
Enhanced activity of H2O2-treated copper(II) oxide nanostructures for the electrochemical evolution of oxygen†
Albertus D.
Handoko‡
a,
Suzi
Deng‡
b,
Yilin
Deng‡
a,
Andy Wing Fai
Cheng
a,
Kuang Wen
Chan
a,
Hui Ru
Tan
c,
Yanlin
Pan
a,
Eng Soon
Tok
b,
Chorng Haur
Sow
b and
Boon Siang
Yeo
*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543 Singapore. E-mail: chmyeos@nus.edu.sg
bDepartment of Physics, National University of Singapore, 2 Science Drive 3, 117542 Singapore
cInstitute of Materials Research and Engineering, A*STAR (Agency for Science, Technology and Research), 3 Research Link, 117602 Singapore
First published on 13th August 2015
Abstract
The successful design and synthesis of earth-abundant and efficient catalysts for the oxygen evolution reaction (OER) will be a major step forward towards the use of electrochemical water splitting as an environmentally-friendly process for producing H2 fuel. Due to their poor activity, copper-based materials have not been considered apt for catalysing OER. In this work, we demonstrate that unique copper(II) oxide nanostructures obtained via hydrothermal synthesis and subsequent hydrogen peroxide treatment exhibit unusually high and sustainable OER activity. In 0.1 M KOH electrolyte, the CuO nanostructures catalyse OER with current densities of 2.6–3.4 mA cm−2 at 1.75 V (vs. RHE). The calculated turnover frequency (per Cu site) of ~2 × 10−3 s−1 for O2 production is markedly higher than that of high-surface area electrodeposited Cu metal nanoparticles by 40–68 times. The OER activity of the CuO nanostructures is also stable, approaching about half of 20% IrOx/Vulcan XC-72 after an hour-long OER. In situ Raman spectroscopy at OER-relevant potentials recorded compelling evidence that CuIII active species may be responsible for the unusual OER activity of the CuO nanostructures, as indicated by its signature vibration at 603 cm−1. This hitherto unobserved peak is assigned, with the aid of the model compound NaCuIIIO2, to the Cu–O stretching vibration of CuIII oxide. This feature was not found on electrodeposited Cu metal, which exhibited correspondingly weaker OER activity. The enhanced catalysis of O2 evolution by the CuO nanostructures is thus attributed to not just their higher surface area, but also the higher population of CuIII active sites on their surface.
Introduction
The lack of cheap and efficient catalysts to reduce the significant overpotential required for the anodic oxygen evolution reaction (OER) is a major bottleneck in the realisation of water splitting as an environmentally friendly process to produce hydrogen gas fuel.1 This critical step occurs in alkaline electrolytes via the reaction 4OH− → 2H2O + 4e− + O2. To date, the most efficacious electrocatalysts for water oxidation are the oxides of ruthenium2 or iridium.3 However, these metals are amongst the rarest and costliest elements on earth and hence are not practical for industrial-scale applications. The OER activities of ruthenium4 and iridium oxides5 are also known to decrease over time in alkaline electrolytes. Promising OER activities have been observed using cobalt-, manganese- and nickel-based compounds.5 However, these metals have relatively high cost as they are used in the synthesis of magnetic materials.6 The toxicity of manganese and nickel also requires extra care during synthesis and handling. For these reasons, there is an intensive ongoing effort to develop earth-abundant, cheap, safe and efficient electrocatalysts for oxygen evolution.Copper is 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× more abundant and 3000× cheaper than iridium.7 It can also be facilely manipulated into various Cu and Cu oxide structures via hydrothermal synthesis8 and electrodeposition.9 This makes it easier to elucidate the fundamental composition–property relationship of Cu catalysts and scale up their production for industry level usage. However, despite these advantages and their low toxicity,10 copper or copper-based compounds or complexes are rarely employed as OER catalysts. This is because copper metal, in itself, exhibits poor OER activity, which is on the same order as gold,11 or approximately two orders of magnitude lower than Co3O4.12 The dissolution of metallic copper in alkaline electrolytes13–15 has also made it an inapt anode material in water electrolysers. Copper oxides are perceived as unfavourable for electrocatalytic oxygen evolution reactions due to their lower conductivity.16 However, more recent works have demonstrated that Cu-based catalysts, especially in their nanocrystalline form, can deliver promising OER performances.17–20 Among the reported Cu-based OER catalysts, only copper–rhodium delafossite (CuRhO2) displays comparable OER performance to Co3O4.19 Unfortunately, the high Rh content in this compound renders it economically unfeasible as a replacement for iridium.
000× more abundant and 3000× cheaper than iridium.7 It can also be facilely manipulated into various Cu and Cu oxide structures via hydrothermal synthesis8 and electrodeposition.9 This makes it easier to elucidate the fundamental composition–property relationship of Cu catalysts and scale up their production for industry level usage. However, despite these advantages and their low toxicity,10 copper or copper-based compounds or complexes are rarely employed as OER catalysts. This is because copper metal, in itself, exhibits poor OER activity, which is on the same order as gold,11 or approximately two orders of magnitude lower than Co3O4.12 The dissolution of metallic copper in alkaline electrolytes13–15 has also made it an inapt anode material in water electrolysers. Copper oxides are perceived as unfavourable for electrocatalytic oxygen evolution reactions due to their lower conductivity.16 However, more recent works have demonstrated that Cu-based catalysts, especially in their nanocrystalline form, can deliver promising OER performances.17–20 Among the reported Cu-based OER catalysts, only copper–rhodium delafossite (CuRhO2) displays comparable OER performance to Co3O4.19 Unfortunately, the high Rh content in this compound renders it economically unfeasible as a replacement for iridium.
Upon electrochemical oxidation, Cu metal has been reported to oxidize progressively to Cu2O, Cu(OH)2, CuO, and CuIII oxides.21–25 Cyclic voltammograms (CV) of copper surfaces show an anodic peak at 1.5 V, prior to oxygen evolution, that has been assigned to the formation of CuIII oxides (all potentials cited in this work are with respect to the reversible hydrogen electrode, RHE).23,26 This finding suggests that highly oxidised Cu species could participate in water oxidation, akin to the role of CoIV ions in the same reaction.27,28 The stronger resistance of Cu oxides towards corrosion in alkalis, as compared to Cu metal,13,29 is also likely to contribute to the stability of Cu-based catalysts during prolonged electrochemical oxygen evolution in water electrolysers.
In this work, we demonstrate that aggregates of CuO nanostructures obtained via hydrothermal synthesis and H2O2 treatment exhibit sustainable and excellent OER activity. The OER activity of these H2O2-treated CuO nanostructures is remarkably higher than that of electrodeposited Cu metal by 40–68 times. The OER current of the CuO nanostructures is also very stable as demonstrated by chronoamperometry. Very interestingly, in situ Raman spectroscopy of the CuO nanostructures revealed the presence of a metastable surface species during OER. This species, which we assign to be CuIII oxide, is proposed to be the active site for catalysing OER. Higher-valence CuIII species have been suspected to play a role in the electrochemical oxygen evolution reaction, although spectroscopic evidence of their presence has never been found.23 The timely discovery of an OER active species on copper oxide surfaces is unprecedented, and a deeper understanding of its chemistry will inspire development of a new class of cheap, safe and earth-abundant OER catalysts.
Results and discussion
Nanostructured CuO powders were prepared by first synthesising Cu2O nanostructured precursors using a hydrothermal method, followed by H2O2 oxidation treatment (ESI† sections 1.1 and 1.2.1). The as-grown CuO nanostructures appear as 300–1000 nm elongated fragments, whose surfaces are densely decorated with ~5–10 nm CuO nanoparticles (Fig. 1a–c). X-ray diffraction analysis confirmed the chemical identity of the sample as CuO with a volume-weighted crystallite size of 12.6 nm (Fig. 1d, ESI† section 1.2.2.). This assignment is corroborated by the Auger Cu LMM peak at a kinetic energy of 917.5 eV and the Cu 2p3/2 peak at a binding energy of 934.1 eV (Fig. 1e, ESI† section 1.3.).30 The formation of CuO nanoparticles on the surface of the nanostructures is attributed to the highly oxidising properties of H2O2 that is capable of inducing the growth of small CuO particles on Cu surfaces.31 Attempts to reproduce these CuO structures by heating the nanostructured Cu2O precursors at 400 °C in air gave only 200–300 nm sized CuO particles (ESI† section 2). This implies that the chemical oxidation of Cu2O by H2O2 is unique in forming CuO nanostructures with the as-observed nanoscale features.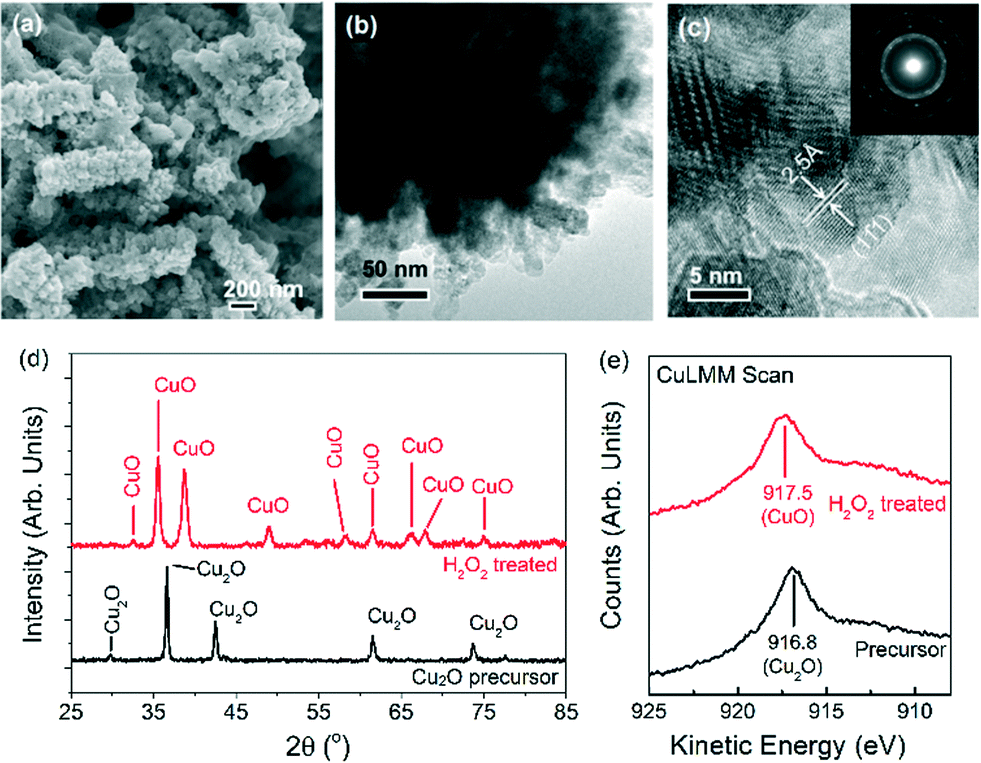 | ||
| Fig. 1 Scanning and transmission electron microscopy images of (a–c) H2O2-treated CuO nanostructures. (d) X-ray diffractograms and (e) Auger Cu LMM signals of Cu2O nanostructures before (black traces) and after H2O2 treatment (red traces). The selected area electron diffraction (SAED) ring patterns and d-spacing shown in (c) match that of CuO. | ||
The oxygen evolution activities of the CuO nanostructures and the Cu2O precursor were assessed by linear sweep voltammetry (LSV) in a 0.1 M KOH electrolyte. For comparison, measurements were also performed on commercially-available Cu2O particles and electrodeposited Cu0 nanoparticulate films (ESI† sections 1.2.3 and 3). Among the Cu catalysts, the H2O2-treated CuO nanostructures exhibited the best OER performance, with a relatively early OER onset potential of ~1.57 V (Fig. 2a). At 1.75 V, a current density of >2.5 mA cm−2 was recorded (Fig. 2a). The untreated Cu2O precursors were comparatively less active, displaying approximately half of the CuO nanostructures' anodic current. The electrodeposited Cu nanoparticles did not display any clear OER activity, and only a weak current density of ~0.15 mA cm−2 was recorded at 1.75 V.
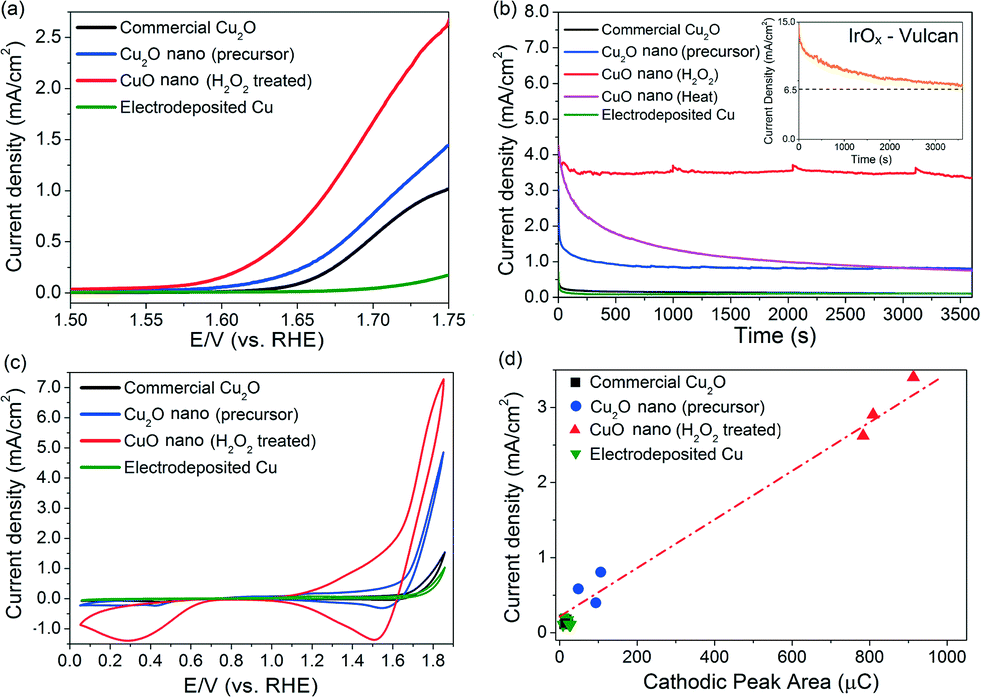 | ||
| Fig. 2 (a) Linear sweep voltammograms and (b) chronoamperograms (at 1.75 V) of copper samples. The chronoamperogram of the 20% IrOx/Vulcan XC-72 catalyst is shown in the insert in (b). (c) Cyclic voltammograms of copper samples. (d) Correlation plot between cathodic peak areas vs. chronoamperometry current densities (measured at 2200 s). A line was drawn in (d) as a visual guide. | ||
Chronoamperometric measurements at 1.75 V for 1 h were made to corroborate the as-observed electrochemical activities and to determine the stabilities of the catalysts (Fig. 2b). Comparing catalysts held under a constant potential is also a more realistic way to assess their operating performance during actual water electrolysis. The current density exhibited by the H2O2-treated CuO nanostructures remained highly stable at ~3.4 mA cm−2 after 1 hour of OER (Fig. 2b). Online mass spectrometry measurements during chronoamperometry confirmed that the gaseous product was O2 (ESI† section 4).
In contrast, the catalytic activity of CuO obtained by heat treating the Cu2O precursor was relatively unstable and decreased by 75% to <1 mA cm−2 after 30 minutes of OER (Fig. 2b). For further reference, we measured the benchmark 20% IrOx/Vulcan XC-72 catalyst under the same electrochemical conditions (insert in Fig. 2b). While its current density is initially ≥2× higher than the H2O2-treated CuO nanostructures, its performance deteriorates rather quickly. This phenomenon, which has been observed in previous OER studies utilising iridium, can be attributed to either carbon corrosion or the dissolution of IrOx to water-soluble IrVI.5
A conservative estimate of the turnover frequency (TOF) of O2 evolution at 1.75 V (η = 520 mV) by the H2O2-treated CuO nanostructures is 2.4 × 10−3 to 2.9 × 10−3 s−1 (assuming all Cu atoms participate in the reaction, ESI† section 5). This value is 4 to 7× higher than that of the as-grown Cu2O nanostructures (4.3 × 10−4 to 6.0 × 10−4 s−1), and 40 to 68× higher than that of the electrodeposited Cu metal (4.3 × 10−5 to 6.0 × 10−5 s−1). This is also approximately two orders of magnitude higher than the values (~10−5 s−1) exhibited by Cu or Cu2O nanoparticles in earlier works.17,18 Furthermore, the TOF value of our CuO nanostructures compares favourably to the TOFs of other reported OER catalysts including CaMn2O5,32 nano-β-MnO2,33 Co3O434,35 and cobalt phosphate.36 Thus, a simple hydrothermal synthesis and a subsequent oxidation process using H2O2 have produced CuO nanostructures that have an OER activity comparable to or even higher than that of many other cobalt- and manganese-based compounds (using the observed current densities and calculated TOFs at fixed overpotentials as figures of merit, Table S6†).17,18,32–36 This is highly significant since manganese and cobalt compounds are among the most promising substitutes for Ir and Ru oxides for electrochemical oxygen evolution.37 We also found that metallic Cu discs oxidised by our H2O2 treatment show improvements in their OER activities. This will facilitate the larger scale use of Cu-based OER catalysts (ESI† section 6).
The surface roughness of the Cu catalysts varied within a factor of three (ESI† section 7). Hence, the enhanced OER activity exhibited by the H2O2-treated CuO nanostructures cannot be totally attributed to the differences in surface area. We hypothesised that their efficacy could have been underpinned by their unique chemical composition during OER. Cyclic voltammetry scans of the catalysts were thus performed in a 0.1 M KOH electrolyte (Fig. 2c) to investigate the cause. A significant cathodic peak at 1.5–1.6 V was found in the CV scans of the copper and copper oxide samples. The cathodic peaks observed at this potential regime have been attributed by Miller, Abd el Haleem and other workers to the reduction of CuIII species.21–23 Interestingly, we found that the amount of charges beneath the cathodic peak of each catalyst is proportional to their OER current density measured during chronoamperometry (Fig. 2d). This correlation suggests that the OER activity of the Cu catalysts is closely related to the surface population of CuIII. To rule out the possibility that the observed cathodic peak at 1.5–1.6 V was from the re-deposition of dissolved Cu ions, repeated LSV scans of the H2O2-treated CuO nanostructures were made for 2 h (ESI† section 8). The measured currents were found to be highly stable.
To elucidate the presence and role of catalytically active Cu species in OER, we performed in situ Raman spectroscopy. For these measurements, Cu2O precursors and H2O2-treated CuO nanostructures were grown on Cu disc substrates (ESI† section 9). The in situ Raman spectra and chronoamperograms of the electrodes were then simultaneously measured in a 0.1 M KOH electrolyte (Fig. 3).
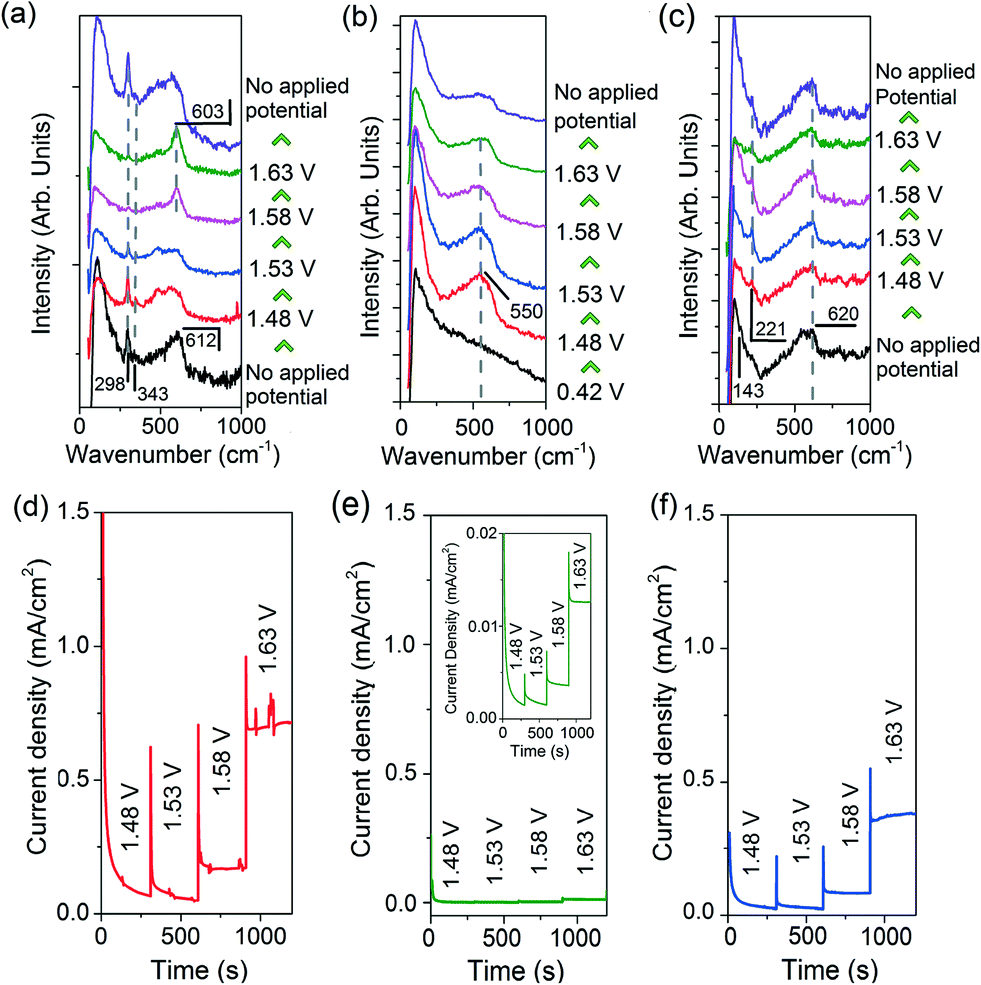 | ||
| Fig. 3 In situ Raman spectra of the (a) H2O2-treated CuO nanostructures, (b) Cu electrode, and (c) Cu2O precursor collected at different applied potentials. The corresponding chronoamperograms are presented below their Raman spectra in (d–f), respectively. The insert in (e) is a magnification of the chronoamperogram of Cu. | ||
The Raman spectra of the H2O2-treated CuO nanostructures are presented in Fig. 3a. Without an applied potential, characteristic CuO bands at 298, 343 and 612 cm−1 (broad) were observed.38 Very interestingly, as the film was subjected to more anodic electrochemical potentials, a sharp peak at 603 cm−1 appears at the expense of the CuO peaks. This feature dominates the spectrum at 1.63 V where OER occurs. However, it proved to be transient as it disappeared as soon as the applied potential was removed (with the restoration of the CuO peaks, Fig. 3a). These observations demonstrate that the band at 603 cm−1 originates from CuO species, and its generation and stabilisation are dependent on the applied potential. To the best of our knowledge, no Raman peak at 603 cm−1 has been observed on copper or copper oxide surfaces with or without electrochemical bias. This peak cannot also be assigned to CuO, Cu(OH)2, Cu2O, or adventitious impurities (ESI† section 9).38–42 Instead, consistent with thermodynamics and with cyclic voltammetry data showing that CuIII oxide is formed at oxygen evolving potentials, we ascribed the 603 cm−1 band to the Cu–O stretching vibration of CuIII oxide.22–24 This assignment is supported by the occurrence of a Raman peak at 603 cm−1 belonging to the CuIII-containing compound, NaCuO2 (ESI† section 10). Furthermore, the CuIII–O stretching vibration of the bis-(μ-oxo)-CuIII2 core of copper complexes is at ~608 cm−1.43
In contrast, only a broad band centred at ~550 cm−1 was observed in the Raman spectra of metallic Cu during OER (Fig. 3b). This feature could be attributed to the formation of amorphous Cu oxides. No trace of CuO was detected even at 1.63 V. This is consistent with a previous report on the anodisation of metallic Cu in a basic electrolyte using stepped voltages in transpassive regions where no CuO could be formed.44 For the Cu2O nanostructures, a broad band at 540–620 cm−1 that could originate from a mixture of amorphous Cu oxides, Cu2O and CuIII oxide was recorded, alongside the Cu2O characteristic peak at 221 cm−1 (Fig. 3c).
The widely accepted reaction mechanism for OER on oxide surfaces consists of a sequence of four consecutive one-electron oxidations (Fig. 4).45,46 The oxygen coupling step produces a –OOH* intermediate, which then dissociates to produce O2. Based on this reaction scheme, it is possible that a higher oxidation state of CuIII could enhance the electrophilicity of the adsorbed O, and hence promote the formation of O–OH*via nucleophilic attack from an incoming OH−. The CuIII is also likely to facilitate the deprotonation of –OOH*, through electron-withdrawing inductive effects, to give molecular O2.11 Our proposition is supported by previous studies which suggest that high-valent metal cations such as FeVI are the active sites for catalysing OER.47 The presence of the intense and sharp Raman peak at 603 cm−1 in the spectra of H2O2-treated CuO nanostructures demonstrates that the OER-active CuIII species is able to form and accumulate easily on the surface (Fig. 3a). Metallic Cu, on the other hand, could not be oxidised directly to CuO by the application of a single anodic potential step.44 Thus, CuIII species could not be generated readily on its surface at high potentials, leading to its weaker OER performance.
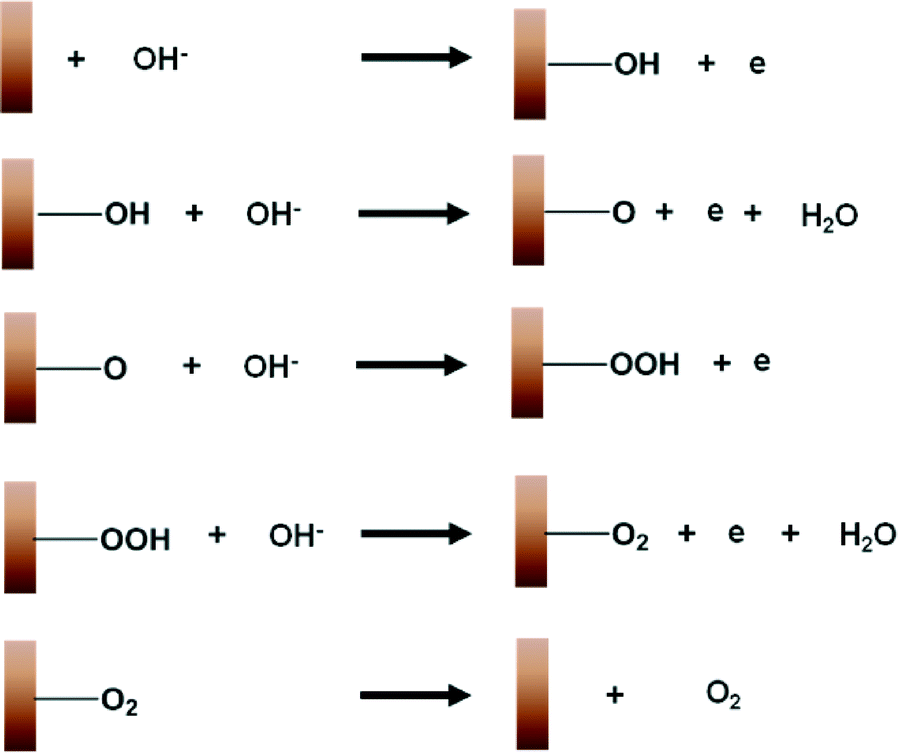 | ||
| Fig. 4 A schematic diagram illustrating the mechanism for the electrochemical oxygen evolution reaction on copper. | ||
A previous OER study using Co, CoO and Co3O4 nanoparticles has shown that their overall activity is independent of their initial oxidation state.28 This is because all three catalysts oxidise to a common species, believed to be CoIV, as oxygen evolving potentials are reached. This work, however, demonstrates that the initial oxidation state of a Cu surface can significantly influence the type of catalytic species formed during OER, and hence its overall O2 production. The use of H2O2 as an oxidant serves to remove the amorphous carbon formed during synthesis,48 which can obscure the active sites on the CuO nanostructures and inhibit their catalytic activities.49 Another important contribution of this work is that expensive metallic dopants such as Rh and Au were not used to enhance the O2-evolving activity of the CuO nanostructures.11,19 Finally, we do note that the performance of our CuO nanostructures still trails that of Ru and Ir oxides. One reason for this could be their relatively low conductivity, which could be addressed by dispersing them in a conductive carbon support or by further reducing their dimensions. Our timely discovery of the active oxidation states of copper during OER is a critical knowledge for other workers to consider when engineering Cu-based compounds with activities that could eventually match those of expensive state-of-the-art catalysts.
Conclusions
In this work, we demonstrate that a simple hydrothermal synthesis and subsequent H2O2 oxidation treatment yield CuO nanostructures that exhibit sustainable and excellent OER activity. The OER activity of these H2O2-treated CuO nanostructures is enhanced such that their activity is 40–68× higher than that of electrodeposited Cu metal, and about half of the 20% IrOx/Vulcan XC-72 commercial OER catalyst. Another notable finding is that the H2O2-treated CuO is catalytically robust as demonstrated by its stable OER current during chronoamperometry. In situ Raman spectroscopy of the Cu catalysts during OER revealed the presence of a transient species that was more readily formed on the surface of H2O2-treated CuO nanostructures than Cu2O nanostructures or metallic Cu. This species, which we assigned to be CuIII oxide, is proposed as an active site for catalysing OER. The discovery of an OER active species on copper oxide surfaces is unprecedented. The knowledge gained in this work will help us to engineer and enhance the OER catalytic activity of Cu-based compounds to a level that is competitive with other more established catalysts.Experimental section
Synthesis
Nanostructured CuO powders were prepared by H2O2 oxidation treatment of Cu2O nanostructured precursors. The latter were grown hydrothermally from an aqueous solution of CuII acetate (≥98%, Sigma Aldrich), graphene oxide and o-anisidine (>99%, Sigma Aldrich) in a Teflon-lined autoclave.50 Oxidation treatment was performed by immersing 10 mg of the nanostructured Cu2O precursors in 1 ml of H2O2 (30 vol%, Scharlau) for 1 hour at 60 °C. This was followed by rinsing 5 times with deionised water and drying overnight at 60 °C. This process was performed five times. Cu2O precursor films for spectroscopy analyses were grown directly on polished 10 mm diameter copper discs (99.99+%, Goodfellow Cambridge Limited) during the hydrothermal process described above. A CuO nanostructured film was obtained by immersing Cu2O precursor films in ~5 ml of H2O2 inside a Teflon-lined autoclave.Preparation of catalyst inks
Catalyst inks were prepared by mixing 2 mg of the as-synthesized catalyst, 0.1 ml of isopropanol (analytical reagent, Fisher), 0.4 ml of deionised H2O (≥18.2 MΩ cm, Barnstead) and 60 μL of Nafion 117 solution (Sigma Aldrich). The suspensions were sonicated at 20 °C for 30 minutes to ensure even mixing. 5 μL of the catalyst ink was then drop-cast onto polished glassy carbon electrodes (3 mm diameter, CH Instruments). 17.9 μg of the catalyst was typically loaded on each electrode. Catalyst inks of 20% Ir-Vulcan XC-72 (Premetek) and Cu2O (99.9%, Alfa Aesar) were similarly prepared.Electrochemical measurements
The electrochemical measurements were performed in a glass H-cell with a 3-electrode configuration. A Ag/AgCl (saturated KCl gel) electrode (Pine Instruments) and a platinum wire were used as reference and counter electrodes, respectively. Aqueous 0.1 M KOH was used as electrolyte (99.98%, Alfa Aesar). A Gamry Reference 600 potentiostat was used for controlling and measuring the potentials/currents. No iR compensation was applied to the measurements because of the low net current and conductive electrolyte. Cyclic and linear sweep voltammetry measurements were conducted at scan rates of 50 and 1 mV s−1, respectively. All electrochemical measurements were conducted at 25 °C. All potentials cited in this work are with respect to the reversible hydrogen electrode (RHE). No trace of Cl− could be detected in the electrolyte before and after electrochemical measurements (using a Cl− ion selective electrode).Materials characterisation
Powder X-ray diffraction was carried out using a Siemens 5005 (Cu Kα radiation with a graphite monochromator) while film samples were scanned using a Bruker Discover D8 GADDS with a general area detector (microfocused Cu Kα radiation). The particle morphology was examined using a scanning electron microscope (JEOL-6701F, secondary electron mode, 5 kV, 10 mA) and a transmission electron microscope (TEM, JEOL JEM-3010). XPS and Auger spectroscopy analyses were carried out using a VG ESCA 220i-XL Imaging XPS. These measurements used monochromatic Al Kα X-ray (hν = 1486.6 eV) with a photoelectron collection angle of 90° with respect to the surface plane.The in situ Raman spectra were collected using an epi-illumination Raman microscope (Horiba Jobin Yvon). The electrochemical cell, designed and constructed in-house, was based on a round Teflon-lined dish.51 Hg/HgO in 0.1 M KOH (CH Instrument) and Pt wire were used as reference and counter electrodes, respectively. The electrochemical potentials were applied by using an Autolab PGSTAT potentiostat from 1.48 V to 1.63 V in 0.05 V increments. Each potential was held constant while the Raman spectrum (acquisition time: 10 seconds × 3 cycles) was acquired. A He–Ne laser (632.8 nm, CVI Melles Griot) was used as the excitation source.
Acknowledgements
This work is primarily supported by a start-up grant (R-143-000-515-133) and an academic research fund (R-143-000-587-112) from the National University of Singapore. We also acknowledge the financial support from the National Research Foundation and Economic Development Board (SPORE, COY-15-EWI-RCFSA/N197-1). SCH and DS acknowledge the support from Singapore National Research Foundation under CRP Award no. NRF-CRP-4-2008-03. ADH is grateful to a research fellowship from the Singapore-Peking Oxford Research Enterprise (R-706-000-100-414).References
- Y. Surendranath, D. K. Bediako and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15617–15621 CrossRef CAS PubMed.
- H. Ma, C. Liu, J. Liao, Y. Su, X. Xue and W. Xing, J. Mol. Catal. A: Chem., 2006, 247, 7–13 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- L. D. Burke, O. J. Murphy, J. F. O'Neill and S. Venkatesan, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 1659–1671 RSC.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- S. Roberts and G. Gunn, in Critical Metals Handbook, ed. G. Gunn, Wiley, West Sussex, UK, ch. 6, 2013 Search PubMed.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, Taylor & Francis, 95th Edition, 2014 Search PubMed.
- Y. Sui, W. Fu, H. Yang, Y. Zeng, Y. Zhang, Q. Zhao, Y. Li, X. Zhou, Y. Leng, M. Li and G. Zou, Cryst. Growth Des., 2009, 10, 99–108 Search PubMed.
- M. J. Siegfried and K.-S. Choi, J. Am. Chem. Soc., 2006, 128, 10356–10357 CrossRef CAS PubMed.
- M. Csuros and C. Csuros, Environmental Sampling and Analysis for Metals, Taylor & Francis, 2002 Search PubMed.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- L. Brossard and B. Marquis, Int. J. Hydrogen Energy, 1994, 19, 231–237 CrossRef CAS.
- M. Biton, G. Salitra, D. Aurbach, P. Mishkov and D. Ilzycer, J. Electrochem. Soc., 2006, 153, B555–B565 CrossRef CAS.
- D. W. Shoesmith, S. Sunder, M. G. Bailey, G. J. Wallace and F. W. Stanchell, J. Electroanal. Chem. Interfacial Electrochem., 1983, 143, 153–165 CrossRef CAS.
- J. S. Halliday, Trans. Faraday Soc., 1954, 50, 171–178 RSC.
- B. Marsan, N. Fradette and G. Beaudoin, J. Electrochem. Soc., 1992, 139, 1889–1896 CrossRef.
- B. Kumar, S. Saha, M. Basu and A. K. Ganguli, J. Mater. Chem. A, 2013, 1, 4728–4735 CAS.
- B. Kumar, S. Saha, A. Ganguly and A. K. Ganguli, RSC Adv., 2014, 4, 12043–12049 RSC.
- R. Hinogami, K. Toyoda, M. Aizawa, S. Yoshii, T. Kawasaki and H. Gyoten, Electrochem. Commun., 2013, 35, 142–145 CrossRef CAS.
- J. Wang, K. Wang, F.-B. Wang and X.-H. Xia, Nat. Commun., 2014, 5, 5285 CrossRef CAS PubMed.
- J. Ambrose, R. G. Barradas and D. W. Shoesmith, J. Electroanal. Chem. Interfacial Electrochem., 1973, 47, 47–64 CrossRef CAS.
- B. Miller, J. Electrochem. Soc., 1969, 116, 1675–1680 CrossRef CAS.
- S. M. Abd el Haleem and B. G. Ateya, J. Electroanal. Chem. Interfacial Electrochem., 1981, 117, 309–319 CrossRef CAS.
- A. M. S. El Din and F. M. A. El Wahab, Electrochim. Acta, 1964, 9, 113–121 CrossRef CAS.
- M. Pourbaix, in Lectures on Electrochemical Corrosion, ed. R. W. Staehle, Springer US, New York, 1st edn., 1973, ch. 7, pp. 297–314, DOI:10.1007/978-1-4684-1806-4_7.
- D. T. Schwartz and R. H. Muller, Surf. Sci., 1991, 248, 349–358 CrossRef CAS.
- M. E. G. Lyons and M. P. Brandon, Int. J. Electrochem. Sci., 2008, 3, 1425–1462 CAS.
- N. H. Chou, P. N. Ross, A. T. Bell and T. D. Tilley, ChemSusChem, 2011, 4, 1566–1569 CrossRef CAS PubMed.
- J. P. Lorimer, T. J. Mason, M. Plattes and D. J. Walton, J. Electroanal. Chem., 2004, 568, 379–390 CrossRef CAS.
- J. F. Moulder and J. Chastain, Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Physical Electronics Division, Perkin–Elmer Corporation, 1992 Search PubMed.
- J.-C. Chen and W.-T. Tsai, Mater. Chem. Phys., 2004, 87, 387–393 CrossRef CAS.
- J. Kim, X. Yin, K.-C. Tsao, S. Fang and H. Yang, J. Am. Chem. Soc., 2014, 136, 14646–14649 CrossRef CAS PubMed.
- M. Fekete, R. K. Hocking, S. L. Y. Chang, C. Italiano, A. F. Patti, F. Arena and L. Spiccia, Energy Environ. Sci., 2013, 6, 2222–2232 CAS.
- C. Iwakura, A. Honji and H. Tamura, Electrochim. Acta, 1981, 26, 1319–1326 CrossRef CAS.
- P. Rasiyah and A. C. C. Tseung, J. Electrochem. Soc., 1983, 130, 365–368 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109–114 RSC.
- L. Debbichi, M. C. Marco de Lucas, J. F. Pierson and P. Krüger, J. Phys. Chem. C, 2012, 116, 10232–10237 CAS.
- D. Reyter, M. Odziemkowski, D. Bélanger and L. Roué, J. Electrochem. Soc., 2007, 154, K36–K44 CrossRef CAS.
- G. Niaura, Electrochim. Acta, 2000, 45, 3507–3519 CrossRef CAS.
- S. T. Mayer and R. H. Muller, J. Electrochem. Soc., 1992, 139, 426–434 CrossRef.
- H. Y. H. Chan, C. G. Takoudis and M. J. Weaver, J. Phys. Chem. B, 1998, 103, 357–365 CrossRef.
- M. J. Henson, M. A. Vance, C. X. Zhang, H.-C. Liang, K. D. Karlin and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 5186–5192 CrossRef CAS PubMed.
- N. Fredj and T. D. Burleigh, J. Electrochem. Soc., 2011, 158, C104–C110 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- M. E. G. Lyons and M. P. Brandon, J. Electroanal. Chem., 2010, 641, 119–130 CrossRef CAS.
- Y. Feng, G. Zhou, G. Wang, M. Qu and Z. Yu, Chem. Phys. Lett., 2003, 375, 645–648 CrossRef CAS.
- Y. C. Kimmel, D. V. Esposito, R. W. Birkmire and J. G. Chen, Int. J. Hydrogen Energy, 2012, 37, 3019–3024 CrossRef CAS.
- S. Deng, V. Tjoa, H. M. Fan, H. R. Tan, D. C. Sayle, M. Olivo, S. Mhaisalkar, J. Wei and C. H. Sow, J. Am. Chem. Soc., 2012, 134, 4905–4917 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394–8400 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: (1) Detailed characterisation of catalysts described in this manuscript, (2) online mass spectrometry data, (3) calculation of turnover frequencies, (4) surface area estimation, (5) stability assessments, (6) details of Raman measurements and peak assignments, and (7) synthesis and characterisation of CuIII-containing NaCuO2. See DOI: 10.1039/c5cy00861a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |