
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Binding of Ru(terpyridine)(pyridine)dipyridophenazine to DNA studied with polarized spectroscopy and calorimetry†
Anna K. F.
Mårtensson
and
Per
Lincoln
*
Department of Chemical and Biological Engineering, Chalmers University of Technology, SE-41296 Gothenburg, Sweden. E-mail: marann@chalmers.se; lincoln@chalmers.se
First published on 29th October 2014
Abstract
Linear and circular dichroism (LD and CD) spectroscopy as well as isothermal titration calorimetry (ITC) have been used to investigate the interaction of Ru(tpy)(py)dppz2+ (tpy = 2,2′:6′,2′′-terpyridyl; py = pyridine; dppz = dipyrido[3,2-a:2′3′-c]phenazine) with DNA, providing detailed information about the DNA binding thermodynamics and binding geometry of the metal complex. Flow LD, CD and isotropic absorption indicate that Ru(tpy)(py)dppz2+ bind to DNA from the minor groove with the dppz ligand intercalated between base pairs, very similar to its chiral structural isomers Δ- and Λ-Ru(bpy)2dppz2+ (bpy = 2,2′-bipyridine). A simple cooperative binding model with one binding geometry provide an excellent fit for calorimetric and absorption titration data. The values of the neighbor interaction thermodynamic parameters for Ru(tpy)(py)dppz2+ suggest that complexes bound contiguously prefer to have their tpy ligands oriented towards the same strand.
Introduction
DNA-binding drugs are small molecules that recognize and interact with specific DNA sites. Many of the chemotherapeutic anticancer agents currently in use fall under this category with cisplatin being the most prevalent example.1 DNA intercalators that unwind DNA in order to π-stack between two base pairs have shown cytotoxicity towards cancerous cells but are often of limited therapeutic use due to their lack of specificity and frequent side effects. After the pioneering work by Barton and coworkers on the selective DNA binding of substitution-inert trisphenanthroline complexes of ruthenium, there has been an increasing interest in octahedral transition metal complexes.2 The discovery of the “light switch” complexes Ru(phen)2dppz2+ and Ru(bpy)2dppz2+ (phen = 1,10-phenanthroline; bpy = 2,2′bipyridine; dppz = dipyrido[3,2-a:2′3′-c]phenazine) lead to the synthesis of many variations of dppz-ruthenium-centered tris-bidentate structures with the potential as biosensors and therapeutic agents.3 Interestingly, the two ruthenium complexes that have reached clinical trials have substitution-labile ligands and a proposed mode of action completely different to DNA intercalation.4Spectroscopic and biophysical methods have established that it is the dppz ligand in tri-bidentate complexes that is intercalated between the base pairs of the DNA, and this has recently been confirmed by several X-ray crystal structures.5 Compared to the intense research on the brightly luminescent bipyridine and phenanthroline complexes, the very low quantum yield at room temperature has led to less interest for ruthenium dppz complexes carrying the tridentate tpy ligand (tpy = 2,2′:6′,2′′-terpyridyl), despite the fact that the absence of a stereocentre at the metal when coordinated to a tridentate ligand eliminates the need of separating Δ and Λ racemic mixtures characteristic of tris-bidentate systems. The main focus has been on tpy-based complexes with reactive oxoruthenium(IV) functionality that cleaves DNA by oxidation of guanine and the 1′-deoxyribose hydrogen.6 However, not until recently has the single free coordination site left on the metal atom been recognized as a potential way of fine-tuning complex–DNA interactions, either by improving the DNA cleaving ability,7 or as a novel light-activated drug delivery system.8 Although some spectrophotometric studies have aimed at the DNA binding mode of tpy-based complexes to DNA,9 a definite proof of the binding geometry has not yet been obtained.
Our group has previously determined that the tris-bidentate complexes affect each other, either cooperatively or anti-cooperatively, when interacting with a DNA-polymer via intercalation.10 A binding model that gives a satisfactory fit to the data needs two distinct binding modes, one symmetrical (perpendicular) and one unsymmetrical (polar). This model with two modes of binding is supported by the crystallographic study by Niyazi et al. (2012) where a symmetric and a non-symmetric intercalation geometry was found for Λ-Ru(phen)2dppz2+.5f
In this study, we have carried out spectrophotometric and calorimetric measurements in order to determine the binding geometry and thermodynamic characteristics of Ru(tpy)(py)dppz2+ (Ru-tpy, see Scheme 1). To simplify the system by avoiding effects from DNA sequence heterogeneity, and to compare with an earlier study,10 we chose to primarily study the interaction of poly(dAdT)2 (AT-DNA). However, since the AT-DNA was too short to orient in the flow cell for the linear dichroism study, we used calf thymus DNA (ctDNA) instead. We wanted to characterize a tpy/dppz ruthenium complex in its simplest form, and for the purpose of comparison, a pyridine (py) ligand was attached to the single coordination site, making the complex an achiral structural isomer of the original “light-switch” complex Ru(bpy)2dppz2+ (Ru-bpy). Once we have gained more understanding in how this mononuclear complex interacts with DNA, the substituents of this single coordination site could be varied to optimize properties that would make it and its binuclear derivatives more suitable as metallo-pharmaceuticals.
 | ||
| Scheme 1 Structures of ruthenium complexes Ru(bpy)2dppz2+ (left) and Ru(tpy)(py)dppz2+ (right). | ||
Results
Absorption
The absorption spectra of ΔRu-bpy and Ru-tpy in the absence and presence of AT-DNA at [base pairs]/[Ru] ratio of 5 are illustrated in Fig. 1. The broad band system centered at about 440 nm for ΔRu-bpy and 475 nm for Ru-tpy is attributed to the metal to ligand charge transfer (MLCT) transitions, which in the presence of AT-DNA show a slight hypochromicity and red shift for both complexes. The characteristic 372 nm band, assigned to the lowest π→π* transitions of the dppz chromophore is almost identical for both Ru-tpy and ΔRu-bpy, and in presence of AT-DNA there is also a very similar pronounced hypochromicity and red-shift (see Fig. 1 and Fig. S1 in ESI†).11 For Ru-tpy, the most intense band outside the overlapping DNA absorption is the band at about 310 nm, which is assigned to the long-axis polarized lowest π→π* transition of the tpy chromophore.12 This peak shows a red-shift of about 5 nm and in contrast to the 372 nm dppz band, only a slight hypochromicity at DNA binding, indicating that of the two large ruthenium ligands in Ru-tpy, the dppz ligand is the one that has the closest interaction with the nucleobases. As shown by titrating a constant concentration of Ru-tpy with AT-DNA (see Fig. S2 in ESI†), the hypochromicity of the dppz-band at 372 nm for Ru-tpy remains virtually constant at ratios [base pairs]/[Ru] > 2, whereas the initial hypochromicity in the 310 nm band is somewhat reduced at higher ratios.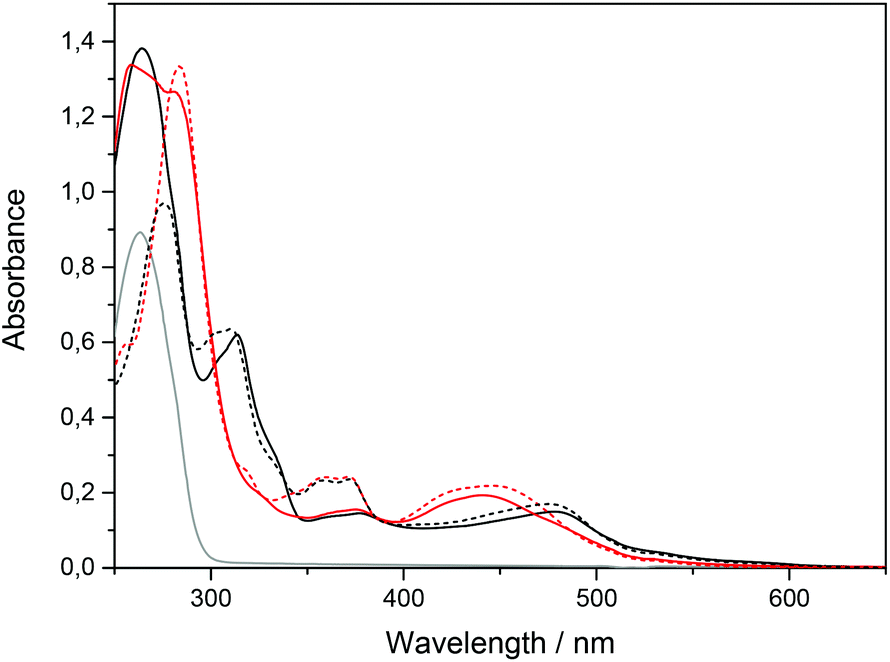 | ||
| Fig. 1 Absorption spectra of Ru-tpy (black) and ΔRu-bpy (red) (14 μM) in 150 mM NaCl solution (dotted line) and in presence of AT-DNA (138 μM nucleotides) (solid line). The gray line shows AT-DNA only. | ||
Circular dichroism
A pure enantiomer of a chiral ruthenium complex, such as Ru-bpy, will show a strong intrinsic CD signal when free in aqueous solution, but Ru-tpy, which is an achiral complex will show zero CD signal under the same conditions. However, for both chiral and achiral molecules, binding to DNA can lead to a proper induced circular dichroism signal (proper ICD) by perturbation of the chromophores of the bound molecule by the chiral arrangement of the nucleobase chromophores. Here the magnitude and the sign of the ICD will mainly be dictated by the distances and angles between the interacting electronic transition dipole moments, and similar geometries are expected to give rise to similar ICD-patterns even for opposite enantiomers. In addition, for chiral molecules, changes merely in position and intensity of the intrinsic CD bands themselves, caused by the interaction with DNA, will add an apparent ICD contribution, but this will be characterized by a mirror-image like pattern for opposite enantiomers with similar binding geometries. Changes in CD were monitored upon addition of ΔRu-bpy, ΛRu-bpy, and Ru-tpy to a [base pairs]/[Ru] ratio of 5. In order to compare the ICD spectra of the complex–DNA interactions, the spectrum of the DNA and the spectrum of the free complex were subtracted from the spectrum in the presence of DNA. Fig. 2 shows the ICD of all three complexes as well as the CD spectrum of the AT-DNA. The general shape of the ICD below 290 nm is quite similar for all three complexes: a negative band between 275 and 290 nm and a positive band at about 260 nm, indicative of a proper ICD mechanism, while the general mirror-image relationship of the ICD curve >290 nm for Δ- and ΛRu-bpy suggests the predominance of an apparent ICD mechanism in this region. Titration of a solution with constant concentration of Ru-tpy shows an almost invariant CD spectrum >300 nm for [base pairs]/[Ru] > 2, similar to the results of the corresponding absorption titration (see Fig. S3 in ESI†).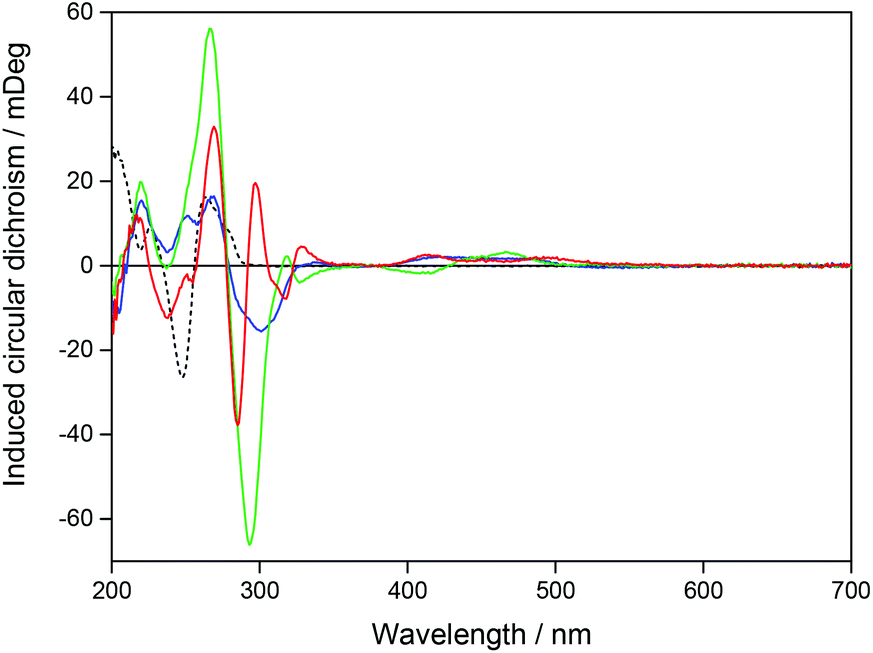 | ||
| Fig. 2 Induced CD for Ru-tpy (blue), ΔRu-bpy (green), and ΛRu-bpy (red) after mixing with AT-DNA. The black dashed line shows the CD signal for AT-DNA in a 150 mM NaCl solution. The concentrations of complex and DNA were 14 and 138 μM respectively. | ||
Linear dichroism
Fig. 3 shows the LD spectra for Ru-tpy in ctDNA solution at [base pairs]/[Ru] ratios 8, 4 and 2. The ctDNA concentration remained constant at 270 μM nucleotides. The reduced LD (LDr), which is the LD divided by the isotropic absorbance, is very similar for all [base pairs]/[Ru] ratios, indicating little or no change in the geometric orientation at higher saturation levels (see Fig. S4 in ESI†). Thus, the conditions were fulfilled for determining the orientation factor S using eqn (5), (8) and (11) (See Theory and methods section below) using the LD spectra of free DNA (L0) and at ratios 8 (L1) and 4 (L2). The relative values S1/S0 = 1.06 and S2/S0 = 1.16 indicate that the DNA with bound Ru-tpy becomes better oriented, as earlier found for Δ- and ΛRu-bpy.11a Finally b, the pure (without DNA contribution) Ru-tpy LD spectrum, at perfect orientation and 10 mm optical path-length, was calculated as b = c2−1(S2−1L1 − S0−1L0). The weights w1 and w2 in eqn (6) were varied manually until the dppz band at 375 nm vanished in component e1 and the sharp tpy absorption band at 310 nm vanished in component envelope spectra e2, as shown in Fig. 4. The optimal weight values were w1 = 3 ± 0.5 and w2 = −1.5 ± 0.3, the theoretical limits for parallel and perpendicular orientation of a transition dipole moment relative to the orientation axis. Thus, the results show that the tpy long-axis (z) is aligned along and the dppz long-axis (x) perpendicular to the DNA helix axis, which, since the dppz ligand lies in the x,y plane, is consistent with intercalation of the dppz ligand in-between the base pairs. | ||
| Fig. 3 Linear dichroism spectra of Ru-tpy in the presence of ctDNA at [base pairs]/[Ru] ratios 8 (blue), 4 (red) and 2 (green) in 10 mM NaCl solution, as well ctDNA alone (dotted). The concentration of ctDNA is 270 μM nucleotides. | ||
 | ||
| Fig. 4 Resolved spectra of the x and y (red) and the z (blue) polarized absorption bands of Ru-tpy bound to ctDNA. The Y-axis units are ε/(1000 M−1 cm−1). The arrows on the molecular structure of the complex show the direction of the x and z transition moments. | ||
Binding isotherms
ITC profiles for the binding of Ru-tpy to AT-DNA at 20, 25, and 30 °C are shown in Fig. 5 and for comparison, the corresponding ITC-profiles for Δ- and ΛRu-bpy from Andersson et al. (2013).10 Ru-tpy shows a similar overall shape with a gradually increasing exothermic enthalpy until a negative maximum is reached at [Ru]/[base pairs] = 0.4–0.5, somewhat higher than for the other two complexes. The initial slope of the ITC profile is in contrast to the initial constant part of the sigmoidal curve expected for the simple binding as indicated above by absorption, circular dichroism and linear dichroism spectroscopies, where the spectra of Ru-tpy in the presence of DNA were found to be practically invariant with binding ratio. This suggests that intermolecular interactions between bound molecules must contribute to the binding enthalpy also for Ru-tpy, as earlier concluded for Δ- and ΛRu-bpy.10 This observation prompted us for a more thorough analysis of the spectroscopic changes, and absorption spectra were collected for addition of Ru-tpy to a constant concentration of AT-DNA at 25 °C. | ||
| Fig. 5 ITC profiles with fitted traces for the binding of Ru-tpy (●), ΔRu-bpy (▲), and ΛRu-bpy (Δ) to AT-DNA in 150 mM NaCl solution at 20, 25, and 30 °C. Symbols indicate the normalized heat absorbed or evolved upon sequential injections (2 μL) of complex into the 206 μL cell containing the DNA. The data has been corrected for heat of complex dilution. The corresponding ITC-profiles for Δ- and ΛRu-bpy are from Andersson et al. (2013).10 | ||
The data (shown in Fig. S5 in ESI†) were analyzed with singular value decomposition (SVD) as described in Theory and methods. The first three (normalized) singular values were s1 = 100, s2 = 2.45 and s3 = 0.23, and the corresponding columns of U and V are plotted in Fig. 6 and 7, respectively. Column 4 in U and V (shown in Fig. S6 and S7 in ESI†) were much less structured and were judged to be insignificant, although the fourth singular value (0.18) was close to the third.
 | ||
| Fig. 6 The U-vectors corresponding to the first three singular values (s1 blue, s2 green, s3 red) from the titration of Ru-tpy to a constant concentration of AT-DNA in 150 mM NaCl at 25 °C. | ||
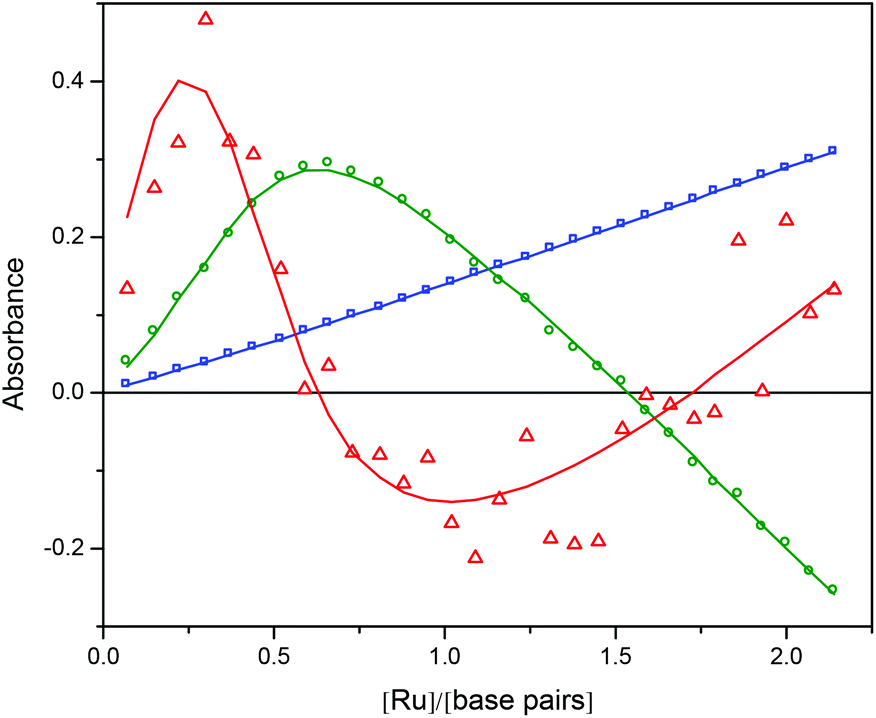 | ||
| Fig. 7 The V-vectors corresponding to the first three singular values (s1 blue squares, s2 green circles, s3 red triangles) from the titration of Ru-tpy to a constant concentration of AT-DNA in 150 mM NaCl at 25 °C. The fit of the model is shown as solid curves. | ||
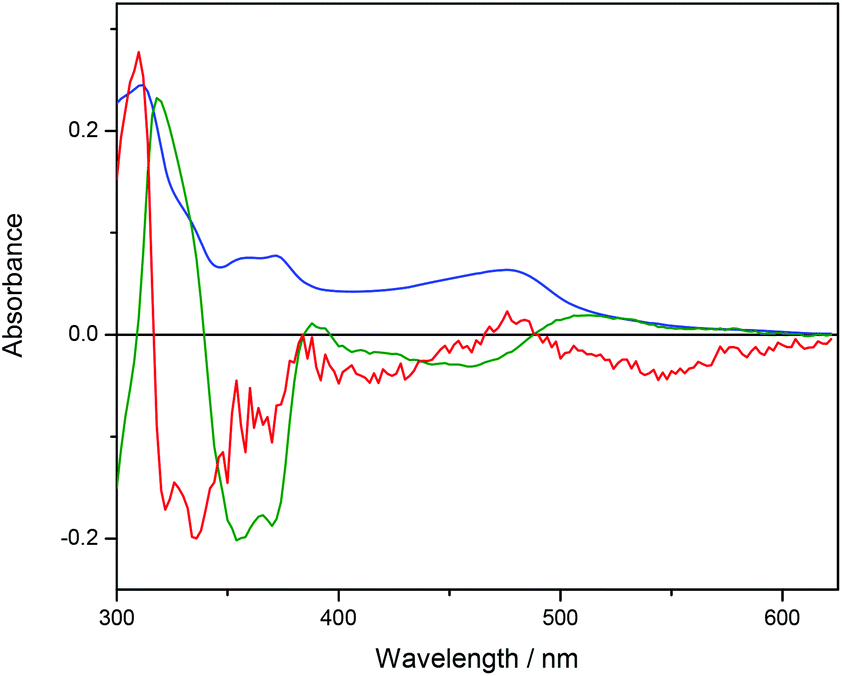
Ru-tpy ITC and absorption data could be excellently globally fitted with the classical McGhee–von Hippel cooperative binding model (see Theory and methods), and the best fit to the data are shown with solid lines in Fig. 5 and 7. Fig. 8 shows the calculated absorption spectra for free complex, bound complex with at most one neighbor and bound complex with at least one neighbor. For comparison, the model was also used to fit the ITC data for ΔRu-bpy and ΛRu-bpy, as shown in Fig. 5. A linear fit to the calculated ΔH° values as a function of temperature is shown in Fig. 9, and Table 1 gives the parameters and derived thermodynamic data.
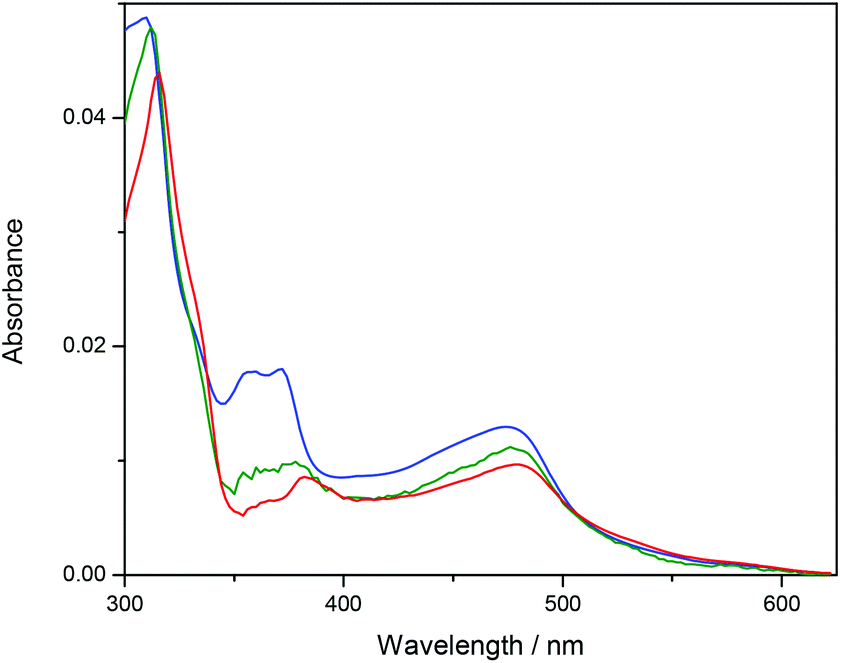 | ||
| Fig. 8 Calculated absorption spectra for unbound complex (blue), bound complex with at most one neighbor (green) and bound complex with at least one neighbor (red). | ||
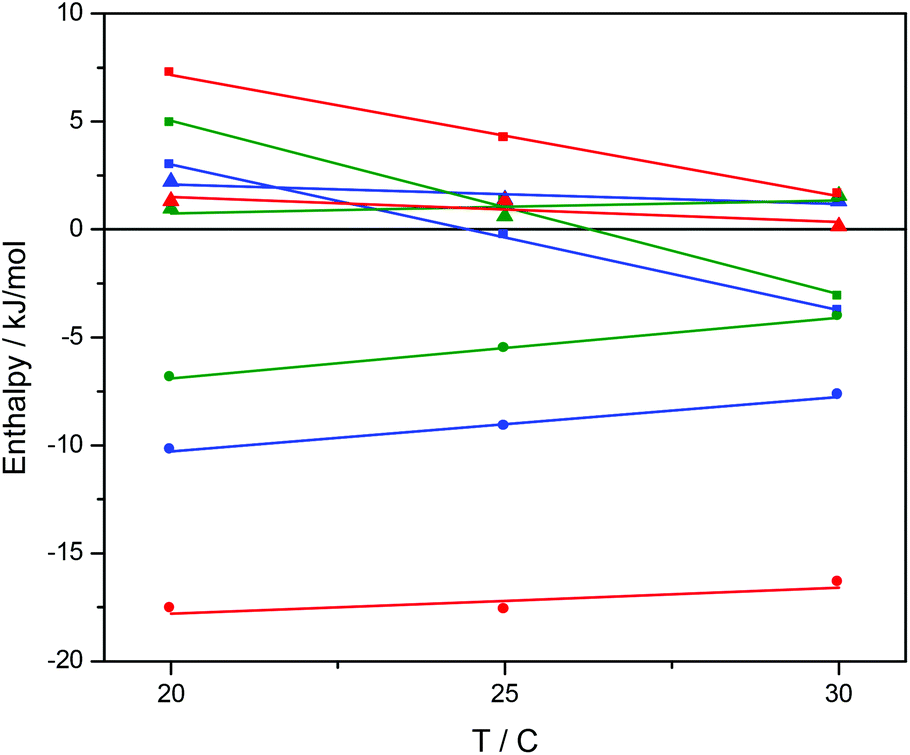 | ||
| Fig. 9 The standard binding enthalpy ΔH°b (■), standard nearest neighbor interaction enthalpy ΔH°nn (●), and ΔH°baseline (▲) for binding of Ru-tpy (blue), ΔRu-bpy (green), and ΛRu-bpy (red) to AT-DNA in 150 mM NaCl. The slopes of the fitted lines correspond to the ΔCp for the reactions. | ||
Discussion
Recently, high-resolution structures and calorimetric studies have highlighted ligand–ligand interactions as an explanation to the complex thermodynamical and photophysical behavior of DNA-bound Ru(L)2dppz complexes.5d–f,10 To further investigate the role of the ancillary L ligands, we chose to study the terpyridine/pyridine Ru-dppz complex Ru-tpy, an achiral isomer of Ru(bpy)2dppz formally made by breaking the pyridine–pyridine bond of one bpy unit and joining it to the second (see Scheme 1).Absorption spectroscopy shows that the spectral changes of the dppz ligand bands are virtually identical for ΔRu-bpy and Ru-tpy upon binding to DNA, giving a first indication that the binding mode of the two isomers are similar. Circular dichroism spectroscopy shows a negative induced CD band in the long-axis polarized tpy band at 300 nm, similar to the negative induced CD shown for Δ- and ΛRu-bpy in their long-axis polarized bpy band at 290 nm, as could be expected for electronic transitions positioned in the minor groove close to parallel to the helix axis.13 Both absorption and CD are practically invariant with binding ratio, although a small perturbation of the tpy-band at 310 nm can be observed at binding ratios close to saturation in both CD and absorption spectra. Likewise, linear dichroism spectra were found to be invariant with binding ratio too, allowing determination of the orientation factor S and a quantitative analysis of the angular binding geometry. In contrast to the Ru(L)2dppz complexes, where major transition moment directions have oblique angles to the plane of the dppz ligand, in Ru-tpy the long-axis polarized tpy transition is perpendicular to the dppz plane. With a weight w1 = +3 ± 0.5 we find that it is almost perfectly parallel oriented to the DNA helix axis, and the dppz long axis polarized transition, with weight w2 = −1.5 ± 0.3, perpendicularly oriented, the geometry expected for intercalation of the dppz ligand in-between the base pairs of DNA. In contrast to the very minute differences in absorption, CD and LD spectra at different binding ratios, the calorimetric titration show strong effects on the heat of binding. We have previously been reported such non-classical ITC curves for Ru(L)2dppz complexes (L = bpy or phen) and attributed them to an additional enthalpy contribution from interaction between neighboring complexes on the DNA.10 In comparison to Δ- and ΛRu-bpy, Ru-tpy showed an ITC profile qualitatively most similar to the latter. Since a satisfactory global fit for the ITC and absorption experimental data of Ru-tpy could be obtained with the classical McGhee–von Hippel model with only one type of binding geometry, for comparison this model was also used to reanalyse our data for Δ- and ΛRu-bpy, which were originally fitted with a symmetrical and a pair of unsymmetrical intercalation geometries.10 In this model (Model 3 in ref. 10), ligands bound with only one nearest-neighbor are assumed to have one distinct binding geometry (unsymmetrical), while ligands bound either isolated or with nearest-neighbors on both sides have a second binding geometry (symmetrical). As noted in our previous study, the fit of the simpler model to the ITC-data alone is excellent, but any attempt to rationalize the observed molar fractions of the short and the long excited state life-time fails. With the simple model, the apparent site sizes for Δ- and ΛRu-bpy were found to be 2.2 and 2.3, consistent with the neglect of the anti-cooperativity which is inherent in Model 3 for which the n parameters were found to be 2.0 and 1.8, respectively.10 Interestingly, the binding site size parameter n was found to be 2.0 for Ru-tpy, indicating that the distinction between symmetrical and unsymmetrical intercalation geometries might be less pronounced for this complex. Although the low emission quantum yield of Ru-tpy bound to DNA precluded time-resolved luminescence measurements, the observation that spectra (absorption, CD, LD) change very little with binding density (see Fig. 3, S2 and S4†) support the conclusion that difference between intercalation geometries is small for Ru-tpy. The simple model has only one cooperativity parameter y, which is found to be 1 for ΔRu-bpy (i.e. non-cooperative binding) and 5.5 for ΛRu-bpy (cooperative binding); for Ru-tpy y = 2.8, in-between the values of the two Ru-bpy enantiomers. The intrinsic binding constant K is 106 M−1 for Ru-tpy, very similar to that of ΔRu-bpy, while K for ΛRu-bpy is almost 50 times smaller. The latter value is about 3 times smaller than that obtained with Model 3, however, since simulated binding isotherms are the most sensitive to the value of the binding constant close to saturation, the best-fit value will normally be quite dependent on the binding model since the influence of the cooperativity parameters will differ.
The intrinsic binding enthalpy ΔH°b is positive for all complexes at 20 °C, i.e. the binding in the absence of neighbor interactions is endothermic. The value of ΔH°b is smaller for Ru-tpy than for either Δ- or ΛRu-bpy, but the temperature dependence is similar as evidenced by the negative ΔCp-values of −680 ± 120 J K−1 M−1. The nearest-neighbor interaction enthalpy ΔH°nn is negative (i.e. exothermic) for all complexes, and exhibits a tendency similar to the cooperativity factor y, namely that the value for Ru-tpy is in-between the values for Δ- and ΛRu-bpy.
However, even if a simple, single binding geometry model is sufficient to model the binding data for Ru-tpy, the lack of a 2-fold axis of symmetry, in contrast to Δ- and ΛRu-bpy, make 2 types of neighbor interaction possible: either the tpy ligands of two consecutive complexes are oriented towards the same strand (TSS, see Fig. 10) or they are oriented towards opposite (alternating) strands (TAS). In comparison to Ru-bpy, neglecting the influence of the single pyridine ring of Ru-tpy, the alternating model (TAS) would have intermolecular contacts resembling alternating Δ–Δ and Λ–Λ contacts, while the same-side model (TSS) would everywhere have intermolecular contacts resembling Δ–Λ, comparable to that found in a recent X-ray crystal structure of Δ- and Λ-Ru(phen)2dppz simultaneously intercalated to a DNA hexamer duplex.5e
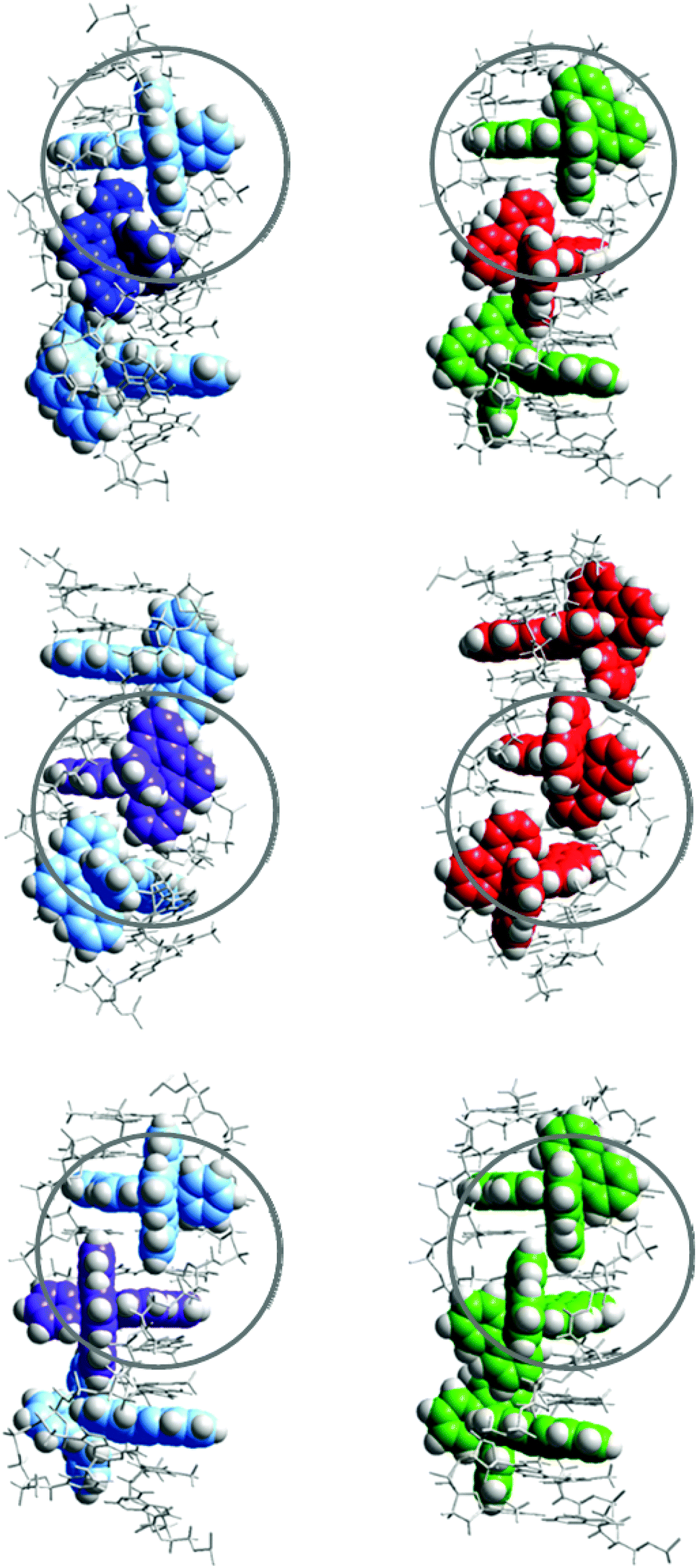 | ||
| Fig. 10 Schematic illustration of the proposed interaction geometries for Ru-tpy (left) and the corresponding geometries of Δ- (green) and Λ- (red) Ru-bpy (right). Top row illustrates the “same-side” (TSS) model while the middle and bottom row illustrate the “alternating side” (TAS) model. Circles indicate similarities in intermolecular contacts. The models where constructed by manual docking and subsequent energy minimization in vacuum, using AMBER 2 force field in the HyperChem 8.0 software package (HyperCube, Inc.). | ||
Since the same-side model TSS has only one type of intermolecular contact (Δ–Λ), it is logically consistent with the simple one-geometry binding model, and also consistent with the assumption that Ru-bpy interactions model those of Ru-tpy, since 7.8, the square of the Ru-tpy y value of 2.8, is larger than the product 5.5 of the corresponding Δ–Δ and Λ–Λ y values for Ru-bpy. However, the values 7.8 and 5.5 are similar enough in magnitude to suggest that both TSS and TAS arrangements will be significant for Ru-tpy, even if the former arrangement with tpy ligands oriented towards the same strand will predominate. Our results indicating a relatively modest cooperativity factor (y = 2.8) for Ru-tpy appears to be in contrast to the case of Ru(phen)2dppz2+, for which a rather substantial Δ–Λ cooperativity can be inferred, since Cardin and co-workers report that the hexamer duplex used in their crystal structure study preferentially binds precisely one Δ- and one Λ-Ru(phen)2dppz in solution.5e
Experimental
Materials
All experiments were performed in aqueous solution (pH = 7.0) containing 150 mM NaCl and 1 mM cacodylate (dimethylarsinic acid sodium salt) except for the LD experiments where 10 mM NaCl was used (pH = 7.0). This was because a lower salt concentration gave a higher signal intensity without affecting the overall shape of the spectra. Stock solutions of calf thymus DNA (ctDNA) (∼5 mM nucleotides) were prepared by dissolving highly polymerized type I sodium salt calf thymus DNA (Sigma-Aldrich) in buffer. A stock solution of poly(dAdT)2 (AT-DNA) (∼5 mM nucleotides) was prepared by dissolving the sodium salt (Sigma-Aldrich) in buffer. The solutions were filtered two times through a 0.7 μm polycarbonate filter. Stock solutions of the complexes (∼1 mM) were prepared by dissolving the chloride salts in buffer. Concentrations were determined spectrophotometric using extinction coefficients: ε258 = 6600 M−1 cm−1 per nucleotide for ctDNA, ε260 = 6600 M−1 cm−1 per nucleotide for AT-DNA, ε371 = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 M−1 cm−1 for Ru-tpy and ε444 = 16100 M−1 cm−1 for Ru(bpy)2dppz2+ (Ru-bpy). For ITC measurements the DNA solution was dialyzed against pure buffer for at least 48 hours at 8 °C. Ruthenium complex solutions of appropriate concentrations were prepared by dilution of the stock solutions in the dialysate. The dialysis membrane used had a molecular weight cut-off of 3.5–5 kDa (Spectra-Por® Float-A-Lyzer® G2, Sigma Aldrich).
900 M−1 cm−1 for Ru-tpy and ε444 = 16100 M−1 cm−1 for Ru(bpy)2dppz2+ (Ru-bpy). For ITC measurements the DNA solution was dialyzed against pure buffer for at least 48 hours at 8 °C. Ruthenium complex solutions of appropriate concentrations were prepared by dilution of the stock solutions in the dialysate. The dialysis membrane used had a molecular weight cut-off of 3.5–5 kDa (Spectra-Por® Float-A-Lyzer® G2, Sigma Aldrich).
Δ- and Λ-[Ru(bpy)2dppz]Cl2 used in this study were prepared as previously reported.11a
Other chemicals were purchased from Sigma-Aldrich and used without purification.
Synthesis of [Ru(tpy)(py)dppz]Cl2
The synthetic route for preparation of [Ru(tpy)(py)dppz]Cl2 is shown in Scheme 2. The procedure for preparation of Ru(tpy)Cl3 and [Ru(tpy)(dppz)Cl]Cl are in accordance to the methods previously reported by Zhou et al. and Leising et al., respectively.7a,14 [Ru(tpy)(dppz)(py)](PF6)2 was synthesized using the method previously described by Zhou et al. with some modifications. A portion of 0.0557 g of [Ru(tpy)(dppz)Cl]Cl and 0.0270 g of AgNO3 were refluxed in 20 mL of ethanol–water (1:1) for 3 h under N2(g). The solution was filtered after cooling and the filtrate was refluxed again under N2 (g) for another 4 h, with 0.0101 g pyridine (py) added. The solution was left in a fridge over night for cooling. The next day the product was precipitated using KPF6 dissolved in MilliQ water, left for a few hours, collected on a filter, and washed with ethanol–ether (1:2). Purification of [Ru(tpy)(dppz)(py)](PF6)2 was done using column chromatography with CH3CN and neutral Al2O3. The eluate containing the pure orange product was collected leaving a dark purple residue layer on top of the column.
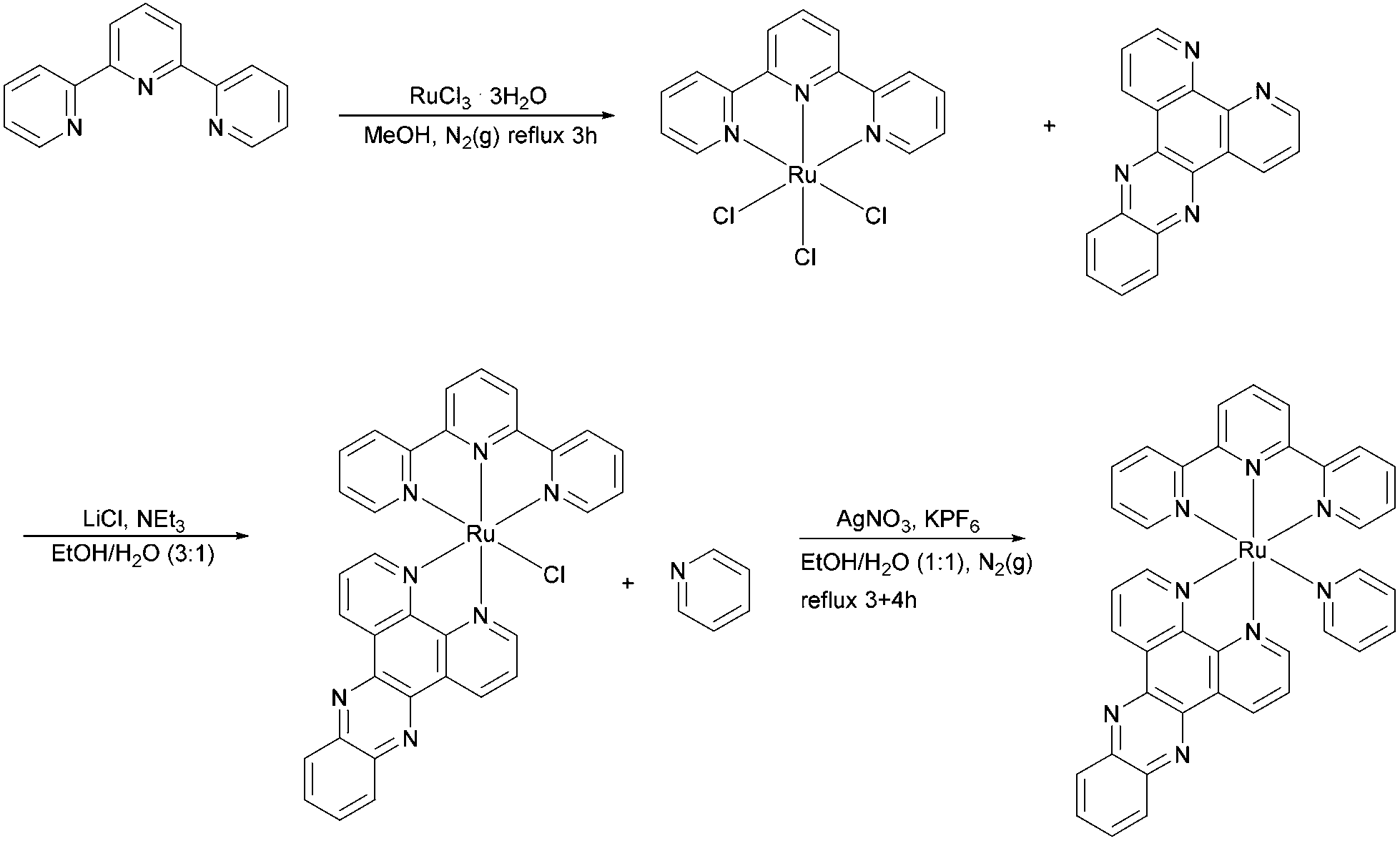 | ||
| Scheme 2 Synthesis of Ru-tpy. | ||
To replace the hexafluorophosphate anion with chloride, the CH3CN solution of [Ru(tpy)(dppz)(py)](PF6)2 was reduced to ∼1 mL evaporating with a stream of N2 under mild heating, where after 0.5 g of ([CH3(CH2)3]4NCl), dissolved in 1 mL of acetone, was added in increasing portions while stirring until the solution was only weakly yellow and the precipitation complete. The product was collected by a sintered glass filter and washed first with acetone and then with diethyl ether to yield [Ru(tpy)(dppz)(py)]Cl2 as a brown powder (36%, calculated from the starting material, Ru(tpy)Cl3). UV/vis (in water; λmax in nm, ε/103 M−1 cm−1 enclosed in parenthesis): 475(12.1), 372(16.9), 310(45.4), 275(69.2). 1H NMR (as PF6 salt, 400 MHz, acetone-d6): δ 10.03 (dd, J = 8.2, 1.3 Hz, 1H), 9.56–9.52 (m, 1H), 9.48 (dd, J = 5.4, 1.4 Hz, 1H), 8.94 (t, J = 8.0 Hz, 2H), 8.84–8.71 (m, 2H), 8.59–8.42 (m, 4H), 8.26–8.11 (m, 9H), 8.04–7.94 (m, 2H), 7.76 (ddd, J = 8.2, 5.5, 0.8 Hz, 1H), 7.49–7.42 (m, 3H). The 1H NMR spectrum of Ru-tpy (see Fig. S8 in ESI†) was in accordance with previous results by Zhou et al. (2009)7a and showed no significant impurities.
Spectroscopy
Absorption spectra were measured on a Varian Cary 4000 UV/vis spectrophotometer (path length = 1 cm). The reverse absorption titration spectra for Ru-tpy were measured with a constant concentration of 5 μM Ru-tpy in buffer. The stock solution of AT-DNA (with 5 μM of Ru-tpy to avoid dilution) added directly in the cuvette (path length = 1 cm) in aliquots up to a concentration of 80 μM nucleotides.Linear dichroism spectra (LD) were measured on a Chirascan LD spectropolarimeter on samples oriented in an outer-rotating Couette flow cell with a 1 mm path length at a rate of 1000 rpm. The spectra of the same samples were recorded without rotation for baseline contribution and were subsequently subtracted from the LD spectra. The concentration of ctDNA used in the measurements was 266 μM nucleotides and mixed with complex solution with appropriate concentration to obtain desired [Ru]/[base pairs] ratios. Circular dichroism (CD) spectra were recorded on a Chirascan CD spectropolarimeter similarly as the absorption spectra. Five CD spectra were averaged for each sample. To be consistent with the absorption and ITC measurements, AT-DNA was used for the CD measurements (∼138 μM nucleotides).
Calorimetric data was obtained using an ITC200 isothermal titration calorimeter (Microcal) controlled by Origin 7.0 software. The ruthenium complexes (∼600 μM) were loaded in a syringe (40 μL) and titrated in 2 μl aliquots into 206 μl of AT-DNA in 150 mM NaCl aqueous solution (∼340 μM nucleotides). By integrating the power required to maintain the reference and sample cells at the same temperature it is possible to obtain a direct measurement of the heat generated or absorbed when complex and DNA interact. The experimental raw data consists of a series of heat flow peaks, and each peak corresponds to one injection of complex. These heat flow spikes are integrated with respect to time, which gives the total heat exchanged per mole injectant, plotted against the ratio [Ru]/[base pairs]. The primary ITC data was corrected for the heat of ligand dilution by subtracting the average heat per injection of complex titrated into buffer. There was negligible heat arising from DNA dilution. The experiments were performed at 20 °C, 25 °C and 30 °C.
1H NMR spectrum of [Ru(tpy)(py)dppz](PF6)2 in acetone-d6 was recorded on an Agilent 400 MHz spectrophotometer.
Theory and methods
Analysis of LD spectra
The isotropic absorption spectrum Aiso of a sample can be considered as a simple sum of component spectral envelopes ei, each such envelope itself being a collecting all electronic transitions with a common polarisation direction relative to the molecular coordinate system: | (1) |
Then, the linear dichroism spectrum LD will be a weighted sum of the same component spectral envelopes:
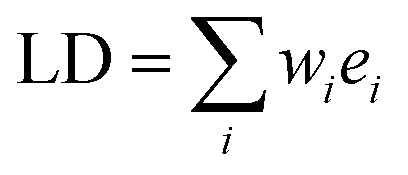 | (2) |
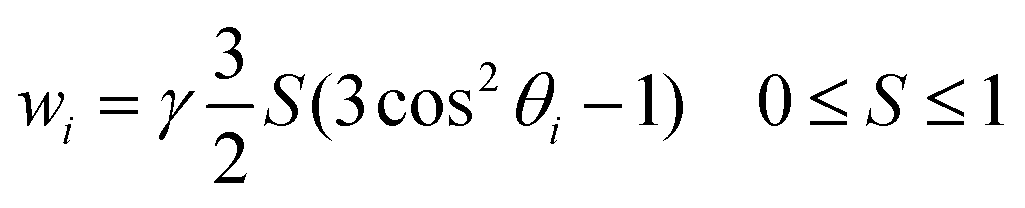 | (3) |
The reduced linear dichroism LDr value at a certain wavelength is defined as the ratio of the linear dichroism value over the isotropic absorbance value:
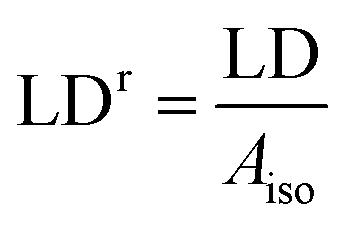 | (4) |
In wavelength regions where a single component spectral envelope ei dominates the absorption spectrum, the LDr curve will be essentially constant and take the value wi, e.g. as observed around the 260 nm band of B-DNA. Since θ for the in-plane polarized π→π* nucleobase transitions here is close to 90°, the orientation factor S0 for ligand-free DNA can readily be calculated from the LDr value.
 | (5) |
However, in most cases, and in particular for 3D-chromophores like ruthenium polypyridyl complexes, component spectral envelopes overlap substantially over the whole range of the spectrum, and the LDr curve will vary strongly with wavelength. Deconvolution of the experimental Aiso and LD spectra into component envelopes, and thereby determining the weights wi, can in favorable cases be accomplished by the TEM-method.5a,15 For a system with two component envelope spectra, having distinct characteristic absorption band features and distinct weights, the TEM-method in matrix notation can be formulated as the solution of the combined eqn (1) and (2):
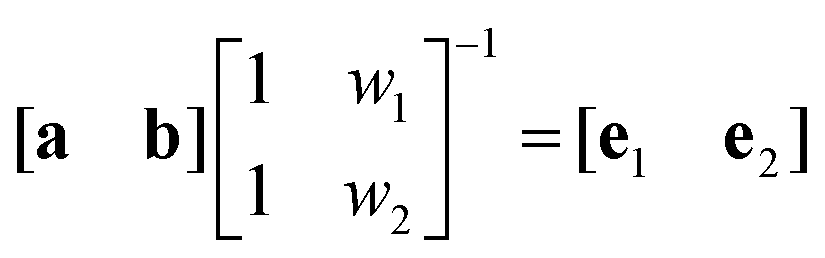 | (6) |
The orientation factor S is required for the angular orientation to be determined from the weights wi. For a dye that (a) is strongly bound to DNA; (b) has significant absorption at wavelengths >300 nm (where the DNA is transparent); and (c) has, within a certain range of [dye]/[DNA] ratios, invariant binding geometry and invariant absorption spectrum; S can readily be obtained as follows:
Let two LD spectra with different [dye]/[DNA] ratios (within the invariant range) be column vectors L1 and L2, and L0 be the LD spectrum of a sample with DNA only. Then, if condition (c) is fulfilled, the three columns are linearly dependent, thus scalars α and β can be found so that αL1 + βL2 = L0; in matrix notation:
| Mx = L0 | (7) |
| (MTM)−1MTL0 = x | (8) |
When condition (a) is fulfilled, practically all added dye can be considered to be bound; and with the DNA concentration being equal in the three samples, the vectors can be written as:
| L0 = γS0d L1 = γS1(d + c1b) L2 = γS2(d + c2b) | (9) |
| αS1 + βS2 = S0 c1αS1 + c2βS2 = 0 | (10) |
Since γS0 can be evaluated from eqn (5), S1 and S2 can be obtained from known quantities after solving the two equations in (10):
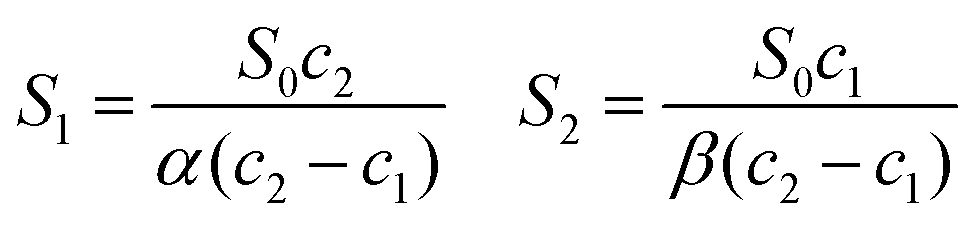 | (11) |
Analysis of binding isotherms
Although global fitting of calorimetric and excited state lifetime data for Δ- and Λ-Ru-bpy and their 1,10-phenanthroline analogues required a fairly complicated binding model comprising a symmetrical and a pair of unsymmetrical intercalation geometries (Model 3), fits to calorimetric data only were found to be satisfactory with a model with a single binding geometry.10 This model, the classical McGhee–von Hippel single ligand cooperative binding model was used also in this work, and involves three adjustable parameters: the thermodynamic binding constant K, the binding site coverage parameter n and the cooperativity factor y.16 Given values of these three parameters (assumed to be constant in the small temperature range used here), and total concentrations of binding sites [B]tot (i.e. base pairs) and DNA-ligand [L]tot (i.e. ruthenium complex) for each step of the titration, the mass-balance equations were solved iteratively with a Newton–Raphson procedure, to give consistent binding densities θ and free ligand concentrations [L]free, as well as the conditional probabilities pij, as previously described.10,17Calorimetric titration
The ITC data obtained at a certain temperature was assumed to be composed of three components:| ITC(i) = (ΔH°b)Δb(i) + (ΔH°nn)Δnn(i) + ΔHbaseline | (12) |
Spectroscopic titration
The absorption spectra were arranged in data matrix M as columns with w elements corresponding to the wavelengths recorded; the columns corresponding to the t titration steps. Singular value decomposition, using the svd command in the MATLAB software, factorized the data matrix into two matrices of orthonormal columns U and V, and a diagonal matrix S with the singular values s1 ≥ s2 ≥ s3⋯ ≥ 0 along the diagonal. Keeping only the m singular values that are significantly larger than zero make it possible to simplify the factorization: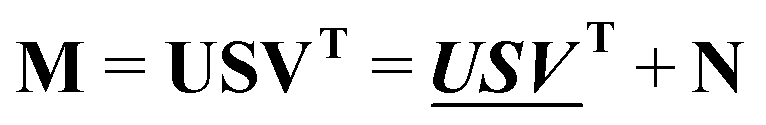 | (13) |
 and
and  are the first m columns of U and V, respectively, and N is a matrix of small elements, ideally corresponding only to the noise in the measurements. Assuming thus that the titration involves m different absorbing species, with absorption spectra in w by m matrix A and concentrations in t by m matrix C, the factorization can now be written as
are the first m columns of U and V, respectively, and N is a matrix of small elements, ideally corresponding only to the noise in the measurements. Assuming thus that the titration involves m different absorbing species, with absorption spectra in w by m matrix A and concentrations in t by m matrix C, the factorization can now be written as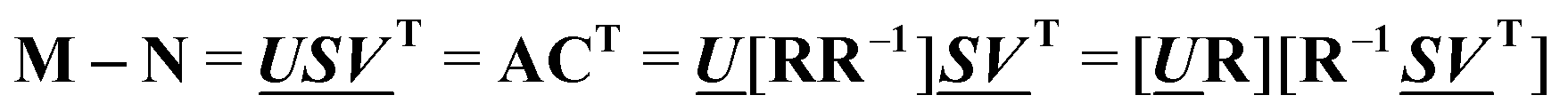 | (14) |
 and
and 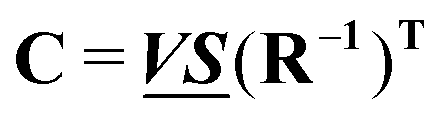 , any theoretical binding model, which can calculate a matrix Cm of concentrations of m species, can be evaluated for consistency with the data by finding the R that is the least square projection of VS on the space spanned by Cm:
, any theoretical binding model, which can calculate a matrix Cm of concentrations of m species, can be evaluated for consistency with the data by finding the R that is the least square projection of VS on the space spanned by Cm: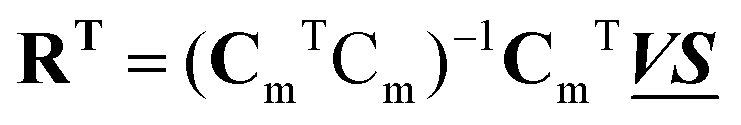 | (15) |
By varying the adjustable parameters of the theoretical model, Cm is varied to minimize the norm of the residual  while keeping the specie spectra in
while keeping the specie spectra in 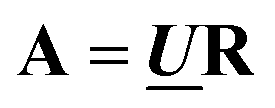 physically reasonable (non-negative in the case of absorption spectra). In the present case, the concentration matrix Cm was constructed with three columns corresponding to free ligand, bound ligand with a neighboring free binding site, calculated as [B]totθ(1 − p22), and bound ligand adjacent to another bound ligand, calculated as [B]totθp22.
physically reasonable (non-negative in the case of absorption spectra). In the present case, the concentration matrix Cm was constructed with three columns corresponding to free ligand, bound ligand with a neighboring free binding site, calculated as [B]totθ(1 − p22), and bound ligand adjacent to another bound ligand, calculated as [B]totθp22.
Conclusion
The angular orientation of the dppz and tpy ligands as determined by LD and CD spectroscopies, strongly indicate that Ru-tpy binds from the minor groove by intercalating the dppz ligand between the base pairs, as has previously been determined for the isomeric complexes Δ- and ΛRu-bpy. The strong hypochromicity in the dppz absorption band is almost identical in magnitude to that of ΔRu-bpy, indicating that the alignment in the intercalation pocket is similar, too.A simple cooperative binding model with one symmetrical binding geometry provide an excellent fit to data for calorimetric and absorption titrations of Ru-tpy into AT-DNA. The intrinsic intercalation has an equilibrium constant of 106 M−1, close to that of Δ-Ru-bpy, and is associated with a small endothermic enthalpy change of +3 kJ M−1. The cooperativity factor is 2.8, and the associated neighbor interaction enthalpy is exothermic, −10.2 kJ M−1; these values being in-between those of Δ- and Λ-Ru-bpy, and consistent with a slight preference of a one-sided arrangement of tpy-ligands of complexes consecutively bound to DNA.
Acknowledgements
The authors want to express their gratitude to Vetenskapsrådet (grant VR 2012-1661) and Chalmers Area of Advance Nano for funding, and COST Action CM1105 for providing a forum for stimulating discussions.Notes and references
- S. M. Cohen and S. J. Lippard, Prog. Nucleic Acid Res. Mol. Biol., 2001, 67, 93–130 CrossRef CAS.
- (a) J. K. Barton, A. T. Danishefsky and J. M. Goldberg, J. Am. Chem. Soc., 1984, 106, 2172–2176 CrossRef CAS; (b) J. K. Barton, J. M. Goldberg, C. V. Kumar and N. J. Turro, J. Am. Chem. Soc., 1986, 108, 2081–2088 CrossRef CAS.
- (a) Y. Jenkins, A. E. Friedman, N. J. Turro and J. K. Barton, Biochemistry, 1992, 31, 10809–10816 CrossRef CAS; (b) C. Hiort, P. Lincoln and B. Nordén, J. Am. Chem. Soc., 1993, 115, 3448–3454 CrossRef CAS; (c) A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef CAS.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbast and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–894 CrossRef CAS PubMed.
- (a) P. Lincoln, A. Broo and B. Nordén, J. Am. Chem. Soc., 1996, 118, 2644–2653 CrossRef CAS; (b) R. Hartshorn and J. Barton, J. Am. Chem. Soc., 1992, 114, 5919–5925 CrossRef CAS; (c) I. Haq, P. Lincoln, D. C. Suh, B. Nordén, B. Z. Chowdry and J. B. Chaires, J. Am. Chem. Soc., 1995, 117, 4788–4796 CrossRef CAS; (d) H. Song, J. T. Kaiser and J. K. Barton, Nat. Chem., 2012, 4, 615–620 CrossRef CAS PubMed; (e) J. P. Hall, D. Cook, S. R. Morte, P. McIntyre, K. Buchner, H. Beer, D. J. Cardin, J. A. Brazier, G. Winter, J. M. Kelly and C. J. Cardin, J. Am. Chem. Soc., 2013, 135, 12652–12659 CrossRef CAS PubMed; (f) H. Niyazi, J. P. Hall, K. O'Sullivan, G. Winters, T. Sorenson, J. M. Kelly and C. J. Cardin, Nat. Chem., 2012, 4, 621–628 CrossRef CAS PubMed.
- (a) H. H. Thorp, J. Inorg. Organomet. Polym., 1993, 3, 41–57 CrossRef CAS; (b) J. G. Goll and H. H. Thorp, Inorg. Chim. Acta, 1996, 242, 219–223 CrossRef CAS; (c) P. J. Carter, C. C. Cheng and H. H. Thorp, J. Am. Chem. Soc., 1998, 120, 632–642 CrossRef CAS; (d) B. T. Farrer and H. H. Thorp, Inorg. Chem., 2000, 39, 44–49 CrossRef CAS; (e) N. Grover, N. Gupta, P. Singh and H. H. Thorp, Inorg. Chem., 1992, 31, 2014–2020 CrossRef CAS.
- (a) Q. X. Zhou, F. Yang, W. H. Lei, J. R. Chen, C. Li, Y. J. Hou, X. C. Ai, J. P. Zhang, X. S. Wang and B. W. Zhang, J. Phys. Chem. B, 2009, 113, 11521–11526 CrossRef CAS PubMed; (b) D. Ossipov, S. Gohil and J. Chattopadhyaya, J. Am. Chem. Soc., 2002, 124, 13416–13433 CrossRef CAS PubMed.
- (a) M. Frasconi, Z. Liu, J. Lei, Y. Wu, E. Strekalova, D. Malin, M. W. Ambrogio, X. Chen, Y. Y. Botros, V. L. Cryns, J.-P. Sauvage and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 11603–11613 CrossRef CAS PubMed; (b) A. Bahreman, B. Limburg, M. A. Siegler, R. Koning, A. J. Koster and S. Bonnet, Chem. – Eur. J., 2012, 18, 10271–10280 CrossRef CAS PubMed; (c) S. Bonnet, B. Limburg, J. D. Meeldijk, R. Gebbink and J. A. Killian, J. Am. Chem. Soc., 2011, 133, 252–261 CrossRef CAS PubMed.
- (a) K. K. Patel, E. A. Plummer, M. Darwish, A. Rodger and M. J. Hannon, J. Inorg. Biochem., 2002, 91, 220–229 CrossRef CAS; (b) C. W. Jiang, H. Chao, H. Li and L. N. Ji, J. Inorg. Biochem., 2003, 93, 247–255 CrossRef CAS; (c) H. Chao, W. J. Mei, Q. W. Huang and L. N. Ji, J. Inorg. Biochem., 2002, 92, 165–170 CrossRef CAS.
- J. Andersson, L. H. Fornander, M. Abrahamsson, E. Tuite, P. Nordell and P. Lincoln, Inorg. Chem., 2013, 52, 1151–1159 CrossRef CAS PubMed.
- (a) P. Lincoln and B. Nordén, J. Phys. Chem. B, 1998, 102, 9583–9594 CrossRef CAS; (b) S. Vasudevan, J. A. Smith, M. Wojdyla, A. DiTrapani, P. E. Kruger, T. McCabe, N. C. Fletcher, S. J. Quinn and J. M. Kelly, Dalton Trans., 2010, 39, 3990–3998 RSC; (c) T. Very, S. Despax, P. Hébraud, A. Monari and X. Assfeld, Phys. Chem. Chem. Phys., 2012, 14, 12496–12504 RSC.
- (a) R. Siebert, F. Schlütter, A. Winter, M. Presselt, H. Görls, U. S. Schubert, B. Dietzek and J. Popp, Cent. Eur. J. Chem., 2011, 9, 990–999 CrossRef CAS PubMed; (b) T. K. Aldridge, E. M. Stacy and D. R. McMillin, Inorg. Chem., 1994, 33, 722–727 CrossRef CAS.
- (a) R. Lyng, A. Rodger and B. Nordén, Biopolymers, 1991, 31, 1709–1720 CrossRef CAS PubMed; (b) R. Lyng, A. Rodger and B. Nordén, Biopolymers, 1992, 32, 1201–1214 CrossRef CAS PubMed.
- R. A. Leising, S. A. Kubow, M. R. Churchill, L. A. Buttrey, J. W. Ziller and K. J. Takeuchi, Inorg. Chem., 1990, 29, 1306–1312 CrossRef CAS.
- J. Michl and E. Thulstrup, Spectroscopy with Polarized Light, VCH Publishers, New York, 1986, p. 120 Search PubMed.
- J. D. McGhee and P. H. V. Hippel, J. Mol. Biol., 1974, 86, 469–489 CrossRef CAS.
- P. Lincoln, Chem. Phys. Lett., 1998, 288, 647–656 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt02642j |
| This journal is © The Royal Society of Chemistry 2015 |