
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAryl-substituted organomolybdenum(II) complexes as olefin epoxidation catalysts†
Lilian
Graser‡
a,
Robert M.
Reich‡
a,
Mirza
Cokoja
b,
Alexander
Pöthig
c and
Fritz E.
Kühn
*ad
aMolecular Catalysis, Department of Chemistry and Catalysis Research Center, Technische Universität München, Lichtenbergstr. 4, D-85747 Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de; Fax: + 49 89 289 13473; Tel: +49 89 289 13096
bDepartment of Chemistry, Technische Universität München, Lichtenbergstr. 4, D-85747 Garching bei München, Germany
cCatalysis Research Center, Technische Universität München, Ernst Otto Fischer-Str. 1, D-85747 Garching bei München, Germany
dChair of Inorganic Chemistry, Department of Chemistry and Catalysis Research Center, Technische Universität München, Lichtenbergstr. 4, D-85747 Garching bei München, Germany
First published on 13th May 2015
Abstract
The epoxidation of selected olefins with a benzyl-substituted organomolybdenum complex and its fluorinated counterpart is described. With hexafluorobenzene (HFB) as solvent, turnover frequencies (TOFs) of >15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 h−1 are achieved in the epoxidation of cyclooctene with tert-butyl hydroperoxide (TBHP) as the oxidant. The fluorinated complex, [CpMo(CO)3BzF5], proved to be superior to the non-fluorinated derivative in activity and selectivity with a variety of substrates. This can be explained via X-ray crystallography analysis and with the help of density functional theory (DFT) calculations. Besides, both compounds were applied in two-phase catalytic reactions. Recycling for multiple catalytic runs is achieved without a significant loss of activity.
500 h−1 are achieved in the epoxidation of cyclooctene with tert-butyl hydroperoxide (TBHP) as the oxidant. The fluorinated complex, [CpMo(CO)3BzF5], proved to be superior to the non-fluorinated derivative in activity and selectivity with a variety of substrates. This can be explained via X-ray crystallography analysis and with the help of density functional theory (DFT) calculations. Besides, both compounds were applied in two-phase catalytic reactions. Recycling for multiple catalytic runs is achieved without a significant loss of activity.
Introduction
Epoxides are important intermediates for a variety of chemical products, e.g. food additives, cosmetics, pharmaceuticals and polymers.1–3 The non-catalytic chlorohydrin process based on the process developed by Wurtz in 18591 and the more environmentally friendly, catalytic Halcon–ARCO process, established in the 1960s, are examples of large scale epoxide production methods.2,3 The Halcon–ARCO process utilizes organometallic compounds, such as [Mo(CO)6], as pre-catalysts that are, according to current textbook knowledge, oxidized to catalytically active oxo-peroxo species. Subsequently many homogeneous and heterogeneous molybdenum catalysts for the epoxidation of olefins have been synthesized and examined.4–13 Among other compound classes, molybdenum complexes featuring an η5-coordinated cyclopentadienyl (Cp) ligand have attracted interest as epoxidation catalysts.Originally, high oxidation state compounds such as [(η5-C5R5)MoO2Cl] (R = H, Me, Bz) were targeted,14 but it turned out that the carbonyl precursor complexes of the type [(η5-C5R5)Mo(CO)3Cl] (R = H, Me, Bz) are usually more easily stored due to their higher stability. They also can be used as catalyst precursors for olefin epoxidation.14–19 As in the Halcon–ARCO process, tert-butyl hydroperoxide (TBHP) proved to be the oxidant of choice, readily oxidizing the carbonyl compounds to active catalyst species and, usually more slowly, olefins to epoxides catalysed by the organometallic species created before.
When alkyl groups R' are attached to the molybdenum centre, a great variability of molecular catalysts is available, e.g. ansa-bridged complexes, which was found to be the benchmark system concerning the activity in the epoxidation of cyclooctene for some time.20 Very recently, the activity of the fluorinated organomolybdenum complex [CpMo(CO)3CF3] was compared to its non-fluorinated counterpart [CpMo(CO)3CH3].21,22 This examination revealed that the substitution of a methyl group in the tricarbonyl compound [CpMo(CO)3CH3] with a fluorinated methyl group leads to a distinct increase of Lewis acidity on the metal center. However, in this case the oxidation of the carbonyl precursor to the active species is very slow, so that only the application of an already oxidized catalyst showing a higher activity for olefin epoxidation was illustrated.22
To obtain a more profound insight into the influence of the electronic environment on the catalytic activity of MoCp compounds, two new aryl-substituted complexes, [CpMo(CO)3Bz] and [CpMo(CO)3BzF5], have been synthesized. DFT calculations were performed and their catalytic properties in olefin epoxidation were examined. The present work aims to compare and investigate the influence of fluorination on the Lewis acidity of the metal centre and the activity in catalysis.
Results and discussion
Synthesis, IR spectroscopy and X-ray crystallography
The reaction of Na[CpMo(CO)3] with benzyl bromide and pentafluorobenzyl bromide, respectively, leads to air- and moisture-stable compounds 1 and 2 in yields of ca. 80% under ambient conditions (Scheme 1).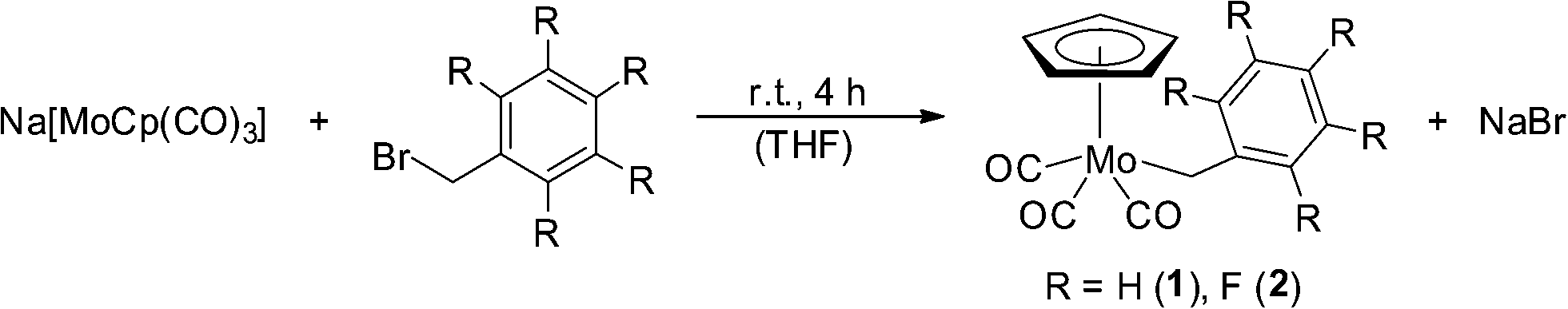 | ||
| Scheme 1 Synthesis of the investigated organomolybdenum complexes 1 and 2. | ||
The formation of the new compounds was confirmed by 1H-NMR with a shift of the Cp signals from 3.84 (THF-d8) to 5.20 and 5.51 ppm (CDCl3), respectively. In the 13C-NMR spectra of both compounds, only two carbonyl signals can be observed at 239.87 ppm and 228.27 ppm for complex 1 and at 238.98 ppm and 227.57 ppm for compound 2.
IR spectroscopic examinations were performed on both complexes, since the vibrational bands of the carbonyl ligand represent a sensitive tool for the determination of the electronic properties at the metal centre.
The infrared spectra of [CpMo(CO)3R]-type complexes are well documented in literature.23–26 The frequencies of the cyclopentadienyl ligand are virtually independent of the ligand R bound to the metal centre. The CO stretching vibrations are, however, very sensitive to the changes of the ligand group R. The characteristic stretching bonds of 1 and 2 are summarized in Table 1. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O symmetric stretching frequency of the CO bond of compound 2 is found to be 20 wavenumbers higher than that of compound 1.
O symmetric stretching frequency of the CO bond of compound 2 is found to be 20 wavenumbers higher than that of compound 1.
| 1 | 2 | |
|---|---|---|
| Stretching frequencies [cm−1] | ||
| CO sym. stretch | 2001 | 2022 |
| CO asym. stretch | 1917 | 1922 |
| CO asym. stretch | 1904 | 1914 |
| Force constants [N cm−1] | ||
| K1 (CO) | 16.18 | 16.52 |
| K2 (CO) | 14.85 | 14.92 |
The observed differences can be explained by the lower electron density at the metal centre in the case of compound 2, generating a weaker metal–ligand interaction. This induces strengthening of the CO bond – reflected in shorter bond distances.
There is a slight difference in the Mo–R bond lengths (2.386(2) Å in 1vs. 2.356(2) Å in 2), reflecting the changed electron density at the metal atom in the solid state. The Mo–R bond length is slightly longer for both compounds than that observed for the previously reported methyl derivative (2.326(3) Å).27 The fluorination of the benzyl moiety does not lead to such a pronounced change of the Mo–R bond distance as observed in the cases of the methyl and the CF3 derivative (2.234(3) Å).21 This can be considered as an indication of a more similar reactivity of compounds 1 and 2 with respect to decarbonylation.
Furthermore, the bond distances between Mo and the carbonyl ligands are very similar for both compounds (see Fig. 1 and 2 and ESI†).
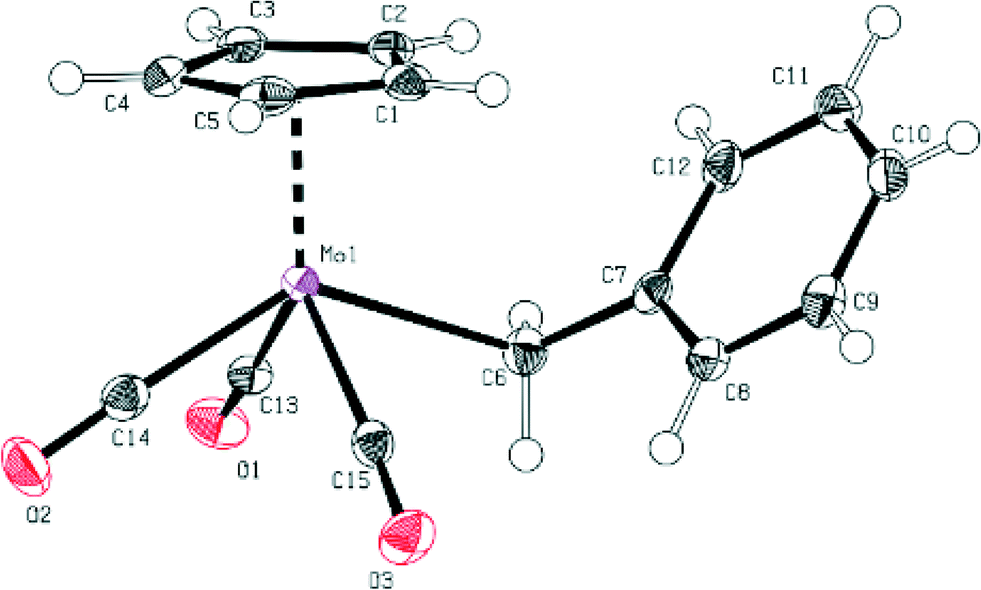 | ||
| Fig. 1 ORTEP-style presentation of the molecular structure of compound 1. Ellipsoids are shown at a 50% probability level. Selected bond lengths (Å) and angles (°): Mo1–C13 1.991(2), Mo1–C14 1.988(2), Mo1–C15 1.987(2), Mo1–C6 2.386(2), C15–O1 1.147(3), C14–O2 1.142(3), C13–O3 1.142(3), C13–Mo1–C14 77.17(9), C14–Mo1–C15 75.42(9), C6–Mo1–C13 74.13(9), C6–Mo1–C15 75.51(8), Mo1–C13–O1 177.2(2), Mo1–C14–O2 178.2(2), Mo1–C15–O3 176.3(2). | ||
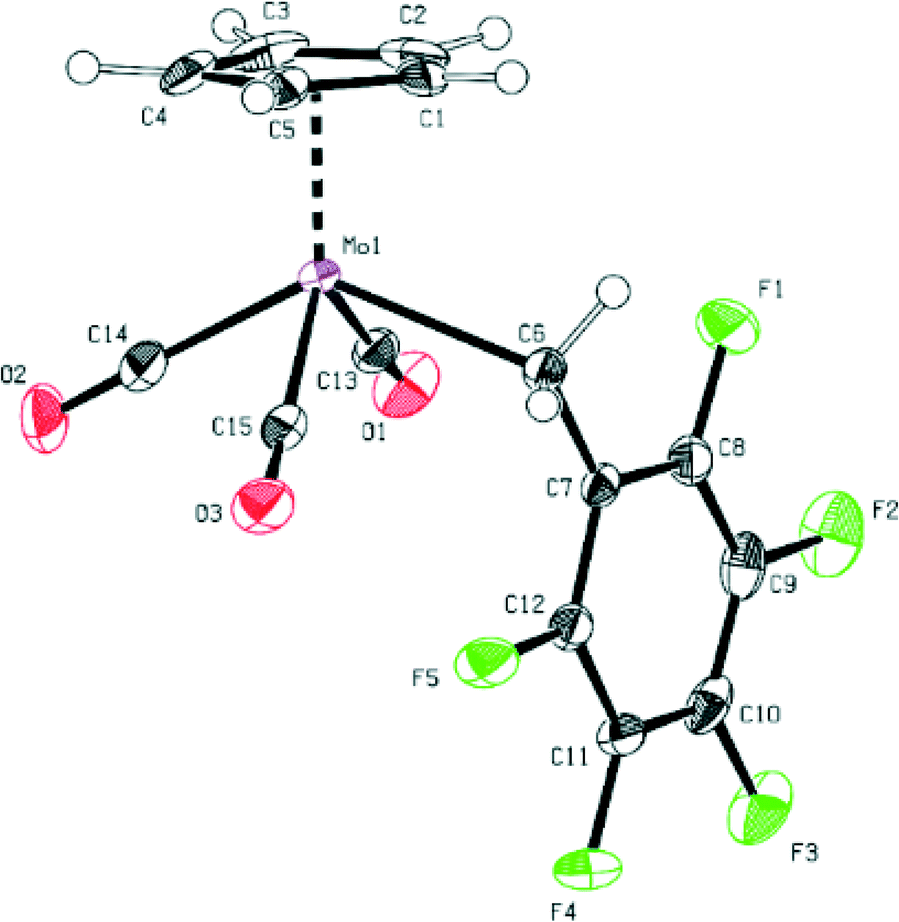 | ||
| Fig. 2 ORTEP-style presentation of the molecular structure of compound 2. Ellipsoids are shown at a 50% probability level. Selected bond lengths (Å) and angles (°): Mo1–C6 2.356(2), Mo1–C13 1.979(2), Mo1–C14 1.988(2), Mo1–C15 2.017(2), C15–O1 1.145(2), C14–O2 1.143(2), C13–O3 1.140(2), C13–Mo1–C14 76.53(7), C14–Mo1–C15 78.17(7), C6–Mo1–C13 78.40(6), C6–Mo1–C15 75.14(6), Mo1–C13–O1 175.1(2), Mo1–C14–O2 178.1(2), Mo1–C15–O3 175.6(2). | ||
DFT examinations
To gain more insight into the electronic properties at the metal centre, DFT calculations of compounds [CpMo(CO)3Bz] (1) and [CpMo(CO)3BzF5] (2) were performed. They reveal that the Lewis acidity of compound 2 at Mo is higher than that of compound 1 (for further details see ESI† S1.1). As 95Mo-NMR is also an indicator of electron deficiency,28 the shifts of 1 (−1588 ppm) and 2 (−1513 ppm) indicate that the metal center of compound 2 is indeed more electron deficient. It had been possible to ascribe the slower decarbonylation, higher stability and shorter Mo–R bond length of the fluorinated compound [CpMo(CO)3CF3] in comparison with those of [CpMo(CO)3CH3] to the differences of the highest occupied molecular orbitals (HOMOs).21 These differences are not observed for compounds 1 and 2 (Fig. 3). Especially, both HOMOs show an Mo–CH2R overlap, which explains the observed similar Mo–R bond lengths for 1 and 2.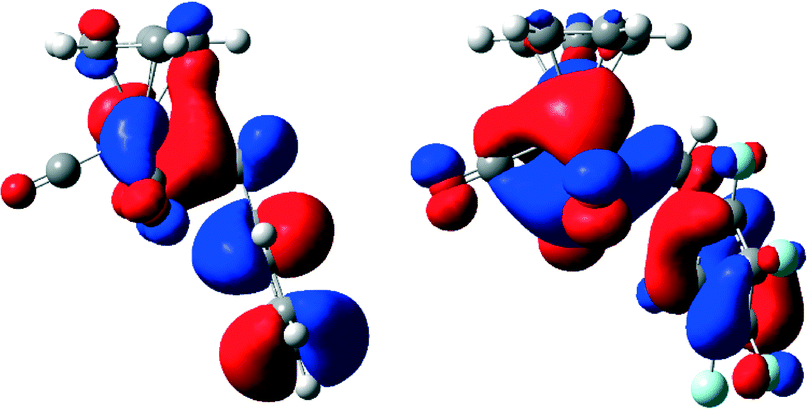 | ||
| Fig. 3 Comparison of the HOMOs of compounds [CpMo(CO)3R] with R = Bz (1) (left), BzF (2) (right) in the gas phase (B3LYP/6-31+G**(d,p) level of theory; for details see the ESI†). | ||
Therefore a similar oxidative decarbonylation rate is to be expected. This is supported by the reaction of compounds 1 and 2 with excess TBHP. After the addition of TBHP, the formation of a dioxo and an oxo-peroxo species (correlating with complete decarbonylation) occurs within 13 min for 1 and 5 min for 2.29 The oxidative decarbonylation was monitored via1H NMR spectroscopy. These observations are in accord with several computational30,31 and experimental32–35 studies which deal with the formation of dioxo and oxo-peroxo species with TBHP as the oxidant. After decarbonylation the dioxo species forms, which can be transformed to the oxo-peroxo species with an excess of oxidant. Both species can take part in the catalytic epoxidation of olefins with TBHP as the oxidant of choice. Detailed mechanistic studies to further examine the active species are under way.29
This difference in the rate of decarbonylation is quite small in comparison with the methyl (20 min) and trifluoromethyl derivatives (>60 min).22
Epoxidation catalysis
Both complexes are well soluble in dichloromethane (DCM), acetonitrile (CH3CN), chloroform (CHCl3), toluene and benzene. For all kinetic experiments and for all applied substrates, one homogeneous phase was obtained.The monomeric complexes 1 and 2 were applied as catalysts for the epoxidation of selected terminal and internal alkenes using tert-butyl hydroperoxide (TBHP, 5.5 M solution in n-decane) as oxidant. To determine the optimal reaction conditions concerning the solvent, temperature, catalyst and oxidant loading, cyclooctene was chosen as substrate. The results obtained for the temperature and solvent screening are summarized in Table 2.
| Entry | Solventa | Yieldb [%] after 4 h/24 h | |||
|---|---|---|---|---|---|
| 1 | 2 | ||||
| r.t. | 55 °C | r.t. | 55 °C | ||
|
a Reaction conditions: reactions were carried out using a molar ratio of catalyst:substrate:oxidant of 1:100:200 in 0.5 mL of solvent.
b Yield determined by GC-MS.
c MeOH: methanol, HFB: hexafluorobenzene.
|
|||||
| 1 | DCM | 74/100 | 87/100 | 100/100 | 100/100 |
| 2 | CHCl3 | 66/100 | 100/100 | 73/100 | 100/100 |
| 3 | Benzene | 75/100 | 99/100 | 100/100 | 100/100 |
| 4 | Hexane | 72/100 | 86/100 | 84/100 | 100/100 |
| 5 | MeOHc | <10/<10 | 58/84 | <10/<10 | 64/87 |
| 6 | HFBc | 77/100 | 100/100 | 100/100 | 100/100 |
| 7 | [C4mim][NTf2] | 72/100 | 84/100 | 79/100 | 100/100 |
| 8 | [C8mim][NTf2] | 74/100 | 92/100 | 86/100 | 100/100 |
A comparison of the two compounds in the catalytic cyclooctene epoxidation reveals the superiority of the fluorinated complex 2. This behaviour is in accord with the more pronounced Lewis acidity of this compound.36,37 Both compounds show the best activity in hexafluorobenzene (HFB) as solvent (Table 2, entry 6). This is in accord with the recent observations for related catalytic systems where the activity of the organomolybdenum complex [CpMo(CO)3CF3] was found to be superior when a solvent able to activate the oxidant, like hexafluoroisopropanol (HFIP), is used.22
To further investigate the difference in activity, all following investigations concerning the catalyst loading were performed in benzene and HFB.
Based on the results summarised in Table 3, it can be stated that catalyst 2 is roughly 20% more active than 1. Furthermore HFB positively influences the catalytic activity of both compounds. When examining a variety of substrates (see Table 4), it can be observed that open chain alkenes such as styrene and 1-octene are epoxidised with good to moderate efficiency. Compared to known MoCp systems, both systems are superior to previously achieved results. For example, the otherwise quite active ansa-complex [Mo(η5-C5H4(CH2-η1-CH)(CO)3)] is inferior with respect to the conversion of 1-octene (52% after 24 h).20
| Entry | Cat. | Cat. conc. [mol %] | Benzene TOF [h−1] | HFB TOF [h−1] |
|---|---|---|---|---|
|
a Reaction conditions: 55 °C, 0.5 mL of solvent, molar ratio cyclooctene:TBHP of 1:2.
|
||||
| 1 | 1 | 1 | 2960 | 4250 |
| 2 | 1 | 0.1 | 6820 | 8330 |
| 3 | 1 | 0.05 | 10430 |
11580 |
| 4 | 2 | 1 | 4300 | 6370 |
| 5 | 2 | 0.1 | 12220 |
13470 |
| 6 | 2 | 0.05 | 15540 |
17820 |
| Entry | Substratea | Catalyst | Conv.b [%] | Sel. [%] |
|---|---|---|---|---|
| a Reaction conditions: 1 mmol of substrate, 2 mmol of TBHP in decane, 0.5 mol% catalyst, 55 °C, 0.5 mL of benzene, 24 h. b Conversion and selectivity were determined via1H-NMR. | ||||
| 1 | 1-Octene | 1 | 73 | 99 |
| 2 | 2 | 77 | 99 | |
| 3 | Styrene | 1 | 57 | 43 |
| 4 | 2 | 61 | 46 | |
| 5 | Trans-β-methylstyrene | 1 | 43 | 99 |
| 6 | 2 | 54 | 99 | |
| 7 | Cis-stilbene | 1 | 52 | 76 |
| 8 | 2 | 57 | 72 | |
| 9 | Trans-stilbene | 1 | 47 | 86 |
| 10 | 2 | 55 | 94 | |
For comparison with [MoCp(CO)3CH3], room-temperature experiments were undertaken. With 38% (1) and 43% (2) conversion after 4 h of reaction time, respectively, both compounds notably surpass the conversion to epoxide of the alkyl-substituted complex (25% conversion after 4 h).22 To explore the influence of the benzyl- and the pentafluorobenzyl ligand on the product stereoselectivity, trans-β-methylstyrene was chosen as the benchmark substrate. 1H-NMR spectroscopy indicates no enantiomeric excess. As shown in Table 2, both compounds proved to be active in imidazolium-based ionic liquids (Table 2, entries 7 and 8). To scrutinize the stability of the catalytically active system and to test the recyclability of a heterogeneous system, recycling experiments for both compounds were undertaken in the ionic liquid [C8mim][NTf2] with cyclooctene as substrate (Fig. 4).
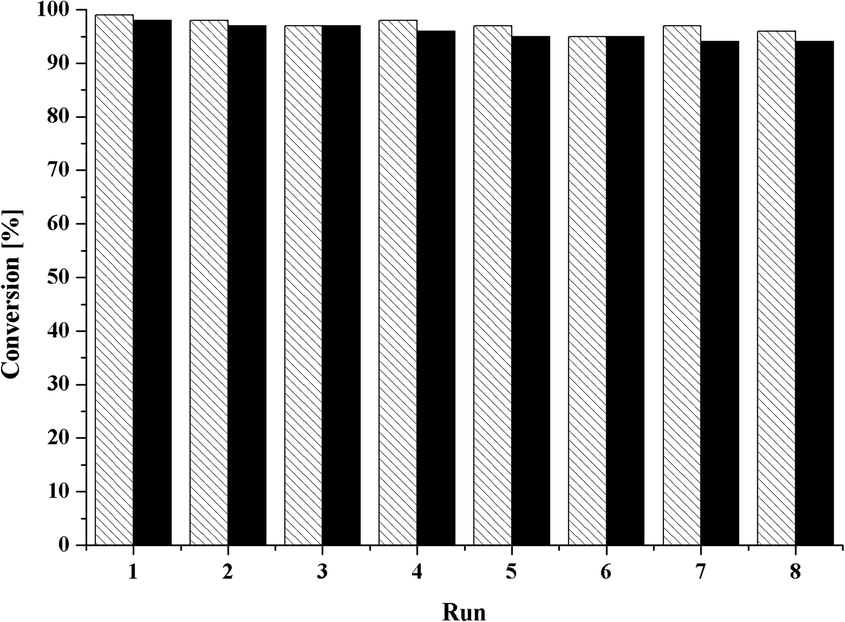 | ||
| Fig. 4 Recycling experiments using compound 1 (dashed line) and compound 2 (black). Reaction conditions: 1 mmol of cis-cyclooctene, 2 mmol of TBHP in n-decane, 0.5 mol% catalyst, 55 °C, 0.5 mL of ionic liquid, 24 h reaction time. The yield was determined via GC (for details see the experimental section). | ||
The stability and reusability of the catalyst is proven by the almost negligible loss of activity after each consecutive run, so that the formed catalytically active species proves to be stable in the reaction media.
Conclusions
A benzyl-substituted Cp molybdenum compound (1) and its fluorinated analogue (2) were synthesised and characterised. They are active and selective catalysts for the epoxidation of olefins using TBHP as an oxidant. Both compounds were examined by DFT calculations, leading to the result that they are much more similar to each other with respect to the HOMOs than the related CH3 and CF3 derivatives. Accordingly, the oxidation of the carbonyls to the active species is more similar. The somewhat enhanced Lewis acidity of 2 explains its higher catalytic activity. This is in accord with the obtained crystal structures and previous studies concerning the influence of Lewis acidity on epoxidation activities. Catalytic results show no formation of diol, underlining the high selectivity of the utilized compounds for standard substrates. Both compounds are highly soluble in common organic solvents in contrast to many previously reported related complexes.With benzene and hexafluorobenzene as solvents, TOFs of up to ca. 18000 h−1 can be achieved in the epoxidation of cyclooctene. Moreover, both compounds are able to epoxidize more demanding substrates such as styrene and stilbenes in acceptable yields applying a 0.5 mol% catalyst. When the precatalysts are dissolved in imidazolium-based ionic liquids, the product phase can be easily separated from the reaction medium and the molecular catalysts can be reused for (at least) 8 consecutive cycles with very low losses of activity.
Experimental section
Material and methods
All experimental synthetic work was carried out using standard Schlenk techniques under argon. Catalytic reactions were performed under laboratory atmosphere. Solvents were dried by standard procedures and stored under argon over molecular sieves. Commercially available chemicals were used as received, unless stated otherwise. High-resolution NMR spectra were measured with a Bruker Avance DPX-400 spectrometer (1H: 400.0 MHz; 13C: 100.6 MHz; 19 F: 376.5 MHz; 95Mo: 26.1 MHz). The signals were referenced to the solvent residual signal (C6D6; 1H: 7.16 ppm; 13C: 128.06 ppm) or external standards in a capillary (95Mo: Mo(CO)6 in C6D6 at −1856 ppm). IR spectra were recorded on a Varian ATR-FTIR instrument. Single crystals suitable for X-ray diffraction were grown by slow diffusion of diethyl ether into a saturated solution of 1 and 2, respectively, in hexane.Synthesis of the complexes
The organomolybdenum complexes [CpMo(CO)3Bz] (1) and [CpMo(CO)3BzF5] (2) were synthesised according to literature procedures.22,35,38(1) [CpMo(CO)3Bz] 1H-NMR (CDCl3, 298 K, 400 MHz): 7.24–7.00 (m, 5H, Bz), 5.20 (s, 5H, Cp), 2.93 (s, 2H, CH2Bz). 13C-NMR (CDCl3, 298 K, 101 MHz): 239.90 (CO), 228.28 (CO), 151.2, 128.05, 127.61, 123.76 (Bz), 94.05 (Cp), 4.98 (CH2Bz). 95Mo-NMR (CDCl3, 298 K, 26 MHz): −1577.28. IR: νCO = 2001.6 cm−1 (vs), 1901.3 cm−1 (s) (84% yield).
(2) [CpMo(CO)3BzF5] 1H-NMR (CDCl3, 298 K, 400 MHz): 5.51 (s, 5H, Cp), 2.48 (s, 2H, CH2BzF5). 13C-NMR (CDCl3, 298 K, 101 MHz): 239.02 (CO), 227.62 (CO), 144.9, 142.59, 138.76, 136.30, 126.07 (BzF5), 93.25 (Cp), −16.12 (CH2BzF5). 95Mo-NMR (CDCl3, 298 K, 26 MHz): −1513.34. 19 F-NMR (CDCl3, 298 K, 376 MHz): −142.34, −163.40, −164.44. IR: νCO = 2020.3 cm−1 (vs), 1914.8 cm−1 (s) (78% yield).
Catalytic reactions
All catalytic reactions were performed under laboratory atmosphere in a reaction vessel equipped with a magnetic stirrer.Cis-cyclooctene: the substrate (1.30 mL, 1 mmol) and catalyst dissolved in 0.5 mL of solvent were added to the reaction vessel and heated to the investigated temperature. The reaction was initiated by the addition of TBHP (3.65 mL, 5.5 M in decane).
Analysis: the course of the reaction was monitored by quantitative GC analysis. Samples taken were treated with MnO2 and MgSO4 to destroy excess peroxide and remove water from the sample. After filtration, the sample was diluted with dichloromethane and an external standard (mixture of 4 mg per 100 mL of p-xylol and 4 mg per 100 mL of indene in isopropanol) was added. The sample was then injected into a GC column and the conversion of cis-cyclooctene to cyclooctene epoxide was calculated from the calibration curves (r2 > 0.999) recorded prior to the start of the reaction.
1-Octene: the substrate (800 mg, 7.3 mmol), dichloroethane (7.3 mmol, internal standard) and the catalyst (0.5 mol%, 36.5 μmol) were added to the reaction vessel and diluted in benzene (0.5 mL). Afterwards, the reaction was initiated by adding TBHP (2.65 mL, 5.5 molar in n-decane).
Cis-stilbene: the substrate (0.291 g, 1.6 mmol), dichloroethane (1.6 mmol, internal standard), and the catalyst (0.5 mol%, 8 μmol) were added to the reaction vessel and diluted in benzene (0.5 mL). The reaction was initiated with the addition of TBHP (0.6 mL, 5.5 molar in n-decane).
Trans-β-methylstyrene: the substrate (0.118 g, 1.0 mmol), dichloroethane (1.0 mmol, internal standard) and the catalyst (0.5 mol%, 0.05 mmol) were added to the reaction vessel and diluted in benzene (0.5 mL). The reaction was initiated by adding TBHP (3.6 mL, 5.5 molar in n-decane).
Analysis: in the case of 1-octene, trans-β-methylstyrene and cis-stilbene, the course of the reaction was monitored by 1H NMR analysis. Samples taken were treated with MgSO4 and MnO2 to remove water and destroy excess peroxide. Afterwards, the sample was diluted with CDCl3, the resulting slurry was filtered and the filtrate was analysed.
Recycling experiments: after the catalytic reaction, the upper phase was removed from the reaction vessel by addition of 5 mL of n-hexane after cooling down to room temperature. The upper phase was removed by means of cannulation. The samples were treated with MgSO4 and MnO2 to remove water and destroy excess peroxide. Afterwards, the sample was diluted with CH2Cl2 and the resulting slurry was filtered. A mixture of 4 mg mL−1 indane and p-xylene in isopropanol (used as external standard) and the filtrate was injected into a GC column. Additionally, oil pump vacuum for 2.5 h at 55 °C allowed the removal of tBuOH from the remaining RTIL (room temperature ionic liquid) phase before further cyclooctene (0.800 g, 7.3 mmol) and TBHP (2.65 mL, 5.5 molar in n-decane) were added.
X-ray single crystal structure determination
Single crystals suitable for X-ray diffraction were grown by slow diffusion of diethyl ether into a saturated solution of 1 and 2, respectively, in hexane.Compound 1: crystal data and details of the structure determination: formula: C15H12MoO3, Mr = 336.19; crystal color and shape: yellow fragment, crystal dimensions = 0.07 × 0.13 × 0.17 mm; crystal system: orthorhombic; space group Pbca (no. 61); a = 16.6109(4), b = 8.7885(2), c = 17.8015(5) Å, V = 2598.75(11) Å3; Z = 8; λ(MoKα) = 1.009 mm−1; ρcalcd = 1.719 g cm−3; θ range = 2.29–25.35°; T = 123 K; data collected: 74161; independent data [Io > 2σ(Io)/all data/Rint]: 2194/2382/0.0368; data/restraints/parameters: 2382/0/220; R1[Io > 2σ(Io)/all data]: 0.0213/0.0237; wR2[Io > 2σ(Io)/all data]: 0.0559/0.0581; GOF = 1.051; ρmax/min: 1.59 (close to Mo)/−0.39 e Å−3.
Compound 2: crystal data and details of the structure determination: formula: C15H7F5MoO3, Mr = 426.15; crystal color and shape: yellow fragment, crystal dimensions = 0.09 × 0.14 × 0.36 mm; crystal system: monoclinic; space group P21/n (no. 14); a = 8.1106(2), b = 14.7261(4), c = 12.4891(3) Å, β = 100.409(1), V = 1467.12(6) Å3; Z = 4; λ(MoKα) = 0.963 mm−1; ρcalcd = 1.929 g cm−3; θ range = 2.77–25.38°; T = 123 K; data collected: 39156; independent data [Io > 2σ(Io)/all data/Rint]: 2466/2689/0.0218; data/restraints/parameters: 2689/0/217; R1[Io > 2σ(Io)/all data]: 0.0153/0.0179; wR2[Io > 2σ(Io)/all data]: 0.0358/0.0369; GOF = 1.083; ρmax/min: 0.27/−0.25 e Å−3.
Computational details
All calculations have been performed with Gaussian03.39 The level of theory contains the hybrid DFT functional B3LYP40,41 and the double zeta 6-31+G**42 basis set for all atoms excluding Mo and the Stuttgart 1997 ECP for molybdenum.43 All obtained geometries have been identified via the number of negative frequencies as minima (NImag = 0). Free energy differences have been calculated for the gas phase at 298.15 K and 1.0 atm.Acknowledgements
L. G. and R. M. R. thank the TUM Graduate School for financial support.Notes and references
- W. F. Richey, Encyclopedia of Chemical Technology, 1994, 140 Search PubMed.
- J. Kollar, Halcon International Inc., Epoxidation process, US Pat., 3,351,635, 1967 Search PubMed.
- M. N Sheng and J. G. Zajacek, J. Am. Chem. Soc., 1968, 418–432 CAS.
- F. E. Kühn, A. M. Santos, A. D. Lopes, J. Gonçalves, E. Herdtweck and C. C. Romåo, J. Organomet. Chem., 2000, 164, 25–38 Search PubMed.
- M. Bagherzadeh, L. Tahsini, R. Latifi and L. Keith Woo, Inorg. Chim. Acta, 2009, 362, 3698–3702 CrossRef CAS PubMed.
- K. R. Jain and F. E. Kühn, Dalton Trans., 2008, 2221–2227 RSC.
- M. Bagherzadeh, L. Tahsini, R. Latifi, A. Ellern and L. K. Woo, Inorg. Chim. Acta, 2008, 361, 2019–2024 CrossRef CAS PubMed.
- C. Freund, W. A. Herrmann, F. E. Kühn, Top. Organomet. Chem., F. Mayer, C. Limberg, Ed. Springer-Verlag, 2007, pp. 39–77.
- F. E. Kühn, A. M. Santos and M. Abrantes, Chem. Rev., 2006, 106, 2455–2475 CrossRef PubMed.
- S. M. Bruno, J. A. Fernandes, L. S. Martins, I. S. Gonçalves, M. Pillinger, P. Ribeiro-Claro, J. Rocha and A. A. Valente, Catal. Today, 2006, 114, 263–271 CrossRef CAS PubMed.
- A. A. Valente, Ž. Petrovski, L. S. C. Branco, C. A. M. Afonso, M. Pillinger, A. D. Lopes, C. C. Romão, C. D. Nunes and I. S. Gonçalves, J. Mol. Catal. A: Chem., 2004, 218, 5–11 CrossRef CAS PubMed.
- K. Kandasamy, H. B. Singh, R. J. Butcher and J. P. Jasinski, Inorg. Chem., 2004, 43, 5704–5713 CrossRef CAS PubMed.
- José A. Brito, M. Gómez, G. Muller, H. Teruel, J.-C. Clinet, E. Duñach and M. A. Maestro, Eur. J. Inorg. Chem., 2004, 2004, 4278–4285 CrossRef PubMed.
- M. K. Trost and R. G. Bergman, Organometallics, 1991, 10, 1172–1178 CrossRef CAS.
- A. Capapé, A. Raith, E. Herdtweck, M. Cokoja and F. E. Kühn, Adv. Synth. Catal., 2010, 352, 547–556 CrossRef PubMed.
- J. Zhao, K. R. Jain, E. Herdtweck and F. E. Kühn, Dalton Trans., 2007, 5567–5571 RSC.
- F. Amor, P. Royo, T. P. Spaniol and J. Okuda, J. Organomet. Chem., 2000, 604, 126–131 CrossRef CAS.
- J. W. Faller and Y. Ma, J. Organomet. Chem., 1989, 368, 45–56 CrossRef CAS.
- J. W. Faller and Y. Ma, J. Organomet. Chem., 1988, 340, 59–69 CrossRef CAS.
- D. Betz, A. Raith, M. Cokoja and F. E. Kühn, ChemSusChem, 2010, 3, 559–562 CrossRef CAS PubMed.
- S. A. Hauser, R. M. Reich, J. Mink, A. Pöthig, M. Cokoja and F. E. Kühn, Catal. Sci. Technol., 2015, 5, 2282–2289 CAS.
- S. A. Hauser, M. Cokoja, M. Drees and F. E. Kühn, J. Mol. Catal. A: Chem., 2012, 363–364, 237–244 CrossRef CAS PubMed.
- D. J. Parker and M. H. B. Stiddard, J. Chem. Soc. A, 1970, 480–490 RSC.
- D. J. Parker, J. Chem. Soc. A, 1970, 1382–1386 RSC.
- R. B. King and L. W. Houk, Can. J. Chem., 1969, 47, 2959–2964 CrossRef CAS.
- R. B. King and M. B. Bisnette, J. Organomet. Chem., 1964, 2, 15–37 CrossRef CAS.
- R. B. King and M. B. Bisnette, J. Organomet. Chem., 1967, 8, 287–297 CrossRef CAS.
- M. Minelli, J. H. Enemark, R. T. C. Brownlee, M. J. O'connor and A. G. Wedd, Coord. Chem. Rev., 1985, 68, 169–278 CrossRef CAS.
- N. Grover, F. E. Kühn (unpublished results).
- M. Drees, S. A. Hauser, M. Cokoja and F. E. Kühn, J. Organomet. Chem., 2013, 748, 36–45 CrossRef CAS PubMed.
- P. J. Costa, M. J. Calhorda and F. E. Kühn, Organometallics, 2010, 29, 303–311 CrossRef CAS.
- N. Grover, A. Pöthig and F. E. Kühn, Catal. Sci. Technol., 2014, 4, 4219–4231 CAS.
- A. Valente, J. Seixas, I. Gonçalves, M. Abrantes, M. Pillinger and C. Romão, Catal. Lett., 2005, 101, 127–130 CrossRef CAS.
- F. E. Kühn, J. Zhao, M. Abrantes, W. Sun, C. A. M. Afonso, L. C. Branco, I. S. Gonçalves, M. Pillinger and C. C. Romão, Tetrahedron Lett., 2005, 46, 47–52 CrossRef PubMed.
- M. Abrantes, A. M. Santos, J. Mink, F. E. Kühn and C. C. Romão, Organometallics, 2003, 22, 2112–2118 CrossRef CAS.
- C. C. Romão, F. E. Kühn and W. A. Herrmann, Chem. Rev., 1997, 97, 3197–3246 CrossRef PubMed.
- W. A. Herrmann and F. E. Kühn, Acc. Chem. Res., 1997, 30, 169–180 CrossRef CAS.
- R. B. King and A. Fronzaglia, J. Am. Chem. Soc., 1966, 88, 709–712 CrossRef CAS.
- M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham,C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, J. A. Pople, Gaussian03, Gaussian, Inc., Wallingford, CT, , 2004 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- Y. W. C. Lee and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- R. D. J. Hehre and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef PubMed.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1055255 (1) and CCDC 1055256 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cy00447k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |