
One-pot synthesis of (R)-2-acetoxy-1-indanone from 1,2-indanedione combining metal catalyzed hydrogenation and chemoenzymatic dynamic kinetic resolution†
Otto
Långvik
a,
Thomas
Sandberg
b,
Johan
Wärnå
c,
Dmitry Yu.
Murzin
c and
Reko
Leino
*a
aLaboratory of Organic Chemistry, Åbo Akademi University, FI-20500 Åbo, Finland. E-mail: reko.leino@abo.fi
bLaboratory of Physical Chemistry, Åbo Akademi University, FI-20500 Åbo, Finland
cLaboratory of Industrial Chemistry and Reaction Engineering, Process Chemistry Centre, Åbo Akademi University, FI-20500 Åbo, Finland
First published on 16th September 2014
Abstract
Combination of the regioselective hydrogenation of the prochiral diketone 1,2-indanedione with chemoenzymatic dynamic kinetic resolution of the resulting rac-2-hydroxy-1-indanone was investigated. A new simple to operate, one-pot reaction sequence provides the valuable building block (R)-2-acetoxy-1-indanone in moderate enantiopurity (86–92% ee) and a competitive isolated yield (39%) compared with a traditional isolated batch reaction approach.
1 Introduction
The development of cost efficient, sustainable chemical processes for preparation of chiral building blocks is of paramount importance in contemporary synthetic and industrial organic chemistry. In recent years, one-pot reaction sequences involving various types of cascade, tandem and multi-component reactions have emerged as a topical research area, providing several advantages over conventional multi-step synthetic methods.1–7 Furthermore, in modern catalysis research, heterogeneous catalysts are receiving increasing attention also in asymmetric fine chemical synthesis with several advantages over their homogeneous counterparts, particularly due to their simpler work-up, catalyst separation and recycling. A particular objective of our ongoing work is the development of selective, one-pot asymmetric synthesis protocols combining heterogeneous metal and enzyme based catalysts separable by filtration, ideally allowing robust access to chiral small molecules.8–10Vicinal hydroxy ketones serve as useful starting materials for various classes of industrially relevant compounds.11–16 As shown recently, the vicinal hydroxy ketone, rac-2-hydroxy-1-indanone (rac-2), can be efficiently obtained by regioselective hydrogenation of the corresponding vicinal dione, 1,2-indanedione (1).9,10 In the absence of asymmetric induction, the hydrogenation reaction produces an equimolar mixture of the two 2-hydroxy-1-indanone enantiomers. In earlier work, we have investigated the chemoenzymatic dynamic kinetic resolution (DKR) of rac-2 by use of heterogeneous catalysts.8 The deprotected chiral reaction product, (R)-2-hydroxy-1-indanone, then serves as a vicinal hydroxy ketone building block for further utilization, e.g., for production of cis-1-amino-2-indanol, a widely utilized intermediate for pharmaceuticals, chiral catalysts, chiral auxiliaries and chiral resolving agents.17–24
The main methodologies traditionally utilized for the preparation of chiral compounds are based on classical racemate resolution, asymmetric synthesis, and kinetic resolution. Enzymatic kinetic resolution of racemic starting materials is a useful tool for obtaining enantiomerically pure products, being, however, limited to the maximum theoretical yield of 50%.25 In order to overcome this drawback, various types of DKR processes have been developed where the slower reacting enantiomer is racemized spontaneously or by a catalytic process.26–29 In DKR reactions, then, theoretical yields of 100% can be achieved. For example, the chemoenzymatic DKR of sec-alcohols typically utilizing enzymes, such as CALB, in combination with homogeneous ruthenium based racemization catalysts have in recent years emerged as a powerful tool for the synthesis of enantiomerically pure esters and, after deprotection, the corresponding chiral parent alcohols.30–34
Examples of heterogeneous or immobilized homogeneous catalytic systems for dynamic kinetic resolution (DKR) processes have, likewise, been reported in the literature.35–38 Nevertheless, one-pot cascade or tandem type operations combining both the hydrogenation and DKR reactions in single vessels are rare, although a few examples for selected ketoximes and carbonyl compounds have been described.39–45 In general, while one-pot and cascade-type reaction approaches ideally result in overall more efficient chemical processes, reducing also the number of work-up stages required, the combination of different types of catalysts, e.g., chemical and enzymatic, into a single reaction vessel often remains challenging. For example, the combination of regioselective hydrogenation with subsequent DKR reaction is particularly demanding, especially in terms of retaining the catalytic activities and selectivities of the different catalysts involved throughout the whole reaction process. Thus, mutually compatible tailored catalyst combinations are required, not undergoing severe catalyst inactivation or poisoning pathways induced by the reaction products, starting materials or intermediates. Ideally, this type of heterogeneous reaction sequences could also further be converted to flow type systems,46–48 receiving increasing interest both in industry and academic laboratories in recent years.49–54
In the present paper, we describe our work on the combination of the two earlier published reactions by us, namely, the regioselective hydrogenation of 1,2-indanedione9 and the chemoenzymatic dynamic kinetic resolution of rac-2 (ref. 8) into a new one-pot reaction sequence (Scheme 1). Initial experiments were also carried out on the potentially regioselective hydrogenation of two other vicinal diketones, 9,10-phenanthrenequinone and 1,2-naphthoquinone. Limitations and scope of the one-pot approach are discussed.
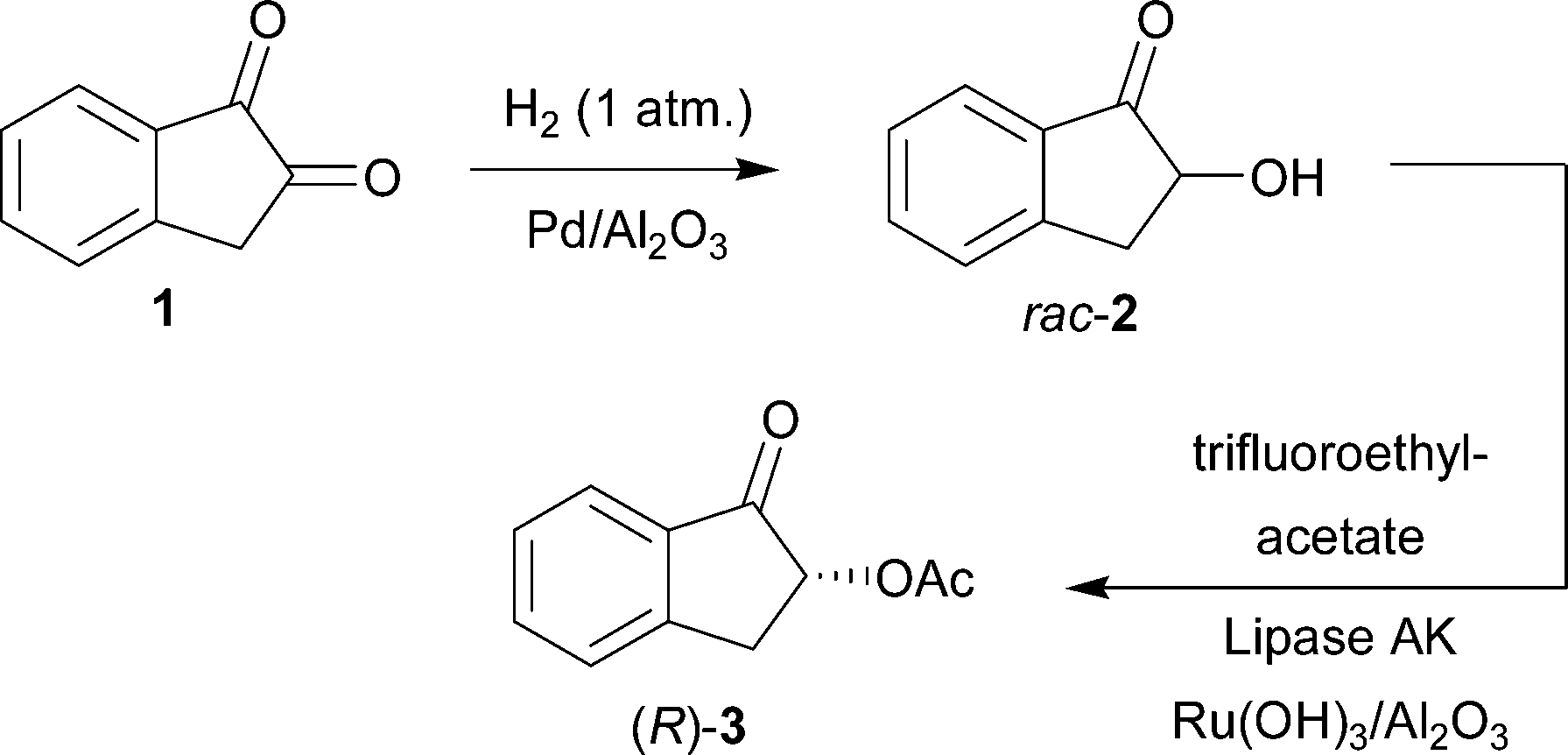 | ||
| Scheme 1 One-pot reaction combining the regioselective hydrogenation of 1,2-indanedione (1) and chemoenzymatic dynamic kinetic resolution of the intermediate rac-2-hydroxy-1-indanone (rac-2) producing (R)-2-acetoxy-1-indanol [(R)-3]. | ||
2 Experimental
2.1 Materials
The starting material 1 was prepared according to literature procedures with 1H and 13C NMR spectroscopic data and melting point identical to those reported previously.55–59 The other diketones, 1,2-naphthoquinone (4) (97%) and 9,10-phenanthrenequinone (5) (95%), were purchased from Sigma-Aldrich and used without further purification. The Pd/Al2O3 catalyst used for the hydrogenation reactions was purchased from Aldrich (product 205710; 5 wt% Pd) and sieved prior to use (<63 μm). The enzyme, lipase AK (from Pseudomonas fluorescens), was purchased from Sigma-Aldrich and used after immobilization on celite and in trial reactions without immobilization. The immobilization of lipase AK on celite was carried out as described in the literature using 10% (w/w) lipase and 5% (w/w) sucrose.60 Ru(OH)3/Al2O3 was prepared according to literature procedures.8,61 The hydrogen and argon gases used were of high quality grade, 99.999% and 99.9999%, respectively (Linde Gas – AGA).2.2 Catalyst characterization
