
Non-noble bimetallic CuCo nanoparticles encapsulated in the pores of metal–organic frameworks: synergetic catalysis in the hydrolysis of ammonia borane for hydrogen generation
Jun
Li
ab,
Qi-Long
Zhu
a and
Qiang
Xu
*ab
aNational Institute of Advanced Industrial Science and Technology (AIST), Ikeda, Osaka, Japan. E-mail: q.xu@aist.go.jp; Fax: (+81)72 751 9629
bGraduate School of Engineering, Kobe University, Nada Ku, Kobe, Hyogo, Japan
First published on 16th September 2014
Abstract
Non-noble bimetallic CuCo alloy nanoparticles (NPs) were successfully encapsulated in the pores of MIL-101 by using the double-solvent method combined with the overwhelming reduction approach. Compared with their monometallic counterparts, the bimetallic CuCo alloy NPs present much higher catalytic activity for hydrolytic dehydrogenation of ammonia borane (AB) to generate a stoichiometric amount of hydrogen at room temperature for chemical hydrogen storage. The synergistic effect between copper and cobalt species plays an important role for the improved performance in the catalytic hydrolysis of AB.
1. Introduction
Hydrogen has been considered as one of the best alternative energy carriers to satisfy the increasing demand for a sustainable and clean energy supply.1 The development of effective hydrogen storage materials is imperative but challenging for establishing a hydrogen-based energy system.2 Ammonia borane (NH3BH3, AB) has become an attractive candidate for chemical hydrogen storage applications due to its high gravimetric hydrogen capacity (19.6 wt%) and low molecular weight (30.86 g mol−1).3 However, one of the major obstacles for the practical application of this system is to develop low-cost and highly efficient catalysts for the improvement of the kinetic properties under moderate conditions.4So far, various catalyst systems have been tested in the hydrolysis of AB and rapid hydrogen generation has been achieved by using noble metals such as Pt, Ru and Rh, although the limited resources restrict their practical applications.5 Alternatively, cost-effective non-noble metals such as Co, Ni, and Fe have been developed.6,7 However, these materials tend to be fairly unstable in solution and air atmospheres due to magnetism-induced agglomeration and easy oxidation. To solve this problem, several strategies, including fabricating non-noble metal-based alloys8 or core–shell9 nanoparticles (NPs) and structured Co–B film catalysts,10 have been explored. Despite much progress, development of high-performance catalysts for AB dehydrogenation with high activity and desired durability simultaneously remains a huge challenge.
In the design and fabrication of highly efficient catalysts for AB dehydrogenation, the dispersion and stabilization of active metal nanoparticles (MNPs) are two key factors. Metal–organic frameworks (MOFs) synthesized by assembling metal ions with organic ligands have emerged as a new class of porous materials due to their high porosity, large surface area and chemical tenability.11 Given the similarity to zeolites, depositing MNPs to MOFs could afford heterogeneous catalysts.12 It is expected that the crystalline porous structures of the MOF would limit the migration and aggregation of the active components and thus enhance the long-term stability. There were some reports of MOF-supported non-noble metal NPs as catalysts for heterogeneous reactions, although it was difficult to completely avoid MNP aggregation on the external surface of MOFs.13 Therefore, it is a big challenge to develop a general and facile method to incorporate fine MNPs within the pores of MOFs. Very recently, we have exploited a strategy via the overwhelming reduction (OWR) approach combined with a double-solvent method (DSM), which can encapsulate the non-noble metal-based NPs (AuNi and AuCo) in the pores of the MOF without MNP aggregation on the external surface of the framework.14
In this work, we report successful preparation and excellent catalytic activity of the ultrafine CuCo alloy NPs encapsulated in the pores of MOF without aggregation on the external surfaces of the host framework by using DSM in combination with the OWR approach, which presents the first example of MOF-supported non-noble bimetallic catalysts for hydrogen generation from the hydrolysis of AB.
2. Experimental
2.1 Chemicals
All chemicals were commercial and were used without further purification. Chromic nitrate nonahydrate (Cr(NO3)3·9H2O, 99%; Sigma-Aldrich), aqueous hydrofluoric acid (HF, 46%; Kishida Chemical Co. Ltd.), ammonium fluoride (NH4F, >97%; Kishida Chemical Co. Ltd.), cobalt(II) chloride hexahydrate (CoCl2·6H2O, >99%; Wako Pure Chemical Industries, Ltd.), copper(II) nitrate trihydrate (Cu(NO3)2·3H2O, >99.5%; Kishida Chemical Co. Ltd.), sodium borohydride (NaBH4, 99%; Aldrich), ammonia borane (NH3BH3, >97%; JSC Aviabor), terephthalic acid (HOOCC6H4COOH, 99%; Tri Chemical Laboratories Inc.), anhydrous n-hexane (Sigma-Aldrich), Vulcan XC-72R (carbon, specific surface area = 240 m2 g−1; Cabot Corp., USA) and ethanol (C2H5OH, >99.8%; Kishida Chemical Co. Ltd.) were used as received. Deionized water with a specific resistance of 18.2 MΩ cm was obtained by reverse osmosis followed by ion exchange and filtration (RFD 250NB; Toyo Seisakusho Kaisha, Ltd., Japan).2.2 Synthesis of MIL-101
MIL-101 was synthesized using the reported procedure.15 Terephthalic acid (1.661 g, 10.0 mmol), Cr(NO3)3·9H2O (4.002 g, 10.0 mmol), aqueous HF (0.5 mL, 46 wt%) and deionized water (70 mL) were placed in a 100 mL Teflon-lined autoclave and heated at 220 °C for 8 h. After cooling, the resulting green powder of MIL-101 with the formula Cr3F(H2O)2O[(O2C)C6H4(CO2)]3·nH2O (n ≈ 25) was doubly filtered off using two glass filters with pore sizes between 40 and 100 μm to remove the unreacted crystals of terephthalic acid, and then further purified by solvothermal treatment in ethanol at 80 °C for 24 h. The resulting green solid was soaked in NH4F (1.0 M) solution at 70 °C for 24 h to eliminate the terephthalic acid inside the pores of MIL-101 and immediately filtered, washed with hot water several times, and finally dried overnight at 150 °C under vacuum for further use.2.3 Preparation of Cu@MIL-101, Co@MIL-101, and CuCo@MIL-101
Encapsulation of Cu2+ and/or Co2+ precursors was carried out by double-solvent method.5f Typically, 100 mg of green MIL-101 powder activated by heating at 150 °C for 12 h under dynamic vacuum, which has a pore volume of 1.72 cm3 g−1 as determined by N2 sorption isotherm, was suspended in 20 mL of dry n-hexane as hydrophobic solvent and the mixture was sonicated for 15 min until it became homogeneous. After stirring for 2 h, 0.15 mL of aqueous Cu(NO3)2·3H2O and/or CoCl2·6H2O solution with the desired concentrations as the hydrophilic solvent was added dropwise over a period of 15 min with constant vigorous stirring. The resulting solution was continuously stirred for 2 h. After filtration, the green powder was dried in air at room temperature. These synthesized samples were further dried at 150 °C under vacuum for 12 h. The molar ratios of Cu2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(Cu2+ + Co2+) were varied resulting in several values (0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.7, 0.8 and 1.00), while the molar contents of (Cu2+ + Co2+) added to 100 mg of MIL-101 matrix were kept at 0.034 mmol. After dehydration of the Cu2+Co2+@MIL-101 with different molar ratios of Cu2+ to (Cu2+ + Co2+), an overwhelming reduction approach14 was employed by using 5 mL of freshly prepared 0.6 M aqueous NaBH4 solution with vigorous stirring, resulting in the generation of catalysts as a dark green suspension. The synthesized samples were collected by centrifugation and used for the catalytic reactions.
:(Cu2+ + Co2+) were varied resulting in several values (0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.7, 0.8 and 1.00), while the molar contents of (Cu2+ + Co2+) added to 100 mg of MIL-101 matrix were kept at 0.034 mmol. After dehydration of the Cu2+Co2+@MIL-101 with different molar ratios of Cu2+ to (Cu2+ + Co2+), an overwhelming reduction approach14 was employed by using 5 mL of freshly prepared 0.6 M aqueous NaBH4 solution with vigorous stirring, resulting in the generation of catalysts as a dark green suspension. The synthesized samples were collected by centrifugation and used for the catalytic reactions.
2.4 Preparation of CuCo/XC-72R and CuCo/MIL-101
For comparison, CuCo/XC-72R and CuCo/MIL-101 catalysts were prepared. 100 mg of XC-72R carbon powder or activated MIL-101 was dispersed in 4.5 mL of aqueous Cu(NO3)2·3H2O and/or CoCl2·6H2O solution. The molar ratio of Cu2+:(Cu2+ + Co2+) was fixed at 0.3, while the molar contents of (Cu2+ + Co2+) added to 100 mg of XC-72R or MIL-101 were kept at 0.034 mmol. The resulted aqueous suspension was further homogenized under sonication for 0.5 h. Then, 20 mg of NaBH4 dissolved in 0.50 mL of water was added to the above obtained solution with vigorous shaking. The mixture was further shaken for 0.5 h at room temperature to fully deposit the metallic nanoparticles onto the support. Finally, the desired catalyst was collected by centrifugation and used for the catalytic reaction.
2.5 Catalytic reaction of AB hydrolysis
The catalytic hydrolysis of AB was performed following our previously reported procedure.4 In general, a mixture of the catalyst (100 mg) and distilled water (5 mL) was placed in a two-necked round-bottomed flask (30 mL), which was placed in a water bath at room temperature under ambient atmosphere. A gas burette filled with water was connected to the reaction flask to measure the volume of hydrogen. The reaction started when aqueous AB solution (1 mmol in 200 μL of water) was injected into the mixture using a syringe. The volume of the evolved hydrogen gas was monitored by recording the displacement of water in the gas burette. The reaction was completed when there was no more gas generation. For the durability test, catalytic reactions were repeated by injecting another aliquot of AB (1.0 mmol in 200 μL of water) into the mixture after the previous cycle. After the reaction, the as-synthesized CuCo@MIL-101 was separated from the reaction solution by centrifugation and dried under vacuum at room temperature for the PXRD and TEM analyses.2.6 Catalyst characterization
Powder X-ray diffraction (PXRD) measurements were performed on a Rigaku Ultima IV X-ray diffractometer with a Cu Kα source (40 kV, 40 mA). X-ray photoelectron spectroscopic (XPS) measurements were carried out on a Shimadzu ESCA-3400 X-ray photoelectron spectrometer using an Mg Kα source (10 kV, 10 mA). The Ar sputtering experiments were carried out under the following conditions: a background vacuum of 3.2 × 10−6 Pa, a sputtering acceleration voltage of 2 kV and a sputtering current of 10 mA. The surface area measurements were performed with N2 adsorption/desorption isotherms at liquid nitrogen temperature (77 K) after dehydration under vacuum at 150 °C for 12 h using automatic volumetric adsorption equipment (Belsorp-max). The pore volume was calculated by a single point method at P/P0 = 0.99. A transmission electron microscope (TEM, TECNAI G2 F20) equipped with an energy-dispersive X-ray detector (EDX) was used to obtain the detailed microstructure and composition information for the prepared samples.3. Results and discussion
3.1 Synthesis and characterization
Chromium(III) terephthalate MIL-101 with a molecular formula of Cr3F(H2O)2O[(O2C)C6H4(CO2)]3·nH2O (n ≈ 25) was chosen as a host matrix for this study to encapsulate metallic particles not only because of its higher stability than most MOFs in water or high temperature but also its large cavity sizes (2.9 to 3.4 nm) and window sizes (1.2 to 1.4 nm), large surface area, and hybrid pore surface, which may facilitate the encapsulation of MNPs and the adsorption of catalytic substrate AB and reductant NaBH4 into the pores.15 The DSM was carried out in this work to impregnate the metal precursors (Cu(NO3)2 and/or CoCl2) into the preactivated MIL-101 in order to avoid the precursors' deposition on the outer surface of MOF.5f This method is based on a hydrophilic solvent containing the metal precursors and a hydrophobic solvent suspending the adsorbent, with the former having a volume set equal to or less than the pore volume of the adsorbent, which can be readily adsorbed within the hydrophilic pores of the adsorbent by capillary force. Then, the OWR approach was used to reduce the precursors after the metal precursor/MOF composite was dried.14 In this approach, a pore volume amount of NaBH4 solution with a high concentration (0.6 M) incorporated into the pores by capillary force can reduce the metal precursors deposited in the pores of MIL-101 completely, and the aggregation of MNPs on the external surfaces of MOF will be avoided.After the impregnation and reduction processes, there was no loss of crystallinity in powder X-ray diffraction (PXRD) patterns for M@MIL-101, suggesting that the framework of MIL-101 was maintained15 (Fig. 1). Moreover, the PXRD patterns of M@MIL-101 did not exhibit the characteristic diffractions for MNPs, indicating the formation of very small MNPs. The BET surface areas of MIL-101, Co@MIL-101, Cu@MIL-101 and CuCo@MIL-101 are 3450, 2154, 2052 and 2125 m2 g−1, respectively. The appreciable decreases in the amount of N2 sorption of M@MIL-101 indicated that MNPs were highly dispersed to the framework of MIL-101 (Fig. 2).
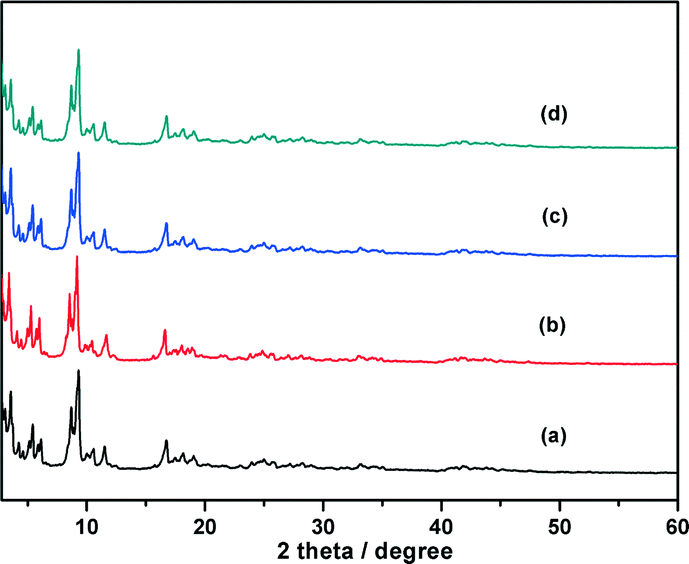 | ||
| Fig. 1 Powder XRD patterns of (a) as-synthesized MIL-101, (b) CuCo@MIL-101 (Cu/Co = 3:7), (c) Co@MIL-101 and (d) Cu@MIL-101. | ||
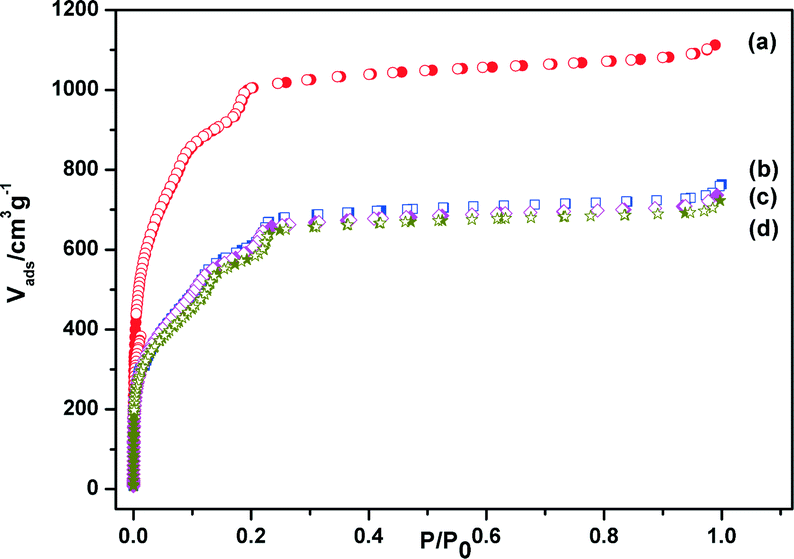 | ||
| Fig. 2 N2 sorption isotherms of (a) MIL-101, (b) Co@MIL-101, (c) CuCo@MIL-101 (Cu/Co = 3:7) and (d) Cu@MIL-101. Filled and open symbols represent adsorption and desorption branches, respectively. | ||
To obtain further insights into the structure–function correlations, X-ray photoelectron spectroscopy (XPS) with Ar etching was applied to Cu@MIL-101, Co@MIL-101 and CuCo@MIL-101 at the Cu 2p and Co 2p levels. Before Ar sputtering, broad peaks of Co 2p3/2 and Co 2p1/2 for Co@MIL-101 and CuCo@MIL-101 were observed with binding energies at 781.2 and 795.8 eV corresponding to Co 2p of CoO (ref. 16), which were caused by oxidation during the exposure of samples to air. After Ar etching for 60 min, well-defined peaks corresponding to both metallic Cu and Co species can be detected, suggesting that both elements are in their zero-valent states. The peaks at 932.6 and 952.4 eV are attributed to Cu 2p3/2 and Cu 2p1/2 of Cu0, and the peaks at 777.9 and 793.0 eV correspond to Co 2p3/2 and Co2p1/2 of Co0.16 The metallic Cu and Co peak intensities changed synchronously during the Ar etching in CuCo@MIL-101, indicating the homogeneity of the CuCo alloy NPs and excluding the possibility of core–shells (Fig. 3).
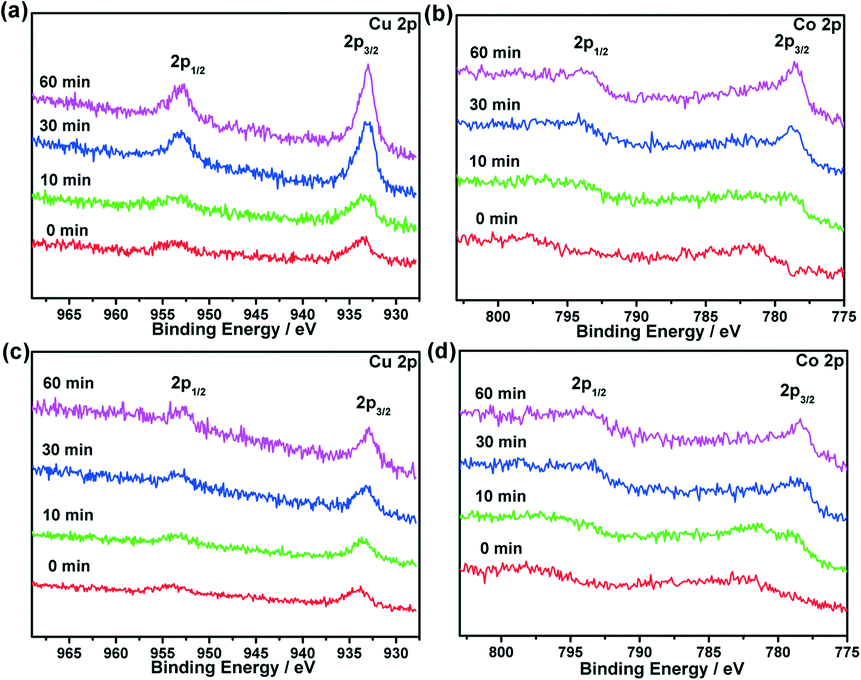 | ||
| Fig. 3 XPS spectra for (a) Cu@MIL-101, (b) Co@MIL-101 and (c, d) CuCo@MIL-101 (Cu/Co = 3:7) before (0 min) and after (10–60 min) Ar etching (Cu 2p3/2 and 2p1/2 peaks, and Co 2p3/2 and 2p1/2 peaks). | ||
The morphologies of MIL-101-immobilized CuCo alloy NPs were further characterized by transmission electron microscopy (TEM), high-angle annular dark-field scanning TEM (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDX) (Fig. 4). Both TEM and HAADF-STEM images showed that the CuCo alloy NPs were highly dispersed. The mean diameters were 2.0 ± 0.4 nm (Fig. 4d), which are small enough to be encapsulated in the two mesoporous cavities of MIL-101. No obvious particle aggregation occurred, which is in good agreement with the PXRD observations.
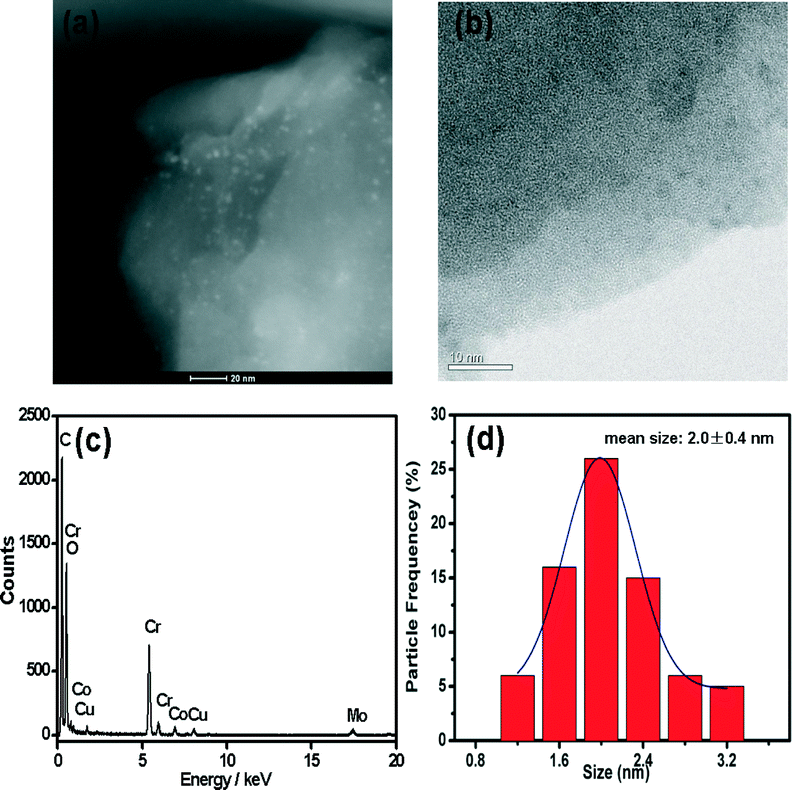 | ||
| Fig. 4 Representative (a) HAADF-STEM and (b) TEM images, (c) EDX spectrum, and (d) CuCo alloy nanoparticle size distribution histogram of CuCo@MIL-101 (Cu/Co = 3:7). The signal of Mo was from the TEM grid. | ||
3.2 Catalytic hydrolysis of ammonia borane
To study the catalytic behavior, the as-synthesized M@MIL-101 composites have been applied as catalysts for hydrolysis of AB. Reaction was initiated by introducing aqueous AB solution into the reaction flask containing a M@MIL-101 catalyst with vigorous shaking at room temperature. The catalytic hydrolysis of AB reaction can be briefly expressed as follows (eqn (1)):| NH3BH3 + 4H2O → NH4+ + B(OH)4− + 3H2 | (1) |
As displayed in Fig. 5, the CuCo@MIL-101 catalyst exhibits much higher catalytic activity for hydrolysis of AB compared with its monometallic counterparts, indicating a synergetic effect between the binary components.17Fig. 6 shows the Cu content dependence of reaction times and the corresponding TOF values for the dehydrogenation of AB. It is clear that, compared to pure Co, the presence of Cu can significantly improve the catalytic activity. For example, when the Cu molar ratio is as low as 0.1, the reaction time decreases from 17.5 min to 7.5 min. Moreover, with the increase in Cu molar ratio, the activity change of the CuCo system is in the shape of a “V”. The CuCo@MIL-101 with a Cu/Co atomic ratio of 3:7 is the most active, over which the AB hydrolysis reaction can be completed (H2/AB = 3.0) in 4.5 min with a 72 mL H2 release at room temperature ((Cu + Co)/AB = 0.034 in molar ratio), giving a turnover frequency (TOF) value of 19.6 molH2 molcat−1 min−1. Reasonably, the remarkably high catalytic activity is attributed to the synergetic effect between Cu and Co. For alloy nanoparticles, synergetic effects may be mainly ascribed to electronic effects, which are important when the metal atoms have significant differences in electronegativity,18 and geometric effects. In the present case, the observed synergetic effects in catalysis might be mainly due to the geometric effects, not the electronic effects, because the electronegativities of Cu (1.9) and Co (1.88) are very close. It is confirmed by the XPS measurements as no significant changes in the electronic states were observed between the CuCo alloy NPs and the monometallic Cu or Co NPs.18 As a control experiment, CuCo/MIL-101 and CuCo/XC-72R (Cu/Co = 3:7) were prepared by a conventional impregnation–reduction method, over which the evolution of 72 mL and 63 mL of hydrogen was completed in 8.5 min and 11.5 min, respectively, under the same reaction conditions (Fig. 7). Moreover, it is noteworthy that the TOF (19.6 molH2 molcat−1 min−1) for CuCo@MIL-101 (Cu/Co = 3:7) is the highest one among the MOF-supported non-noble metal catalysts ever reported for this reaction.4a,19
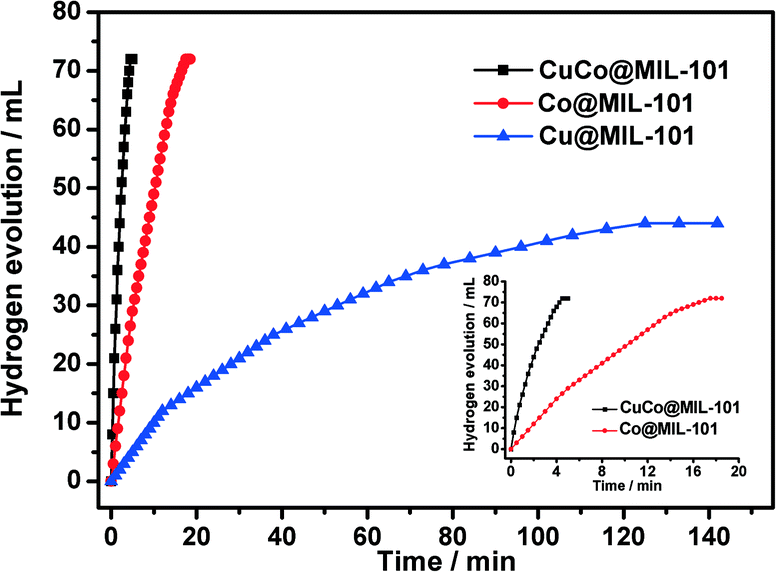 | ||
| Fig. 5 Plots of time versus volume of hydrogen generated from AB (1 mmol in 5 mL of water) hydrolysis at room temperature catalyzed by the Cu@MIL-101, Co@MIL-101, and CuCo@MIL-101 (Cu/Co = 3:7) catalysts (100 mg, (Cu + Co)/AB (molar ratio) = 0.034). | ||
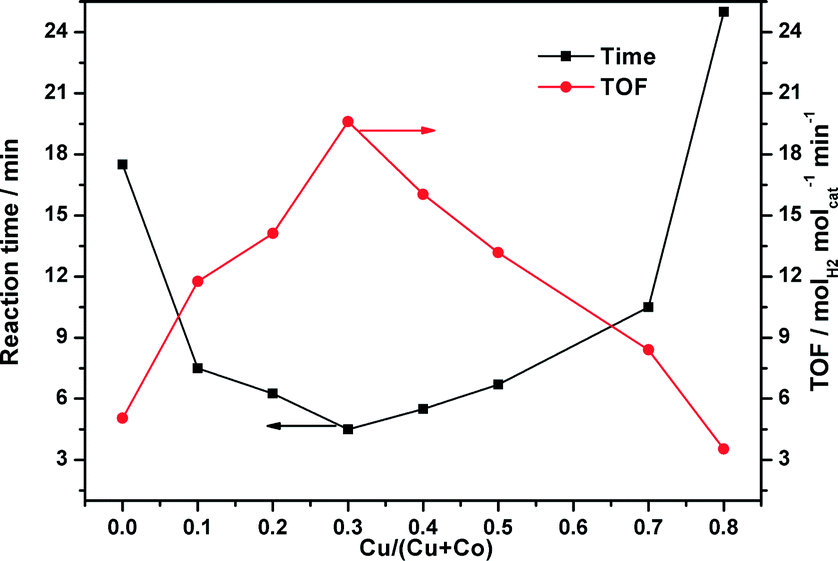 | ||
| Fig. 6 Plots of time for reaction completion and the corresponding TOF value versus Cu molar content (Cu/(Cu + Co)) in CuCo alloy NPs ((Cu + Co)/AB = 0.034). | ||
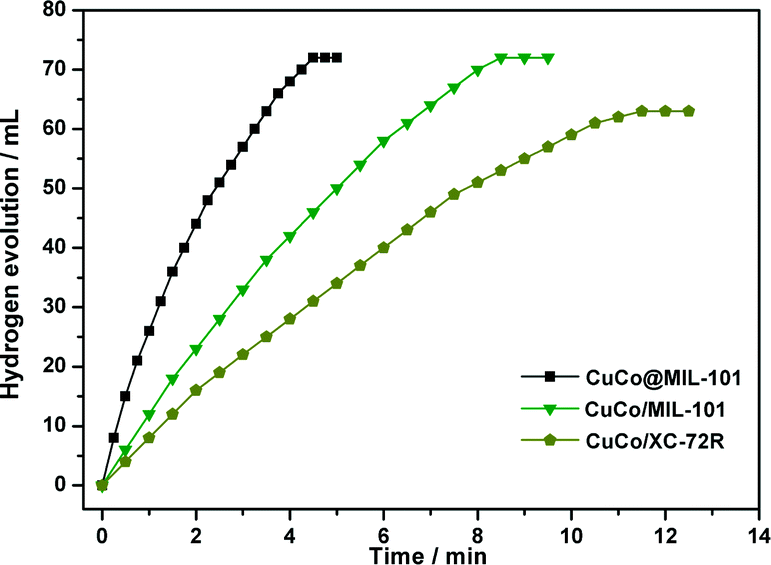 | ||
| Fig. 7 Plots of time versus volume of hydrogen generated from AB (1 mmol in 5 mL of water) hydrolysis at room temperature catalyzed by CuCo@MIL-101, CuCo/MIL-101, and CuCo/XC-72R (Cu/Co = 3:7) catalysts (100 mg, (Cu + Co)/AB (molar ratio) = 0.034). | ||
The durability/stability is the key point for the practical application of catalysts. In this sense, the durability of the CuCo@MIL-101 catalyst was tested by adding additional aliquots (1.0 mmol, 200 μL) of AB under ambient atmosphere. As shown in Fig. 8, the activity of the as-prepared CuCo@MIL-101 catalyst has no significant decrease after a five-time recycle test, which denotes its high durability in AB hydrolysis. The long durability of the catalyst should be attributed to the fact that the ultrafine CuCo alloy NPs have been effectively encapsulated in the pores of MIL-101 for avoiding aggregation. PXRD and TEM measurements of CuCo@MIL-101 after catalysis revealed no obvious changes in the morphologies of CuCo NPs with retention of the MIL-101 framework (Fig. 9), which further confirms the advantage of the confinement effect of the MOF matrices.
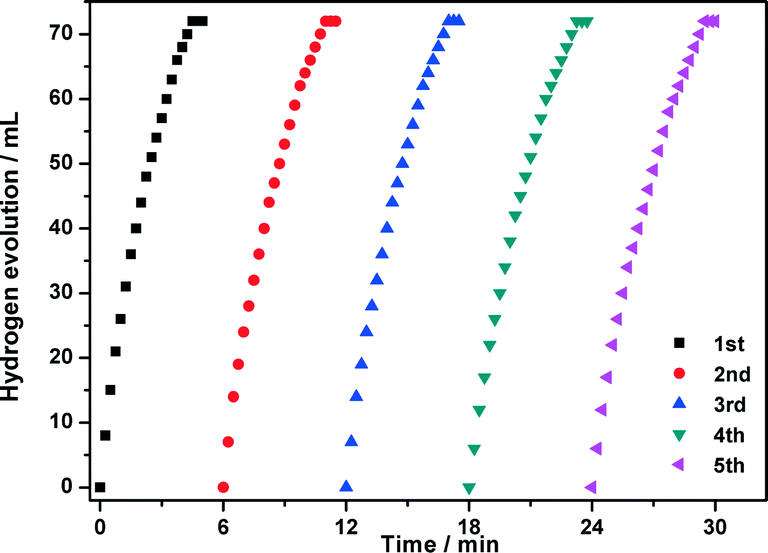 | ||
| Fig. 8 Durability characterization of CuCo@MIL-101 in five runs for hydrogen generation from hydrolysis of aqueous AB solution (1.0 mmol, Cu/Co = 3:7, (Cu + Co)/AB = 0.034). | ||
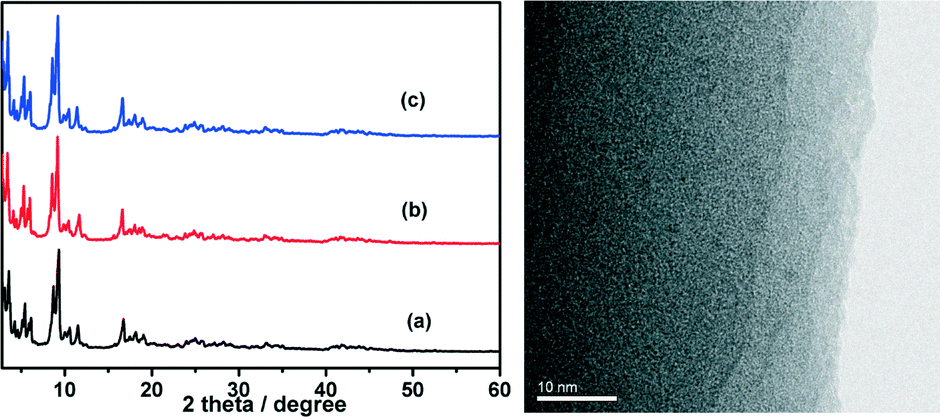 | ||
| Fig. 9 Left: powder XRD patterns of (a) as-synthesized MIL-101 and CuCo@MIL-101 (Cu/Co = 3:7) (b) before and (c) after the catalytic hydrolysis of AB. Right: TEM image of CuCo@MIL-101 after AB hydrolysis (Cu/Co = 3:7). | ||
4. Conclusions
In summary, the ultrafine CuCo alloy NPs have been successfully encapsulated in the pores of MIL-101 by using the DSM in combination with the OWR approach, which display excellent catalytic activity for hydrolytic dehydrogenation of aqueous AB, surpassing their monometallic Cu and Co counterparts. This is the first example of MOF-supported non-noble bimetallic catalysts for hydrogen generation from hydrolysis of AB. The highly efficient catalyst represents a promising step toward the practical applications of MOF-supported non-noble metal catalysts in the catalytic hydrolysis reaction system.Acknowledgements
We acknowledge the reviewers for their valuable suggestions, Dr. Takeyuki Uchida for TEM measurements and AIST and Kobe University for financial support.Notes and references
- (a) P. P. Edwards, V. L. Kuznetsov, W. I. David and N. P. Brandon, Energy Policy, 2008, 36, 4356 CrossRef PubMed; (b) C. W. Hamilton, R. T. Baker, A. Staubitzc and I. Manners, Chem. Soc. Rev., 2009, 38, 279 RSC; (c) H. M. Chen, C. K. Chen, R.-S. Liu, L. Zhang, J. Zhang and D. P. Wilkinson, Chem. Soc. Rev., 2012, 41, 5654 RSC.
- (a) L. Schlapbach and A. Züttel, Nature, 2001, 414, 353 CrossRef CAS PubMed; (b) R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844 CrossRef CAS PubMed; (c) Z.-G. Huang and T. Autrey, Energy Environ. Sci., 2012, 5, 9257 RSC; (d) Q.-L. Zhu, N. Tsumori and Q. Xu, Chem. Sci., 2014, 5, 195 RSC.
- (a) A. Gutowska, L. Li, Y. Shin, C.-M. Wang, X.-S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578 CrossRef CAS PubMed; (b) M. Chandra and Q. Xu, J. Power Sources, 2006, 156, 190 CrossRef CAS PubMed; (c) Q. Xu and M. J. Chandra, J. Alloys Compd., 2007, 446, 729 CrossRef PubMed; (d) S.-K. Kim, W.-S. Han, T.-J. Kim, T.-Y. Kim, S. W. Nam, M. Mitoraj, L. Piekos, A. Michalak, S. J. Hwang and S. O. Kang, J. Am. Chem. Soc., 2010, 132, 9954 CrossRef CAS PubMed; (e) U. Sanyal, U. B. Demirci, B. R. Jagirdar and P. Miele, ChemSusChem, 2011, 4, 1731 CrossRef CAS PubMed; (f) S.-K. Kim, T.-J. Kim, T.-Y. Kim, G. Lee, J. T. Park, S. W. Nam and S. O. Kang, Chem. Commun., 2012, 48, 2021 RSC; (g) M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698 RSC.
- (a) H.-L. Jiang and Q. Xu, Catal. Today, 2011, 170, 56 CrossRef CAS PubMed; (b) Q. Xu and M. Chandra, J. Power Sources, 2006, 163, 364 CrossRef CAS PubMed.
- (a) N. Mohajeri, A. T-Raissi and O. Adebiyi, J. Power Sources, 2007, 167, 482 CrossRef CAS PubMed; (b) S. Basu, A. Brockman, P. Gagare, Y. Zheng, P. V. Ramachandran, W. N. Delgass and J. P. Gore, J. Power Sources, 2009, 188, 238 CrossRef CAS PubMed; (c) F. Durap, M. Zahmakiran and S. Ökar, Int. J. Hydrogen Energy, 2009, 34, 7223 CrossRef CAS PubMed; (d) S. Karahan, M. Zahmakiran and S. Özkar, Chem. Commun., 2012, 48, 1180 RSC; (e) D.-C. Zhong, K. Aranishi, A. K. Singh, U. B. Demirci and Q. Xu, Chem. Commun., 2012, 48, 11945 RSC; (f) A. Aijaz, A. Karkamkar, Y. J. Choi, N. Tsumori, E. Rönnebro, T. Autrey, H. Shioyama and Q. Xu, J. Am. Chem. Soc., 2012, 134, 13926 CrossRef CAS PubMed.
- (a) J.-M. Yan, X.-B. Zhang, S. Han, H. Shioyama and Q. Xu, Angew. Chem., Int. Ed., 2008, 47, 2287 CrossRef CAS PubMed; (b) T. Umegaki, J.-M. Yan, X.-B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, Int. J. Hydrogen Energy, 2009, 34, 3816 CrossRef CAS PubMed; (c) Ö. Metin, V. Mazumder, S. Özkar and S. H. Sun, J. Am. Chem. Soc., 2010, 132, 1468 CrossRef PubMed; (d) P.-Z. Li, A. Aijaz and Q. Xu, Angew. Chem., Int. Ed., 2012, 51, 6753 CrossRef CAS PubMed.
- (a) J.-M. Yan, X.-B. Zhang, H. Shioyama and Q. Xu, J. Power Sources, 2010, 195, 1091 CrossRef CAS PubMed; (b) T. Umegaki, J.-M. Yan, X.-B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, J. Power Sources, 2010, 195, 8209 CrossRef CAS PubMed.
- (a) X. H. Zhou, Z. X. Chen, D. H. Yan and H. B. Lu, J. Mater. Chem., 2012, 22, 13506 RSC; (b) K. Aranishi, A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 2248 CrossRef CAS.
- (a) J.-M. Yan, X.-B. Zhang, S. Han, H. Shioyama and Q. Xu, J. Am. Chem. Soc., 2009, 131, 2778 CrossRef PubMed; (b) H.-L. Jiang, T. Akita and Q. Xu, Chem. Commun., 2011, 47, 10999 RSC.
- (a) N. Patel, R. Fernandes, G. Guella, A. Kal, A. Miotello, B. Patton and C. Zanchetta, J. Phys. Chem. C, 2008, 11, 6968 CrossRef; (b) N. Patel, R. Fernandes, G. Guella and A. Miotello, Appl. Catal., B, 2010, 95, 137 CrossRef CAS PubMed.
- (a) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed; (b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed; (c) J.-P. Zhang, X.-C. Huang and X.-M. Chen, Chem. Soc. Rev., 2009, 38, 2385 RSC; (d) G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380 RSC; (e) B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115 CrossRef CAS PubMed; (f) H.-L. Jiang and Q. Xu, Chem. Commun., 2011, 47, 3351 RSC; (g) D. Zhao, D. J. Timmons, D. Yuan and H.-C. Zhou, Acc. Chem. Res., 2011, 44, 123 CrossRef CAS PubMed.
- (a) A. Henschel, K. Gedrich, R. Kraehnert and S. Kaskel, Chem. Commun., 2008, 4192 RSC; (b) S. Proch, J. Herrmannsdörfer, R. Kempe, C. Kern, A. Jess, L. Seyfarth and J. Senker, Chem. – Eur. J., 2008, 14, 8204 CrossRef CAS PubMed; (c) H.-L. Jiang, T. Akita, T. Ishida, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2011, 133, 1304 CrossRef CAS PubMed; (d) X. Gu, Z.-H. Lu, H.-L. Jiang, T. Akita and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11822 CrossRef CAS PubMed; (e) G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310 CrossRef CAS PubMed; (f) A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262 RSC; (g) C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211 CrossRef CAS PubMed; (h) S.-L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656 RSC; (i) Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468 RSC.
- (a) S. Hermes, M.-K. Schröter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237 CrossRef CAS PubMed; (b) M. Müller, O. I. Lebedev and R. A. Fischer, J. Mater. Chem., 2008, 18, 5274 RSC; (c) F. Schröder, S. Henke, X. Zhang and R. A. Fischer, Eur. J. Inorg. Chem., 2009, 3131 CrossRef; (d) Y. K. Park, S. B. Choi, H. J. Nam, D. Jung, H. C. Ahn, K. Choi, H. Furukawa and J. Kim, Chem. Commun., 2010, 46, 3086 RSC; (e) M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. Van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 3701 CrossRef CAS; (f) J. Hermannsdörfer, M. Friedrich, N. Miyajima, R. Q. Albuquerque, S. Kümmel and R. Kempe, Angew. Chem., Int. Ed., 2012, 51, 11473 CrossRef PubMed; (g) H. R. Moon, D.-W. Limb and M. P. Suh, Chem. Soc. Rev., 2013, 42, 1807 RSC; (h) A. Aijaz and Q. Xu, J. Phys. Chem. Lett., 2014, 5, 1400 CrossRef CAS.
- (a) Q.-L. Zhu, J. Li and Q. Xu, J. Am. Chem. Soc., 2013, 135, 10210 CrossRef CAS PubMed; (b) J. Li, Q.-L. Zhu and Q. Xu, Chem. Commun., 2014, 50, 5899 RSC.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef PubMed.
- C. D. Wagner, W. N. Riggs, L. E. Davis and J. F. Moulder, Handbook of X-Ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Use In X-Ray Photoelectron Spectroscopy, ed. G. E. Muilenberg, Physical Electronics Division Perkin-Elmer Corp., Minnesota, 1978 Search PubMed.
- (a) H.-L. Jiang and Q. Xu, J. Mater. Chem., 2011, 21, 13705 RSC; (b) A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 652 CrossRef CAS.
- (a) B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71 CrossRef CAS; (b) R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845 CrossRef CAS PubMed.
- P.-Z. Li, K. Aranishi and Q. Xu, Chem. Commun., 2012, 48, 3173 RSC.
| This journal is © The Royal Society of Chemistry 2015 |