
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic combinatorial chemistry: a tool to facilitate the identification of inhibitors for protein targets
Milon
Mondal
and
Anna K. H.
Hirsch
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands. E-mail: a.k.h.hirsch@rug.nl
First published on 23rd February 2015
Abstract
Dynamic combinatorial chemistry (DCC) has emerged as a powerful strategy to identify ligands for biological targets given that it enables the target to direct the synthesis and amplification of its strongest binder(s) from the library of interconverting compounds. Since the first report of DCC applied to the discovery of binders for a protein, this elegant tool has been employed on a range of protein targets at various stages of medicinal-chemistry projects. A series of suitable, reversible reactions that are biocompatible have been established and the portfolio of analytical techniques is growing. Despite progress, in most cases, the libraries employed remain of moderate size. We present here the most recent advances in the field of DCC applied to protein targets, paying particular attention to the experimental conditions and analytical methods chosen.
 Milon Mondal | Milon Mondal obtained his Bachelor of Science in 2008 from the University of Calcutta, Kolkata. In 2010, he graduated from the Indian Institute of Technology, Bombay (IIT Bombay) with a Master's degree in Chemistry and pursued his Master's project under the supervision of Professor M. Ravikanth. Later he joined the group of Professor C. P. Rao at IIT Bombay as a Junior Research Fellow. Since May 2011, he has been working towards his PhD in the Hirsch group on a structure-based design project, in which he combines de novo design with dynamic combinatorial chemistry. |
 Anna K. H. Hirsch | Anna K. H. Hirsch read Natural Sciences at the University of Cambridge. After an exchange at the Massachusetts Institute of Technology, she carried out her Master's project in the group of Prof. Steven Ley at the University of Cambridge. She pursued her PhD at the ETH Zurich in 2008 under the supervision of Prof. François Diederich. She joined the group of Prof. Jean-Marie Lehn in Strasbourg as a post-doctoral researcher before taking up her current position as assistant professor at the University of Groningen in 2010. Her work focuses on rational approaches to drug design, exploiting dynamic combinatorial chemistry. |
Introduction
Dynamic combinatorial chemistry (DCC) allows for the reversible combination of molecular building blocks via covalent or non-covalent bonds,1–4 affording dynamic combinatorial libraries (DCLs) of potentially complex and interchanging products in an efficient manner. Given that the reaction between the building blocks is reversible, the product distribution is dictated by the thermodynamic stability of the compounds formed. A DCL has the advantage over a classical combinatorial library that it is responsive to external stimuli such as the addition of a target, which causes the composition of the library to re-equilibrate upon selection and binding of the library members with the strongest affinity for the target.2 Ultimately, this leads to amplification of the best binders, which can be identified circumventing the need for synthesis, purification and characterization of every individual library member. Protein-templated DCC was used for the first time in the late 1990's, leading to the identification of its own inhibitors.5 Protein-templated DCC thus offers an efficient and powerful approach for the acceleration of the identification and optimization of novel ligands for biological targets, and therefore holds an enormous potential for drug discovery (Scheme 1).6–10 In short, protein-templated DCC combines the synthesis of inhibitors and screening for affinity for the drug target in a single operation where the target selects its own inhibitors. We will discuss recent applications of DCC for the identification of protein ligands according to the type of reversible reaction used over the past decade.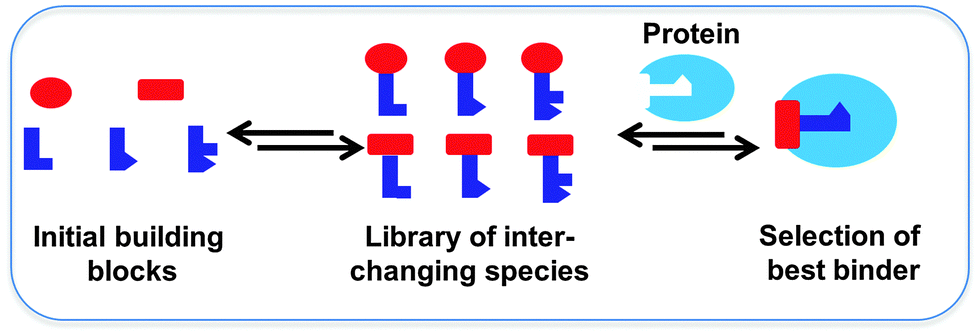 | ||
| Scheme 1 Schematic representation of the fundamental concept of protein-templated DCC. | ||
General features of DCC applied to protein targets
Several reversible reactions have been used in protein-templated DCC where the target is a protein (Scheme 2).11–18 Most of these reversible reactions can be carried out in aqueous media, which makes them biocompatible. The reversible reaction chosen for DCL formation should equilibrate fast enough under the conditions where the protein is stable, usually an aqueous medium in a certain pH and temperature window. In the case of sluggish reactions, use of a catalyst such as aniline might be required, which can be used as a nucleophilic catalyst for acylhydrazone formation.19,20 The reactions should be chemoselective to avoid cross-reactivity with functional groups of the building blocks or the target.5,21 DCL formation can be carried out in the presence (adaptive DCC) or absence (pre-equilibrated DCC) of the target. The former set-up is certainly preferred as it endows the system with true adaptability. In the latter case, the protein is added to the pre-equilibrated DCL, which is necessary for targets that are unstable under the conditions required for DCL generation.21 To ensure formation of an unbiased DCL, building blocks of comparable reactivity and energy should be used. If mass spectrometry (MS) is to be used as an analytical tool, selection of building blocks should avoid having products or building blocks of similar or identical molecular weight. Furthermore, all building blocks and products need to be soluble to preclude that the DCL is biased by precipitation of some of its members. Solubility of organic ligands is usually ensured by the use of an organic co-solvent such as DMSO. The concentration of organic co-solvent needs to be chosen such that it maximizes the solubility of the organic components whilst not affecting the protein target. A DCL should be “frozen” in the presence of the target protein for convenient analysis and to ensure its composition is fixed. There are multiple ways of achieving this: subsequent irreversible functionalization such as reduction of the reversible imines to amines,5 pH change from basic to acidic pH for disulfide exchange or removal or inactivation of the catalyst.21 It is important to note that the parameters used for DCL generation will also affect the equilibrium composition and amplification factors (e.g., building-block and target concentration, respectively). Finally, the protocol and analytical method need to be chosen based on how readily available the protein is. Saturation-transfer difference (STD)-NMR spectroscopy, for instance, only works if the ligand is used in excess, therefore requiring significantly less protein than liquid chromatography-mass spectrometry (LC-MS).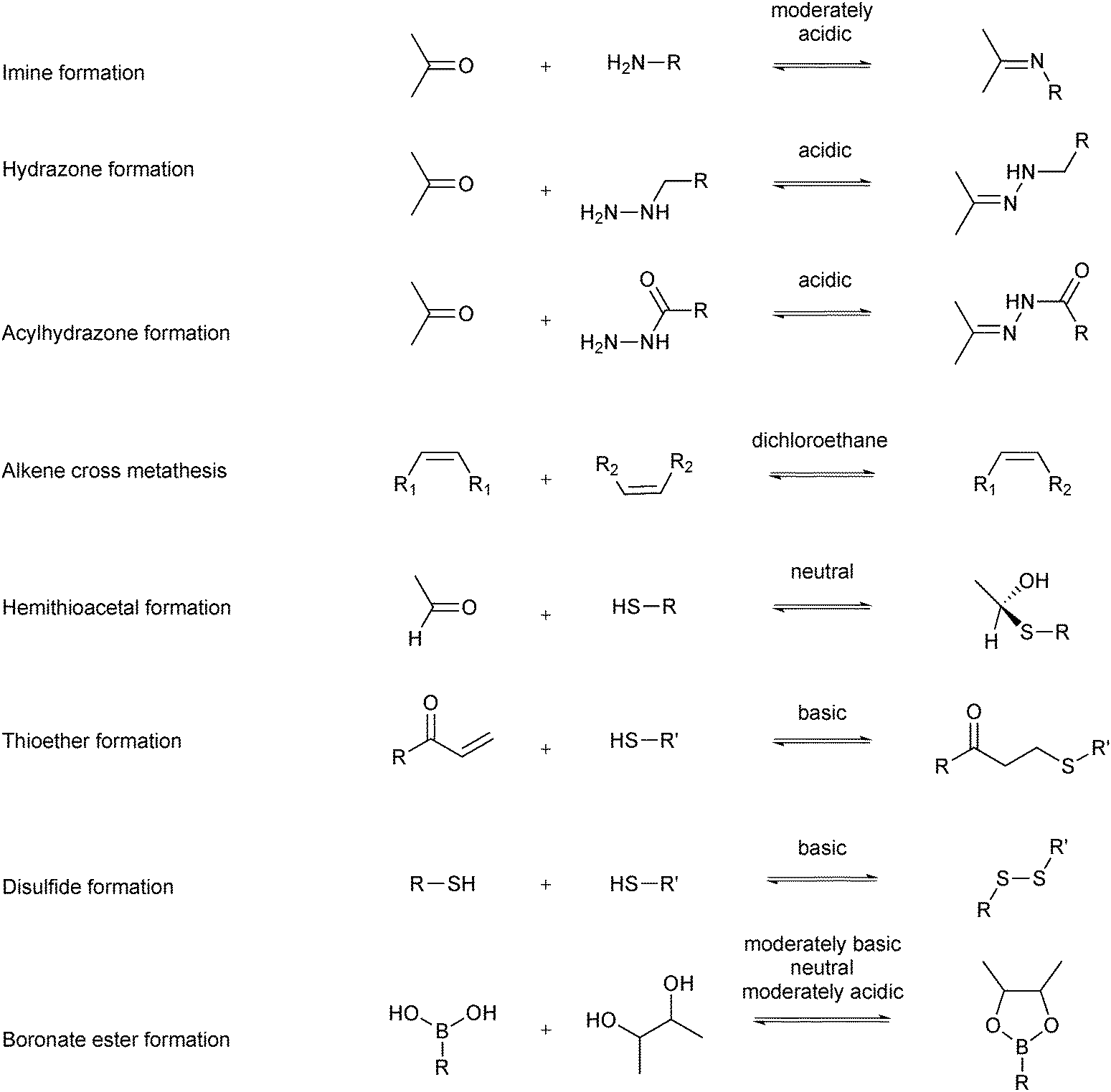 | ||
| Scheme 2 Reversible reactions used in protein-templated DCC to generate bioactive compounds. | ||
Reversible covalent bond formation for protein targets
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) N bond formation
N bond formation
In 2008, the Rademann group demonstrated that dynamic ligation screening (DLS), in which a reversibly formed bioactive ligation product from the DCL competes with a fluorogenic reporter substrate for a target enzyme, facilitates site-directed and sensitized detection of low-affinity fragments that are difficult to detect with normal DCC or conventional fragment-based drug design (FBDD) approaches.17 By detecting the fragments using an enzymatic reaction, very small amounts of protein are sufficient, making the screen amenable to a high-throughput format. SARS coronavirus SARS-CoV Mpro, a cysteine protease that is crucial for virus replication inside the host cell and responsible for severe acute respiratory syndrome, was used as a protein target to pioneer the DLS approach.
To do so, a fluorescence-based assay was developed incorporating substrate 1.28 A peptide aldehyde inhibitor 2 was chosen as a directing probe for DLS that mimics the native protease substrate and addresses the enzyme's S1, S2 and S3 pockets. Given that aliphatic aldehydes are less reactive towards imine formation in aqueous media than aromatic aldehydes, it was envisaged that the ligation products of the peptide aldehyde 2 and the nucleophiles are only formed when bound to the protein surface enabling detection by substrate competition. For this study, a library of 234 amines, thiols and hydrazines were selected as nucleophiles. Aldehyde 2 was incubated with an eightfold excess of one nucleophile per well in the presence of an enzyme. Subsequently, fluorescence was recorded upon addition of reporter substrate 1.
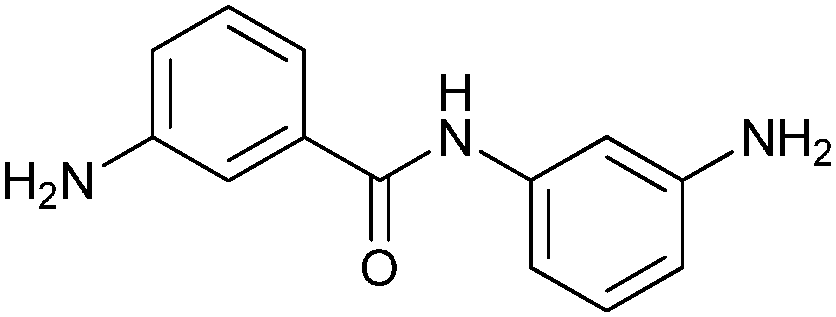
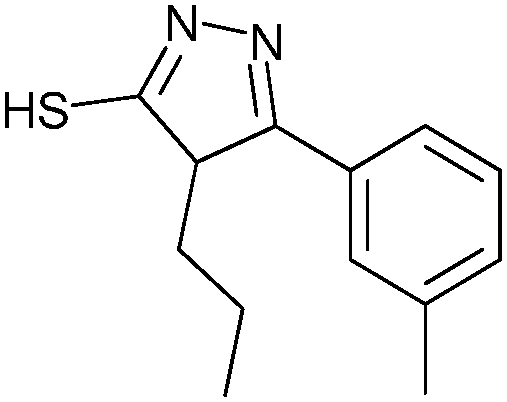
None of the individual fragments showed inhibition against the SARS-CoV Mpro, but in the presence of seven nucleophiles 3a–g, aldehyde 2 showed strong inhibitory activity (Table 1). To validate the specific binding modes of the hits identified, the most active amine 3a was selected for imine formation with the aldehyde 2. The corresponding reductive amination product 4 was tested and displays an inhibition constant (Ki) of 50.3 μM (Fig. 1). Truncating the amine moiety 3a in compound 4 afforded amine 5, which is inactive (Ki > 500 μM), confirming the directing effect of peptide aldehyde 2 and validating that amine 3a binds in the S1′ pocket, which was further corroborated by modeling studies. Further evidence of the specific binding of amine 3a in the S1′ pocket was provided by synthesizing and testing the aldehyde analogues of amine 3a, which were designed to interact with the active-site cysteine residue of the protease. Biochemical evaluation showed that aldehydes 6a–d are active inhibitors of SARS-CoV Mpro and thus validated the binding mode of the fragments identified by DLS to be indeed in the S1′ pocket of SARS-CoV Mpro.
| Aldehyde | Nucleophile | ν 0 (μM min−1) | |
|---|---|---|---|
| — | — | 5.5 ± 0.2 | |
| 2 | 2.8 ± 0.1 | ||
| 2 |
|
3a | 1.0 ± 0.1 |
| 2 |
|
3b | 1.0 ± 0.1 |
| 2 |
|
3c | 1.6 ± 0.1 |
| 2 |
|
3d | 1.9 ± 0.1 |
| 2 |
|
3e | 2.1 ± 0.1 |
| 2 |
|
3f | 2.2 ± 0.1 |
| 2 |
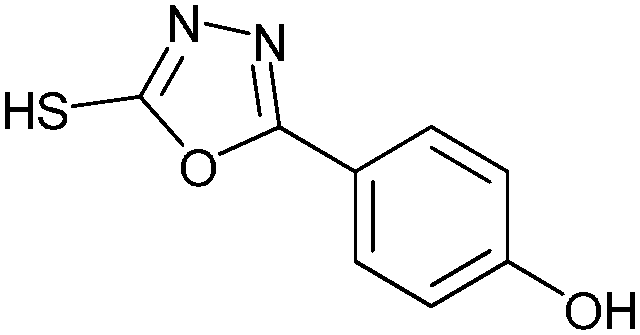
|
3g | 2.2 ± 0.1 |
| 6d |
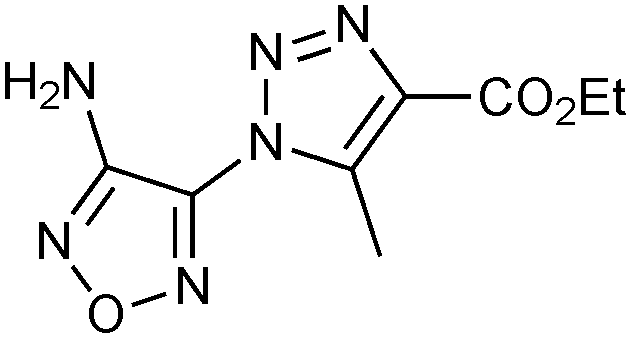
|
3h | 2.0 ± 0.05 |
| 6d |
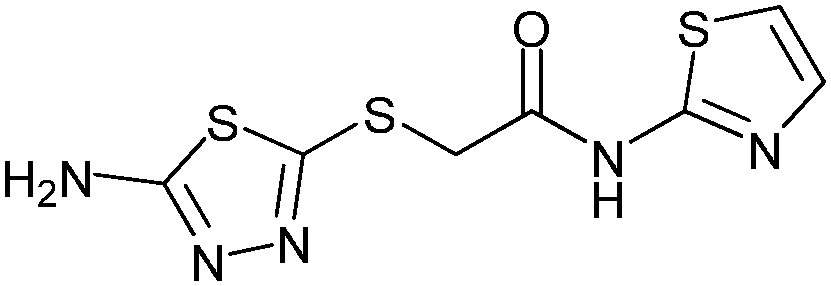
|
3i | 2.5 ± 0.05 |
| 6d |
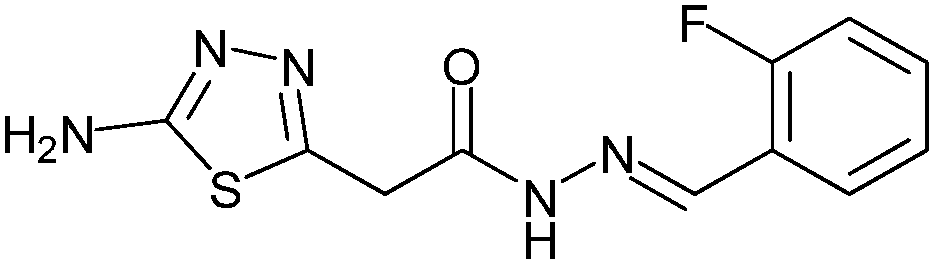
|
3j | 3.7 ± 0.1 |
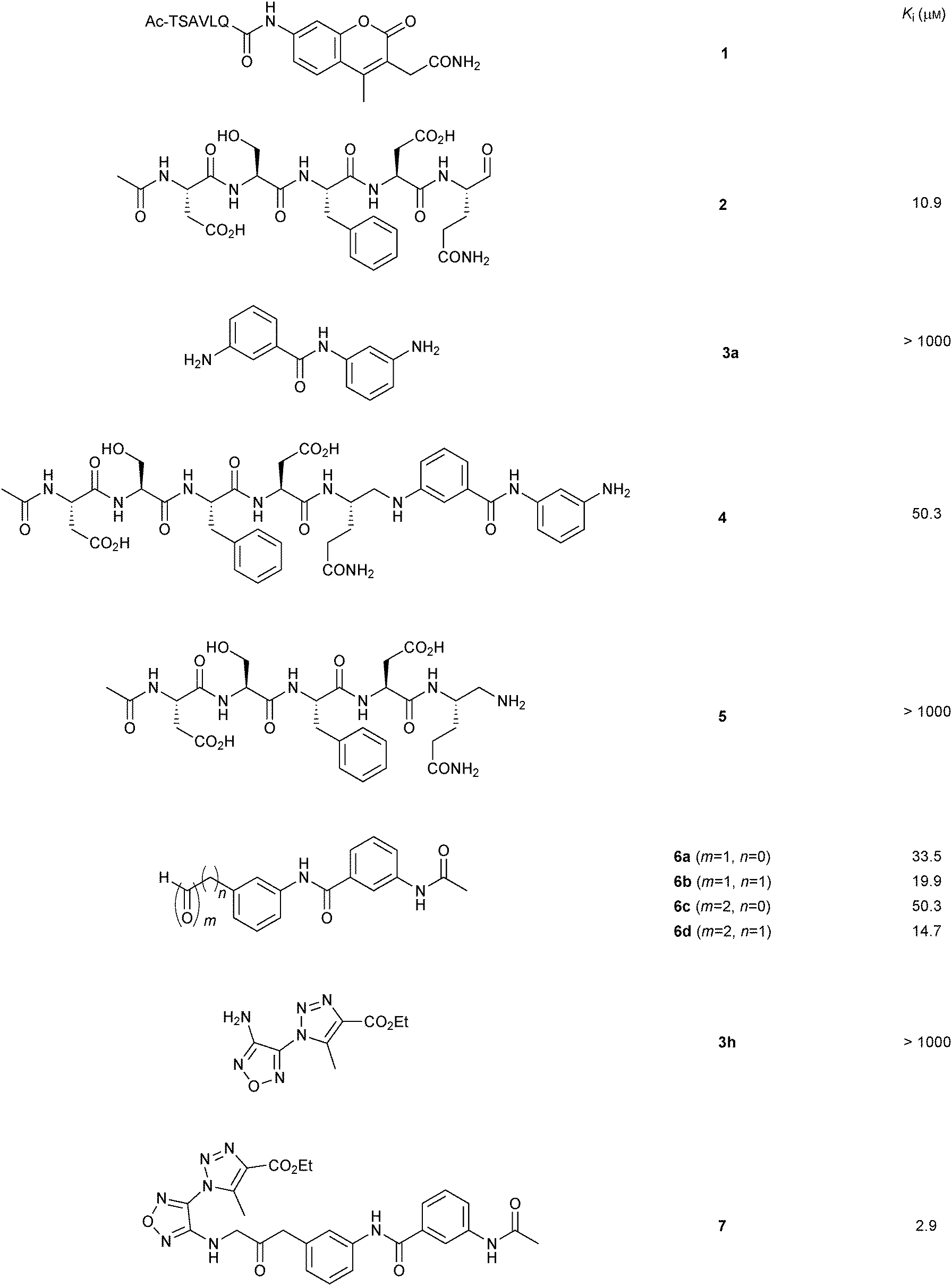 | ||
| Fig. 1 Structures and Ki values of non-peptidic SARS-CoV Mpro inhibitors identified by imine-based dynamic combinatorial chemistry and subsequent optimization.17 | ||
To obtain a non-peptidic inhibitor of SARS-CoV Mpro targeting both the S1 and the S1′ pockets, a second round of DLS was carried out in an inverted way using the aldehyde 6d, which preferentially binds to the S1′ pocket. A library of 110 amines were selected as nucleophiles. Site-directed DLS experiments were performed as previously and, three nucleophiles 3h–j were identified as active fragments in the presence of aldehyde 6d (Table 1). The amine 3h appeared to be the most active fragment, which was then used for the reductive amination with aldehyde 6d to afford amine 7, which displays a Ki value of 2.9 μM. This proof-of-concept study shows that DLS can be used iteratively to first identify a peptidic inhibitor, which was subsequently transformed into a non-peptidic inhibitor with a low micromolar inhibition constant. DLS requires low amounts of protein and is compatible with an HTS format. This approach holds the potential to be readily extended to other enzyme classes as well as protein–protein interactions.
In 2009, the groups of Barboiu and Supuran exploited DCC based on imine interconversion for the identification of inhibitors of human carbonic anhydrase (hCA II).25 Carbonic anhydrases are zinc metalloenzymes, which are responsible for numerous physiological and pathological processes, by catalyzing the hydration of carbondioxide to bicarbonate.29 To demonstrate the use of reversible imine formation, it was applied for the DCC-mediated identification of isozyme-specific inhibitors of the pharmacologically most relevant isoform hCA II. A library of 20 imines that should have affinity for the hCA II isozyme were selected. To generate the imine-based DCL, a set of four aldehydes 8a–d and a ten-fold excess of five amines 9a–e were employed to ensure full conversion to the corresponding imines (Scheme 3). Aromatic amines and aldehydes were used given that they form stable Schiff's bases at pH 6.5, which are able to interact with different CAs, affording access to potentially effective inhibitors. The aromatic amines were selected so as to contain a sulfonamide, a carboxylic and a carboxymethyl group, which are able to bind zinc strongly, weakly and not at all, respectively.
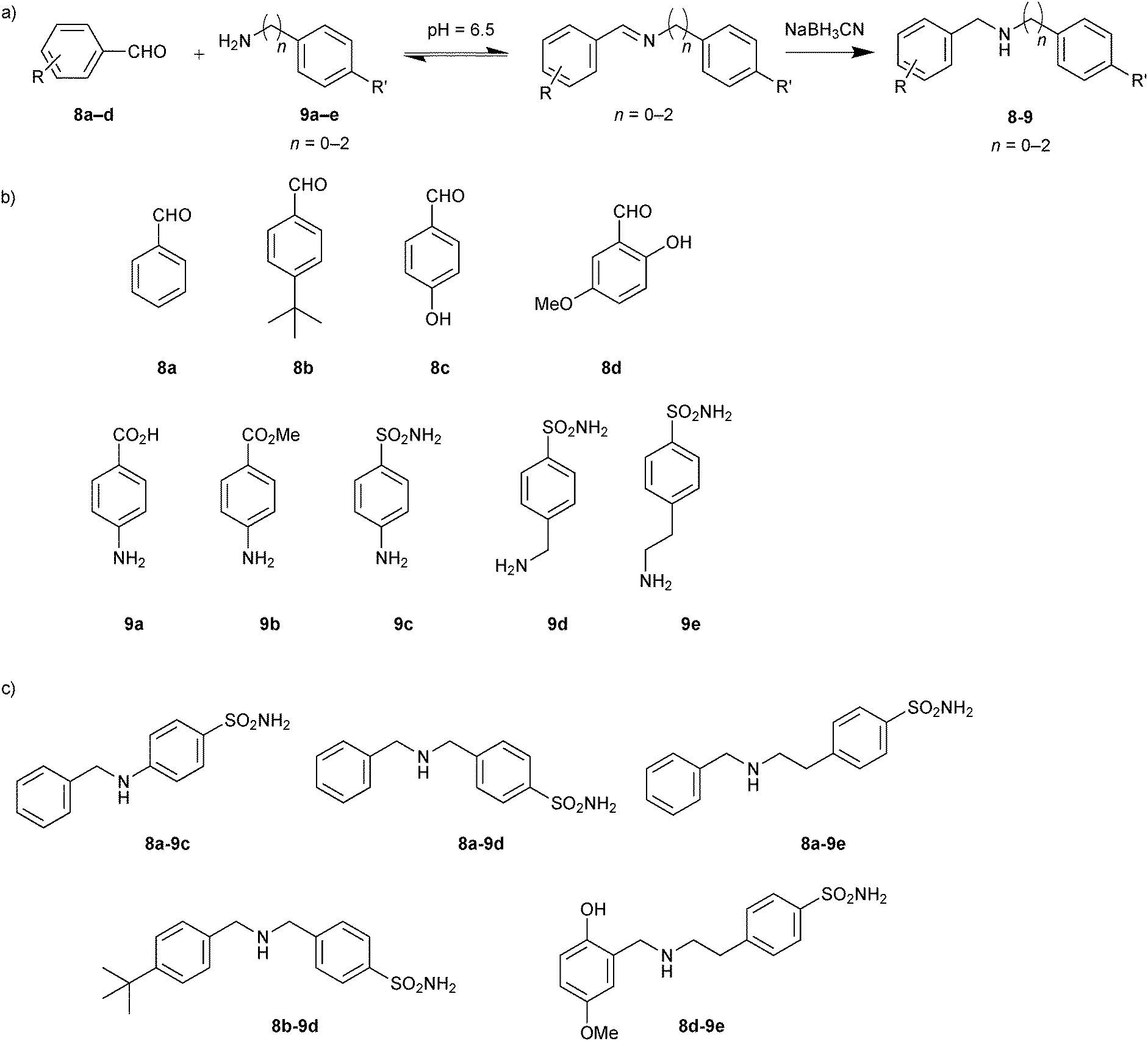 | ||
| Scheme 3 (a) Reversible formation of DCLs based on imine formation followed by reduction to the corresponding amines. (b) Initial aldehydes 8a–d and amine building blocks 9a–e. (c) Inhibitors (8-9) identified from screening experiments with human carbonic anhydrase II.25 | ||
Various aromatic aldehydes were chosen to explore the hydrophobic pocket adjacent to the active site of hCA II. Addition of protein altered the initial equilibrium distribution of the DCL, which was analyzed in the absence and presence of protein by HPLC. Having frozen the equilibrium by reducing the imines to the corresponding amines with NaBH3CN, the reduced amines were subsequently identified by comparing the retention time with pure compounds. Some of the imines were not formed, which might be ascribed to the weak reactivity of the aromatic amines with electron-withdrawing substituents in the para-position.30 All possible 20 amines were synthesized and inhibition constants (Ki) were determined against hCA II (Table 2). Generally speaking, the DCL screening results correlate with the inhibition studies. Five amines (8a-9c, 8a-9d, 8a-9e, 8b-9d and 8d-9e) were amplified in presence of hCA II and three of them (8a-9c, 8a-9d and 8b-9d) showed amplification factors of more than 1.4 and very good inhibitory potency confirming the importance of the sulfonamide moiety of 9c and 9d in combination with hydrophobic aldehydes 8a and 8b. The poor correlation observed for the significantly longer ethylamine 8a-9e, which displayed weak inhibition but strong amplification in the DCL screening, might be due to the strong affinity of the sulfonamide group for the zinc cation. Amine 8d-9e, which showed strong amplification in the DCL screening as well as good inhibition of hCA II, seems to constitute a good balance of unfavorable entropic loss and favorable enthalpic gain imparted by the longer ethylamine chain of 9e and the hydrophobic and hydrogen-bonding interactions of aldehyde 8d, respectively. This study illustrates the power of imine-based DCC for the identification of enzyme inhibitors and sets the stage for subsequent optimization of isozyme-specific inhibitors.
Building on these encouraging results, imine-based DCC was used to optimize the lead compounds into highly specific and selective inhibitors of hCA I and hCA II isozymes.26 The DCLs of six components were generated using two amines 10a and 10b bearing specific zinc-binding groups and a set of three hydrophobic aldehydes 11a–c to interact with the hydrophobic pocket of the enzyme (Scheme 4). The structural variability of the various substituents on the aromatic ring such as a sulfonamide and a sulfonic acid moiety as a strong and relatively weak zinc-binding group, respectively, were used to generate a library of diverse affinity for the enzyme. Reaction between amines 10a and 10b and aldehydes 11a–c led to the formation of six imines 10-11im in equilibrium with the building blocks. Addition of hCA I and hCA II shifted the equilibrium towards the strong binders, which were identified just like in the preceding report after NaBH3CN reduction.
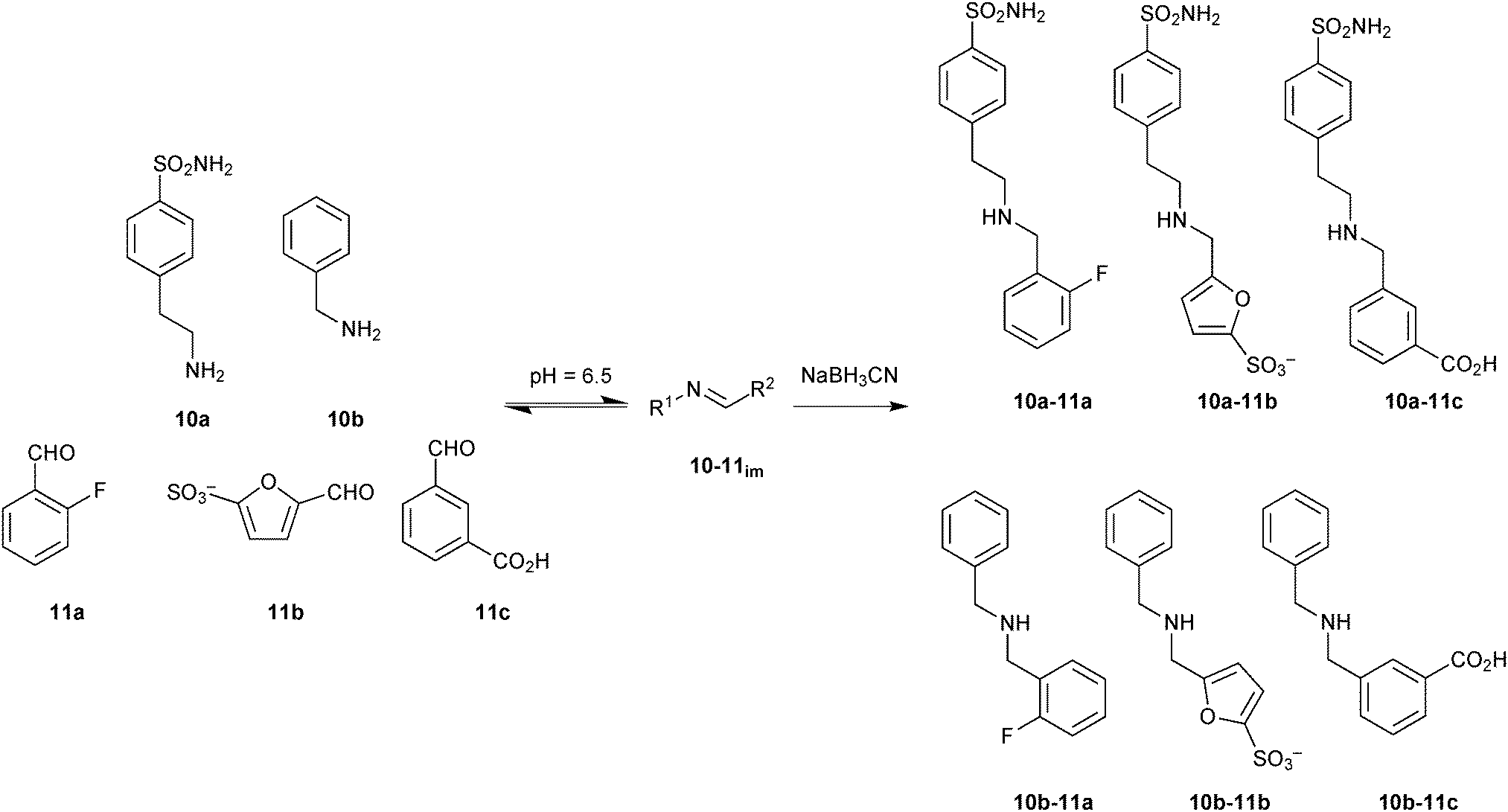 | ||
| Scheme 4 Generation of imine-based dynamic combinatorial libraries of amines 10a and 10b and aldehydes 11a–c for templating by human carbonic anhydrase I and II, followed by reduction using NaBH3CN.26 | ||
In parallel, all possible six imines (10-11im) and their corresponding amines (10-11) were synthesized separately and tested for their inhibition (Ki) against hCA I and hCA II. The biochemical results correlate with the DCL-screening results (Table 3). Only two amines 10a-11c and 10b-11b were amplified in presence of hCA I. Amine 10a-11c showed a relative peak area of 4.5 as well as very good inhibition against hCA I, confirming the strong inhibitory power of the sulfonamide group in combination with hydrophobic and H-bonding effects of the aromatic carboxyl group. The presence of 2-fluoro benzaldehyde (11a) led to a decrease of enzyme inhibition as noticed in the case of amine 10a-11a. The amine 10a-11b, which showed similar inhibitory potency to 10a-11c, was not amplified in presence of hCA I, which might be due to the weak reactivity of furansulfonic aldehyde 11b. In presence of hCA II, three amines 10a-11a–c were amplified presumably owing to a better steric fit into the active site compared to amines 10b-11a–c. Amine 10a-11a, which showed the same inhibition as 10a-11b against hCA II, was amplified more than 10a-11b, which could be due to the presence of fluorine in the aromatic ring. The presence of a carboxylic acid group in 10a-11c led to ten-fold weaker inhibitory activity against hCA II. These results demonstrate the adaptive behavior of the DCL towards two target isozymes hCA I and hCA II. This concept holds the promise to be extended to enzyme families with similar active-site features such as kinases.
| Inhibitor | K i (nM) | Relative peak areaa | ||
|---|---|---|---|---|
| hCA I | hCA II | hCA I | hCA II | |
| a Relative peak area is the experimental peak area in presence of enzyme in comparison to the reaction in absence of enzyme. | ||||
| 10a-11aim | 620 | 7.2 | ||
| 10a-11a | 260 | 6.9 | 0.6 | 5.2 |
| 10a-11bim | 35 | 4.9 | ||
| 10a-11b | 39 | 7.6 | 4.4 | |
| 10a-11cim | 65 | 51 | ||
| 10a-11c | 39 | 57 | 4.5 | 3.5 |
| 10b-11aim | >100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
>100000 |
||
| 10b-11a | >100000 |
>100000 |
||
| 10b-11bim | 8960 | >100000 |
||
| 10b-11b | 3490 | 8025 | 2.8 | 0.7 |
| 10b-11cim | >100000 |
>100000 |
||
| 10b-11c | >100000 |
>100000 |
||
In 2013, the group of Guo demonstrated a novel protocol based on size-exclusion chromatography (SEC) and MS detection for the direct identification of binders from a DCL through isolation of ligand–target complexes.27 For the proof-of-principle study, hen egg-white lysozyme (HEWL) was chosen as the protein target, which has an important role in immune defense by degrading and attacking bacterial cell walls. A library of amines were designed based on the imines derived from a set of four amines 12a–d and eight aldehydes 13a–h (Scheme 5). The pre-equilibrated DCL was reduced to the corresponding amines using NaBH3CN and incubated with the protein target HEWL, before being passed through a suitable SEC column to retain all non-binders. After denaturation of the eluted protein–ligand complexes using acetonitrile to release binders, MS analysis indicated the presence of three amines 12a-13b, 12a-13d and 12a-13f as binders of HEWL, which were confirmed using a control DCL generated in the absence of the protein.
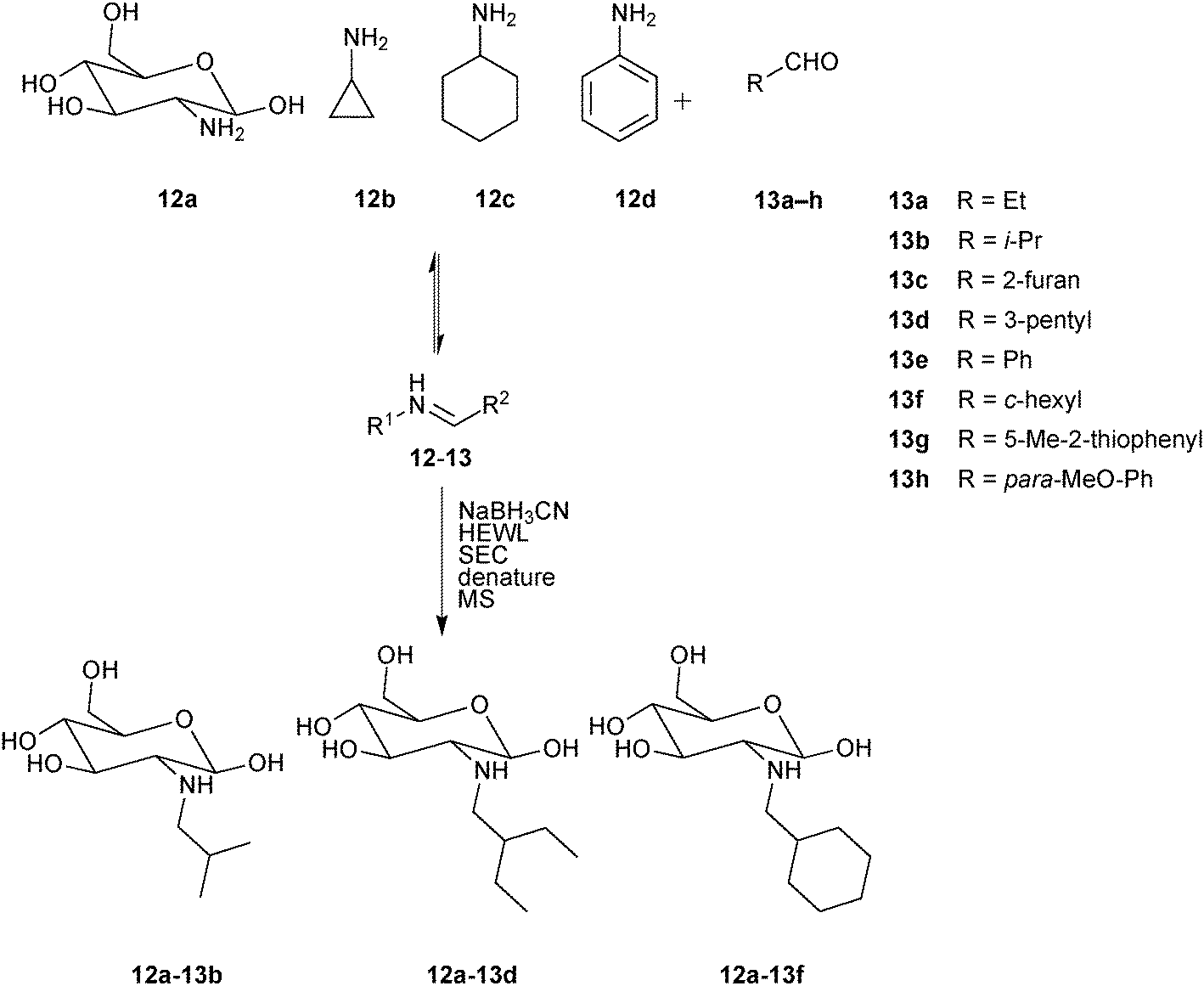 | ||
| Scheme 5 Generation of pre-equilibrated imine-based dynamic combinatorial libraries of amines 12a–d and aldehydes 13a–h, followed by reduction using NaBH3CN, incubation with hen egg-white lysozyme (HEWL) and separation of protein–ligand complexes by size-exclusion chromatography (SEC).27 | ||
In a control experiment set up without the reducing agent, no imines were detected, suggesting that amines rather than imines are the ligands of HEWL. To validate this hypothesis and quantify binding affinity towards HEWL, these amines were synthesized and tested for their effect on the lysis rate of Micrococcus lysodeikticus.31 The inhibition results (Km values (Michaelis constants) are reported given that determination of Ki values was impossible due to incomplete inhibition) indicate that the binding affinities for HEWL decrease in the following order 12a-13f > 12a-13d ≈ 12a-13b (Table 4). A drawback of this SEC-MS protocols is the limited capacity of the SEC column. Consequently, the concentration of building blocks for DCL generation is limited to the micromolar or even nanomolar range, leading to long equilibration times. This novel analytical protocol for the analysis of DCLs using SEC separation of the protein–ligand complexes followed by denaturation of the complexes and MS analysis has the advantage of circumventing the need for resolution of all library members by LC, which has been one of the major bottlenecks, restricting access to significantly larger library sizes. As a result, this proof-of-concept study sets the stage for application in early-stage drug discovery programs with protein targets of therapeutic interest.