The metal loading of the Ru catalyst was determined by using an inductively coupled plasma optical emission spectrometer (ICP–OES; PerkinElmer, Optima 5300 DV) working at λ = 240.272 nm. The ICP sample (16 mg) was applied in a teflon bomb together with aqua regia (2.5 mL) and HF (0.5 mL). The sample was digested in a microwave oven (Anton Paar, Multiwave 3000) and diluted to 100 mL volume with deionized water (18 MΩ) prior to analysis. The Ru catalyst samples for high resolution transmission electron spectroscopy (HRTEM) analyses were prepared as a suspension in ethanol, and for calculating the diameter of particles, more than 100 particles for each sample were taken (in one or more picture). The HRTEM measurements were performed with LEO 912 Omega, voltage 120 kV. The metal dispersion of the Pd catalyst was determined by applying CO pulse chemisorption method using Micromeritics Auto-Chem 2900 apparatus. The Pd dispersion was calculated assuming an adsorption stoichiometry of 1/1 for CO/Pd.62 The specific surface area of the Pd/Al2O3 catalyst was measured by nitrogen physisorption using an automatic physisorption apparatus (Sorptomatic 1900, Carlo Erba Instruments). BET method was used for calculation of the surface area. The catalyst was degassed prior to the surface area measurement in vacuum at 150 °C. Temperature programmed reduction (TPR) of the Ru(OH)3/Al2O3 catalyst was performed with an AutoChem Micromeritics apparatus by using the following temperature ramp: 10 °C min−1 to 400 °C. X-Ray photoelectron spectroscopy (XPS) was conducted with Kartos Axis Ultra electron spectrometer equipped with a delay line detector. The binding energy scale was referenced to the C 1s first line of aliphatic carbon, set at 285.0 eV. Processing of the spectra was accomplished with the Kratos software.2.3 Quantum mechanical calculations
The TURBOMOLE program package63,64 version 6.1 was used for conducting the quantum mechanical calculations determining the optimized structure of 1. The calculations were performed by applying density functional theory65 with the B3LYP hybrid exchange-correlation functional66–68 in combination with the MARI-J approximation69–71 and the TZVP basis set72 for all atoms, as implemented in the TURBOMOLE program package. For determining the charge delocalization in 1, the GAMESS software73 was applied using the Hartree–Fock (HF) theory74 with the basis set 6-31G*![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 75–77 obtaining corresponding electrostatic potential fit (ESP) charges.
75–77 obtaining corresponding electrostatic potential fit (ESP) charges.
2.4 Reaction setup
The hydrogenation and one-pot reactions were conducted in a five-necked 100 mL round-bottom flask equipped with a mechanical stirrer (gas tight adaptor), gas inlet (7 μm gas disperser merged in the solvent), in situ thermocouple recording the temperature, funnel (with degassing capability), rubber septa for sampling and condenser further connected to an oil bubbler for gas outlet. The one-pot reactions were performed using hydrogen gas under atmospheric pressure at 40 °C. The Pd catalyst for hydrogenation was pretreated under H2 flow (1 atm.) for 2 h at elevated temperature (250 °C) in order to ensure that all active metal is in metallic form (oxidation state zero).9,43 Efficient stirring (500 rpm) and small hydrogenation catalyst particle size (<63 μm) were employed to obtain data in the kinetic regime without external and internal mass transfer limitations. After Pd-catalyst pretreatment, the reaction vessel was cooled down and the degassed (three vacuum-argon cycles) Ru(OH)3/Al2O3, lipase AK and 4 Å molecular sieves were introduced.2.5 Hydrogenation of 1,2-naphthoquinone and 9,10-phenanthrenequinone
The hydrogenations of 1,2-naphthoquinone (4) and 9,10-phenanthrenequinone (5) were performed by dissolving 1.2 mmol of 4 or 5 in 60 mL ethyl acetate (EA). The hydrogenation reactions were started by introducing the deoxygenated solution into the reaction vessel with H2-flow atmosphere (1 atm.) containing the Pd/Al2O3 catalyst (51.1 mg, 2 mol% Pd). After 2.5 h reaction time at 40 °C, an in situ derivatization was applied by addition of 1 mL acetic anhydride and 1 mL pyridine whereafter stirring was continued for 20 min. The products were separated after filtration of the catalyst, by traditional column chromatography using EA:hexane mixture as the eluent. The isolated yields for 1,2-diacetoxynaphthalene (6) was 85% and for 9,10-diacetoxyphenanthrene (7) 78%.2.6 One-pot reaction sequence
In the one-pot reaction sequence 1,2-indanedione (175 mg, 1.2 mmol) and dodecane as an internal standard (68 μL, 0.3 mmol) were dissolved in ethyl acetate (EA), 60 mL, and transferred to a funnel directly connected to the hydrogenation vessel. Deoxygenation of the solution was carried out in the funnel by bubbling with H2 (15 min) shortly before reaction. The reaction was started by introducing the deoxygenated solution into the reaction vessel with H2 atmosphere containing the Pd catalyst (51.2 mg, 2 mol%), lipase (125 mg), Ru-catalyst (85 mg, 1.5 mol%) and pre-dried molecular sieves (30 mg), ensuring dry reaction conditions. Trifluoroethyl acetate (545 μL, 4.8 mmol, 4 equivalents) was introduced to the reaction mixture in the reactor 15 min after the reaction had started in five portions with 30 min intervals through the septa using a syringe. It should be noted, however, that for the kinetic experiments, in order to simplify the experimental procedure, the full amount of the acyl donor was added directly in the beginning of the reactions. This data, then, was used for plotting of the concentration profiles (Fig. 1–4) and calculation of the kinetic parameters (Table 3). The hydrogen flow rate during the reactions was 25–35 mL min−1 and the reaction mixtures were kept at 40 °C using a PEG-bath. Prior to product isolation, the catalysts were removed by filtration through Celite. Chromatographic separation was performed using automated flash system (CombiFlash Companion) equipped with a 4 g (20–40 μm) silica column using EA:hexane as the eluent system (gradient program). Yield of the product (R)-2-acetoxy-1-indanol according to GC chromatogram was 51% (peak area) and the isolated yield was 39% (89 mg, 0.47 mmol), colorless oil.2.7 Product analyses
Samples (0.15 mL) from the reactor were taken through a gas tight septum, filtered using a 0.2 μm syringe filter and diluted with three parts of the same solvent after which 1 μL of the solution was injected to the gas chromatograph. The product distribution was monitored by GC or GC/MS. The GC apparatus used was Agilent Technologies 6850 GC equipped with a Varian CP-7502 column (25.0 m × 250 μm × 0.25 μm), 1/50 split, FID detector and He was applied as a carrier gas. The chiral-GC analyses were conducted using the following temperature program: injector 230 °C, detector 280 °C, oven Tinitial = 130 °C (0 min), rate 2.2° min−1, Tfinal = 185 °C (10 min). The GC/MS apparatus used was Agilent Technologies 7890 A GC equipped with 5975 C MS detector (EI), HP-5MS column (30 m × 250 μm × 0.25 μm), 1/50 split and He as a carrier gas. The NMR spectra of the isolated compounds were recorded on a Bruker Avance 600 MHz NMR spectrometer equipped with a BBI-5 mm-Zgrad-ATM probe at 25 °C operating at 600.13 MHz for 1H and 150.92 MHz for 13C.3 Results and discussion
3.1 Catalyst characterization
Based on the ICP–OES results, the heterogeneous Ru(OH)3/Al2O3 catalyst contains a 1.9% (w/w) metal loading. The mean Ru-particle size, based on high resolution TEM, was determined to 1.3 ± 0.4 nm. The TPR of the catalyst confirms that no reduction takes place at temperatures below 70 °C in H2 atmosphere and that the maxima for the H2 uptake is at 137, 244, and 338 °C.8 The XPS data displayed binding energy peaks at 463.5 and 485.7 eV defining the oxidation state of the ruthenium species to +III, i.e., Ru(OH)3/Al2O3.8The values for the metal dispersion and the corresponding average metal particle sizes of the Pd-catalyst were determined to be 22% and 5.1 nm, respectively. The BET specific surface area of the Pd catalyst is, according to nitrogen physisorption measurements, 174 m2 g−1.
3.2 Hydrogenation
The utilization of 1 as a starting material for further synthetic applications is attractive. This compound is readily available, in part due to its application as a forensic fingerprint reagent.78,79 Our recently reported regioselective hydrogenation of 1 is an efficient and practical method for preparation of rac-2 (Scheme 2)9 compared with the traditional literature route starting from 1-indanone, providing rac-2 after hydrolysis of the corresponding 2-diazo-1-indanone, in turn obtained by oxidation of the 2-oximino-1-indanone intermediate.55,58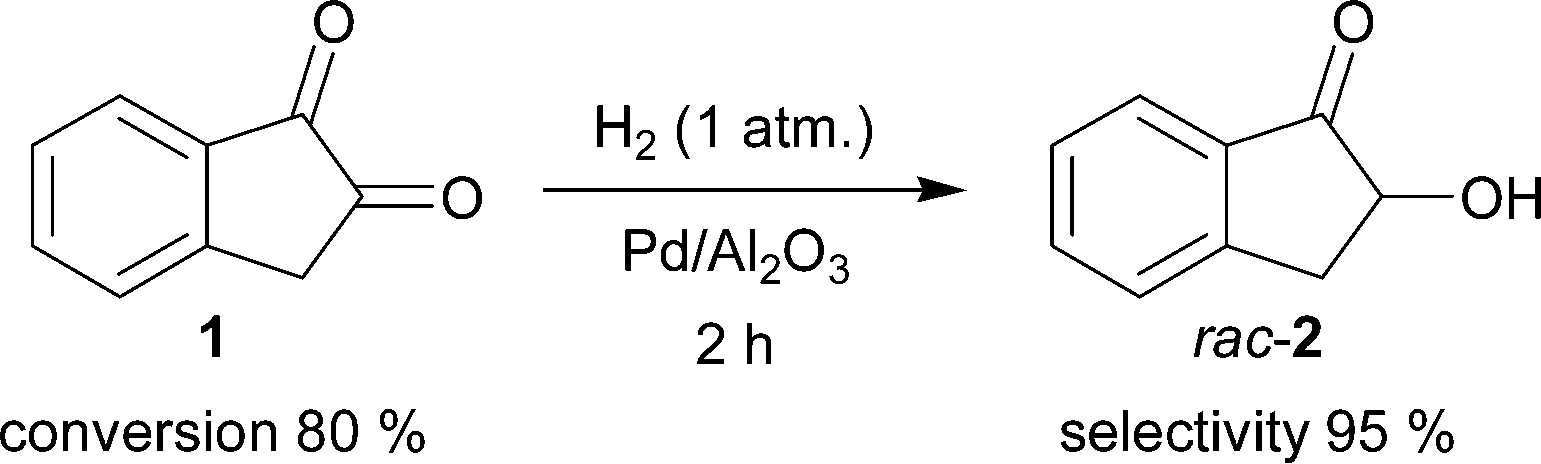 | ||
| Scheme 2 Hydrogenation of 1 to rac-2 with H2 using Pd/Al2O3 (2 mol%) as the catalyst. | ||
| Entry | Conversion of 1 [%] | Selectivity towards rac-2 [%] |
|---|---|---|
| 1 | 56 | 99 |
| 2 | 84 | 92 |
| 3 | 87 | 87 |
| 4 | 90 | 42 |
As the tendency to alternation of bond lengths is one of the characteristics possessed by antiaromatic compounds, the bond lengths of 1 were evaluated here using quantum mechanical calculations with the TURBOMOLE program package (for details, see experimental section). The calculations performed clearly indicate the unmodified structure 1 to be less antiaromatic compared to its enolic counterpart with the calculated bond lengths of 1 not significantly differing from the expected ones, also not indicating any significant Jahn–Teller distortions (Fig. S3, ESI†).
As the benzylic charge delocalization likely results in different atomic charges on the carbonyl groups in position 1 and 2, and is likely to influence the chemical behaviour of compound 1 in the hydrogenation reaction, the atomic charges were calculated (for details, see Experimental section). While the difference between the atomic charges of the two carbonyl carbons is moderate, the results obtained support the earlier suggested property of delocalization of the benzylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond to the aromatic region and thus resulting in the relatively higher reactivity of the carbonyl group in position 2 (Fig. S4, ESI†). However, in order to fully explain the behaviour of 1 in the heterogeneously catalyzed hydrogenation reactions, more thorough calculations would be needed, including calculations of the different adsorption modes of the substrate to the catalyst surface.
O double bond to the aromatic region and thus resulting in the relatively higher reactivity of the carbonyl group in position 2 (Fig. S4, ESI†). However, in order to fully explain the behaviour of 1 in the heterogeneously catalyzed hydrogenation reactions, more thorough calculations would be needed, including calculations of the different adsorption modes of the substrate to the catalyst surface.
Conclusively, as indicated by the results obtained herein, the non-ionized form of 1 shows only one antiaromatic feature, namely the small band gap between the HOMO and LUMO, −0.246 Eh and −0.110 Eh, respectively.
3.3 One-pot hydrogenation and esterification of 1,2-naphthoquinone and 9,10-phenanthrenequinone
In order to further develop and apply the regioselective hydrogenation of vicinal diketones to other potential structures, preliminary hydrogenation studies were carried out by using 4 and 5 as the starting materials. These hydrogenations were initially carried out under similar reaction and pretreatment conditions as used for 1. While investigating the hydrogenation of 5, it became evident, however, that the conditions used for 1 were not applicable for this compound. In the GC analysis, no product was observed although during the hydrogenation reaction a clear color change from yellow to colorless, characteristic for diones, was observed. The disappearance of the product was then shown to result from back-oxidation of the product to starting material in air atmosphere prior to analysis. The reduction-oxidation equilibrium shifts rapidly towards the oxidized form upon exposure to air after venting of the hydrogenation reactor. An in situ derivatization, by acylation, was applied in order to avoid oxidation of the products and also enabling easier separation and increasing the isolated yield. This sequence provided, for the two different starting materials, the corresponding diacetates 6 and 7 in isolated yields of 85% and 78%, respectively (Schemes 3 and 4). The isolated diacetate products are likely formed by rearrangement of the hydroxy ketones by proton transfer and subsequent aromatization (Schemes 3 and 4). | ||
| Scheme 3 Hydrogenation (a) and subsequent esterification (b) of 4 to 6 with H2 (1 atm.) and Pd/Al2O3 as catalyst. | ||
 | ||
| Scheme 4 Hydrogenation (a) and subsequent esterification (b) of 5 to 7 with H2 (1 atm.) and Pd/Al2O3 as catalyst. | ||
The hydrogenation and derivatization or protection of the diols formed, i.e., reductive acetylation, is a well-known transformation for quinone type structures.85–88 Hydrogenations of compounds 4 and 5 initially proceed similar to that of 1, possibly forming hydroxy ketones as intermediates, which then are rapidly and spontaneously rearranged to form more stable and energetically favored aromatic diols, or alternatively are easily oxidized back to the starting material. Notably, because of the symmetric structure of 5 the hydrogenation cannot proceed in a regioselective manner. Nevertheless, the successful in situ derivatizations of the reaction products indicate that the envisioned conceptually related one-pot hydrogenation and esterification sequence should be feasible.
3.4 Dynamic kinetic resolution
Lipase catalyzed kinetic resolution is a powerful tool for production of enantiomerically pure compounds. Prerequisites for a viable resolution process include the correct selection of a suitable enzyme, active acyl donor, low water content in the reaction media and rapid in situ racemization of the starting material. The most efficient sec-alcohol racemization catalysts developed to date are based on soluble half-sandwich ruthenium complexes.89–91 For these homogeneous catalysts, strictly anhydrous and oxygen-free reaction conditions are often required. Moreover, efficient procedures for separation and recycling of the homogeneous catalysts are seldom available.Heterogeneous catalysts, in general, enable easier recycling by simple filtration. Several examples of heterogeneous DKR processes have been reported in literature.35,92–96 The majority of the heterogeneously catalyzed DKRs utilize transition metal, Brönsted or Lewis acid based racemization catalysts.35,92–96 Furthermore, some of these heterogeneously catalyzed DKR reactions have been converted into flow type reaction applications.46–48 In the literature, the reported one-pot catalyst systems suffer, however, from often reduced catalytic activities. The racemization catalysts may be inhibited to various degrees by the starting materials or products.8,45 Activity of the enzyme may, in turn, be inhibited by acidic catalysts used for racemization.92,93
During our development of the chemoenzymatic DKR of rac-2,8 several potential racemization catalysts were investigated. In addition to the catalysts studied in the earlier paper, we have now additionally investigated VOSO4 for racemization of (S)-2. Racemizations of various benzylic alcohols using VOSO4 have been reported earlier.94 Racemization of (S)-2 (0.02 M) at 40 °C using EA as the solvent in the presence of 0.02 M VOSO4 proved, however, unsuccessful and any significant racemization could not be detected during the course of a 5 h reaction. The previously developed heterogeneously catalyzed DKR,8 converting rac-2 into (R)-1-oxoindan-2-yl-butanoate (Scheme 5) in 85–90% conversion and up to 92% ee was, therefore, considered sufficiently efficient for combining the DKR with hydrogenation experiments.
 | ||
| Scheme 5 DKR of rac-2 using Ru(OH)3/Al2O3 as the racemization catalyst in combination with lipase AK yielding (R)-1-oxo-indan-2-yl-butanoate with high ee 92% and 90% conversion.8 | ||
3.5 One-pot combination of hydrogenation and DKR
The primary aim of the present work was to combine the earlier described regioselective hydrogenation of 1 and the DKR of rac-2 into a one-pot reaction sequence (Scheme 6). The results are presented in Table 2. An interesting observation during the earlier reported DKR (Scheme 5) was the minor formation of 1 as an undesired by-product.8 The aim here was to employ reductive reaction conditions and to utilize catalytic hydrogenation of compound 1, a more benign and accessible starting material compared to rac-2, further to rac-2 producing after DKR the desired chiral product (R)-3. | ||
| Scheme 6 One-pot reaction sequence including: i) hydrogenation of 1; ii) racemization of (S)-2 and (R)-2; iii) acylation of the formed (R)-2 with acyl donor forming (R)-3. | ||
| Entry | Solvent | Acyl donora(equivalents) | Yieldb[%] | ee , [%] |
|---|---|---|---|---|
| a Number of equivalents acyl donor related to the amount starting material 1. b According to GC chromatogram. c ee = 100·([(R)-3]-[(S)-3])/([(R)-3] + [(S)-3]). d Added in portions. | ||||
| 1 | MTBE | 8 (3,7) | 52 | 28 |
| 2 | EA | — | 34 | 90 |
| 3 | EA | 9 (1) | 33 | 91 |
| 4 | EA | 9 (2.5) | 40 | 86 |
| 5 | EA | 9 (4)d | 51 | 86 |
The initial one-pot experiments combining the hydrogenation and DKR were conducted by switching the reaction atmosphere from hydrogen gas to argon during the reaction, similar to the earlier reported, conceptually related one-pot type reaction sequence of Bäckvall and co-workers.41 Thus, in the first experiments performed here, the gas feed was changed from hydrogen to argon after two hours of reaction. In this time, the earlier reported regioselective hydrogenation proceeds rapidly providing reasonable conversion.9 In this work it became evident, however, that the introduction of argon did not enhance the conversion or selectivity of the one-pot reaction sequence. Therefore, in further experiments only hydrogen gas was used. For selection of solvent, methyl-tert-butyl ether (MTBE) and EA were considered. While MTBE was initially used with good results for the DKR, as reported earlier,8EA was here found more suitable for the one-pot sequence with better dissolution properties for starting material 1 compared to MTBE. The ether required surprisingly long time to dissolve 1 at the desired concentrations and at higher concentrations resulted in slightly hazy solutions, also observed earlier in the hydrogenation reaction.9
In the one-pot sequence reactions using EA as the solvent and trifluoroethyl butyrate (8) as the acyl donor, we observed, not surprisingly, both the corresponding acetate and butanoate products in 1.7:1, product ratios, respectively. In previous work, Park and co-workers have reported the utilization of EA in a dual role, both as an acetyl donor and a solvent, in lipase catalyzed esterification reactions.97–99 Generally, a significant molar excess of the solvent acting as the acetyl donor compared to the amount of substrate drives the reaction equilibrium heavily to the right without a substantial backward reaction. In the one-pot sequence reactions investigated here, the use of EA as the acetyl donor also proved feasible at first, retaining the high product ee (>90% after 21 h) value throughout the whole reaction (Table 2, entry 2) in the absence of other acyl donors. For increasing the conversion and yield, an activated acetyl donor was desirable. Therefore, trifluoroethyl acetate (9) was added to the one-pot sequence reaction mixture. By carrying out one-pot reactions with further increased amounts of acyl donor, the yield consequently increased as expected (Table 2, entries 3–5).
To further study the advantages and limitations of the one-pot reaction sequence, reactions with different enzyme amounts (93, 130, and 163 mg) were performed. Changes in the enzyme amount were expected to influence the reaction rate of the lipase AK catalyzed resolution of the rac-2 intermediate to the acetylated end product (kA, Scheme 6). In order to produce an efficient DKR type reaction, the ee value should be kept as low as possible for compound 2. Therefore, the change in the acetylation rate was consequently assumed to influence the (R)-2:(S)-2 ratio (kA:kRS2). The rate by which the acetylated end product was formed (kA) was, nevertheless, influenced only slightly by the variation of the enzyme amount (Fig. 1). Subsequently, the concentrations of the components in the three separate one-pot reactions are similar. One example of the kinetic curves obtained is shown in Fig. 2 (130 mg lipase AK). Conclusively, these results demonstrate that the enzyme catalyzed resolution in the one-pot sequence is not the rate limiting step and good kinetic resolution is accomplished with the reaction conditions applied.
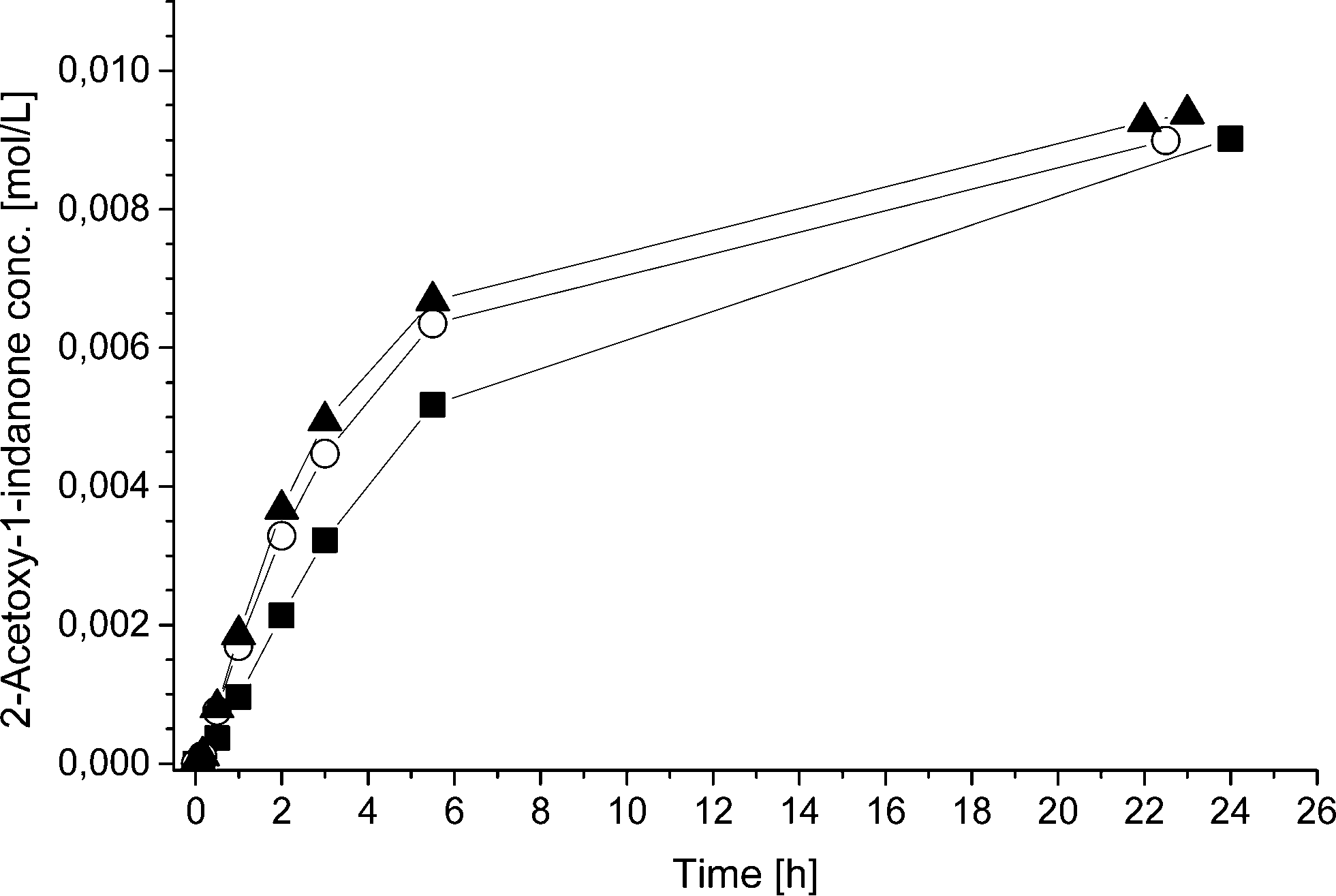 | ||
| Fig. 1 2-Acetoxy-1-indanone concentration during the one-pot reactions using ■ 93 mg, ○ 130 mg and ▲ 163 mg of lipase AK, 51.6–51.9 mg Pd/Al2O3, 89.5–90.1 mg Ru(OH)3/Al2O3 and 4 equivalents (550 μL) of the acyl donor 9 at 40 °C. | ||
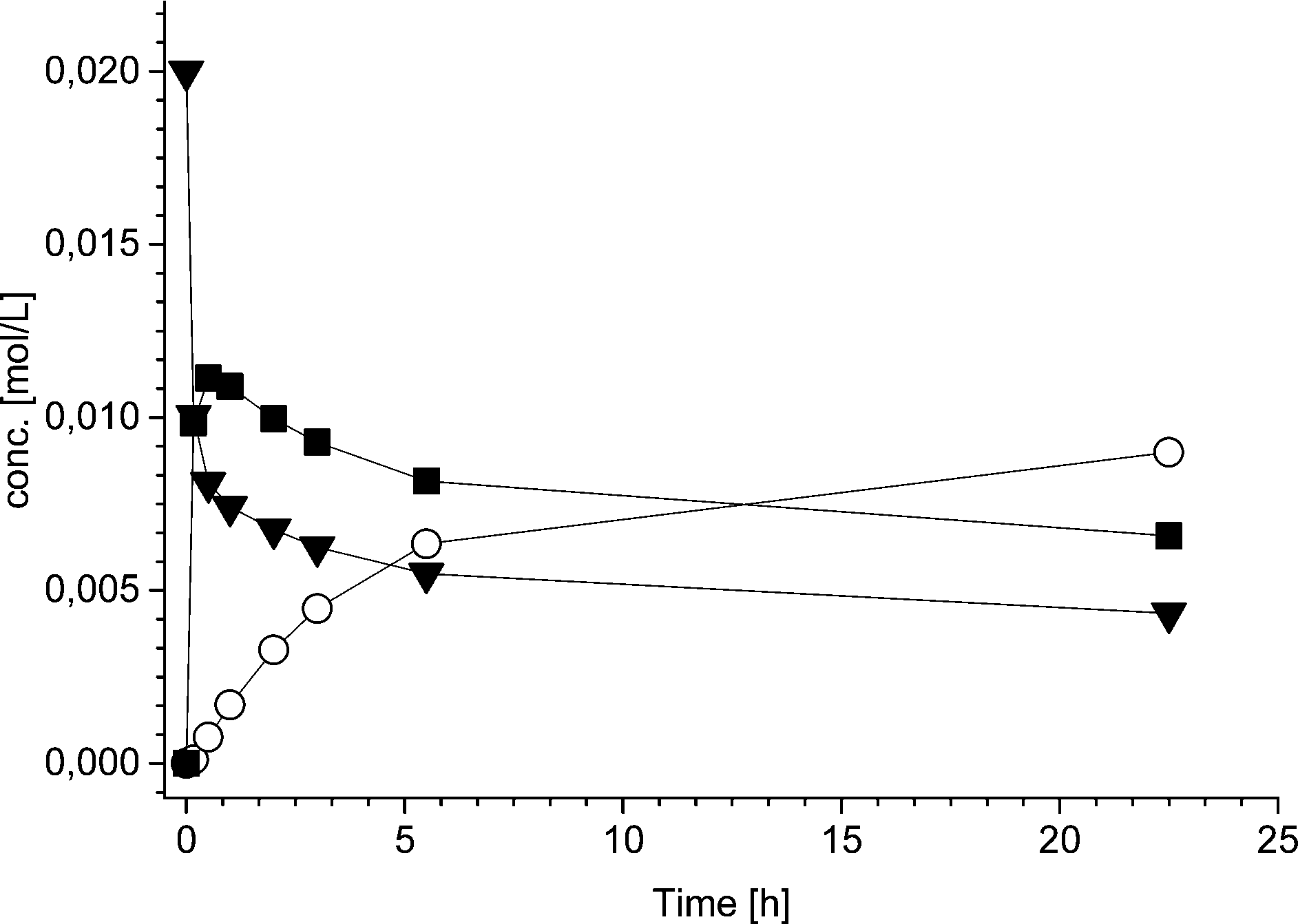 | ||
| Fig. 2 Concentration profiles of ▼ 1,2-indanedione, ■ 2-hydroxy-1-indanone, ○ 2-acetoxy-1-indanone for one-pot reaction using 51.6 mg Pd/Al2O3, 89.5 mg Ru(OH)3/Al2O3, 130 mg lipase AK and 4 equivalents (550 μL) of the acyl donor 9 at 40 °C. | ||
The limiting step in the one-pot reaction appears to be, at least partially, the hydrogenation of compound 1 to rac-2. This was somewhat unexpected, as in the earlier work on regioselective hydrogenation,9 a long reaction time (20 h) resulted in high (>85%) conversion of 1. The high conversion (>85%) in turn resulted in significantly decreased selectivity (less than 40%) due to the consecutive hydrogenation of rac-2 to diols.9 During the development of the one-pot reaction sequence described here, the 2 mol% loading of Pd was considered to be sufficiently high for obtaining satisfactory reaction rates while avoiding the diol formation from over hydrogenation. Suppression of the consecutive hydrogenation to diols is particularly essential in the one-pot approach where, in the presence of an enzyme, lipase catalyzed acetylation of the isomeric alcohols would result in undesired mixtures of acetylated products.
Additionally, also the racemization rate (rRS2) decreases under the applied one-pot conditions resulting in increased ee of the 2-hydroxy-1-indanone intermediate (Fig. 3). The compounds present in the one-pot reaction sequence, particularly the esterified product, appear to inhibit the Ru(OH)3/Al2O3 racemization catalyst as shown in Fig. 3(A), where ee of the intermediate compound 2vs. the concentration of the 2-acetoxy-1-indanone end product exhibit linear dependence. However, the amount of the enzyme catalyst is not influencing the racemization rate. Similar observations have been made in earlier studies, suggested to be a consequence of the adsorption of the esters present in the reaction mixture on the heterogeneous Ru catalyst surface active sites.8,43
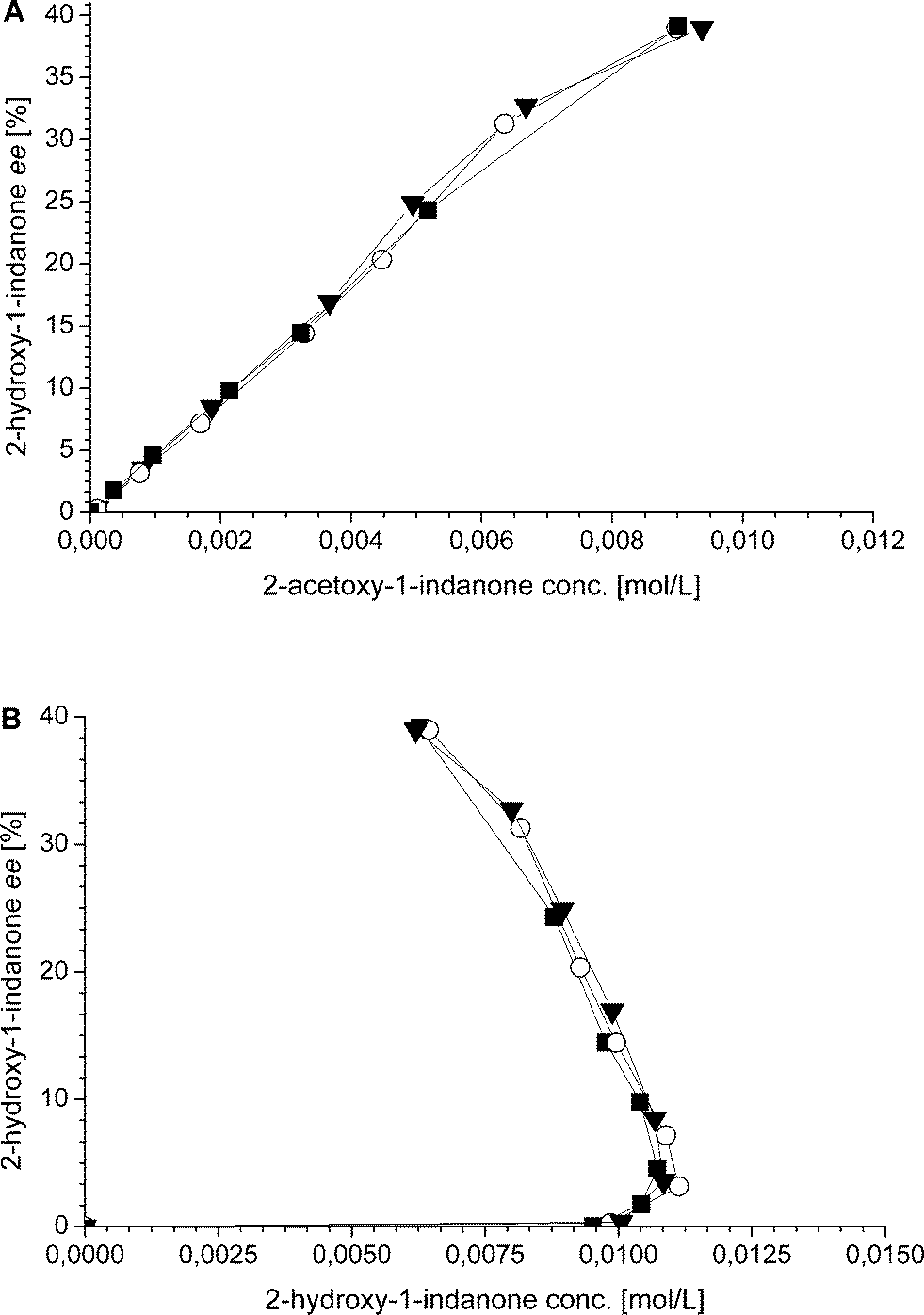 | ||
| Fig. 3 (A) Enantiomeric excess (ee = 100·([(R)-3]-[(S)-3])/([(R)-3] + [(S)-3])) of 2-hydroxy-1-indanone versus concentration of 2-acetoxy-1-indanone in one-pot reactions using ■ 93 mg, ○ 130 mg and ▼ 163 mg lipase AK; (B) enantiomeric excess (ee) of 2-hydroxy-1-indanone versus 2-hydroxy-1-indanone concentration in one-pot reactions using ■ 93 mg, ○ 130 mg and ▼ 163 mg lipase AK. | ||
Hence, in order to improve the conversion and yield and to decrease the influence of the acyl donor (9) on the racemization catalyst activity, the acetyl donor was added in five portions during the first two hours of the reaction, providing the end product in 51% yield (according to GC). The stepwise addition of the acyl donor also further enhanced the efficiency of the racemization catalyst Ru(OH)3/Al2O3 to some degree. Despite of this, the intermediate product 2 in the one-pot sequence did not remain fully racemic throughout the reaction.
Thus, when the one-pot reaction sequence was carried out according to the method described in the Experimental section (see: 2.6 One-pot reaction sequence), acceptable yields and high ee values of the product (Table 1) (R)-3 were obtained. Overall, at least conceptually, combination of the regioselective hydrogenation of 1 yielding rac-2 with the subsequent DKR producing (R)-3 enables an efficient reaction operation without the need for separation of the intermediate hydrogenation product. Furthermore, the reductive reaction conditions employed here enhance the efficiency of the DKR of rac-2, eliminating the formation of the oxidized DKR byproduct reported previously.8
For better understanding of the reaction network, kinetic modelling of the one–pot process was performed with three different concentrations of the enzyme.
The following rate equations were used:
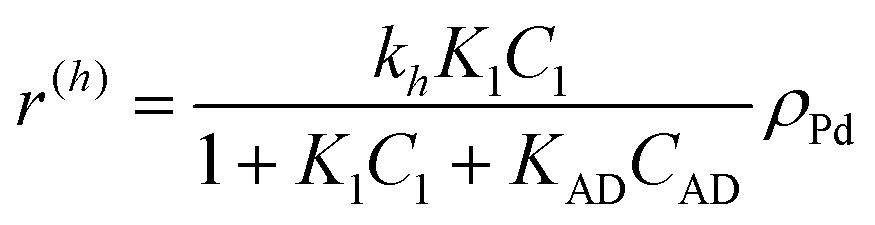 | (1) |
 | (2) |
 | (3) |
 | (4) |
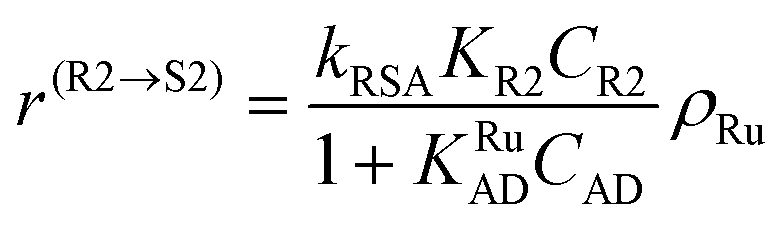 | (5) |
In eqn (1)kh is a lumped constant containing also the hydrogen concentration, C1 and K1 denote concentrations of compound 1 and its adsorption coefficient on Pt. Besides the reactant, also the acylation agent was assumed to be adsorbed on the platinum surface with the adsorption coefficient KAD. This was included in the model due to the nature of the acyl donor, which can block the catalyst surface, thereby decreasing the catalyst activity. The enzymatic reactions r(R2→R3) and r(S2→S3) were assumed to proceed by binding of both reagents in a mechanism conceptually similar to sequential bisubstrate reactions. Finally, for the racemization reactions, adsorptions of (R)-2 and (S)-2 were considered to be inferior to the much stronger adsorption of the acyl donor. In eqn (1)–(5)ρRu, ρpd, ρEnetc., correspond to the bulk densities of ruthenium, palladium and lipase, respectively.
The generation rates for the compounds can be subsequently written
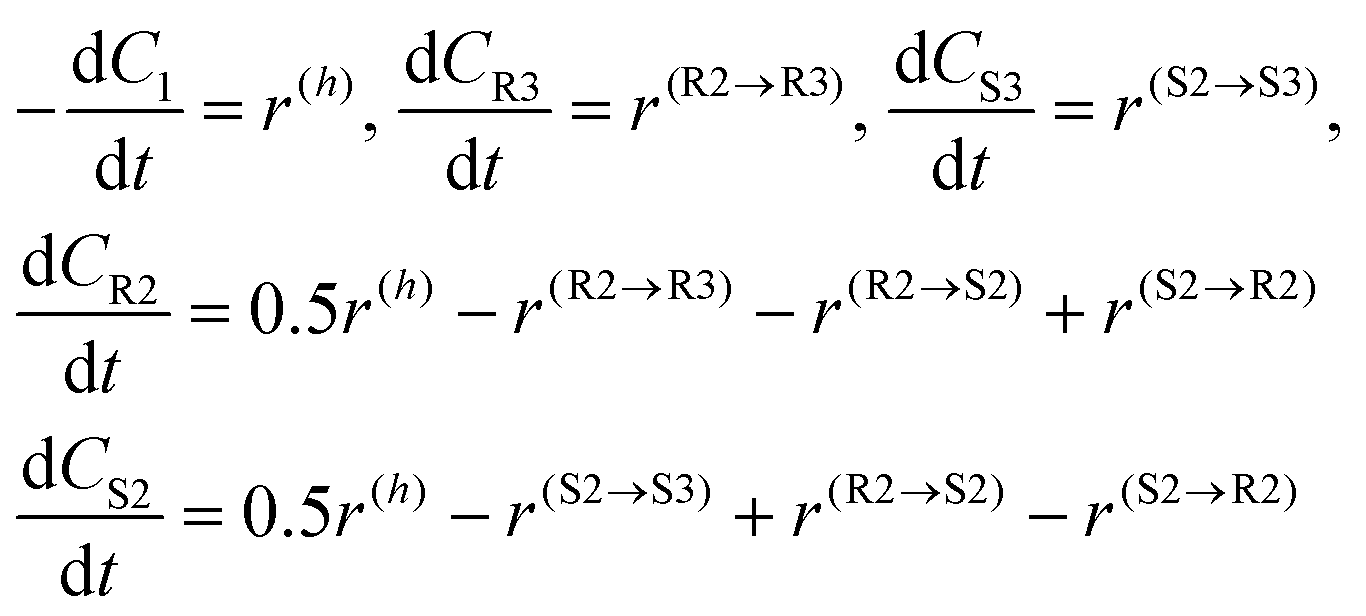 | (6) |
The kinetic modelling was performed for all reaction rates and three reaction sets together. For the parameter estimation, a set of differential equations describing the changes in the concentration profiles of the reagents and products with time was solved by means of ModEst software.100 Using Levenberg–Marquardt simplex method, the target function, which was defined as incompliance between the experimental and calculated values of concentrations was used to solve the system. The sum of the residual squares between the model and the experimental data was minimized using the following objective function;
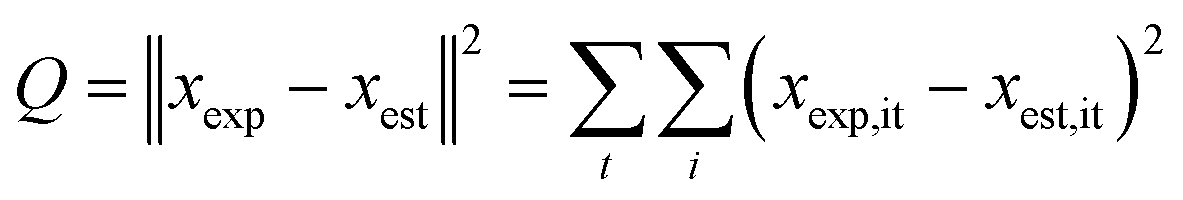 | (7) |
 | (8) |
Preliminary calculations demonstrated that the description of the hydrogenation reaction is mediocre due to strong catalyst deactivation, which could not be sufficiently well described assuming only adsorption of the acyl donor on Pd surface acting as a catalyst poison.
In previous work,42 where also hydrogenation and chemoenzymatic DKR were combined in a one-pot fashion, the hydrogenation of acetophenone on palladium was found to undergo strong deactivation, similar to the current case where palladium is inhibited in the hydrogenation of compound 1. For the sake of modelling, an empirical time dependent function for the catalyst activity was proposed.42 Since a physical meaning in such dependence is unclear it was decided to use a more mechanistically based assumption.
The expression for hydrogenation was modified
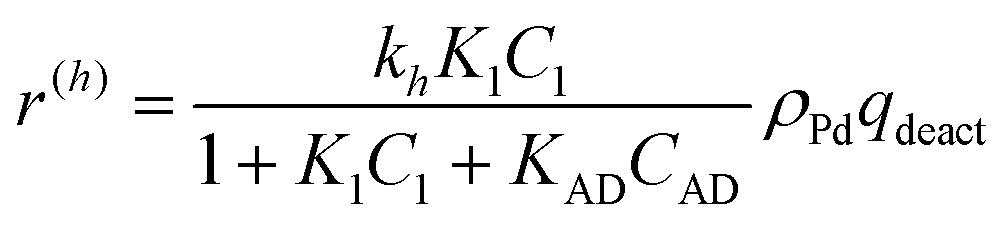 | (9) |
 | (10) |
The deactivation function, used in earlier work101 to explain the activity profile in the hydrogenation of cyclohexene, is based on the assumption that a decline in catalyst activity is proportional to the amount of product formed and could be related to catalyst fouling.
Calculations made with eqn (2)–(5) and (9) allowed further simplifications but neglecting adsorption terms in denominators of all rate expressions. The final expressions for the reaction rates are
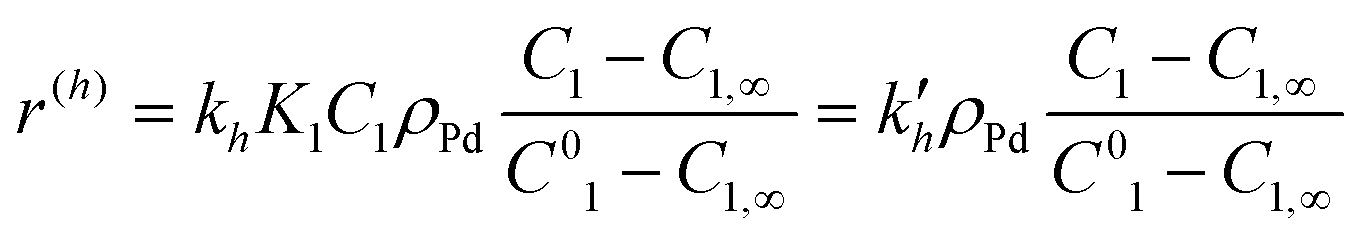 | (11) |
| r(R2→R3) = kR3KR2KEADCR2CADρEn = k′R3CR2CADρEn | (12) |
| r(S2→S3) = − kS3KS2KEADCS2CADρEn = k′S3CS2CADρEn | (13) |
| r(S2→R2) = kRSAKS2CS2ρRu = k′RSACS2ρRu | (14) |
| r(R2→S2) = kRSAKR2CR2ρRu = k′RSACR2ρRu | (15) |
The estimated values of the parameters as well as the relative standard errors (in %) are presented in Table 3. The fit of the kinetic model to the experimental data for the three different reactions is displayed Fig. 4.
| Parameter | Value | Relative standard error [%] |
|---|---|---|
| k ′ h [h−1] | 3.22 | 8.1 |
| k ′R3 [h−1] | 2.09 × 10−3 | 7.0 |
| k ′RSA [h−1] | 2.56 × 10−2 | 17.2 |
| k ′S3 [h−1] | 3.52 × 10−5 | 50.9 |
| C 1,∞ [mol L−1] | 6.37 × 10−3 | 2.5 |
 | ||
| Fig. 4 Comparison between the experimental data and the model predictions for the reactants in the three one-pot reactions using (A) 93 mg, (B) 130 mg and (C) 163 mg lipase AK. Solid lines represent the estimated data and circles the experimental data. The different compounds are represented by the following colors: blue compound 1, red (S)-2, green, (R)-2, cyan (R)-3, pink (S)-3. The degree of explanation 92.94% and the standard errors of the parameters are acceptably low. | ||
4 Conclusions
Combination of the regioselective heterogeneously catalysed hydrogenation of the prochiral diketone 1,2-indandione with dynamic kinetic resolution of the resulting rac-2-hydroxy-1-indanone using a heterogeneous Ru catalyst for racemization and an enzyme, lipase AK, for acylation, has been investigated. A new, simple to operate one-pot reaction sequence provides the valuable building block (R)-2-acetoxy-1-indanone in moderate enantiopurity (86–92%) in a reasonable reaction time (20 h). In contrast to a multi-step reaction approach in separate reactors, the one-pot, heterogeneously catalysed reaction sequence ideally enables simple recovery of the catalysts by filtration facilitating product isolation. Overall, the readily available 1,2-indanedione can be transferred to a useful chiral building block with high regioselectivity and moderate stereoselectivity using economically viable heterogeneous catalysts in a practical catalytic operation.Acknowledgements
The authors thank Alexey Kirilin (Laboratory of Industrial Chemistry and Reaction Engineering, Åbo Akademi University) for preparation of the Ru(OH)3/Al2O3 catalyst and temperature programmed reduction analyses. The authors also thank Sten Lindholm (Laboratory of Analytical Chemistry, Åbo Akademi University) for ICP analyses, Päivi Mäki-Arvela (Laboratory of Industrial Chemistry and Reaction Engineering, Åbo Akademi University) for the CO pulse chemisorption analyses, Atte Aho (Laboratory of Industrial Chemistry and Reaction Engineering, Åbo Akademi University) for nitrogen physisorpiton analyses and Krisztian Kordas and Anne-Riikka Leino (Microelectronics and Materials Physics Laboratories, Department of Electrical and Information Engineering, University of Oulu) for the HRTEM analyses. The Magnus Ehrnrooth foundation, Makarna Olins Stipendiefond at the Student Union of Åbo Akademi University and Rector of Åbo Akademi University are gratefully acknowledged for the financial support to OL.Notes and references
- M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS PubMed.
- A. Bruggink, R. Schoevaart and T. Kieboom, Org. Process Res. Dev., 2003, 7, 622–640 CrossRef CAS.
- T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS PubMed.
- C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390–2431 CrossRef CAS PubMed.
- K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed.
- H.-C. Guo and J.-A. Ma, Angew. Chem., Int. Ed., 2006, 45, 354–366 CrossRef CAS PubMed.
- D. Yu. Murzin and R. Leino, Chem. Eng. Res. Des., 2008, 86, 1002–1010 CrossRef CAS.
- O. Långvik, T. Saloranta, A. Kirilin, A. Liljeblad, P. Mäki-Arvela, L. T. Kanerva, D. Y. Murzin and R. Leino, ChemCatChem, 2010, 2, 1615–1621 CrossRef.
- O. Långvik, P. Mäki-Arvela, A. Aho, T. Saloranta, D. Yu. Murzin and R. Leino, Catal. Lett., 2013, 143, 142–149 CrossRef.
- I. Busygin, M. Rosenholm, E. Toukoniitty, D. Yu. Murzin and R. Leino, Catal. Lett., 2007, 117, 91–98 CrossRef CAS.
- T. Tanaka, M. Kawase and S. Tani, Bioorg. Med. Chem., 2004, 12, 501–505 CrossRef CAS PubMed.
- O. B. Wallace, D. W. Smith, M. S. Deshpande, C. Polson and K. M. Felsenstein, Bioorg. Med. Chem. Lett., 2003, 13, 1203–1206 CrossRef CAS PubMed.
- Q. K. Fang, Z. Han, P. Grover, D. Kessler, C. H. Senanayake and S. A. Wald, Tetrahedron: Asymmetry, 2000, 11, 3659–3663 CrossRef CAS.
- W. Adam, M. Lazarus, C. R. Saha-Möller and P. Schreier, Acc. Chem. Res., 1999, 32, 837–845 CrossRef CAS.
- F. A. Davis and B. C. Chen, Chem. Rev., 1992, 92, 919–934 CrossRef CAS.
- P. Hoyos, J.-V. Sinisterra, F. Molinari, A. R. Alcántara and P. D. de María, Acc. Chem. Res., 2010, 43, 288–299 CrossRef CAS PubMed.
- C. H. Senanayake, Aldrichimica Acta, 1998, 31, 3–16 CAS.
- A. K. Ghosh, S. Fidanze and C. H. Senanayake, Synthesis, 1998, 937–961 CrossRef CAS.
- Y. Hong, Y. Gao, X. Nie and C. M. Zepp, Tetrahedron Lett., 1994, 35, 6631–6634 CrossRef CAS.
- J. Y.-T. Soh and C.-H. Tan, J. Am. Chem. Soc., 2009, 131, 6904–6905 CrossRef CAS PubMed.
- S. Liu and C. Wolf, Org. Lett., 2008, 10, 1831–1834 CrossRef CAS PubMed.
- X. L. Liu, Z. J. Wu, X. L. Du, X. M. Zhang and W. C. Yuan, J. Org. Chem., 2011, 76, 4008–4017 CrossRef CAS PubMed.
- M. Delamare, S. Belot, J. C. Caille, F. Martinet, H. B. Kaagn and V. Hentyon, Tetrahedron Lett., 2009, 50, 1702–1704 CrossRef CAS.
- Y. Kobayashi, F. Morisawa and K. Saigo, J. Org. Chem., 2005, 71, 606–615 CrossRef PubMed.
- W. Adam, M. T. Díaz, R. T. Fell and C. R. Saha-Möller, Tetrahedron: Asymmetry, 1996, 7, 2207–2210 CrossRef CAS.
- O. Pàmies and J.-E. Bäckvall, Chem. Rev., 2003, 103, 3247–3262 CrossRef PubMed.
- B. Martín-Matute and J.-E. Bäckvall, Curr. Opin. Chem. Biol., 2007, 11, 226–232 CrossRef PubMed.
- J. H. Lee, K. Han, M.-J. Kim and J. Park, Eur. J. Org. Chem., 2010, 999–1015 CrossRef CAS.
- K. Szőri, G. Szöllősi and M. Bartók, Adv. Synth. Catal., 2006, 348, 515–522 CrossRef.
- B. Stewart, J. Nyhlen, B. Martín-Matute, J.-E. Bäckvall and T. Privalov, Dalton Trans., 2013, 42, 927–934 RSC.
- M. Päiviö, D. Mavrynsky, R. Leino and L. T. Kanerva, Eur. J. Org. Chem., 2011, 1452–1457 CrossRef.
- O. Verho, E. V. Johnston, E. Karlsson and J.-E. Bäckvall, Chem. – Eur. J., 2011, 17, 11216–11222 CrossRef CAS PubMed.
- M. C. Warner, O. Verho and J.-E. Bäckvall, J. Am. Chem. Soc., 2011, 133, 2820–2823 CrossRef CAS PubMed.
- D. Mavrynsky, L. T. Kanerva, R. Sillanpää and R. Leino, Pure Appl. Chem., 2011, 83, 479–487 CAS.
- A. N. Parvulescu, J. Janssens, J. Vanderleyden and D. De Vos, Top. Catal., 2010, 53, 931–941 CrossRef CAS.
- A. N. Parvulescu, P. A. Jacobs and D. E. De Vos, Adv. Synth. Catal., 2008, 350, 113–121 CrossRef CAS.
- N. Kim, S.-B. Ko, M. S. Kwon, M.-J. Kim and J. Park, Org. Lett., 2005, 7, 4523–4526 CrossRef CAS PubMed.
- J.-S. Im, S.-H. Ahn and Y.-H. Park, Chem. Eng. J., 2013, 234, 49–56 CrossRef CAS.
- Y. K. Choi, M. J. Kim, Y. Ahn and M.-J. Kim, Org. Lett., 2001, 3, 4099–4101 CrossRef CAS PubMed.
- N. Ravasio, F. Zaccheria, A. Fusi and R. Psaro, Phys. Rev. A: At., Mol., Opt. Phys., 2006, 315, 114–119 CAS.
- R. Millet, A. M. Träff, M. L. Petrus and J.-E. Bäckvall, J. Am. Chem. Soc., 2010, 132, 15182–15184 CrossRef CAS PubMed.
- S. Sahin, J. Wärnå, P. Mäki-Arvela, T. Salmi and D. Yu. Murzin, J. Chem. Technol. Biotechnol., 2010, 85, 192–198 CAS.
- A. Kirilin, P. Mäki-Arvela, M. Rupp, E. Toukoniitty, N. Kumar, K. Kordas, L. M. Kustov, T. Salmi and D. Yu. Murzin, Res. Chem. Intermed., 2010, 36, 193–210 CrossRef CAS.
- A. Kirilin, P. Mäki-Arvela, K. Kordas, A.-R. Leino, A. Shchukarev, D. Boström, J.-P. Mikkola, L. M. Kustov, T. O. Salmi and D. Yu. Murzin, Kinet. Catal., 2011, 52, 72–76 CrossRef CAS.
- A. Kirilin, P. Mäki-Arvela, K. Kordas, A.-R. Leino, A. Shchukarev, D. Boström, J.-P. Mikkola, L. M. Kustov, T. O. Salmi and D. Yu. Murzin, Kinet. Catal., 2011, 52, 77–81 CrossRef CAS.
- S. Sahin, P. Mäki-Arvela, M. Kangas, K. Eränen, J. Wärnå, T. Salmi and D. Yu. Murzin, Kinet. Catal., 2012, 53, 673–683 CrossRef CAS.
- P. Lozano, T. de Diego, D. Carrie, M. Vaultier and J. L. Iborra, Chem. Commun., 2002, 692–693 RSC.
- H. R. Hobbs, B. Kondor, P. Stephenson, R. A. Sheldon, N. R. Thomas and M. Poliakoff, Green Chem., 2006, 8, 816–821 RSC.
- M. Hartmann and D. Jung, J. Mater. Chem., 2010, 20, 844–857 RSC.
- R. L. Hartman, J. P. Mc Mullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 2–20 CrossRef PubMed.
- N. G. Anderson, Org. Process Res. Dev., 2012, 16, 852–869 CrossRef CAS.
- S. Strompen, M. Weiß, H. Gröger, L. Hilterhaus and A. Liese, Adv. Synth. Catal., 2013, 355, 2391–2399 CrossRef CAS.
- R. Porcar, V. Sans, N. Ríos-Lombardía, V. Gotor-Fernández, V. Gotor, M. I. Burguete, E. García-Verdugo and S. V. Luis, ACS Catal., 2012, 2, 1976–1983 CrossRef CAS.
- S. Mascia, P. L. Heider, H. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans, T. F. Jamison, K. F. Jensen, A. S. Myerson and B. L. Trout, Angew. Chem., Int. Ed., 2013, 52, 12359–12363 CrossRef CAS PubMed.
- M. P. Cava, R. L. Litle and D. R. Napier, J. Am. Chem. Soc., 1958, 80, 2257–2263 CrossRef CAS.
- O. Petrovskaia, B. M. Taylor, D. B. Hauze, P. J. Carroll and M. M. Joullie, J. Org. Chem., 2001, 66, 7666–7675 CrossRef CAS PubMed.
- S. K. Gupta and S. A. Marathe, J. Pharm. Sci., 1976, 65, 134–135 CrossRef CAS PubMed.
- P. K. Banerjee, D. Mukhopadhyay and D. N. Chaudhury, J. Indian Chem. Soc., 1965, 42, 115–120 CAS.
- Y. Chiang, A. J. Kresge, O. Sadovski, X. Zeng and Y. Zhu, Can. J. Chem., 2005, 83, 1202–1206 CrossRef CAS.
- L. T. Kanerva and O. Sundholm, J. Chem. Soc., Perkin Trans. 1, 1993, 2407–2410 RSC.
- K. Yamaguchi, T. Koike, J. W. Kim, Y. Ogasawara and N. Mizuno, Chem. – Eur. J., 2008, 14, 11480–11487 CrossRef CAS PubMed.
- L. M. Neal, S. D. Jones, M. L. Everett, G. B. Hoflund and H. E. Hagelin-Weaver, J. Mol. Catal. A: Chem., 2010, 325, 25–35 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- M. Von Arnim and R. Ahlrichs, J. Comput. Chem., 1998, 19, 1746–1757 CrossRef CAS.
- O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652–660 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 CrossRef CAS.
- M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 18, 9136–9148 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- W. J. Hehre, L. Radom, P. von R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, John Wiley&Sons, New York, 1986 Search PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, D. J. DeFrees, J. A. Pople and M. S. Gordon, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- D. E. Bicknell and R. S. Ramotowski, J. Forensic Sci., 2008, 53, 1108–1116 CrossRef CAS PubMed.
- S. Wiesner, E. Springer, Y. Sasson and J. Almog, J. Forensic Sci., 2001, 46, 1082–1084 CAS.
- Compendium of Chemical Terminology - Gold Book Version 2.3.2; [Online], International Union of Pure and Applied Chemistry (IUPAC), Posted, 2012, pp. 97–98, http://goldbook.iupac.org/ (accessed Dec. 2, 2013) Search PubMed.
- N. Tyutyulkov, M. Tasseva and E. Georgiev, J. Prakt. Chem., 1987, 329, 780–786 CrossRef CAS.
- G. A. Russel, C. L. Myers, P. Bruni, F. A. Neugebauer and R. Blankespoor, J. Am. Chem. Soc., 1970, 92, 2762–2769 CrossRef.
- E. T. Strom, J. Org. Chem., 1995, 60, 5686–5689 CrossRef CAS.
- E. T. Strom, E. G. Janzen and J. L. Gerlock, Mol. Phys., 1970, 19, 577–580 CrossRef CAS.
- L. F. Fieser, J. Am. Chem. Soc., 1948, 70, 3165–3174 CrossRef CAS PubMed.
- K. L. Platt and F. Oesch, J. Org. Chem., 1983, 48, 265–268 CrossRef CAS.
- P. Ciuffreda, S. Casati and E. Santaniello, Tetrahedron, 2000, 56, 317–321 CrossRef CAS.
- C. Ran, Q. Dai, Q. Ruan, T. M. Penning, I. A. Blair and R. G. Harvey, J. Org. Chem., 2007, 73, 992–1003 CrossRef PubMed.
- D. Mavrynsky, D. Yu. Murzin and R. Leino, ChemCatChem, 2013, 5, 2436–2445 CrossRef CAS.
- B. Martín-Matute, M. Edin, K. Bogár, F. B. Kaynak and J.-E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 8817–8825 CrossRef PubMed.
- M. C. Warner and J.-E. Bäckvall, Acc. Chem. Res., 2013, 46, 2545–2555 CrossRef CAS PubMed.
- P. Ödman, L. A. Wessjohan and U. T. Bornscheuer, J. Org. Chem., 2005, 70, 9551–9555 CrossRef PubMed.
- S. Wuyts, K. De Temmerman, D. E. De Vos and P. A. Jacobs, Chem. – Eur. J., 2005, 11, 386–397 CrossRef PubMed.
- S. Wuyts, J. Wahlen, P. A. Jacobs and D. E. De Vos, Green Chem., 2007, 9, 1104–1108 RSC.
- K. Engström, M. Shakeri and J.-E. Bäckvall, Eur. J. Org. Chem., 2011, 1827–1830 CrossRef.
- F. Zaccheria, N. Ravasio, R. Psaro and A. Fusi, Chem. – Eur. J., 2006, 12, 6426–6431 CrossRef CAS PubMed.
- K. Yamaguchi, T. Koike, M. Kotani, M. Matsushita, S. Shinachi and N. Mizuno, Chem. – Eur. J., 2005, 11, 6574–6582 CrossRef CAS PubMed.
- H. M. Jung, J. H. Koh, M.-J. Kim and J. Park, Org. Lett., 2000, 2, 2487–2490 CrossRef CAS PubMed.
- M.-J. Kim, W.-H. Kim, K. Han, Y. K. Choi and J. Park, Org. Lett., 2007, 9, 1157–1159 CrossRef CAS PubMed.
- H. Haario, ModEst 6.0, User Guide, Helsinki, 2010 Search PubMed.
- D. Yu. Murzin, N. V. Kul'kova and M. I. Temkin, Proceeding of the USSR conference on catalyst deactivation, Book of Abstracts, Ufa, 1989, vol. 2, pp. 85–86 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR spectra for (R)-3, 6 and 7, DFT calculations results, chiral GC chromatograms and Ru catalyst characterization (HR-TEM, TPR, XPS). See DOI: 10.1039/c4cy01099j |
| This journal is © The Royal Society of Chemistry 2015 |