fragment ions, mainly consisting of w and d ions, meaning that a larger proportion of spectral peaks corresponded to sequence-informative fragments. The fragments assigned indicate two different binding locations for the bound {(η6-bip)Os(en)} complex. Fragmentation analysis clearly shows the T1–A2 and T4–A12 regions are free from osmium modification, while the G3–A12 region is modified by the osmium modification, which clearly indicated the osmium modification was bound to the guanine (G3) nucleotide. Analysis also showed a second product in which the T1–A9 region was free from osmium modification, however the T1–C10 and T1–T11 regions were Os1-modified, clearly showing the osmium complex was bound to cytosine (C10) nucleotide. Unlike CAD MS/MS scans shown above, EDD cleaved the A2–G3 bond and narrowed down the exact site of the modification to one nucleotide. A peak corresponding to the unmodified oligonucleotide was produced during the EDD experiment; [DNA12-5H]5− at m/z ∼ 727 indicating that cleavage of the metallodrug from the DNA is only a minor fragmentation channel during EDD for this species. However, unlike CAD, further fragmentation of the DNA12 ion was not observed, a CRS at sufficient intensity for the DNA12 ion was not present, indicating that after dissociation of the metallodrug, the species did not undergo enough further electron capture to cause further dissociation and affect the peak intensities of fragments. The lack of further fragmentation of the unmodified [DNA12-xH]x− ions indicated that use of unmodified fragments for fragment analysis was viable and allowed the determination of the two reaction products in two discrete locations. The loss of the biphenyl ligand from the metallodrug complex was also observed, indicating the influence of OsII on the electron dissociation process, though MS/MS analysis was not hindered in this instance.
MS/MS experiments were then conducted on the di-osmiated species at m/z ∼ 740 [DNA12 + 2Os1-10H]6−. Effective CAD was achieved at a slightly higher value of 12 V, which could indicate a stabilising effect from the osmium complex in the gas phase. Fig. 4a shows the CAD MS/MS spectrum obtained. The fragments assigned are summarised in the fragmentation map in Fig. 4a (and Table S5X in ESI†). The osmiated fragments were more numerous than for the mono-osmiated DNA12 MS/MS described earlier, and indicated binding to the A2–G3 region (correlating with the previous CAD result) and to the terminal 3 nucleotides at the 3′ end of DNA12 (C10–T11–A12). The results correlate well with the CAD and EDD results obtained for the mono-osmiated species, and are consistent with discrete G and C binding, with CAD showing poor fragmentation directly next to the osmiated nucleotide. The lack of fragmentation adjacent to the osmiated nucleotide via slow heating dissociation might indicate interactions between the osmium complex and its neighbouring nucleotides/bases; for instance, the biphenyl ligand might interact with the A2, hindering dissociation of the A2–G3 bond, and likewise for the thymine residue neighbouring the osmiated cytosine at position 10. Since CAD MS/MS of the unmodified species produced such extensive fragmentation, analysis of the differences compared to the osmiated product may be indicative of interactions with neighbouring groups.
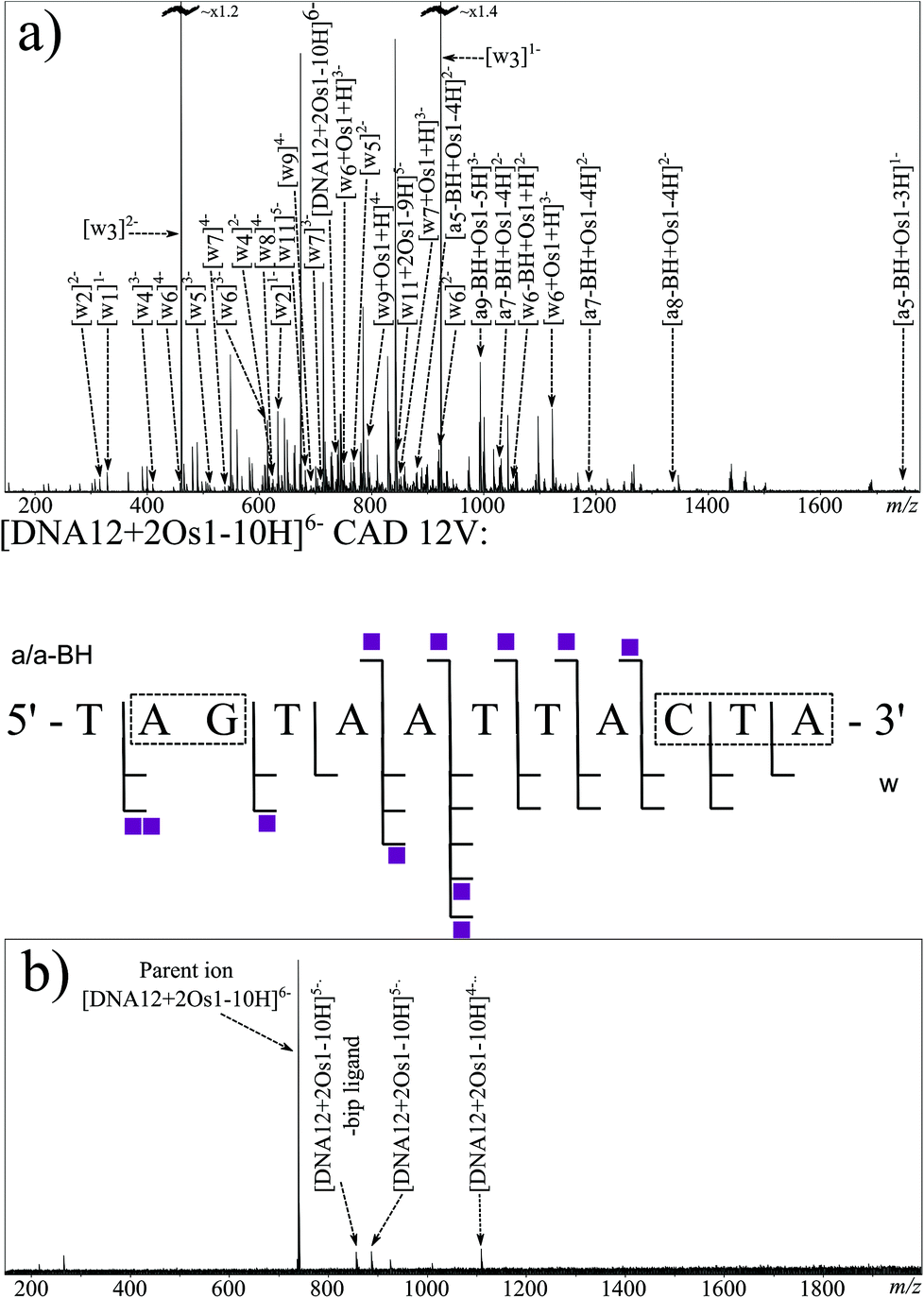 | ||
| Fig. 4 FT-ICR MS/MS spectra of the [DNA12 + 2Os1-10H]6− species fragmented by (a) CAD, (b) EDD, along with the corresponding fragmentation map for CAD (centre), no backbone dissociation was observed via EDD for the (possibly cyclic) species presumably due to non-covalent interactions preventing gas phase fragmentation. Shaded squares indicate presence and number of osmium complex modifications bound to observed fragments. | ||
EDD MS/MS was also attempted on the [DNA12 + 2Os1-10H]6− species using the same EDD parameters as before. The resulting spectrum, shown in Fig. 4b, shows consistent electron capture events as before, with the observation of the [DNA12 + 2Os1-10H]5− and [DNA12 + 2Os1-10H]4−species. Loss of the bip ligand was also observed as before. Unfortunately very little fragmentation of the DNA backbone was observed during EDD MS/MS of the di-osmiated species, which was unexpected compared to the 100% sequence coverage observed for the unmodified species and mono-osmiated oligonucleotide described above. Lack of fragmentation during electron-based dissociation such as this is usually the result of electron quenching/capture (e.g. by electron traps such as metal ions) with no resulting fragmentation thereafter, otherwise it is usually the indication of cyclisation by either covalent or non-covalent interactions, with dissociation of the backbone producing two fragments which would stay bound together by the interaction elsewhere in the macrocycle, hindering sequence-informative fragmentation. Since the osmium complex {(η6-bip)Os(en)}2+ was shown to bind to different nucleotides during the EDD MS/MS of the mono-osmiated species above, and the mass of the precursor clearly showed retention of all but the chloride ligand of Os1-Cl, it can be concluded that the osmium modifications at discrete locations at either end of the oligonucleotide (as shown by the EDD MS/MS) hinder the electron based fragmentation when both binding sites are occupied simultaneously (as shown by the EDD here). These observations could be explained by the osmium arene complexes causing folding of the oligonucleotide, most likely involving π–π stacking of two biphenyl ligands, one from each osmium complex, meaning when the oligonucleotide backbone fragments, the resulting fragments remain bound non-covalently, in a similar fashion to a poly(amino acid) CRS during electron based dissociations.45
Conclusions
The organometallic half-sandwich 'piano-stool’ osmium(II) complex Os1-Cl, is thought to exert its anticancer activity partly through binding to DNA.28 The clinical anticancer drug cisplatin also has DNA as a target site, causing deformation of the DNA structure and, eventually, leading to apoptosis. However, since Os1-Cl is not cross-resistant with cisplatin, the mode of interaction with DNA would be expected to be different. Indeed Os1-Cl is monofunctional and cannot readily crosslink DNA bases, in contrast to bifunctional cisplatin. The base specificity of Os1-Cl may also be different from cisplatin. Prior to the current work, previous studies had indicated that guanine N7 is a preferred binding site for Os1-Cl on DNA as it is for cisplatin.In this work, [(η6-biphenyl)Os(en)Cl]+ was reacted with a 12 mer DNA oligonucleotide containing single guanine and cytosine sites under conditions (low ionic strength) in which it was single-stranded. Using ultra-high resolution-FT-ICR MS up to two osmium complexes were shown to bind to DNA12. Each reaction product was fragmented via tandem mass spectrometry (MS/MS) and the identity and binding location of the modifications were elucidated.
{(η6-Biphenyl)Os(en)}2+ was shown to bind to both the guanine and cytosine residues, both independently in the mono-osmiated [oligonucleotide + [(η6-biphenyl)Os(en)] − (n + 2)H]n− species, and in the di-osmiated [oligonucleotide + 2[(η6-biphenyl)Os(en)] − (n + 4)H]n− species.
Though Collision Activated Dissociation (CAD) produced extensive fragmentation and excellent sequence coverage with the unmodified oligonucleotide, the metallodrug modification dissociated from the mono-osmiated oligonucleotides. With the retention of the complex in only a fraction of the fragments, CAD was able to narrow the location of the binding site of the modification to the A2–G3–T4 region of the sequence. However Electron Detachment Dissociation (EDD) allowed unambiguous identification of both guanine (G3) and cytosine (C10) binding from assignment of fragmentation patterns. Fragmentation of the di-osmiated DNA12 species proved more difficult, since the two bound osmium complexes appeared to stabilise the oligonucleotide toward EDD fragmentation. Such stabilisation toward EDD fragmentation observed here for DNA12 − 2Os1 species may result from folding of DNA12 due to π–π stacking interactions between the two biphenyl ligands from separate guanine- and cytosine-bound osmium complexes (as previously characterised by NMR for related guanine bound Ru arene-oligonucleotide adducts),46 creating a cyclic gas-phase species and hindering fragmentation via using electron detachment. Collisional dissociation methods were able to dissociate the species due to a multiple collision/slow heating mechanism giving providing results in agreement to those for the mono-osmiated species studied.
This work demonstrates the flexibility and aptitude of FT-ICR mass spectrometry for Top–Down fragmentation of DNA-metallodrug reaction products, and shows how a multi-platform fragmentation approach is often needed to fully characterise metallodrug binding to biomolecules of interest. FT-ICR MS, provides the highest resolving power and mass accuracy performance of any mass spectrometry platform available, and offers the highest possible reliability and confidence in mass spectral assignment, coupled with the largest range of fragmentation techniques. FT-ICR MS is therefore uniquely suited to Top–Down fragmentation of metallodrug-biomolecule adducts.
Acknowledgements
We thank Warwick Centre for Analytical Science (EPSRC grant no. EP/F034210/1), the European Research Council (grant no. 247450), Bruker Daltonics, Warwick Collaborative Postgraduate Research Scholarships (WCPRS), the Key International (Regional) Joint Research Program of NSF (grant no. 2014456) and National Science Foundation (NSF) (project 21171095) for their support for this work, and members of EU COST Action CM1105 for stimulating discussions. This study was also funded by grant number EP/J000302 from the Engineering and Physical Sciences Research Council.References
- P. J. Dyson and G. Sava, Dalton Trans., 2006, 1929–1933 RSC.
- S. Betanzos-Lara, L. Salassa, A. Habtemariam, O. Novakova, A. M. Pizarro, G. J. Clarkson, B. Liskova, V. Brabec and P. J. Sadler, Organometallics, 2012, 31, 3466–3479 CrossRef CAS.
- J. M. Hearn, I. Romero-Canelon, B. Qamar, Z. Liu, I. Hands-Portman and P. J. Sadler, ACS Chem. Biol., 2013, 8, 2345 CrossRef CAS.
- M. Strianese, A. Basile, A. Mazzone, S. Morello, M. C. Turco and C. Pellecchia, J. Cell. Physiol., 2013, 228, 2202–2209 CrossRef CAS PubMed.
- A. M. Funston, C. Cullinane, K. P. Ghiggino, W. D. McFadyen, S. S. Stylli and P. a. Tregloan, Aust. J. Chem., 2005, 58, 206 CrossRef CAS.
- A. F. A. Peacock, A. Habtemariam, R. Fernández, V. Walland, F. P. A. Fabbiani, S. Parsons, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Am. Chem. Soc., 2006, 128, 1739–1748 CrossRef CAS PubMed.
- F. S. Mackay, J. a. Woods, H. Moseley, J. Ferguson, A. Dawson, S. Parsons and P. J. Sadler, Chemistry, 2006, 12, 3155–3161 CrossRef CAS PubMed.
- R. A. Alderden, M. D. Hall and T. W. Hambley, J. Chem. Educ., 2006, 83, 728–734 CrossRef CAS.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed.
- J. Reedijk, Eur. J. Inorg. Chem., 2009, 10, 1303–1312 CrossRef.
- A. M. Pizarro and P. J. Sadler, Biochimie, 2009, 91, 1198–1211 CrossRef CAS PubMed.
- C. Centerwall and K. Tacka, Mol. Pharmacol., 2006, 348–355 CAS.
- H. Li, T. Lin, S. L. Van Orden, Y. Zhao, M. P. Barrow, A. M. Pizarro, Y. Qi, P. J. Sadler and P. B. O'Connor, Anal. Chem., 2011, 84, 9507–9515 CrossRef PubMed.
- N. J. Farrer, P. Gierth and P. J. Sadler, Chemistry, 2011, 17, 12059–12066 CrossRef CAS PubMed.
- V. Calderone, A. Casini, S. Mangani, L. Messori and P. L. Orioli, Angew. Chem., Int. Ed., 2006, 45, 1267–1269 CrossRef CAS PubMed.
- Y. Cho, A. Ahmed, A. Islam and S. Kim, Mass Spectrom. Rev., 2014, 1–16 Search PubMed.
- F. W. McLafferty, Int. J. Mass Spectrom., 2001, 212, 81–87 CrossRef CAS.
- K. R. Jennings, Int. J. Mass Spectrom. Ion Phys., 1968, 1, 227–235 CrossRef CAS.
- R. Zubarev, J. Am. Chem. Soc., 1998, 7863, 3265–3266 CrossRef.
- J. E. P. Syka, J. J. Coon, M. J. Schroeder, J. Shabanowitz and D. F. Hunt, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9528–9533 CrossRef CAS PubMed.
- D. P. Little, J. P. Speir, M. W. Senko, P. B. O'Connor and F. W. McLafferty, Anal. Chem., 1994, 66, 2809–2815 CrossRef CAS.
- J. J. Wolff, T. N. Laremore, H. Aslam, R. J. Linhardt and I. J. Amster, J. Am. Soc. Mass Spectrom., 2008, 19, 1449–1458 CrossRef CAS PubMed.
- W. D. Bowers, S. Delbert, R. L. Hunter and R. T. Mciver, J. Am. Chem. Soc., 1984, 7288–7289 CrossRef CAS.
- B. Budnik, K. Haselmann and R. Zubarev, Chem. Phys. Lett., 2001, 342, 299–302 CrossRef CAS.
- I. Amster, J. Mass Spectrom., 1996, 31, 1325–1337 CrossRef CAS.
- a. G. Marshall, C. L. Hendrickson and G. S. Jackson, Mass Spectrom. Rev., 1998, 17, 1–35 CrossRef CAS.
- H. Li, J. J. Wolff, S. L. Van Orden and J. a. Loo, Anal. Chem., 2014, 86, 317–320 CrossRef CAS PubMed.
- F. W. McLafferty, K. Breuker, M. Jin, X. Han, G. Infusini, H. Jiang, X. Kong and T. P. Begley, FEBS J., 2007, 274, 6256–6268 CrossRef CAS PubMed.
- A. V. Tolmachev, E. W. Robinson, S. Wu, L. Paša-Tolić and R. D. Smith, Int. J. Mass Spectrom., 2009, 281, 32–38 CrossRef PubMed.
- T. L. Quenzer, M. R. Emmett, C. L. Hendrickson, P. H. Kelly and a. G. Marshall, Anal. Chem., 2001, 73, 1721–1725 CrossRef CAS.
- P. Caravatti and M. Allemann, Org. Mass Spectrom., 1991, 26, 514–518 CrossRef CAS.
- L.-K. Zhang, D. Rempel, B. N. Pramanik and M. L. Gross, Mass Spectrom. Rev., 2005, 24, 286–309 CrossRef CAS.
- L. Konermann, E. Ahadi, A. D. Rodriguez and S. Vahidi, Anal. Chem, 2012, 85, 2–9 CrossRef PubMed.
- J. Wu and S. a. McLuckey, Int. J. Mass Spectrom., 2004, 237, 197–241 CrossRef CAS PubMed.
- D. R. Phillips and J. a. McCloskey, Int. J. Mass Spectrom. Ion Processes, 1993, 128, 61–82 CrossRef CAS.
- J. Tromp and S. Schürch, Raipd Commun. Mass Spectrom., 2006, 2348–2354 CrossRef CAS PubMed.
- A. Nyakas, L. C. Blum, S. R. Stucki, J.-L. Reymond and S. Schürch, J. Am. Soc. Mass Spectrom., 2013, 24, 249–256 CrossRef CAS PubMed.
- J. Reedijk, Chem. Rev., 1999, 99, 2499–2510 CrossRef CAS PubMed.
- Y. Qi, Z. Liu, H. Li, P. J. Sadler and P. B. O'Connor, Rapid Commun. Mass Spectrom., 2013, 27, 2028–2032 CrossRef CAS PubMed.
- Z. Xu, J. B. Shaw and J. S. Brodbelt, J. Am. Soc. Mass Spectrom., 2013, 24, 265–273 CrossRef CAS PubMed.
- N. Leymarie, C. E. Costello and P. B. O'Connor, J. Am. Chem. Soc., 2003, 125, 8949–8958 CrossRef CAS PubMed.
- J. Zhang, M. J. Guy, H. S. Norman, Y.-C. Chen, Q. Xu, X. Dong, H. Guner, S. Wang, T. Kohmoto, K. H. Young, R. L. Moss and Y. Ge, J. Proteome Res., 2011, 10, 4054–4065 CrossRef CAS PubMed.
- H. Li, Y. Zhao, H. I. a. Phillips, Y. Qi, T.-Y. Lin, P. J. Sadler and P. B. O'Connor, Anal. Chem., 2011, 83, 5369–5376 CrossRef CAS PubMed.
- M. Taucher and K. Breuker, J. Am. Soc. Mass Spectrom., 2010, 21, 918–929 CrossRef CAS PubMed.
- J. J. Wolff, I. J. Amster, L. Chi and R. J. Linhardt, J. Am. Soc. Mass Spectrom., 2007, 18, 234–244 CrossRef CAS PubMed.
- H.-K. Liu and P. J. Sadler, Unpublished work.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt03819c |
| This journal is © The Royal Society of Chemistry 2015 |