
When catalyst meets reactor: continuous biphasic processing of xylan to furfural over GaUSY/Amberlyst-36
Christof
Aellig
,
David
Scholz
,
Pierre Y.
Dapsens
,
Cecilia
Mondelli
and
Javier
Pérez-Ramírez
*
Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, CH-8093 Zurich, Switzerland. E-mail: jpr@chem.ethz.ch; Fax: +41 44 6331405; Tel: +41 44 6337120
First published on 1st September 2014
Abstract
The multi-step conversion of xylan hemicellulose and xylose to furfural is investigated in a continuous-flow biphasic fixed-bed reactor over a catalytic bed constituted of a physical mixture of a Lewis acid gallium-containing USY zeolite for xylose isomerisation and a Brønsted acid ion-exchanged resin, Amberlyst-36, for hemicellulose hydrolysis and xylulose dehydration. The water-soluble substrates are converted in the aqueous phase into intermediates which promptly transform into the desired furan product due to the high degree of contact between the solid catalysts. The large interfacial area developed in the reactor through the Taylor flow of liquids enables efficient extraction of furfural to the organic phase, minimising side reactions. The diminished contact of the GaUSY catalyst with water controls its instability against metal leaching, leading to stable operation for 24 h on stream. Optimisation of the variables, including the catalyst ratio, the nature of the extracting phase, the contact time, and the feed concentration, enables to attain furfural yields of 72% from xylose and 69% from xylan. The latter value is the highest reported in the literature for hemicellulose processing over a heterogeneous catalytic system. These results highlight the potential of concerted catalyst and reactor design strategies towards the realisation of more efficient and intensified processes for the sustainable production of bio-based chemicals and fuels.
1 Introduction
Over the past decade, the finiteness of fossil fuel reserves has promoted increasing research efforts aimed towards the development of effective strategies to valorise abundant saccharide feedstocks.1 Along with a few other compounds, furfural and 5-hydroxymethylfurfural (HMF) have been identified as the most promising intermediates in future bio-based value chains for chemical and fuel production.2 They can be derived from xylan hemicellulose and cellulose through hydrolysis followed by either Brønsted acid-catalysed dehydration or Lewis and Brønsted acid-catalysed isomerisation–dehydration of the xylose and glucose obtained. Upon the initial investigation of the conversion of the latter C5 and C6 sugars in an aqueous medium mainly over homogeneous catalysts,3 furfural and HMF yields have not exceeded 30% due to the occurrence of multiple side and consecutive reactions.4 The alternative use of organic solvents (e.g. DMSO, gamma-valerolactone) with heterogeneous catalysts or of ionic liquids with homogeneous catalysts has proved to significantly boost the furan yields (up to 80 or 95%, respectively).5 Still, these media are impractical at a large scale. The high boiling point of the former implies a high energy input for downstream purification procedures, while the latter are costly and relatively unstable against the water formed as a by-product. As a more attractive option for enhancing the selectivity, Dumesic et al.6 have proposed to process sugars in a biphasic medium comprising water and a low-boiling point organic solvent such as methyl isobutyl ketone. In this system, an extractive reaction occurs, i.e. the carbohydrates are transformed in the aqueous phase and the organic phase extracts and accumulates the desired furan product as soon as it is formed, limiting the occurrence of further unwanted reactions.6,7 The benefits of this approach have been demonstrated by the enhanced HMF yield (55%) attained upon HCl-catalysed fructose dehydration.6 Following this concept, Huber et al.8 have reported furfural yields >90% from hemicellulose using HCl or H2SO4 as a catalyst in a mixture of NaCl-containing water and THF.In addition to a high process selectivity, the use of heterogeneous rather than homogenous catalysts, not imposing energy-intensive separation steps,9 and the application of continuous-flow instead of batch reactors, ensuring higher productivities, are most prominent and complementary aspects to enable a prospective efficient large-scale conversion of sugars. With respect to the use of solid catalysts, Davis et al.10 have evaluated the batch biphasic isomerisation–dehydration of different C6 sugars using the Lewis acidic Sn-beta zeolite in combination with diluted HCl, obtaining an HMF yield of 60%. Fully heterogeneous catalytic systems based on TiO2, metal phosphates, supported Cs-containing heteropolyacids or zeolites have also been assessed in biphasic media and have led to limited HMF (40%) and furfural (54%) yields.11 The highest furfural yield (74%) was achieved with a composite material consisting of zeolite beta nanocrystals embedded in a siliceous TUD-1 mesoporous matrix.12 Concerning the employment of flow reactors, continuous stirred biphasic tank reactors6 and tubular reactors operated with a segmented flow of the aqueous and organic phases13 have been explored. The second configuration allows for superior mass transfer properties, since the organic phase forms a thin wall around the slugs of the aqueous phase, leading to a 4-fold higher interfacial area.14 Thus, Brasholz et al.15 have been able to convert fructose to HMF in the presence of HCl with a yield of 74% at a contact time of 15 min. When using zirconia and titania as catalysts, McNeff et al.16 have obtained HMF from glucose with a maximum yield of only 29% at a contact time of 2 min. The limited relevance of the catalytic results of the latter isolated effort to combine a continuous-flow reactor with a heterogeneous solid is likely due to the suboptimal acid characteristics of the materials applied.
Herein, we design a highly efficient process for the transformation of C5 sugars into furfural under a Taylor flow of water and an organic phase over a bifunctional catalytic bed (Scheme 1). The Lewis acid solid comprises a gallium-containing USY zeolite prepared via the facile alkaline-assisted metallation method, which was already demonstrated to be suitable to introduce highly selective Lewis acid centres in (alumino)silicates.17 Its activity, selectivity, and stability were first evaluated in the aqueous-phase conversion of xylose to xylulose. Thereafter, it was complemented by Brønsted acid Amberlyst-36 to yield furfural. Due to the consecutive nature of the reactions involved and thus the crucial role of an intimate mixing between the two catalysts to maximise the furfural yield, a well-homogenised physical mixture of the solids was used. The influence of key variables including the nature of the extracting phase, H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :organic solvent ratio, temperature, and catalyst proportion on the process was determined in order to enhance the furfural yield and the stability of the catalysts. Catalytic tests at variable particle sizes and temperatures and calculations were performed to verify a kinetic controlling regime and to gain insight into the characteristics of the Taylor flow in the packed bed. Finally, our optimised technology was extrapolated to the conversion of the more complex xylan hemicellulose substrate to furfural, attaining successful results.
:organic solvent ratio, temperature, and catalyst proportion on the process was determined in order to enhance the furfural yield and the stability of the catalysts. Catalytic tests at variable particle sizes and temperatures and calculations were performed to verify a kinetic controlling regime and to gain insight into the characteristics of the Taylor flow in the packed bed. Finally, our optimised technology was extrapolated to the conversion of the more complex xylan hemicellulose substrate to furfural, attaining successful results.
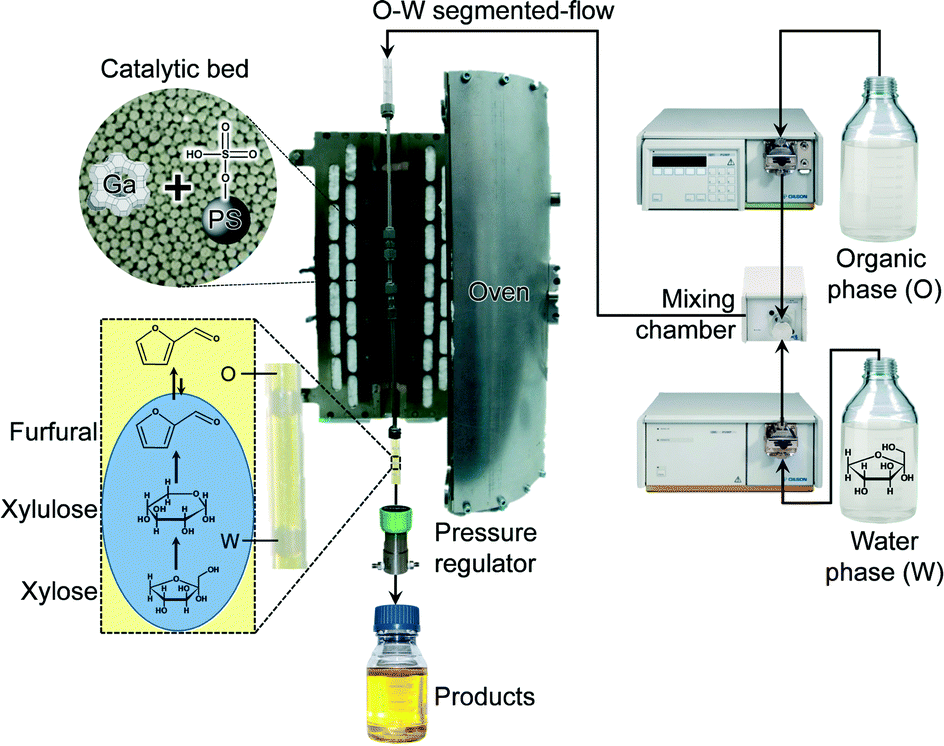 | ||
| Scheme 1 Continuous-flow biphasic reactor used for the conversion of C5 carbohydrates to furfural. | ||
2 Experimental
2.1 Catalysts
Two different commercially available USY zeolites were used in this study. CBV720 (bulk Si/Al = 15, denoted as USY-1) was purchased from Zeolyst, whereas HSZ390-HUA (bulk Si/Al = 385, denoted as USY-2) was supplied by Tosoh. Gallium was introduced in the zeolites via post-synthetic alkaline-assisted metallation, as previously reported.17 Thus, the as-received samples were treated in NaOH solutions (0.2 M, 30 cm3 per gram of dried zeolite) containing 0.04 or 0.05 M Ga(NO3)3·H2O (ABCR, 99.9%) in the case of USY-1 or USY-2, respectively, for 30 min at 338 K in an EasyMax™ 102 reactor (Mettler Toledo). The resulting materials were converted into their protonic forms by three consecutive ion exchanges in an aqueous solution of ammonium nitrate (0.1 M NH4NO3, 6 h, 298 K, 100 cm3 per gram of dried zeolite), followed by calcination in static air at 823 K (5 K min−1) for 5 h. The Ga-containing catalysts are referred to as GaUSY-1 and GaUSY-2. Amberlyst-36 (acid site concentration ≥ 5.40 eq kg−1, SBET = 33 m2 g−1) was purchased from Dow Chemicals.2.2 Characterisation
The gallium content in the fresh and used materials was evaluated by inductively coupled plasma-optical emission spectroscopy (ICP-OES) using a Horiba Ultra2 instrument. Prior to ICP measurements, the materials were digested in an acidic mixture (HCl–HNO3–HF) under heating (343 K, overnight). The amount of carbon deposited on the catalysts after reaction was determined by elemental analysis using a LECO CHN-9000 instrument. Powder X-ray diffraction (XRD) was measured using a PANalytical X'Pert Pro-MPD diffractometer with Ni-filtered Cu Kα radiation (λ = 0.1541 nm). Data were recorded in the 5–70° 2θ range with an angular step size of 0.05° and a counting time of 7 s per step. Nitrogen isotherms at 77 K were collected using a Quantachrome Quadrasorb SI analyser. Prior to measurements, samples were degassed under vacuum (10−1 mbar) at 573 K for 3 h. Fourier transform infrared (FTIR) spectroscopy of adsorbed pyridine was performed using a Bruker IFS 66 spectrometer equipped with a liquid N2-cooled mercury cadmium telluride (MTC) detector. Self-supporting zeolite wafers (20 mg, 5 ton cm−2, 1 cm2) were pretreated at 10−3 mbar and 693 K for 4 h. After cooling down to room temperature, the samples were saturated with pyridine vapour and then evacuated at room temperature for 15 min and subsequently at 473 K for 30 min. Spectra were recorded in the 650–4000 cm−1 range (4 cm−1 resolution) by co-addition of 32 scans. High-resolution magic angle spinning 71Ga nuclear magnetic resonance (MAS-NMR) spectroscopy was conducted using a Bruker AVANCE 700 NMR spectrometer equipped with a 4 mm probe head and 4 mm ZrO2 rotors at 213.5 MHz. Spectra were acquired using a spinning speed of 10 kHz, 20000 accumulations, 1 μs pulses, a recycle delay of 0.02 s and Ga(NO3)3·H2O as the reference.
Catalytic tests
Catalytic tests were performed using a homemade continuous-flow reactor setup (Scheme 1) composed of (i) a dual-pump system (Gilson-305 and Gilson-306) equipped with a mixing chamber (Gilson-811D) and a manometric module (Gilson-806), (ii) a stainless steel tubular reactor with a precolumn (Swagelok SS-T4-S-035, o.d. = 1/4 inch, i.d. = 4.6 mm), both heated in a tubular oven, and (iii) a backpressure regulator (Swagelok, LH2981001). The reactor was loaded with GaUSY-1, GaUSY-2 (0.18–0.54 g in both cases) or Amberlyst-36 (0.72 g, sieve fraction = 0.18–0.25, 0.25–0.36 or 0.6–0.7 mm), or a physical mixture of the zeolite- and resin-based catalysts (1.08 g in total), diluted with quartz in a 1:3 ratio (sieve fraction = 0.25–0.36 mm) and inserted in a tubular oven heated at 393–413 K. Thereafter, the liquid feed was admitted. The latter was composed of a Taylor flow of an aqueous phase containing 5 wt.% xylose (Sigma-Aldrich, 99%), or 2.5 wt.% of xylan (from beech wood, Sigma-Aldrich, 90%), and an extracting organic phase consisting of either methyl isobutyl ketone (MIBK, ABCR, 99%), toluene (Fluka, ≥99.7%) or dichloroethane (DCE, Fluka, ≥99.5%) in a 10:90 or 20:80 H2O:organic solvent volume ratio. To prevent solvent loss by evaporation, the system was pressurised to 25 bar prior to heating. Samples were periodically collected at the outlet. The aqueous and organic phases were separated after decantation. Xylose, xylulose and lyxose were isolated by high-performance liquid chromatography (HPLC) using an Agilent 1260 Infinity system equipped with a Biorad Aminex HPX-87C column heated at 353 K and a refractive index detector (Agilent G1362 A) set at 303 K using Millipore water (0.50 cm3 min−1) as the eluent. Quantification was done by integration of their respective peaks. Xylose and lyxose (Sigma-Aldrich, 99%) were employed as references. In the case of xylulose, the yield was calculated using the response factor of xylose. Furan derivatives present in the organic phase were analysed using a gas chromatograph (GC, HP 6890) equipped with an HP-5 capillary column and a flame ionisation detector. Furfural (Sigma-Aldrich, 99%) was employed as the reference. He was used as the carrier gas (flow rate = 1 cm3 min−1, pressure = 0.4 bar, split ratio = 35) and an injection volume of 2 μL was applied. The initial temperature of 353 K was held for 1 min before heating to 523 K (10 K min−1). The yield of furfural was determined using biphenyl (ABCR, 99%) as the internal standard.
Results and discussion
GaUSY catalysts for sugar isomerisation
In order to select the most suitable GaUSY isomerisation catalyst, alkaline-assisted galliation was performed on two different parent zeolites, the Al-rich USY-1 and the Al-lean USY-2.While galliated USY-1 has been reported to be highly active and stable for the conversion of dihydroxyacetone to ethyl lactate,17 post-synthetic gallium introduction in USY-2 has never been attempted so far. For the sake of comparison, the quantity of Ga(NO3)3 used in the metallation procedure was adapted for each sample in order to generate catalysts containing similar gallium content (ca. 6 wt.%). The compositional, structural and porous properties of parent and modified samples are reported in Table 1. The crystallinity only decreased by ca. 20% upon galliation of both USY-1 and USY-2, confirming the retention of the zeolitic structure upon the treatment. This was further supported by the negligible changes in the micropore volume (Vmicro), which remained at the levels expected for the large-pore USY zeolites (0.25 and 0.29 cm3 g−1 for GaUSY-1 and GaUSY-2, respectively). The external surface area was hardly altered by the treatment, in agreement with our previous observation that the alkaline-assisted metallation process heals vacancies.17
| Catalyst | Ga loadinga (wt.%) | Crystallinityb (%) | V pore (cm3 g−1) | V micro (cm3 g−1) | S meso (m2 g−1) |
|---|---|---|---|---|---|
| a Determined by ICP-OES. b Derived from XRD. c Volume adsorbed at p/p0 = 0.99. d Determined by the t-plot method. | |||||
| USY-1 | — | 100 | 0.56 | 0.29 | 128 |
| USY-2 | — | 100 | 0.58 | 0.30 | 105 |
| GaUSY-1 | 5.8 | 78 | 0.49 | 0.25 | 119 |
| GaUSY-2 | 5.9 | 77 | 0.57 | 0.29 | 116 |
The modified materials were evaluated in the continuous-flow isomerisation of xylose to xylulose at 403 K (Fig. 1a). While USY-1 did not show any activity, thus excluding a possible contribution of Lewis acidic extraframework aluminium (EFAl), GaUSY-1 and GaUSY-2 exhibited a xylose conversion of 14% and 21%, respectively. Xylulose was the dominant product (61 and 69% selectivity, respectively), while only traces of lyxose were detected (3% selectivity in both cases). As no additional signal was visualised in the chromatogram, it was assumed that the remaining portion of xylose converted was transformed into insoluble humins. In view of elucidating this difference in performance, the zeolites were studied by IR spectroscopy of adsorbed pyridine as well as 71Ga MAS NMR spectroscopy. The concentration of Lewis acid sites for GaUSY-2 was slightly higher than that for GaUSY-1 (134 μmol g−1versus 101 μmol g−1). Additionally, differences were observed in the structure of the metal sites introduced (Fig. 2a). While both spectra of GaUSY-1 and of GaUSY-2 featured a peak at 174 ppm, usually associated with tetrahedral (Td) gallium species, and a signal at 0 ppm, likely associated with octahedral (Oh) or distorted tetrahedral (dTh) gallium species, the latter contribution was much stronger and sharper for GaUSY-2, indicating the presence of gallium species with a more uniform structure. These results suggest that the higher activity of the GaUSY-2 material is due to either a slightly higher acidity or the presence of sites with more suitable characteristics or a combination of both factors. The role of hydrophobicity, which is well known to relate to the Al content in the zeolite, should also not be excluded, as the USY-1 and USY-2 matrices have substantially different Si/Al ratios (15 versus 385). Indeed, hydrophobicity plays a pivotal role in catalyst stability and activity in the upgrading of biomass in aqueous and biphasic mixtures, as recently demonstrated by Resasco et al.18
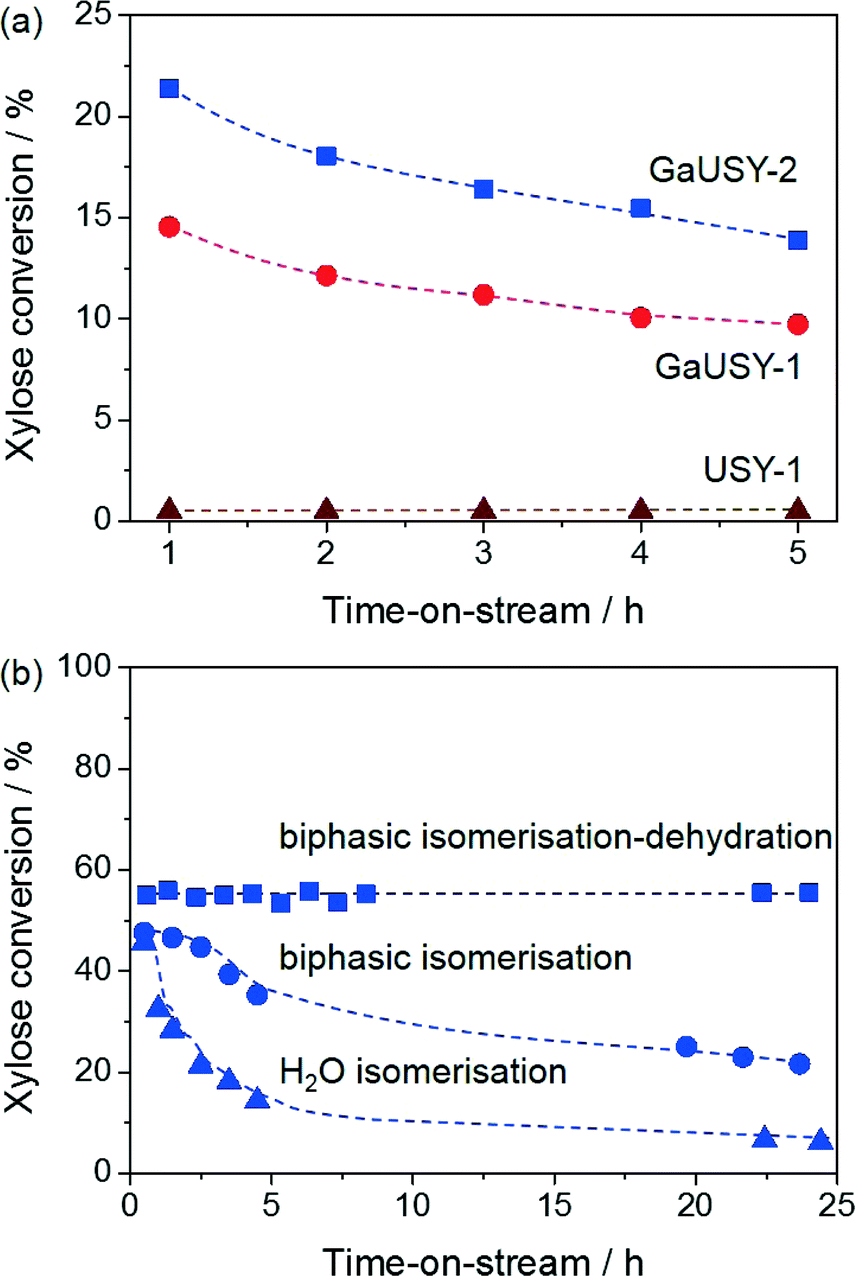 | ||
| Fig. 1 (a) Xylose isomerisation over Lewis acid USY zeolites in water. Conditions: xylose content in H2O = 5 wt.%, Wzeolite = 0.36 g, T = 403 K, τr = 6.8 min. (b) Xylose isomerisation over GaUSY-2 in water and H2O:MIBK (20:80) and isomerisation–dehydration over GaUSY-2/Amberlyst-36 in H2O:MIBK (20:80). Conditions: xylose content in H2O = 5 wt.%, Wzeolite = 0.36 g, Wresin = 0.72 g, T = 403 K, τr = 6.8 min. | ||
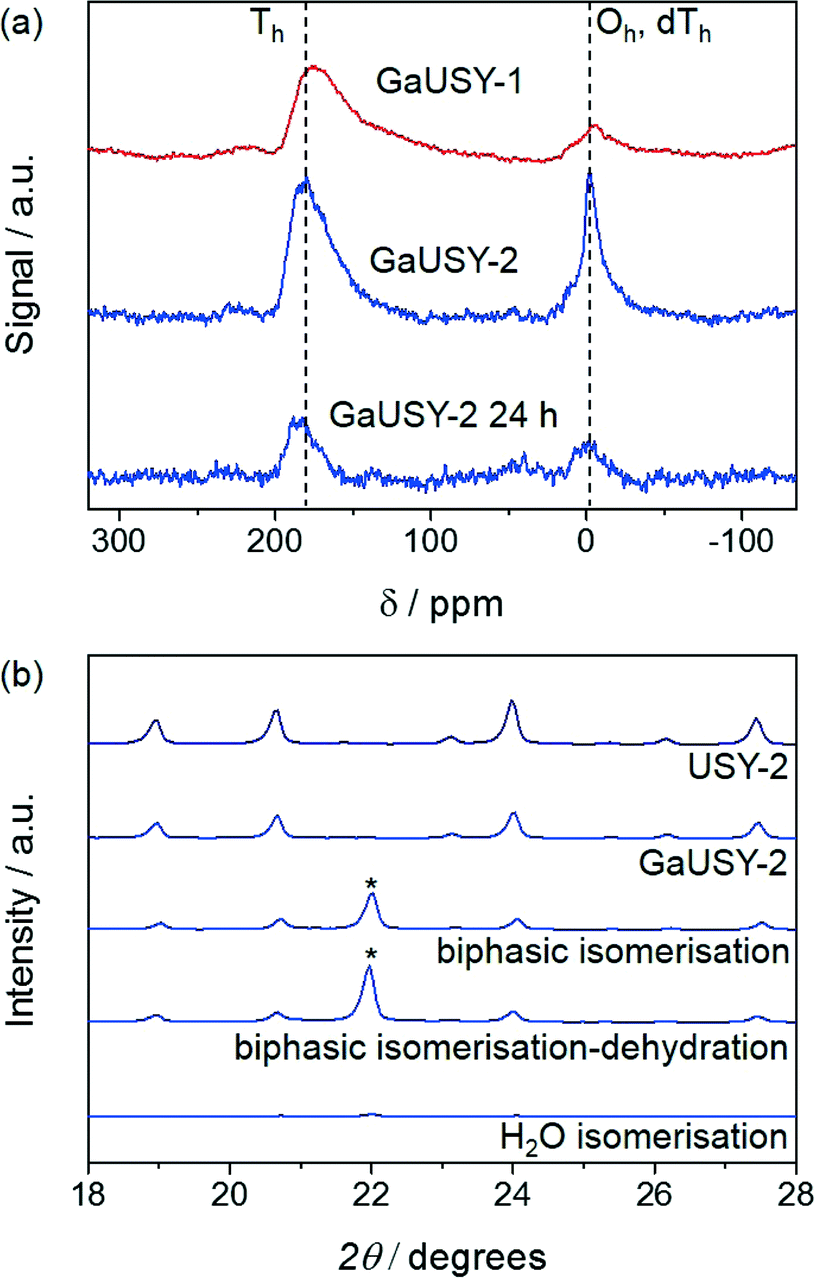 | ||
| Fig. 2 (a) 71Ga MAS-NMR spectra of galliated USY zeolites prior to and after 24 h of use in isomerisation–dehydration and (b) XRD patterns of USY-1 and its galliated counterpart prior to and after use in isomerisation or isomerisation–dehydration. The reflections marked by the asterisk are due to the quartz that was unavoidably mixed with the catalyst upon unloading of the reactor. Conditions: xylose content in H2O = 5 wt.%, Wzeolite = 0.36 g, Wresin = 0.72 g, H2O:MIBK = 20:80, T = 403 K, τr = 6.8 min. | ||
The activity of GaUSY-2 was monitored for 24 h on stream in the presence of (i) pure water, (ii) a mixture of water and MIBK and (iii) a mixture of water, MIBK and the dehydration catalyst Amberlyst-36 (Fig. 1b). In order to more easily identify differences, the catalyst amount was doubled to reach higher conversion levels. A strong decrease in xylose conversion (from 46% to 6%) was observed in pure water after 24 h on stream (Fig. 1b). XRD, ICP-OES and elemental composition analyses of the spent catalysts were performed to shed light on the origin of this activity loss. The lack of diffraction lines in the pattern of the used catalyst evidenced the full collapse of the zeolite structure upon reaction in pure water (Fig. 2b), which was accompanied by significant gallium leaching (from 5.9 to 0.27 wt.%) and deposition of C-based species on the catalyst surface (4.1 wt.%). Xylose isomerisation was then conducted in the presence of a mixture of water and MIBK in a 20:80 vol.% ratio, mimicking the liquid environment later applied to the full process (Fig. 1b). Under these conditions, gallium leaching was better controlled (from 5.9 to 2.1 wt.%) and the crystallinity of the zeolite was preserved to a much greater extent (Fig. 2b). Remarkably, in a biphasic medium and in the copresence of Amberlyst-36, the xylose conversion remained constant along the duration of the run (Fig. 1b). Analysis of the gallium content after 6 and 24 h of reaction still evidenced metal depletion in the first few hours of the test (from 5.9 to 2.3 wt.%), but the catalyst composition remained substantially unaltered upon further time on stream (1.9 wt.%). Since gallium loss was analogous in the presence or absence of Amberlyst-36 and the 71Ga MAS NMR spectra did not show any considerable changes in the structure of the gallium sites upon reaction (Fig. 2a), the unstable behaviour of GaUSY-2 in the biphasic isomerisation was likely due to the accumulation of xylulose and the consequent generation of by-products easily adsorbed on the catalyst surface. Unfortunately, it was impossible to completely separate GaUSY-2 from Amberlyst-36 after the reaction for CHN analysis to support this interpretation.
In order to prove the truly heterogeneous nature of our catalytic system, the pH of the outlet stream was analysed, as leaching of sulfonic groups from the resin would render the mixture acidic. No deviation from pH 7 was found either during the first or the last 5 h of the continuous test. Additionally, it was demonstrated that the reactor effluent was not active in converting freshly added xylose (5 wt.%) to either xylulose or furfural during a 5 h batch experiment at 403 K.
Reaction optimisation
Relevant reaction parameters were systematically varied in order to maximise the furfural yield and minimise the reaction temperature, which are beneficial to reduce the amount of waste produced and the energy input for the reaction and the downstream processing. Firstly, the extraction efficiency of furfural in the organic phase was considered as this plays a critical role in the suppression of side and consecutive reactions. Different organic solvents were tested, i.e. MIBK, toluene and DCE (Fig. 3a). A comparable furfural yield (ca. 40%) was observed at the same contact time (13.6 min) when the reaction was performed in H2O:MIBK (20:80) and H2O:DCE (20:80). However, as the conversion was lower when MIBK was used as the extraction solvent compared to that when DCE was used (55 versus 77%, respectively), the former solvent allowed for a better selectivity. The use of toluene led to the worst conversion. The superiority of MIBK is in line with the preference for this extrinsic phase in biphasic sugar processing in slurry reactors.6b Furthermore, the selectivity trend MIBK > DCE > toluene appears to correlate with the solubility of the three solvents in H2O, decreasing from 17 to 8.7 and to 0.5 g L−1, respectively. Catalyst deactivation was observed using DCE as well as toluene, while the activity could be fully retained using MIBK over 24 h (Fig. 3a). The efficient removal of furfural and humins deposited on the catalyst surface by MIBK has already been reported by Ordomsky et al.4c Further screening was therefore conducted using this solvent.
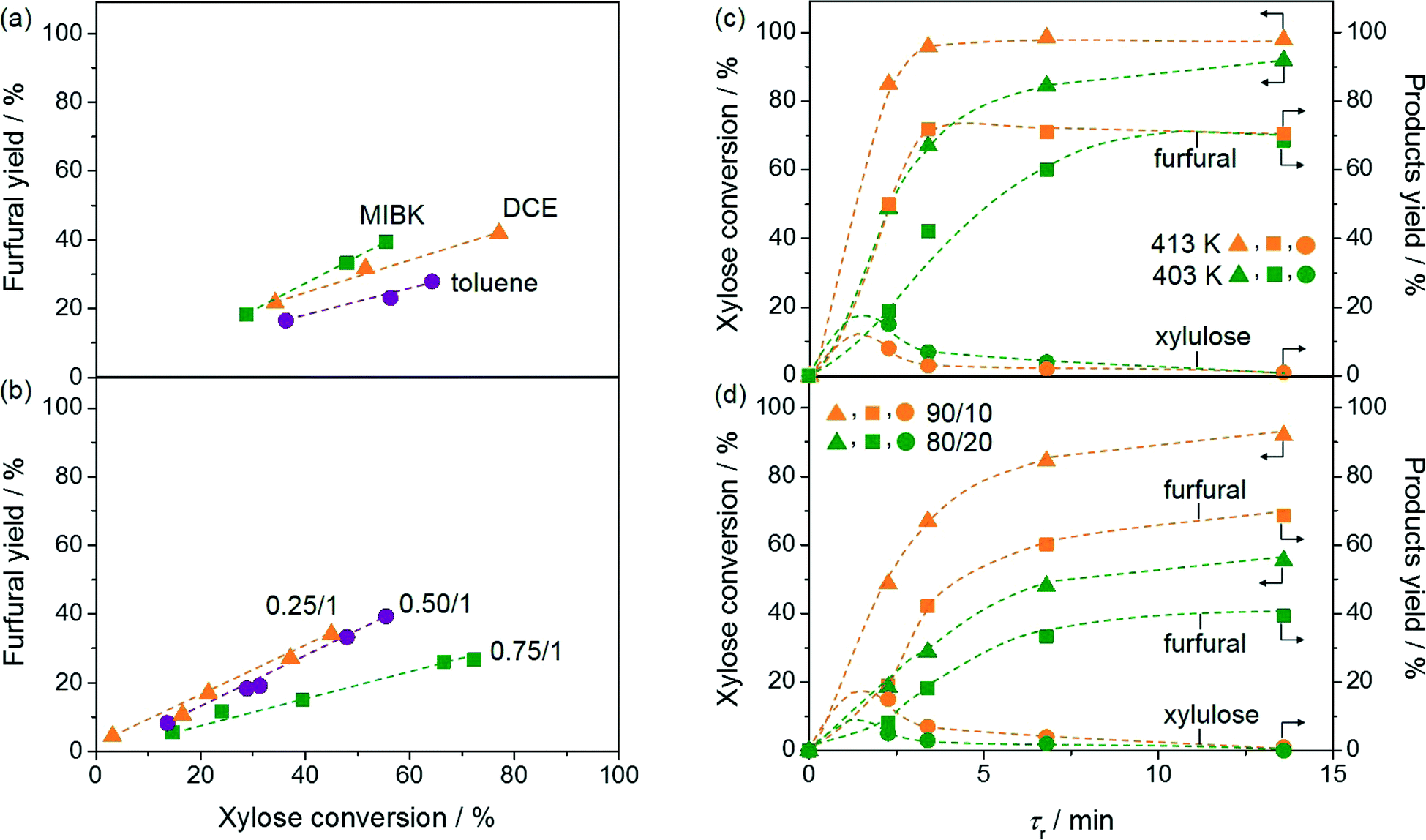 | ||
| Fig. 3 Screening of different parameters in the biphasic isomerisation–dehydration of xylose to furfural. Conditions: a) xylose content in H2O = 5 wt.%, Wzeolite = 0.36 g, Wresin = 0.72 g, T = 403 K, H2O:organic solvent = 20:80; b) xylose content in H2O = 5 wt.%, Wzeolite = 0.18, 0.36, or 0.54 g, Wresin = 0.72 g (corresponding to Wzeolite:Wresin ratio of 0.25:1, 0.50:1, or 0.75:1) , T = 403 K, H2O:MIBK = 20:80; c) xylose content in H2O = 5 wt.%, Wzeolite = 0.36 g, Wresin = 0.72 g, T = 403 or 413 K, H2O:organic solvent = 20:80; d) xylose content in H2O = 5 wt.%, Wzeolite = 0.36 g, Wresin = 0.72 g, T = 403 K, H2O:MIBK = 10:90 or 20:80. | ||
As isomerisation and dehydration occur consecutively, the ratio of GaUSY-2:Amberlyst-36 also has crucial implications on the process selectivity. Three catalyst ratios (on a mass basis) were thus tested (Fig. 3b). Similar results were obtained with the 0.25:1 and 0.5:1 ratios in terms of furfural selectivity, but the reaction proceeded more rapidly in the second case, leading to a process with improved overall productivity. For the ratio 0.75:1, the furfural selectivity was lower, as confirmed by the dark brown colour of the outlet solution. Xylulose accumulation due to the insufficient dehydration ability of this catalyst composition clearly enhanced humin formation.
The reaction was also tested at different temperatures (Fig. 3c). Only traces of furfural were observed at 293 K. The highest furfural yield, 72%, was obtained at 98% conversion at 413 K and a contact time of only 3.4 min, but the catalyst deactivated over time on stream. Remarkably, a similar yield (70%) was reached at the same conversion at 403 K after an only moderately longer contact time (13.6 min) and the catalyst remained stable. At both temperatures, only traces of xylulose and lyxose (2% selectivity) could be detected. These furfural yields are only slightly inferior to those obtained with the best heterogeneous catalysts in batch experiments, i.e. a composite material in a water–toluene mixture (74%) and H-mordenite in gamma-valerolactone (80%).5b,21
Apart from the nature of the organic solvent, the ratio of the aqueous and organic phases strongly impacts the extraction efficiency and thus the furfural selectivity (Fig. 3d). The best performance was obtained at a H2O:MIBK ratio of 10:90 with a furfural yield of 70% at a xylose conversion of 92%. The maximum yield obtained at H2O:MIBK = 20:80 was 40% at 55% xylose conversion.
Kinetic and flow analysis
The profiles of the xylulose and furfural yields versus contact time presented in Fig. 3b and c, respectively, display the typical volcano and S shapes observed for the intermediate and end products of a consecutive reaction. This evidence represents the first indication that the reaction is kinetically controlled and indicates xylulose dehydration as the rate-determining step. In order to support the kinetic regime, catalytic tests were conducted in which the particle size of the resin was varied, while keeping all other parameters (including the volume of the catalytic bed and the mass of the resin) constant (Fig. 4a). Based on the virtually unchanged furfural yield, the presence of both intra- and extra-particle diffusion limitations was excluded. Additionally, the dependence of the reaction rate on the temperature was evaluated, complementing the catalytic tests previously conducted at 403 and 413 K with a measurement at 393 K. As shown in Fig. 4b, the Arrhenius plot obtained could be fitted very well by a linear regression line and the activation energy estimated (54 kJ mol−1) was found to be in the range of values already reported in the literature for this reaction. Indeed, while activation energies of 111–125 kJ mol−1 have been associated with the activation energy of purely Brønsted acid-catalysed dehydration of xylose to furfural in water,19 the addition of a Lewis acid catalyst (i.e. the isomerisation–dehydration pathway herein studied) has been shown to lower these values by up to 60%.20 This further corroborates a kinetically controlled reaction.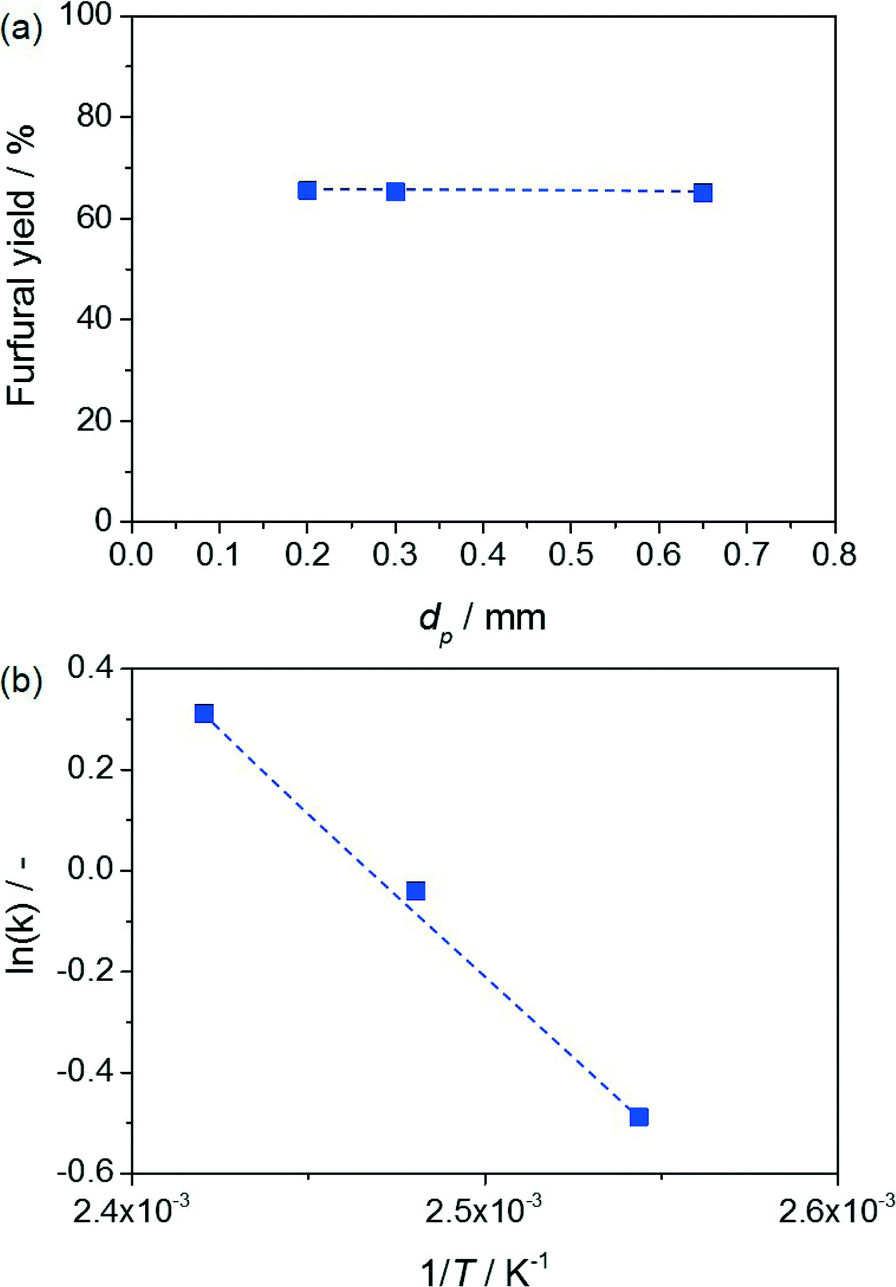 | ||
| Fig. 4 a) Furfural yield from xylose versus particle size of the resin. Conditions: xylose content in H2O = 5 wt.%, Wzeolite = 0.36 g, Wresin = 0.72 g, H2O:MIBK = 10:90, T = 403 K, τr = 6.8 min. b) Arrhenius plot for conversion of xylose to furfural. Conditions: xylose content in H2O = 5 wt.%, Wzeolite = 0.36 g, Wresin = 0.72 g, H2O:MIBK = 10:90, T = 293–413 K, τr = 2.26 min. | ||
While it would certainly be interesting to relate the good mass transfer properties of the system to the characteristics of the Taylor flow in the packed bed, this is a very challenging task. A visual inspection of the contacting patterns of fluids and solids is impossible since, due to the temperature and pressure requirements of the reaction, the steel reactor cannot be replaced by an identical unit made of quartz or Teflon. However, we gathered two strong hints that the aqueous and organic slugs would only marginally intermix in the packed bed, thus preserving the large interface and avoiding channelling effects. Firstly, a Taylor flow was still observed right at the outlet of the packed bed and the slugs exhibited similar sizes and frequency to those of the inlet Taylor flow. Secondly, the Reynolds number calculated using the highest flow rate is extremely low (0.1), indicating laminar flow.
Extrapolation to xylan conversion
The hydrolysis–isomerisation–dehydration reaction of xylan was performed in the continuous-flow biphasic reactor under the optimised conditions determined for the conversion of xylose to furfural. Remarkably, at a contact time of 13.6 min, a furfural yield of 69% and a xylose yield of 8% were obtained (Fig. 5). Only a few other heterogeneous materials have been shown to enable hemicellulose conversion into furfural, the best system exhibiting a furfural yield of 63%.5b,21 Compared to this latter case, our reaction proceeds ca. 30 times faster, implying an enhanced space time yield for the overall process. As xylulose was still detected in the reaction mixture and its yield features the characteristic volcano dependence on the contact time (Fig. 5), xylulose dehydration also appears to be the rate-determining step in xylan processing. Relevantly, our catalyst displayed stable performance over 24 h on stream (Fig. 6) despite the higher complexity of the reaction in terms of number of steps and substrate bulkiness. Probably due to the lower saccharide concentration, the Ga loss during this test (5.9 to 2.7 wt.%) was even moderately less pronounced compared to the case of xylose conversion.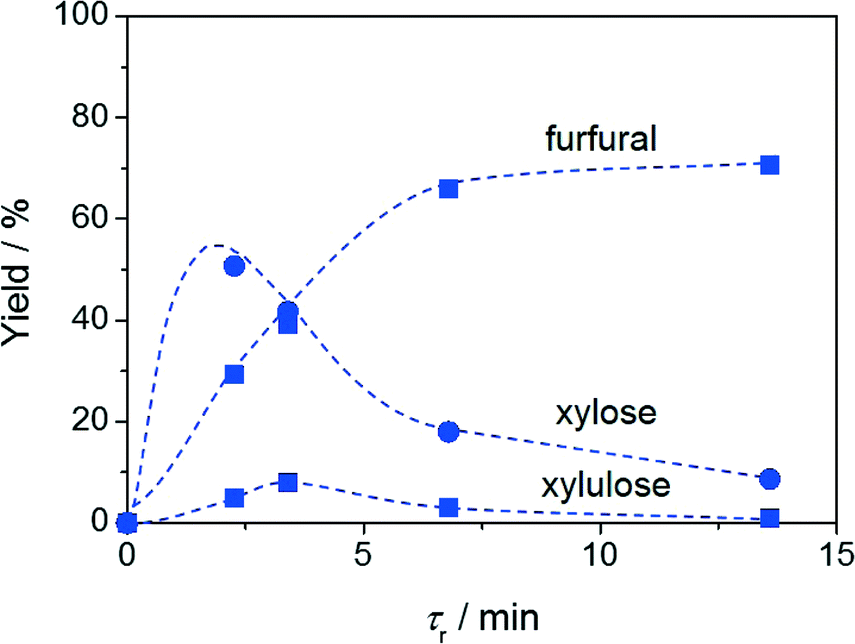 | ||
| Fig. 5 Furfural and xylose yields upon continuous-flow biphasic processing of xylan versus contact time. Conditions: xylan content in H2O = 2.5 wt.%, Wzeolite = 0.36 g, Wresin = 0.72 g, T = 403 K, H2O:MIBK = 10:90. | ||
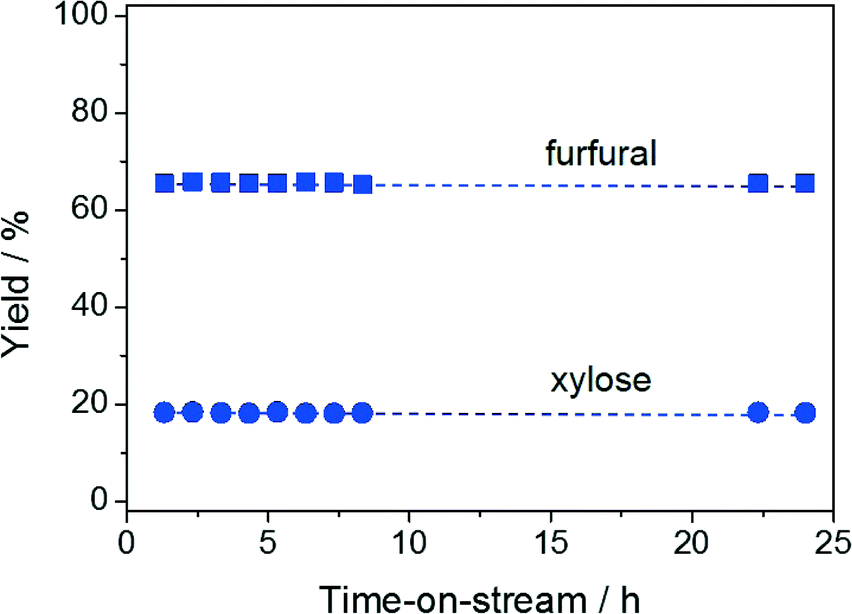 | ||
| Fig. 6 Furfural and xylose yields versus time-on-stream upon xylan conversion in H2O/MIBK (10:90). Conditions: xylan content in H2O = 2.5 wt.%, Wzeolite = 0.36 g, Wresin = 0.72 g, T = 403 K, τr = 6.8 min. | ||
Conclusions
An efficient continuous conversion of C5 carbohydrates was attained over a physical mixture of a galliated USY zeolite for the isomerisation of xylose to xylulose and of Amberlyst-36 for the hydrolysis of xylan and dehydration of the xylulose generated to furfural under a Taylor flow of an aqueous and organic phase. Galliated Al-lean zeolites proved to be more active than analogous Al-rich materials. The stability of this catalyst in terms of metal leaching and amorphisation appeared to strongly depend on the solvent composition and the products formed, being poor in water upon single isomerisation and promising under biphasic operation upon isomerisation–dehydration. Operating parameters such as extraction solvent, catalyst ratio, temperature, and H2O:organic solvent ratio were optimised to maximise the furfural yield from xylose. The best value was 72%. Remarkably, xylan hemicellulose could be converted with very similar efficacy too. These results indicate that the simultaneous exploitation of suitable catalyst formulation and adequate reactor engineering concepts possess great potential to lead to processes with enhanced value. Performing reaction and separation in a single-unit operation is not only beneficial in terms of the selectivity of the transformation but also in terms of process intensification. This approach should be explored more broadly in future biomass-based applications.
Acknowledgements
This work was supported by the Swiss National Science Foundation (project number 200021-140496).Notes and references
- (a) Y. C. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68 Search PubMed; (b) D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075 Search PubMed.
- (a) B. Kamm, P. R. Gruber and M. Kamm, Biorefineries - Industrial Processes and Products, Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2012 Search PubMed; (b) T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Vol. I - Results of Screening for Potential Candidates from Sugars and Synthesis Gas, U. S. D. o. Energy, 2004 Search PubMed.
- (a) T. Deng, X. Cui, Y. Qi, Y. Wang, X. Hou and Y. Zhu, Chem. Commun., 2012, 48, 5494 Search PubMed; (b) P. Carniti, A. Gervasini, S. Biella and A. Auroux, Catal. Today, 2006, 118, 373 Search PubMed; (c) R. L. De Souze, H. Tu, F. Rataboul and N. Essayem, Challenges, 2012, 3, 2012 Search PubMed; (d) F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2006, 45, 2163 Search PubMed; (e) V. Choudhary, A. B. Pinar, S. I. Sandler, D. G. Vlachos and R. F. Lobo, ACS Catal., 2011, 1, 1724 Search PubMed.
- (a) X. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Green Chem., 2009, 11, 1327 Search PubMed; (b) B. Saha and M. Abu-Omar, Green Chem., 2014, 16, 24 Search PubMed; (c) V. V. Ordomsky, V. L. Sushkevich, J. C. Shouten, J. van der Schaaf and T. A. Nijhuis, J. Catal., 2013, 300, 37 Search PubMed.
- (a) T. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548 Search PubMed; (b) E. I. Gürbüz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 1270 Search PubMed.
- (a) E. I. Gürbüz, S. G. Wettstein and J. A. Dumesic, ChemSusChem, 2012, 5, 383 Search PubMed; (b) Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933 Search PubMed.
- S. Dutta, S. De, B. Saha and M. I. Alam, Catal. Sci. Technol., 2012, 2, 2025 CAS.
- R. Xing, W. Qi and G. W. Huber, Energy Environ. Sci., 2011, 4, 2193 CAS.
- (a) S. Hu, Z. Zhang, Y. Zhou, B. Han, H. Fan, W. Li, J. Song and Y. Xie, Green Chem., 2008, 10, 1280 Search PubMed; (b) S. De, S. Dutta and B. Saha, Green Chem., 2011, 13, 2859 Search PubMed; (c) Y. J. Pagán-Torres, T. Wang, J. M. R. Gallo, B. H. Shanks and J. A. Dumesic, ACS Catal., 2012, 2, 930 Search PubMed.
- E. Nikolla, Y. Román-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408 CrossRef CAS.
- (a) S. Dutta, S. De, A. K. Patra, M. Sasidharan, A. Bahumik and S. Saha, Appl. Catal., A, 2011, 409, 133 CrossRef PubMed; (b) S. De, S. Dutta, A. K. Patra, B. S. Rana, A. K. Sinha, B. Saha and A. Bhaumik, Appl. Catal., A, 2012, 435, 197 Search PubMed; (c) V. V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijhuis, ChemSusChem, 2013, 6, 1697 Search PubMed; (d) F. Yang, Q. Liu, M. Yue, X. Baia and Y. Du, Chem. Commun., 2011, 47, 4469 Search PubMed; (e) I. Jiménez-Morales, A. Teckchandani-Ortiz, J. Santamaría-González, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2014, 144, 22 Search PubMed; (f) M. M. Antunes, S. Lima, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Appl. Catal., A, 2012, 417, 243 CrossRef PubMed; (g) J. Zhang, J. Zhuang, L. Lin, S. Liu and Z. Zhang, Biomass Bioenergy, 2012, 39, 75 Search PubMed; (h) S. Lima, A. Fernandes, M. M. Antunes, M. Pillinger, F. Ribeiro and A. A. Valente, Catal. Lett., 2010, 135, 41 Search PubMed; (i) A. S. Dias, S. Lima, M. Pillinger and A. A. Valente, Carbohydr. Res., 2006, 341, 2946 Search PubMed; (j) S. Lima, M. Pillinger and A. A. Valente, Catal. Commun., 2008, 9, 2144 Search PubMed.
- S. Lima, M. M. Antunes, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Appl. Catal., A, 2010, 388, 141 Search PubMed.
- D. M. Fries, T. Voitl and P. R. von Rohr, Chem. Eng. Technol., 2008, 31, 1182 Search PubMed.
- M. N. Kashid, Y. M. Harshe and D. W. Agar, Ind. Eng. Chem. Res., 2007, 46, 8420 Search PubMed.
- M. Brasholz, K. V. Känel, C. H. Hornung, S. Saubern and J. Tsanaktsidis, Green Chem., 2011, 13, 1114 Search PubMed.
- C. V. McNeff, D. T. Nowlan, L. C. McNeff, B. Yan and R. L. Fedie, Appl. Catal., A, 2010, 384, 65 Search PubMed.
- P. Y. Dapsens, M. J. Menart, C. Mondelli and J. Pérez-Ramírez, Green Chem., 2014, 16, 589 Search PubMed.
- P. A. Zapata, J. Faria, M. P. Ruiz, R. E. Jentof and D. E. Resasco, J. Am. Chem. Soc., 2012, 134, 8570 Search PubMed.
- (a) R. Weingarten, J. Cho, W. Curtis Conner Jr. and G. W. Huber, Green Chem., 2010, 12, 1423 Search PubMed; (b) J. Qi and L. Xiuyang, Chin. J. Chem. Eng., 2007, 15, 666 Search PubMed; (c) E. R. Garrett and B. H. Dvorchik, J. Pharm. Sci., 1969, 58, 813 Search PubMed; (d) P. J. Oefner, A. H. Lanziner, G. Bonn and O. Bobleter, Monatsh. Chem., 1992, 123, 547 Search PubMed.
- I. Agirrezabal-Telleria, C. García-Sancho, P. Maireles-Torres and P. Luis Arias, Chin. J. Catal., 2013, 34, 1402 Search PubMed.
- P. Bhaumik and P. L. Dhepe, ACS Catal., 2013, 3, 2299 Search PubMed; R. Sahu and P. L. Dhepe, ChemSusChem, 2012, 5, 751 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |