
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selectivity and kinetics of methyl crotonate hydrogenation over Pt/Al2O3†
Chaoquan
Hu
ab,
Derek
Creaser
*b,
Henrik
Grönbeck
ac,
Houman
Ojagh
ab and
Magnus
Skoglundh
ad
aCompetence Centre for Catalysis, Chalmers University of Technology, SE-412 96 Göteborg, Sweden
bDivision of Chemical Engineering, Department of Chemical and Biological Engineering, Chalmers University of Technology, SE-41296 Göteborg, Sweden. E-mail: derek.creaser@chalmers.se
cDepartment of Applied Physics, Chalmers University of Technology, SE-412 96 Göteborg, Sweden
dApplied Surface Chemistry, Department of Chemical and Biological Engineering, Chalmers University of Technology, SE-412 96 Göteborg, Sweden
First published on 12th December 2014
Abstract
The hydrogenation of gas-phase methyl crotonate (MC) over Pt/Al2O3 was investigated with the aim to understand C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogenation in unsaturated methyl esters. Three Pt/Al2O3 catalysts with different Pt dispersions were prepared by varying calcination temperature and evaluated for MC hydrogenation. The main products were found to be methyl butyrate (MB) and methyl 3-butenoate (M3B), resulting from hydrogenation and shift of the CC bond in MC, respectively. The measured activity for both hydrogenation and shift of the CC in MC was found to depend on the Pt dispersion where higher Pt dispersion favors the CC hydrogenation reaction. The effect of reactant concentrations on the activity and selectivity for MC hydrogenation over the Pt/Al2O3 catalyst was examined in detail. Under the investigated conditions, the CC hydrogenation was found to have a negative reaction order with respect to MC concentration but a positive H2 order. Further understanding of the MC hydrogenation was provided from H2 chemisorption experiments over the catalyst with and without pre-adsorbed MC and from transient experiments using alternating MC and H2 feeds. Based on the present experimental results, a reaction pathway was proposed to describe gas-phase MC hydrogenation over Pt/Al2O3. In order to gain more insight into the reaction, a kinetic analysis of MC hydrogenation was performed by fitting a power-law model to the kinetic data, moreover, dissociative H2 adsorption on the catalyst was found to be the rate-determining step by comparing the power-law model with the overall rate expressions derived from mechanistic considerations.
C hydrogenation in unsaturated methyl esters. Three Pt/Al2O3 catalysts with different Pt dispersions were prepared by varying calcination temperature and evaluated for MC hydrogenation. The main products were found to be methyl butyrate (MB) and methyl 3-butenoate (M3B), resulting from hydrogenation and shift of the CC bond in MC, respectively. The measured activity for both hydrogenation and shift of the CC in MC was found to depend on the Pt dispersion where higher Pt dispersion favors the CC hydrogenation reaction. The effect of reactant concentrations on the activity and selectivity for MC hydrogenation over the Pt/Al2O3 catalyst was examined in detail. Under the investigated conditions, the CC hydrogenation was found to have a negative reaction order with respect to MC concentration but a positive H2 order. Further understanding of the MC hydrogenation was provided from H2 chemisorption experiments over the catalyst with and without pre-adsorbed MC and from transient experiments using alternating MC and H2 feeds. Based on the present experimental results, a reaction pathway was proposed to describe gas-phase MC hydrogenation over Pt/Al2O3. In order to gain more insight into the reaction, a kinetic analysis of MC hydrogenation was performed by fitting a power-law model to the kinetic data, moreover, dissociative H2 adsorption on the catalyst was found to be the rate-determining step by comparing the power-law model with the overall rate expressions derived from mechanistic considerations.
1. Introduction
The catalytic hydrogenation of CC bonds is an important and useful reaction in the pharmaceutical and fine chemical industry. In most cases, the main purpose of hydrogenation is to reduce the degree of unsaturation or completely saturate the CC bonds in organic compounds. For instance, partial CC hydrogenation is desirable in the field of edible oil, while cyclohexane production requires saturation of the benzene ring. CC hydrogenation is also of importance for improving the stability of biodiesel, which is considered as a renewable energy carrier. Biodiesel is commonly produced by hydrogenation of unsaturated vegetable oils (HVOs) or by transesterification between vegetable oil or animal fats and methanol.1–3 Partial CC hydrogenation can considerably improve the stability of biodiesel against oxidation.4
With respect to CC hydrogenation, extensive work has been performed using various model molecules to understand the corresponding reaction mechanism. The simplest probe molecule for studying CC hydrogenation is ethylene, which only contains a CC bond without other functional groups. From the results of ethylene hydrogenation, the Horiuti–Polanyi mechanism was proposed to elucidate the CC hydrogenation.5 In this mechanism, two hydrogen atoms are assumed to be sequentially incorporated into ethylene with the formation of an alkyl intermediate. In some cases, this mechanism can be used to interpret the experimental observations from various surface analysis techniques.6–8 Besides ethylene, some small molecules, e.g., acrolein and crotonaldehyde, have been used as probe molecules to investigate selective hydrogenation.9–20 The interest in these molecules with conjugated CC and CO groups has mainly lied in the issue of selectivity, i.e., controlling hydrogenation of either the CC or the CO bond, rather than the fundamental aspects of CC hydrogenation, such as adsorption mode and rate-determining step.
Besides the small molecules, studies have thus far also investigated the hydrogenation of large methyl ester molecules, e.g., methyl oleate and methyl linoleate, over supported metal catalysts.21–35 Recently, we have reviewed this subject and summarized the progresses for hydrogenation of CC in unsaturated fatty acid methyl esters.36 Surprisingly, some fundamental issues about hydrogenation of CC in methyl esters are still under debate in spite of intensive work in this field. For instance, Jonker et al. showed that the rate expression derived from the Langmuir–Hinshelwood–Hougen–Watson (LHHW) mechanism based on two types of adsorption sites could be used to fit the kinetic data for hydrogenation of methyl oleate and elaidate.37 However, Cabrera and Grau demonstrated that the models based on a competitive or semi-competitive adsorption mode provided a better fit than the non-competitive model to the kinetic data of methyl oleate hydrogenation over Ni/Al2O3.38,39 Obviously, more work is required to understand hydrogenation of unsaturated methyl esters.
Our strategy for understanding CC hydrogenation of unsaturated methyl esters will be from simple to complicated molecules. The purpose is not limited to understanding the underlying mechanisms of CC hydrogenation, but also provide valuable information for future theoretical modelling to bridge the gap between hydrogenation of small simple molecules and larger ones with functional groups. To this end, we have studied hydrogenation of methyl crotonate (MC) in gas-phase over Pt/Al2O3 catalysts with different Pt dispersions. Herein, MC is chosen as the starting molecule because of its simple chemical structure compared to other larger methyl esters. The combustion characteristics of MC as a typical methyl ester in air have been investigated in previous studies.40,41 Furthermore, MC can be produced from the decomposition and oxidation of large methyl esters.42 In addition, it should be noted that hydrogenation of MC will provide insight into the special kinetics of a CC bond conjugated with the CO bond in a methyl ester. In this aspect, a correlation of the hydrogenation behavior of larger methyl esters and the above mentioned small conjugated molecules may be built if the product selectivity and reaction kinetics are similar.
In this study, hydrogenation of methyl crotonate over Pt/Al2O3 is systematically investigated by varying reactant concentrations at steady state and under transient conditions. A reaction pathway is proposed to understand MC hydrogenation on the catalyst and some characteristics of this catalyzed reaction are revealed from the reaction kinetics. Moreover, the hydrogenation behaviors of MC, ethylene, and crotonaldehyde over Pt catalysts are compared considering product selectivity and reaction kinetics.
2. Experimental
2.1. Catalyst preparation and characterization
All the neat compounds used in this study, namely, MC (98% purity), methyl butyrate (MB, ≥98% purity), and methyl 3-butenoate (M3B, 95% purity), were purchased from Sigma-Aldrich and used as received without further purification. The catalyst containing 5 wt.% of Pt on Al2O3 was prepared by wet impregnation. Typically, 0.3230 g of Pt(NO3)2 solution (Heraeus GmbH 15.46 wt.%) was diluted in distilled water to form an aqueous solution of 2.70 g. Then 0.95 g of Al2O3 (Puralox SBa-200 Sasol) after thermal treatment at 700 °C for 4 h under a static air atmosphere was added to the above Pt-containing solution under strong and constant stirring for 1 h at room temperature. The resulting paste-like slurry was dried at 105 °C for about 12 h. The obtained powder was divided into three portions and calcined at different temperatures (500, 600, and 700 °C) for 4 h. The heating rate was 2 °C min−1 for the calcination treatment, while in the drying process a constant temperature of 105 °C was used. The sample calcined at a defined temperature is denoted as Pt/Al2O3-T, for instance, Pt/Al2O3-500 means a calcination temperature of 500 °C. Note that the minimum calcination temperature chosen was 500 °C because at this temperature all possible nitrate species could be removed from the platinum nitrate. Evidence for this conclusion was obtained by thermal decomposition of the platinum precursor on the prepared Pt/Al2O3 samples using differential scanning calorimetry (DSC) technique (ESI†).The thermal decomposition behavior of the catalyst precursor, as well as temperature-programmed reduction with hydrogen (H2-TPR) and CO or H2 chemisorption, was performed by using a calorimeter (Setaram Sensys DSC) equipped with a quadrupole mass spectrometer (MS, Hiden Analytic Inc., HPR-20). The calorimeter consists of two quartz tubes: one is used as a reference while the other one contains the sample which is placed on a sintered quartz bed. For measurement of the thermal decomposition and H2-TPR, 20 mg of the sample was loaded into the sample tube and the ramping rate was set to 10 °C min−1. For the H2-TPR experiment, the sample was exposed to 2% H2/Ar with a flow rate of 20 ml min−1. In the case of CO and H2 chemisorption, 50 mg sample was used and pre-reduced at 400 °C for 1 h. After the sample temperature decreased to 25 °C, 200 ppm CO or 150 ppm H2 in Ar was introduced to the sample tube until reaching a stable state of the corresponding outlet gas concentration determined by the MS analysis. Two CO pulses were performed to determine the chemisorbed amount of CO on the sample. The first CO pulse was considered to involve the adsorption of both strongly and weakly adsorbed CO, while the second CO pulse only resulted in adsorption and desorption of the weakly bonded CO. The difference between the two CO pulses thus represents the amount of chemisorbed CO (nCO) on the Pt/Al2O3 sample. Taking into account the possible adsorption sites for CO on Pt, a factor of 0.8 was used in calculation of the Pt dispersion (D), namely, D = 0.8 × nCO/nPt.43 Assuming the Pt to be in the form of spherical particles, the average particle size (d) of Pt was estimated according to the equation: d = 1.13/D.44
As expected, the Pt dispersion decreased with increasing calcination temperature, namely, from 16% for the Pt/Al2O3-500 sample to 12% for the Pt/Al2O3-600 and further to 8.8% for the sample calcined at 700 °C. The average Pt particle size for the three samples was estimated to be 7.1, 9.4, and 12.8 nm, respectively. The obtained Pt dispersion for the sample calcined at 500 °C in the present study is considerably lower than the dispersion reported by Auvray et al.43 but basically in agreement with the value (16%) for 5 wt.% Pt/Al2O3 calcined at 600 °C for 1 h.45 Besides the calculation method, the large differences in Pt dispersion on Al2O3 should be related to the high Pt loading (5 wt.%), which increases the possibility for sintering of Pt in the calcination process and hence results in larger Pt particles.
2.2. Hydrogenation activity measurement
The hydrogenation measurements were performed in a tubular reactor with a catalyst bed diameter of 4 mm under atmospheric pressure. The gas pipelines for transportation of the organic compounds were heated to about 150 °C using heating tapes. In each test run, a certain amount (≤20 mg) of catalyst was placed at the center of the reactor and pretreated in a stream 5% H2/Ar at 400 °C for 1 h prior to the MC hydrogenation measurements. Note that the sample before the activity test was reduced twice: the first reduction after the calcination of the sample at 500–700 °C was to remove NOx, as shown in H2-TPR results (ESI†), and the second reduction was to remove possible oxygen species adsorbed due to exposure to air during storage of the catalyst. The liquid feed was vaporized and injected into a 100 ml min−1 flow of H2/Ar prior to feeding to the reactor. The concentration of gas-phase MC was controlled by adjusting the feeding rate from a syringe pump (NE-1000, New Era Pump Systems, Inc.). The temperature of the catalyst bed was measured with a thermocouple mounted axially in the reactor tube. The products were analyzed by a calibrated GC (Scion 456-GC, BRUKER) equipped with a FID detector for organic species and a TCD detector for H2 and COx. Three columns (Hayesep Q 80/100 mesh, Molsieve 5A 60/80 mesh, and BR-SWax) and a back flush mode were used in the GC analysis to separate the components in the gas sample. The initial oven temperature in the GC was 40 °C and increased at 5 °C min−1 to 80 °C and then increased at 30 °C min−1 to 150 °C where it was held constant for 1 min.2.3. Products analysis and calculation method
There are several possible reaction pathways, e.g., thermal decomposition, hydrogenation of CO or CC, and CC double bond shift, during MC hydrogenation. A reaction network is shown in Fig. 1. Also the reactions involving the removal of oxygen from MC may occur according to hydrodeoxygenation, decarboxylation and decarbonylation (not shown in Fig. 1). Under the present experimental conditions, the product detected by the GC analysis contained the following species: H2, CO, CO2, C3H6, MC, MB, M3B and several minor unidentified species. A typical GC spectrum of the FID is included in the ESI.† The characteristics of the GC peaks of the species depend on the experimental conditions. Under atmospheric pressure and temperatures between 60 and 200 °C, the predominant components formed from MC hydrogenation were MB and M3B without detection of alcohols except methanol, indicating the absence of CO hydrogenation. Though it is not possible to precisely quantify the unidentified species, their concentrations should be lower than 2 ppm if CH4, or heptane, or MC was used as the reference. Since the minimum concentration of MC used in this study is 0.31%, the selectivity for these unknown species was calculated to be less than 0.5%. In addition, it was found that the outlet concentrations of CH3OH and C3H6 were enhanced with increasing the reaction temperature probably due to the promotion of cracking reactions or decarbonylation or decarboxylation at higher temperatures. The concentrations of the other minor gaseous products, however, did not show any apparent variation with the experimental conditions. It is thus possible that some of these minor species were impurities in the MC feedstock. For instance, the GC peak area of the species at the retention time of about 3 min remained constant for the reaction temperatures from 60 to 350 °C.
 | ||
| Fig. 1 Possible reaction pathways for MC hydrogenation excluding hydrodeoxygenation, decarboxylation, and decarbonylation. | ||
Taking into account the concentrations of the minor species, only CH3OH, which can also indicate the percentage of MC cracking, was considered in the calculation of selectivity and reaction rate. In this study, the conversion of MC and the selectivity were defined as follows:
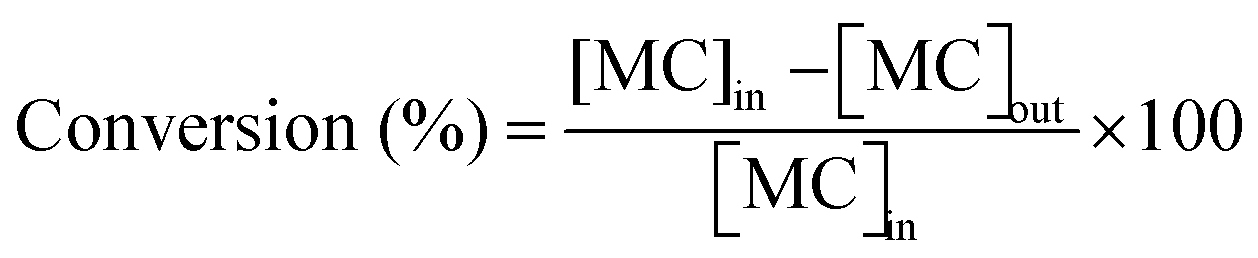
where [MC]in, [MC]out, [product]out represent the inlet concentration of MC and the outlet concentrations of MC and product, respectively. Assuming a differential operation of the tubular reactor at MC conversions lower than 20%, the reaction rates for MC hydrogenation and the turnover frequency (TOF) were calculated according to:

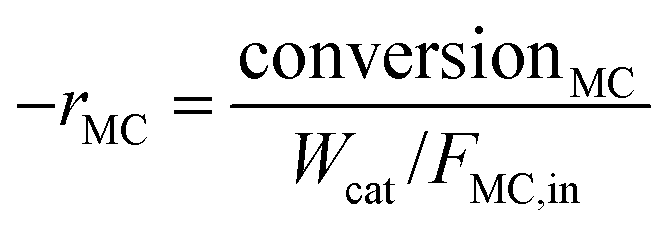
| rMB = (−rMC) × SelMB |
| rM3B = (−rMC) × SelM3B |

| TOFMB = TOFMC × SelM3B |
| TOFM3B = TOFMC × SelM3B |
3. Results and discussion
3.1. Catalyst performance and selectivity in MC hydrogenation
Fig. 2 shows the MC conversion and the selectivity to MB and M3B, which were the main species from MC hydrogenation, over the three Pt/Al2O3 catalysts calcined at different temperatures. The carbon balance, as well as the GC peak areas of the observed minor species, is included in the ESI.† As shown in Fig. 2a, over the investigated temperature range, the Pt/Al2O3-500 sample showed the highest MC conversion among the three samples investigated. At temperatures below 230 °C, the MC conversion increased with the reaction temperature for all samples. Over the Pt/Al2O3-500 sample, the MC conversion reached about 95% at 210 °C and further increase in the reaction temperature to 250 °C only resulted in a minor change in MC conversion, probably due to mass transfer limitations at high MC conversion levels. However, for the samples calcined at 600 and 700 °C, the MC conversion first decreased slightly with increasing reaction temperature from 220 to 300 °C and then sharply decreased when further increasing the reaction temperature to 350 °C. For instance, the MC conversion decreased from 61 to 28% over the Pt/Al2O3-600 catalyst when increasing the reaction temperature from 300 to 350 °C. This phenomenon might be the result of catalyst poisoning by carbonaceous deposits at high reaction temperatures. Direct evidence for this was the observation of coke on the reactor wall after the activity evaluation at 350 °C. However, this effect was not observed for reaction temperatures lower than 200 °C. This appears to be consistent with the previous studies showing that coking is one of the main reasons for deactivation of metal-based catalysts in dehydrogenation or hydro-treating processes at high temperatures.46,47 In addition, the presence of Lewis acidic sites on the Al2O3 surface could also result in coke formation.48 However, further investigations are required to clarify the nature of the deactivation. Obviously, high reaction temperature is one factor that causes side-reactions during methyl ester hydrogenation and should thus be avoided during selective CC hydrogenation.
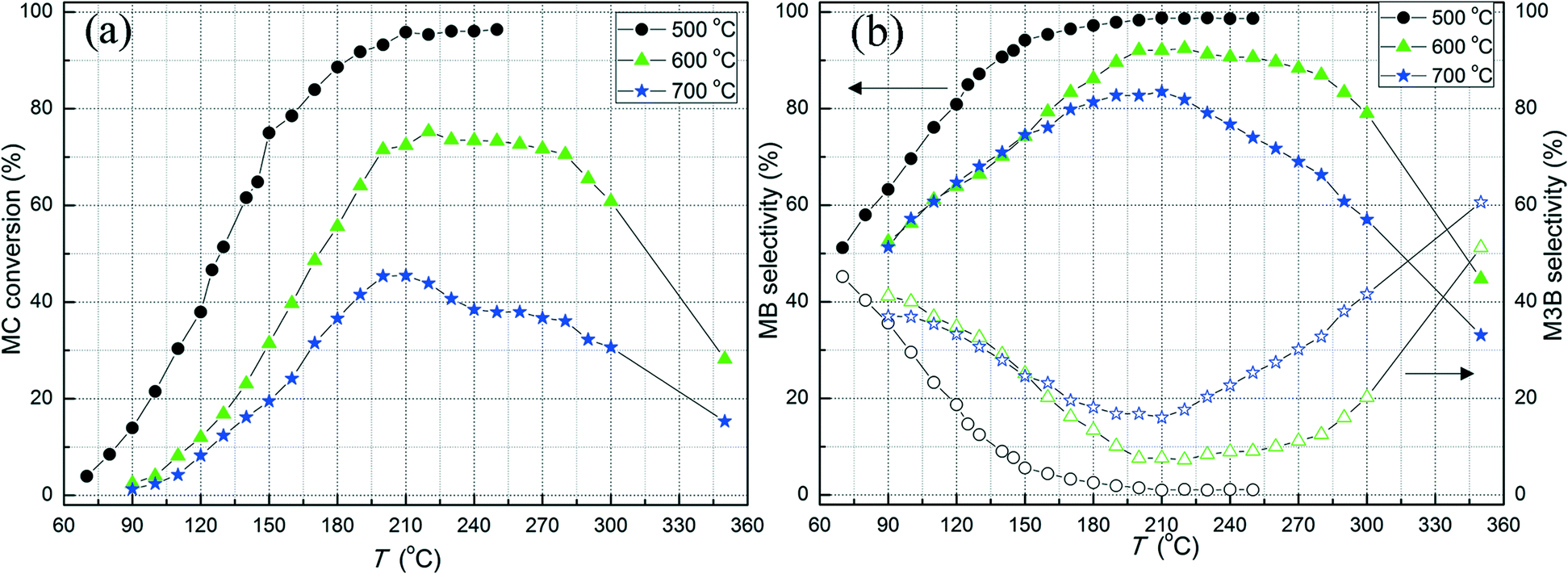 | ||
Fig. 2 (a) MC conversion and (b) selectivity as a function of reaction temperature for MC hydrogenation over the Pt/Al2O3 catalysts calcined at different temperatures. Experimental conditions: 20 mg catalyst, pMC = 0.63%, pH2 = 4.60% and GHSV = ~240![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. 000 h−1. | ||
The selectivity as a function of the reaction temperature for MC hydrogenation over the three samples is displayed in Fig. 2b. For all samples, the selectivity to MB as a result of CC hydrogenation in MC, increased as the reaction temperature increased and was always higher than that for M3B under the present conditions. This is similar to the selectivity during liquid-phase 1-butene hydrogenation over Pt/alumina,49 which also showed a higher selectivity towards CC hydrogenation than the CC shift. Furthermore, the Pt/Al2O3-500 sample exhibited the highest selectivity to MB and hence lowest selectivity to M3B among the three samples. It appears that higher dispersion, i.e. smaller Pt particle size, is favorable for MC hydrogenation.
To gain more insight into the activity of the three samples, the TOF was calculated and the comparison is displayed in Fig. 3. Herein the experimental data at 90 and 120 °C were used for the calculation of TOFs in order to minimize possible mass transfer limitations and hence more approach observations of intrinsic activity. As shown in Fig. 3, the overall TOFs of MC over the Pt/Al2O3-500 sample at the two reaction temperatures was more than two times higher than those of the other two samples, which exhibited similar overall TOFs. At 90 °C (Fig. 3a), the TOF of MB over the Pt/Al2O3-500 sample was more than 5 times higher than those for the other two samples, while the TOF of M3B was about 4 times higher. When the reaction temperature was increased to 120 °C (Fig. 3b), as expected, both the TOFs of MB and M3B increased. However, large differences in the TOFs of MB over the three samples were still observed, while the TOFs of the selectivity towards M3B were similar. This may indicate that the Pt/Al2O3-500 sample is more selective for direct hydrogenation of MC to MB, however it is also possible that a larger amount of M3B was hydrogenated to MB in a series process due to the higher conversion of MC for the Pt/Al2O3-500 sample at 120 °C since the high conversion (40%) of MC deviates from differential conditions. From the comparison of the TOFs, it can be said that the CC hydrogenation contributed more than the shift of CC bond to the activity of the samples, moreover, the higher activity of Pt/Al2O3-500 compared to the other two samples mainly came from the hydrogenation rate.
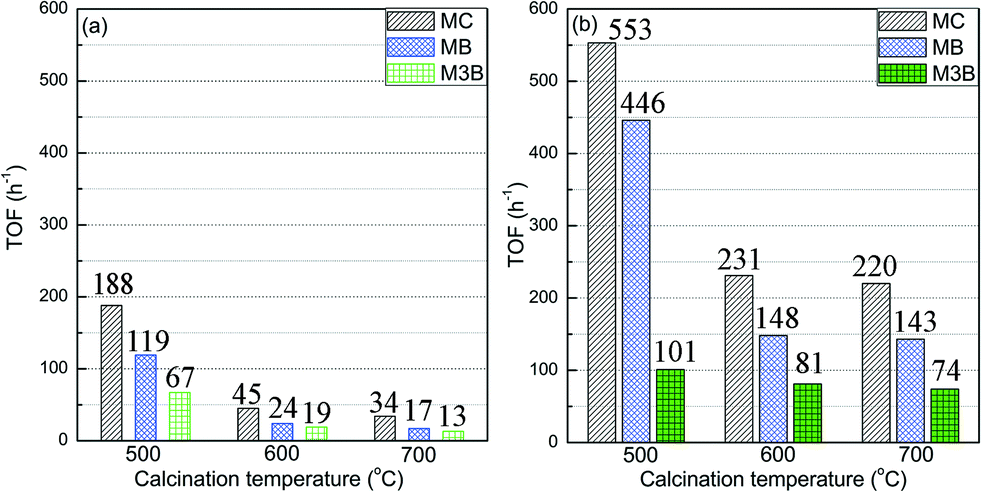 | ||
| Fig. 3 Comparison of TOFs of the three Pt/Al2O3 catalysts towards MC hydrogenation at (a) 90 and (b) 120 °C. | ||
Note that the samples treated at 600 and 700 °C did not exhibit any significant difference in the TOFs. This could suggest that the TOF of MC hydrogenation has only a minor dependence on Pt dispersion for values lower than 12%. Similar phenomenon has been observed for various chemical reactions, depending on the type of reaction employed and the support used. For instance, the total oxidation of alkanes increased with Pt particle size up to 4 nm and did not change significantly when further increasing the particle size of Pt.50 Haneda et al. found that the TOFs of propene oxidation over Pt/Al2O3 remained almost constant for Pt dispersions exceeding 0.2.51 The hydrogenation of crotonaldehyde over Pt catalysts was also reported to be sensitive to the particle size of Pt.12 However, the invariance of TOF with higher Pt dispersion was not observed in the study probably due to the narrow range of dispersion used.
3.2. Effect of MC and H2 concentration on performance of Pt/Al2O3-500
The effect of the concentration of MC on the hydrogenation of MC was investigated for the Pt/Al2O3-500 catalyst. Note that the carbon balances for these measurements were acceptable (ESI†) for the steady state conditions. As shown in Fig. 4a, when the MC concentration was raised from 0.31 to 0.70%, the conversion of MC decreased at the same reaction temperature. Correspondingly, the selectivity towards hydrogenation also decreased (Fig. 4b) when keeping the H2 concentration constant. The results herein indicate that MC acts as a self-inhibiting reactant for the hydrogenation of CC in MC. The negative effect of MC concentration on hydrogenation also suggests that the adsorption of MC on the catalyst is strong. To quantify the effect of MC concentration, the general power-law model was used to fit the reaction rates based on the MC conversion lower than 20%. As shown in Fig. 4c, the power-law model provides a good fit to the rates of MC conversion, hydrogenation (MB), and CC bond shift (M3B). The estimated reaction orders with respect to MC for the overall reaction rate and the production rates of MB and M3B were −0.24, −0.58, and 0.11, respectively. The negative reaction order with respect to MC indicates that MC is strongly adsorbed on the catalyst surface and that the number of vacant adsorption sites decreases with increasing MC concentration to hinder H2 adsorption. If MC hydrogenation is assumed to proceed according to the LHHW kinetics with a surface reaction between H2 and MC both adsorbed, the negative reaction order of MC suggests that a competitive adsorption between H2 and MC over a common adsorption site may be the relevant mechanism. Note however, that the M3B formation rate had a small positive reaction order with respect to MC, indicating the possible influence of non-competitive adsorption between H2 and MC.
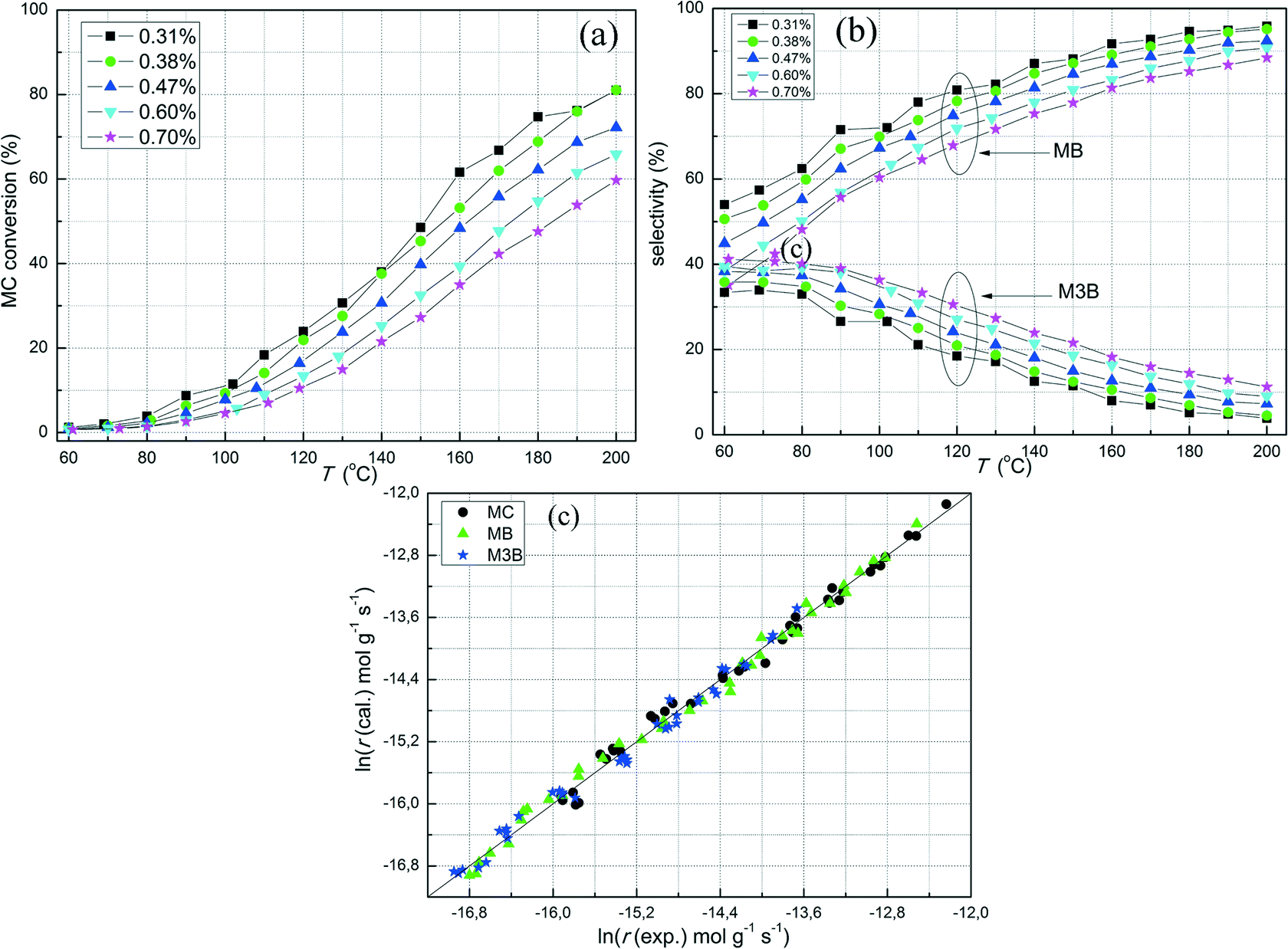 | ||
| Fig. 4 (a) MC conversion and (b) selectivity of MC hydrogenation over the Pt/Al2O3-500 catalyst at various MC concentrations. (c) Experimental reaction rates of MC hydrogenation on the catalyst versus the calculated values by using the power-law model. Experimental conditions: 20 mg, pH2 = 2.58% and GHSV = ~240000 h−1. | ||
The effect of H2 concentration on MC conversion and selectivity over the Pt/Al2O3-500 catalyst was also studied. Contrary to the above observed effect of MC concentration, the MC conversion (Fig. 5a) as well as the selectivity towards hydrogenation (Fig. 5b) increased with increasing the H2 concentration from 2.78 to 33.5%. For instance, at the MC conversion of about 20%, the selectivity towards the hydrogenation product (MB) was about 71 and 80% for the H2 concentration of 2.78 and 9.00%, respectively. Thus, it can be concluded that high H2 feed favors the CC hydrogenation and the formation of MB even at constant MC conversion. Without H2, MC did not show any detectable conversion to neither MB nor M3B (Fig. 5a), indicating that the formation of both MB and M3B are related to H2. It allows us to conclude that the intermediate in MC hydrogenation should involve the activation of CC in MC by a hydrogen atom, which can be further hydrogenated to MB or lose a hydrogen atom to form M3B. Obviously, the formation of M3B and MB can occur as parallel reactions originating from a common adsorbed intermediate species. A consecutive path involving re-adsorption of M3B and its hydrogenation to MB also likely occurs, but should be more important at higher MC conversions. However, the carbon balance (Fig. 5c) in this case became unacceptable for steady state measurements when the H2 concentration exceeded 4.60% at higher temperatures. Further, the obtained carbon in the product was always higher than the nominal value, suggesting that some carbon-containing adsorbed species as intermediates or reactant or product species were stored on the catalyst and subsequently released at higher temperatures. Note that, as a result, the fitting of the reaction rates at varying H2 concentrations using the power-law model was not performed and the reaction order with respect to H2 was instead determined from dedicated kinetic data in section 3.6.2 below.
 | ||
| Fig. 5 (a) MC conversion, (b) selectivity and (c) carbon balance of MC hydrogenation on the Pt/Al2O3-500 catalyst at different H2 concentrations. Experimental conditions: 20 mg catalyst, PMC = 0.60% and GHSV = ~240000 h−1. | ||
3.3. Transient experiments
The excess of carbon at high H2 concentrations motivated us to investigate the MC hydrogenation using transient response experiments, intended to gain more insights into MC hydrogenation on the Pt surface. The procedure was conducted as follows. First, MC with a partial pressure of 0.46% in the absence of H2 passed through the catalyst bed in the reactor. After reaching a stable state, the MC feed was stopped and H2/Ar was introduced to the reactor. The concentrations of different products were measured by the GC. The outlet gas concentrations from several successive cycles each with increasing H2 partial pressures are shown in Fig. 6. At reaction time 0 the starting feed was MC in the absence of H2. Without H2 in the feed, no formation of MB or M3B was observed. This is in agreement with the above conclusion that H2 is required to activate MC to form an intermediate which can produce MB and M3B. When hydrogen concentration was increased to 2.58%, MC and MB were detected while M3B was almost negligible, indicating that MC was stored on the surface of the catalyst. The introduction of H2 competed with the MC for adsorption on the catalyst and hence resulted in the release of MC as well as the hydrogenation of CC in MC. The absence of M3B in this case could be due to the limited initial amount produced or its further hydrogenation to MB under a H2 atmosphere without MC. When the feed was switched back to 0.46% MC in Ar, M3B was detected while MB was negligible under the specific conditions. The formation of M3B may result from the activation of MC by the diminishing quantity of hydrogen atoms left on the Pt surface from the previous cycle. At this stage the absence of MB suggests a weaker adsorption of MB than MC on the Pt surface. In the following successive cycles at H2 partial pressures ranging from 9.2 to 26.3%, similar phenomena to the case with H2 partial pressure of 2.58% were observed. As expected, larger amounts of MC were released and more MB was formed in presence of higher H2 concentrations. These results indicate that an H2-rich environment is favorable for CC hydrogenation, while H-lean conditions on the Pt surface likely promote the formation of M3B. This is consistent with the above effect of H2 concentration at steady state conditions (Fig. 5b), namely, more H2 results in higher selectivity to CC hydrogenation.
 | ||
| Fig. 6 Transient experiments over the Pt/Al2O3-500 catalyst under isothermal conditions at 190 °C by switching the gas feed between 0.46% MC and different H2 partial pressures. | ||
3.4. H2 adsorption behavior
From the results of the transient experiments, it was apparent that MC can be stored on the catalyst and H2 competes for adsorption. Thus, the adsorption of H2 on the Pt/Al2O3-500 catalyst without and with pre-adsorbed MC was further investigated. Without MC on the catalyst, there was a complete uptake of H2ca. 400 s after the feed of H2 was started (Fig. 7). After this period, a breakthrough of H2 was observed and then the concentration of H2 reached a stable value in a short time. In the corresponding heat flow profile, there is a clear exotherm with constant heat flow observed which subsequently drops upon the H2 breakthrough. Based on the stable H2 uptake during the initial period, the adsorption energy of H2 on the catalyst was calculated to be 97.4 kJ mol−1, which is comparable to a previously reported value (116 kJ mol−1).52 For comparison of the H2 adsorption behavior on the catalyst with pre-adsorbed MC, the catalyst was first exposed to 0.46% MC in Ar at 100 °C for about 1 h until saturation as confirmed by determination of the MC concentration in the outlet gas analyzed by the GC. As shown in Fig. 7, after saturation with MC the catalyst exhibited significantly different adsorption behavior towards H2. Both the adsorption of H2 on the catalyst and the corresponding heat flow decreased considerably, indicating a notable decrease in hydrogen adsorption on the Pt surface. This could be related to the high coverage of strongly pre-adsorbed MC on the Pt surface and hence the available sites for H2 dissociation were limited. It is also possible that the MC on the Pt surface may modify the electronic structure of Pt and thus decrease the adsorption strength between H2 and the Pt surface. | ||
| Fig. 7 H2 chemisorption on the Pt/Al2O3-500 catalyst without and with MC at 25 °C. | ||
Also the significant decrease in H2 adsorption on the catalyst with MC suggests that a high partial pressure of H2 is required for hydrogenation of CC in MC. This is consistent with the results in the previous section 3.2 where higher H2 partial pressure was more favorable for CC hydrogenation. Actually, the dependence of CC hydrogenation on H2 partial pressure was observed in some previous studies about methyl ester hydrogenation over noble metals.21–23,53 The positive effect of H2 on hydrogenation of CC in these liquid-phase systems was attributed to the increased H2 availability on catalyst surface from the viewpoint of mass transfer rather than H2 dissociation since H2 is believed to readily dissociate on most noble metals due to the high adsorption energies. However, the present study clearly shows that the high H2 partial pressures benefits H2 dissociation on Pt/Al2O3 with pre-adsorbed MC. Thus, it can be claimed that besides the H2 availability on the catalyst surface the adsorption and dissociation of H2 on metals should be another important factor for methyl ester hydrogenation under liquid-phase conditions.
3.5. Proposed reaction pathway
Before deriving the reaction pathway for MC hydrogenation, a brief summary of the above experimental results is necessary.• Under the present reaction conditions, no fatty alcohols were observed as reaction products and thus hydrogenation of CO did not occur over the Pt/Al2O3 catalyst. It can be further concluded that MC is adsorbed on the Pt surface via the CC bond rather than the other groups in the molecule. If the other groups, e.g., CO, were significantly chemisorbed on the surface, the CC bond would be away from the catalyst surface and the products should include aldehydes or alcohols.
• The negative reaction order with respect to MC for the production rate of MB indicates a competitive adsorption between MC and H2 on Pt. This could be further confirmed by the transient experiments where the introduction of H2 led to the release of MC from the catalyst.
• H2 is necessary to activate MC since neither MB nor M3B were formed in the absence of H2. It can be considered that the formation of M3B and MB can be parallel reactions that originate from a common adsorbed intermediate species.
• In the transient experiments, the absence of MB when switching gas feed from H2 to MC indicates that MB is more weakly adsorbed than MC on Pt.
With these points in mind, we propose the reaction pathway for MC hydrogenation on Pt/Al2O3 as shown in Fig. 8. Gas-phase MC and H2 molecules approach and compete for adsorption sites on the Pt surface. Due to the stronger adsorption of MC on Pt, MC is more preferably adsorbed. The adsorbed MC and H from H2 dissociation form a semi-hydrogenated intermediate which can be further hydrogenated into MB or lose one proton to produce M3B. It is also likely that the generated M3B can be re-adsorbed and hydrogenated to form MB according to the semi-hydrogenated intermediate pathway. From the above reaction pathway, it can be said that the MC hydrogenation over Pt/Al2O3 follows the Horiuti–Polanyi mechanism despite the presence of conjugated multi-functional groups in MC compared to olefins.
 | ||
| Fig. 8 Proposed reaction pathway for MC hydrogenation to form MB and M3B over the Pt/Al2O3 catalyst. | ||
3.6. Reaction kinetics
Besides determination of the reaction pathway for MC hydrogenation, another additional objective is the identification of the rate-determining step of the reaction. This can aid in understanding the activity difference among metals but also provide possible means to modify or design catalysts towards improved hydrogenation. Despite the fact that the reaction pathway may be largely explained by the Horiuti–Polanyi mechanism, the rate-determining step in the reaction pathway is still not clear. To better understand MC hydrogenation over the Pt/Al2O3 catalyst, we performed catalytic kinetics over two catalysts with different Pt dispersions. | ||
| Fig. 9 MC conversions over the (a) Pt/Al2O3-500 and (b) Pt/Al2O3-700 catalysts at different MC and H2 partial pressures and reaction temperatures. | ||
| Sample | Species | ln(A/mol g−1 s−1) | E a (kJ mol−1) | n(MC) | n(H2) | R 2 |
|---|---|---|---|---|---|---|
a The power-law model used for the fitting is as follows:  , where yi is the mole fraction of the reactant (i). The method used for the fitting is a combination of the standard Levenberge Marquardt (LM) and the Universal-Global-Optimization (UGO) method. , where yi is the mole fraction of the reactant (i). The method used for the fitting is a combination of the standard Levenberge Marquardt (LM) and the Universal-Global-Optimization (UGO) method.
|
||||||
| Pt/Al2O3-500 | MC | 4.27 | 50.4 | −0.17 | 0.72 | 0.96 |
| MB | 6.35 | 61.1 | −0.53 | 1.02 | 0.98 | |
| M3B | 1.02 | 40.9 | 0.30 | 0.30 | 0.94 | |
| Pt/Al2O3-700 | MC | −7.44 | 36.0 | −1.28 | 0.81 | 0.96 |
| MB | −6.91 | 44.3 | −1.75 | 1.10 | 0.98 | |
| M3B | −9.96 | 28.4 | −0.76 | 0.31 | 0.93 | |
 | ||
| Fig. 10 Experimental reaction rates versus calculated values by using different models: (a and b) the power-law model over the Pt/Al2O3-500 and the Pt/Al2O3-700 sample, respectively. (c and d) LHHW model with step (ii) and step (iv) as rate determining for MB formation, respectively. | ||
The obtained apparent kinetic parameters for the two samples had similarities but also clear differences. As shown in Table 1, all reaction rates showed positive reaction orders with respect to H2 but negative MC reaction orders except that of M3B production rate over the Pt/Al2O3-500 catalyst. For the two samples, the H2 reaction orders for hydrogenation and shift of CC in MC are about 1 and 0.3, respectively. This further confirms that both the hydrogenation and the shift of CC bond in MC over Pt/Al2O3 are related to the availability of adsorbed hydrogen on the catalyst.
Although the H2 reaction order was similar over the two samples, the reaction orders with respect to MC showed larger differences. Note that the reaction temperature used to measure the kinetics for the Pt/Al2O3-500 sample is in the range of 60–110 °C, while that of the other sample is between 60–150 °C. It may be argued that a direct comparison cannot be made between the two samples due to the different temperature ranges over which the kinetic data were gathered. If only the reaction temperature should explain the different kinetic parameters for the two samples, it would be expected that the reaction orders for MC at higher temperature should be less negative, since at higher temperature less MC should be adsorbed and it thus would have less of an inhibiting effect. However, as discussed below, the reaction order with respect to MC is more negative for the Pt/Al2O3-700 sample for which the temperature range was wider and higher. Thus, the differences observed in the following kinetic parameters should be more related to the nature of the catalyst rather than the temperature range used.
As shown in Table 1, the MC reaction orders over Pt/Al2O3-700 are more negative than those for the Pt/Al2O3-500 sample, indicating a more negative dependence on MC surface coverage on larger Pt particles. Furthermore, over the Pt/Al2O3-500 sample the reaction rate of M3B had a positive MC reaction order of 0.3, while the other sample showed a negative reaction order for M3B formation. If a completely competitive adsorption between MC and H2 is assumed, the formation of M3B is more likely to display near zero or a negative reaction order with respect to MC. This indicates the existence of non-competitive sites that are available for H2 but not for MC adsorption. However, these sites should be quite limited and hence the H coverage from these sites was low. This would cause an H-lean condition on the Pt surface and be favorable for M3B formation, according to the transient experiments. The amount of non-competitive sites for H2 adsorption may be related to the Pt particle size and the MC orientation on the surface. According to the above product analysis, it is proposed that MC adsorbs on Pt via the CC group while the CO group is not in direct contact with the surface of the catalyst. It is imaginable there are some adsorption sites, especially those that are located vertical to the CO group, which cannot further adsorb MC due to space repulsive MC–MC interactions. However, repulsive interactions between MC and H2 may be alleviated since H2 is considerably smaller than MC. Thus, it is understandable that there are a limited number of non-competitive adsorption sites accessible for H2, resulting in a positive MC reaction order for the formation of M3B. Based on the above analysis, we propose that the adsorption of MC and H2 on Pt/Al2O3 is partially competitive, though the number of non-competitive sites are likely quite limited.
Contrary to the approximately equal reaction order with respect to H2, significant differences in the pre-exponential factors for the two Pt/Al2O3 samples were obtained. It suggests that the number of collisions that result in reaction were higher for Pt/Al2O3-500 than for the Pt/Al2O3-700 catalyst. One possible reason is that the former catalyst had a smaller particle size of Pt and hence more unsaturated coordination Pt sites were available for the reaction. However, this can be ruled out from the comparison of TOFs, as shown in Fig. 3. Another possibility is a higher coverage of MC on the Pt/Al2O3-700 sample since it had a more negative order with respect to MC than the Pt/Al2O3-500 sample (Table 1). This would lower the H coverage on the Pt/Al2O3-700 catalyst and hence reduce the possibility for reaction collisions. It is also possible that the non-competitive sites for H2 adsorption on Pt/Al2O3-700 were relatively less than that on the Pt/Al2O3-500 sample. This can lead to a significant decrease in the number of collisions.
Besides the higher rate constants, the activation energies towards the reactions were higher over Pt/Al2O3-500 than for the other sample. This may also be related to the smaller Pt particle size, which leads to different relative amounts of adsorption sites, e.g., facets, edges, and corners. The combination of reactions on these sites could lead to a higher apparent activation energy for a catalytic reaction since these sites may have different reactivities towards a reaction.
C double bond are assumed to occupy two vacant sites though the exact number is not known. In addition, the negative reaction order with respect to MC suggests that the adsorption of MC on Pt should be stronger than H2 and cannot be the rate-determining step. Hence, the adsorption of MC (step (i)) is assumed to be quasi-equilibrated.
| Elementary stepa | Rate expressionb | H2 reaction order | ||||
|---|---|---|---|---|---|---|
| a * represents a free surface site and X* is an adsorbed X species. b K i, ki and θ are the equilibrium constants, the forward rate constant parameters, and the coverage, respectively. | ||||||
| i | MC + 2* ↔ *MC* |
|
— | |||
| ii | H2 + 2* ↔ 2H* |
|
≤ 1 | |||
| iii | *MC* + H* ↔ MCH* + 2* |
|
≤ 0.5 | |||
| iv | MCH* + H* ↔ MB + 2* |
|
≤ 1 | |||
| v | MCH* + 2* ↔ *M3B* + H* |
|
≤ 0.5 | |||
| vi | *M3B* ↔ M3B + 2* |
|
≤ 0 | |||

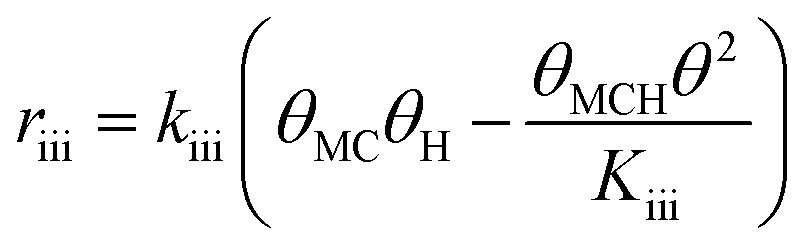
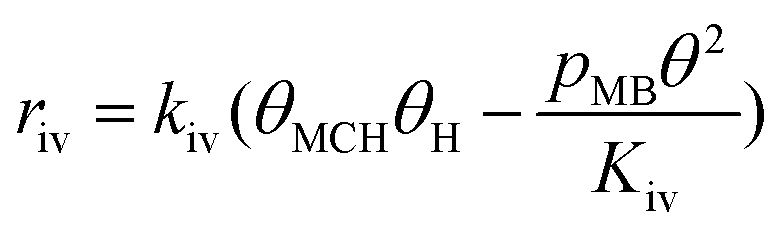


Since a good fit to the kinetic data was given by the power-law model, we focus on comparison between possible LHHW rate expressions derived from the reaction pathway and the power-law model. As shown in the proposed reaction pathway (Table 2), the formation of MB involves the steps (i), (ii), (iii), and (iv), while the selectivity to M3B is related to the steps (i), (ii), (iii), (v), and (vi). The formation of MB and M3B share the first three elementary steps (i–iii). The possible H2 reaction orders for each of the elementary steps are also included in Table 2. Since the reaction order with respect to H2 was about 1 for MB and 0.3 for M3B for both catalysts, the rate-determining step for MB could be step (ii) or (iv), while that for M3B could be (ii), or (iii), or (v). If the shared elementary step (ii) is the rate determining step for the formation of MB, the observed activation energy for M3B should be similar to that of MB. This is against the results obtained using the power-law model, from which the apparent activation energy of M3B is always lower than that of MB over the two catalysts. Thus, it can be concluded that the rate-determining step for the formation of MB should be the step (iv), namely, the surface reaction between the intermediate and one proton. In this case, the proceeding steps (i), (ii), and (iii), involving the formation of MB can be assumed at equilibrium:
 | (7) |
 | (8) |
By combining the eqn (1), (7), and (8) and assuming that the adsorption of MB on the catalyst surface was negligible according to the transient experiments, the rate expression of MB can be obtained.
| rMB = riv = kivθMCHθH = kivKiKiiKiiipH2pMCθ2 | (9) |
The fraction of vacant sites, θ, can usually be expressed as a function of the partial pressures of reactants with an inverse dependence on reactant pressure. If the reaction order of H2 is 1, the fractional coverage θ in the rate expression (eqn (9)) should be independent of the hydrogen partial pressure. It indicates that the adsorption of H2 on the catalyst surface should be negligible compared to the adsorption of MC. The site balance gives
| θ = 1 − θMC − θH − θMCH − θM3B | (10) |
Since M3B had a low partial pressure in the product for the experiments considered here, θM3B could be assumed to be negligible. Then eqn (10) can be further simplified:
| θ = 1 − θMC − θMCH | (11) |
Using the eqn (1), (7), and (8) to give θMC and θMCH in terms of θ, we obtain
 | (12) |
Solving for θ gives
 | (13) |
After substituting θ in the rate expression for MB, we find
 | (14) |
From the above MB rate expression, it can be found that the reaction order with respect to MC is not negative, but instead near zero order. This conflicts with the results obtained using the power-law model where the MB reaction rate had a negative MC reaction order. This inconsistency between the rate expression derived from the above assumptions and the experimental results indicates that the fully competitive Horiuti–Polanyi mechanism does not capture all of the characteristics of MC hydrogenation over Pt/Al2O3.
Therefore, the initial proposed reaction pathway needs to be modified to be fully consistent with the kinetics. Another possibility is that the existence of partially competitive adsorption between MC and H2 on Pt/Al2O3, as mentioned above. In this case, the hydrogenation and the CC bond shift could come from partially different H sources. For the partially competitive adsorption mode, it is still desirable to answer which step could possibly be the rate-determining step. Revisiting all the data may be helpful to understand this. As evidenced by the H2 adsorption behavior discussed in section 3.6 above, the coverage of H should be quite limited when MC is pre-adsorbed on the catalyst surface even considering a non-competitive pathway for H2 and MC. Hence, the coverages of the main species involved in the reaction pathway would still follow eqn (11). If the surface reaction is assumed to be the rate-determining step, the relationship between the MB formation rate and the MC partial pressure should remain similar to eqn (14) even in the presence of non-competitive sites for H2 adsorption. Hence, the elementary step (iv) cannot be considered to be the rate-determining step. Considering the fact that the hydrogenation rate displayed a first-order dependence on the H2 partial pressure, the only possible rate-determining step is thus the H2 dissociation (step (ii)).
To further confirm the suitability of these steps as rate determining, LHHW rate expressions based on steps (ii) or (iv) were fitted to the reaction rates of MB formation. In this case only the kinetic data for the Pt/Al2O3-700 catalyst was included since the reaction with respect to MC was of higher negative order and thus less likely influenced by a possible non-competitive reaction pathway. To derive the LHHW rate expressions, the MB, H2, and M3B coverages were again assumed to be negligible compared to that of MC on the Pt surface. Note that the rate expression and site balance for step (iv) as rate determining in Table 3 are identical to eqn (9) and (12), respectively, except that the parameters are lumped. Table 3 shows the rate expressions and the resulting kinetic parameter values obtained by a nonlinear regression. Parity plots for the fitted overall rate expressions are shown in Fig. 10(c) and (d). From the values of the correlation coefficients in Table 3 and the parity plots, it is evident that the rate expression based on step (ii) as the rate-determining step provides a better fit to the kinetic hydrogenation data. The apparent orders with respect to H2 and MC from the model with step (ii) as the rate-determining step are 1.0 and −1.0 respectively, while those of the model derived from step (vi) are 0.9 and −0.1. The former model with step (ii) as the rate-determining step is basically consistent with the power-law model. This is because its rate expression depends directly on H2 but not on the partial pressure of MC, whereas the fraction of vacant sites was inversely dependent on the MC partial pressure. This leads to a negative order dependence on the partial pressure of MC. Furthermore, for the experimental conditions the ranges of coverages for MC and MCH were estimated to be 0.92–0.93 and 0.001–0.026, respectively. As a result the coverages for the other species were negligible compared to that of MC. This is consistent with the previous assumption which is used to derive the overall LHHW rate expression. As for the LHHW rate expression based on step (iv) as rate determining, a near zero order dependence on MC partial pressure is obtained, as predicted above and thus it shows a poorer fit to the kinetic data.
| Rate determining step | Rate expression | Parameter values | R 2 | ||
|---|---|---|---|---|---|
| ii | r MB = kPH2θ2 | k | A | 1.13 × 1012 mol g−1 s−1 Pa−1 | 0.96 |
| E | 36.3 kJ mol−1 | ||||
| with site balance: θ = 1 − θMC + θMCH | K 1 | A | 5.02 × 108 Pa−1 | ||
| E | −6.3 kJ mol−1 | ||||

|
K 2 | A | 3.91 × 10−6 Pa−1 | ||
| E | −49.2 kJ mol−1 | ||||
| iv | r MB = kPMCPH2θ2 | k | A | 6.25 × 1013 mol g−1 s−1 Pa−2 | 0.85 |
| E | 27.6 kJ mol−1 | ||||
| with site balance: θMC + θMCH + θ = 1 | K 1 | A | 4.76 × 108 Pa−1 | ||
| E | −10.5 kJ mol−1 | ||||

|
K 2 | A | 6.97 × 109 Pa−1.5 | ||
| E | −10.2 kJ mol−1 | ||||
3.7. Comparison to related catalytic hydrogenations
Hydrogenation of CC is an important reaction in heterogeneous catalysis and has been widely studied. Considering the absence of fatty alcohol in the products, it is believed that the functional group on the catalyst surface should be the CC rather than the CO in MC. In this sense, it is of interest to make a comparison of hydrogenation between MC and ethylene, which is a probe molecule in the field of CC hydrogenation. Actually the Horiuti–Polanyi mechanism was originally proposed for ethylene hydrogenation.5 However, the adsorption mode of ethylene and H2 on the catalyst was not identified in the study since the hydrogenation rate was not observed to decrease with increasing ethylene partial pressure. Microkinetic studies of ethylene hydrogenation on supported Pt catalysts showed that a non-competitive adsorption mode was dominant at low temperatures while a competitive pathway was proposed at relatively higher temperatures.55 This is different from the present MC hydrogenation which has negative orders at the studied temperature range. Another kinetic study found that the ethylene hydrogenation was structure insensitive over Pt supported on silica and on Pt wire.56 This is also different from the MC hydrogenation which is sensitive to Pt dispersion. It appears to be difficult to build a direct relationship between MC and ethylene hydrogenation on the Pt surface probably due to the nature of the differences of the two molecules. However, from the viewpoint of kinetics, there are still similar hydrogenation characteristics between MC and ethylene. A direct common characteristic for ethylene and MC hydrogenation on Pt is the observation of first order dependence on H2. This indicates that both ethylene and MC hydrogenation on a Pt surface is affected by the availability of adsorbed hydrogen. Another similar characteristic is that the combination of competitive and non-competitive Horiuti–Polanyi mechanism is required to interpret the hydrogenation kinetics on Pt.
One can also consider how MC and crotonaldehyde (CH3CHCHCHO) hydrogenation compare over supported Pt catalysts. The two molecules have two same bonds, i.e., CC and CO, but different group (methoxy vs. H) attached to the CO group. Englisch et al. showed that hydrogenation of crotonaldehyde was sensitive to the particle size of Pt supported on SiO2 and TiO2.12 In this respect, it appears that the two molecules could have similar hydrogenation behaviors over the Pt surface. However, the selectivity and the main reaction pathways of the hydrogenation of the two molecules over Pt are different. In the case of crotonaldehyde hydrogenation, at comparable reaction conditions, the reaction pathways mainly involve the hydrogenation of CC, CO, decarbonylation, and complete saturation of double bonds. The corresponding products are butyraldehyde from CC hydrogenation, crotylalcohol via CO hydrogenation, propylene, and saturated alcohol (butanol), respectively. Vannice and Sen reported 100% selectivity to the hydrogenation of the CC bond of crotonaldehyde over the Pt supported on Al2O3 or SiO2 at around 45 °C.57 Recent kinetic studies from Kliewer et al. demonstrated that the selectivity of crotonaldehyde hydrogenation over Pt(111) was related to the reaction temperature.58 In the temperature range between 30 and 120 °C, the selectivity to CC hydrogenation increased and that to CO hydrogenation decreased with increasing reaction temperature. At the same time, the selectivity to decarbonylation remained almost constant. From these results, the dominant reaction pathways of crotonaldehyde hydrogenation over Pt surface are the hydrogenation of the CC and the CO group. However, in the present study MC hydrogenation involves hydrogenation and shift of the CC bond without the CO hydrogenation. More importantly, the two reaction pathways in this study share a common adsorbed intermediate species, while those in crotonaldehyde hydrogenation over Pt surface are parallel with different adsorbed intermediate species. Obviously, the correlation between MC and crotonaldehyde hydrogenation cannot be built. The difference could be related to the steric hindrance due to the presence of methoxy group on the side of the CO group.
4. Conclusions
In summary, this study has investigated gas-phase MC hydrogenation over Pt/Al2O3 as a probe reaction to understand CC hydrogenation of a methyl ester. The hydrogenation and the shift of the CC bond in MC were identified as the main reactions during the MC hydrogenation over the catalyst. It is revealed that the formation of MB and M3B from MC hydrogenation are parallel reactions originating from a common semi-hydrogenated surface intermediate species. The activity for both hydrogenation and shift of CC bond in MC was found to decrease with decreasing Pt dispersion. The availability of hydrogen on the catalyst determines the selectivity of the MC hydrogenation over the catalyst, namely, a hydrogen-rich environment is favorable for CC hydrogenation, while hydrogen-lean conditions promote the CC bond shift. The hydrogenation of the CC bond in MC over Pt/Al2O3 appears to proceed via the Horiuti–Polanyi mechanism with a more preferable adsorption of MC than H2. Furthermore, the strongly adsorbed MC on the Pt surface impedes adsorption and dissociation of H2 on the catalyst since in this case the H2 adsorption decreased significantly compared to that on the clean Pt/Al2O3 catalyst. However, the kinetic analysis indicates that the overall LHHW rate expressions derived from the fully competitive Horiuti–Polanyi mechanism are inconsistent with the results obtained using a power-law model which provided a good fit to the kinetic data. This is particularly the case for the catalyst with relatively higher Pt dispersion, whereas a fully-competitive mechanism is more appropriate for the catalyst with low Pt dispersion. A combination of competitive and non-competitive adsorption between H2 and MC is more reasonable to explain the reaction kinetics over the two samples, though no other direct evidence except the kinetic analysis supports the existence of a non-competitive adsorption mechanism. If the partially competitive adsorption between MC and H2 is considered, the rate-determining step for MC hydrogenation would be the dissociation of H2 on the Pt/Al2O3 catalyst. The information in this study contributes to a deepened understanding of hydrogenation of unsaturated methyl esters and application of the Horiuti–Polanyi mechanism.
Acknowledgements
This work has been performed within the Competence Centre for Catalysis, which is hosted by Chalmers University of Technology and financially supported by the Swedish Energy Agency and the member companies AB Volvo, ECAPS AB, Haldor Topsøe A/S, Scania CV AB, Volvo Car Corporation AB and Wärtsilä Finland Oy.Notes and references
- E. Santacesaria, G. Martinez Vicente, M. Di Serio and R. Tesser, Catal. Today, 2012, 195, 2 CrossRef CAS PubMed.
- T. Issariyakul, M. G. Kulkarni, L. C. Meher, A. K. Dalai and N. N. Bakhshi, Chem. Eng. J., 2008, 140, 77 CrossRef CAS PubMed.
- B. R. Moser, Fuel, 2012, 92, 231–238 CrossRef CAS PubMed.
- O. Falk and R. Meyer-Pittroff, Eur. J. Lipid Sci. Technol., 2004, 106, 837 CrossRef CAS.
- M. Polanyi and J. Horiuti, Trans. Faraday Soc., 1934, 30, 1164 RSC.
- A. M. Doyle, Sh. K. Shaikhutdinov and H.-J. Freund, J. Catal., 2004, 223, 444 CrossRef CAS PubMed.
- A. M. Doyle, Sh. K. Shaikhutdinov, S. D. Jackson and H.-J. Freund, Angew. Chem., Int. Ed., 2003, 42, 5240 CrossRef CAS PubMed.
- B. Brandt, J. H. Fischer, W. Ludwig, J. Libuda, F. Zaera, S. Schauermann and H. J. Freund, J. Phys. Chem. C, 2008, 112, 11408 CAS.
- X.-X. Wang, H.-Y. Zheng, X.-J. Liu, G.-Q. Xie, J.-Q. Lu, L.-Y. Jin and M.-F. Luo, Appl. Catal., A, 2010, 388, 134 CrossRef CAS PubMed.
- G. Kennedy, L. R. Baker and G. A. Somorjai, Angew. Chem., Int. Ed., 2014, 53, 3405 CrossRef CAS PubMed.
- G. Budroni, S. A. Kondrat, S. H. Taylor, D. J. Morgan, A. F. Carley, P. B. Williamsy and G. J. Hutchings, Catal. Sci. Technol., 2013, 3, 2746 CAS.
- M. Englisch, A. Jentys and J. A. Lercher, J. Catal., 1997, 166, 25 CrossRef CAS.
- I. Kun, G. Szöllösi and M. Bartók, J. Mol. Catal. A: Chem., 2001, 169, 235–246 CrossRef CAS.
- F. Coloma, J. Llorca, N. Homs, P. R. de la Piscina, F. Rodríguez-Reinoso and A. Sepúlveda-Escribano, Phys. Chem. Chem. Phys., 2000, 2, 3063 RSC.
- M. Abid, V. Paul-Boncour and R. Touroude, Appl. Catal., A, 2006, 297, 48 CrossRef CAS PubMed.
- C. E. Volckmar, M. Brona, U. Bentrup, A. Martin and P. Claus, J. Catal., 2009, 261, 1 CrossRef CAS PubMed.
- C. Hoang-Van and O. Zegaoui, Appl. Catal., A, 1997, 164, 91 CrossRef CAS.
- H. Wei, C. Gomez, J. Liu, N. Guo, T. Wu, R. Lobo-Lapidus, C. L. Marshall, J. T. Miller and R. J. Meyer, J. Catal., 2013, 298, 18 CrossRef CAS PubMed.
- M. S. Ide, B. Hao, M. Neurock and R. J. Davis, ACS Catal., 2012, 2, 671 CrossRef CAS.
- X. Yang, A. Wang, X. Wang, T. Zhang, K. Han and J. Li, J. Phys. Chem. C, 2009, 113, 20918 CAS.
- M. B. Macher, J. Högberg, P. Møller and M. Härröd, Fett/Lipid, 1999, 101, 301 CrossRef CAS.
- E. Ramírez, F. Recasens, M. Fernández and M. A. Larrayoz, AIChE J., 2004, 50, 1545 CrossRef.
- E. Santacesaria, P. Parrella, M. snm Di Serio and G. Borrelli, Appl. Catal., A, 1994, 116, 269 CrossRef CAS.
- B. S. Souza, D. M. M. Pinho, E. C. Leopoldino, P. A. Z. Suarez and F. Nome, Appl. Catal., A, 2012, 433–434, 109 CrossRef CAS PubMed.
- M. B. Fernández, M. J. F. Sánchez, G. M. Tonetto and D. E. Damiani, Chem. Eng. J., 2009, 155, 941 CrossRef PubMed.
- M. J. F. Sánchez, D. E. Boldrini, G. M. Tonetto and D. E. Damiani, Chem. Eng. J., 2011, 167, 355 CrossRef PubMed.
- A. F. Pérez-Cadenas, M. M. P. Zieverink, F. Kapteijn and J. A. Moulijn, Carbon, 2006, 44, 173 CrossRef PubMed.
- A. F. Pérez-Cadenas, F. Kapteijn, M. M. P. Zieverink and J. A. Moulijn, Catal. Today, 2007, 128, 13 CrossRef PubMed.
- A. F. Pérez-Cadenas, M. M. P. Zieverink, F. Kapteijn and J. A. Moulijn, Catal. Today, 2005, 105, 623 CrossRef PubMed.
- N. Numwong, A. Luengnaruemitchai, N. Chollacoop and Y. Yoshimura, Appl. Catal., A, 2012, 441–442, 72 CrossRef CAS PubMed.
- A. Philippaerts, S. Paulussen, S. Turner, O. I. Lebedev, G. V. Tendeloo, H. Poelman, M. Bulut, F. D. Clippel, P. Smeets, B. Sels and P. Jacobs, J. Catal., 2010, 270, 172 CrossRef CAS PubMed.
- A. J. Dijkstra, Eur. J. Lipid Sci. Technol., 2006, 108, 249 CrossRef CAS.
- I. V. Deliy, I. L. Simakova, N. Ravasio and R. Psaro, Appl. Catal., A, 2009, 357, 170 CrossRef CAS PubMed.
- S. McArdle, S. Girish, J. J. Leahy and T. Curtin, J. Mol. Catal. A: Chem., 2011, 351, 179 CrossRef CAS PubMed.
- F. Zaccheria, N. Ravasio, C. E. Chan-Thaw, N. Scotti and P. Bondioli, Top. Catal., 2012, 55, 631 CrossRef CAS.
- C. Hu, D. Creaser, S. Siahrostami, H. Grönbeck, H. Ojagh and M. Skoglundh, Catal. Sci. Technol., 2014, 4, 2427 CAS.
- G. H. Jonker, J.-W. Veldsink and A. A. C. M. Beenackers, Ind. Eng. Chem. Res., 1997, 36, 1567 CrossRef CAS.
- M. I. Cabrera and R. J. Grau, J. Mol. Catal. A: Chem., 2006, 260, 269 CrossRef CAS PubMed.
- M. I. Cabrera and R. J. Grau, J. Mol. Catal. A: Chem., 2008, 287, 24 CrossRef CAS PubMed.
- S. M. Sarathy, S. Gaïl, S. A. Syed, M. J. Thomson and P. Dagaut, Proc. Combust. Inst., 2007, 31, 1015 CrossRef PubMed.
- H. Bennadij, L. Coniglio, F. Billaud, R. Bounaceur, V. Warth, P.-A. Glaude and F. Battin-Leclerc, Int. J. Chem. Kinet., 2011, 43, 204 CrossRef.
- R. Grana, A. Frassoldati, A. Cuoci, T. Faravelli and E. Ranzi, Energy, 2012, 43, 124 CrossRef CAS PubMed.
- X. Auvray, T. Pingel, E. Olsson and L. Olsson, Appl. Catal., B, 2013, 129, 517 CrossRef CAS PubMed.
- A. J. Plomp, H. Vuori, A. O. I. Krause, K. P. de Jong and J. H. Bitter, Appl. Catal., A, 2008, 351, 9 CrossRef CAS PubMed.
- C. M. Jeong, G. W. Park, J. Choi, J. W. Kang, S. M. Kim, W.-H. Lee, S. I. Woo and H. N. Chang, Int. J. Hydrogen Energy, 2011, 36, 7505 CrossRef CAS PubMed.
- P. Forzatti and L. Lietti, Catal. Today, 1999, 52, 165 CrossRef CAS.
- P. M. Mortensen, J.-D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1 CrossRef CAS PubMed.
- G. de la Puente, A. Gil, J. J. Pis and P. Grange, Langmuir, 1999, 15, 5800 CrossRef CAS.
- J. P. Boitiaux, J. Cosyns and E. Robert, Appl. Catal., 1987, 35, 193 CrossRef CAS.
- A. M. Gololobov, I. E. Bekk, G. O. Bragina, V. I. Zaikovskii, A. B. Ayupov, N. S. Telegina, V. I. Bukhtiyarov and A. Y. Stakheeva, Kinet. Catal., 2009, 50, 830 CrossRef CAS.
- M. Haneda, T. Watanabe, N. Kamiuchi and M. Ozawa, Appl. Catal., B, 2013, 142–143, 8 CrossRef CAS PubMed.
- M. A. Vannice, L. C. Hasselbring and B. Sen, J. Phys. Chem., 1985, 89, 2972 CrossRef CAS.
- A. J. Dijkstra, Int. News Fats, Oils Relat. Mater., 1997, 8, 1150 Search PubMed.
- M. P. Latusek, B. P. Spigarelli, R. M. Heimerl and J. H. Holles, J. Catal., 2009, 263, 306 CrossRef CAS PubMed.
- J. E. Rekoske, R. D. Cortright, S. A. Goddard, S. B. Sharma and J. A. Dumesic, J. Phys. Chem., 1992, 96, 1880 CrossRef CAS.
- R. D. Cortright, S. A. Goddard, J. E. Rekoske and J. A. Dumesic, J. Catal., 1991, 127, 342 CrossRef CAS.
- M. Albert Vannice and B. Sen, J. Catal., 1989, 115, 65 CrossRef.
- C. J. Kliewer, M. Bieri and G. A. Somorjai, J. Am. Chem. Soc., 2009, 131, 9958 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Thermal decomposition of the Pt precursor, H2-TPR profiles of the Pt/Al2O3 catalysts, typical GC spectrum, GC peak areas of the minor species with reaction temperature, and carbon balance. See DOI: 10.1039/c4cy01470g |
| This journal is © The Royal Society of Chemistry 2015 |