
Controlled synthesis of Pd–NiO@SiO2 mesoporous core–shell nanoparticles and their enhanced catalytic performance for p-chloronitrobenzene hydrogenation with H2†
Hongmei
Liu
ab,
Kai
Tao
b,
Chunrong
Xiong
a and
Shenghu
Zhou
*b
aCollege of Materials and Chemical Engineering, Hainan University, Haikou, Hainan 570228, PR China
bNingbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, Zhejiang 315201, PR China. E-mail: zhoush@nimte.ac.cn; Fax: +86 574 86685043; Tel: +86 574 86696927
First published on 28th August 2014
Abstract
In this work, Pd–NiO@SiO2 core–shell mesoporous nanocatalysts with ~4 nm Pd–NiO heteroaggregate nanoparticle cores and ~17 nm mesoporous silica shells were successfully synthesized by a sol–gel method. The surfactant-capped PdNi alloy nanoparticles were coated with SiO2 through hydrolysis of tetraethylorthosilicate to obtain PdNi@SiO2 nanoparticles, and the mesoporous Pd–NiO@SiO2 core–shell nanocatalysts were formed after removal of surfactants by calcination at 500 °C and subsequent H2 reduction at 200 °C. The characterization results by XRD, TEM and BET revealed that Pd–NiO@SiO2 nanocatalysts were highly stable with the maintenance of intact core–shell structures under high-temperature thermal treatments. The Pd–NiO@SiO2 nanocatalysts illustrated a superior catalytic performance for p-chloronitrobenzene hydrogenation with H2 to the control Pd@SiO2 nanocatalysts. The catalytic performance enhancement of Pd–NiO@SiO2 nanocatalysts is ascribed to the strong interaction between Pd and NiO in the cores, where the interfaces may be beneficial for hydrogenation reactions.
1. Introduction
As a new kind of multifunctional composite nanomaterials, core–shell structured nanomaterials have been intensively studied in recent decades.1–4 After coating with shell materials, not only the stability and dispersion of core materials are significantly enhanced, but physicochemical properties are also improved, resulting in a large number of new characteristics, such as good monodispersity, regulated core–shell ratio, stable physicochemical properties and so on.5 Due to their unique properties, core–shell nanomaterials have broad applications specially in the fields of energy materials,6 optoelectronic technology,7 biology,8 and catalysis.9,10Core–shell nanoparticles (NPs) using monometallic NPs as cores and oxides as shells have been extensively studied in recent years.11–16 By coating active metals with mesoporous shells, the heterogeneous catalysts could be easily separated and recycled. The mesoporous shells could protect the active metal cores from sintering and allow reactants to reach the surface of the active metal and allow products to diffuse out the cores, where the reaction occurs. Typically, SiO2,17 TiO2,18 Al2O3,19 Fe2O3,20 and SnO2 (ref. 21) are the common shell materials. Among the abovementioned shell materials, SiO2 is the most widely adopted material due to its high thermal stability, controllable morphology and desirable surface area.5 The Stöber method22 and sol–gel process23 were mostly used to encapsulate metal NPs with a mesoporous SiO2 shell. The thickness of the mesoporous SiO2 shell can be controlled by adjusting the pH value of the solution and the tetraethoxysilane (TEOS)/water ratio. The resultant mesoporous core–shell structures after removal of templates by calcination allow catalytic reactions to occur on the surface of the metal cores. Yin and co-workers reported mesoporous Au@SiO2 core–shell NPs by the sol–gel method, and the corresponding CO oxidation over the core–shell nanocatalysts was also studied.24 In our previous work, Pd@SiO2 mesoporous nanocatalysts were successfully prepared in a water system using tetradecyl trimethyl ammonium bromide (TTAB) as a template for hydrolysis of TEOS,25 and the thickness of the silica shells can be well controlled by changing the synthetic parameters. The obtained mesoporous Pd@SiO2 nanocatalysts after calcination and following H2 treatment illustrated superior catalytic and thermal stability.
Bimetallic NPs illustrate enhanced catalytic performance to their monometallic counterparts.26–28 However, in some cases, the catalytic selectivity enhancement over bimetallic NPs is obtained at the expense of catalytic activity.29 To obtain high activity and high selectivity at the same time, strong metal–support interaction catalysts have been developed for various catalytic reactions. Corma and co-workers reported high-temperature hydrogen-reduced Pt/TiO2 catalysts for hydrogenation of substituted nitrobenzenes.30 The high-temperature H2 treatment causes the Pt NPs to be covered by a thin layer of partially reduced TiOx, and the strong interaction between Pt and TiOx stabilized Pt NPs and produced a highly active interface for catalytic hydrogenation. Zhou et al.31,32 developed highly active Au–NiO/SiO2 catalysts for low-temperature CO oxidation, where the strong interaction was generated by in situ transformation of bimetallic AuNi alloy NPs to close contact Au–NiO heteroaggregate NPs. Similarly, enhanced catalytic performance of Pt–MxOy/Al2O3 (M = Ni, Fe, Co) heteroaggregate nanocatalysts by transformation of PtM alloy NPs was also reported.33
Here, we reported a successful synthesis of Pd–NiO@SiO2 mesoporous nanocatalysts, where the strong metal–oxide interaction and mesoporous core–shell structures coexist. Catalytic hydrogenation of p-chloronitrobenzene with H2 was used to investigate the catalytic performance of Pd–NiO@SiO2. In this work, TTAB-capped PdNi alloy NPs were synthesized by co-reduction of Pd and Ni precursors using NaBH4 as a reducing agent. SiO2 shells were then used to coat the PdNi alloy NPs to obtain PdNi@SiO2 core–shell NPs through the hydrolysis of TEOS. The mesoporous Pd–NiO@SiO2 or PdNi@SiO2 nanocatalysts were obtained after calcination and following H2 reduction under different conditions. The Pd–NiO@SiO2 nanocatalysts illustrated a superior catalytic performance for catalytic p-chloronitrobenzene (p-CNB) hydrogenation to the control Pd@SiO2 mesoporous nanocatalysts synthesized by a similar method, and their reusability was also studied. The enhanced catalytic performance over Pd–NiO@SiO2 is ascribed to core–shell structures and the strong interaction between Pd and NiO. Scheme 1 demonstrates the synthetic procedure for Pd–NiO@SiO2 mesoporous nanocatalysts.
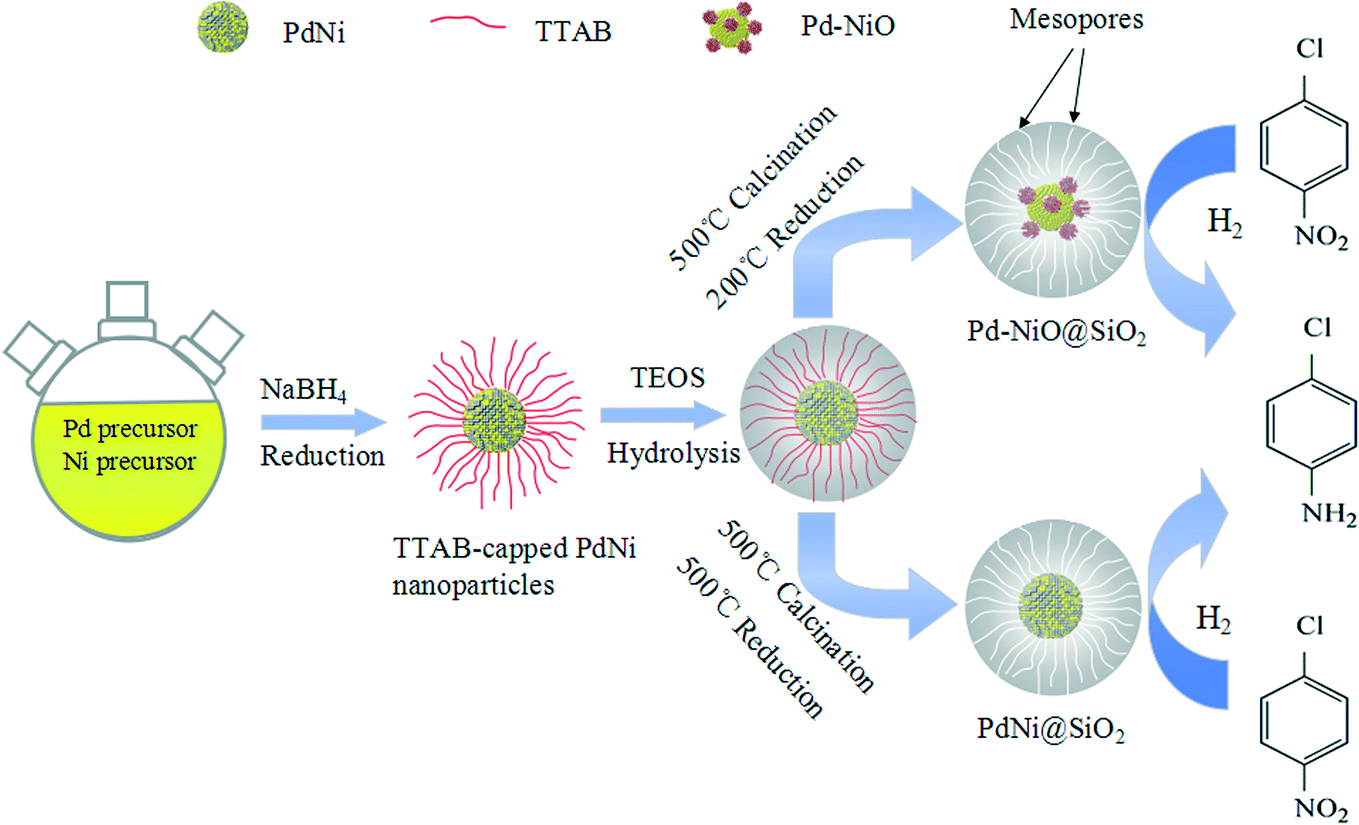 | ||
| Scheme 1 Schematic demonstration of synthetic procedures of Pd–NiO@SiO2 and PdNi@SiO2 mesoporous core–shell nanocatalysts. | ||
2. Experimental section
2.1. Chemicals
Potassium tetrachloropalladate(II) (K2PdCl4), nickel(II) acetylacetonate (Ni(acac)2, 95%), sodium borohydride (NaBH4, 98%), ammonium hydroxide (NH4OH, AR), tetraethylorthosilicate (TEOS, AR), and tetradecyl trimethyl ammonium bromide (TTAB, AR) were purchased from Aladdin. p-Chloronitrobenzene (p-CNB, AR) was purchased from Shanghai Chemical Reagent Company. Deionized water was purchased from local sources. All reagents were used without further purification.2.2. Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were synthesized by a modified sol–gel process.25 The abovementioned as-synthesized PdNi colloids (24.0 mL, 0.0072 mmol Pd and 0.0082 mmol Ni) were mixed with 120 mL of deionized water in a 250 mL three-necked round-bottomed flask at room temperature with magnetic stirring. A certain amount of concentrated ammonia aqueous solution (25–28 wt%) was then added to the solution to adjust the pH value of the solution to 11.6. TEOS (600 μL, 2.69 mmol) was then added into the system, and the reaction system was stirred for 1.5 hours at room temperature. The gray powdery PdNi@SiO2 products were obtained by centrifugation and dried at 60 °C for 10 hours for future use.
:1 were prepared according to the literature.25 Briefly, K2PdCl4 aqueous solution (7.5 mL, 10.0 mM), TTAB (39.0 ml, 100 mM) aqueous solution and 90 mL of deionized water were mixed in a 250 mL three-necked round-bottomed flask. The system was stirred for ~20 minutes. Ice-cold NaBH4 (9.0 mL, 250 mM) aqueous solution was then injected into the system using a syringe. The syringe was kept in place to release the gas generated in the reaction, and then the flask was sealed to make a closed system. The obtained reaction mixture was further stirred for 15 hours at a low speed to obtain Pd colloids with an average Pd particle size of 4.2 nm. To coat the Pd cores with silica shells, the abovementioned as-synthesized Pd colloids (24.0 mL, 0.0125 mmol Pd) were mixed with 120 mL of deionized water in a 250 mL three-necked round-bottomed flask at room temperature with magnetic stirring, and a few drops of concentrated ammonia solution (25–28 wt%) were then added to adjust the pH of the solution to 10.6. TEOS (642 μL, 2.88 mmol) was then introduced into the solution after 10 minutes of stirring. The resultant mixture was further stirred for 1 hour at room temperature. The collection procedure and treatment process of Pd@SiO2 core–shell nanocatalysts were the same as those of Pd–NiO@SiO2.
:1 were synthesized by a modified sol–gel process.25 The abovementioned as-synthesized PdNi colloids (24.0 mL, 0.0072 mmol Pd and 0.0082 mmol Ni) were mixed with 120 mL of deionized water in a 250 mL three-necked round-bottomed flask at room temperature with magnetic stirring. A certain amount of concentrated ammonia aqueous solution (25–28 wt%) was then added to the solution to adjust the pH value of the solution to 11.6. TEOS (600 μL, 2.69 mmol) was then added into the system, and the reaction system was stirred for 1.5 hours at room temperature. The gray powdery PdNi@SiO2 products were obtained by centrifugation and dried at 60 °C for 10 hours for future use.
:1 were prepared according to the literature.25 Briefly, K2PdCl4 aqueous solution (7.5 mL, 10.0 mM), TTAB (39.0 ml, 100 mM) aqueous solution and 90 mL of deionized water were mixed in a 250 mL three-necked round-bottomed flask. The system was stirred for ~20 minutes. Ice-cold NaBH4 (9.0 mL, 250 mM) aqueous solution was then injected into the system using a syringe. The syringe was kept in place to release the gas generated in the reaction, and then the flask was sealed to make a closed system. The obtained reaction mixture was further stirred for 15 hours at a low speed to obtain Pd colloids with an average Pd particle size of 4.2 nm. To coat the Pd cores with silica shells, the abovementioned as-synthesized Pd colloids (24.0 mL, 0.0125 mmol Pd) were mixed with 120 mL of deionized water in a 250 mL three-necked round-bottomed flask at room temperature with magnetic stirring, and a few drops of concentrated ammonia solution (25–28 wt%) were then added to adjust the pH of the solution to 10.6. TEOS (642 μL, 2.88 mmol) was then introduced into the solution after 10 minutes of stirring. The resultant mixture was further stirred for 1 hour at room temperature. The collection procedure and treatment process of Pd@SiO2 core–shell nanocatalysts were the same as those of Pd–NiO@SiO2.
2.3. Characterization
X-ray diffraction (XRD) measurements were carried out using a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation in the 2θ range from 10° to 80°. Transmission electron microscopy (TEM) images were obtained using a JEOL 2100 transmission electron microscope. X-ray photoelectron spectroscopy (XPS) spectra were acquired by using an AXIS ULTRADLD multifunctional X-ray photoelectron spectroscope with an Al source. The data processing was performed by the CasaXPS software. The actual metal loadings of the nanocatalysts were determined by using a PE Optima 2100DV inductively coupled plasma optical emission spectrometer (ICP-OES). H2 temperature-programmed reduction (H2-TPR) was carried out using a FINESORB3010 instrument with a thermal conductivity detector (TCD). The infrared (IR) spectra of the nanocatalysts were recorded in transmission mode using a Bruker Tensor 27 spectrophotometer. The thermal property of the samples was measured by using a Pyris Diamond thermogravimetric analyzer (TGA). During the test, the samples were heated from 50 °C to 500 °C at a heating rate of 5 °C min−1 in an air atmosphere with a gas flow rate of 50 mL min−1. Brunauer–Emmett–Teller (BET) surface area, pore size distribution and the adsorption/desorption isotherms of the nanocatalysts were measured by N2 adsorption at 77 K, using a Micromeritics ASAP-2020 M automatic specific surface area and porous physical adsorption analyzer.2.4. Catalytic hydrogenation of p-chloronitrobenzene with H2
In this study, the hydrogenation of p-CNB with H2 at ambient pressure was carried out to investigate the catalytic performance of different Pd-containing core–shell nanocatalysts with ~17 nm thick SiO2 shells. Pd–NiO@SiO2 mesoporous nanocatalysts (0.19 g) with actual Pd loading of 0.36 wt% and 1.0 g of p-CNB were mixed with 24 mL of absolute ethanol in a 50 mL three-necked round-bottomed flask at room temperature. The system was purged with high-purity H2 to remove air prior to reaction. The mixture was then heated to 30 °C and maintained at 30 °C for a certain reaction time under hydrogen flow with vigorous magnetic stirring. The liquid products were collected by centrifugation to remove the solid catalysts and were analyzed online using a gas chromatograph equipped with a flame ionization detector (FID). For control experiments, 0.10 g of Pd@SiO2 mesoporous nanocatalysts with actual Pd loading of 0.68 wt% was used to maintain the same amount of Pd as that of Pd–NiO@SiO2 nanocatalysts, and other conditions were the same as those of Pd–NiO@SiO2 catalytic experiments.To study the durability of Pd–NiO@SiO2 nanocatalysts, cycle-to-cycle experiments were carried out. Pd–NiO@SiO2 nanocatalysts (0.19 g) with an actual Pd loading of 0.36 wt% and 1.0 g of p-CNB were mixed in 24 mL of absolute ethanol in the first cycle. The catalytic reactions were carried out at 30 °C and ambient H2 pressure. After each reaction run, the solid nanocatalysts were collected by centrifugation, washed twice with absolute ethanol, and dried in oven at 60 °C for the next reaction cycle. Because of the loss of catalysts during the recovery process, lesser amounts of nitrobenzene and ethanol in the next reaction cycle were used to keep the same ratio of p-CNB/catalysts and p-CNB/solvent as those of the first cycle. Due to the loss of catalysts, 5 cycles of catalytic reactions were carried out.
3. Results and discussion
In this study, a PdNi alloy colloid was synthesized through co-reduction of Ni(acac)2 and K2PdCl4 by NaBH4 using TTAB as a capping agent. The molar ratio of Ni(acac)2/K2PdCl4 was set to 1.15:1.00 to ensure the formation of 1:1 PdNi alloy NPs by XRD measurement. SiO2 shells were then used to coat the PdNi alloy cores to form PdNi@SiO2 NPs by a sol–gel process through the hydrolysis of TEOS, and the thickness of SiO2 shells was controlled by adjusting the pH value of the solution and the TEOS/Pd ratios. The TTAB surfactants on PdNi alloy surfaces were removed by calcination at 500 °C, and the obtained PdO–NiO@SiO2 NPs were then reduced by H2 reduction at 200 °C and 500 °C to obtain Pd–NiO@SiO2 and PdNi@SiO2 nanocatalysts, respectively. For comparison of catalytic activity, the Pd@SiO2 nanocatalysts with the same SiO2 shell thickness were also prepared by a similar process. The actual Pd and Ni loadings of corresponding nanocatalysts were determined by ICP-OES.
TEM images of various NPs are shown in Fig. 1. As shown in Fig. 1a, TTAB-capped PdNi NPs demonstrated irregular shapes with an average particle size of 4.2 nm, and a lattice spacing of 0.217 nm in the HRTEM image was clearly visible, which is in line with the lattice spacing of an alloyed PdNi (111) plane. After encapsulation of PdNi NPs with SiO2 shells, the PdNi cores and the SiO2 shells were clearly observed in PdNi@SiO2 NPs as shown in Fig. 1b–f. The synthetic conditions were found to have a profound effect on the structures of PdNi@SiO2 NPs, especially the pH value. When the pH values were adjusted to 11.5, 11.6, 11.7 and 11.8, the corresponding thicknesses of the SiO2 shells in PdNi@SiO2 NPs were ~14 nm, ~17 nm, ~23 nm and ~28 nm, respectively. The correlation between pH value and the SiO2 thickness suggested that slow hydrolysis of TEOS at a pH value of 11.5 resulted in small NPs with some percentage of NPs without PdNi cores, while fast hydrolysis at 11.8 resulted in large NPs with several PdNi NPs encapsulated into one silica particle, which is consistent with the finding in the synthesis of Pd@SiO2 NPs.25
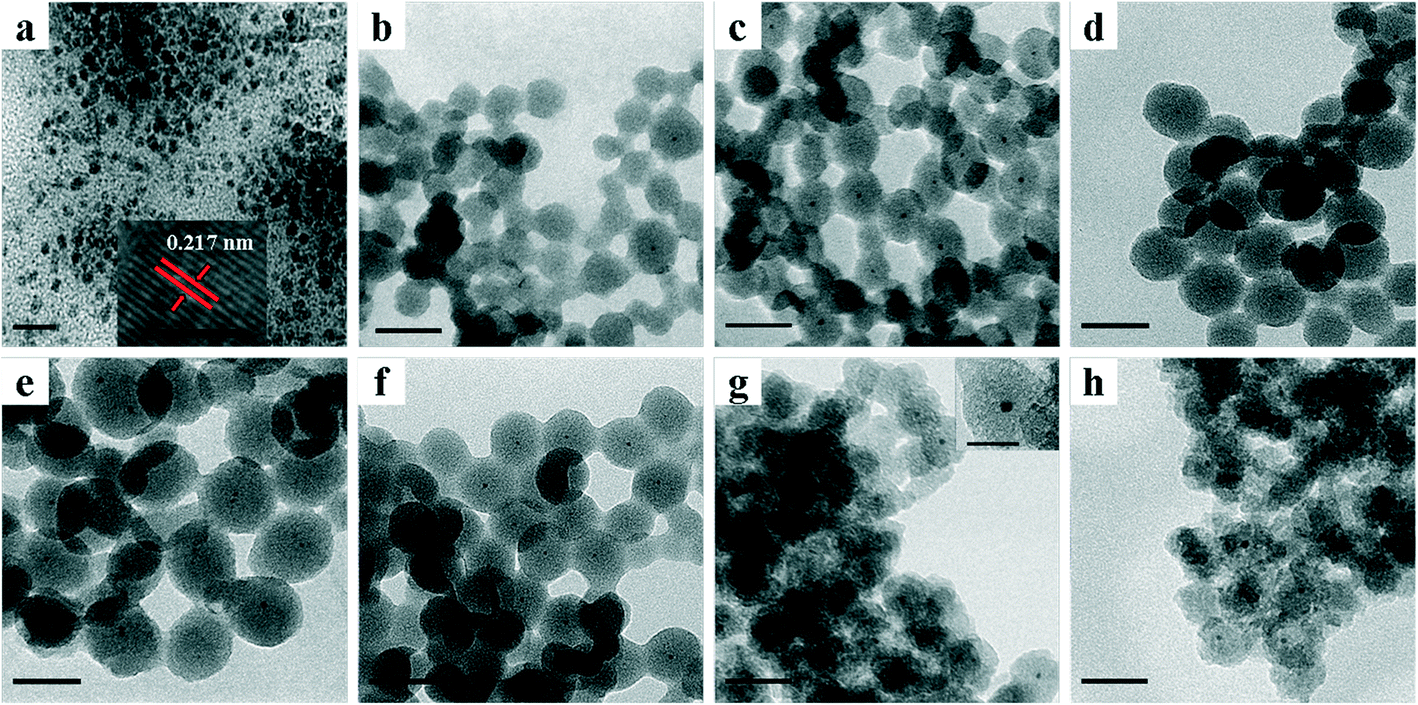 | ||
| Fig. 1 TEM images of different NPs showing a) TTAB-capped PdNi NPs (the inset is an HRTEM image of one nanoparticle showing a lattice spacing of 0.217 nm); b) PdNi@SiO2 NPs synthesized at a pH value of 11.5 and a TEOS/Pd ratio of 370:1; c) PdNi@SiO2 NPs synthesized at a pH value of 11.6 and a TEOS/Pd ratio of 370:1; d) PdNi@SiO2 NPs synthesized at a pH value of 11.7 and a TEOS/Pd ratio of 370:1; e) PdNi@SiO2 NPs synthesized at a pH value of 11.8 and a TEOS/Pd ratio of 370:1; f) PdNi@SiO2 NPs synthesized at a pH value of 11.6 and a TEOS/Pd ratio of 490:1; g) Pd–NiO@SiO2 nanocatalysts obtained after 500 °C calcination and following 200 °C H2 reduction of PdNi@SiO2 synthesized at a pH value of 11.6 and a TEOS/Pd ratio of 370:1; h) PdNi@SiO2 nanocatalysts obtained after 500 °C calcination and following 500 °C H2 reduction of PdNi@SiO2 synthesized at a pH value of 11.6 and a TEOS/Pd ratio of 370:1. Scale bar of a)—20 nm; scale bar of the inset in a)—2 nm; scale bars of b)–h)—50 nm; scale bar of the inset in g)—20 nm. | ||
The thickness of the SiO2 shell of PdNi@SiO2 NPs was controlled by the TEOS/Pd ratio. As shown in Fig. 1c and f, when the molar ratio of TEOS/Pd increased from 370:1 to 490:1, the thickness of the SiO2 shells increased from ~17 nm to ~20 nm at the same pH value of 11.6. In this study, the synthetic conditions under a pH value of 11.6 and a TEOS/Pd ratio of 370:1 are optimum since PdNi@SiO2 NPs synthesized under these conditions have perfect core–shell structures and thin SiO2 shells. Therefore, the PdNi@SiO2 NPs prepared under these conditions will be treated under different conditions and tested for catalytic hydrogenation of p-CNB with H2. The size distributions of PdNi NPs and PdNi cores in PdNi@SiO2 NPs are illustrated in Fig. 2a and b. It was clearly demonstrated that PdNi cores maintained the same size as that of as-prepared PdNi bimetallic NPs.
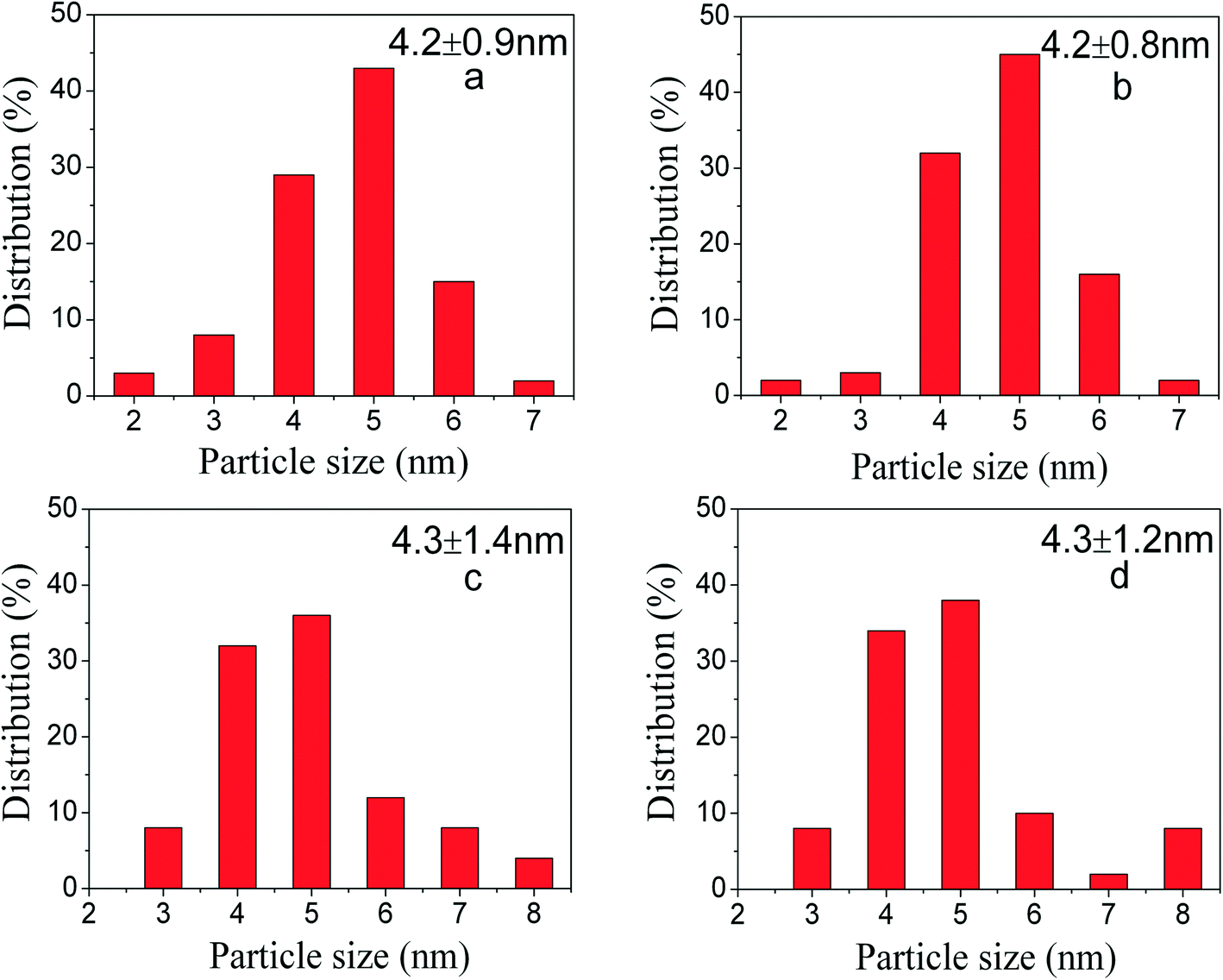 | ||
| Fig. 2 Size distribution of Pd-containing particles in different systems showing a) PdNi NPs capped with TTAB; b) PdNi@SiO2 NPs synthesized at a pH value of 11.6 and a TEOS/Pd ratio of 370:1; c) Pd–NiO@SiO2 nanocatalysts obtained after 500 °C calcination and following 200 °C H2 reduction; d) PdNi@SiO2 nanocatalysts obtained after 500 °C calcination and following 500 °C H2 reduction. | ||
XRD patterns of PdNi bimetallic NPs and as-prepared PdNi@SiO2 NPs with a SiO2 shell thickness of ~17 nm are shown in Fig. 3. As shown in Fig. 3a, the PdNi bimetallic NPs showed a broad diffraction at 2θ of 41.6° without any diffractions of monometallic Pd and Ni, which is consistent with the diffraction of (111) planes of PdNi alloy and the lattice spacing of 0.217 nm in the inset of Fig. 1a. The weak XRD diffractions of PdNi alloy NPs and lack of diffractions at high 2θ values were consistent with their small sizes around 4.2 nm. After coating PdNi alloy NPs with SiO2, the diffractions of PdNi alloy NPs (Fig. 3b) became invisible due to the low weight percentage of PdNi in PdNi@SiO2 NPs, and only broad XRD diffraction of amorphous SiO2 at 2θ of 21.9° was present.
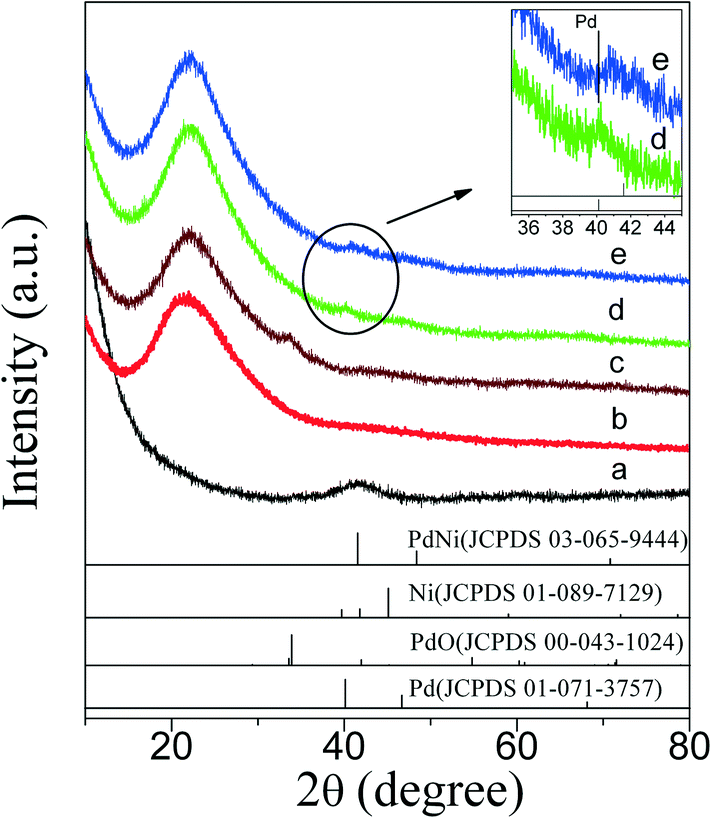 | ||
| Fig. 3 XRD patterns of NPs showing a) as-prepared PdNi alloy NPs; b) as-prepared PdNi@SiO2 NPs; c) PdO–NiO@SiO2 NPs obtained by 500 °C calcination of PdNi@SiO2 NPs; d) Pd–NiO@SiO2 nanocatalysts obtained by 500 °C calcination and following 200 °C reduction of PdNi@SiO2 NPs; e) PdNi@SiO2 nanocatalysts obtained by 500 °C calcination and following 500 °C reduction of PdNi@SiO2 NPs. All PdNi@SiO2 samples before treatments were synthesized under the conditions of a pH value of 11.6 and a TEOS/Pd ratio of 370:1. Vertical lines indicate PdNi (JCPDS 03-065-9444), Ni (JCPDS 01-089-7129), PdO (JCPDS 00-043-1024), and Pd (JCPDS 01-071-3757). | ||
To obtain the oxidation state of PdNi alloy nanoparticles, XPS studies were carried out. As shown in Fig. 4a, four peaks with binding energies of 335.1, 336.1, 340.4 and 341.7 eV were observed. According to the literature,34,35 the binding energies of 335.1 and 340.4 eV were ascribed to 3d5/2 and 3d3/2 of zero-valent Pd, while the binding energies of 336.1 and 341.7 eV were consistent with the binding energies of 3d5/2 and 3d3/2 of oxidized Pd2+, respectively. The appearance of Pd2+ species is due to the oxidation of surface Pd atoms by O2 in the air, and a Pd2+/Pd0 ratio of 29.1:70.9 by XPS curve fitting36 was observed. The XPS spectrum of Ni is shown in Fig. 4b. The binding energies of 855.3, 860.9, 872.9 and 878.9 eV were assigned to 2p3/2, 2p3/2 satellite, 2p1/2, and 2p1/2 satellite of Ni2+,37–39 respectively. A very weak peak with a binding energy of 851.7 eV was also observed and can be assigned to 2p3/2 of zero-valent Ni species. It is well known that Ni is easily oxidized in air, and the literature has reported that Ni metallic NPs are fully transformed into NiO NPs after their exposure to air.40 Since XPS is a surface-sensitive technology and surface Ni atoms are more easily oxidized, the dominant Ni2+ species in PdNi alloy NPs by XPS measurement is reasonable.
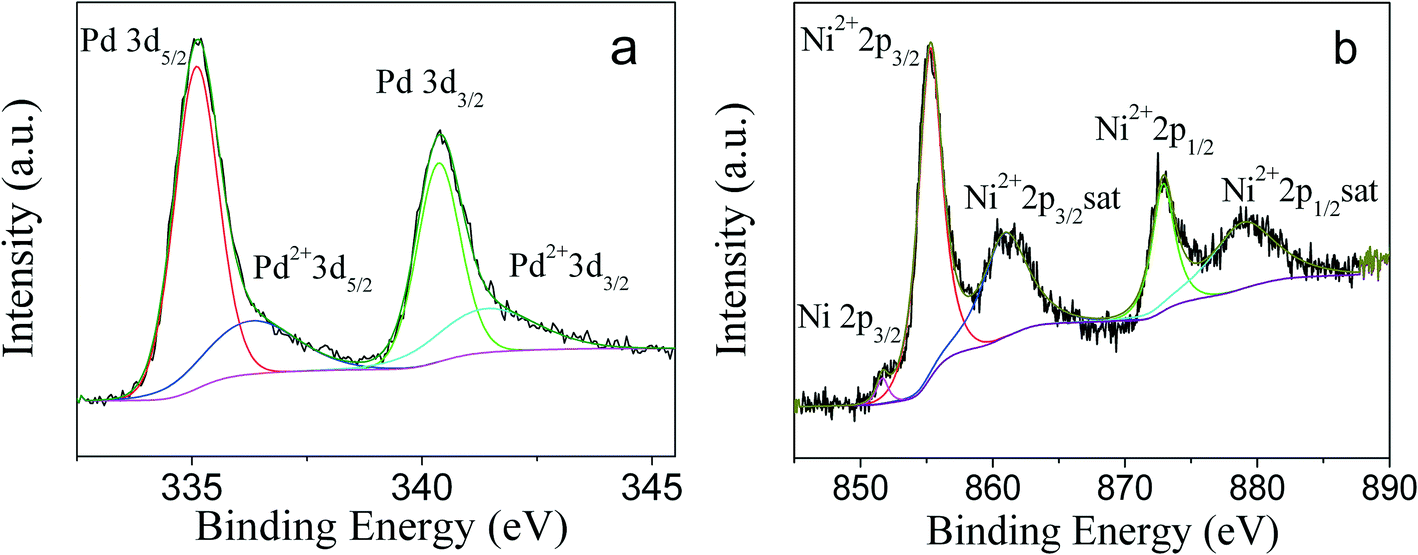 | ||
| Fig. 4 XPS spectra of PdNi alloy showing a) Pd and b) Ni. | ||
For catalytic applications, the remaining TTAB in PdNi@SiO2 NPs should be removed by calcination to open the mesoporous channels of the SiO2 shell so that reactants can contact with the reactive metal cores and the products can diffuse out through the mesoporous channels. Therefore, the thermal treatments of PdNi@SiO2 NPs were investigated. The TG/DTA profiles of the as-synthesized PdNi@SiO2 NPs with a shell thickness of ~17 nm are illustrated in the left panel of Fig. 5. The TTAB surfactants began to be removed at 150 °C, and the complete removal of TTAB was observed around 400 °C. The FT-IR spectra of PdNi@SiO2 with a shell thickness of ~17 nm before and after 500 °C calcination are shown in the right panel of Fig. 5. The absorption peaks at 2926 cm−1 and 2852 cm−1 before calcination were ascribed to antisymmetric and symmetric stretching modes of methylene of TTAB,41,42 respectively. However, the absorption peaks of methylene disappeared after calcination at 500 °C, indicating the complete removal of TTAB at 500 °C.
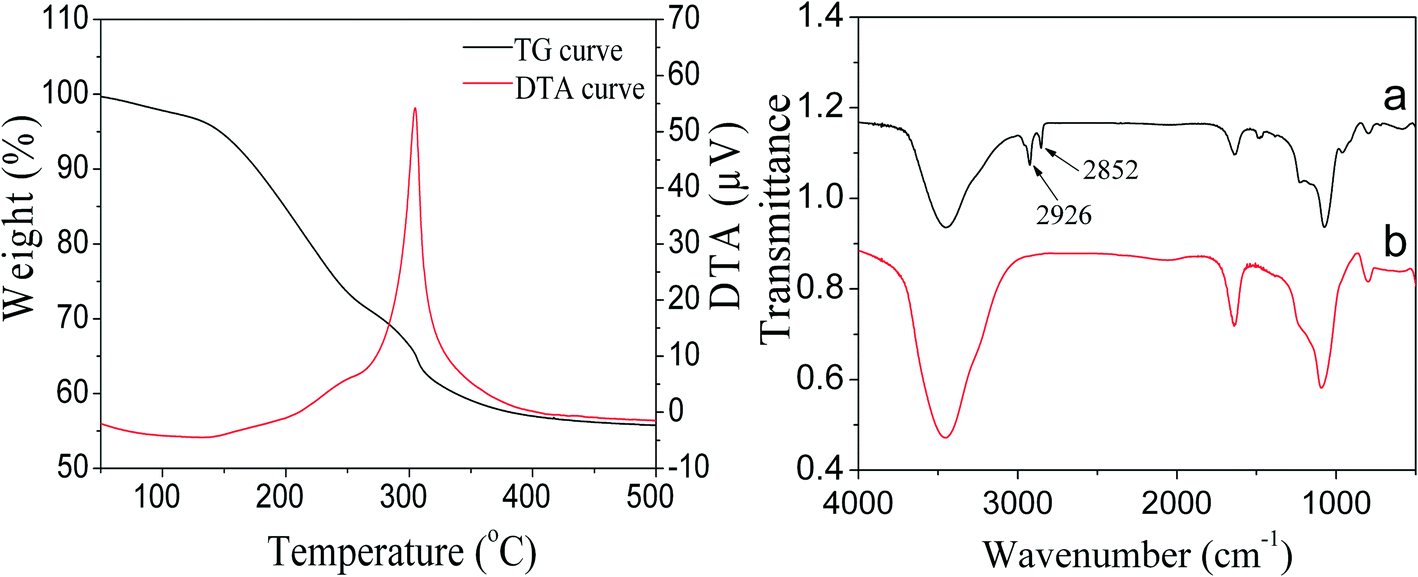 | ||
| Fig. 5 TG/DTA curves of as-synthesized PdNi@SiO2 NPs (left panel) and FT-IR spectra of PdNi@SiO2 NPs before and after 500 °C calcination (right panel). | ||
The XRD diffractions (Fig. 3c) at 2θ of 33.8° of PdNi@SiO2 NPs after calcination at 500 °C confirmed the formation of PdO. The NiO diffraction is absent due to the formation of amorphous NiO or the low weight percentage of NiO, and this phenomenon has been recorded in the Au–NiO/SiO2 system.32 The presence of Ni in the PdO–NiO@SiO2 NPs obtained by calcination of PdNi@SiO2 NPs was evidenced by ICP-OES measurement. Since the active component for catalytic hydrogenation is zero-valent Pd species, the PdO–NiO@SiO2 NPs were subjected to H2 reaction to reduce PdO. Fig. 6 illustrates the H2-TPR curve of PdO–NiO@SiO2 NPs. The two H2 consumption peaks at 106 °C and 123 °C were ascribed to the reduction of PdO, while the broad peak around 300–500 °C was ascribed to the reduction of NiO.43,44
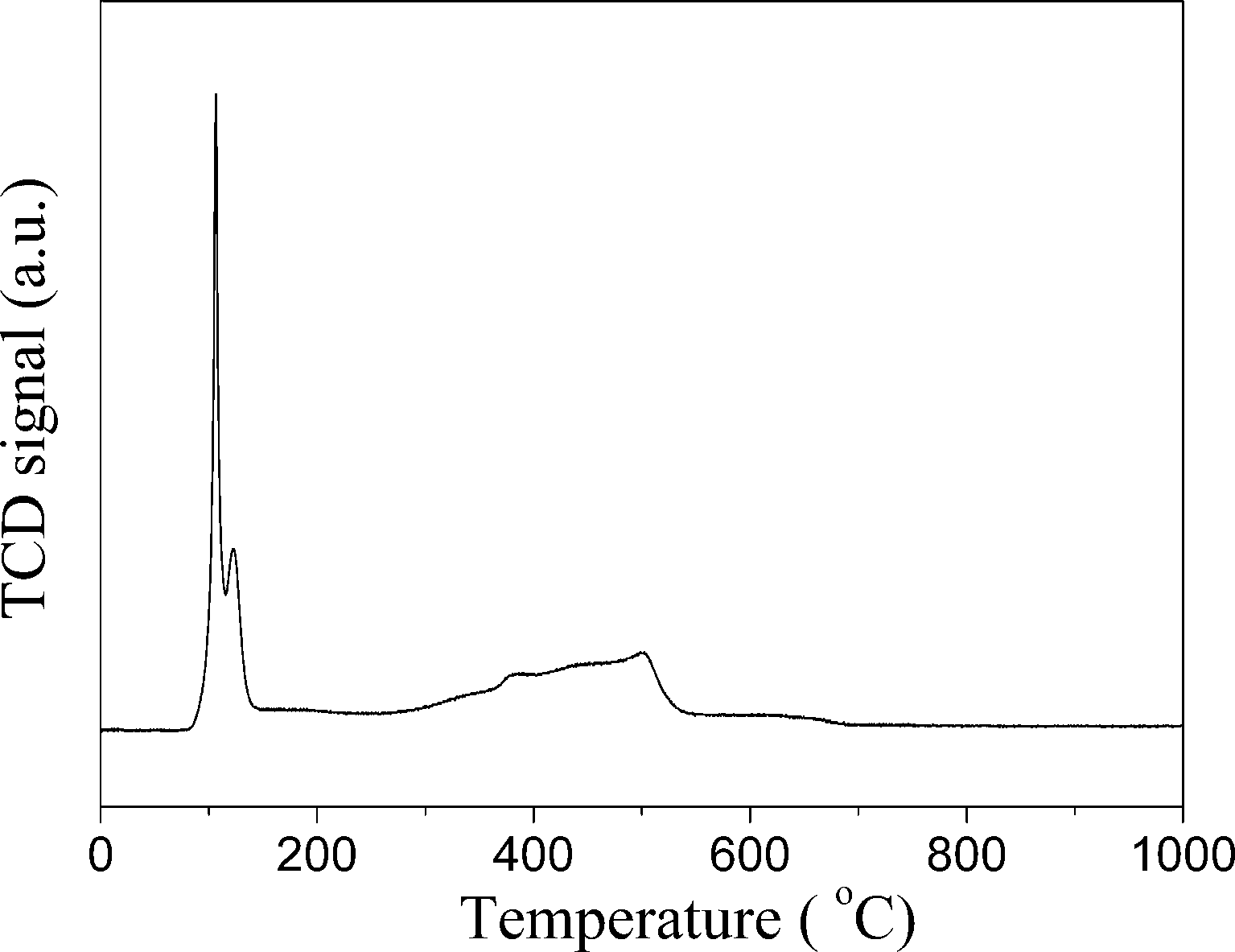 | ||
| Fig. 6 H2-TPR curves of PdO–NiO@SiO2 NPs. | ||
According to the H2-TPR result of PdO–NiO@SiO2 NPs in Fig. 6, the PdO–NiO@SiO2 NPs were reduced by H2 at 200 °C and 500 °C to obtain Pd–NiO@SiO2 and bimetallic PdNi@SiO2 nanocatalysts, respectively. The XRD patterns of Pd–NiO@SiO2 nanocatalysts in Fig. 3d confirmed the formation of Pd by the presence of weak Pd (111) diffractions at 2θ of 40.1°. The corresponding TEM images of Pd–NiO@SiO2 nanocatalysts are shown in Fig. 1g. It was clearly observed that the core–shell structure remained intact after calcination at 500 °C and following H2 reduction at 200 °C, and the inset clearly demonstrated a porous structure of Pd–NiO@SiO2 NPs. The XRD patterns and TEM images of PdNi@SiO2 nanocatalysts are illustrated in Fig. 3e and 1h, respectively. The absence of individual Pd and individual Ni diffractions in Fig. 3e suggested the presence of bimetallic PdNi NP cores in PdNi@SiO2 nanocatalysts, and the weak diffractions at 2θ of 40.8° is due to the formation of PdNi alloy NPs with concentration gradients inside the NPs possibly by surface segregation. The corresponding TEM images in Fig. 1h illustrated porous intact core–shell structures, showing the high thermal stability of PdNi@SiO2 nanocatalysts. Moreover, the size analysis of cores of Pd–NiO@SiO2 (Fig. 2c) and PdNi@SiO2 (Fig. 2d) nanocatalysts revealed nearly the same average size before and after thermal treatment, further confirming the high thermal stability of the materials.
The porosity of PdO–NiO@SiO2 NPs, Pd–NiO@SiO2 and PdNi@SiO2 nanocatalysts was examined by N2 adsorption experiments. Adsorption–desorption isotherms and the pore size distribution are shown in Fig. 7. The BET surface areas, pore volumes and pore sizes of different samples are summarized in Table 1. Typical type IV isotherms in Fig. 7 according to IUPAC nomenclature45 were observed for all samples, indicating the formation of mesopores by removal of TTAB surfactants through calcination at 500 °C. The mesopores with a mean pore size of around 2.2 nm were found. The BET surface areas of PdO–NiO@SiO2 NPs, Pd–NiO@SiO2 and PdNi@SiO2 nanocatalysts were around 771–791 m2 g−1, suggesting that the following H2 treatment at 200 °C and 500 °C did not significantly change the surface area and that this kind of core–shell material was highly thermally stable.
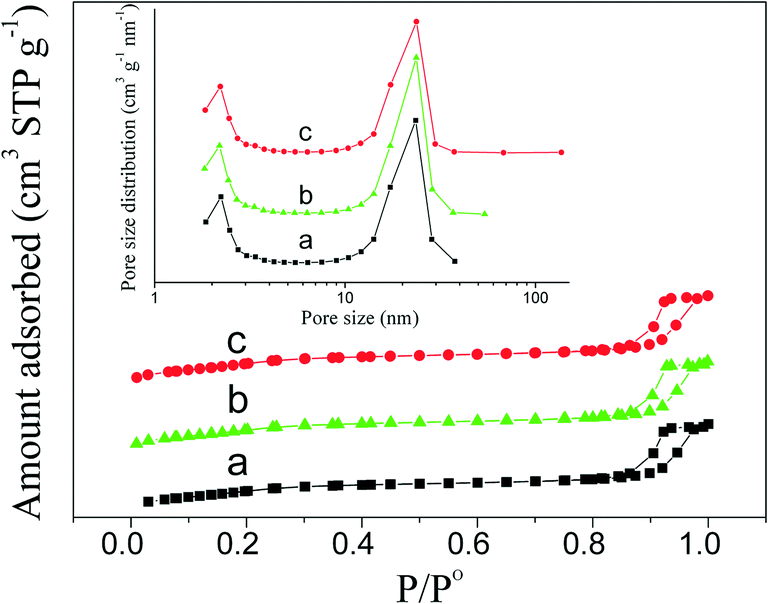 | ||
| Fig. 7 N2 adsorption–desorption isotherms and pore size distribution (inset) showing a) PdO–NiO@SiO2 NPs, b) Pd–NiO@SiO2 nanocatalysts, and c) PdNi@SiO2 nanocatalysts. | ||
| Samples | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|
| PdO–NiO@SiO2 | 771 | 1.1 | 2.2 |
| Pd–NiO@SiO2 | 791 | 1.1 | 2.2 |
| PdNi@SiO2 | 773 | 1.1 | 2.2 |
The elemental compositions of Pd–NiO@SiO2, PdNi@SiO2 and the control Pd@SiO2 nanocatalysts were analyzed by ICP-OES. Table 2 summarizes the real loadings of Pd and Ni in Pd-containing core–shell nanocatalysts. The control Pd@SiO2 mesoporous nanocatalysts were synthesized according to the literature,25 and the detailed characterization is presented in the ESI.† The real Pd loadings of Pd@SiO2, Pd–NiO@SiO2 and PdNi@SiO2 nanocatalysts are 0.68 wt%, 0.36 wt%, and 0.36 wt%, respectively, and the molar ratios of Pd/Ni in the Pd–NiO@SiO2 and PdNi@SiO2 nanocatalysts are close to 1:1.
| Sample | Pd loading (wt%) | Ni loading (wt%) |
|---|---|---|
| Pd@SiO2 | 0.68 | — |
| Pd–NiO@SiO2 | 0.36 | 0.23 |
| PdNi@SiO2 | 0.36 | 0.24 |
The hydrogenation of p-chloronitrobenzene (p-CNB) to p-chloroaniline (p-CAN) is accompanied by side reactions such as dechlorination to aniline (AN). The supported Pd catalysts exhibited moderate p-CAN selectivity,46 and the dechlorination over Pd catalysts was more severe than that of Pt-based catalysts.47 The literature has also reported that the application of supported Pd catalysts was very active in dechlorination reactions, where the dechlorination products are desired.48 In this study, to investigate the influence of elemental compositions of cores on dechlorination reactions, the Pd–NiO@SiO2, PdNi@SiO2 and control Pd@SiO2 mesoporous nanocatalysts were tested for catalytic hydrogenation of p-chloronitrobenzene with H2 at 30 °C and ambient H2 pressure. To ensure the same mole number of Pd in each nanocatalyst for catalytic performance comparison, different amounts of Pd-containing core–shell nanocatalysts with the same thickness of SiO2 shells were used since the Pd loadings in different catalysts were different. Table 3 summarizes the catalytic performance of Pd–NiO@SiO2, PdNi@SiO2 and Pd@SiO2 nanocatalysts. Indeed, the control Pd@SiO2 mesoporous nanocatalysts with a SiO2 shell thickness of ~17 nm showed a p-CNB conversion of 98.7% with a poor p-chloroaniline (p-CAN) selectivity of 62.9%, showing a significant formation of the by-product aniline produced by dechlorination reactions. The p-aminophenol (p-AP) formation was also found in this reaction. The literature has reported the formation of p-AP during catalytic hydrogenation of nitrobenzene with H2,49,50 and p-AP in this study could be originated from the hydrogenation of nitrobenzene produced by dechlorination of p-CNB and/or the dechlorination of p-CAN. In contrast, the PdNi@SiO2 illustrated an enhanced p-CAN selectivity of 80.9% with a decreased conversion of 68.5%. The Pd–NiO@SiO2 nanocatalysts demonstrated a superior catalytic performance to Pd@SiO2 and PdNi@SiO2 nanocatalysts. At the conversion of 100%, the selectivity of p-CAN over Pd–NiO@SiO2 is 82.7%, which is significantly higher than that of control Pd@SiO2 nanocatalysts.
| Catalyst | Conversion (%) | Selectivity (%) | |||
|---|---|---|---|---|---|
| p-CAN | AN | p-AP | Others | ||
| Reaction conditions: reaction temperature—30 °C; ambient H2 pressure; p-CNB—1.0 g; reaction time—2.5 hours; ethanol—24 mL; Pd@SiO2—0.100 g (Pd loading—0.68 wt%); PdNiO@SiO2—0.190 g (Pd loading—0.36 wt%); PdNi@SiO2—0.190 g (Pd loading—0.36 wt%); speed of agitation—500 rpm. | |||||
| Pd@SiO2 | 98.7 | 62.9 | 22.8 | 14.3 | 0.0 |
| PdNi@SiO2 | 68.5 | 80.9 | 5.4 | 13.6 | 0.1 |
| Pd–NiO@SiO2 | 100.0 | 82.7 | 12.5 | 4.8 | 0.0 |
Supported Pd catalysts were active for hydrogenation reactions51 but showed moderate p-CAN selectivity for catalytic p-CNB hydrogenation. By alloying with Ni, the p-CAN selectivity over PdNi@SiO2 is enhanced at the expense of activity, which is due to dilution of Pd by Ni on the surface. The same phenomenon had been well stated in the literature.30 However, the transformation of PdNi alloy NPs to close contact Pd–NiO heteroaggregate NPs via proper treatment could induce the strong Pd–NiO interaction, which is possibly beneficial for hydrogenation reactions as suggested in the literature.30–33 In this study, the selectivity enhancement of p-CAN over Pd–NiO@SiO2 is ascribed to the strong Pd–NiO interaction, and the Pd–NiO interface facilitates hydrogenation reactions.
The influence of reaction time on catalytic performance over Pd@SiO2 and Pd–NiO@SiO2 nanocatalysts at 30 °C under ambient H2 pressure was examined, and the result is shown in Fig. 8. The conversion of p-CNB increased from 44% at 0.5 hours to 100% at 2.5 hours for both Pd@SiO2 and Pd–NiO@SiO2 nanocatalysts and remained 100% with further increasing reaction time. The p-CAN selectivity of Pd–NiO@SiO2 was slightly higher than that of Pd@SiO2 at the beginning of the reactions, but the p-CAN selectivity gap between Pd–NiO@SiO2 and Pd@SiO2 nanocatalysts became significant when the reaction time was increased. The influence of reaction temperatures on the catalytic performance over Pd–NiO@SiO2 nanocatalysts is shown in Fig. 9. It was found that the conversion of p-CNB increased from 86% at 20 °C to 100% at 30 °C. On the contrary, the selectivity of p-CAN firstly increased from 65% at 20 °C to 82% at 30 °C and then decreased to 60% at 60 °C. The result suggested that when the temperature exceeded 30 °C, the formation of a by-product is much severe, resulting in decreased p-CAN selectivity.
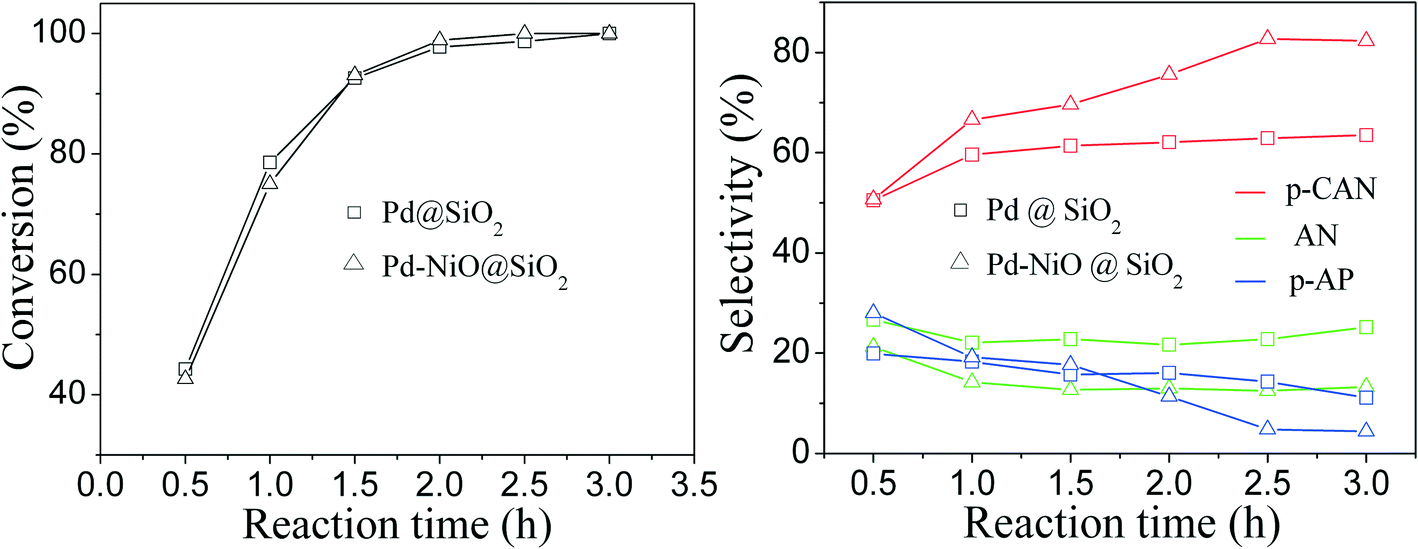 | ||
| Fig. 8 Effect of reaction time on p-CNB conversion (left panel) and selectivity of p-CAN, AN and p-AP (right panel) for catalytic p-CNB hydrogenation over Pd@SiO2 and Pd–NiO@SiO2 nanocatalysts. Reaction conditions: reaction temperature—30 °C; ambient H2 pressure; Pd@SiO2—0.100 g; Pd–NiO@SiO2—0.190 g; p-CNB—1.0 g; 24 mL of ethanol; speed of agitation—500 rpm. | ||
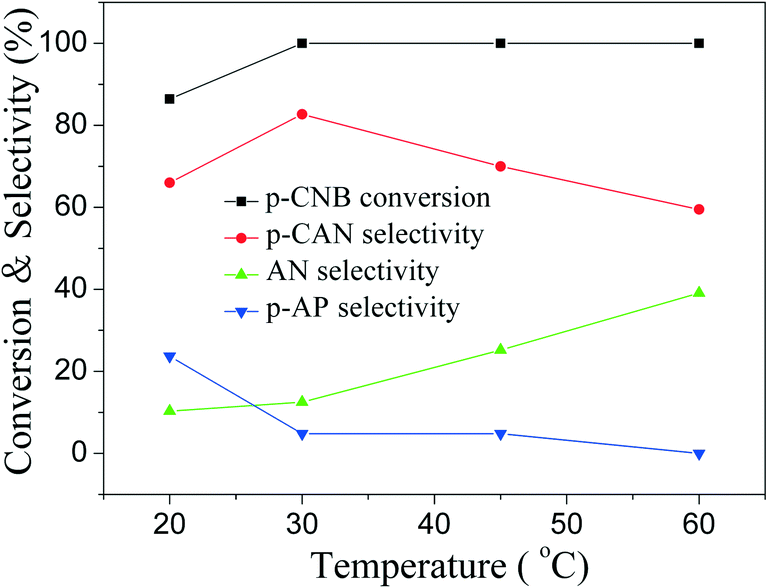 | ||
| Fig. 9 Effect of temperature on p-CNB conversion and selectivity of different products for catalytic p-CNB hydrogenation over Pd–NiO@SiO2 nanocatalysts. Reaction conditions: ambient H2 pressure; Pd–NiO@SiO2 catalysts—0.190 g; p-CNB—1.0 g; 24 mL of ethanol; reaction time—2.5 hours; speed of agitation—500 rpm. | ||
Cycle-to-cycle catalytic reactions were carried out to investigate the catalytic stability of Pd–NiO@SiO2 and Pd@SiO2 nanocatalysts, and the results are listed in Table 4. Because of the loss of catalysts during the recovery process after each reaction cycle, the amounts of p-CNB and ethanol in the next cycle were decreased accordingly to maintain the ratios of p-CNB/catalysts and p-CNB/solvent identical in each reaction run. In these cycle-to-cycle reactions, the conversion of p-CNB for Pd@SiO2 and Pd–NiO@SiO2 was basically maintained and approached 95.9% and 99.4% after five cycles of reactions, respectively. The p-CAN selectivity over Pd–NiO@SiO2 nanocatalysts in each cycle was higher than that of Pd@SiO2 nanocatalysts and was stabilized around 66.9% after five cycles of reactions. The average p-CAN selectivity over Pd–NiO@SiO2 nanocatalysts during five cycles of reactions was 72.6%, which is significantly higher than that over Pd@SiO2 nanocatalysts (61.0%), illustrating the catalytic selectivity enhancement over Pd–NiO@SiO2 nanocatalysts. The decrease of p-CAN selectivity over Pd–NiO@SiO2 catalysts might be caused by a slight dissolution of NiO since the reaction system is acidic due to the dechlorination side reaction. Although the selectivity decreased from the first run to the second run, the selectivity of p-CAN was stabilized in the range of 66.9% to 69.4%, which was still higher than that of Pd@SiO2.
| Cycle index | Pd–NiO@SiO2 catalyst | Pd@SiO2 catalyst | ||||||
|---|---|---|---|---|---|---|---|---|
| Weight (g) | p-CNB (g) | Conversion (%) | p-CAN selectivity (%) | Weight (g) | p-CNB (g) | Conversion (%) | p-CAN selectivity (%) | |
| Reaction conditions: reaction temperature—30 °C; p-CNB—1.0 g; reaction time—2.5 hours; atmospheric H2 pressure; speed of agitation—500 rpm; 24 mL of ethanol was used as solvent in the first cycle, and the ratio of ethanol/p-CNB in the following cycles was kept the same as that of the first cycle. | ||||||||
| 1 | 0.190 | 1.000 | 100.0 | 82.7 | 0.100 | 1.000 | 98.7 | 62.9 |
| 2 | 0.170 | 0.885 | 99.9 | 68.5 | 0.086 | 0.858 | 97.8 | 61.7 |
| 3 | 0.151 | 0.800 | 99.4 | 67.5 | 0.072 | 0.722 | 98.5 | 59.6 |
| 4 | 0.129 | 0.681 | 98.9 | 69.4 | 0.057 | 0.574 | 96.4 | 61.5 |
| 5 | 0.106 | 0.557 | 97.6 | 68.6 | 0.045 | 0.447 | 98.4 | 60.7 |
| 6 | 0.085 | 0.447 | 99.4 | 66.9 | 0.032 | 0.319 | 95.9 | 59.7 |
The activity sequence of p-CNB hydrogenation over Pd@SiO2, PdNi@SiO2 and Pd–NiO@SiO2 is in the following order: Pd–NiO@SiO2 > Pd@SiO2 > PdNi@SiO2. The catalytic p-CAN selectivity sequence of p-CNB hydrogenation is in the following order: Pd–NiO@SiO2 > PdNi@SiO2 > Pd@SiO2. In this study, the p-CAN selectivity of PdNi@SiO2 is higher than that of Pd@SiO2, while the activity of PdNi@SiO2 is lower than that of Pd@SiO2, reflecting that the adjustment of electronic state by metal–metal interaction in bimetallic alloy NPs enhances the p-CAN selectivity but at the expense of activity. However, both catalytic activity and selectivity over Pd–NiO@SiO2 nanocatalysts are higher than those of Pd@SiO2, suggesting that the interfaces between Pd and NiO in the cores of Pd–NiO@SiO2 nanocatalysts can enhance the catalytic activity and selectivity at the same time.
4. Conclusions
In this work, Pd–NiO@SiO2 and PdNi@SiO2 core–shell mesoporous nanocatalysts were successfully synthesized by a sol–gel process. PdNi alloy NPs were firstly synthesized by co-reduction of K2PdCl4 and Ni(acac)2 by NaBH4 using TTAB as a capping agent. The SiO2 shells were then used to coat the PdNi NPs by hydrolysis of TEOS, and the thickness of SiO2 could be tuned by adjusting the pH value of the solution and the TEOS/Pd ratios. The resultant PdNi@SiO2 NPs were subjected to calcination at 500 °C and subsequent H2 reduction at 200 °C or 500 °C to obtain mesoporous Pd–NiO@SiO2 or PdNi@SiO2 nanocatalysts, respectively. The Pd–NiO@SiO2 nanocatalysts illustrated a superior catalytic performance for p-chloronitrobenzene hydrogenation with H2 to the PdNi@SiO2 and Pd@SiO2 core–shell nanocatalysts at the same amount of Pd and the same thickness of mesoporous silica shells. The catalytic performance enhancement is assigned to the strong interaction between Pd and NiO, where the interfaces between Pd and NiO promote catalytic activity and selectivity for p-chloronitrobenzene hydrogenation with H2. The developed Pd–NiO@SiO2 heteroaggregate nanocatalysts demonstrated enhanced catalytic selectivity while maintaining high catalytic activity at the same time, which is in contrast to the selectivity enhancement at the expense of activity for bimetallic alloy catalysts. This work provides a new method to enhance catalytic selectivity for catalytic hydrogenation reactions. The synthetic method developed in this study could be extended to diverse core–shell mesoporous nanocatalysts with metal–oxide heteroaggregate NP cores and mesoporous shells for wide catalytic applications.Acknowledgements
S. Zhou would like to thank the Ministry of Science and Technology of China (grant no. 2012DFA40550) for financial support. H. Liu would like to thank Hainan University for financial support to the development plan of outstanding graduate thesis.References
- Y. W. Lee, M. Kim, Z. H. Kim and S. W. Han, J. Am. Chem. Soc., 2009, 131, 17036–17037 CrossRef CAS PubMed.
- L. J. Lauhon, M. S. Gudiksen, C. L. Wang and C. M. Lieber, Nature, 2002, 420, 57–61 CrossRef CAS PubMed.
- F. Tao, M. E. Grass, Y. W. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932–934 CrossRef CAS PubMed.
- L. Zhang, H. Jing, G. Boisvert, J. Z. He and H. Wang, ACS Nano, 2012, 6, 3514–3527 CrossRef CAS PubMed.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS PubMed.
- Y. Tak, S. J. Hong, J. S. Lee and K. Yong, J. Mater. Chem., 2009, 19, 5945–5951 RSC.
- T. Gao, Q. H. Li and T. H. Wang, Chem. Mater., 2005, 17, 887–892 CrossRef CAS.
- V. Salgueirino-Maceira and M. A. Correa-Duarte, Adv. Mater., 2007, 19, 4131–4144 CrossRef CAS PubMed.
- S. Alayoglu and B. Eichhorn, J. Am. Chem. Soc., 2008, 130, 17479–17486 CrossRef CAS PubMed.
- S. Alayoglu, A. U. Nilekar, M. Mavrikakis and B. Eichhorn, Nat. Mater., 2008, 7, 333–338 CrossRef CAS PubMed.
- T. Li, J. Moon, A. A. Morrone, J. J. Mecholsky, D. R. Talham and J. H. Adair, Langmuir, 1999, 15, 4328–4334 CrossRef CAS.
- H. J. Cha, Y. H. Kim, Y. C. Kang and Y. S. Kang, IEEE NMDC 2006: IEEE. Nanotechnology Materials and Devices Conference 2006, Proceedings, 2006, pp. 670–671 Search PubMed.
- N. Zhang, S. Q. Liu, X. Z. Fu and Y. J. Xu, J. Phys. Chem. C, 2011, 115, 9136–9145 CAS.
- X. G. Lu, G. Y. Liang, Z. B. Sun and W. Zhang, Mater. Sci. Eng., B, 2005, 117, 147–152 CrossRef PubMed.
- G. H. Wang and A. Harrison, J. Colloid Interface Sci., 1999, 217, 203–207 CrossRef CAS PubMed.
- N. J. Tang, L. Y. Lu, W. Zhong, C. T. Au and Y. W. Du, Sci. China, Ser. G: Phys., Mech. Astron., 2009, 52, 31–34 CrossRef CAS PubMed.
- S. H. Joo, J. Y. Park, C. K. Tsung, Y. Yamada, P. D. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed.
- I. Lee, J. B. Joo, Y. Yin and F. Zaera, Angew. Chem., Int. Ed., 2011, 50, 10208–10211 CrossRef CAS PubMed.
- T. Montini, A. M. Condo, N. Hickey, F. C. Lovey, L. De Rogatis, P. Fornasiero and M. Graziani, Appl. Catal., B, 2007, 73, 84–97 CrossRef CAS PubMed.
- H. Yin, Z. Ma, M. Chi and S. Dai, Catal. Today, 2011, 160, 87–95 CrossRef CAS PubMed.
- X. Zhang, H. Zhu, Z. Guo, Y. Wei and F. Wang, Int. J. Hydrogen Energy, 2010, 35, 8841 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- T. Liu, D. Li, Y. Zou, D. Yang, H. Li, Y. Wu and M. Jiang, J. Colloid Interface Sci., 2010, 350, 58 CrossRef CAS PubMed.
- C. Z. Wu, Z. Y. Lim, C. Zhou, W. G. Wang, S. H. Zhou, H. F. Yin and Y. J. Zhu, Chem. Commun., 2013, 49, 321 Search PubMed.
- Y. B. Hu, K. Tao, C. Z. Wu, C. Zhou, H. F. Yin and S. H. Zhou, J. Phys. Chem. C, 2013, 117, 8974–8982 CAS.
- M. Koussathana, D. Vamvouka, H. Economou and X. Verykios, Appl. Catal., 1991, 77, 283–301 CrossRef CAS.
- P. K. Cheekatamarla and A. M. Lane, Int. J. Hydrogen Energy, 2005, 30, 1277–1285 CrossRef CAS PubMed.
- N. Lingaiah, P. S. S. Prasad, P. K. Rao, L. E. Smart and F. J. Berry, Appl. Catal., A, 2001, 213, 189–196 CrossRef CAS.
- Y. Li, Z. G. Li and R. X. Zhou, J. Mol. Catal. A: Chem., 2008, 279, 140–146 CrossRef CAS PubMed.
- A. Corma, P. Serna, P. Concepcion and J. J. Calvino, J. Am. Chem. Soc., 2008, 130, 8748–8753 CrossRef CAS PubMed.
- S. Zhou, Z. Ma, H. F. Yin, Z. L. Wu, B. Eichhorn, S. H. Overbury and S. Dai, J. Phys. Chem. C, 2009, 113, 5758–5765 CAS.
- S. Zhou, H. F. Yin, V. Schwartz, Z. L. Wu, D. Mullins, B. Eichhorn, S. H. Overbury and S. Dai, ChemPhysChem, 2008, 9, 2475–2479 CrossRef CAS PubMed.
- X. D. Wang, H. B. Yu, D. Y. Hua and S. Zhou, J. Phys. Chem. C, 2013, 117, 7294–7302 CAS.
- E. Lundgren, G. Kresse, C. Klein, M. Borg, J. N. Andersen, M. De Santis, Y. Gauthier, C. Konvicka, M. Schmid and P. Varga, Phys. Rev. Lett., 2002, 88, 246103 CrossRef CAS.
- D. Zemlyanov, B. Aszalos-Kiss, E. Kleimenov, D. Teschner, S. Zafeiratos, M. Havecker, A. Knop-Gericke, R. Schlögl, H. Gabasch, W. Unterberger, K. Hayek and B. Klözer, Surf. Sci., 2006, 600, 983–994 CrossRef CAS PubMed.
- J. M. Conny and C. J. Powell, Surf. Interface Anal., 2000, 29, 856–872 CrossRef CAS.
- C. P. Li, A. Proctor and D. M. Hercules, Appl. Spectrosc., 1984, 38, 880–886 CrossRef CAS.
- G. R. Gavalas, C. Phichitkul and G. E. Voecks, J. Catal., 1984, 88, 54–64 CrossRef CAS.
- T. H. Gardner, J. J. Spivey, E. L. Kugler, A. Campos, J. C. Hissam and A. D. Roy, J. Phys. Chem. C, 2010, 114, 7888–7894 CAS.
- Y. L. Hou and S. Gao, J. Mater. Chem., 2003, 13, 1510–1512 RSC.
- T. Noguchi and M. Sugiura, Biochemistry, 2003, 42, 6035–6042 CrossRef CAS PubMed.
- C. Aliaga, J. Y. Park, Y. Yamada, H. S. Lee, C. K. Tsung, P. D. Yang and G. A. Somorjai, J. Phys. Chem. C, 2009, 113, 6150–6155 CAS.
- D. Karthikeyan, N. Lingappan, B. Sivasankar and N. J. Jabarathinam, Ind. Eng. Chem. Res., 2008, 47, 6538–6546 CrossRef CAS.
- K. W. Wang, S. R. Chung and C. W. Liu, J. Phys. Chem. C, 2008, 112, 10242–10246 CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- C. A. Marques, M. Selva and P. Tundo, J. Org. Chem., 1993, 58, 5256–5260 CrossRef CAS.
- L. Prati and M. Rossi, Appl. Catal., B, 1999, 23, 135–142 CrossRef CAS.
- Y. P. Zhang, S. J. Liao and Y. Xu, Tetrahedron Lett., 1994, 35, 4599–4602 CrossRef CAS.
- C. V. Rode, M. J. Vaidya and R. V. Chaudhari, Org. Process Res. Dev., 1999, 3, 465–470 CrossRef CAS.
- S. F. Wang, Y. H. Ma, Y. J. Wang, W. Xue and X. Q. Zhao, J. Chem. Technol. Biotechnol., 2008, 83, 1466–1471 CrossRef CAS PubMed.
- S. Dutta, S. Sarkar, C. Ray, A. Roy, R. Sahoo and T. Pal, ACS Appl. Mater. Interfaces, 2014, 6, 9134–9143 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy00996g |
| This journal is © The Royal Society of Chemistry 2015 |