
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCan we beat the biotin–avidin pair?: cucurbit[7]uril-based ultrahigh affinity host–guest complexes and their applications
Dinesh
Shetty
a,
Jayshree K.
Khedkar
a,
Kyeng Min
Park
ad and
Kimoon
Kim
*abc
aCenter for Self-assembly and Complexity (CSC), Institute for Basic Science (IBS), Pohang 37673, Republic of Korea. E-mail: kkim@postech.ac.kr
bDepartment of Chemistry, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea
cDivision of Advanced Materials Science, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea
dDepartment of Nanomaterials Science and Engineering, University of Science and Technology (UST), Pohang 37673, Republic of Korea
First published on 5th October 2015
Abstract
The design of synthetic, monovalent host–guest molecular recognition pairs is still challenging and of particular interest to inquire into the limits of the affinity that can be achieved with designed systems. In this regard, cucurbit[7]uril (CB[7]), an important member of the host family cucurbit[n]uril (CB[n], n = 5–8, 10, 14), has attracted much attention because of its ability to form ultra-stable complexes with multiple guests. The strong hydrophobic effect between the host cavity and guests, ion–dipole and dipole–dipole interactions of guests with CB portals helps in cooperative and multiple noncovalent interactions that are essential for realizing such strong complexations. These highly selective, strong yet dynamic interactions can be exploited in many applications including affinity chromatography, biomolecule immobilization, protein isolation, biological catalysis, and sensor technologies. In this review, we summarize the progress in the development of high affinity guests for CB[7], factors affecting the stability of complexes, theoretical insights, and the utility of these high affinity pairs in different challenging applications.
 Dinesh Shetty | Dinesh Shetty received his MS degree from Mangalore University, India and PhD in chemistry from Seoul National University, Korea (2011; advisor: Prof. Jae Min Jeong). After two years of postdoctoral work at Emory University, USA (advisor: Prof. Hyunsuk Shim) he joined Center for Self-assembly and Complexity (CSC), Institute for Basic Science (IBS), POSTECH as a research fellow in 2013. His current research focuses on the development of novel materials for biomedical applications. |
 Jayshree K. Khedkar | Jayshree K. Khedkar was born in Ahmednagar, India in 1985. She obtained both her MSc (2008) and PhD (2012) in chemistry under Prof S. P. Gejji at the University of Pune, India. Later she worked as a research associate in the group of Prof. C. P. Rao, at the Indian Institute of Technology Bombay, India. Since 2014, she has been working as a Research Fellow at CSC, IBS. Her research interests focus on computational and supramolecular chemistry. |
 Kyeng Min Park | Kyeng Min Park received his PhD degree in supramolecular chemistry from POSTECH in 2009. After 2 years of postdoctoral work at Harvard University, he returned to Korea and worked for semiconductor R&D center of Samsung electronics as a senior researcher until 2014. Then, he joined CSC, IBS as a research fellow and served as an adjunct Professor of the University of Science and Technology (UST). His current research focuses on the development of novel supramolecular materials and exploring their applications. |
 Kimoon Kim | Kimoon Kim studied chemistry at SNU (BS, 1976), Korea Advanced Institute of Science and Technology (MS, 1978), and Stanford University (PhD, 1986). After two-years of postdoctoral work at Northwestern University, he joined POSTECH where he is now a Distinguished University Professor. Recently, he has been appointed as director of CSC, IBS. His current research focuses on developing novel functional materials and systems based on supramolecular chemistry. |
1. Introduction
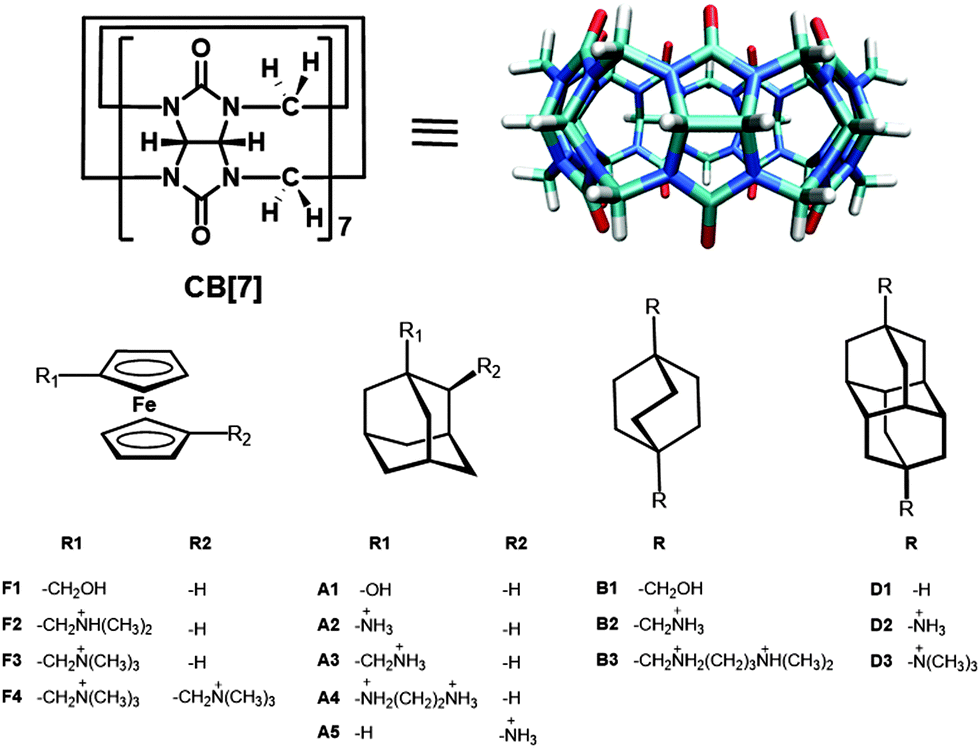
The high affinity and fidelity in molecular recognition and transformation is a key component of the most common biomolecules (e.g. between protein and ligand, antigen and antibody, sugar and lectin, RNA and ribosome) in the generation of life-like complexity and function. Apart from understanding the complexity of life, high affinity host–guest pairs have tremendous values in both biotechnology and chemical applications. The strongest and best known natural noncovalent interaction pair is the biotin–avidin (or streptavidin) system with a binding constant in the range of 1013–1015 M−1.1,2 This affinity pair has been exploited in many applications including enzyme-linked immunosorbent assays, protein and nucleic acid detection and purification, and immobilization of biomolecules on surfaces.3–9 However, this natural robust binding pair suffers some downsides such that the avidin–biotin complex cannot be easily dissociated (although there has been a considerable amount of work on the site-directed mutagenesis of avidin to temper its affinity for biotin)10–14 unless operated in elevated temperatures, which causes the denaturation of the proteins. Also, the interaction between avidin and biotin can be undesirably affected by bio-systems, thus limiting their biological applications.15–17 In order to mimic some of the complexity of natural systems, researchers have been trying to create high affinity synthetic receptor–ligand complexes. In this regard, the central goal has been the development of host–guest systems with binding affinities as high as those reached in biological systems, which often permit the selective binding at concentrations in the nanomolar or picomolar regimes. However, the number of synthetic host families that display biomolecular range affinity (Kd < 1 μM, Ka > 106 M−1) and selectivity in aqueous solution is limited to certain cyclophanes,18 self-assembled receptors,19–24 and cucurbiturils.25–29Among them, cucurbit[n]uril (CB[n], n = 5–8, 10, 14), a family of synthetic macrocyclic host molecules composed of n glycoluril units with a hydrophobic cavity and two identical carbonyl-fringed portals, attracted tremendous interest, which is depicted in regular reviews from different perspectives.30–39 Isaacs, Kim, and others reported that this host family forms exceptionally stable host–guest complexes with specific guest molecules.40–44 This complexation mechanism contrasts with that found in other cavitands, such as cyclodextrins (CDs), in which the hydrophobic interactions alone are believed to dominate the complexation process.45 Consequently, the multiple binding forces involved in the complex formation of CB[n] with different guests give rise to highly stable inclusion complexes in solution as well as in the solid state.
CB[7], an important member of the CB[n] family with an internal cavity volume of 279 Å3, which is close to that of β-CD (262 Å3), has drawn much attention in host–guest systems because of its ability to complex with a variety of guest molecules with high binding affinity and selectivity.40–44,46 There are some unique features that separate CB[7] from its other family members. Unlike other extensively studied members – CB[6] and CB[8] – which show poor solubility in common solvents (<10−5 M), CB[7] has a moderate solubility in water (20–30 mM). More importantly, in contrast to the other synthetic receptors, CB[7] commonly displays binding affinities to its guests between 107 M−1 and 1017 M−1. These binding constant values are equal to or greater than that of the natural biotin–avidin complex. In 2005, the ultrahigh affinity of CB[7] toward ferrocene40,41 and adamantane41 derivatives was first measured by Isaacs using 1H NMR competition experiments and independently measured by our group in collaboration with Inoue and Kaifer by competition ITC methods. In the subsequent years, researchers explored new high affinity guest scaffolds such as bicyclo[2.2.2]octane43 and diamantane derivatives,44 while computational scientists demonstrated the importance of new parameters such as the release of high-energy water molecules during the formation of CB[7]–guest complexes.47–50 Considering the importance of high affinity host–guest pairs, this review first summarizes and presents the development of high affinity guest molecules for CB[7]. In the later parts, we describe the origins of high-affinity binding and the details about enthalpy/entropy compensation, hydrophobic effects, and their contributions to the unusual binding phenomenon. At the end, we discuss the applications of these high-affinity binding pairs in different fields of research.
2. High-affinity host–guest pairs
Even though CB homologues share characteristic features such as a hydrophobic cavity and polar carbonyl groups surrounding the portals, their varying cavity and portal sizes lead to molecular recognition properties different from each other. In general, CB[6] forms very stable complexes with protonated diaminoalkanes whereas CB[7] forms 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with larger guest molecules including ferrocene (and cobalotocene),26,40–42,46,51 adamantane,41,43 diamantane,44,52 and bicyclooctane derivatives,43,53 which are too large to be included in CB[6] (Fig. 1). The reported affinities of such molecules can approach or even exceed those of the strongest noncovalent interaction pairs found in nature.3,54 On the other hand, CB[8] has a cavity large enough to accommodate two guest molecules to form 1:2 or 1:1:1 complexes. In general, the affinity of CB[7] complexes in water is higher than buffer solutions because of the ability of metal ions (such as Na+) to bind at the carbonyl portals of CB[7] which reduces the observed values of association equilibrium constants (Ka) by competition.55,56
:1 complexes with larger guest molecules including ferrocene (and cobalotocene),26,40–42,46,51 adamantane,41,43 diamantane,44,52 and bicyclooctane derivatives,43,53 which are too large to be included in CB[6] (Fig. 1). The reported affinities of such molecules can approach or even exceed those of the strongest noncovalent interaction pairs found in nature.3,54 On the other hand, CB[8] has a cavity large enough to accommodate two guest molecules to form 1:2 or 1:1:1 complexes. In general, the affinity of CB[7] complexes in water is higher than buffer solutions because of the ability of metal ions (such as Na+) to bind at the carbonyl portals of CB[7] which reduces the observed values of association equilibrium constants (Ka) by competition.55,56
 | ||
| Fig. 1 Molecular structures of CB[7] and its high affinity guests. | ||
Host–guest properties of CBs have often been compared with those of CDs because of the similarity in their cavity sizes. However, molecular composition and guest binding properties of these two families of molecular containers are quite different from each other. In general, CBs bind guest molecules with higher affinity and higher selectivity than CDs do. For example, the Ka values for β-CD rarely get over 106 M−1 whereas the corresponding member of the CB family, CB[7] displays much higher binding affinity up to 1017 M−1. Furthermore, CBs show strong affinity toward cationic guests, moderate affinity toward neutral guests, and little affinity toward anionic guests,40 while CDs show moderate affinity to hydrophobic guests regardless of their charges, which can be explained by the electrostatic potential of the host families (Fig. 2). In CBs, the regions around the carbonyl oxygens are found to be quite negative (red colored) and the inner surface of the cavity is also significantly negative while the outer surface is somewhat positive (blue colored). On the other hand, the portal and cavity of CDs are almost neutral. We should also note that the high structural rigidity of CBs in comparison with CDs allows highly selective recognition processes for the former. Furthermore, the inner cavity of CB[7] has extremely low polarizability, which is closer to the gas-phase than to that of any other known solvents,57,58 and a very high quadruple moment,59 which allows higher-order electrostatic interactions.
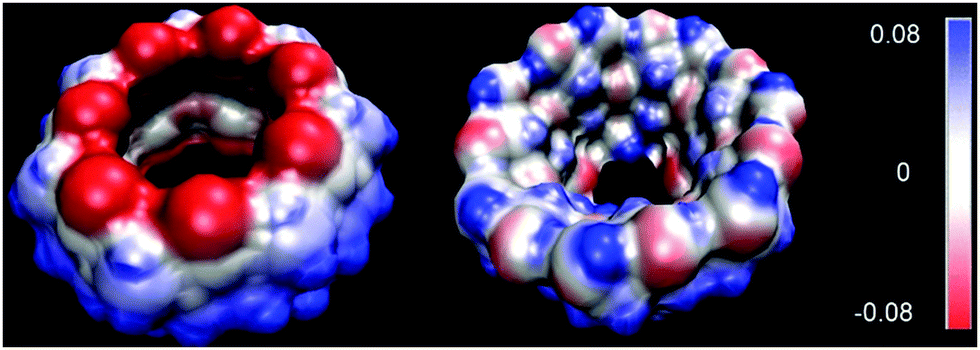 | ||
| Fig. 2 The surface electrostatic potential for CB[7] (left) and β-CD (right). The color ranges from negative (red) to positive (blue). | ||
High-affinity CB[7]–ferrocene complexes
In 2003, the formation of a stable complex between CB[7] and ferrocene26 or cobaltocene46 derivatives was observed by us and Kaifer, respectively. In 2005, a parallel effort by Isaacs41 and our group40 in collaboration with Inoue and Kaifer revealed that all neutral (F1) and cationic ferrocene derivatives (F2 and F3) (Fig. 1) form highly stable inclusion complexes with CB[7] with Ka values of ∼109 M−1 and ∼1012 M−1, respectively (Table 1). The binding affinities of the ferrocene derivatives toward CB[7] drop by two or three orders of magnitude upon oxidation of the ferrocene core.60 On the other hand, quite surprisingly, anionic ferrocene derivatives with a similar size and structure do not bind to CB[7].40 These results are in sharp contrast to the fact that the Ka values of β-CD for the same guests are only in the range of 103–104 M−1 regardless of the charge on the guests. It was surprising to observe that the simple guest molecules bind to CB[7] with such a high binding affinity. In fact, it was the tightest binding observed for any monovalent synthetic host–guest systems then. Even though this phenomenon was difficult to understand completely at that time, the much higher guest binding affinity of CB[7] compared to β-CD was attributed at least in part, to the contribution of ion–dipole interactions, which are not favored in the case of β-CD as suggested by the electrostatic potential profiles (Fig. 2). Also, the highly negative regions around the two cavity entrances (Fig. 2) of CB[7] lead to the rejection of negatively charged ferrocene guests. The importance of ion–dipole interactions between the host and guest molecules for the high affinity binding process was further supported by the enhancement of binding affinity (∼103) upon the replacement of the hydroxymethylene group of F1 (Ka = ∼109 M−1) with a positively charged ammonium methylene unit (F2 and F3; Ka = ∼1012 M−1). In our observations it was clear that the ferrocene moiety is nicely located inside the CB[7] cavity through hydrophobic interactions, while the positive charge on the substituents interacts with the negatively charged portals of CB[7] by electrostatic interactions.| Guest |
|
|
K (Μ−1) |
|---|---|---|---|
| a From ref. 43. b From ref. 44. c Due to the extremely slow equilibrium, it was difficult to determine the error by the NMR competition technique. | |||
| F1 | −21.5 ± 0.5 | 8.6 ± 0.5 | (3.2 ± 0.5) × 109 |
| F2 | −21.0 ± 0.5 | 4.1 ± 0.5 | (2.4 ± 0.8) × 1012 |
| F3 | −21.5 ± 0.5 | 4.3 ± 0.4 | (4.1 ± 1.0) × 1012 |
| F4 | −21.5 ± 0.2 | 0.5 ± 0.5 | (3.0 ± 1.0) × 1015 |
| A1 | −19.0 ± 0.4 | 4.9 ± 0.4 | (2.3 ± 0.8) × 1010 |
| A2 | −19.3 ± 0.4 | −0.1 ± 0.5 | (1.7 ± 0.8) × 1014 |
| A3 , | −21.9 ± 0.4 | 1.7 | 7.7 × 1014 |
| A4 , | −20.1 ± 0.4 | −1.4 | 5 × 1015 |
| A5 | −19.5 ± 0.4 | 0.4 ± 0.5 | (1.0 ± 0.3) × 1014 |
| B1 | −15.8 ± 0.2 | 2.4 ± 0.2 | (6.1 ± 0.5) × 109 |
| B2 | −15.6 ± 0.4 | −3.9 ± 0.5 | (2.0 ± 0.5) × 1014 |
| B3 | −16.3 ± 0.4 | −4.3 ± 0.5 | (1.2 ± 0.5) × 1015 |
| D1 | — | — | 4.0 × 109 |
| D2 | — | — | (1.4 ± 0.3) × 1011 |
| D3 | — | — | 7.2 × 1017 |

Intrigued by the exceptionally high affinity of CB[7] toward F2 and F3, we decided to extend this work to develop even tighter binding guests for CB[7] in collaboration with Inoue, Kaifer, Isaacs, and Gilson. We thought that the addition of a second ammonium unit to the ferrocene unit in F3, which can interact with the other portal of CB[7], would further increase the affinity of the guest to CB[7]. Indeed, the binding affinity of CB[7] toward F4 having two ammonium units further increased to 3 × 1015 M−1, as measured by a 4-step competition ITC experiment.42 The X-ray crystal structure shows that each of the trimethylammoniomethyl arms protrudes through the CB[7] cavity and consequently the dicationic ferrocene guest resides inside CB[7] symmetrically. The tilting of the ferrocenyl residue allows the ideal positioning of each of the trimethylammonium groups to maximize ion–dipole interactions with the carbonyl rims on each of the host portals. The tight fit of F4 inside the CB[7] cavity was also evident in NMR spectroscopic experiments; the much shorter spin–lattice relaxation time (T1) observed for the ferrocene core protons of F4@CB[7] compared to F4 indicate that the rotational motion of the ferrocene is restricted due to the tight fit of the guest upon complexation. Apart from extensive use of NMR, ITC, electrochemical analysis, and crystallography techniques for the interrogation of these complexes, Raman spectroscopy was also used recently to investigate the solid state structural aspects.51 The Raman spectra of ferrocene@CB[7] showed significant frequency shifts of specific vibrational modes localized on different parts of the complex due to the structural and electronic interactions between the host and guest components, which confirmed the presence of the ferrocene moiety inside the CB[7] cavity.
Another interesting observation was the enthalpy and entropy changes associated with the guest binding. Unexpectedly, the addition of methylammonium groups onto the ferrocene core does not affect the enthalpy change (ΔH° = ∼−21 kcal mol−1) associated with the complexation; instead, it causes an increase in the entropy change by 3.8–4.3 kcal mol−1, which results in a thousand fold enhancement in the corresponding binding constant. The change in solvation entropy ( ) upon complexation is more favorable for the strong binding dication F4 (18 kcal mol−1) compared to monocationic or neutral guests (11 kcal mol−1 for F3 and 9 kcal mol−1 for F1). This observation suggests that the enthalpy–entropy compensation effect, which is commonly observed in other molecular recognition systems,45,61,62 is not being obeyed in this case. Fig. 3 presents the data points for different high affinity guests along with those for the binding of CDs with a variety of guest molecules, which greatly deviate from the enthalpy–entropy compensation plot for CD–guest complexation.
) upon complexation is more favorable for the strong binding dication F4 (18 kcal mol−1) compared to monocationic or neutral guests (11 kcal mol−1 for F3 and 9 kcal mol−1 for F1). This observation suggests that the enthalpy–entropy compensation effect, which is commonly observed in other molecular recognition systems,45,61,62 is not being obeyed in this case. Fig. 3 presents the data points for different high affinity guests along with those for the binding of CDs with a variety of guest molecules, which greatly deviate from the enthalpy–entropy compensation plot for CD–guest complexation.
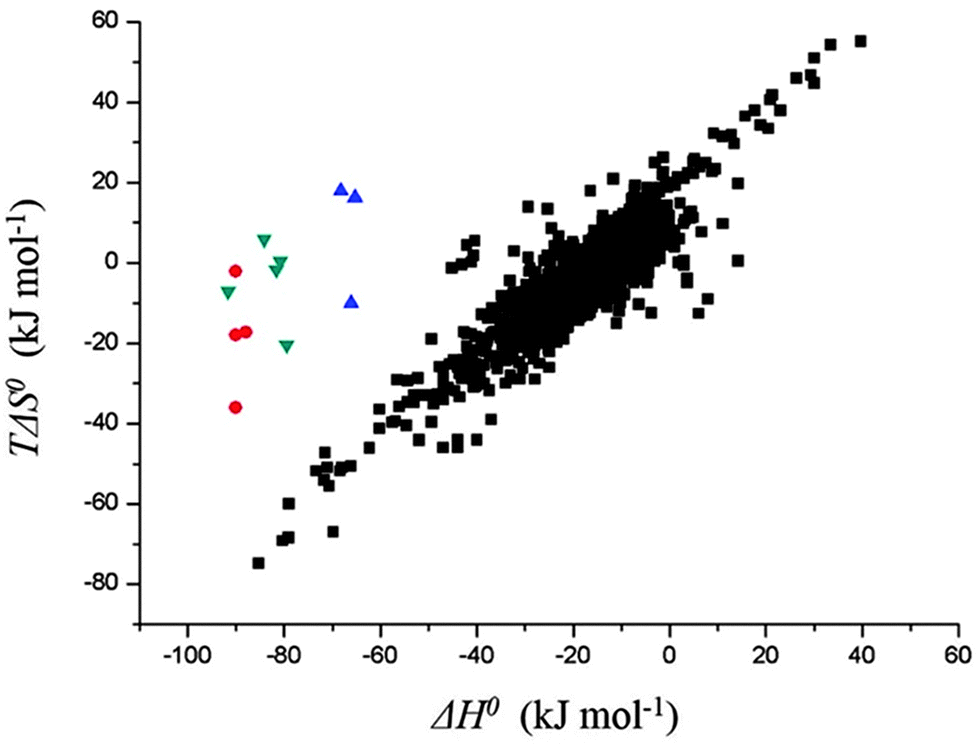 | ||
| Fig. 3 The thermodynamic data obtained for complexation of ferrocene (●), adamantane (▼), and bicyclo[2.2.2]octane (▲) derivatives with CB[7] which are significantly deviated from the enthalpy–entropy compensation plot for cyclodextrin–guest complexation (■). Data reproduced from ref. 43 and 45. | ||
High-affinity complexes of CB[7] with bicyclo[2.2.2]octane and adamantane guests
Right after the successful isolation of CB[7], we observed the formation of a stable host–guest complex between protonated adamantylamine (A2) and CB[7].26 Isaacs and coworkers measured the binding constant for the first time and found that the binding between CB[7] and A2 is indeed very strong and the binding constant to be 4.2 × 1012 M−1.41 The authors studied the binding properties of CB[7] toward multiple mono-, di-, and tri-substituted adamantanes to probe the size limits and functional group preferences of CB[7]. Among the monosubstituted compounds, cationic ammonium compounds were preferred by CB[7] (104-fold preference) relative to an anionic compound, irrespective of the nature of the ammonium group (NH3+, quaternary or pyridinium). They also found that the bulkiness of the substituents on the adamantane core in the case of di- or tri-substituted derivatives has a significant effect on the affinity of the guest molecules toward CB[7]. In 2009, Inoue, Gilson and co-workers designed a new series of high affinity guest molecules for CB[7] through theoretical studies.63 The designed molecules essentially replace the ferrocene moiety with a bicyclo[2.2.2]octane moiety, which is similar to the former in terms of size and hydrophobic character. Two years later, the same group tested their blinded calculations on designed molecules by experimental methods and found that bicyclo[2.2.2]octane derivatives indeed have high affinity toward CB[7] (Table 1).43 They synthesized and tested a series of bicyclo[2.2.2]octane and adamantane derivatives to further probe the high-affinity binding regime of CB[7]. These new guests having a rigid and aliphatic core are chosen to fit well into the cavity of CB[7] and outfitted with cationic groups positioned to interact favorably with one or both CB[7] portals. These metal-free guest molecules also show ultrahigh binding affinity toward CB[7], with a binding constant in the range of 1014–1015 M−1. Similar to our previous observations for the ferrocene derivatives, the neutral bicyclo[2.2.2]octane guest (B1) showed less affinity toward CB[7] compared to those having ammonium groups (B2 and B3). Remarkably, the binding affinity of mono-cationic adamantane derivatives (A2 and A3) match with those of F4 or B3 having two cationic substituents. However, in these series of guest molecules, the increase in overall charge of the molecules resulted in only a modest increment in binding affinities (Table 1). These observations suggest that a random charge increment to the guest molecule does not warrant a drastic increase in affinity and hence a proper design of guest molecules in accordance with the size of the CB[7] cavity and substitutions at proper positions is more important to achieve the ultra-affinity toward CB[7]. Similar to ferrocene, the remarkably high affinities of these guests are attributed in part to the fact that they lose an unusually small amount of entropy on binding in proportion to their enthalpic changes. A very recent work by Gilson suggests that dispersion forces contribute strongly to the computed binding enthalpies of these complexes, partly due to the fact that their packing density is greater than that of water.64Ultrahigh affinity CB[7]–diamantane complexes
Very recently, Isaacs, Glaser, Mlinarić-Majerski, and co-workers reported an attomolar-affinity host–guest complex between CB[7] and diamantane quaternary diammonium ion derivative D3 (Ka = 7.2 × 1017 M−1 in pure D2O), which is the tightest binding small molecule host–guest complex ever measured for a monovalent molecular recognition to date.44,52 They studied two diamantane derivatives (D2 and D3) and found that D3 has much higher binding affinity toward CB[7] than D2, though the backbone of the molecules remained the same. The X-ray crystal structure of D3@CB[7] shows that the core D3 is complementary to the cavity of CB[7] and the quaternary ammonium groups of D3 allow the guest to form optimal ion–dipole interactions with CB[7]. In addition, the authors speculated that the highest binding constant is a combinational outcome of the desolvation of the carbonyl portals within the D3@CB[7] complex and the hydrophobic nature of the diamantane core relative to the adamantane and ferrocene cores. It is interesting to note that D2@CB[7] has a packing coefficient (79%)39 much higher than the ideal packing coefficient (55%) for host–guest systems,21 which may be reflected in its relatively low binding constant (Ka = 1.3 × 1011 M−1) compared to that of A2@CB[7] (Ka = 4.23 × 1012 M−1; packing coefficient = 61%). Nevertheless, the ultrahigh binding affinity of D3 for CB[7] suggests that the unfavorable effect due to the high packing density may be overcompensated by the positive effect caused by the two quaternary ammonium groups. This work illustrates a possibility of developing synthetic systems whose functions match or exceed their biological counterparts.The overall binding constants for CB[7] in Table 1 can be generally understood by two common features: (1) the hydrophobic cores of the guest molecules are rigid and complementary to the cavity of CB[7] in size and shape, which contributes to the hydrophobic effect and (2) the ammonium groups of the guest molecules are properly positioned, ideally centro-symmetrically, to enable close polar interactions with the CB[7] portals, which results in ion–dipole interactions. The recent experimental and computational thermodynamic analysis give a clearer picture of the factors accounting for the exceptional binding affinities of selected guests toward CB[7]. The removal of all high-energy water molecules and the desolvation of the guests depending on their hydrophobic surface area can greatly contribute to the binding affinity. In general, the hydrogen bonding interactions between the host and guest molecules are not a dominant force in the guest binding in water, which is evident from the fact that a quaternary trimethylammonium group gives a better binding motif than a tertiary or primary ammonium group. In contrast to other supramolecular recognition systems, the CB[n] family shows a unique thermodynamic trend by not obeying enthalpy–entropy compensation.
3. Theoretical findings on high stability
The extensive theoretical analysis based on density functional theory (DFT) calculations and molecular dynamics (MD) simulations describes the factors responsible for the high-affinity binding phenomenon of CB[7]–guest pairs.39,50,65 Among such factors, the one related to solvent and thermodynamic relationships are highly considered due to their potential use in elucidating the complexation mechanism. The ITC data (Table 1) suggest that the unusually high affinities of CB[7] with ferrocene, bicyclooctane, and adamantane derivatives can be attributed to their ability to overcome the usual pattern of the enthalpy–entropy compensation.40,42,43 The molecular modeling studies using the second-generation mining minima algorithm (M2) method by Gilson well supported these experimental observations. According to these studies, the positive solvation entropy (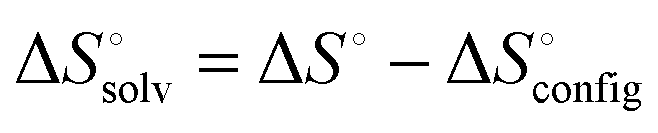 ) gained by dehydration from the CB portals and cavity as well as guests is canceled by the negative entropy arising from the increased conformational restriction of the host and guest upon complexation. This unique thermodynamic behavior avoids the large entropic penalty expected on the basis of enthalpy–entropy compensation and produces the extraordinarily high affinity as in F4@CB[7]. Thus, in addition to the size-shape complementarity between the host and guest, the release of water molecules to the bulk solvent upon guest binding is also suggested as an equally important factor for high binding affinity.42 According to Gilson,63 in the case of binding of cationic guests toward CB[7], despite the electrostatic complementarity among the guests and the electronegative carbonyl portals of CB[7], these interactions do not provide a significant net driving force for the high binding. Since the strong Coulombic attractions between the host and guests are precisely balanced by the energetic cost of stripping solvating water molecules upon tight binding through ion–dipole interactions and hence, the free energy contributions are almost entirely entropic in nature.63 Such near-cancelation of electrostatic terms leaves the van der Waals energy component as the main net contributor to the high-affinities among CB[7] complexes. With the central role of solvent entropy in determining the variations in binding free energy across the high-affinity binding systems, recently another theory demonstrating the importance of the removal of high-energy water molecules has also been put-forth. Using MD simulations, De Simone and co-workers,47 reviving the earlier hypothesis for CDs,66,67 demonstrated that the major driving force for complexation of hydrophobic residues with CB[n] originates from the release of high-energy water molecules (enthalpically and entropically unfavorable) in the cavities (Fig. 4). The unfavorable water–water interactions in CB[n] (n = 5–7) cavities than the bulk phase are evident from the calculated potential energy for water molecules and hydrogen bond count; with increase in the cavity size, the energy of the individual encapsulated water molecules decreases, since the larger cavities allow for the formation of more stable H-bond networks. The potential energy of release of all internal water molecules was noted to be maximum for CB[7], since the size (diameter) of the CB[7] cavity does not allow for the cavity water molecules (7 to 8) to arrange in an energetically stable H-bond network and gains maximum energy by a complete removal of all water molecules from the cavity. This potential energy is higher than that in CB[6] (a smaller cavity) on account of more than twice as large the number of water molecules released upon binding. Also, the larger cavity of CB[8] with 13–14 water molecules optimizes the stable H-bond network to a degree that is structurally similar to the bulk phase and consequently the resulting energetic gain per released water molecule, compared to CB[7], is less for CB[8]. The complementary sizes between the host and the guest are equally necessary to remove the high energy water molecules in the cavity after complexation that would result in a large enthalpic gain as observed in high-affinity CB[7]–ferrocene derivatives.42 Meanwhile, Gilson and coworkers also reported a related study on the unfavorable energetic and entropic properties of the confined water molecules in the interior toroidal region of the non-polar cavity of CB[7] that contribute to the unusually high affinity binding.48
) gained by dehydration from the CB portals and cavity as well as guests is canceled by the negative entropy arising from the increased conformational restriction of the host and guest upon complexation. This unique thermodynamic behavior avoids the large entropic penalty expected on the basis of enthalpy–entropy compensation and produces the extraordinarily high affinity as in F4@CB[7]. Thus, in addition to the size-shape complementarity between the host and guest, the release of water molecules to the bulk solvent upon guest binding is also suggested as an equally important factor for high binding affinity.42 According to Gilson,63 in the case of binding of cationic guests toward CB[7], despite the electrostatic complementarity among the guests and the electronegative carbonyl portals of CB[7], these interactions do not provide a significant net driving force for the high binding. Since the strong Coulombic attractions between the host and guests are precisely balanced by the energetic cost of stripping solvating water molecules upon tight binding through ion–dipole interactions and hence, the free energy contributions are almost entirely entropic in nature.63 Such near-cancelation of electrostatic terms leaves the van der Waals energy component as the main net contributor to the high-affinities among CB[7] complexes. With the central role of solvent entropy in determining the variations in binding free energy across the high-affinity binding systems, recently another theory demonstrating the importance of the removal of high-energy water molecules has also been put-forth. Using MD simulations, De Simone and co-workers,47 reviving the earlier hypothesis for CDs,66,67 demonstrated that the major driving force for complexation of hydrophobic residues with CB[n] originates from the release of high-energy water molecules (enthalpically and entropically unfavorable) in the cavities (Fig. 4). The unfavorable water–water interactions in CB[n] (n = 5–7) cavities than the bulk phase are evident from the calculated potential energy for water molecules and hydrogen bond count; with increase in the cavity size, the energy of the individual encapsulated water molecules decreases, since the larger cavities allow for the formation of more stable H-bond networks. The potential energy of release of all internal water molecules was noted to be maximum for CB[7], since the size (diameter) of the CB[7] cavity does not allow for the cavity water molecules (7 to 8) to arrange in an energetically stable H-bond network and gains maximum energy by a complete removal of all water molecules from the cavity. This potential energy is higher than that in CB[6] (a smaller cavity) on account of more than twice as large the number of water molecules released upon binding. Also, the larger cavity of CB[8] with 13–14 water molecules optimizes the stable H-bond network to a degree that is structurally similar to the bulk phase and consequently the resulting energetic gain per released water molecule, compared to CB[7], is less for CB[8]. The complementary sizes between the host and the guest are equally necessary to remove the high energy water molecules in the cavity after complexation that would result in a large enthalpic gain as observed in high-affinity CB[7]–ferrocene derivatives.42 Meanwhile, Gilson and coworkers also reported a related study on the unfavorable energetic and entropic properties of the confined water molecules in the interior toroidal region of the non-polar cavity of CB[7] that contribute to the unusually high affinity binding.48
 | ||
| Fig. 4 Schematic representation elucidating the release of high-energy water molecules from the CB[7] cavity upon binding of a hydrophobic guest. | ||
A recent report on attomolar-affinity CB[7] and diamantane complexes by Isaacs et al.44 is the best example that explains the importance of both favorable solvent entropy and the release of high-energy water molecules for the ultrahigh affinity complexation along with the direct interactions between the host and the guest. Although the inclusion of either ferrocene, adamantane, or diamantane derivatives in the CB[7] cavity results in the release of high energy water molecules, the highest binding constant of D3@CB[7] complexes is a result of additional solvating water molecule release due to the larger and more hydrophobic core of D3 (14 C atoms), which may subsequently give an additional entropic driving force. Overall, multiple studies have demonstrated the importance of various factors driving the high-affinity complexation in CB[7]. The theoretical calculations point that high-affinity binding is a combined effect of removal of all high energy water molecules from cavity, desolvation of guest molecules, and the direct interactions between the host and the guest. With the mentioned factors molecular recognition studies are aiming to control the selectivity and affinity of these host–guest systems.
4. Applications of high-affinity CB[7] complexes
Brief discussion on functionalized CB[7]
Even though the CB[n] family has proved great potential in molecular recognition, self-assembly, and nanotechnology, their practical applications were limited mainly because of their lack of solubility in common organic solvents, and relatively inert internal or external molecular surfaces. Ideally, if the CB[n] family could be modified to improve their solubility and to provide different functional groups that allow further derivatization, the range of potential applications would be significantly expanded. Over the years, thus, significant efforts have been devoted to the functionalization of CB[n] not only to improve their solubility but also to synthesize tailor-made derivatives for various applications.28 The functionalization was achieved either by use of substituted glycoluril derivatives in CB[n]-forming reactions or by direct substitution at the “equator” positions of CB[n].68–78 In a major breakthrough in the CB chemistry, we discovered a direct functionalization method to produce perhydroxylated products of CB[n] through oxidation with K2S2O8 in water.77 This finding represented the first covalent derivatization reactions of the CB[n] family and allowed us to synthesize a wide variety of CB derivatives having many potential applications.79–95 The reaction efficiency is reasonably good for CB[5] and CB[6] (yield ∼ 40–45%) but the higher homologues CB[7] and CB[8] give a low yield (5% or less) presumably due to instability of the perhydroxylated products. These substituted hydroxy groups at the periphery of the CB[n] frameworks do not alter the portal and cavity sizes of the parent molecule and hence the host–guest chemistry pertaining these molecules remains the same. In the subsequent years, by controlling the stoichiometry of the oxidizing agent, we were also able to synthesize multihydroxy77,80 and monohydroxy-CB[7] derivatives,96 which are soluble in organic solvents such as DMSO and DMF, which allows further functionalization via conventional organic transformation routes including acylation and alkylation (Scheme 1). For example, it can be alkylated with allyl bromide or propargyl bromide to yield mono or multi substituted allyl or propargyl derivatives from corresponding mono or multihydroxyl substituted CB[7], which turned out to be a useful precursor for further transformation and used in many applications that we discussed in this review. We did further modification of allyl substituted CB[7] through photo-reaction with suitably designed thiol compounds.28 In addition, the multihydroxy CB[7] derivative found to be useful in reducing non-specific binding in biological applications as discussed below.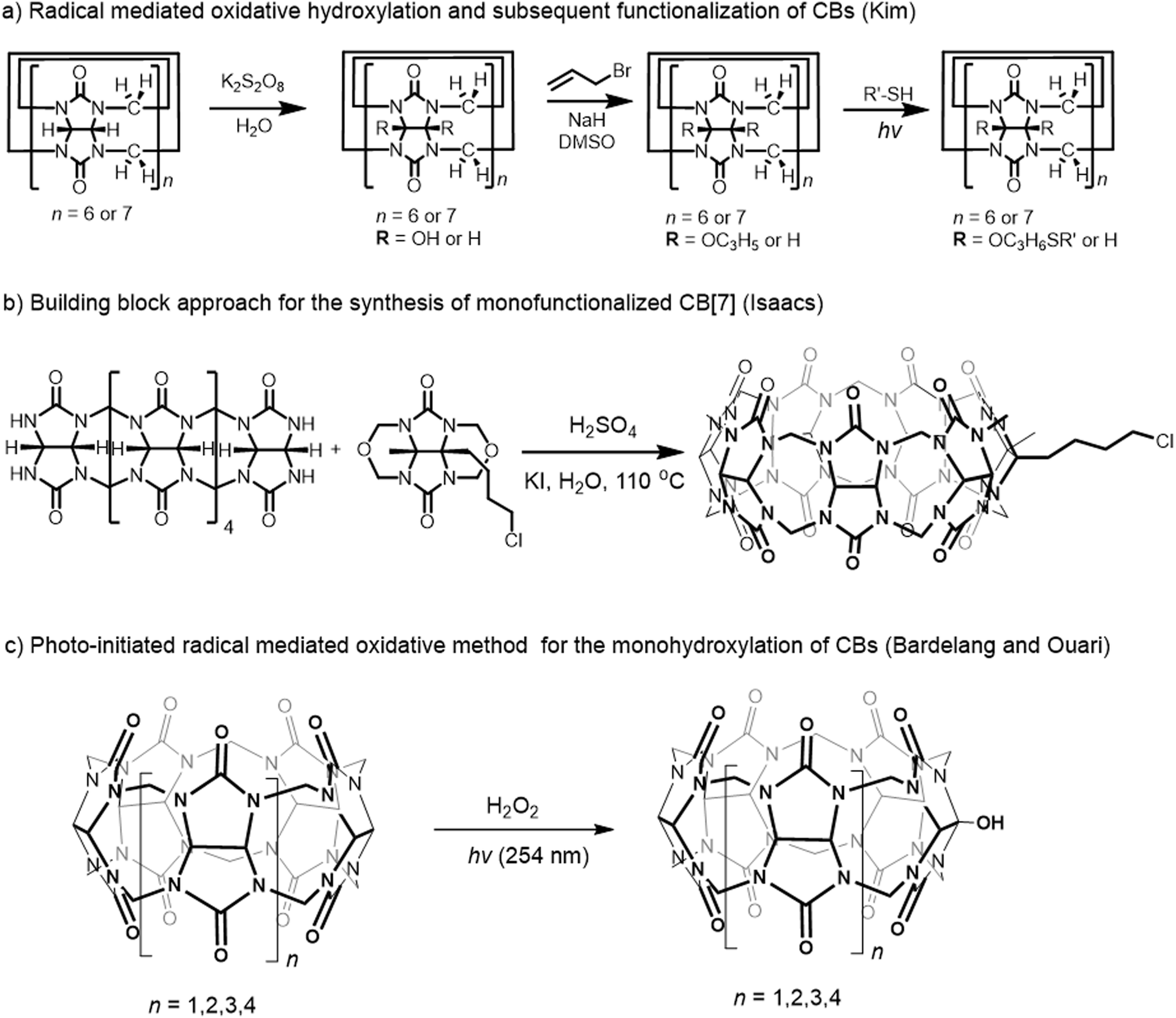 | ||
| Scheme 1 Synthetic methods developed for the functionalization of CBs. | ||
Until recently, the method introduced by our group has been the only direct route to obtain functionalized CB[7] compounds. However, Isaacs and co-workers recently reported a building-block approach to obtain monofunctionalized CB[7] derivatives (Scheme 1).97 This approach involves the condensation of an acyclic glycoluril hexamer with glycoluril bis(cyclic ethers) to produce monofunctionalized CB[7] which can be further transformed into tailor-made CB[7] derivatives for various applications. In addition, very recently Bardelang, Ouari, and co-workers have reported another oxidative method to synthesize high purity monohydroxy-CBs in good yield using hydrogen peroxide and UV light.98 This method relying on a controlled generation of hydroxyl radicals from hydrogen peroxide seems useful in the rapid generation of monohydroxy derivatives of the main members of the CB[n] family in high yield (>90%). By taking the advantage of progress in functionalization of CB[7], a number of applications of CB[7] and its functional derivatives are reported which will be covered in the following sections.
Immobilization of biomolecules on a solid surface
Developing a robust yet convenient method to link biomolecules to predefined locations on a reactive surface is of great importance in biotechnology and biochemical applications.99–101 Site-specific chemical strategies, as opposed to random attachment, provide precise control over immobilizing biomolecules in homogeneously oriented layers, and yield improved performance of biochips.102–107 For the immobilization of structurally sensitive biomolecules, mild reaction conditions compatible with physiological conditions, and rapid and quantitative conversion are essential. Biotin–streptavidin pair is one of the most widely used tools for such applications because of their strong and specific interaction. However, it suffers some shortcomings including denaturation by organic solvents or elevated temperatures. Right after the measurement of the exceptionally high affinity of ferrocenemethylammonium (F3) toward CB[7] by Isaacs41 and our group,40 we immediately realized that the ultrastable host–guest complex can be utilized as a replacement for the biotin–avidin pair in many applications where the latter has been widely used. The synthetic system has advantages including resistance to harsh conditions such as elevated temperature or unwanted enzymatic degradation, easiness to handle and manipulate, long-term durability due to the usage of stable synthetic molecules, and redox-active nature of the ferrocene unit, which allows modulation of binding strength.With this idea in mind, we first decided to develop a novel method to immobilize proteins on gold using the ultrastable F3@CB[7] (FA@CB[7]) complexes, and explore their applications as a biosensor.80 The surface immobilization of CB[7] was achieved by olefin metathesis reaction between allyloxy CB[7] and a vinyl-terminated mixed self-assembled monolayer (SAM) on gold. Ferrocene-conjugated proteins can directly interact with CB[7] moieties on the surface (Fig. 5). We chose glucose oxidase (GOx), one of the most well-known redox proteins extensively studied for glucose sensor applications, as a model protein and the “ferrocenylated” GOx (FA-GOx) was prepared by simple coupling reaction. The protein was immobilized on the CB[7]-anchored gold substrate by direct immersion of the substrate in a solution of FA-GOx. The surface plasmon resonance (SPR) technique confirmed that FA-GOx was anchored with a surface density of 1.1 × 1012 mol cm−2, which is approximately 5.5 times higher than that of GOx nonspecifically bound on gold. The protein immobilized on the surface was utilized as a glucose sensor with millimolar sensitivity as the catalytic current increases linearly with increasing analyte concentration.
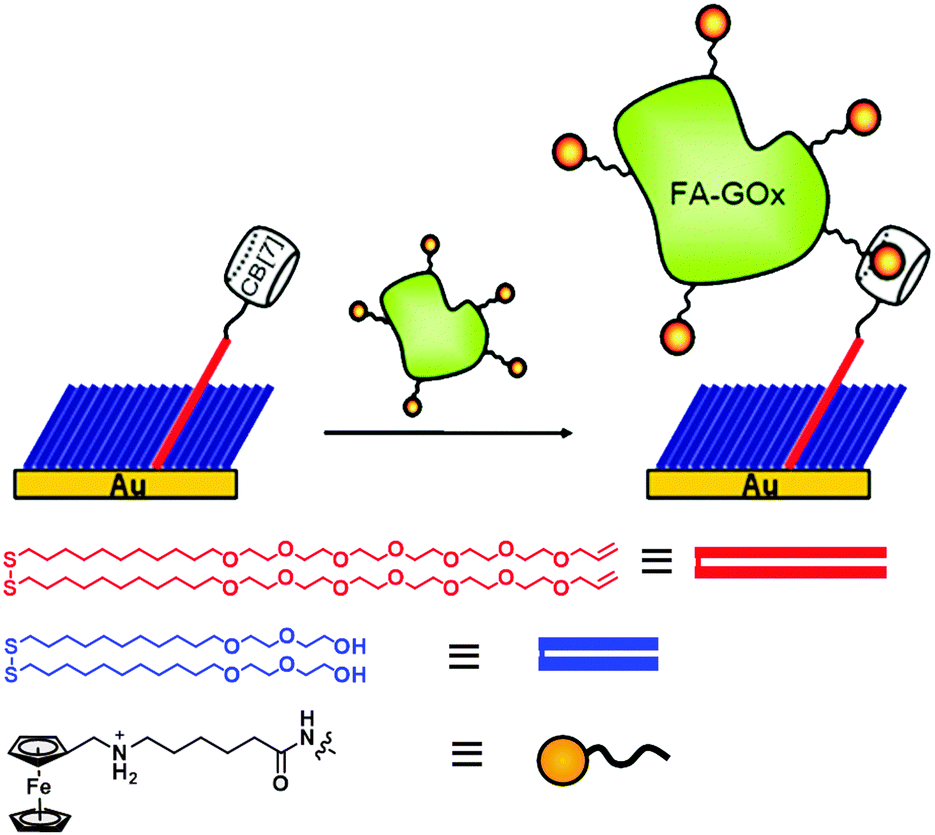 | ||
| Fig. 5 Immobilization of ferrocenylated proteins on a gold surface using an ultrastable synthetic binding pair system. Reprinted with permission from ref. 80. Copyright 2015 American Chemical Society. | ||
More recently, we extended this platform utility to immobilize aptamers for sensor applications.108 By taking advantage of the fact that CB[7] readily forms a self-assembled monolayer (CB[7]SAM) on gold,109 we developed a new SPR-based aptasensor chip which can be easily prepared by simple addition of FA-aptamers to CB[7]SAM (Fig. 6). A concentration dependent sensor response was observed when a target protein (thrombin) was injected into a channel of the sensor chip; a nanomolar (sub-ppm) level of sensitivity to thrombin was achieved. Brunsveld and others utilized the same strategy to immobilize different proteins and tested their applications (Fig. 6).110–112 Their method allowed us to print stable protein monolayers in well-defined formats with controlled protein orientation and its release from the surface by treating an excess amount of a competitive guest such as (ferrocenylmethyl)trimethyl-ammonium iodide. There is a recent report on supramolecular control of endothelial cell adhesion via synthetic integrin binding RGD peptide-ferrocene (Fc-cRGD) conjugates that were immobilized onto CB[7] coated gold surfaces.111 Supramolecular coating with Fc-cRGD resulted in efficient adhesion and growth of human umbilical vein endothelial cells (hUVECs) on the surfaces as evidenced by the increase in the surface coverage and cell density after 24 h. They also achieved patterned adhesion of hUVECs using glass substrates patterned with gold arrays. In general, these reports suggest that CB[7]–FA pairs can be applied to reversible immobilization of different biomolecules on solid surfaces.
 | ||
| Fig. 6 General strategy for the immobilization of biomolecules on a self-assembled monolayer surface using CB[7]. Reprinted with permission from ref. 108. Copyright 2015 The Royal Society of Chemistry. | ||
Plasma membrane protein fishing
The isolation and characterization of plasma membrane proteins (PMPs) and their functions are critical for understanding cell signaling and the effects of stimulation with various signal molecules. However, it is highly challenging because of PMPs inherent hydrophobicity and heterogeneity.113 A well-known method for isolation of membrane proteins is via the use of affinity enrichment, where cell surface membrane proteins are biotinylated on amino acid residues located in exterior domains and subsequently enriched using streptavidin beads.16,114,115 However, this method has some intrinsic drawbacks, including low yield, contamination with endogenously biotinylated proteins, and interference by streptavidin detached from beads.15–17,116–118 There have been new approaches for protein purification using synthetic host–guest pairs but they suffered low binding affinities (K < 105 m−1) and poor selectivity.119,120 To address these issues, we developed a new supramolecular fishing method utilizing ultrastable and highly selective synthetic binding pair, a CB[7]–FA system (Fig. 7).90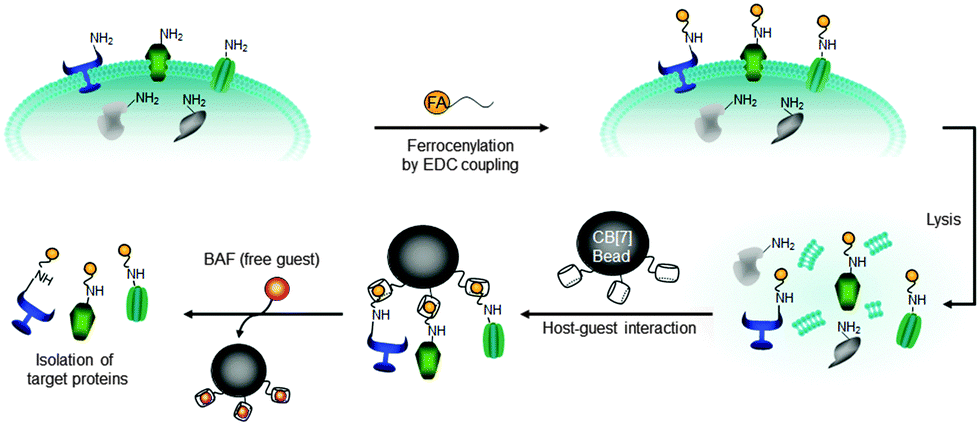 | ||
| Fig. 7 Strategy for the isolation of plasma membrane proteins using an ultrastable synthetic host–guest binding pair system. Reprinted with permission from ref. 90. Copyright 2011, Nature Publishing Group. | ||
CB[7]-conjugated sepharose beads (CB[7] beads) were utilized to capture FA-labelled proteins selectively from heterogeneous protein mixtures of various mammalian cells including rat fibroblast (Rat-1) and HEK 293 cells. The capture efficiency for plasma membrane proteins (EGFR, InsR, caveolin and integrin alpha 6) is much higher compared to cytoplasmic proteins (histone H3, cyclin B, Cpn60 and AMPK). In addition, the CB[7] system is not affected by the contaminations from endogenously biotinylated proteins and the captured proteins were easily recovered from the beads at room temperature by treating a strong competitor such as 1′,1-bis(trimethylammoniomethyl)ferrocene (BAF), which cannot be achieved by the streptavidin–biotin-based method. These results demonstrated another promising biological application of the ultrastable host–guest pair system – efficient isolation and purification of proteins. This work may be extended to the selective isolation of cytoplasmic proteins, which is currently under investigation.
Supramolecular Velcro system
Another interesting application of high affinity host–guest pairs is the development of an underwater adhesive system. There are growing needs for new adhesion systems that have high strength in water, repeated attachment and switchability on demand for biological applications such as surgery sutures and repairing living tissue. Previous efforts to create underwater adhesives have largely focused on biomimetic polymers that are secreted by marine organisms such as mussels, sandcastle worms, barnacles, and sea urchins.121–125 However, these efforts require curing agents and/or covalent crosslinking that irreversibly crosslink the residues to the surfaces. Another significant problem in developing underwater adhesive surfaces is how to expel the water molecules from the region between the two interacting surfaces. Since the formation of extremely stable complexes between CB[7] and hydrophobic guests is strongly associated with the transfer of water molecules to the bulk solution,47 we utilized the ultrastable host–guest system to develop functionalized surfaces with strong underwater adhesion properties, which work like a hook and loop fastener, Velcro®. Our system comprises of two complementary silicon wafer surfaces: a ‘loop’ surface that is functionalized with CB[7]; and a ‘hook’ surface that is covered with FA molecules (Fig. 8).96 | ||
| Fig. 8 Supramolecular Velcro® or hook-and-loop strategy for underwater adhesion based on CB[7]- and FA-modified surfaces (above). Reprinted with permission from ref. 96. Copyright 2013 Wiley-VCH Verlag. | ||
We deposited a layer of branched polyethyleneimine (PEI) on epoxy-functionalized silicon surfaces to generate an amine-terminated surface, which in turn reacts with ferrocenecarboxaldehyde to form ferrocenylmethylamino (FA) groups. A complementary surface was prepared by reaction of thiol groups on surface with monoallyloxy-CB[7] via thiol–allyl click reaction. When the two complementary surfaces are pushed together under water, the FA moiety binds tightly inside the CB[7] cavity which is enough to “glue” them together with enough strength to lift a 2 kg weight. The lap shear adhesion strength in air is much higher than that of commercially available adhesives, including double-sided tapes and traditional Velcro®. Interestingly, the adhesive strength can be tuned by decreasing the surface density of FA on one of the interacting surfaces. Unlike existing adhesives, our supramolecular Velcro is not only mechanically reversible but also chemically switchable. The binding affinity of the FA@CB[7] complex can be modulated by the oxidation of the ferrocenyl group.126 The surfaces can be either mechanically peeled apart or separated chemically with sodium hypochlorite. Exposure to oxidizing agents decreases the adhesive strength between the two surfaces. However, after exposure to a reducing agent, ascorbic acid, the adhesive strength is mostly recovered, although it does not reach the original level, probably because of the partial decomposition of the ferrocenyl groups in the oxidized state. The redox properties of the CB[7]–FA complex afford an interesting method to modulate the strength of adhesion, which is not available in any other adhesive systems. However, our material lost some of its adhesion strength after repeated cycles of binding and chemical unbinding. We think that an electrochemical method may offer a more efficient alternative to a chemical redox agent for switchable underwater adhesion. Recently, Chinese researchers reported another underwater adhesive based on a similar principle using the FA and β-CD systems.127 Overall, our work translates a molecular recognition event into macroscopic adhesion between surfaces that function in water. The non-cytotoxic nature of these host and guest molecules permits potential biological applications, but an in vivo adhesion test is required to strengthen our claims.
Development of a high-affinity host–guest FRET pair and its application in single vesicle content mixing assays
Fluorescence resonance energy transfer (FRET) has been widely used as a spectroscopic technique in various areas such as structural elucidation of biological molecules and their interactions, in vitro assays, and in vivo monitoring in cellular research. Among a series of FRET systems, host–guest FRET pairs using β-CD have recently attracted considerable attention as chemical sensors and biosensors.128–130 However, because of the moderate affinity between β-CD and guest molecules, it is not desired to use these FRET pairs as a single molecule analysis probe. Therefore, developing a host–guest FRET system with an exceptional affinity may greatly enhance the chances of exploring these pairs in single molecule analysis. Very recently, we developed a high-affinity host–guest FRET pair by utilizing the ultrastable CB[7]–adamantylamine complex.131 We synthesized the fluorophore-tagged CB[7]-Cy3 and Ad-Cy5, found that these dye-conjugated CB[7]-Ad pairs produced high FRET signals in aqueous solution upon host–guest binding (Fig. 9). | ||
| Fig. 9 Synthesis and characterization of CB[7]-Cy3 and Ad-Cy5 high affinity FRET pair. Reprinted with permission from ref. 131. Copyright 2015 American Chemical Society. | ||
We utilized this host–guest FRET pair to study the challenging SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptors) mediated membrane fusion. Protein-mediated membrane fusion is a common biological process and plays a principal role in a variety of cellular activities, such as vesicle trafficking, viral infection, exocytosis and neurotransmitter.132,133 A majority of vesicle fusion is mediated by SNAREs which act as molecular tethers between the two fusing bodies. Single vesicle in vitro fusion assays using SNARE-reconstituted vesicles have played a vital role in elucidating the mechanisms of SNARE-mediated vesicle fusion by allowing controlled observations which are not possible by in vivo studies.134–137 However, the existing assays have limitations such as low sensitivity (signal to noise ratio) and limited ability to detect transient fusion pore openings.
We thus decided to develop a novel FRET-based single-vesicle content assay using our host–guest FRET pair, which combines the benefits of small molecular probes with the high signal-to-noise ratio benefits of FRET based techniques, to study the mechanism of synaptic vesicle fusion mediated by SNARE proteins (Fig. 10).131 We first encapsulated CB[7]-Cy3 and Ad-Cy5 in SNARE protein reconstituted donor and accepter vesicles (v-vesicle and t-vesicle, respectively) to produce CB[7]-Cy3@v-vesicle and Ad-Cy5@t-vesicle, respectively. We then performed a single-vesicle content mixing assay with these vesicles using a total internal reflection fluorescence microscope (TIRF). The acceptor vesicle, Ad-Cy5@t-vesicle, was immobilized on a quartz surface and a donor vesicle, CB[7]-Cy3@v-vesicle, was injected into the flow chamber of our analysis set-up. Docking of a donor vesicle onto the acceptor vesicle occurred as evidenced by the sudden appearance of Cy3 emission in the time trace. The pore opening led to mixing of the content molecules of the two opposite vesicles to form a strong host–guest complex, which generated the FRET signal of Cy5 emission. The high stability of the CB[7]-Ad complex allowed our technique to be compatible with a wide range of reaction conditions during both vesicle preparation and fusion.
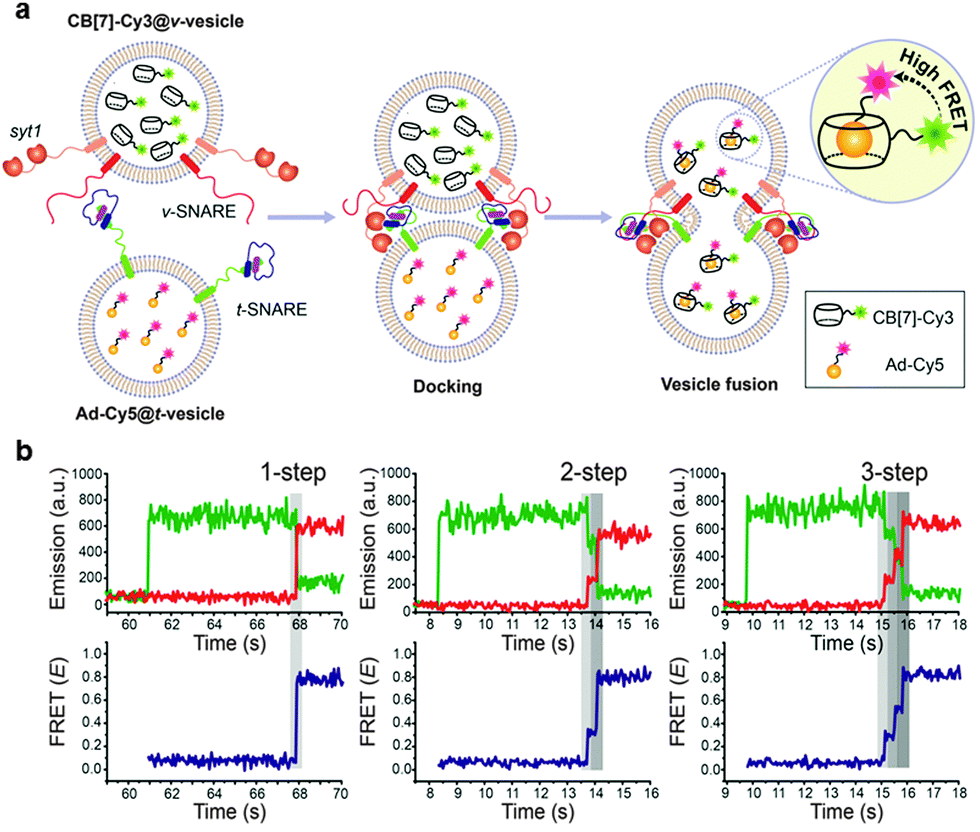 | ||
| Fig. 10 Schematic illustration of the SNARE-mediated content mixing using a high affinity host–guest binding FRET pair (a) and observation of multi-step fusion pore opening and closure dynamics (b). Reprinted with permission from ref. 131. Copyright 2015 American Chemical Society. | ||
The most important finding of our assay is the direct detection of multistep fusion flickering events, for the first time, in an in vitro content mixing assay. It is generally believed that SNARE fusion proceeds through intermediates, which may lead directly to the formation of a fusion pore followed by expansion of the pore or it can open and close in a transient manner (flickering). The merits of our system allowed us to observe the fusion pore flickering events and the intermediate steps involved in the fusion process, which was unsuccessful with existing techniques.137,138 In major traces, full fusion was observed with the appearance of a plateau corresponding to the FRET efficiency of complete content mixing without any intermediates. In other cases, however, the initial pore formation state is followed by one or more additional plateaus with a FRET efficiency that is intermediate between initial docking and full fusion. This is a characteristic behavior of flickering in which a transient fusion pore opens and closes quickly and hence does not allow full mixing of the contents at once.
This work demonstrates the power of the synthetic system in the field of complex biochemical analysis. Furthermore, this powerful tool may allow us to investigate further to clarify the roles of individual components in the membrane fusion, which is difficult to achieve at the cellular level. This synthetic “host–guest FRET pair” with extremly strong binding affinity and specificity may be applied to various chemical and biological studies as a supramolecular beacon.
Recognition-mediated activation of therapeutic gold nanoparticles inside cells
Apart from the utility of high affinity host–guest pairs in the separation of proteins and vesicle fusion, Rotello, Isaacs and co-workers exploited it as a trigger in therapeutic systems. Their system consists of complementary diaminohexane-terminated gold nanoparticles (AuNP–NH2) and CB[7] to form a non-toxic nanoparticle assembly that is readily taken up by cells (Fig. 11).139 Intracellular removal of CB[7] from the nanoparticle surface using an orthogonal competitor guest A2 results in the endosomal escape of AuNP–NH2, which activates the cytotoxicity of the amine-tethered gold nanoparticles and thereby causes cell death. This strategy for triggering therapeutic systems has some advantages in dosage control and potential utility in dual-targeting therapies. The authors used ∼2.5 nm sized AuNP–NH2 as a therapeutic component of the system, which features a self-assembled monolayer of diaminohexane-terminated thiol ligands. It is believed that the terminal diaminohexane moiety renders cytotoxicity to the nanoparticle and also serves as a recognition unit for the formation of a host–guest inclusion complex with CB[7]. Cells treated with AuNP–NH2 and CB[7]–AuNP–NH2 alone exhibited 34% and 100% cell viability after 24 h of incubation, respectively. The addition of A2 resulted in the formation of more favorable A2@CB[7] complexes, which triggered the release of CB[7] from the nanoparticles. After 24 h of incubation, AuNP–NH2 was dispersed in the cytosol whereas CB[7]–AuNP–NH2 remained in the endosome. Remarkably, the incubation of CB[7]–AuNP–NH2 treated cells with A2 resulted in the escape of a substantial number of nanoparticles into the cytosol from the endosome. This result demonstrates the use of synthetic host–guest chemistry to provide triggered activation of a therapeutic system by competitive complexation and provides a potential strategy for the construction of synthetic host–guest systems using CB[7], which has an ability to form a strong complex within living cells. This is an interesting engineering of host–guest systems for the regulation of therapeutics that can function inside the cell. Based on the same principle, the authors also reported the regulation of exocytosis of nanoparticles using CB[7]-based host–guest chemistry.140 This approach provides a potential strategy for prolonged retention of drug carriers within endosomes, enabling a sustained therapeutic effect of the carriers.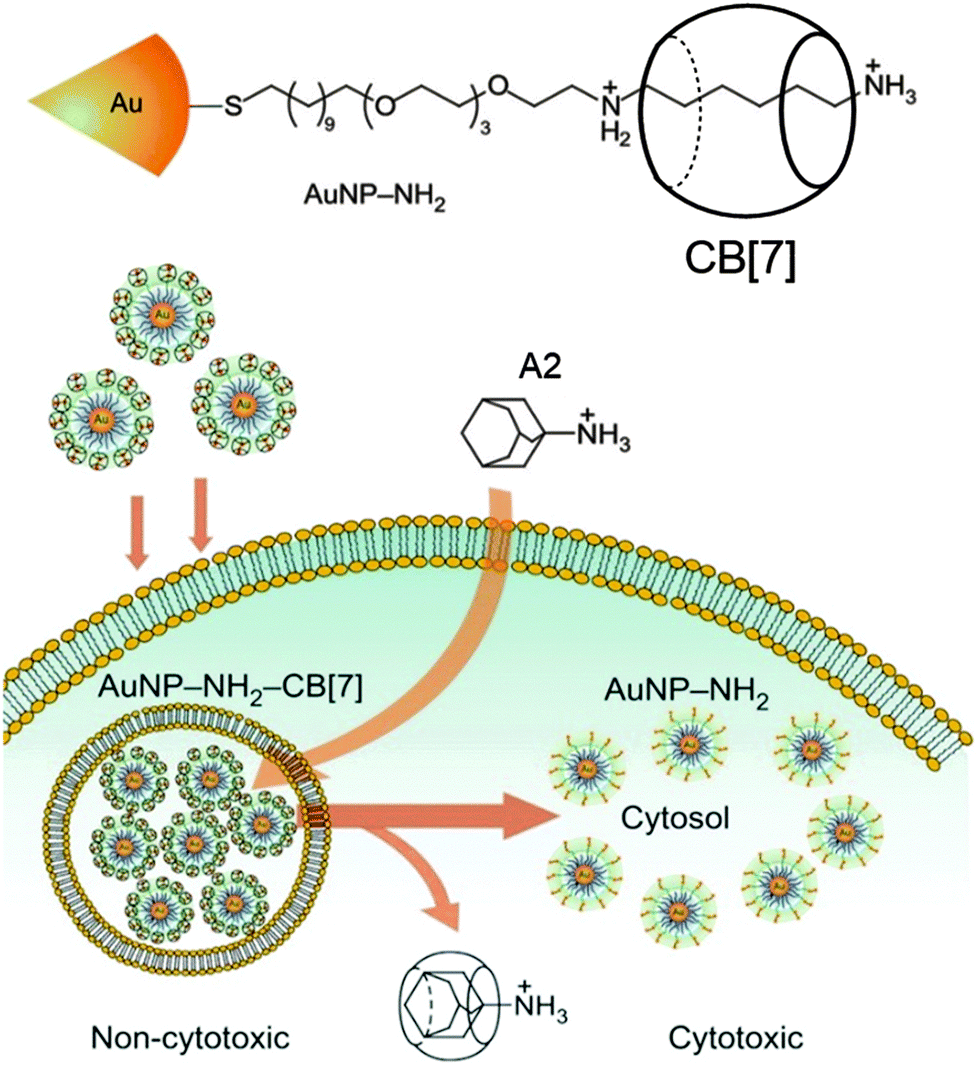 | ||
| Fig. 11 Schematic illustration of the use of intracellular host–guest complexation to trigger gold nanoparticle (AuNP–NH2) cytotoxicity. Reprinted with permission from ref. 139. Copyright 2010, Nature Publishing Group. | ||
Biological catalysis regulated by CB[7]
It is important to note that the high affinity and selectivity of CB[7] towards selected guest molecules can be comparable to those typically observed in the interaction of proteins and antibodies toward their cognate ligands.141 By taking this merit into consideration, Isaacs and co-workers demonstrated the influence of CB[7] on the behavior of biological receptors toward their guests and thereby change in their biological activity (Fig. 12).142 They explored the self-sorting capability of two-faced guests for the regulation of the catalytic activity of bovine carbonic anhydrase (BCA). The active site of the enzyme was initially occupied by a two-faced inhibitor that contains benzenesulfonamide (enzyme inhibitor unit) and trimethylsilylmethylammonium group (CB[7] binding unit). The initial enzymatic activity was recovered by the addition of CB[7], which forms a 1:1 host–guest complex with trimethylsilylmethylammonium part (Ka ∼ 109 M−1) of the two-faced guests to release the inhibitor from the enzyme. However, the enzyme activity was inhibited again by the addition of adamantaneammonium derivative, which triggered the dissociation of the CB[7]·enzyme inhibitor resulting in the release of the free two-faced guest, which then re-occupied the catalytic site of the enzyme. Addition of CB[7] and A2 in an alternating sequence turned the enzymatic activity off and on over several additional cycles. One of the key controlling factors in this process is the relative binding affinity of the two-faced inhibitors toward enzyme versus CB[7]. The interesting aspect of this work is the ability of CB[7] to enhance the rate of dissociation of the enzyme·inhibitor by the transient formation of the enzyme·inhibitor·CB[7] ternary complexes. This strategy differs from the typical allosteric regulation of enzyme activity, in that direct competition between two receptors (enzyme and CB[7]) is involved. Overall, this finding demonstrates the advantage of CB[7] selectivity towards specific guest molecules and hence it may be possible to use this receptor to compete with and thereby control the activity of specific portions of the biological interaction networks.
 | ||
| Fig. 12 Schematic illustration of the control of enzymatic activity using CB[7] and an adamantane derivative. Reprinted with permission from ref. 142. Copyright 2010 American Chemical Society. | ||
Very recently, Rotello and co-workers have also explored the benefit of high affinity CB[7]-Ad pairs for the supramolecular regulation of bioorthogonal catalysis in cells.143 They developed a protein-sized bioorthogonal nanozyme through the encapsulation of hydrophobic transition metal catalysts into the monolayer of water-soluble gold nanoparticles. The activity of the catalyst was reversibly controlled by using CB[7] as the ‘gate-keeper’ onto the monolayer surface which provides a biomimetic control mechanism that mimics the allosteric regulation of natural enzymes.
5. Conclusions
This review summarizes the high affinity host–guest interactions between CB[7] and various guest molecules, as well as their applications in constructing various supramolecular systems. We have described the evolution of different guest molecules that bind CB[7] to water with affinities equivalent to or higher than those of the highest-affinity protein-small molecule systems. In general, the guest binding affinity and selectivity of CB[7] are higher than those of any other synthetic receptors reported to date. These high affinity pairs offer numerous advantages including an attomolar dissociation constant and tunable complexation kinetics over other conventional macrocycles. CB[7] shows remarkable binding characteristics based on the guest size, shape, length, and chemical functionalities. The positive entropy changes from the extensive dehydration of the host and guest upon complexation and thereby overcoming the enthalpy–entropy compensation critically assists in achieving high binding affinity. The ultrastable host–guest complexes are unique as they rely on monovalency to achieve such high binding affinity. While there have been continuing efforts to develop new high affinity guest molecules, their implementation in challenging applications has rapidly unfolded during the past decade. The inherent high affinity of CB[7] paves its way to deal with challenging applications in materials science, biology, and medicine. With exceptional stability, chemical robustness, biocompatibility, and easy handling, these complexes may replace the biotin–(strept)avidin system in diverse areas of research and therefore the application perspectives of these complexes are looking very bright. Overall, CB[7] brings synthetic receptor systems very close to their natural counterparts with new hope in the development of novel synthetic systems which function equally well or better than their biological counterparts. Furthermore, the recent progress in this field clearly suggests that the strong host–guest complexes are making the transition from a simple research interest to modular components of chemical systems with important practical applications.Acknowledgements
We gratefully appreciate the financial support from the Institute for Basic Science (IBS) [IBS-R007-D1]. We thank all the past and present co-workers who have contributed to this research as cited. We also thank Professors Y. Inoue, A. E. Kaifer, L. Isaacs, and M. K. Gilson for fruitful collaboration over the years.References
- M. J. Swamy, Biochem. Mol. Biol. Int., 1995, 36, 219–225 CAS.
- B. Kuhn and P. A. Kollman, J. Am. Chem. Soc., 2000, 122, 3909–3916 CrossRef CAS.
- N. M. Green, Methods Enzymol., 1990, 184, 51–67 CAS.
- M. Wilchek and E. A. Bayer, Methods Enzymol., 1990, 184, 14–45 CAS.
- T. J. Park, K. B. Lee, S. J. Lee, J. P. Park, Z. W. Lee, S. K. Choi, H. C. Jung, J. G. Pan, S. Y. Lee and I. S. Choi, J. Am. Chem. Soc., 2004, 126, 10512–10513 CrossRef CAS PubMed.
- J. Ladd, C. Boozer, Q. Yu, S. Chen, J. Homola and S. Jiang, Langmuir, 2004, 20, 8090–8095 CrossRef CAS PubMed.
- S. C. Tao and H. Zhu, Nat. Biotechnol., 2006, 24, 1253–1254 CrossRef CAS PubMed.
- O. H. Laitinen, H. R. Nordlund, V. P. Hytonen and M. S. Kulomaa, Trends Biotechnol., 2007, 25, 269–277 CrossRef CAS PubMed.
- G. N. Ostojic and M. C. Hersam, Small, 2012, 8, 1840–1845 CrossRef CAS PubMed.
- A. Chilkoti, P. H. Tan and P. S. Stayton, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 1754–1758 CrossRef CAS.
- O. H. Laitinen, K. J. Airenne, A. T. Marttila, T. Kulik, E. Porkka, E. A. Bayer, M. Wilchek and M. S. Kulomaa, FEBS Lett., 1999, 461, 52–58 CrossRef CAS.
- A. T. Marttila, O. H. Laitinen, K. J. Airenne, T. Kulik, E. A. Bayer, M. Wilchek and M. S. Kulomaa, FEBS Lett., 2000, 467, 31–36 CrossRef CAS.
- O. H. Laitinen, H. R. Nordlund, V. P. Hytonen, S. T. Uotila, A. T. Marttila, J. Savolainen, K. J. Airenne, O. Livnah, E. A. Bayer, M. Wilchek and M. S. Kulomaa, J. Biol. Chem., 2003, 278, 4010–4014 CrossRef CAS PubMed.
- M. Howarth, D. J. Chinnapen, K. Gerrow, P. C. Dorrestein, M. R. Grandy, N. L. Kelleher, A. El-Husseini and A. Y. Ting, Nat. Methods, 2006, 3, 267–273 CrossRef CAS PubMed.
- S. R. Jaffrey, H. Erdjument-Bromage, C. D. Ferris, P. Tempst and S. H. Snyder, Nat. Cell Biol., 2001, 3, 193–197 CrossRef CAS PubMed.
- W. Zhang, G. Zhou, Y. Zhao, M. A. White and Y. Zhao, Electrophoresis, 2003, 24, 2855–2863 CrossRef CAS PubMed.
- R. Takechi, A. Taniguchi, S. Ebara, T. Fukui and T. Watanabe, J. Nutr., 2008, 138, 680–684 CAS.
- W. L. Mock, in Comprehensive Supramolecular Chemistry, Molecular Recognition: Receptors for Molecular Guests, ed. F. Vögtle, Pergamon Press, Oxford, UK, 1996, vol. 2, pp. 477–493 Search PubMed.
- C. L. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2004, 126, 11408–11409 CrossRef CAS PubMed.
- A. V. Davis and K. N. Raymond, J. Am. Chem. Soc., 2005, 127, 7912–7919 CrossRef CAS PubMed.
- J. Rebek, Jr., Angew. Chem., Int. Ed., 2005, 44, 2068–2078 CrossRef PubMed.
- K. Takaoka, M. Kawano, M. Tominaga and M. Fujita, Angew. Chem., Int. Ed., 2005, 44, 2151–2154 CrossRef CAS PubMed.
- R. Custelcean, Chem. Soc. Rev., 2014, 43, 1813–1824 RSC.
- F. L. Dickert, Sensors, 2014, 14, 22525–22531 CrossRef CAS PubMed.
- W. L. Mock, Top. Curr. Chem., 1995, 175, 1–24 CrossRef CAS.
- J. W. Lee, S. Samal, N. Selvapalam, H. J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267–279 RSC.
- X. J. Cheng, L. L. Liang, K. Chen, N. N. Ji, X. Xiao, J. X. Zhang, Y. Q. Zhang, S. F. Xue, Q. J. Zhu, X. L. Ni and Z. Tao, Angew. Chem., Int. Ed., 2013, 52, 7252–7255 CrossRef CAS PubMed.
- A. E. Kaifer, W. Li and S. Yi, Isr. J. Chem., 2011, 51, 496–505 CrossRef CAS PubMed.
- E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, RSC Adv., 2012, 2, 1213–1247 RSC.
- J. Lü, J.-X. Lin, M.-N. Cao and R. Cao, Coord. Chem. Rev., 2013, 257, 1334–1356 CrossRef PubMed.
- X. L. Ni, X. Xiao, H. Cong, L. L. Liang, K. Cheng, X. J. Cheng, N. N. Ji, Q. J. Zhu, S. F. Xue and Z. Tao, Chem. Soc. Rev., 2013, 42, 9480–9508 RSC.
- L. Isaacs, Acc. Chem. Res., 2014, 47, 2052–2062 CrossRef CAS PubMed.
- X. L. Ni, X. Xiao, H. Cong, Q. J. Zhu, S. F. Xue and Z. Tao, Acc. Chem. Res., 2014, 47, 1386–1395 CrossRef CAS PubMed.
- L. Peng, A. Feng, M. Huo and J. Yuan, Chem. Commun., 2014, 50, 13005–13014 RSC.
- N. Vallavoju and J. Sivaguru, Chem. Soc. Rev., 2014, 43, 4084–4101 RSC.
- H. Yang, B. Yuan, X. Zhang and O. A. Scherman, Acc. Chem. Res., 2014, 47, 2106–2115 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- W. S. Jeon, K. Moon, S. H. Park, H. Chun, Y. H. Ko, J. Y. Lee, E. S. Lee, S. Samal, N. Selvapalam, M. V. Rekharsky, V. Sindelar, D. Sobransingh, Y. Inoue, A. E. Kaifer and K. Kim, J. Am. Chem. Soc., 2005, 127, 12984–12989 CrossRef CAS PubMed.
- S. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 15959–15967 CrossRef CAS PubMed.
- M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737–20742 CrossRef CAS PubMed.
- S. Moghaddam, C. Yang, M. Rekharsky, Y. H. Ko, K. Kim, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2011, 133, 3570–3581 CrossRef CAS PubMed.
- L. Cao, M. Šekutor, P. Y. Zavalij, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef CAS PubMed.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1918 CrossRef CAS PubMed.
- W. Ong and A. E. Kaifer, Organometallics, 2003, 22, 4181–4183 CrossRef CAS.
- F. Biedermann, V. D. Uzunova, O. A. Scherman, W. M. Nau and A. De Simone, J. Am. Chem. Soc., 2012, 134, 15318–15323 CrossRef CAS PubMed.
- C. N. Nguyen, T. K. Young and M. K. Gilson, J. Chem. Phys., 2012, 137, 044101 CrossRef PubMed.
- F. Biedermann, M. Vendruscolo, O. A. Scherman, A. De Simone and W. M. Nau, J. Am. Chem. Soc., 2013, 135, 14879–14888 CrossRef CAS PubMed.
- F. Biedermann, W. M. Nau and H. J. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11158–11171 CrossRef CAS PubMed.
- Y. Chen, A. Klimczak, E. Galoppini and J. V. Lockard, RSC Adv., 2013, 3, 1354–1358 RSC.
- M. Šekutor, K. Molčanov, L. Cao, L. Isaacs, R. Glaser and K. Mlinarić-Majerski, Eur. J. Org. Chem., 2014, 2533–2542 CrossRef PubMed.
- C. Marquez and W. M. Nau, Angew. Chem., Int. Ed., 2001, 40, 4387–4390 CrossRef CAS.
- Y. Pazy, T. Kulik, E. A. Bayer, M. Wilchek and O. Livnah, J. Biol. Chem., 2002, 277, 30892–30900 CrossRef CAS PubMed.
- C. Márquez, R. R. Hudgins and W. M. Nau, J. Am. Chem. Soc., 2004, 126, 5806–5816 CrossRef PubMed.
- W. Ong and A. E. Kaifer, J. Org. Chem., 2004, 69, 1383–1385 CrossRef CAS PubMed.
- C. Marquez and W. M. Nau, Angew. Chem., Int. Ed., 2001, 40, 4387–4390 CrossRef CAS.
- W. M. Nau, A. Hennig and A. L. Koner, Fluorescence of Supermolecules, Polymers, and Nanosystems, Springer, 2008, pp. 185–211 Search PubMed.
- W. M. Nau, M. Florea and K. I. Assaf, Isr. J. Chem., 2011, 51, 559–577 CrossRef CAS PubMed.
- D. Sobransingh and A. E. Kaifer, Org. Lett., 2006, 8, 3247–3250 CrossRef CAS PubMed.
- M. Rekharsky and Y. Inoue, J. Am. Chem. Soc., 2000, 122, 4418–4435 CrossRef CAS.
- C.-E. Chang and M. K. Gilson, J. Am. Chem. Soc., 2004, 126, 13156–13164 CrossRef CAS PubMed.
- S. Moghaddam, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2009, 131, 4012–4021 CrossRef CAS PubMed.
- A. T. Fenley, N. M. Henriksen, H. S. Muddana and M. K. Gilson, J. Chem. Theory Comput., 2014, 10, 4069–4078 CrossRef CAS PubMed.
- S. J. C. Lee, J. W. Lee, H. H. Lee, J. Seo, D. H. Noh, Y. H. Ko, K. Kim and H. I. Kim, J. Phys. Chem. B, 2013, 117, 8855–8864 CrossRef CAS PubMed.
- W. Saenger, Angew. Chem., Int. Ed., 1980, 19, 344–362 CrossRef PubMed.
- H. J. Schneider, Angew. Chem., Int. Ed., 1991, 30, 1417–1436 CrossRef PubMed.
- A. Flinn, G. C. Hough, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed., 1992, 31, 1475–1477 CrossRef PubMed.
- A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, J. Org. Chem., 2001, 66, 8094–8100 CrossRef CAS PubMed.
- J. Zhao, H. J. Kim, J. Oh, S. Y. Kim, J. W. Lee, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 4233–4235 CrossRef CAS.
- A. I. Day, R. J. Blanch, A. Coe and A. P. Arnold, J. Inclusion Phenom. Macrocyclic Chem., 2002, 43, 247–250 CrossRef CAS.
- H. Isobe, S. Sato and E. Nakamura, Org. Lett., 2002, 4, 1287–1289 CrossRef CAS PubMed.
- C. A. Burnett, J. Lagona, A. Wu, J. A. Shaw, D. Coady, J. C. Fettinger, A. I. Day and L. Isaacs, Tetrahedron, 2003, 59, 1961–1970 CrossRef CAS.
- C. A. Burnett, D. Witt, J. C. Fettinger and L. Isaacs, J. Org. Chem., 2003, 68, 6184–6191 CrossRef CAS PubMed.
- A. I. Day, A. P. Arnold and R. J. Blanch, Molecules, 2003, 8, 74–84 CrossRef CAS PubMed.
- K. Jansen, A. Wego, H.-J. Buschmann, E. Schollmeyer and D. Döpp, Des. Monomers Polym., 2003, 6, 43–55 CrossRef CAS PubMed.
- S. Y. Jon, N. Selvapalam, D. H. Oh, J. K. Kang, S. Y. Kim, Y. J. Jeon, J. W. Lee and K. Kim, J. Am. Chem. Soc., 2003, 125, 10186–10187 CrossRef CAS PubMed.
- S. Sasmal, M. K. Sinha and E. Keinan, Org. Lett., 2004, 6, 1225–1228 CrossRef CAS PubMed.
- E. R. Nagarajan, D. H. Oh, N. Selvapalam, Y. H. Ko, K. M. Park and K. Kim, Tetrahedron Lett., 2006, 47, 2073–2075 CrossRef CAS PubMed.
- I. Hwang, K. Baek, M. Jung, Y. Kim, K. M. Park, D. W. Lee, N. Selvapalam and K. Kim, J. Am. Chem. Soc., 2007, 129, 4170–4171 CrossRef CAS PubMed.
- D. Kim, E. Kim, J. Kim, K. M. Park, K. Baek, M. Jung, Y. H. Ko, W. Sung, H. S. Kim, J. H. Suh, C. G. Park, O. S. Na, D. K. Lee, K. E. Lee, S. S. Han and K. Kim, Angew. Chem., Int. Ed., 2007, 46, 3471–3474 CrossRef CAS PubMed.
- J. Kim, Y. Ahn, K. M. Park, Y. Kim, Y. H. Ko, D. H. Oh and K. Kim, Angew. Chem., Int. Ed., 2007, 46, 7393–7395 CrossRef CAS PubMed.
- K. M. Park, K. Suh, H. Jung, D. W. Lee, Y. Ahn, J. Kim, K. Baek and K. Kim, Chem. Commun., 2009, 71–73 RSC.
- K. Baek, Y. Kim, H. Kim, M. Yoon, I. Hwang, Y. H. Ko and K. Kim, Chem. Commun., 2010, 46, 4091–4093 RSC.
- D. Kim, E. Kim, J. Lee, S. Hong, W. Sung, N. Lim, C. G. Park and K. Kim, J. Am. Chem. Soc., 2010, 132, 9908–9919 CrossRef CAS PubMed.
- E. Kim, D. Kim, H. Jung, J. Lee, S. Paul, N. Selvapalam, Y. Yang, N. Lim, C. G. Park and K. Kim, Angew. Chem., Int. Ed., 2010, 49, 4405–4408 CrossRef CAS PubMed.
- S. K. Kim, K. M. Park, K. Singha, J. Kim, Y. Ahn, K. Kim and W. J. Kim, Chem. Commun., 2010, 46, 692–694 RSC.
- K. M. Park, D. W. Lee, B. Sarkar, H. Jung, J. Kim, Y. H. Ko, K. E. Lee, H. Jeon and K. Kim, Small, 2010, 6, 1430–1441 CrossRef CAS PubMed.
- H. Jung, K. M. Park, J. A. Yang, E. J. Oh, D. W. Lee, K. Park, S. H. Ryu, S. K. Hahn and K. Kim, Biomaterials, 2011, 32, 7687–7694 CrossRef CAS PubMed.
- D. W. Lee, K. M. Park, M. Banerjee, S. H. Ha, T. Lee, K. Suh, S. Paul, H. Jung, J. Kim, N. Selvapalam, S. H. Ryu and K. Kim, Nat. Chem., 2011, 3, 154–159 CrossRef CAS PubMed.
- K. M. Park, J. A. Yang, H. Jung, J. Yeom, J. S. Park, K. H. Park, A. S. Hoffman, S. K. Hahn and K. Kim, ACS Nano, 2012, 6, 2960–2968 CrossRef CAS PubMed.
- K. Baek, G. Yun, Y. Kim, D. Kim, R. Hota, I. Hwang, D. Xu, Y. H. Ko, G. H. Gu, J. H. Suh, C. G. Park, B. J. Sung and K. Kim, J. Am. Chem. Soc., 2013, 135, 6523–6528 CrossRef CAS PubMed.
- G. Yun, Z. Hassan, J. Lee, J. Kim, N. S. Lee, N. H. Kim, K. Baek, I. Hwang, C. G. Park and K. Kim, Angew. Chem., Int. Ed., 2014, 53, 6414–6418 CrossRef CAS PubMed.
- J. Kim, K. Baek, D. Shetty, N. Selvapalam, G. Yun, N. H. Kim, Y. H. Ko, K. M. Park, I. Hwang and K. Kim, Angew. Chem., Int. Ed., 2015, 54, 2693–2697 CrossRef CAS PubMed.
- J. Yeom, S. J. Kim, H. Jung, H. Namkoong, J. Yang, B. W. Hwang, K. Oh, K. Kim, Y. C. Sung and S. K. Hahn, Adv. Healthcare Mater., 2015, 4, 237–244 CrossRef CAS PubMed.
- Y. Ahn, Y. Jang, N. Selvapalam, G. Yun and K. Kim, Angew. Chem., Int. Ed., 2013, 52, 3140–3144 CrossRef CAS PubMed.
- B. Vinciguerra, L. Cao, J. R. Cannon, P. Y. Zavalij, C. Fenselau and L. Isaacs, J. Am. Chem. Soc., 2012, 134, 13133–13140 CrossRef CAS PubMed.
- M. M. Ayhan, H. Karoui, M. Hardy, A. Rockenbauer, L. Charles, R. Rosas, K. Udachin, P. Tordo, D. Bardelang and O. Ouari, J. Am. Chem. Soc., 2015, 137, 10238–10245 CrossRef CAS PubMed.
- M. Dufva and C. B. Christensen, Expert Rev. Proteomics, 2005, 2, 41–48 CrossRef CAS PubMed.
- S. F. Kingsmore, Nat. Rev. Drug Discovery, 2006, 5, 310–320 CrossRef CAS PubMed.
- D. Weinrich, P. Jonkheijm, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2009, 48, 7744–7751 CrossRef CAS PubMed.
- M. B. Soellner, K. A. Dickson, B. L. Nilsson and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 11790–11791 CrossRef CAS PubMed.
- P. C. Lin, S. H. Ueng, M. C. Tseng, J. L. Ko, K. T. Huang, S. C. Yu, A. K. Adak, Y. J. Chen and C. C. Lin, Angew. Chem., Int. Ed., 2006, 45, 4286–4290 CrossRef CAS PubMed.
- T. Govindaraju, P. Jonkheijm, L. Gogolin, H. Schroeder, C. F. Becker, C. M. Niemeyer and H. Waldmann, Chem. Commun., 2008, 3723–3725 RSC.
- L. S. Wong, F. Khan and J. Micklefield, Chem. Rev., 2009, 109, 4025–4053 CrossRef CAS PubMed.
- D. Weinrich, P. C. Lin, P. Jonkheijm, U. T. Nguyen, H. Schroder, C. M. Niemeyer, K. Alexandrov, R. Goody and H. Waldmann, Angew. Chem., Int. Ed., 2010, 49, 1252–1257 CrossRef CAS PubMed.
- Y. X. Chen, G. Triola and H. Waldmann, Acc. Chem. Res., 2011, 44, 762–773 CrossRef CAS PubMed.
- D. W. Lee, K. M. Park, B. Gong, D. Shetty, J. K. Khedkar, K. Baek, J. Kim, S. H. Ryu and K. Kim, Chem. Commun., 2015, 51, 3098–3101 RSC.
- Q. An, G. Li, C. Tao, Y. Li, Y. Wu and W. Zhang, Chem. Commun., 2008, 1989–1991 RSC.
- J. F. Young, H. D. Nguyen, L. Yang, J. Huskens, P. Jonkheijm and L. Brunsveld, ChemBioChem, 2010, 11, 180–183 CrossRef CAS PubMed.
- P. Neirynck, J. Brinkmann, Q. An, D. W. van der Schaft, L. G. Milroy, P. Jonkheijm and L. Brunsveld, Chem. Commun., 2013, 49, 3679–3681 RSC.
- D. Wasserberg, D. A. Uhlenheuer, P. Neirynck, J. Cabanas-Danes, J. H. Schenkel, B. J. Ravoo, Q. An, J. Huskens, L. G. Milroy, L. Brunsveld and P. Jonkheijm, Int. J. Mol. Sci., 2013, 14, 4066–4080 CrossRef CAS PubMed.
- D. Josic and J. G. Clifton, Proteomics, 2007, 7, 3010–3029 CrossRef CAS PubMed.
- Y. Zhao, W. Zhang, Y. Kho and Y. Zhao, Anal. Chem., 2004, 76, 1817–1823 CrossRef CAS PubMed.
- G. Elia, Proteomics, 2008, 8, 4012–4024 CrossRef CAS PubMed.
- L. Zhang, X. e. Wang, X. Peng, Y. Wei, R. Cao, Z. Liu, J. Xiong, X. Ying, P. Chen and S. Liang, J. Proteome Res., 2007, 6, 34–43 CrossRef CAS PubMed.
- M. Fonovic, S. H. Verhelst, M. T. Sorum and M. Bogyo, Mol. Cell. Proteomics, 2007, 6, 1761–1770 CAS.
- C. E. Chivers, E. Crozat, C. Chu, V. T. Moy, D. J. Sherratt and M. Howarth, Nat. Methods, 2010, 7, 391–393 CrossRef CAS PubMed.
- T. Nguyen, N. S. Joshi and M. B. Francis, Bioconjugate Chem., 2006, 17, 869–872 CrossRef CAS PubMed.
- J. A. Chung, J. W. Wollack, M. L. Hovlid, A. Okesli, Y. Chen, J. D. Mueller, M. D. Distefano and T. A. Taton, Anal. Biochem., 2009, 386, 1–8 CrossRef CAS PubMed.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS PubMed.
- R. Santos, G. da Costa, C. Franco, P. Gomes-Alves, P. Flammang and A. V. Coelho, Mar. Biotechnol., 2009, 11, 686–698 CrossRef CAS PubMed.
- R. J. Stewart and C. S. Wang, Biomacromolecules, 2010, 11, 969–974 CrossRef CAS PubMed.
- Q. Ye, F. Zhou and W. Liu, Chem. Soc. Rev., 2011, 40, 4244–4258 RSC.
- C. Zhong, T. Gurry, A. A. Cheng, J. Downey, Z. Deng, C. M. Stultz and T. K. Lu, Nat. Nanotechnol., 2014, 9, 858–866 CrossRef CAS PubMed.
- W. Li and A. E. Kaifer, Langmuir, 2012, 28, 15075–15079 CrossRef CAS PubMed.
- J. Guo, C. Yuan, M. Guo, L. Wang and F. Yan, Chem. Sci., 2014, 5, 3261–3266 RSC.
- T. Ogoshi and A. Harada, Sensors, 2008, 8, 4961–4982 CrossRef CAS PubMed.
- P. Li, Y. Liu, X. Wang and B. Tang, Analyst, 2011, 136, 4520–4525 RSC.
- L. Feng, C. Tong, Y. He, B. Liu, C. Wang, J. Sha and C. Lü, J. Lumin., 2014, 146, 502–507 CrossRef CAS PubMed.
- B. Gong, B.-K. Choi, J.-Y. Kim, D. Shetty, Y. H. Ko, N. Selvapalam, N. K. Lee and K. Kim, J. Am. Chem. Soc., 2015, 137, 8908–8911 CrossRef CAS PubMed.
- R. Jahn and T. C. Sudhof, Annu. Rev. Biochem., 1999, 68, 863–911 CrossRef CAS PubMed.
- S. Martens and H. T. McMahon, Nat. Rev. Mol. Cell Biol., 2008, 9, 543–556 CrossRef CAS PubMed.
- J. A. McNew, F. Parlati, R. Fukuda, R. J. Johnston, K. Paz, F. Paumet, T. H. Sollner and J. E. Rothman, Nature, 2000, 407, 153–159 CrossRef CAS PubMed.
- S. Martens, M. M. Kozlov and H. T. McMahon, Science, 2007, 316, 1205–1208 CrossRef CAS PubMed.
- T. Ohya, M. Miaczynska, U. Coskun, B. Lommer, A. Runge, D. Drechsel, Y. Kalaidzidis and M. Zerial, Nature, 2009, 459, 1091–1097 CrossRef CAS PubMed.
- M. Kyoung, A. Srivastava, Y. Zhang, J. Diao, M. Vrljic, P. Grob, E. Nogales, S. Chu and A. T. Brunger, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E304–E313 CrossRef CAS PubMed.
- J. Diao, Z. Su, Y. Ishitsuka, B. Lu, K. S. Lee, Y. Lai, Y. K. Shin and T. Ha, Nat. Commun., 2010, 1, 54 Search PubMed.
- C. Kim, S. S. Agasti, Z. Zhu, L. Isaacs and V. M. Rotello, Nat. Chem., 2010, 2, 962–966 CrossRef CAS PubMed.
- C. Kim, G. Y. Tonga, B. Yan, C. S. Kim, S. T. Kim, M.-H. Park, Z. Zhu, B. Duncan, B. Creran and V. M. Rotello, Org. Biomol. Chem., 2015, 13, 2474–2479 CAS.
- K. N. Houk, A. G. Leach, S. P. Kim and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 4872–4897 CrossRef CAS PubMed.
- S. Ghosh and L. Isaacs, J. Am. Chem. Soc., 2010, 132, 4445–4454 CrossRef CAS PubMed.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y.-C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |