
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protein ruthenation and DNA alkylation: chlorambucil-functionalized RAPTA complexes and their anticancer activity
Alexey A.
Nazarov
*ab,
Samuel M.
Meier
c,
Olivier
Zava
a,
Yulia N.
Nosova
b,
Elena R.
Milaeva
b,
Christian G.
Hartinger
*d and
Paul J.
Dyson
*a
aInstitute of Chemical Sciences and Engineering, Swiss Federal Institute of Technology (EPFL), CH-1015, Lausanne, Switzerland. E-mail: paul.dyson@epfl.ch; Fax: +41-21-6939885; Tel: +41-21-6939854
bM. V. Lomonosov Moscow State University, Leninskie gory 1/3, 119991 Moscow, Russia. E-mail: alexey.nazarov@me.com
cUniversity of Vienna, Institute of Analytical Chemistry, Waehringer Str. 38, 1090 Vienna, Austria
dUniversity of Auckland, School of Chemical Sciences, Private Bag 92019, Auckland 1142, New Zealand. E-mail: c.hartinger@auckland.ac.nz; Fax: +64-9-3737 599 ext. 87422; Tel: +64-9-3737 599 ext. 83220
First published on 30th October 2014
Abstract
Chemotherapeutics for the treatment of tumorigenic conditions that feature novel modes of action are highly sought after to overcome the limitations of current chemotherapies. Herein, we report the conjugation of the alkylating agent chlorambucil to the RAPTA scaffold, a well-established pharmacophore. While chlorambucil is known to alkylate DNA, the RAPTA complexes are known to coordinate to amino acid side chains of proteins. Therefore, such a molecule combines DNA and protein targeting properties in a single molecule. Several chlorambucil-tethered RAPTA derivatives were prepared and tested for their cytotoxicity, stability in water and reactivity to protein and DNA substrates. The anticancer activity of the complexes is widely driven by the cytotoxicity of the chlorambucil moiety. However, especially in the cisplatin-resistant A2780R cells, the chlorambucil-functionalized RAPTA derivatives are in general more cytotoxic than chlorambucil and also a mixture of chlorambucil and the parent organoruthenium RAPTA compound. In a proof-of-principle experiment, the cross-linking of DNA and protein fragments by a chlorambucil–RAPTA derivative was observed.
Introduction
Alkylating agents have dominated cancer chemotherapy for many years, and among many examples of alkylating agents that have been approved for cancer treatment, the nitrogen mustard, chlorambucil (Chart 1), is one of the classic anticancer drugs.1–3 Chlorambucil is administered orally4 and has been mainly used in the treatment of chronic lymphocytic leukemia, but may also be used in the therapy of tumors, such as certain types of non-Hodgkin lymphoma, trophoblastic neoplasms, and ovarian carcinoma. This compound class exhibits its mode of action through the alkylation of the N7 atoms of guanine or adenine as well as N3 of adenine in double helical DNA, which consequently inhibits cell proliferation.5,6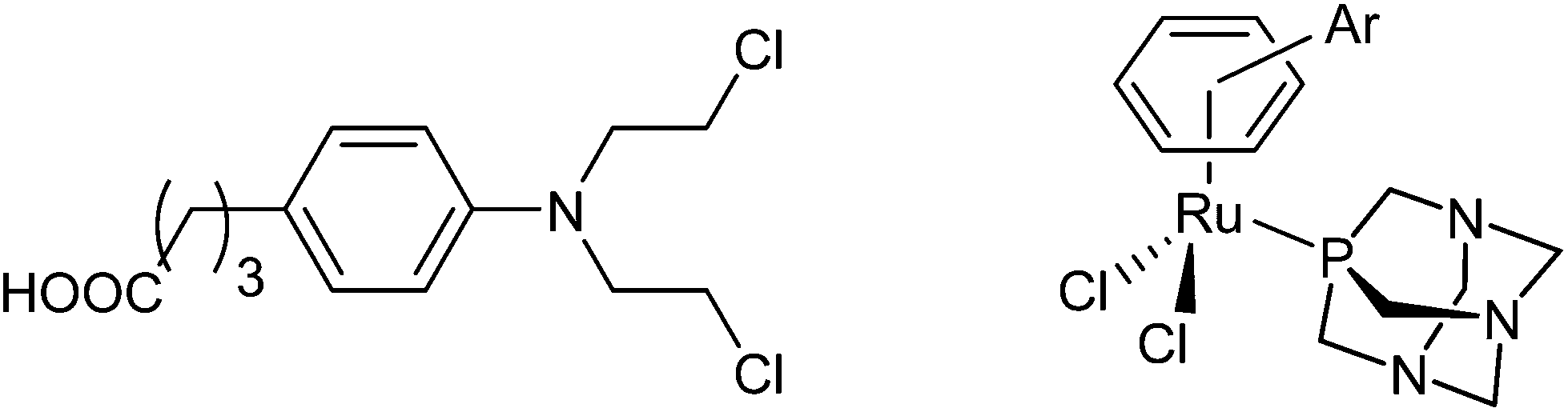 | ||
| Chart 1 The structures of chlorambucil (left) and the RAPTA scaffold (right). | ||
Platinum complexes are often also considered as alkylating agents as they form coordination bonds to DNA nucleobases.2,7 The potency of platinum complexes in chemotherapy is unprecedented and they are used to treat many types of tumors.8–11 However, their disadvantages, in particular, resistance development and side effects, have driven the development of metal complexes comprising metal centers other than platinum.12–14 Ruthenium compounds take a prominent position in this development process, as two compounds have entered clinical trials.15–17 Organometallic RAPTA complexes are among the most promising ruthenium-based compounds in preclinical development and although they are essentially non-cytotoxic in cancer cell lines, they have demonstrated anticancer activity in vivo.17–20 The RAPTA framework (Chart 1) consists of a ruthenium center, an η6-coordinated arene, typically p-cymene (RAPTA-C) or toluene (RAPTA-T), the 1,3,5-triaza-7-phosphaadamantane (PTA) ligand and two chlorido leaving groups.19,21 The RAPTA scaffold can be equipped with functionalities that provide specific properties. For example, derivatization of the arene ligand with ethacrynic acid resulted in efficient glutathione-S-transferase inhibitors.22 The mode of action appears to involve the enzymatic cleavage of the enzyme-inhibiting moiety from the metal fragment allowing the metal moiety to target other biomolecules. This represents an example of a multi-targeted anticancer agent with components of the same molecule interacting with different targets.23 Such drugs are designed to act against several individual molecular targets rather than a single enzyme or DNA as the target for platinum anticancer drugs.24 This approach offers a number of advantages such as tuneable pharmacological properties and anticancer activity, and altered metabolism and resistance development.23,25 An alternative approach for developing ruthenium-arene multitargeted compounds is through the coordination of a bioactive ligand to the metal center as shown for RuII(cym) (cym = η6-p-cymene) complexes of 3-hydroxyflavones,26–28 lapachol,29 paullone,30 and lonidamine31 derivatives and other examples.32 Such an “intramolecular” combination therapy may also help to overcome disadvantages and limitations of antineoplastic alkylating agents.
Therefore, we have designed a new compound class that features the RAPTA framework conjugated with chlorambucil. Such compounds should be able to alkylate DNA and at the same time coordinate to proteins through the ruthenium center while maintaining the structure of the RAPTA pharmacophore. This approach is supported by the observation that RAPTA binds preferentially to proteins through coordination, as shown in crystallographic experiments using the nucleosome core particle (NCP) in which the RAPTA scaffold was found predominantly at the histone proteins.33,34 The synthesis of alkylating RAPTA derivatives was complemented by studies on the cytotoxic activity in human cancer cell lines and mass spectrometric investigations with 9-ethylguanine (EtG) and L-histidine (His), L-methionine (Met) and ubiquitin (Ub) as DNA and protein models, respectively.
Experimental
Materials
Solvents were purified and degassed prior to use.35 All compounds were obtained from Sigma-Aldrich except for L-histidine which was obtained from Merck; formic acid was from Fluka; 9-ethylguanine and tetramethylammonium acetate were from TCI Europe. Methanol (HPLC grade, Fisher) and Millipore water (18.2 MΩ, Advantage A10, 185 UV ultrapure water, Millipore) were used for ESI-MS studies.Instrumentation
1H and 31P{1H} NMR spectra were recorded on a Bruker Avance II 400 spectrometer at room temperature and were referenced to the 1H signal of the NMR solvent or to H3PO4 as an external reference for 31P{1H} NMR spectroscopy. ESI mass spectra of the compounds were obtained in MeOH on a ThermoFinnigan LCQ Deca XP Plus quadrupole ion-trap instrument operated in the positive ion mode over m/z 150–1000. The ionization energy was set at 3.5 kV (capillary voltage) and the capillary temperature at 150 °C. Melting points were determined with a Stuart Scientific SMP3 apparatus and are uncorrected. The flash chromatography system Varian 971-FP was used for purification. Elemental analyses were carried out by the microanalytical laboratory at the EPFL.Cell lines and culture conditions
Human A2780 and A2780cisR ovarian carcinoma cell lines were obtained from the European Centre of Cell Cultures (ECACC, Porton Down, Salisbury, UK). Human MCF-7 breast carcinoma cells were obtained from the American Type Culture Collection (ATCC). These cells were routinely grown in Dulbecco's modified Eagle medium containing 4.5 g L−1 glucose and glutamax, and were supplemented with 10% heat-inactivated fetal bovine serum and antibiotics. Cultures were maintained at 37 °C under a humidified atmosphere containing 6% CO2. All cell culture reagents were purchased from Gibco-BRL, Basel, Switzerland. For cell viability experiments tests, cells were grown on 96-well plates (Costar, Integra Biosciences, Cambridge, MA, USA) as monolayers for 24 h in complete medium with 10% FCS to reach sub-confluence. Then fresh complete medium with 5% FCS was added together with the drugs, and the culture was continued for another 72 h. The compounds were pre-dissolved at 20 mM in DMSO and then added to the cell culture medium at the required concentration with a maximum DMSO content of 0.5%, v/v, to be incubated for 72 h. At these concentrations, DMSO has no effect on cell viability. Cell viability was determined using the MTT assay, which quantifies the mitochondrial activity in metabolically active cells, essentially as reported previously.36 Briefly, following drug exposure, MTT (Sigma, final concentration 0.2 mg ml−1) was added to the cell culture medium for the final 2 h, and then the culture medium was aspirated and the violet formazan precipitate dissolved in 0.1 M HCl in 2-propanol. The optical density, which is directly proportional to the number of surviving cells, was quantified at 540 nm using a multiwell plate reader (iEMS Reader MF, Labsystems, USA) and the percentage of surviving cells was calculated from the absorbance of untreated cells.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). Yield: 4.8 g (78.3%), mp: 98–99 °C, elem. anal. calcd (%) for C21H28Cl2N2O: C 63.80, H 7.14, N 7.09; found: C 64.08, H 7.21, N 7.23. 1H NMR (400.13 MHz, CDCl3): δ = 7.09 (d, J = 8.3 Hz, 2H; Har), 6.64 (d, J = 8.3 Hz, 2H, Har), 5.72 (s, 2H; CH), 5.60 (s, 1H; CH), 5.42 (brs, 1H; NH), 3.81 (d, J = 5.4 Hz, 2H; CH2NHC(O)), 3.72 (m, 4H; NCH2CH2Cl), 3.64 (m, 4H; NCH2CH2Cl), 2.71 (m, 2H; CH2C6H4), 2.65–2.57 (m, 4H, CH2diene), 2.12 (tr, 2H, CH2CH2C(O)), 1.96 (m, 2H, CH2CH2C(O)) ppm. MS (ESI+): m/z 395 [M + H]+.
:2). Yield: 4.8 g (78.3%), mp: 98–99 °C, elem. anal. calcd (%) for C21H28Cl2N2O: C 63.80, H 7.14, N 7.09; found: C 64.08, H 7.21, N 7.23. 1H NMR (400.13 MHz, CDCl3): δ = 7.09 (d, J = 8.3 Hz, 2H; Har), 6.64 (d, J = 8.3 Hz, 2H, Har), 5.72 (s, 2H; CH), 5.60 (s, 1H; CH), 5.42 (brs, 1H; NH), 3.81 (d, J = 5.4 Hz, 2H; CH2NHC(O)), 3.72 (m, 4H; NCH2CH2Cl), 3.64 (m, 4H; NCH2CH2Cl), 2.71 (m, 2H; CH2C6H4), 2.65–2.57 (m, 4H, CH2diene), 2.12 (tr, 2H, CH2CH2C(O)), 1.96 (m, 2H, CH2CH2C(O)) ppm. MS (ESI+): m/z 395 [M + H]+.
Protein and DNA binding studies
Complex 6 (25 μM) was incubated at 37 °C in 1% DMSO–water or in 1 mM tetramethylammonium acetate with 9-ethylguanine (EtG) or ubiquitin (Ub) at metal-to-biomolecule ratios of 1:2 and 1:1, respectively. Complex 6 was also incubated with both EtG and Ub at molar ratios of 1:2:1 and 2:8:1 giving a final protein concentration of 25 μM. Aqueous solutions of 6 were analyzed for comparison purposes under the same conditions as the biomolecule-containing reaction mixtures. In a further set of experiments, histidine (His) and methionine (Met) were separately preincubated with 6 for 1 h in buffered solution prior to the addition of EtG giving a final molar ratio of 2:1:4. Prior to analysis, the incubation mixtures were diluted with water 1:5 in the case of the low-molecular weight biomolecules and 1:1 for ubiquitin-containing samples, corresponding to 5 μM complex and 12.5 μM protein, respectively. Protein samples were analyzed under non-denaturing conditions.
To characterize the adducts formed between 6 and the biomolecules, full mass spectra were recorded in the m/z range of 70–2500 on a Bruker Maxis UHR qTOF (capillary −4500 V, dry gas 6.0 l min−1, nebulizer 0.6 bar, dry temperature 180 °C), and collision-induced MSn experiments were conducted on a Bruker Amazon SL ion trap under similar source conditions. Data analysis was done with the Bruker data analysis software ESI Compass 1.3 and DataAnalysis 4.0 (build 234). Protein mass spectra were deconvoluted using the maximum entropy deconvolution algorithm with automatic data point spacing and 30000 resolving power.
Results and discussion
We decided to combine the RAPTA pharmacophore, which has a ruthenium(II) center able to bind to histone proteins,33,34 with the alkylating moiety found in the anticancer drug chlorambucil 1, which is known to act as a DNA alkylating agent.5,6 The chlorambucil group was conjugated to the RAPTA scaffold by derivatization of the arene ring with chlorambucil via an amide bond to the piano-stool Ru complex. The PTA co-ligand was also varied with 3,7-diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]nonane (DAPTA) and glucose phosphite ligands. The synthetic route used is similar to the one reported earlier for the preparation of RAPTA compounds carrying ethacrynic acid,22 a naphthalimide intercalator39 or a fluorescent tag.40 Chlorambucil was converted into the acid chloride by oxalyl chloride–DMF and reacted with 1,4-cyclohexadiene-1-methylamine or 1,4-cyclohexadiene-1-ethylamine to yield 4-(p-(bis(2-chloroethyl)amino)phenyl)-N-(cyclohexa-1,4-dien-1-ylmethyl)butanamide and 4-(p-(bis(2-chloroethyl)amino)phenyl)-N-(cyclohexa-1,4-dien-1-ylethyl)butanamide, respectively. These dienes were reacted with RuCl3 in ethanol under reflux to afford the respective RuII(arene) dimers as orange solids (Scheme 1). Finally, the dimers were treated with PTA, DAPTA or 3,5,6-bicyclophosphite-1,2-O-isopropylidene-α-D-glucofuranoside to yield complexes 6–9. The compounds were characterized by NMR spectroscopy, mass spectrometry and elemental analysis. 31P{1H} NMR spectroscopy was used to monitor the formation of complexes 6–9. In these spectra a shift from ca. −100 to −33 ppm, from −78 to −17 ppm and from 119 to 133 ppm was observed for the 31P nuclei in the PTA, DAPTA and phosphite ligands, respectively, upon coordination to the ruthenium center. In addition, characteristic shifts in the 1H NMR spectra were observed, as reported for similar types of complexes,40,41 confirming the proposed structures of the complexes. Compounds 6–9 are stable in CDCl3 and DMF solution and no decomposition or release of the arene moiety was observed.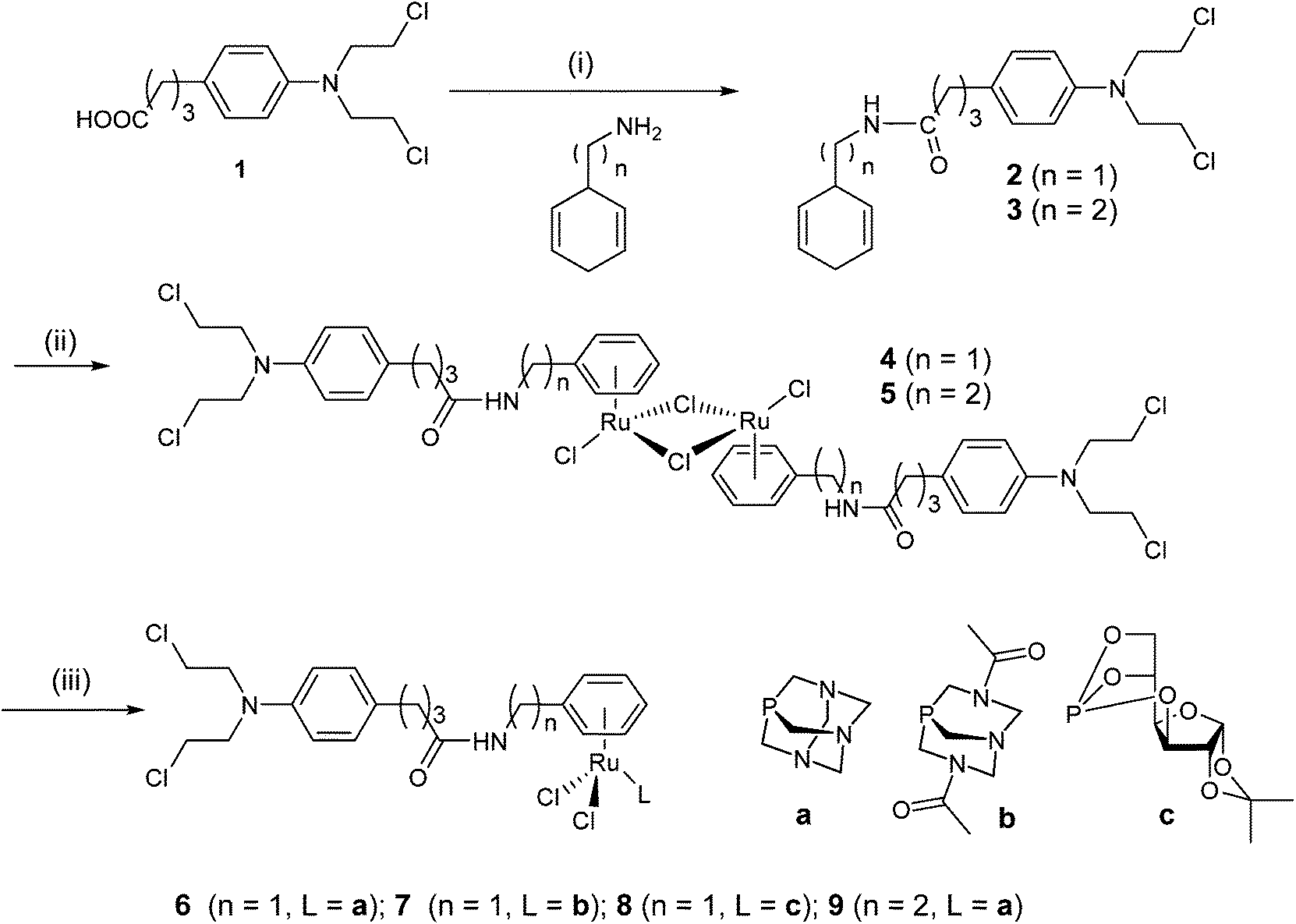 | ||
| Scheme 1 Synthetic route to the chlorambucil-functionalized ruthenium complexes with amphiphilic phosphorus-based co-ligands. | ||
The cytotoxicity of the chlorambucil-functionalized RAPTA-type complexes (6–9) was evaluated on human ovarian cancer (A2780) cells, their cisplatin-resistant variant (A2780R) and a human breast cancer (MCF7) cell line. In addition, studies mixing chlorambucil and RAPTA-T at a molar ratio of 1:1, as the closest structural analogue to 6–9, were conducted and compared to RAPTA-T as the parent compound (Table 1). In general, most of the compounds were more active in the ovarian cancer cell lines than in MCF7. While RAPTA-T is not cytotoxic under the applied conditions in the three cell lines, the chlorambucil-functionalized complexes are active in the low μM range. Both chlorambucil 1 and a 1:1 mixture of RAPTA-T with 1 show similar cytotoxicity profiles to complexes 6–9 suggesting that the alkylating agent determines the cytotoxicity of the complexes. Notably, 1 and a 1:1 mixture of RAPTA-T with 1 show virtually identical IC50 values in the cell lines. This is not surprising given the fact that RAPTA compounds are not cytotoxic. Furthermore, the nature of the phosphorus donor co-ligand is of low importance with the exception of 7 in MCF7 cells. The cytotoxicity decreases in all cell lines for 6 over 9. This may be related to 9 having a longer linker between the chlorambucil moiety and the arene moiety and being therefore more flexible than 6 and 9. In the A2780 and MCF7 cell lines there does not appear to be any advantage in conjugating 1 with a RAPTA fragment, whereas in the cisplatin-resistant cell line A2780R, complexes 6–8 are considerably more potent than 1 and the mixture of 1 with RAPTA-T. This observation suggests that conjugation of 1 to Ru(arene) compounds, irrespective of the phosphorus donor co-ligand, provides a means to overcome cisplatin-induced resistance mechanisms in cancer cells.
:1) in the human ovarian cancer cells A2780, their cisplatin-resistant variant A2780R and MCF7 breast cancer cells
| Compound | IC50 (μM) | ||
|---|---|---|---|
| A2780 | A2780R | MCF7 | |
| 1 | 5.2 ± 1.6 | 25 ± 4 | 22 ± 7 |
| RAPTA-T | >200 | >200 | >200 |
|
1 + RAPTA-T (1:1) |
5.2 ± 1.6 | 25 ± 3 | 25 ± 9 |
| 6 | 8.3 ± 1.3 | 10 ± 3 | 12 ± 4 |
| 7 | 9.4 ± 2.2 | 7.0 ± 0.8 | 42 ± 7 |
| 8 | 8.2 ± 2.3 | 8.7 ± 0.9 | 14 ± 1 |
| 9 | 12 ± 4 | 45 ± 17 | 37 ± 9 |
Compound 6 was selected for stability studies and subsequent experiments to demonstrate the cross-linking ability of biomolecules as it possesses the optimum cytotoxicity properties of the compound series and also reasonably soluble in aqueous solution. Furthermore, the experiments are not interfered by the instability of the DAPTA ligand in 7 or the complicated hydrolysis pathways of the glucose-derived phosphite in 8.41
Initially, the hydrolysis behavior of 6 was studied after dissolving the compound in 1% DMSO–water. Immediate analysis showed the presence of a single peak in the ESI mass spectrum at m/z 688.0891, which was assigned to the ion [M − Cl]+ (see Table 2 for experimental and simulated mass signals; Scheme 2). The same mixture was analyzed over a period of 2 days revealing that 6 starts to hydrolyze, to some extent, after incubation for 1.5 h as indicated by the appearance of hydroxide-containing mass signals, corresponding to [6 − 2Cl + OH]+ (Fig. 1A). Simultaneously, 6 starts to release the activated aziridinium chlorambucil-arene ligand [L − Cl]+. After 21 h, the ligand release is quantitative and is accompanied by the formation of Ru(PTA)Cl species, which appear to be stabilized by DMSO, e.g., [Ru(PTA)(DMSO)nCl]+ (n = 2, 3). Within the same time frame, the activated chlorambucil is converted completely into the dihydroxyl species L2OH as indicated by the change in the isotopic pattern and a small mass shift from m/z 357.1711 to 357.2180 (Fig. 1B). There was no further change in the mass spectrum from 21 to 72 h. In order to characterize this species further, collision-induced tandem mass spectrometric experiments were conducted using L2OH at m/z 357.1 as the precursor ion. A monohydroxyl LOH ion was detected at m/z 340.1, together with an amide-cleaved product ion at m/z 250.1 and neutral loss products at m/z 232.1 (–H2O) and 214.0 (–2H2O), all underlining the identity of the parent mass signal.
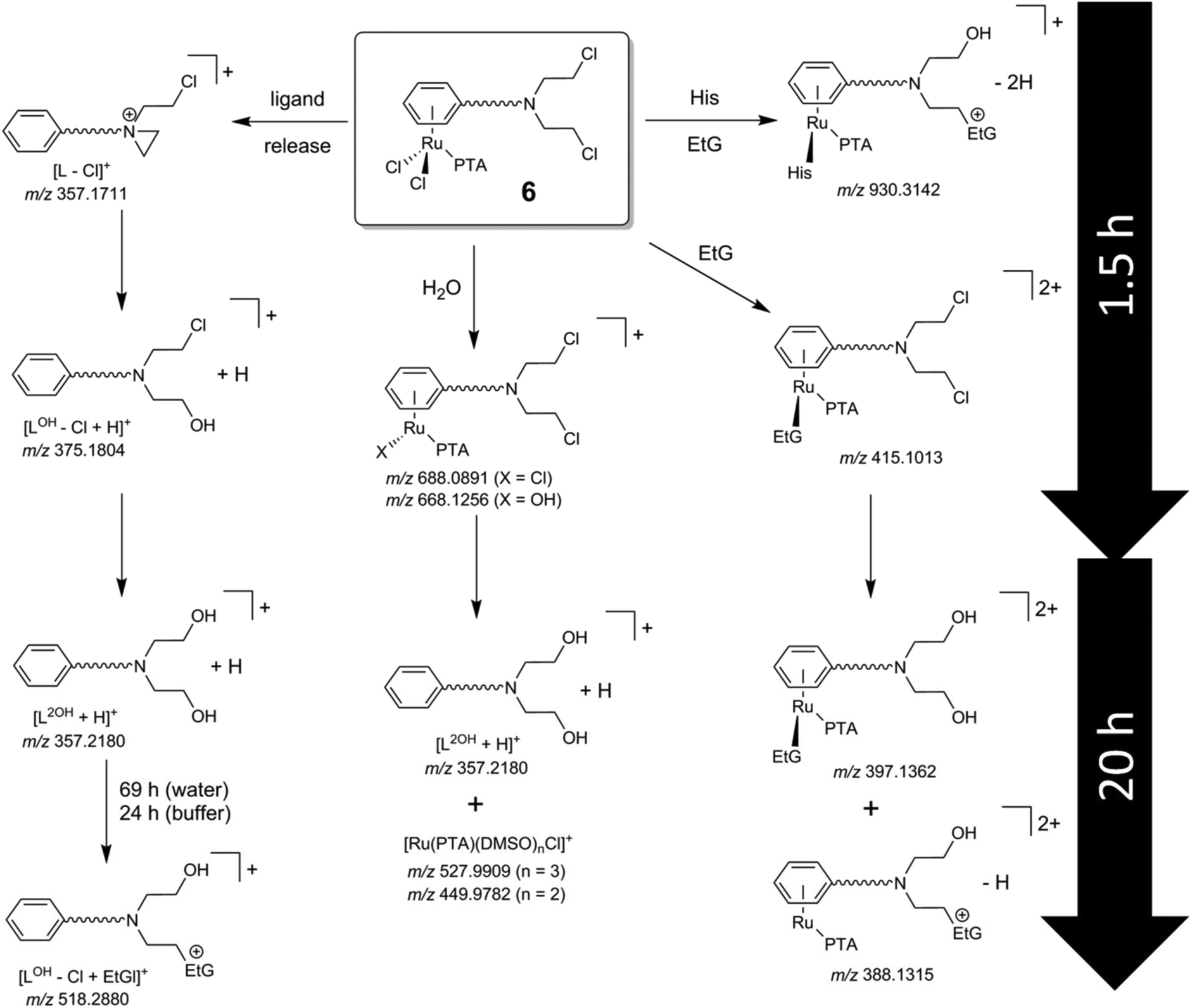 | ||
| Scheme 2 Identified species from the reaction pathways of 6 with small molecules. The compound was initially dissolved in aqueous 1% DMSO and samples were diluted with water prior to analysis. Hydrolysis of the metal–chloride bonds is fast with respect to alkylation of the DNA model 9-ethylguanine (EtG). The hydrolysis of chlorambucil can be well followed. The species identified in 1% DMSO/H2O and due to the ligand release were also observed in incubations with EtG and His/EtG. | ||
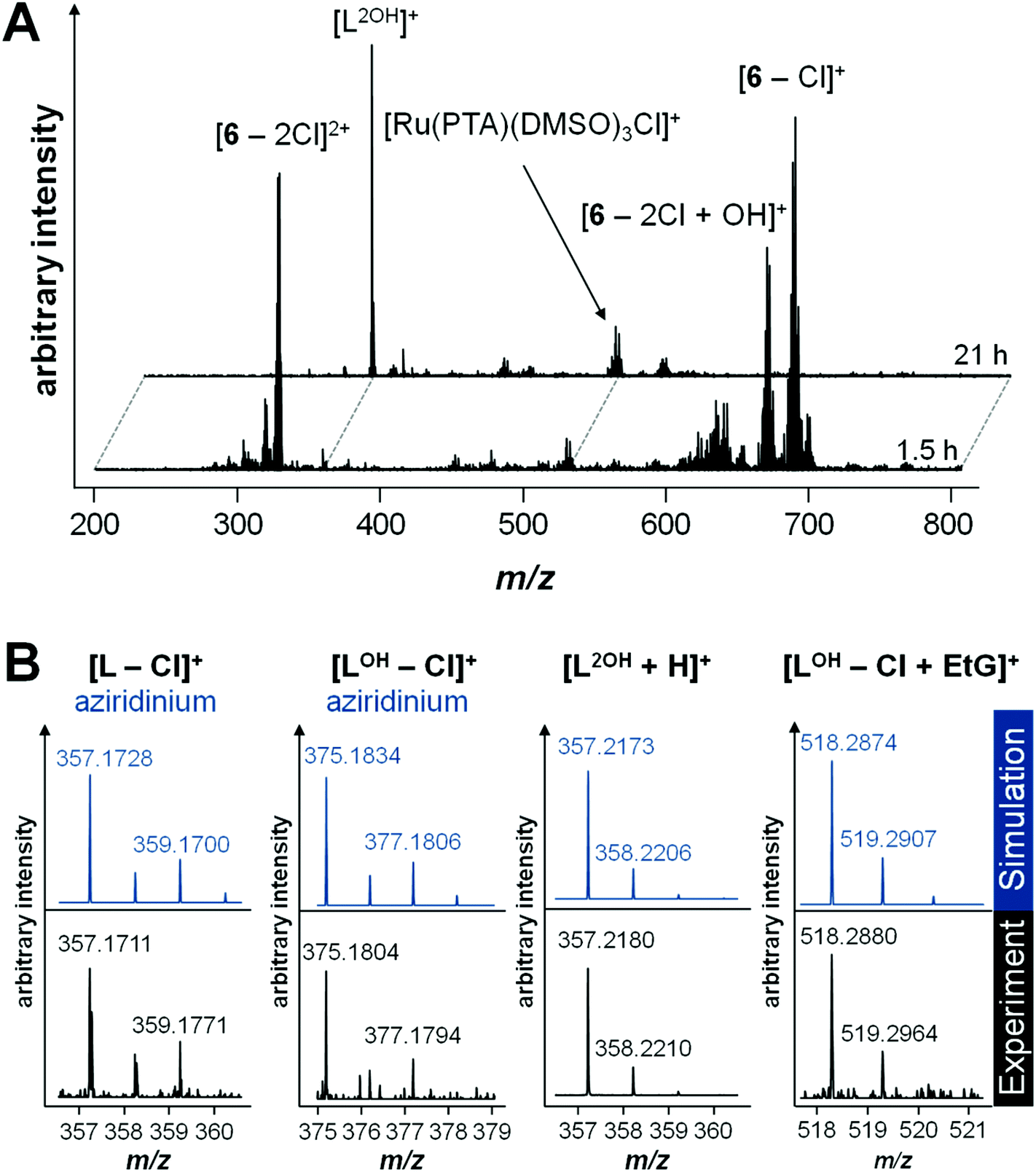 | ||
| Fig. 1 (A) Mass spectra recorded during hydrolysis experiments of 6 in water after 1.5 and 21 h. Compound 6 was dissolved in DMSO and further diluted with water before analysis. (B) Experimental and simulated isotopic patterns of the different released chlorambucil-arene species in unbuffered solution: the aziridinium chlorambucil-arene and aziridinium hydroxyl ligands were detected after 1.5 h, the dihydroxyl ligand after 21 h and the alkylated 9-ethylguanine (EtG) hydroxyl ligand after 69 h. A higher pH value also increases the kinetics for EtG alkylation. | ||
| Species | m acc | m ex | Δppm | |
|---|---|---|---|---|
| Compound | [(LCl)Ru(PTA)(His)(EtG) + DMSO + H2O − 2H]+ | 1044.3333 | 1044.3138 | 18.7 |
| [(LCl)Ru(PTA)(Met)(EtG) + DMSO + H2O − 2H]+ | 1038.3129 | 1038.2953 | 17.0 | |
| [(LOH)Ru(PTA)(His) + EtG − 2H]+ | 930.3142 | 930.3235 | 10.0 | |
| [6 − Cl + EtG]+ | 867.1720 | 867.1698 | 2.5 | |
| [6 − 2Cl + His + CH3COO]+ | 865.1925 | 865.2060 | 15.6 | |
| [6 − 2Cl − H + OH + EtG]+ | 847.2059 | 847.2066 | 0.8 | |
| [6 − 2Cl + EtG − H]+ | 829.1921 | 829.1961 | 4.8 | |
| [6 − 2Cl + His − H]+ | 805.1725 | 805.1848 | 15.3 | |
| [6 − 2Cl + Met − H]+ | 799.1547 | 799.1663 | 14.5 | |
| [(LOH)Ru(PTA) + EtG + OH − H]+ | 793.2534 | 793.2645 | 14.0 | |
| [(LOH)Ru(PTA) + EtG − 2H]+ | 775.2537 | 775.2539 | 0.3 | |
| [6 − 2Cl + Ac]+ | 712.1332 | 712.1355 | 3.2 | |
| [6 − Cl]+ | 688.0891 | 688.0900 | 1.3 | |
| [6 − 2Cl + OH]+ | 668.1256 | 668.1258 | 0.3 | |
| [(L2OH)Ru(PTA)Cl]+ | 650.1573 | 650.1799 | 34.8 | |
| [Ru(PTA)(DMSO)2 + EtG − H]+ | 593.0883 | 593.0817 | 11.1 | |
| [Ru(PTA)(DMSO)3Cl]+ | 527.9909 | 527.9912 | 0.6 | |
| [Ru(PTA)(DMSO)2 + CH3COO]+ | 474.0238 | 474.0220 | 3.8 | |
| [Ru(PTA)(DMSO)2Cl]+ | 449.9782 | 449.9772 | 2.2 | |
| [6 − 2Cl + EtG]2+ | 415.1013 | 415.1017 | 1.0 | |
| [(L2OH)Ru(PTA) + EtG]2+ | 397.1362 | 397.1359 | 0.8 | |
| [(LOH)Ru(PTA) + EtG − H]2+ | 388.1315 | 388.1306 | 2.3 | |
| [Ru(PTA)(DMSO) + 2EtG]2+ | 347.5747 | 347.5780 | 9.5 | |
| [6 − 2Cl]2+ | 326.5623 | 326.5608 | 4.6 | |
| [Ru(PTA) + 2EtG]2+ | 308.5734 | 308.5710 | 7.8 | |
| Ligand | [LOH − Cl + EtG]+ | 518.2880 | 518.2874 | 1.2 |
| [LOH − Cl + H]+ | 375.1804 | 375.1834 | 8.0 | |
| [L2OH − H]+ | 357.2180 | 357.2173 | 2.0 | |
| [L − Cl]+ (aziridinium) | 357.1711 | 357.1728 | 4.8 | |
Hydrolysis of 6 in tetramethylammonium acetate buffer is similar to that observed in water. Hydrolysis products of intact 6 are observed after 10 min, including an additional acetate adduct [6 − 2Cl + CH3COO]+ from the buffer as the most abundant peak. Similarly, intact 6 disappeared completely due to release of the chlorambucil-arene ligand within 20 h, which was detected in the dihydroxyl form L2OH. One additional mass signal with a ruthenium isotope pattern was observed at m/z 474.0238, which was attributed to [Ru(PTA)(DMSO)2 + CH3COO]+. The mass shifts are also accompanied by a change in isotope pattern due to the absence of chloride in the detected species. It should be noted that the arene ligand in other RAPTA derivatives is strongly bound to the Ru center.34
Compound 6 was designed with the aim to provide a compound able to alkylate DNA and simultaneously coordinate to proteins. In order to demonstrate the ability of 6 to cross-link, its reactivity was investigated with the DNA model nucleobase EtG and His and Met as protein building blocks. Thus, 6 was incubated with EtG (1:2), His or Met (1:2) and with both (6:EtG:His/Met = 1:4:2) and the solutions were analyzed by ESI-TOF mass spectrometry.
Incubation of 6 with EtG in water resulted immediately in the detection of several EtG–ruthenium adducts besides the mass signals caused by hydrolysed 6. Accordingly, mono-chlorido, mono-hydroxido and EtG coordination adducts were observed for hydrolysed 6, e.g. [6 − Cl + EtG]+ and [6 − 2Cl − H + EtG]+. After about 16 h, the arene ligand was cleaved to some extent and underwent again Cl/OH substitution (Scheme 2), as indicated by the signals at m/z 357, which became the most intense signal in the mass spectrum. However, there were still coordination adducts of EtG detectable, such as [(L2OH)Ru(PTA) + EtG]2+, although at low relative abundance. Continuing the incubation for another 48 h did not result in significant changes to the mass spectra. Although arene release is observed to some extent in parallel with the experiments in pure water, the addition of EtG seems to stabilize the arene coordination.
In general, the arene ligand steadily dissociates with time and a species detected at m/z 375.1804 after 1.5 h, which indicates hydrolysis of the aziridinium forming [L − Cl + OH]+. Finally, after three days of incubation with EtG, a chlorambucil–EtG adduct is observed corresponding to [LOH − Cl + EtG]+ at m/z 518.2880 (Fig. 1C). Therefore, it seems that the DNA-alkylation reaction with the chlorambucil fragment is much slower than the hydrolysis and complexation reactions of the ruthenium ion. Based on this hypothesis it is possible to distinguish EtG complexation from EtG alkylation.
In tetramethylammonium acetate buffer (1 mM), the peak assigned to [6 − 2Cl − H + EtG]+ was detected after 15 min in 54% relative abundance. In light of the observations made in water, this peak is assumed to be due to the coordination of EtG to the ruthenium center rather than an alkylation product. Unreacted and hydrolyzed 6 were also detected in the forms of [6 − 2Cl + CH3COO]+, [6 − Cl]+, [6 − 2Cl + OH]+ and [6 − 2Cl]2+. After 20 h, the most abundant signal in the mass spectrum besides the acetate adduct [Ru(PTA)(DMSO)2 + CH3COO]+ corresponded to cleavage of the arene. Moreover, the alkylated EtG species [LOH − Cl + EtG]+ was already detected after this time period. Increasing the pH seems to result in an increased rate of alkylation, in accordance with the mode of action of chlorambucil.5,6 Additionally, an EtG alkylation product was detected at m/z 775.2537 corresponding to [(LOH)Ru(PTA) + EtG − 2H]+.
In an attempt to saturate the hydrolyzed coordination sites at the ruthenium center in 6 prior to incubation with EtG in buffer, the compound was preincubated with 2 equivalents of His or Met for 1 h. Analysis of the reaction mixtures revealed the formation of [6 − 2Cl + AA − H]+ (Scheme 2), where AA = His or Met, and additionally [6 − 2Cl + His + CH3COO]+. Interestingly, the mass spectra of these reactions recorded 19 h after the addition of 4 equivalents of EtG were very similar to each other except for a mass shift of 6 Da (His 155.2 g mol−1; Met 149.2 g mol−1) indicating that both amino acids coordinated tightly to the complex and stabilized the hydrolyzed complex against loss of the arene (Fig. 2). Two peaks that were not shifted are the ones with m/z 755 and 357, corresponding to the alkylated EtG adduct [(LOH)Ru(PTA) + EtG − 2H]+ and free L2OH, respectively, which were also found during the experiments of 6 with EtG alone. Of note are the mass signals at m/z 1038 and 1044 for Met and His containing reaction mixtures, respectively. The isotopic pattern of both mass signals is indicative of molecules containing one chloride and one ruthenium. Fragmentation of these mass signals in MSn experiments on an ion trap instrument by collision-induced dissociation yielded very similar product spectra again featuring 6 Da differences suggesting that the amino acids remain coordinated to the smallest fragments. The MS2 spectra are very similar to the full mass spectra and, consequently, these peaks may be tentatively attributed to [(LCl)Ru(PTA)(EtG) + AA + DMSO + H2O − H]+, where LCl is a chloride-containing arene and AA is either His or Met. The DMSO and water adducts may be caused by clustering, which is underlined by very broad signals in the full mass spectra. Furthermore, in the reaction mixture containing His, a signal at m/z 670.05 corresponds to [Ru(PTA)(His)(EtG) + DMSO − H]+ and indicates that in this case, EtG is not alkylated by the chlorambucil ligand, but is rather coordinated to the metal. Accordingly, the mass signals were attributed to identical species containing Met.
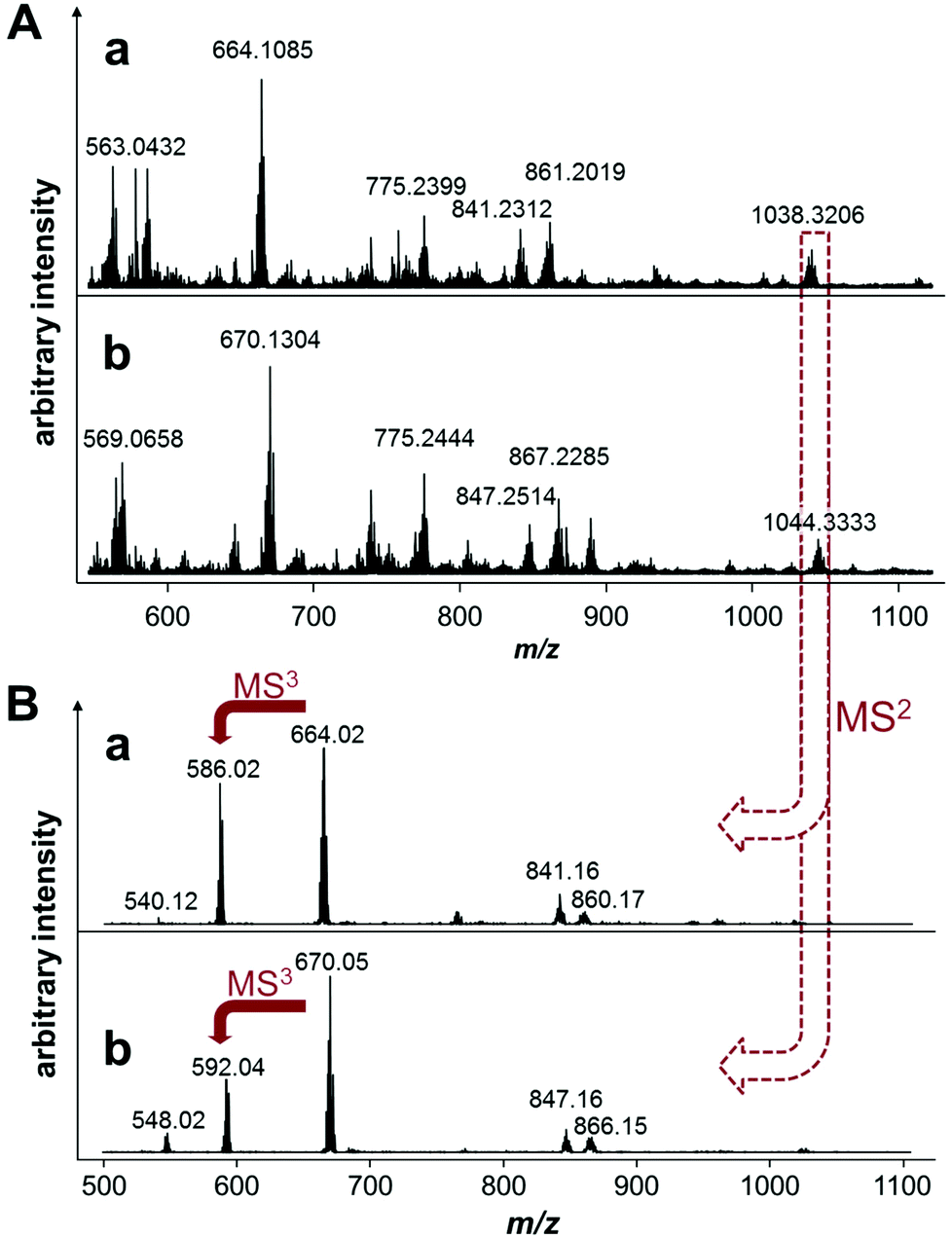 | ||
| Fig. 2 (A) Mass spectra of 6 preincubated in tetramethylammonium acetate with Met (a) or His (b) at a molar ratio of 1:2 for 1 h and then exposed to 4 equivalents of EtG for 19 h and (B) tandem mass spectrometric studies of the Met (a) and His (b) containing EtG adducts of 6, i.e., [(LCl)Ru(PTA)(amino acid)(EtG) + DMSO + H2O − 2H]+. The mass shift of 6 Da between the His and Met mass spectra suggests that these mass signals may be attributed to amino acid adducts. | ||
Finally, a mass signal corresponding to an alkylated EtG adduct containing coordinated His [(LOH)Ru(PTA)(His) + EtG − 2H]+ was observed in the TOF mass spectra at m/z 930.3142 in low abundance. However, this species was not detected in the MSn fragmentation spectra. Thus, it appears that amino acid coordination and simultaneous EtG alkylation is possible, but only to minor extents.
Hydrolysis of the metal–chloride bonds and coordination of biomolecules take place within several hours whereas alkylation requires longer reaction times. Therefore, stabilization of the metal–arene coordination is crucial for a synergistic antiproliferative activity. It appears that the presence of small nucleophilic molecules, e.g. EtG, His or Met, may stabilize the coordination of the arene to the ruthenium and the formed products were stable over several days. Moreover, the PTA remains tightly coordinated as well, so that the RAPTA core remains intact. Increased pH conditions also seem to increase the reaction kinetics for both hydrolysis/coordination and alkylation.
Complex 6 was also incubated with ubiquitin (2:1) or with a mixture of EtG and Ub (2:4:1) in water and tetramethylammonium acetate (1 mM). Ubiquitin is a low molecular weight protein well-suited for MS experiments and ruthenium arene complexes are known to coordinatively bind to it.42 Incubation of 6 with Ub showed a similar hydrolysis behavior to that observed in the stability studies and only very minor adducts were observed even after 69 h of incubation, mainly corresponding to Ru(PTA)Cl and Ru(PTA)(DMSO) adducts. Fewer 6–Ub adducts were observed in the presence of 2 equivalents of EtG in the reaction mixture. However, after 69 h, an adduct between hydrolyzed 6 and EtG is observed at m/z 829 which was also detected during incubation with EtG alone. The detected Ub adducts are similar to those observed during the incubation without EtG. Of note is a +10 charged protein mass signal at m/z 1904.19, which was observed at low abundance after 69 h. This mass signal suggests the presence of a ruthenium cross-linked Ub dimer, tentatively attributed to [Ub + Ru(DMSO) + (LOH)Ru(PTA)(EtG)]210+ (Fig. 3), showing that the compound may cross-link a DNA base with a protein, albeit in low yield.
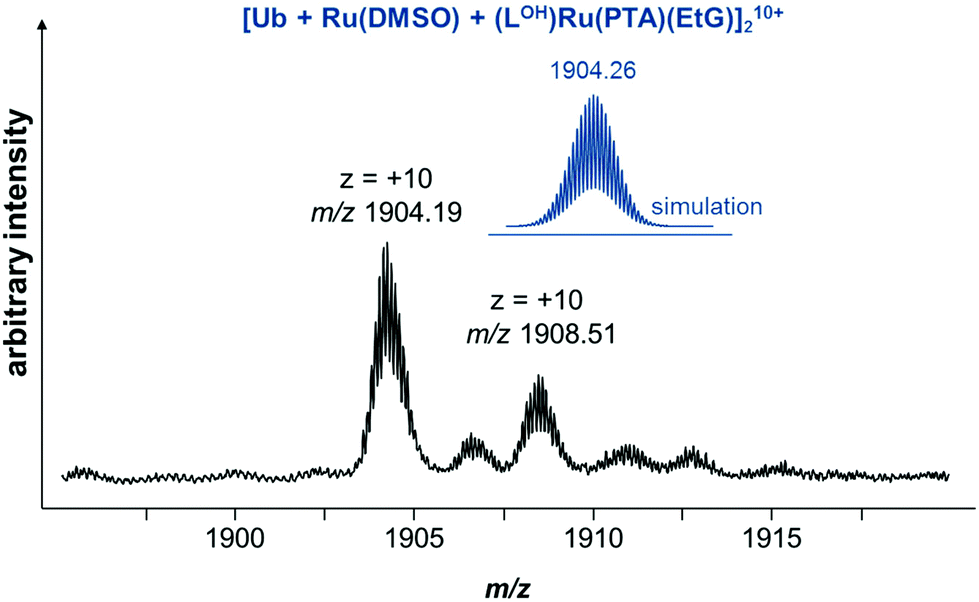 | ||
| Fig. 3 Excerpt of a mass spectrum recorded for a reaction mixture of 6, Ub and EtG (1:1:2) in water after 69 h. | ||
The reaction of 6 with Ub in tetramethylammonium acetate proceeds similarly to water. However the reaction appears to be more selective. In the presence of EtG, adduct formation is similar, yielding the Ru(PTA)(DMSO) adduct as the most prevalent species and after 44 h a mass signal corresponding to [LOH − Cl + EtG]+ is clearly observed.
The molar ratio of EtG was increased to 8 equivalents with respect to 6 during the reaction with Ub in buffered water. This did not influence the extent and the type of adducts. However, several small molecules formed featuring the characteristic ruthenium isotope pattern. Among these were several DMSO-stabilized species with up to two coordinated EtG, which underwent release of the arene, and accordingly, free L2OH was detected. Furthermore, [LOH − Cl + EtG]+ was observed after 24 h and slightly increased with time. Of note is an additional mass signal, which was found at m/z 793. This species may correspond to [(LOH)Ru(PTA) + EtG + OH − H]+ and appears to further underline the alkylation of EtG. The mass-to-charge ratio suggests again that this species would be +3 charged, which is decreased to +1 by the addition of hydroxide and loss of a proton.
Conclusions
The combination of more than one pharmacophore has been shown to be a useful strategy for improving the anticancer activity of compounds with modes of action different from those of established drug compounds. Herein, we described the modification of a RAPTA complex with chlorambucil and the variation of the co-ligands and the distance between the Ru moiety and the alkylating agent chlorambucil. This strategy led to compounds with in vitro anticancer activity in the low μM range, being superior in cisplatin resistant cancer cells to chlorambucil, RAPTA-T or a mixture of both. Mass spectrometry studies showed that the η6-arene in 6 slowly cleaves in solution, but is sufficiently stable to result in a different cytotoxic effect as compared to the combination of chlorambucil and RAPTA-T. Moreover, the coordination of EtG to 6 following hydrolysis proceeds more rapidly than alkylation of EtG by the chlorambucil fragment. Due to the instability of the ruthenium–arene bond, alkylation of EtG is mainly observed for the free ligand and is detected as an [LOH − Cl + EtG]+ ion, but this reaction also takes place with 6, as peaks that may be assigned to [(LOH)Ru(PTA) + EtG − 2H]+ and [(LOH)Ru(PTA)(OH) + EtG − H]+ are observed. Peaks assigned to cross-linked species indicate that the design strategy for such a compound is feasible. However, further refinement of the compound class is needed to increase the stability of the Ru–arene bond to obtain a higher extent of simultaneous protein ruthenation and DNA alkylation.Acknowledgements
We thank the University of Auckland, Genesis Oncology Trust (GOT-1263-RPG), the Royal Society of New Zealand, the EPFL, the Swiss National Science Foundation and COST CM1105 for financial support. The authors are grateful to the University of Vienna for the access to the Mass Spectrometry Core Facilities at the Faculty of Chemistry. Y. N. N. and E. R. M. thank the Russian Science Foundation for support (no. 14-13-00483).References
- C. H. Takimoto and E. Calvo, in Cancer Management: A Multidisciplinary Approach, ed. R. Pazdur, L. D. Wagman, K. A. Camphausen and W. J. Hoskins, 2008 Search PubMed.
- A. Caley and R. Jones, Surgery, 2012, 30, 186–190 Search PubMed.
- World Health Organization , WHO Model List of Essential Medicines, accessed 22 April 2014.
- S. Weingart, E. Brown, P. Bach, K. Eng, S. Johnson, T. Kuzel, T. Langbaum, R. Leedy, R. Muller and L. Newcomer, J. Natl. Compr. Cancer Network, 2008, 6, S1–14 Search PubMed.
- B. B. Bank, D. Kanganis, L. F. Liebes and R. Silber, Cancer Res., 1989, 49, 554–559 CAS.
- M. Di Antonio, K. I. E. McLuckie and S. Balasubramanian, J. Am. Chem. Soc., 2014, 136, 5860–5863 CrossRef CAS PubMed.
- M. Galanski, V. B. Arion, M. A. Jakupec and B. K. Keppler, Curr. Pharm. Des., 2003, 9, 2078–2089 CrossRef CAS.
- J. Reedijk, Chem. Rev., 1999, 99, 2499–2510 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- F. Arnesano and G. Natile, Coord. Chem. Rev., 2009, 253, 2070–2081 CrossRef CAS PubMed.
- P. J. Dyson and G. Sava, Dalton Trans., 2006, 1929–1933 RSC.
- I. Ott and R. Gust, Arch. Pharm., 2007, 340, 117–126 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2010, 54, 3–25 CrossRef PubMed.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5(10), 2140–2155 CAS.
- E. A. Hillard and G. Jaouen, Organometallics, 2011, 30, 20–27 CrossRef CAS.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- P. J. Dyson, Chimia, 2007, 61, 698–703 CrossRef CAS.
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC.
- A. A. Nazarov, C. G. Hartinger and P. J. Dyson, J. Organomet. Chem., 2014, 751, 251–260 CrossRef CAS PubMed.
- W. H. Ang, L. J. Parker, A. De Luca, L. Juillerat-Jeanneret, C. J. Morton, M. Lo Bello, M. W. Parker and P. J. Dyson, Angew. Chem., Int. Ed., 2009, 121, 3912–3915 CrossRef.
- G. R. Zimmermann, J. Lehár and C. T. Keith, Drug Discovery Today, 2007, 12, 34–42 CrossRef CAS PubMed.
- A. M. Pizarro, A. Habtemariam and P. J. Sadler, in Medicinal Organometallic Chemistry, Springer, 2010, pp. 21–56 Search PubMed.
- D. Y. Q. Wong, C. H. F. Yeo and W. H. Ang, Angew. Chem., Int. Ed., 2014, 53, 6752–6756 CrossRef CAS PubMed.
- A. Kurzwernhart, W. Kandioller, S. Bächler, C. Bartel, S. Martic, M. Buczkowska, G. Mühlgassner, M. A. Jakupec, H.-B. Kraatz and P. J. Bednarski, J. Med. Chem., 2012, 55, 10512–10522 CrossRef CAS PubMed.
- A. Kurzwernhart, W. Kandioller, C. Bartel, S. Bächler, R. Trondl, G. Mühlgassner, M. A. Jakupec, V. B. Arion, D. Marko and B. K. Keppler, Chem. Commun., 2012, 48, 4839–4841 RSC.
- A. Kurzwernhart, W. Kandioller, É. A. Enyedy, M. Novak, M. A. Jakupec, B. K. Keppler and C. G. Hartinger, Dalton Trans., 2013, 42, 6193–6202 RSC.
- W. Kandioller, E. Balsano, S. M. Meier, U. Jungwirth, S. Goschl, A. Roller, M. A. Jakupec, W. Berger, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2013, 49, 3348–3350 RSC.
- V. B. Arion, A. Dobrov, S. Goschl, M. A. Jakupec, B. K. Keppler and P. Rapta, Chem. Commun., 2012, 48, 8559–8561 RSC.
- A. A. Nazarov, D. Gardini, M. Baquie, L. Juillerat-Jeanneret, T. P. Serkova, E. P. Shevtsova, R. Scopelliti and P. J. Dyson, Dalton Trans., 2013, 42, 2347–2350 RSC.
- S. M. Meier, M. Hanif, Z. Adhireksan, V. Pichler, M. Novak, E. Jirkovsky, M. A. Jakupec, V. B. Arion, C. A. Davey, B. K. Keppler and C. G. Hartinger, Chem. Sci., 2013, 4, 1837–1846 RSC.
- B. Wu, M. S. Ong, M. Groessl, Z. Adhireksan, C. G. Hartinger, P. J. Dyson and C. A. Davey, Chem. – Eur. J., 2011, 17, 3562–3566 CrossRef CAS PubMed.
- Z. Adhireksan, G. E. Davey, P. Campomanes, M. Groessl, C. M. Clavel, H. Yu, A. A. Nazarov, C. H. F. Yeo, W. H. Ang, P. Dröge, U. Rothlisberger, P. J. Dyson and C. A. Davey, Nat. Commun., 2014, 5 Search PubMed.
- W. L. F. Armarego and C. Chai, Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, 5th edn, 2003 Search PubMed.
- C. M. Clavel, O. Zava, F. Schmitt, B. Halamoda Kenzaoui, A. A. Nazarov, L. Juillerat-Jeanneret and P. J. Dyson, Angew. Chem., Int. Ed., 2011, 50, 7124–7127 CrossRef CAS PubMed.
- J. H. Billman and G. R. Roehrig, J. Pharm. Sci., 1974, 63, 1487–1489 CrossRef CAS.
- W. H. Ang, E. Daldini, L. Juillerat-Jeanneret and P. J. Dyson, Inorg. Chem., 2007, 46, 9048–9050 CrossRef CAS PubMed.
- K. J. Kilpin, C. M. Clavel, F. Edafe and P. J. Dyson, Organometallics, 2012, 31, 7031–7039 CrossRef CAS.
- A. A. Nazarov, J. Risse, W. H. Ang, F. Schmitt, O. Zava, A. Ruggi, M. Groessl, R. Scopelitti, L. Juillerat-Jeanneret, C. G. Hartinger and P. J. Dyson, Inorg. Chem., 2012, 51, 3633–3639 CrossRef CAS PubMed.
- I. Berger, M. Hanif, A. A. Nazarov, C. G. Hartinger, R. O. John, M. L. Kuznetsov, M. Groessl, F. Schmitt, O. Zava, F. Biba, V. B. Arion, M. Galanski, M. A. Jakupec, L. Juillerat-Jeanneret, P. J. Dyson and B. K. Keppler, Chem. – Eur. J., 2008, 14, 9046–9057 CrossRef CAS PubMed.
- C. G. Hartinger, M. Groessl, S. M. Meier, A. Casini and P. J. Dyson, Chem. Soc. Rev., 2013, 42, 6186–6199 RSC.
| This journal is © The Royal Society of Chemistry 2015 |