
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First principles static and dynamic calculations for the transition metal hydride series MH4L3 (M = Fe, Ru and Os; L = NH3, PH3 and PF3)†
Nicolas
Sieffert
ab,
Thomas
Kendrick
c,
Davide
Tiana
d and
Carole A.
Morrison
*c
aUniv. Grenoble Alpes, DCM, F-38000 Grenoble, France
bCNRS, DCM, F-38000, Grenoble, France
cSchool of Chemistry, and EaSTCHEM Research School, University of Edinburgh, The King's Buildings, West Mains Road, Edinburgh, EH9 3JJ, UK. E-mail: c.morrison@ed.ac.uk
dDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 20th January 2015
Abstract
We present a first principles static and dynamical study of the transition metal hydride series MH4L3 (M = Fe, Ru and Os; L = NH3, PH3 and PF3), with a view to arriving at an understanding of how the variation in the electronic properties of the metal sites and ligands can influence the dynamics of the resulting complexes. A broad range of behaviour was observed, encompassing stable classical minima (M = Os, L = NH3 and M = Ru, L = PH3) to stable η2-H2 non-classical minima (M = Fe, L = PF3 and M = Ru, L = PH3 or PF3), with the other structures exhibiting dynamical behaviour that spontaneously converted between the classical and non-classical states during the molecular dynamics simulations. The importance of a small Laxial–M–Laxial angle in stabilising the non-classical state is highlighted, as is a short η2-H2⋯Hcis distance in non-classical complexes that spontaneously convert to the classical form. We also investigated the changes in the electronic structure of the complex FeH4(PH3)3 during a η2-H2 bond breaking/bond making reaction and observed direct evidence of the ‘cis effect’, whereby a neighbouring hydride ligand acts to stabilise the intermediate classical state.
Introduction
Transition metal (TM) hydrides are at the heart of many chemical reactions, such as hydrogenation,1 dehydrogenation,2,3 decarbonylation4 and hydrogen-transfer.1,5 Of these, dehydrogenation reactions have found particular interest in catalysis,6 for they create the potential to generate molecular hydrogen from low molecular weight organic molecules, which is of great interest in the context of the hydrogen economy.7 Reaction profiles all involve the formation of several [M]–H bonds followed by hydrogen evolution at the metal centre(s). Early studies pointed out the fluxional behaviour of a wide range of TM hydrides,8 where classical hydrides (B; see Scheme 1) are in “fast” equilibrium with non-classical forms (A, C and A′), such that two hydride (H−) ligands combine to create a single H2 molecule coordinated in a η2 fashion to the metal. The relative stabilities of the two states, and the height of the associated activation barrier, are governed by electronic effects that mainly depend on a subtle balance between a forward-bonding interaction (involving a σ donation from the σg orbital of H2 to the metal dz2 orbital) and a back-bonding interaction (from the dxz orbital of the metal to the σu* orbital of H2 if the ligand lies perpendicular to the equatorial plane, dyz if it is parallel; see Scheme 1). Thus TM-η2-H2 bonding exhibits a clear analogy with the well-known Dewar–Chatt–Duncanson model for metal-olefin π bonding.9–12 | ||
| Scheme 1 Top: schematic representation of the two investigated pathways: rotation of the H2 ligand and H2 scrambling, and the model systems considered herein. Bottom: Schematic representation of the forward- and back-bonding interactions involved in the binding of H2 (adapted from ref. 13). | ||
Numerous studies have revealed that the underlying motions for the passage from one state to another are complex and involve so-called “secondary topological changes”, in which structural reorganization of the ligands lead to positional changes for the hydrogen atoms that reduces the energies of the transition states.8 The underlying mechanisms are found to depend on multiple factors (e.g. the number of hydride ligands, the nature of the metal,14 the geometry of the complexes,15 the nature of the ligands,8 the temperature and the environment16). Drawing general conclusions on common mechanisms for different complexes is therefore difficult, and a careful case-by-case investigation is required. However, two general ‘channels’ on the potential energy surface are widely recognised: the first involves rotation of the η2-H2 ligand, resulting in an exchange of hydrogen atoms H1 and H2; the second is the η2-H2 ↔ 2H scrambling motion, defining the classical/non-classical- hydride equilibrium (see “rotation” and “scrambling” in Scheme 1).
It is known that of the hundred or so reported complexes that support dihydrogen bonding, most tend to exist in a low-spin d6 pseudo-octahedral geometry, thus presenting a fully occupied t2g orbital set to be utilised for the metal-H2 back-bonding interaction and a vacant dz2 (eg) orbital for the forward-bonding interaction.17 3rd Row transition metals are unlikely to support a dihydrogen ligand, as relativistic effects lead to an enhance core screening effect, thus rendering the valence d-orbitals more available to participate in back-donation, which in turn leads to cleavage of the dihydrogen bond. All ligands have a role to play, with those in the axial and equatorial positions either pushing or pulling electron density onto or from the metal, and thus altering the availability of the metal orbitals for bonding to H2. From early experimental and computational studies it was shown that the length of the dihydrogen bond depends directly on the nature of the ligand trans to the H2 unit, with σ-donor ligands such as hydrogen (as used in this study) generally resulting in very short H–H distances of around 0.9 Å.10,11 This arises due to a trans labilizing effect, which reduces σ-donation from H2 thus weakening M–H2 binding, causing the H–H distance to contract.18 Moreover, the barrier to H2 rotation can be lowered by the presence of a hydride cis to H2, in a phenomenon known as the ‘cis-effect’, whereby a two-electron interaction between the orbitals σM–Hcis and σ* H2 results in a preferential conformation for the H2 ligand that eclipses the M–Hcis bond.19 This overrides the conformational setting dictated by the back-donation orbital interaction, which would otherwise position the H2 ligand perpendicular to the equatorial plane.20
Among the various examples of highly fluxional hydrides, the [MH4(L)3] (M = Fe, Ru and Os; L = phosphines) family has gained particular attention in the literature. For instance, a rotation motion has been reported in the case of the OsH4L3 (L = PMe2Ph) complex,19 possibly accompanied by a significant deformation of the OsL3 fragment within the transition structure.8 The η2-H2 ↔ 2H scrambling motion has also been investigated and is shown to involve the breaking of the dihydrogen bond to afford a transient classical tetrahydride. The whole process is expected to occur with the four hydrogen atoms remaining in the equatorial plane. Relatively small activation barriers (in the range of ca. 8 kcal mol−1) have been determined experimentally.8
In the most part, where data have been obtained from computational studies, the structures reported are those of the most stable geometries, as obtained from “static” optimizations. A more realistic picture can be gained by explicitly considering the dynamics of the complexes, i.e. considering their intrinsic vibrational motions and permitting the effects of temperature to be included. We recently applied density functional theory molecular dynamics (DFT-MD) simulations to study the dynamical behaviour of a series of [MH4(PH3)3] complexes (M = Fe, Ru and Os), focusing on the description of the anharmonic vibrational features encountered in these complexes. However, we also showed in this study that a dynamical exchange between classical and non-classical isomers can be directly observed within the course of a simulation (in a 30 ps timescale).21 Others have since also reported the direct observation of a classical/non-classical transition for transition metal hydride complexes.15 Recently, hydride migration around low-coordinate metal centres containing N-heterocyclic carbenes has also been investigated by DFT-MD simulations.22
Herein, we build on our first study,21 to further investigate the dynamical mechanisms involved in TM hydrides by means of DFT-MD simulations. Our approach involves the screening of several metals and companion ligands and aims to arrive at an understanding of how the variation in their electronic properties can influence the dynamics of the resulting complexes. In selected cases, clear dynamical equilibria corresponding to the rotation and scrambling pathways (as presented in Scheme 1) can be observed during simulations (i.e. many events occur within 30 ps), a feature that therefore allows us to provide statistical pictures of the behaviour of the complexes by plotting correlation diagrams between selected geometrical parameters. Correlations between geometrical and electronic parameters (namely energies of molecular orbitals) are also presented. All data taken together allow us to better understand how the bonding system evolves under the effect of (an elevated) temperature (350 K, i.e. in a case where hydrogen tunnelling should be negligible; vide infra).
As target systems, we chose a series of complexes conforming to the formula [MH4L3] where the metal M is Fe, Ru, or Os, and the ligands are L = NH3, PH3, and PF3, such that different combinations of M and L permit the “fine-tuning” of the electronic structure of the complexes. All metals can be considered to be in a low-spin d6 configuration and in a pseudo-octahedral environment, thus presenting full occupation of the t2g orbitals to support M–H2 back-bonding. The availability of those orbitals increases when going from Fe to Os due to increased core screening brought on by relativistic effects. For the ligands, NH3 was selected as a small ligand that acts as a pure σ-electron donating ligand (from the nitrogen lone pair). This ligand should therefore increase the ability of the metal centres to back-donate, which should result in a less stable dihydrogen bond. Conversely, PH3 and PF3 are generally considered to be simultaneously σ-donating (from the phosphorous lone pair) and π-accepting ligands (into the P-H/F σ* orbital). Overall, these ligands can therefore withdraw more electron density from the metal centres, and thus should lessen the ability of the metal to backdonate into the H2 σ* orbital, resulting in a more stable η2-H2 unit.
For all complexes, static optimizations have first been performed in order to characterise the nature of the stationary points (minima or transition states) of the different isomers (A–C, Scheme 1) and to get insights into the relative kinetic barriers and driving forces of the rotation and scrambling pathways. Next, we performed extensive DFT-MD simulations to gather statistical information on the geometric parameters involved in making and breaking the dihydrogen bonds in these complexes. Analysis of the time evolution of the electronic structure for the complex FeH4(PH3)3 has also been performed, a posteriori, using snapshots extracted from the DFT-MD simulation.
Computational methods
| PBEa | CCSD(T)b | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| M | L | Isomerc | Typed | α/° | ϕ/° | η2-H2⋯Hcis/Å | ΔErel | ΔGrel | ΔErel |
| a Geometries and thermochemistry corrections are computed at the PBE/SDD/6-31G** level (at 350 K and 1 atm). Defined energies obtained from single points at the PBE/SDD/6-311+G** level. b CCSD(T)/SDD/aug-cc-pVTZ single points on PBE/SDD/6-31G** optimized geometries. c See Scheme 2 for schematic representation of the complexes. d Indicates whether the structure is a minimum (“Min.”) or a transition state (T.S.). e Optimization leads to the classical isomer. | |||||||||
| Fe | NH3 | Non-classical | Min. | 171.9 | 0.1 | 1.76 | 0.0 | 0.0 | 0.0 |
| Non-class. perp | T.S. | 161.9 | 90.2 | 2.23 | 6.0 | 7.2 | 6.7 | ||
| Classical | Min. | 175.5 | 0.1 | 1.49 | 1.3 | 1.8 | 0.7 | ||
| TS scrambling | T.S. | 174.1 | 0.0 | 1.59 | 1.2 | 1.5 | 0.8 | ||
| Fe | PH3 | Non-classical | Min. | 156.8 | 0.2 | 1.89 | 0.0 | 0.0 | 0.0 |
| Non-class. perp | T.S. | 148.0 | 90.4 | 2.19 | 1.2 | 1.2 | 1.3 | ||
| Classical | T.S. | 166.0 | 0.0 | 1.77 | 5.1 | 4.1 | 5.9 | ||
| Fe | PF3 | Non-classical | Min. | 146.8 | 0.4 | 1.86 | 0.0 | 0.0 | 0.0 |
| Non-class. perp | Min. | 140.2 | 90.5 | 2.14 | 0.4 | −1.1 | 0.3 | ||
| TS rotation | T.S. | 141.1 | 74.6 | 2.03 | 0.4 | 0.1 | 0.2 | ||
| Classical | T.S. | 159.4 | 0.0 | 1.87 | 7.7 | 6.8 | 6.9 | ||
| Ru | NH3 | Non-classical | →classicale | ||||||
| Non-class. perp | T.S. | 163.1 | 92.8 | 2.32 | 5.4 | 4.9 | 9.4 | ||
| Classical | Min. | 175.2 | 0.0 | 1.66 | 0.0 | 0.0 | 0.0 | ||
| Ru | PH3 | Non-classical | Min. | 161.7 | 0.0 | 2.10 | 0.0 | 0.0 | 0.0 |
| Non-class. perp | T.S. | 154.8 | 90.8 | 2.38 | 2.3 | 1.9 | 2.6 | ||
| Classical | Min. | 168.5 | 0.0 | 1.83 | 3.0 | 2.0 | 0.0 | ||
| TS scrambling | T.S. | 166.4 | 0.0 | 1.92 | 3.3 | 1.5 | 1.1 | ||
| Ru | PF3 | Non-classical | Min. | 155.5 | 0.1 | 2.09 | 0.0 | 0.0 | |
| Non-class. perp | T.S. | 145.7 | 90.5 | 2.37 | 1.7 | 0.9 | |||
| TS scrambling | T.S. | 163.9 | 9.6 | 1.90 | 7.2 | 6.4 | |||
| Classical | Min. | 164.9 | 0.0 | 1.87 | 7.0 | 5.6 | |||
| Os | NH3 | Non-classical | →classicale | ||||||
| Non-class. perp | T.S. | 157.1 | 90.2 | 2.30 | 9.0 | 9.1 | |||
| Classical | Min. | 175.7 | 0.0 | 1.71 | 0.0 | 0.0 | |||
| Os | PH3 | Non-classical | Min. | 165.2 | 0.0 | 2.09 | 1.9 | 1.5 | 5.2 |
| Non-class. perp | T.S. | 157.1 | 90.7 | 2.39 | 5.4 | 5.3 | 9.9 | ||
| Classical | Min. | 169.4 | 0.0 | 1.86 | 0.0 | 0.0 | 0.0 | ||
| TS scrambling | T.S. | 165.8 | 0.0 | 2.06 | 2.0 | 1.2 | 4.7 | ||
| Os | PF3 | Non-classical | Min. | 160.7 | 0.0 | 2.12 | 0.0 | 0.0 | |
| Non-class. perp | T.S. | 152.1 | 90.7 | 2.39 | 2.7 | 1.5 | |||
| Classical | Min | 166.2 | 0.0 | 1.88 | 1.6 | 1.0 | |||
| TS scrambling | T.S. | 164.8 | 0.0 | 1.99 | 2.5 | 1.5 | |||
 | ||
| Fig. 1 Correlation frequency maps between the dihedral angle H1⋯H2⋯H3⋯H4 (ϕ) and d(H1⋯H2)–d(H3⋯H4) (Δd) for complexes MH4(L)3. | ||
Importance of quantum mechanical tunnelling
Quantum mechanical effects on the dynamical behaviour of TM hydrides are well recognized but are known to occur only at rather low temperature (i.e. up to 150–200 K).33 An estimate of the importance of tunnelling can be obtained from the crossover temperature34Tc (in K), defined as follows:where h is the Planck constant, ν is the absolute value of the imaginary frequency corresponding to the transition mode (in Hz) and kB is the Boltzmann constant. Tc marks the temperature (in K) where tunnelling and thermally activated barrier crossing are roughly equally important (as an example, Tc is about room temperature when ν = 1300i cm−1). Imaginary frequencies for all transition states are provided in ESI† (Table S1). The latter range from 49i cm−1 (rotation pathway in [OsH4(NH3)3]) to 520i cm−1 (scrambling pathway in [OsH4(PF3)3]) and therefore correspond to Tc generally below 100 K (namely, ranging from 11 K to 119 K), i.e. well below the temperature considered herein (T = 350 K). The corresponding Wigner correction factors35,36 are generally small (between 1.00 and 1.10, and somewhat larger in the case of [OsH4(PF3)3] where it amounts to 1.19; see Table S1†). When two transition states were located for a given complex (for rotation and scrambling paths), we note that Tc and Wigner correction factors are very similar, in such a way that the relative rate of rotation vs. scrambling is little affected by the quantum correction (see rate constants in Table S1†). Herein, we therefore considered that quantum mechanical tunnelling does not influence the conclusions of our study and this effect was not accounted for (neither in our static calculations based on classical transition state theory nor in our DFT-MD simulations where nuclei are described as classical particles).
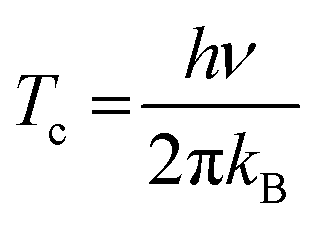
Finally, the H–H bond breaking event in FeH4(PH3)3 has been analysed with the quantum theory of atoms in molecules (QTAIM)37 using the Promolden code.38 The electronic structures were calculated using a standard 6-31G* basis set for p-block elements and the phosphine hydrogen atoms, coupled to the PBE level of theory; a second polarisation function was added to this basis set description for a higher accuracy treatment of the hydrogen atoms attached to the metal centre. Hay-Wadt small core potential with its relative basis set was employed to describe Fe,39 and the metal core shell was reconstructed a posteriori.40 Snapshots from the MD trajectory have been extracted and used to calculate the charges and the delocalisation indices (DI) δ.41
Performing a topological analysis of the electron charge density ρ, allows the space to be partitioned into atomic basins, Ω, which defines the atoms within the QTAIM approach. Integrating ρ over Ω therefore provides the atomic charge qA, since:


It should be noted that DFT calculations do not provide a proper 2nd order density matrix, and for that reason the values reported for δ are approximate only. However, it has been found that δDFT values are reasonable, and tend to be in good agreement with pure wavefunction values.43–45
Results and discussion
Static calculations
We first discuss the results from the static geometry optimisations of the MH4L3 complexes (M = Fe, Ru and Os; L = NH3, PH3 and PF3), as presented in Table 1. Definitions of all of the stationary point structures obtained on the potential energy landscapes can be found in Scheme 2, and a table listing more selected geometric parameters is given in the ESI† (see Table S2), along with Cartesian coordinates of selected optimised geometries. | ||
| Scheme 2 Competitive scrambling (a) and rotation (b and c) pathways. The rotation profile can involve either one (b) or two transition states (c). | ||
The relative energies (and the thermodynamically-corrected free energies) of the optimised structures obtained on the potential energy surfaces show the trends we expected from varying M and L, with the trends observed in the DFT calculations mirrored in the CCSD(T) calculations (see Table 1). When M = Fe the non-classical state (with the η2-H2 ligand sitting parallel to the equatorial plane) is always preferred to the classical, and as the electron withdrawing properties of L increases, the energy separation between the two states increases (from 1.2, to 5.1 to 7.7 kcal mol−1 for L = NH3, PH3 or PF3, respectively). The non-classical state where the η2-H2 ligand sits perpendicular to the equatorial plane is a high-energy transition state structure when L = NH3, but as L becomes more electron withdrawing the relative energy of this stationary point lowers until for L = PF3 it is an almost isoenergetic minimum with the non-classical parallel state. With reference to the molecular orbital diagram shown in Scheme 1, this suggests that electron withdrawing properties of the auxiliary ligands have all but ‘switched off’ the π-back-donating orbital exchange, resulting in a more freely rotating η2-H2 ligand and very short H–H bond lengths (0.99, 0.90 and 0.88 Å for L = NH3, PH3 and PF3, respectively, see Table S2†). Interestingly, the Laxial–Fe–Laxial angle (angle α) also becomes more bent as the ligand becomes more electron withdrawing [171.9° (L = NH3), 156.8° (L = PH3) and 146.8° (L = PF3)], and with the barrier to the η2-H2 ligand sitting perpendicular to the equatorial plane dropping, we see an increase in the η2-H2⋯Hcis distance, from 1.76 Å (for L = NH3) to 1.89 Å (L = PH3) to 1.86 and 2.14 Å for the near iso-electronic L = PF3 parallel and perpendicular minima, respectively. Thus as the ability of Fe to π-back donate electrons into the σ* H2 orbital is reduced the population of the classical structure becomes increasingly unfavourable.
The geometry optimisation calculations for the M = Fe series therefore suggest in total three different pathways, which are summarised pictorially in Scheme 2. When L = NH3 the complex should be able to switch between the classical and non-classical states via the TS-scrambling pathway (route ‘a’ in Scheme 2), while the rotational pathway (where there are two routes – ‘b’ and ‘c’) are blocked. Switching L for PH3 suppresses the TS-scrambling pathway, while opening up the rotational pathway, which proceeds via a transition state where the η2-H2 ligand is perpendicular to the equatorial plane (route ‘b’ in Scheme 2). Finally switching L for PF3 further suppresses the TS-scrambling pathway and renders the perpendicular η2-H2 ligand geometry a minimum on the potential energy surface (route ‘c’ in Scheme 2).
Switching M to Ru now stabilises the classical form, provided L = NH3. According to the molecular orbital diagram drawn in Scheme 1, with relativistic effects rendering the t2g orbital set more available for bonding to H2, an electron donating ligand will encourage π-back donation to σ* H2 and thus sever the H–H bond, such that the classical state is the only one observed. This suggests that route ‘a’ in Scheme 2 should now be suppressed. The η2-H2 ligand lies parallel to the equatorial plane, with the barrier to H2 rotation lying at least 5 kcal mol−1 above the minimum energy structure, also shutting down routes ‘b’ and ‘c’ for this system. These results therefore suggest that complex M = Ru, L = NH3 is effectively trapped in one well on the potential energy surface, corresponding to the classical structure. As the electron withdrawing properties of L increase the non-classical structure once again becomes the more stable state; for L = PH3 the rotational and scrambling pathways are both accessed via transition state structures that are similar in energy, suggesting routes ‘a’ and ‘b’ are now open, while for L = PF3 the barrier to the rotational pathway is significantly lower, suggesting route ‘b’ as the lowest energy pathway. Turning now to geometrical considerations, as L becomes more electron withdrawing angle α reduces, as with the Fe series, however a clear-cut pattern in the η2-H2⋯Hcis distances is now less obvious. The classical minimum for the L = NH3 complex results in a very short value for bond distance η2-H2⋯Hcis (1.66 Å), while the non-classical structures return much longer values, at 2.10 and 2.09 Å, for L = PH3 and PF3, respectively.
Finally when M = Os, and L = NH3 we again see a preference for the classical state. With the absence of a minimum for the non-classical state and a predicted high barrier to η2-H2 rotation, this complex should be trapped in one well on the potential energy surface, as with the Ru analogue. A similar story exists when L = PH3, but now a stationary point for the “in plane” (ϕ = 0°) non-classical form could be located, and only an extremely low barrier (ca. ΔΔErel = 0.1 kcal mol−1 at the PBE level) prevents its optimization towards the most stable classical structure. We note that the non-classical [OsH4(PH3)3] complex exhibits the longest H1–H2 distance (1.02 Å) of the MH4L3 series (see Table S2†), and its structure is found to be very close to the TS-scrambling transition state (consistent with the very similar energies values obtained for the two structures in Table 1). Note the barrier to the rotational pathway is high, shutting down routes ‘b’ and ‘c’. When L = PF3, the non-classical state is again a minimum, only slightly less stable than the classical global minimum (<2 kcal mol−1, see Table 1), with the scrambling and rotational pathways accessed via barriers of similar height (<3 kcal mol−1, see Table 1). Geometry considerations again indicate that angle α decreases with increasing electron withdrawing capability of L; no clear pattern emerges for the bond distance η2-H2⋯Hcis.
Molecular dynamics
For characterisation of the spontaneous molecular motions observed during the production run trajectories, the following geometrical descriptors have been considered: (i) the difference in distances between d(H1–H2) and d(H3⋯H4) (labelled Δd, in Å), (ii) the dihedral angle involving H1⋯H2⋯H3⋯H4 (ϕ, in degrees), (iii) the Laxial–M–Laxial angle (α, in degrees), and the η2-H2⋯Hcis distance [i.e. d(H2⋯H3)] (See Scheme 1 for atom numbering). Correlation plots between these parameters are depicted in Fig. 1–3.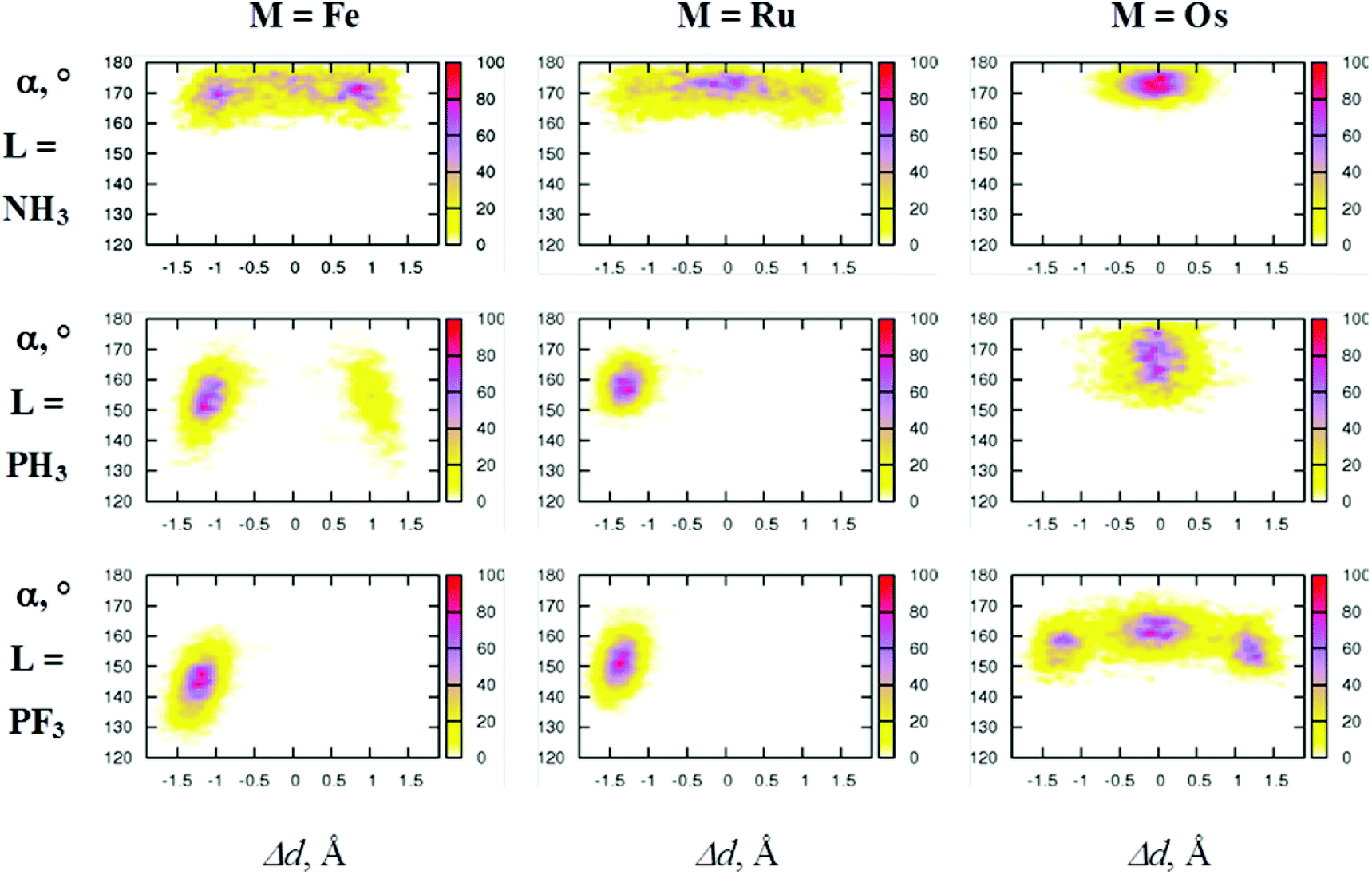 | ||
| Fig. 2 Correlation frequency maps between the angle Laxial–M–Laxial (α) and d(H1⋯H2)–d(H3⋯H4) (Δd) for complexes MH4(L)3. | ||
 | ||
| Fig. 3 Correlation between the non-classical/classical hydride exchange and the H2–H3 (d23) distance. Frequency maps for complexes of series 1 and 2 (see Scheme 1). Note for complexes [FeH4(PH3)3] and [OsH4(PF3)3], which show dynamic η2-H2 ↔ 2H behaviour, the identities of atom pairs (H1, H2) and (H3, H4) switch during the simulation, so some of the data presented in these plots relates to the longer distance d(H1⋯H4). | ||
It is immediately obvious from Fig. 1 and 2 that the dynamical behaviour of the series of complexes depends strongly on the choice of the metals and the ligands. If a complex accesses the scrambling pathway (route ‘a’, Scheme 2), where it interconverts between the classical and non-classical states, large variations in Δd are expected; if it accesses the rotational pathway (route ‘b’, Scheme 2) large variations in ϕ are expected. Starting with [FeH4(NH3)3], the geometry optimisation calculations suggested that route ‘a’ only was to be expected. This is borne out in the MD results, where many exchanges between classical and non-classical forms occur within the 30 ps MD trajectory. Note the conformations with the largest population occur when Δd ca. ± 1.5 Å. Classical hydrides are also significantly populated (for which Δd is close to zero) and a continuous motion from one form to the other is observed. The ϕ dihedral angle always remains close to 0° (Fig. 1), clear evidence that the scrambling pathway is the one invoked for the η2-H2 ↔ 2H exchange reaction. The η2-H2 ligand sitting parallel to the equatorial is accompanied by an α angle which remains close to linear throughout the simulation (170 ± 10°, Fig. 2, cf. 171.9° for the geometry optimised structure). The correlation plots for parameter η2-H2⋯Hcis indicate a rather short average distance, of around 1.8 Å (Fig. 3) for the non-classical state. We note that the correlation plots are very symmetric, indicating that good sampling of the configurations of the system has been obtained.
When we switch to the less electron-donating ligand PH3 we observe a dramatic change in behaviour. The geometry optimisation calculations suggested that the barrier to the rotational pathway should be significantly lower than for the scrambling pathway, and this is supported by the MD results. The non-classical [FeH2(H1–H2)(PH3)3] complex (Δd = −1.0 Å and ϕ = 0°) is overwhelmingly favoured. Rotation of the H2 moiety occurs spontaneously in the course of the dynamics, as indicated in the spread of values for ϕ from 0° to ±180° (Fig. 1), however the more populated regions correspond to ϕ = 0 and ±180°, indicating that the four hydride atoms lie flat in the equatorial plane for the majority of the time. Both Δd = ±1.0 Å regions are populated, but as the scrambling pathway was accessed only once during the dynamical trajectory, their probabilities are asymmetric. The static calculations suggested a higher activation barrier for the scrambling process (the classical structure was a transition state), which is borne out by the MD data. We note that d(H1–H2) elongates noticeably when the ligand lies in the equatorial plane, possibly due to a η2-H2⋯Hcis interaction. Angle α fluctuates around 155 ± 15° (see Fig. 2), similar to the geometry optimised value 156.8°, and again we see a population basin around 1.8 Å for η2-H2⋯Hcis (Fig. 3).
When L = PF3, the geometry optimisation calculations predicted that the rotational pathway would be favoured over the scrambling pathway, and this is indeed observed in the MD simulation, with the Fe complex remaining in its non-classical state during the whole 30 ps of MD. The probability distribution plot shown in Fig. 1 suggesting an almost freely rotating η2-H2 ligand, commensurate with the drop in barrier height predicted by the optimisation calculations, and d(H1–H2) elongating slightly when the ligand lies in the equatorial plane. Angle α has dropped further, to around 145 ± 15° (see Fig. 2, c.f. optimised structure 146.8°), while the population basis around 1.8 Å for η2-H2⋯Hcis is reduced (Fig. 3), which presumably can be directly linked to the absence of an η2-H2 ↔ 2H exchange reaction for this complex.
Switching the metal from Fe to Ru and Os, we again see that changing the auxiliary ligands NH3 → PH3 → PF3 induces a shift from the scrambling pathway to the rotational one, with the switch occurring later in the case of Os (Fig. 1). This switch in pathway we attribute to the reduction in π-back-donation between the metal t2g orbital and the ligand σ*; the later switchover for Os can presumably be tied into the premise that relativistic effects will render the metal t2g orbital more closely involved in the frontier orbital set, and thus more strongly electron withdrawing ligands are required to induce the switchover.17 This change in pathway is also observed in the α angles (Fig. 2), with a close to linear configuration observed when the scrambling pathway is accessed, and an increasingly bent configuration supporting an increasingly more freely rotating η2-H2 ligand. The parameter density maps reveal interesting differences compared to the Fe analogues (Fig. 1). Whereas the non-classical forms are more populated for [FeH4(NH3)3], for [RuH4(NH3)3] and [OsH4(NH3)3] it is now the classical states that are preferred. The geometry optimisation calculations predicted these structures were essentially trapped in a single well, with a steeper exit barrier for the Os system, a result mirrored in the MD probability plots. For [OsH4(PF3)3], both the scrambling and rotational pathways are accessed, with the α angle widening for the classical state, commensurate with the near identical barriers for both routes predicted from the geometry optimisation calculations (Table 1). Concerning the η2-H2⋯Hcis probability plots, for those structures that support an η2-H2 structure and no η2-H2 ↔ 2H scrambling (i.e. [RuH4(PH3)3] and [RuH4(PF3)3]) we observe that d(H2⋯H3) > 2.0 Å. For [OsH4(PF3)3], which does support scrambling the data is less clear cut, as the rapid fluxional η2-H2 ↔ 2H behaviour means this plot is a combination of d(H2⋯H3) and d(H1⋯H4) data which affects the statistics of the data binning process, however it is apparent that a significant population of d(H2⋯H3) ca. 2.0 Å is present.
Thus, to summarise, the results obtained from the MD trajectories and static geometry optimisation calculations point towards various key structural parameters that dictate whether the resulting complex adopts a classical, non-classical, or a dynamic η2-H2 ↔ 2H structure. To support a non-classical hydride, it is clear that the angle α must be quite small, in agreement with the findings by Mitoraj et al. on molybdocene trihydride complexes.15 This can be achieved by the presence of auxiliary ligands that are strongly electron withdrawing, which interferes with the ability for the metal centre to π-back donate to the H2 ligand, resulting in a freely rotating η2-H2 unit with a short H–H bond distance. Conversely, if the axillary ligands are electron donating, π-back donation is favoured; angle α widens and the classical state is favoured. To encourage dynamic η2-H2 ↔ 2H behaviour the barrier height to the TS-scrambling state must be minimised, and the evidence points towards fluxional complexes having a short η2-H2⋯Hcis distance in the non-classical state.
Time evolution of molecular orbitals, geometric parameters and topological analysis during η2-H2 ↔ 2H scrambling reaction
We can use the MD trajectories to build up a representation of the changes in the properties and energies of the molecular orbitals (MOs) involved in the dihydrogen bond making/breaking process. This is done by sampling single-point energy calculations for geometries obtained from snapshots taken from the MD trajectories, and then plotting the resulting orbital energies and isosurfaces as the chemical reaction proceeds, or performing a more in-depth topological analysis such as QTAIM.We have performed this analysis for [FeH4(PH3)3], which undergoes just one η2-H2 ↔ 2H scrambling reaction during the 30 ps MD trajectory. In Fig. 4 we present a timeline for the energies of the molecular orbitals in FeH4(PH3)3 (from HOMO−17 to LUMO+32) over a 180 fs period, with t = 0 marking the point at which Δd = 0 (i.e. the classical structure) is obtained. From Fig. 1 and 2 we observe that this complex exhibits nearly-free η2-H2 rotation and that Δd is minimised when angle α widens, which coincides with the η2-H2 ligand rotating into the equatorial plane. Thus on Fig. 4 we denote that the H2 ligand is first oriented perpendicular to the equatorial plane and at t = −42 fs it has rotated to adopt a parallel orientation. By t = 90 fs the non-classical hydride, with the η2-H2 parallel to the equatorial plane, is observed.
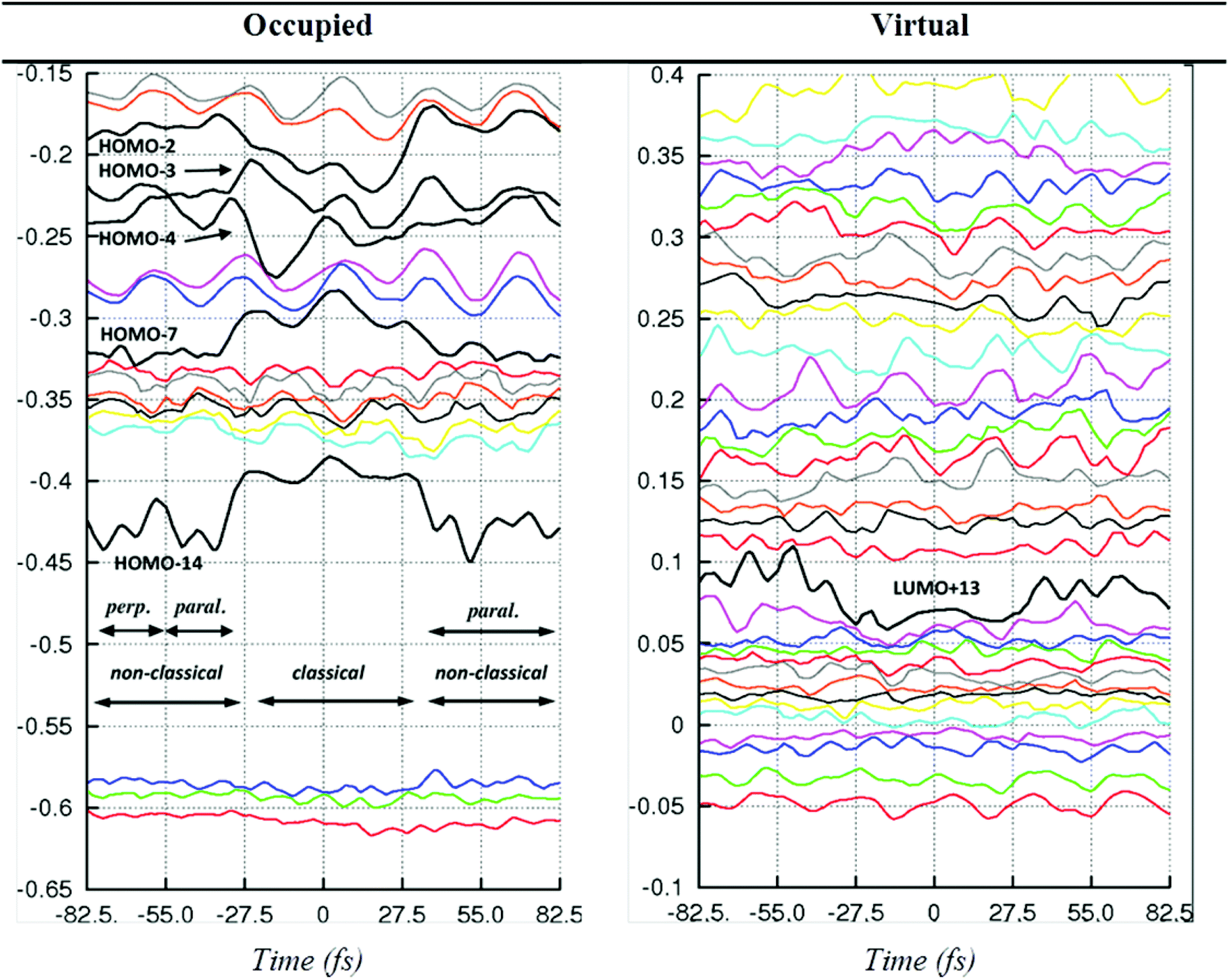 | ||
| Fig. 4 Time evolution of the eigenvalues (in Hartree) of the molecular orbitals in [FeH4(PH3)3] during the scrambling process. “perp.” and “paral.” denote a perpendicular (ϕ = 90°) and parallel (ϕ = 0°) orientation of the H2 ligand with respect to the equatorial plane. Since HOMO−3 and HOMO−4 cross during the rotation of H2 (at t = −55 fs), we labelled the molecular orbitals according to their order from t > 55 fs. | ||
While large oscillations in the energies of the MOs are observed (ca. 0.02 Hartrees), which are due to the effects of temperature in the simulation, some orbitals are clearly seen to undergo significant change in energy (ca. 0.05 Hartrees) as the reaction proceeds. These are HOMO−14, HOMO−7, HOMO−4, HOMO−3, HOMO−2 and LUMO+13. The identities of these orbitals have been manually assigned through careful inspection of isosurface plots. We thus assign HOMO−14 to the σ-donation Fe(dz2)–H2(σ) bonding orbital, HOMO−4 and HOMO−3 are orbitals which involve the Hcis and Htrans ligands and the Fe metal centre, while HOMO−2 represents the π-back-donation Fe(dxz/dyz, depending on whether the H2 unit sits perpendicular or parallel to the equatorial plane)-H2(σ*) bonding orbital. HOMO−7 is a complex mix involving the p-orbitals from the phosphorous atoms, the Fe d-orbitals and a component from the H2(σ) bonding orbital; LUMO+13 is the antibonding partner to HOMO−2. All of these orbitals are stabilized in the classical form (t = 0 fs), with the exceptions of HOMO−14 and HOMO−7; these orbitals are both bonding with respect to the η2-H2 ligand, which is destroyed when the classical structure is formed. The same information from Fig. 4 is portrayed as a scatter plot with reference to the parameter classical/non-classical switch parameter Δd in Fig. S2,† along with images of the orbitals mentioned, and how they evolve as the complex undergoes the η2-H2 ↔ 2H scrambling reaction. The orbital timeline plots therefore appear to support the well documented ‘cis-effect’,20 whereby orbitals on a neighbouring hydride ligand act to stabilise the classical state.
From the orbital timeline plots in Fig. 4, we observe that the onset in change in energies occur ca. 50 fs prior to the t = 0 crossover point. This also marks the point where geometrical changes occur in the H⋯H distances, as shown in Fig. 5(a). Here we observe an oscillating η2-H2⋯Hcis distance (H2⋯H3), which shortens to around 1.75 Å, while the old η2-H2 distance (H1⋯H2) begins to lengthen and the new η2-H2 distance (H3⋯H4) begins to shorten. The topological analysis, performed on the same MD frames allows us to observe that the atomic charges on the η2-H2 atoms [H1 and H2 in Fig. 5(b)] build up around the same time, indicating that charge is flowing into the old η2-H2 ligand from the metal site, which echoes results obtained by Mitoraj for molybdocene trihydride complexes.15 Charge begins to drain from the hydride ligands (H3, H4) from around t = −40 fs. The delocalisation indices for the Fe–H bonds [Fig. 5(c)] allow us to rationalise the change in relative strengths of these bonds during the scrambling process: the results indicate that around t = −50 fs the Fe–H(1,2) bonds strengthen quite dramatically; the Fe–H(3,4) bonds weaken, but only marginally during this time. Finally, we discuss the delocalisation indices for the H⋯H bonds themselves, which are observed to be of equal strength at the t = 0 classical structure [Fig. 5(d)]. By −50 fs the old η2-H2 distance (H1⋯H2) weakens rapidly, matching the trace from the atomic charges plot [Fig. 5(b)], while a spike on the new η2-H2 distance (H3⋯H4) trace is observed at −30 fs, matching a contraction to 1.5 Å observed on the geometry plot [Fig. 5(a)]. From the H2⋯H3 trace on Fig. 5(d) we again observe some direct evidence of the ‘cis effect’, with the delocalisation index rising for this neighbouring interaction as the H1⋯H2 distance begins to weaken. Once the classical structure has been reached, the new η2-H2 distance (H3⋯H4) forms rapidly (<30 fs), as evidenced by all data shown in Fig. 5.
 | ||
| Fig. 5 Time evolution of (a) H⋯H distances, (b) H atomic charges, (c) Fe–H delocalisation indices (d) H⋯H delocalisation indices for [FeH4(PH3)3] during the scrambling process. Atomic labelling (prior to scrambling) is shown in inset in (a). | ||
Conclusions
In this study we have performed a series of static optimisation calculations and molecular dynamic simulations on the family of compounds [MH4L3], where M = Fe, Ru and Os, and L = NH3, PH3 and PF3, with a view to arriving at an understanding of how the variation in the electronic properties of the metal and ligands influence the dynamics of the resulting complexes. The results point towards various key structural parameters that dictate whether the resulting compound adopts a classical, non-classical or a dynamic η2-H2 ↔ 2H structure. To support a non-classical hydride it is clear that the angle α must be quite small, in agreement with the findings of Mitoraj et al.15 This can be achieved by the presence of auxiliary ligands that are strongly electron withdrawing, which interferes with the ability for the metal centre to π-back donate to the H2 ligand, resulting in a freely rotating η2-H2 unit with a short H–H bond distance. Conversely, if the axillary ligands are electron donating, π-back donation is favoured; angle α widens and the classical state is favoured. To encourage dynamic η2-H2 ↔ 2H behaviour the barrier height to the TS-scrambling state must be minimised, and the evidence points towards fluxional complexes having a short mean η2-H2⋯Hcis distance in the non-classical state. Of the set of nine compounds investigated, four were found to have η2-H2 ↔ 2H dynamical behaviour: [FeH4(NH3)3], [FeH4(PH3)3], [RuH4(NH3)3] and [OsH4(PF3)3].To investigate the changes in electronic structure during the dynamic scrambling reaction, geometric snapshots were taken from the MD trajectory for [FeH4(PH3)3], to plot the change in eigenvalues for the frontier orbital set, as well as to undertake a full topological analysis during the spontaneous η2-H2 → 2H reaction. From this direct evidence of the well-documented ‘cis effect’ emerged, as orbitals involving the neighbouring hydride were stabilised and the η2-H2⋯Hcis distance gained in strength in the timeline leading up to the appearance of the transition state classical structure.
Acknowledgements
This work was carried out under the HPC-EUROPA2 project (project number: 228398) with the support of the European Commission Capacities Area – Research Infrastructures Initiative. This work made use of the facilities of HECToR, the UK's national high-performance computing service, which is provided by UoE HPCx Ltd at the University of Edinburgh, Cray Inc and NAG Ltd, and funded by the Office of Science and Technology through EPSRC's High End Computing Programme. This work was also granted access to the HPC resources of IDRIS under the allocation 2013-i2011086670 made by GENCI (Grand Equipement National de Calcul Intensif). N.S. thanks the Univ. Grenoble Alpes, the CNRS and the ICMG FR 2607 for financial support, CIMENT for allocation of CPU time on the “ceciccluster” and “froggy” platforms (project “liqsim”), and Chris Johnson for technical support.References
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201 CrossRef CAS PubMed.
- D. Morton and D. J. Cole-Hamilton, J. Chem. Soc., Chem. Commun., 1988, 1154 RSC.
- H. Junge, B. Loges and M. Beller, Chem. Commun., 2007, 522 RSC.
- L. S. Van der Sluys, G. J. Kubas and K. G. Caulton, Organometallics, 1991, 10, 1033 CrossRef CAS.
- A. Aranyos, G. Csjernyik, K. J. Szabó and J.-E. Bäckvall, Chem. Commun., 1999, 351 RSC.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578 CrossRef CAS PubMed.
- T. C. Johnson, D. J. Morris and M. Wills, Chem. Soc. Rev., 2010, 39, 81 RSC.
- D. G. Gusev and H. Berke, Chem. Ber., 1996, 129, 1143 CrossRef CAS.
- Y. Jean, O. Eisenstein, F. Volatron, B. Maouche and F. Sefta, J. Am. Chem. Soc., 1986, 108, 6587 CrossRef CAS.
- G. J. Kubas, J. Organomet. Chem., 2001, 635, 37 CrossRef CAS.
- G. J. Kubas, Chem. Rev., 2007, 107, 4152 CrossRef CAS PubMed.
- D. Tiana, E. Francisco, M. A. Blanco, P. Macchi, A. Sironi and A. Martin Pendas, J. Chem. Theory Comput., 2010, 6, 1064 CrossRef CAS.
- J. C. Green, M. L. H. Green and G. Parkin, Chem. Commun., 2012, 48, 11481 RSC.
- R. H. Crabtree and D. G. Hamilton, J. Am. Chem. Soc., 1986, 108, 3124 CrossRef CAS.
- Ł. Piękoś and M. P. Mitoraj, J. Comput. Chem., 2012, 34, 294 CrossRef PubMed.
- D. G. Gusev, A. B. Vymenits and V. I. Bakhmutov, Inorg. Chim. Acta, 1991, 179, 195 CrossRef CAS.
- G. S. McGrady and G. Guilera, Chem. Soc. Rev., 2003, 32, 383 RSC.
- R. H. Morris, Can. J. Chem., 1996, 74, 1907 CrossRef CAS.
- J. W. Bruno, J. C. Huffman, M. A. Green, J. D. Zubkowski, W. E. Hatfield and K. G. Caulton, Organometallics, 1990, 9, 2556 CrossRef CAS.
- L. S. Van der Sluys, J. Eckert, O. Eisenstein, J. H. Hall, J. C. Huffman, S. A. Jackson, T. F. Koetzle, G. J. Kubas, P. J. Vergamini and K. G. Caulton, J. Am. Chem. Soc., 1990, 112, 4831 CrossRef CAS.
- C. A. Morrison and A. M. Reilly, Dalton Trans., 2010, 39, 5527 RSC.
- M. A. Ortuño, P. Vidossich, S. Conejero and A. Lledós, Angew. Chem., Int. Ed., 2014, 53, 13158 CrossRef PubMed.
- J. A. Pople, et al., Gaussian 09, Revision A.02, Gaussian 09, Gaussian, Inc., Pittsburgh, PA, 2009 Search PubMed (the full reference is given in ESI†).
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 181, 1477 CrossRef CAS PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103 CrossRef CAS PubMed.
- For more information on CP2K, see: http://www.cp2k.org (last accessed: January 2015).
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter, 1996, 54, 1703 CrossRef CAS.
- S. Nosé, J. Chem. Phys., 1984, 81, 511 CrossRef PubMed.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695 CrossRef.
- See for instance the case of the prototypical Kubas complex in: J. Eckert, G. J. Kubas and A. J. Dianoux, J. Chem. Phys., 1988, 88, 466 CrossRef CAS PubMed.
- M. J. Gillan, J. Phys. C: Solid State Phys., 1987, 20, 3621 CrossRef.
- E. Wigner, Phys. Rev., 1932, 40, 749 CrossRef CAS.
- R. P. Bell, Trans. Faraday Soc., 1959, 55, 1 RSC.
- R. F. W. Bader, Atoms in Molecules, Oxford University Press, Oxford, 1990 Search PubMed.
- M. A. Blanco, A. Martín Pendás and E. Francisco, J. Chem. Theory Comput., 2005, 1, 1096 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS PubMed.
- D. Tiana, E. Francisco, M. A. Blanco and A. Martin Pendas, J. Phys. Chem. A, 2009, 113, 7963 CrossRef CAS PubMed.
- R. F. W. Bader and M. E. Stephens, J. Am. Chem. Soc., 1975, 97, 7391 CrossRef CAS.
- The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design, ed. C. F. Matta and R. J. Boyd, John Wiley and Sons, 2007 Search PubMed.
- Y.-G. Wang and N. H. Werstiuk, J. Comput. Chem., 2003, 24, 379 CrossRef CAS PubMed.
- R. Ponec, G. Yuzhakov and R. Carbó-Dorca, J. Comput. Chem., 2003, 24, 1829 CrossRef CAS PubMed.
- D. Tiana, E. Francisco, M. A. Blanco, P. Macchi, A. Sironi and A. M. Pendas, Phys. Chem. Chem. Phys., 2011, 13, 5068 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt02475c |
| This journal is © The Royal Society of Chemistry 2015 |