
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mono- and dinuclear metal complexes containing the 1,5,9-triazacyclododecane ([12]aneN3) unit and their interaction with DNA†
Alfredo
Medina-Molner
,
Melanie
Rohner
,
Devaraj
Pandiarajan
and
Bernhard
Spingler
*
University of Zürich, Winterthurerstr. 190, CH 8057 Zürich, Switzerland. E-mail: spingler@chem.uzh.ch; Fax: +41 44 635 68 03; Tel: +41 44 635 46 56
First published on 5th December 2014
Abstract
The ability of mononucleating and dinucleating macrocylic polyamines and their novel nickel, copper and zinc complexes to induce the left-handed form of poly d(GC) was evaluated. The influence of the nuclearity, the presence or absence of metals ions, the linker length in the case of dinucleating ligands and the metal ion was determined. Almost all dinuclear metal complexes efficiently induced Z-DNA, the zinc ones being the least and the copper ones the most efficient ones. Additionally, the X-ray structures of three dinuclear metal complexes and one partially protonated ligand could be determined.
Introduction
Left-handed Z-DNA is a high-energy form of DNA, it can only be formed by alternating purine–pyrimindine sequences and when additional factors like supercoiling or multiple charged cations are present.1 In plasmids, formation of Z-DNA depends upon torsional stress that is induced by negative supercoiling.2–4 The twin-supercoiled domain model suggests that the movement of an RNA polymerase along the DNA double helix induces positive supercoil waves ahead of, and negative supercoil waves behind the transcription ensemble.4 As a consequence, Z-DNA is formed after the passing of the polymerase as the kinetics of the B- to Z-DNA transition is relatively rapid compared to the cellular processes such as transcription. Z-DNA was shown to play a functional role in gene regulation,5 positioning of the nucleosome6 and chromatin remodelling.7 Several research groups have studied the possible correlation between chromosomal breakpoints in human tumours with potential Z-DNA forming sequences.8 Z-DNA-GC repeats were shown to cause large-scale genome deletions in mammalian cells9 but only small changes in E. Coli.10 These variations were explained by different DNA repair mechanisms.11 Vásquez observed that the deletions or rearrangements generated in mammalian cells are mainly replication independent and are likely to be initiated by repair processing cleavages surrounding the Z-DNA-forming sequence. It was proposed that these types of genomic alterations are related to the chromosome breaks and gene translocations that map near Z-DNA-forming sequences in human leukaemia and lymphoma.12 Recently, the group of Dröge employed the Z-DNA binding protein ADAR1 as a probe together with a chromatin affinity precipitation. This procedure revealed almost 200 genomic Z-DNA hotspots in human A549 tumor cells.13 A forth of the hotspots were located in centromeres, but only two in promoter regions.In contrast with B-DNA, Z-DNA is highly immunogenic and some antibodies can recognise this conformation and these antibodies are not species-specific.14 For instance, patients with lupus erythematosis produce – among other nuclear components – antibodies with high specificity for Z-DNA.3 Lipps carried out an experiment where Z-DNA antibodies were used to stain ciliated protozoa that have both macronucleus and micronucleus.15 The micronucleus is only needed for genetic reproduction, whereas the transcription occurs exclusively in the macronucleus. The Z-DNA antibody was only staining the macronucleus, not the micronucleus. This experiment indirectly connected Z-DNA and transcription.
Antibodies were also used in metabolically active mammalian nuclei to detect Z-DNA. The experiments showed a relationship between the presence of Z-DNA and the negative torsional strain of DNA. As for the findings of Lipps’ group, the amount of Z-DNA increased considerably as transcription occurred, but was unaffected during DNA replication.16 De la Torre used immune-detection of Z-DNA as a marker for active transcription.14 She proposed this mechanism of detecting transcription as a cheaper and quicker method than the alternative in situ assay for active RNA polymerases.
The goal of our project was to design and study small molecules that are able to induce Z-DNA by strong and selective binding to this structural motive. The presence of Z-DNA has been linked to gene regulation in general and transcription in particular.1,17 Therefore, Z-DNA is an attractive new target for cancer therapy. Since Z-DNA is distinguished from the other conformations of DNA by its own very characteristic geometry, the recognition must be mainly based on geometrical features and not so much on the sequence of the nucleobases. In Z-DNA, the bases are pointing away from the helix axis. As a consequence, the bases are not as protected from the environment as in B-DNA; for example nitrogen N7 of the purines is highly exposed to the solvent.18,19
Previously, we have studied the ability of mononuclear complexes to induce Z-DNA.19,20 Furthermore, we have chosen to explore mono- and dinuclear metal complexes based upon 1,5,9-triazacyclododecane ([12]aneN3), which are expected to coordinate to N7 of neighbouring guanosine nucleobases.21 Linking two [12]aneN3 rings creates dinucleating ligands that are unable to form sandwich complexes in contrast to their smaller [9]aneN3 analogues.22 Lu and co-worker have systematically substituted hydrogen atoms of one or two N–H groups by methyl group(s) of dinucleating [12]aneN3 ligands.23 An increasing number of methyl groups dramatically decreased the ability of the corresponding dizinc complexes to hydrolyse RNA model compounds. This observation was explained by steric effects, though it remains unclear, whether the missing hydrogen donor ability of these methyl substituted complexes had also an influence.
A preliminary description of our results with one dinucleating [12]aneN3 ligand L3 and its metal complexes has recently appeared.24 Here, we discuss the chosen system in detail with an emphasis on the influence of the linker length between the two [12]aneN3 macrocycles.
Results and discussion
In this publication, we study the interaction of poly d(GC) with the mononucleating ligands L, LEt and the dinucleating ligands L2, L3, L4 as well as their Ni, Zn and Cu complexes (Scheme 1).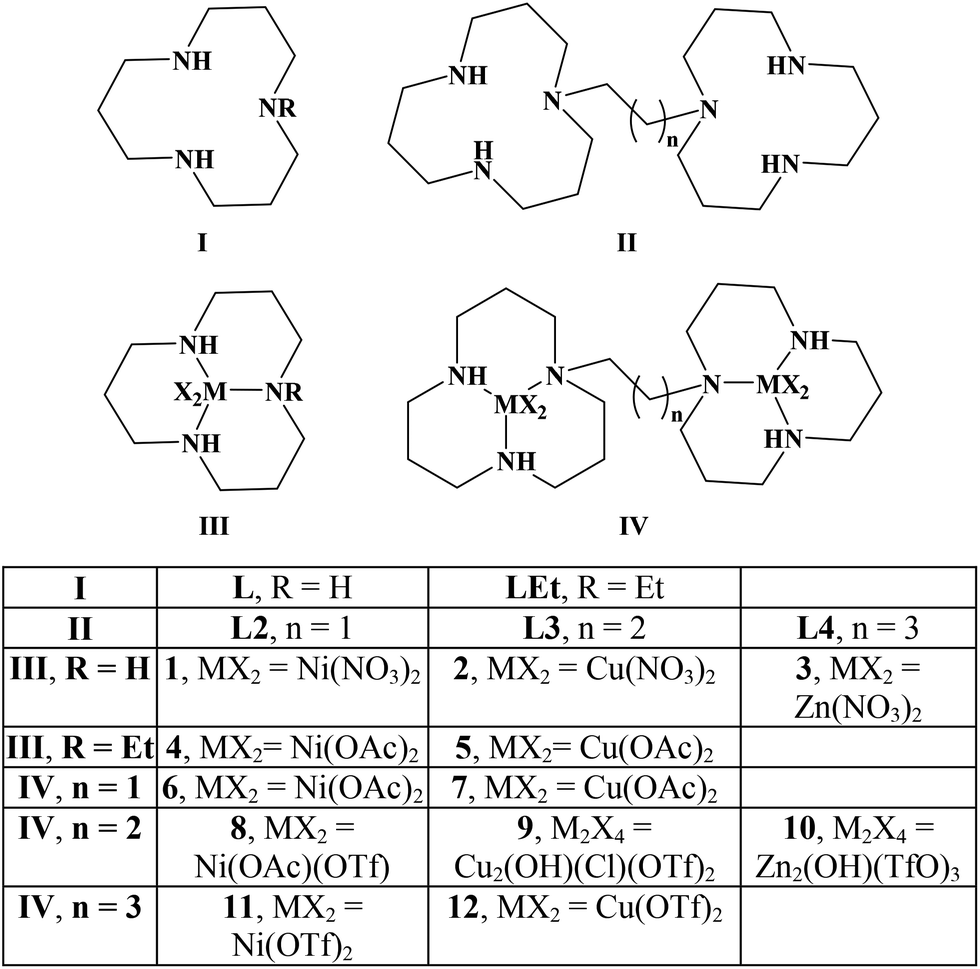 | ||
| Scheme 1 Studied compounds. | ||
Originally, we started working on the bis-[12]aneN3 ligand L3 that has its two [12]aneN3 rings joined together by a propylene linker.24 When comparing the two X-ray structures of the copper complexes L3Cu2(NO3)2(TfO)2 (Fig. 1) and 9,24 they revealed the high flexibility of the chosen ligand system. Both complexes were synthesized starting from ligand L3, in the latter case with copper(II) chloride to give the μ-hydroxo,μ-chlorido bridged complex 9 with a copper–copper distance of 3.2350(9) Å. In the former case, reaction of L3 with two equivalents of copper(II) nitrate gave the dinuclear complex L3Cu2(NO3)2(TfO)2 that does not show any copper–copper interaction (Cu(1)–Cu(2) distance: 8.5803(10) Å, Fig. 1). These findings were the reason why the linker length in the dinucleating ligand was varied in order to study the effect of increasing and reducing the flexibility of the system on the B- to Z-DNA transition.
 | ||
| Fig. 1 ORTEP representation of L3Cu2(NO3)2(TfO)2·(CH3OH)0.7(H2O)0.3 at 50% probability. Non-acidic hydrogen atoms and solvent molecules were omitted for clarity. | ||
In order to synthesize the known 1,4-bis(triazacyclododecyl)-butane (L4), we did not follow the route described in the literature25 but chose an alternative way that we already had employed for the synthesis of 1,4-bis(triazacyclododecyl)-propane.24 Our method led to an improvement of the overall yield of 1,4-bis(triazacyclododecyl)butane L4 from 40%25 to 74%. The synthesis of the corresponding precursor 1,4-butylenebistriflate could also be improved from 63%26 to 98%. 1,4-butylenebistriflate was added to two equivalents of 1,5,9-triazatricyclo[7.3.1.0]tridecane at room temperature to yield the desired product 13 in 86% yield. Compound 13 was then refluxed in triflic acid for 18 hours in order to obtain the hexaprotonated ligand L4 in 86% yield (Scheme 2). Deprotonation by sodium hydroxide and finally ion exchange column quantitatively yielded the free ligand L4.
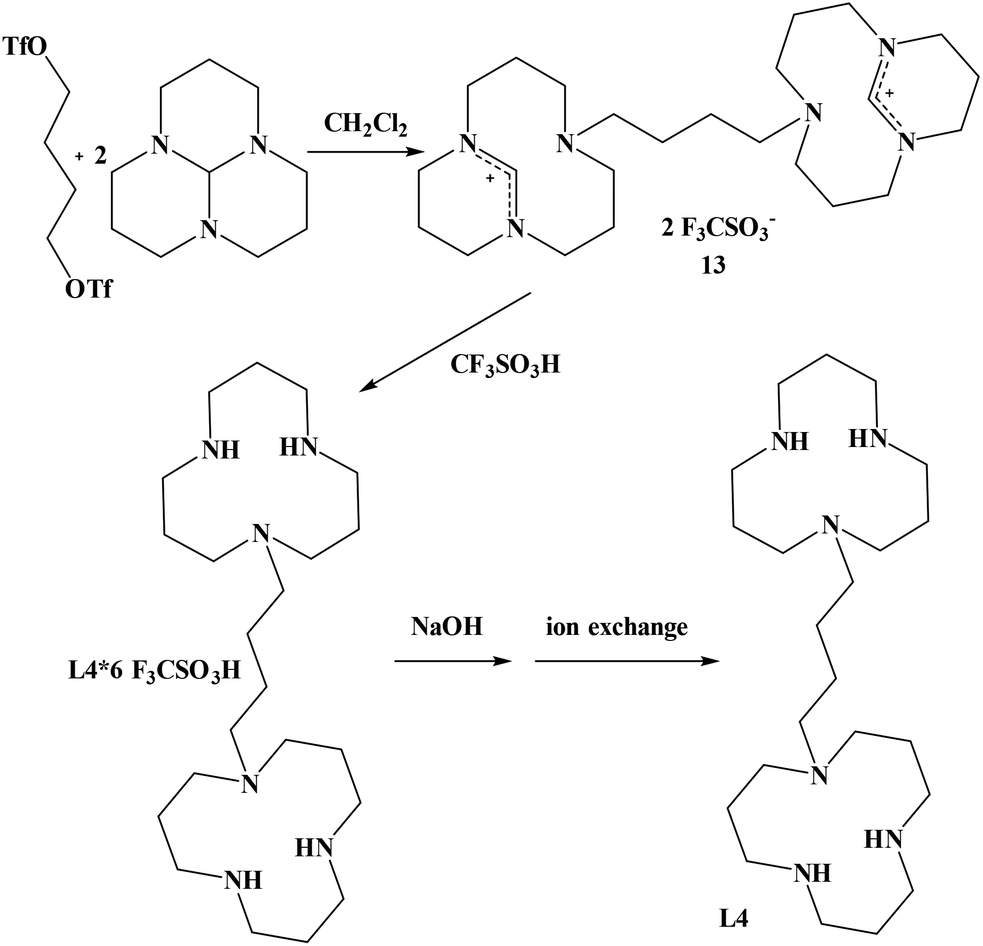 | ||
| Scheme 2 Synthesis of 1,4-bis(triazacyclododecyl)butane (L4). | ||
Crystals of 1,4-bis(triazacyclododecyl)butane·4TfOH could be obtained from a partially neutralised ethanolic solution of the hexaprotonated ligand L4, simply by concentrating the sample and storing it at 4 °C (Fig. S1†). In the crystal structure of L4·4TfOH, the asymmetric unit consists of half of a molecule – the other half being generated by a centre of inversion. All four NH protons of one [12]aneN3 ring were localized and, not surprisingly, all are involved in hydrogen bonding: The proton of N1 forms an internal bridge across the ring to N2 (2.693(2) Å), while the remaining 3 protons all bind to triflate anions with N–O distances between 2.732(3) Å (N3–O31_ x + 0.5, −y + 0.5, z − 0.5) to 2.934(3) Å (N2–O30). The synthesis of the ligand 1,2-bis(triazacyclododecyl)ethane (L2) has already been described by us before.27
The synthesis and isolation of the nickel (1), copper (2) and zinc (3) complexes has been achieved from ligand L in methanol and the corresponding nitrate salts. The synthesis of 1 with hydroxide and perchlorate as the anions has been reported together with the stability constant of L·Cu(II).28 A μ-chloro-μ-hydroxo dimer of Cu(II)[12]aneN3 with perchlorates as remaining anions has been characterised.29 The in situ generation of 3, but not its characterisation, was reported.30
Complex L4Ni2(AcO)4 was obtained when ligand L4 was mixed with two equivalents of Ni(CH3CO2)2. Since crystals could not be grown of the reaction product, an excess of NaB(C6H5)4 was added to a solution of complex L4Ni2(AcO)4 in water. The resulting precipitate was filtered off and dissolved in acetonitrile. Crystals could finally be grown by vapour diffusion of tetrahydropyrane (THP) to the acetonitrile solution.31 The nickel centre of L4Ni2(OAc)2(B(C6H5)4)2(NCCH3)2 presents a distorted octahedral coordination centre (Fig. 2). The triazamacrocycle binds to the nickel cation in a facial tridentate fashion; the remaining coordination sites are occupied by a bidentate acetate and an acetonitrile. The intramolecular nickel–nickel distance is 9.6723(7) Å. The asymmetric unit is composed of half a molecule, the other half of the complex is generated by a centre of inversion. A disordered THP molecule was found in the crystal lattice as well.
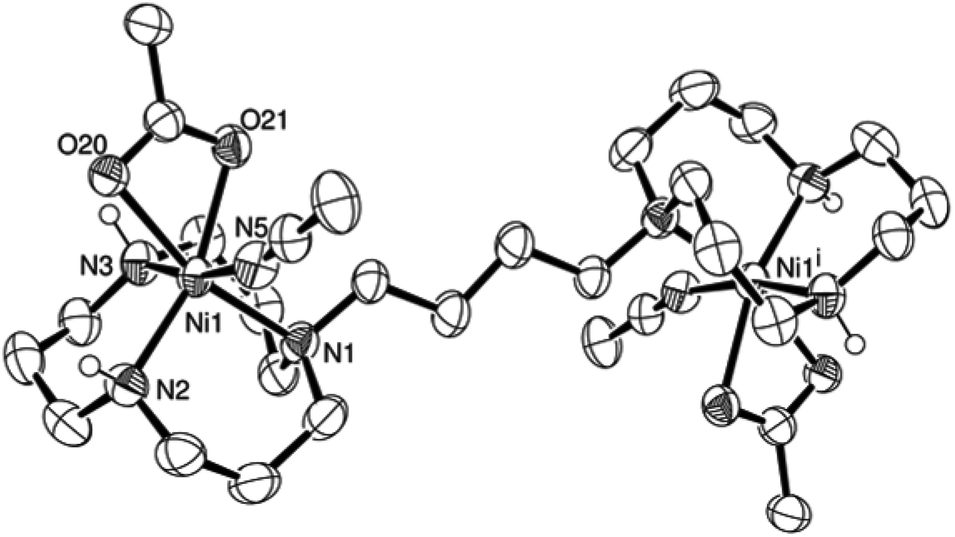 | ||
| Fig. 2 ORTEP representation of [L4Ni2(AcO)2(BPh4)2(NCCH3)2]·THP at 50% probability. Non-acidic hydrogen atoms, non-coordinating anions and non-coordinating solvent molecules were omitted for clarity. | ||
Ligand L2 was reacted with two equivalents of nickel acetate in a mixture of methanol–acetonitrile to yield a blue solution. The resulting nickel complex (6) was crystallised by vapour diffusion of pentane into the corresponding complex solution in dichloromethane. The nickel centre presents a slightly distorted octahedral coordination. The nickel–nickel distance is with 6.542(1) Å 3.13 Å shorter than in the butylene bridged case. The water molecule coordinated to the nickel centre is in hydrogen bonding with the non-coordinating oxygen atoms of both acetate groups (Fig. 3).
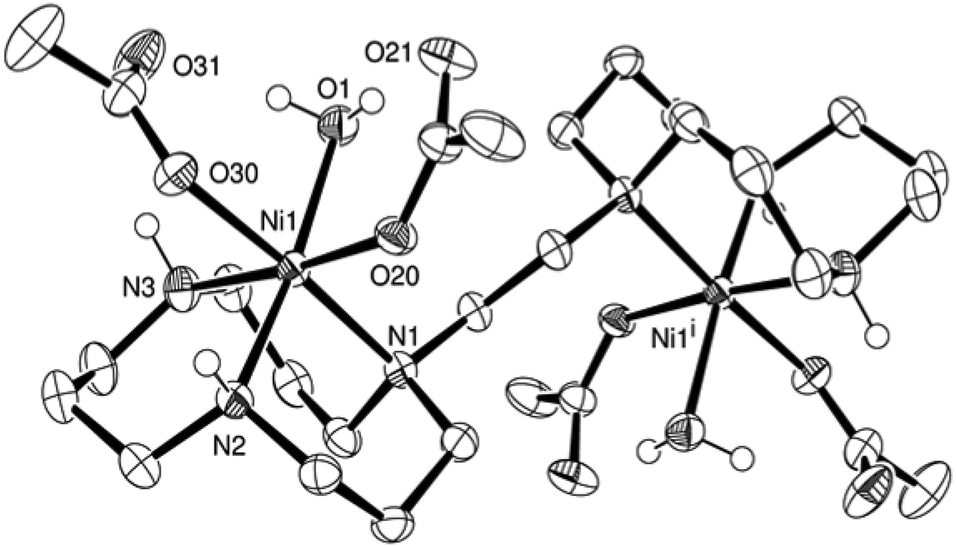 | ||
| Fig. 3 ORTEP representation of 6·2H2O at 50% probability. Non-acidic hydrogen atoms were omitted for clarity. | ||
Ligand L3 was used to synthesise dinuclear nickel (8), copper (9) and zinc (10) complexes. All three metal complexes could induce Z-DNA (Fig. 4). These results showed for the very first time an opposite behaviour between mononuclear and dinuclear metal complexes. Thereby, our original hypothesis was confirmed that dinuclear metal complexes with the appropriate distance between the two metal centres would be more efficient Z-DNA inducers than mononuclear ones.32 However, our experimental studies showed a much more dramatic result, since the mononuclear complexes 1, 2 and 3 (Fig. S2, pink; Fig. S3, blue; Fig. S3,† pink) did not induce Z-DNA at all, or even induced denaturation (complex 5), whereas their equivalent dinuclear complexes are one of the best Z-DNA inducers described so far (Table 1).
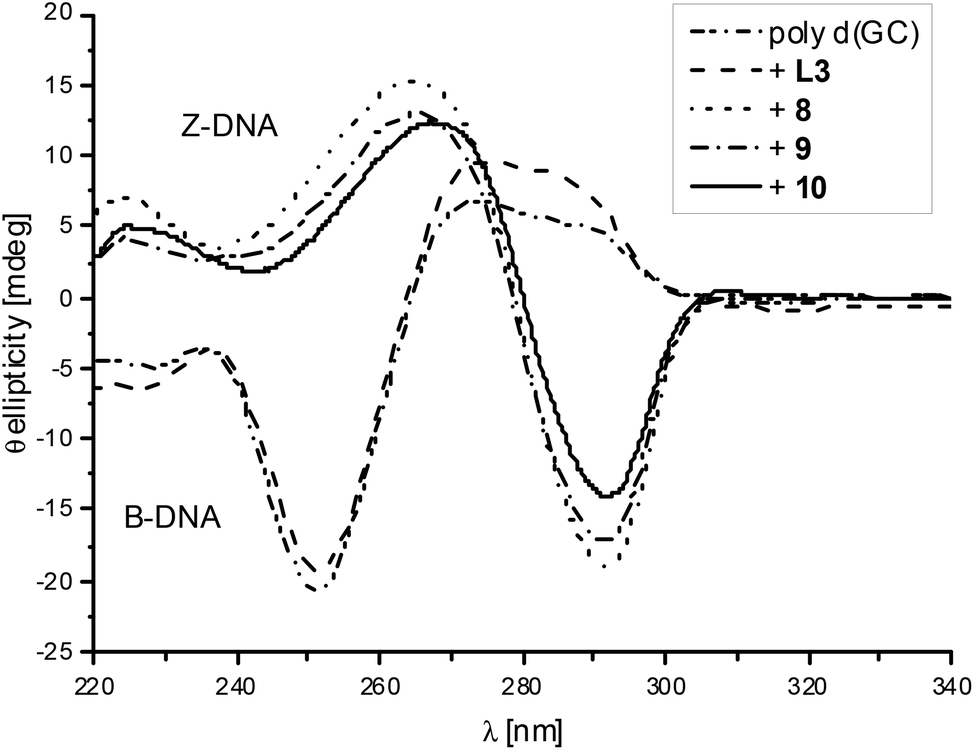 | ||
| Fig. 4 CD spectra of poly d(GC) and with added L3, 8, 9 or 10. The spectra are shown after the possible transition has been completed. | ||
The nickel complex 8 induced the midpoint of the B- to Z-DNA transition at 0.21 equivalents of metal (0.11 equivalents of metal complex). The zinc complex 10 was the worst Z-DNA inducer among the three successful complexes having a midpoint at 0.30 equivalents of zinc (0.15 equivalents of complex, Fig. 5). The best Z-DNA inducer was the copper complex 9, even though it contained chloride which is retarding the B- to Z-DNA transition.33 The midpoint of this transition happened at 0.16 equivalents of copper (0.08 equivalents of complex, Fig. 5). As the zinc complex 10 was the worst Z-DNA inducer (0.30 equivalent metals needed versus DNA phosphate) among the metal series of nickel, copper and zinc, we decided not to further study the other members of the homologous series of the zinc complexes.
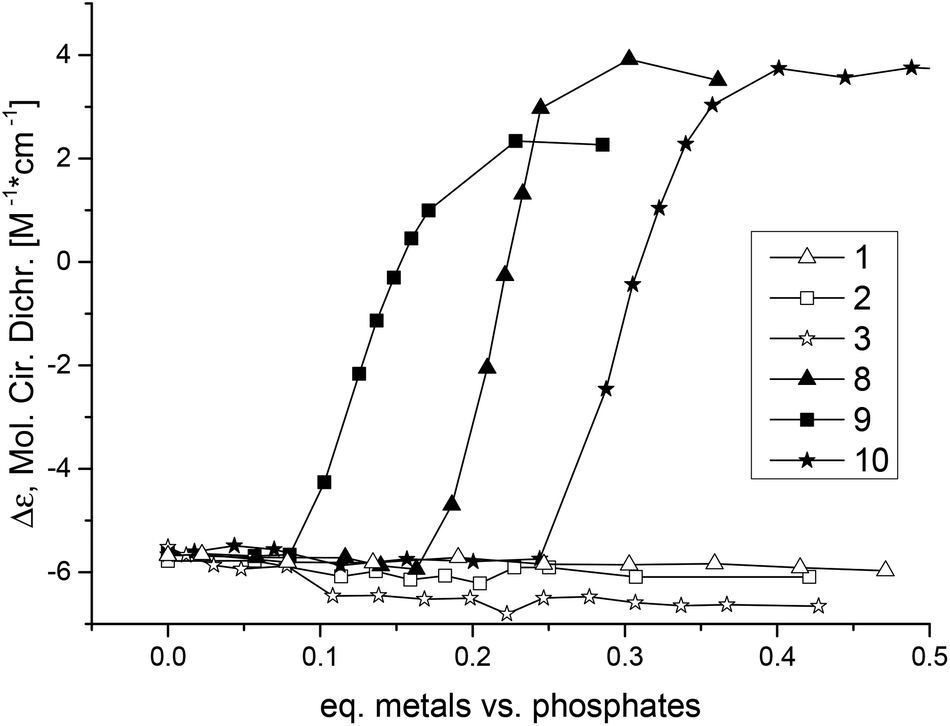 | ||
| Fig. 5 CD titration at 255 nm of poly d(GC) with added 1, 2, 3, 8, 9, or 10. The metal content of the complexes and not the equivalents of complexes is given in the x-axis (in order to compensate for the fact that dinuclear complexes have twice as much metal ions as the mononuclear ones). | ||
The linker between the two units in ligand L3 was first increased from three to four carbon atoms to give ligand L4, which is expected to have a higher flexibility and to generate a longer distance between the two metal centres. Nickel (11) and copper (12) complexes induced Z-DNA at a slightly higher concentration than their equivalent complexes with ligand L3. Thus increasing the flexibility of the system had only a slightly negative if any effect on the ability of the complexes to induce Z-DNA (Fig. S4 and S5†).
Since increasing the flexibility of the system did not help to improve the efficiency of Z-DNA induction, a more rigid ligand was synthesised by reducing the length of the linker to two carbon atoms in ligand L2. Nickel (6) and copper (7) complexes were synthesised and tested for their ability to promote the B- to Z-DNA transition. The nickel complex (6) was slightly more efficient in inducing Z-DNA (Fig. S4†) than the other dinickel complexes. However, the copper complex (7) was not able to induce Z-DNA (Fig. S5†), demonstrating a dramatic difference when the flexibility of the ligand was reduced.
The mononucleating ligand LEt was synthesised in order to generate a mononuclear system that resembles a halved dinuclear complex, in which the ethyl substituent mimics the steric and electronic influences of the linker in the dinuclear systems. A similar behaviour as for nickel complex 1 was observed when the nickel complex (4, Fig. S6,† blue) with ligand LEt was tested in the B- to Z-DNA transition. However a different result was obtained with the copper complex 5 that contained ligand LEt. Denaturation of the DNA was induced after the addition of 0.3 equivalents of the copper complex 5 (Fig. S6,† pink). The denaturation is more easily seen by either looking at the whole CD or UV/vis spectra at different equivalents of the copper complex (Fig. S7†). In summary it can be said that neither the nickel 4 nor the copper 5 complexes induced Z-DNA.
As a control, the mononuclear ligand LEt·3TfOH and dinuclear butylene bridged ligand L4·6TfOH were also tested for their ability to induce Z-DNA (Fig. S8†). Ligand LEt·3TfOH did not have any effect on DNA, however the dinucleating ligand L4·6TfOH induced condensation of DNA.34 This condensation started at around 0.5 equivalents of ligand versus DNA phosphates and lead to a strong increase of the absorption at 255 nm. This aggregation of DNA did not have an intermediate Z-DNA transition as observed in Zacharias’ work, instead of that the condensation occurred directly from B-DNA as in Chaires studies34,35 (Fig. S9†). Zacharias suggested that the hydrophobic part of the acetate group could be decisive for the condensation.34 In our system, 6 triflates per ligand are present; the CF3 group of the triflates could be the hydrophobic tale needed for the condensation. However, LEt·3TfOH and L3·6TfOH with the same ratio of nitrogen atoms versus triflates did not induce condensation of DNA.
EDTA was used to study the reversibility of the transition metal induced B- to Z-DNA transformation.32,36 The action of EDTA alone upon poly d(GC) was studied first. As expected, EDTA did not induce Z-DNA but rather surprisingly, at high concentration caused condensation of DNA (Fig. S10†). Further addition of EDTA lowered the positive CD band at 275 nm. Then, the effect of EDTA upon the complexes 6 (Ni)/9 (Cu) and poly d(GC) were investigated (Fig. S11†). In both cases, the DNA essentially converted back to the B-form after the addition of two equivalents of EDTA, and a full conversion back to B-DNA was found after 3 equivalents of EDTA versus metal ions.
Conclusions
This study aimed to determine the factors that favour the induction of Z-DNA by metal complexes. Our original hypothesis was that sterically demanding dinuclear metal complexes with a metal to metal distance of about 5–7 Å would be more efficient inducers of the left-handed form than other dinuclear and mononuclear complexes. This idea was inspired by looking at the geometry of Z-DNA with its solvent exposed atom N7 of guanine and the interstrand N7–N7 distance between two guanine of a GC base pair step. We soon realized that for the 1,5,9-triazadodecane system, the reality was even better than our hypothesis: Only the dinuclear copper and nickel complexes of 1,3-bis-triazadodecyl-propane were inducing the left-handed form of poly d(GC) but not the corresponding N-ethyl-triazadodecane mononuclear metal complexes.24 In the current publication, we have substantially expanded the set of studied compounds by including dinucleating ligands whose rings are either linked by an ethylene or butylene group. As a first conclusion, we can say that none of the studied ligands were inducing the Z-form. L4·6TfOH was causing condensation of the DNA, but apart from this result no effects were observed. This is interesting because various linear polyamines were found to be powerful Z-DNA inducers.37 To the best of our knowledge, no macrocyclic polyamines were ever reported to induce Z-DNA.38 Summarising the results of the extended set of mononuclear metal complexes, none of them induce Z-DNA. On the other hand, almost all of the dinuclear complexes do induce the left-handed form with the notable exception of the di-copper complex of 1,2-bis-triazadodecyl-ethane. At the moment, we still do not know, what causes this exception. Comparing the relative ability of the various metal ions, copper seems to be always better than nickel and zinc. This trend essentially follows the Irving–Williams series39 as discussed by Barone et al.40 Concerning the influence of the linker length, we can note that the differences between ethylene, propylene and butylene linker are very subtle, with the propylene linker being the most efficient among them. This indifference of the system towards the linker length might be caused by the nature of the ligands that lack an anionic central attachment point for the metal ions41 and the flexibility of the chosen aliphatic linker.As an outlook, one could imagine similar complexes having high affinity for these specific forms of DNA that could be functionalised with fluorescence markers or damaging moieties for future applications in a chemical detection of the transcription or in an antitumour therapy.
Experimental section
Instrumentation and materials
All chemicals were purchased from Sigma-Aldrich (Buchs, CH) and used without further purification. The synthesis of compounds LEt, L3, 4, 5, 8, 924 and L227 has already been described by us previously. The synthesis of 1,4-butylenebistriflate was done slightly differently than reported in the literature26 (see ESI† for details). All the reactions were performed under a nitrogen atmosphere. The reactions were monitored by HPLC or thin layer chromatography (TLC). TLC was carried out on 0.25 mm Merck silica gel aluminium plates (60 F254) or aluminium oxide pre-coated plastic sheets (alox N/UV254) using UV light or Schlittler's reagent42 as a visualising agent. Column chromatography was performed on silica gel (particle size 0.040–0.063 mm) or aluminium oxide (basic, 0.05–0.15 mm, pH 9.5 ± 0.5). Ion exchange chromatography was performed on DOWEX 2 X8 20–50 (Fluka) or AMBERLYST A26-OH, macromolecular ion-exchange resin (ABCR). Elemental analyses were performed on a Leco CHNS-932 elemental analyser. Electrospray ionisation (ESI) mass spectra were recorded on a Merck Hitachi M-8000 spectrometer or a Bruker Daltonics HCT 6000 mass spectrometer. UV/Vis spectra were recorded on a Varian Cary 50 spectrometer and IR spectra on a Perkin Elmer, Spectrum Two spectrometer equipped with a Golden Gate ATR. NMR spectra were acquired on a Varian Mercury 200 MHz or Gemini 300 MHz, and on a Bruker DRX 400 or 500 MHz spectrometer. The chemical shifts are relative to residual solvent protons as reference. Circular dichroism measurements were performed using a Jasco J-810 spectropolarimeter equipped with a Jasco PFD-4255 Peltier temperature controller. A 0.1 mM poly d(GC) solution in 1 mM sodium cacodylate buffer (pH = 7) was titrated with various aliquots of 1 mM solutions of the ligands or metal complexes in the same buffer. The samples were warmed to 60 °C for 5 minutes and then cooled down to 25 °C for the CD measurements. The CD spectra were smoothed by adjacent averaging. Crystallographic data were collected at 183(2) K on a Stoe IPDS (Mo Kα1 radiation, λ = 0.71073 Å) diffractometer using a graphite-monochromated radiation. Suitable crystals were covered with Paratone N oil, mounted on top of a glass fibre and immediately transferred to the diffractometer. A maximum of eight thousand reflections distributed over the whole limiting sphere were selected by the program SELECT and used for unit cell parameter refinement with the program CELL.43 Data were collected for Lorentz and polarisation effects as well as for absorption (numerical). Structures were solved with direct methods using SHELXS-9744 or SIR9745 and were refined by full-matrix least-squares methods on F2 with SHELXL-97.46 Crystals with low symmetry space groups measured on the one-circle Stoe IPDS diffractometer have an intrinsically low completeness of their data sets. CCDC 1021588–1021591 contain the supplementary crystallographic data for this paper.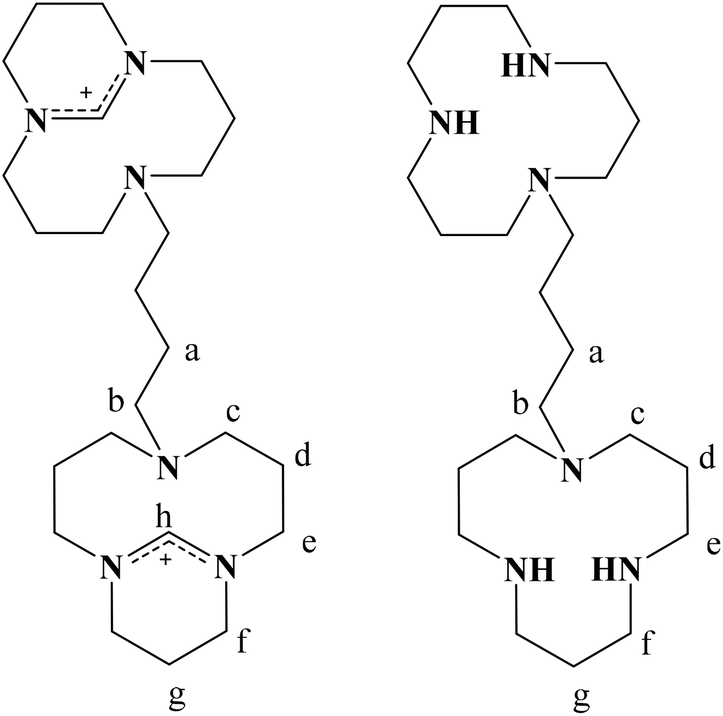 | ||
| Scheme 3 Atom numbering for 13 (left) and L4 (right). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6) (10.11 g, 86%). Elemental analysis: C28H54F18N6O18S6: calc. C (25.93%), H (4.20%), N (6.48%); exp. C (25.81%), H (4.23%), N (6.43%). MS (ESI): m/z (%) = 547 [M + TfOH + H]+ (100), 697 [M + 2(TfOH) + H]+ (7), 847 [M + 3(TfOH) + H]+ (4). IR (Golden Gate ATR):
:6) (10.11 g, 86%). Elemental analysis: C28H54F18N6O18S6: calc. C (25.93%), H (4.20%), N (6.48%); exp. C (25.81%), H (4.23%), N (6.43%). MS (ESI): m/z (%) = 547 [M + TfOH + H]+ (100), 697 [M + 2(TfOH) + H]+ (7), 847 [M + 3(TfOH) + H]+ (4). IR (Golden Gate ATR): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3045 (NH2+, w), 2830 (CH2, w), 1212 (C–F, s), 1165 (SO3, s), 1021 (S
= 3045 (NH2+, w), 2830 (CH2, w), 1212 (C–F, s), 1165 (SO3, s), 1021 (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, s) cm−1. 1H-NMR (400 MHz, D2O): δ = 1.93 (broad s, 4 H, H(a)), 2.30–2.33 (m, 12 H, H(d) + H(g)), 3.37 (broad s, 4 H, H(b)), 3.41–3.46 (m, 16 H, H(e) + H(f)) and 3.53 ppm (broad s, 8 H, H(c)). 13C-NMR (100 MHz, D2O): δ = 17.88 (C(d)), 20.96 (C(g)), 21.47 (Ca(a)), 41.39 (C(f)), 42.46 (C(e)), 47.57 (C(c)), 54.60 (C(b)) and 120.21 (q, J = 317.0 Hz, CF3). Crystals could be obtained from a partially neutralised ethanolic solution of the originally hexaprotonated ligand, simply by concentrating the sample and storing at 4 °C.
= 3197 (NH, w), 2935 (CH2, w), 1487 (CH2, m), 1385 (NO3−, s) cm−1.
= 3233 (NH, w), 1421 (NO3−, m), 1340 (NO3−, s), 1007 (Cu–N, w) cm−1.
= 3192 (NH, m), 2934 (CH2, w), 1490 (CH2, m), 1457 (NO3−, m), 1324 (NO3−, s), 1009 (Zn–N, m) cm−1.
= 2912 (CH2, w), 1568 (AcO−, s), 1407 (AcO−, s), 877 (Ni–N, m) cm−1.
= 2962 (CH2, w), 1557 (AcO−, m), 1393 (AcO−, m), 1015 (Cu–N, m) cm−1.
:10:1), yielding a bright blue solution with some solid material. The suspension was filtered while still hot and dried under reduced pressure obtaining a green oil in 90% yield (11). Elemental analysis: C26H48F12N6Ni2O12S4·(CH3CN): calc. C (29.21%), H (4.46%) N (8.52%); exp. C (29.67%), H (4.80%), N (8.58%). IR (Golden Gate ATR): = 3446 (NH2+, w), 2879 (CH2, w), 1224 (C–F, s), 1160 (SO3, s), 1026 (SO, s) cm−1.
O, s) cm−1. 1H-NMR (400 MHz, D2O): δ = 1.93 (broad s, 4 H, H(a)), 2.30–2.33 (m, 12 H, H(d) + H(g)), 3.37 (broad s, 4 H, H(b)), 3.41–3.46 (m, 16 H, H(e) + H(f)) and 3.53 ppm (broad s, 8 H, H(c)). 13C-NMR (100 MHz, D2O): δ = 17.88 (C(d)), 20.96 (C(g)), 21.47 (Ca(a)), 41.39 (C(f)), 42.46 (C(e)), 47.57 (C(c)), 54.60 (C(b)) and 120.21 (q, J = 317.0 Hz, CF3). Crystals could be obtained from a partially neutralised ethanolic solution of the originally hexaprotonated ligand, simply by concentrating the sample and storing at 4 °C.
= 3197 (NH, w), 2935 (CH2, w), 1487 (CH2, m), 1385 (NO3−, s) cm−1.
= 3233 (NH, w), 1421 (NO3−, m), 1340 (NO3−, s), 1007 (Cu–N, w) cm−1.
= 3192 (NH, m), 2934 (CH2, w), 1490 (CH2, m), 1457 (NO3−, m), 1324 (NO3−, s), 1009 (Zn–N, m) cm−1.
= 2912 (CH2, w), 1568 (AcO−, s), 1407 (AcO−, s), 877 (Ni–N, m) cm−1.
= 2962 (CH2, w), 1557 (AcO−, m), 1393 (AcO−, m), 1015 (Cu–N, m) cm−1.
:10:1), yielding a bright blue solution with some solid material. The suspension was filtered while still hot and dried under reduced pressure obtaining a green oil in 90% yield (11). Elemental analysis: C26H48F12N6Ni2O12S4·(CH3CN): calc. C (29.21%), H (4.46%) N (8.52%); exp. C (29.67%), H (4.80%), N (8.58%). IR (Golden Gate ATR): = 3446 (NH2+, w), 2879 (CH2, w), 1224 (C–F, s), 1160 (SO3, s), 1026 (SO, s) cm−1.
Ligand L4 (61 mg, 0.15 mmol) in dichloromethane was added to a stirred solution of nickel acetate (76.6 mg, 0.31 mmol) in EtOH–MeOH, obtaining a pale green solution. Part of this solution was treated with a saturated solution of NaBPh4 in water in order to precipitate the dinickel complex of L4. Suitable crystals for X-ray analysis could be grown by slow vapour diffusion of tetrahydropyrane into a solution of the nickel complex in acetonitrile.31
= 3251 (NH2+, w), 2883 (CH2, w), 1220 (C–F, s), 1158 (SO3, s), 1022 (SO, s) cm−1.
Acknowledgements
We thank the Swiss National Science Foundation, the Eidgenössische Stipendien Kommission für ausländische Studierende and the Department of Chemistry, University of Zurich for financial support.Notes and references
- L. Yang, S. Wang, T. Tian and X. Zhou, Curr. Med. Chem., 2012, 19, 557–568 CrossRef CAS.
- C. K. Singleton, J. Klysik, S. M. Stirdivant and R. D. Wells, Nature, 1982, 299, 312–316 CrossRef CAS.
- A. Herbert and A. Rich, J. Biol. Chem., 1996, 271, 11595–11598 CrossRef CAS PubMed.
- L. F. Liu and J. C. Wang, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7024–7027 CrossRef CAS.
- S. Rothenburg, F. Koch-Nolte, A. Rich and F. Haag, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8985–8990 CrossRef CAS PubMed; D.-B. Oh, Y.-G. Kim and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16666–16671 CrossRef PubMed.
- B. Wong, S. Chen, J.-A. Kwon and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2229–2234 CrossRef CAS PubMed.
- H. Liu, N. Mulholland, H. Fu and K. Zhao, Mol. Cell. Biol., 2006, 26, 2550–2559 CrossRef CAS PubMed.
- M. Steinmetz, D. Stephan and K. F. Lindahl, Cell, 1986, 44, 895–904 CrossRef CAS; A. Weinreb, D. R. Katzenberg, G. L. Gilmore and B. K. Birshtein, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 529–533 CrossRef.
- G. Wang, S. Carbajal, J. Vijg, J. DiGiovanni and K. M. Vasquez, J. Natl. Cancer Inst., 2008, 100, 1815–1817 CrossRef CAS PubMed.
- A. M. Freund, M. Bichara and R. P. P. Fuchs, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 7465–7469 CrossRef CAS.
- D. T. Kha, G. Wang, N. Natrajan, L. Harrison and K. M. Vasquez, J. Mol. Biol., 2010, 398, 471–480 CrossRef CAS PubMed.
- G. Wang, L. A. Christensen and K. M. Vasquez, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2677–2682 CrossRef CAS PubMed.
- H. Li, J. Xiao, J. Li, L. Lu, S. Feng and P. Dröge, Nucleic Acids Res., 2009, 37, 2737–2746 CrossRef CAS PubMed.
- A. Cerna, A. Cuadrado, N. Jouve, S. M. Diaz de la Espina and C. De la Torre, Eur. J. Histochem., 2004, 48, 49–55 CAS.
- H. J. Lipps, A. Nordheim, E. M. Lafer, D. Ammermann, B. D. Stollar and A. Rich, Cell, 1983, 32, 435–441 CrossRef CAS.
- B. Wittig, T. Dorbic and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 2259–2263 CrossRef CAS.
- S. Rothenburg, F. Koch-Nolte and F. Haag, Immunol. Rev., 2001, 184, 286–298 CAS.
- R. R. Sinden, DNA: Structure and Function, Academic Press, San Diego, 1994 Search PubMed.
- B. Spingler, in Metal Ions and Nucleic Acids, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Springer Science+Business Media, 2012, vol. 10, pp. 103–118 Search PubMed.
- B. Spingler and C. Da Pieve, Dalton Trans., 2005, 1637–1643 RSC; M. G. Santangelo, A. Medina-Molner, A. Schweiger, G. Mitrikas and B. Spingler, J. Biol. Inorg. Chem., 2007, 12, 767–775 CrossRef CAS PubMed; B. Spingler, C. Da Pieve, A. Medina-Molner, P. M. Antoni and M. G. Santangelo, Chimia, 2009, 63, 153–156 CrossRef; M. G. Santangelo, P. M. Antoni, B. Spingler and G. Jeschke, ChemPhysChem, 2010, 11, 599–606 CrossRef PubMed.
- B. Spingler, F. Zobi, P. M. Antoni, A. Medina-Molner and R. Alberto, Chimia, 2005, 59, 826–831 CrossRef CAS.
- R. Haidar, M. Ipek, B. DasGupta, M. Yousaf and L. J. Zompa, Inorg. Chem., 1997, 36, 3125–3132 CrossRef CAS PubMed; B. Graham, L. Spiccia, A. M. Bond, M. T. W. Hearn and C. M. Kepert, J. Chem. Soc., Dalton Trans., 2001, 2232–2238 RSC; T. Moufarrej, K. Bui and L. J. Zompa, Inorg. Chim. Acta, 2002, 340, 56–64 CrossRef.
- Y. Song, J. Zan, H. Yan, Z.-L. Lu and R. Wang, Org. Biomol. Chem., 2012, 10, 7714–7720 CAS.
- A. Medina-Molner and B. Spingler, Chem. Commun., 2012, 48, 1961–1963 RSC.
- G. R. Weisman, D. J. Vachon, V. B. Johnson and D. A. Gronbeck, J. Chem. Soc., Chem. Commun., 1987, 886–887 RSC.
- R. W. Alder, D. D. Ellis, R. Gleiter, C. J. Harris, H. Lange, A. G. Orpen, D. Read and P. N. Taylor, J. Chem. Soc., Perkin Trans. 1, 1998, 1657–1668 RSC.
- A. Medina-Molner, O. Blacque and B. Spingler, Org. Lett., 2007, 9, 4829–4831 CrossRef CAS PubMed.
- L. J. Zompa, Inorg. Chem., 1978, 17, 2531–2536 CrossRef CAS.
- P. G. Graham, D. C. Weatherburn, F. C. March and W. T. Robinson, Inorg. Chim. Acta, 1990, 178, 227–232 CrossRef CAS.
- V. M. Shelton and J. R. Morrow, Inorg. Chem., 1991, 30, 4295–4299 CrossRef CAS.
- B. Spingler, S. Schnidrig, T. Todorova and F. Wild, CrystEngComm, 2012, 14, 751–757 RSC.
- B. Spingler and P. M. Antoni, Chem. – Eur. J., 2007, 13, 6617–6622 CrossRef CAS PubMed.
- B. Spingler, Inorg. Chem., 2005, 44, 831–833 CrossRef CAS.
- W. Zacharias, J. C. Martin and R. D. Wells, Biochemistry, 1983, 22, 2398–2405 CrossRef CAS.
- J. B. Chaires and J. M. Sturtevant, Biopolymers, 1988, 27, 1375–1387 CrossRef CAS PubMed.
- J. H. van de Sande and T. M. Jovin, EMBO J., 1982, 1, 115–120 CAS; W. C. Russell, B. Precious, S. R. Martin and P. M. Bayley, EMBO J., 1983, 2, 1647–1653 Search PubMed; M. Govindaraju, F. S. Monica, R. Berrocal, K. R. S. Sambasiva Rao and K. S. Rao, Curr. Trends Biotechnol. Pharm., 2012, 6, 204–209 Search PubMed.
- M. Behe and G. Felsenfeld, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 1619–1623 CrossRef CAS.
- A. Parkinson, M. Hawken, M. Hall, K. J. Sanders and A. Rodger, Phys. Chem. Chem. Phys., 2000, 2, 5469–5478 RSC.
- H. Irving and R. J. P. Williams, Nature, 1948, 162, 746–747 CrossRef CAS.
- G. Barone, A. Terenzi, A. Lauria, A. M. Almerico, J. M. Leal, N. Busto and B. Garcia, Coord. Chem. Rev., 2013, 257, 2848–2862 CrossRef CAS PubMed.
- A. L. Gavrilova and B. Bosnich, Chem. Rev., 2004, 104, 349–383 CrossRef CAS PubMed.
- E. Schlittler and J. Hohl, Helv. Chim. Acta, 1952, 35, 29–45 CrossRef.
- STOE-IPDS Software package, STOE & Cie, GmbH, Darmstadt, Germany, 1998 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467–473 CrossRef.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. Da Pieve, A. Medina-Molner and B. Spingler, Synthesis, 2007, 679–682 CAS.
- E. Kimura, T. Koike, M. Shionoya and M. Shiro, Chem. Lett., 1992, 21, 787–790 CrossRef.
- A. A. Neverov, Z.-L. Lu, C. I. Maxwell, M. F. Mohamed, C. J. White, J. S. W. Tsang and R. S. Brown, J. Am. Chem. Soc., 2006, 128, 16398–16405 CrossRef CAS PubMed; Z.-L. Lu, C. T. Liu, A. A. Neverov and R. S. Brown, J. Am. Chem. Soc., 2007, 129, 11642–11652 CrossRef PubMed; S. E. Bunn, C. T. Liu, Z.-L. Lu, A. A. Neverov and R. S. Brown, J. Am. Chem. Soc., 2007, 129, 16238–16248 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of 1,4-butylenebistriflate, ORTEP representation of L4·4TfOH, crystallographic table and various CD spectra/titrations of poly d(GC) with of poly d(GC) with LEt, L3, L4, 1, 2, 3, 4, 5, 6, 7, 11, 12, EDTA, 6 and EDTA, 9 and EDTA. CCDC 1021588–1021591. For ESI see DOI: 10.1039/c4dt02713b |
| This journal is © The Royal Society of Chemistry 2015 |