| Inhibitor | Concentration (μM) | K m (mg mL−1) |
|---|---|---|
| a K m values determined using non-linear regression. | ||
| — | — | 0.104 ± 0.018 |
| 12a-13b | 300 | 0.163 ± 0.022 |
| 12a-13d | 300 | 0.162 ± 0.063 |
| 12a-13f | 300 | 0.203 ± 0.028 |
In 2003, Congreve and co-workers at Astex Technology demonstrated for the first time that X-ray crystallography can be used to detect binders directly from a DCL by exposing protein crystals to the library.16 The cyclin-dependent kinase 2 (CDK2), which is involved in a number of human cancers, was used for this proof-of-concept study.36,37 A library of potential hydrazone inhibitors that would display a variety of functional groups in the hydrophobic regions of the ATP-binding site was chosen taking inspiration from the known oxindole inhibitor series for CDK2. Based on these hydrazones, a range of hydrazines 14a–f and isatins 15a–e were selected as the building blocks for the DCL (Table 5), giving rise to potentially 30 hydrazones. Having established the hydrazone chemistry in aqueous media, the reactions were performed in presence of protein crystals.
16
| 14 × 15 |
14ab |
14b R1 = Cl | 14c R2 = Cl | 14d R3 = Cl | 14e R3 = SO2NH2 | 14f R1 = Cl; R3 = SO2NH2 | |
|---|---|---|---|---|---|---|---|
| a Values indicate the extent to which the reaction occurred in aqueous solution after 2 days at room temperature as measured by percentage purity using peak area of the product by LC-MS (10–25%, 30–50%, or 60–95% of total peaks excluding solvent front). b R groups = H, unless indicated otherwise. | |||||||
| 15a | R5 = NO2 | 10–25 | 60–95 | 60–95 | 30–50 | 60–95 | 30–50 |
| 15b | R5 = Cl | 60–95 | 60–95 | 60–95 | 60–95 | 60–95 | 30–50 |
| 15c | R5 = SO3H | 10–25 | 60–95 | 30–50 | 10–25 | 60–95 | 30–50 |
| 15d | R7 = CF3 | 30–50 | 60–95 | 60–95 | 60–95 | 60–95 | 30–50 |
| 15e | R5 = OCF3 | 30–50 | 60–95 | 60–95 | 60–95 | 60–95 | 10–25 |
|
|||||||
Soaking experiments with CDK2 crystals and mixtures of the hydrazine 14e and each of the isatins (15a–e), led to electron density in the ATP-binding pocket implying that the corresponding hydrazones had bound to the protein crystal except for one combination (14e + 15d). Biochemical evaluation of these bound hydrazones showed that all of them are potent inhibitors of CDK2 with IC50 values of 30 nM except for 14e-15d, which was not observed by protein crystallography and was found to be inactive against CDK2 (Table 6).
| Mixture composition | Product | IC50 (nM) |
|---|---|---|
| 14e + 15a | Yes | 30 |
| 14e + 15b | Yes | 30 |
| 14e + 15c | Yes | 30 |
| 14e + 15d | No | Inactive |
| 14e + 15e | Yes | 30 |
| 14a–d + 15b | No | — |
| 14a–f + 15b | Yes (14e-15b) | — |
| 14a–f + 15a–e | Yes (14e-15b) | — |
Increasing the level of complexity, this method was used to analyze the ligands from DCLs. Soaking experiments of two DCLs containing 14a–d + 15b, 14a–f + 15b and CDK2 crystals only led to electron density in the ATP-binding pocket for the latter case corresponding to the potent hydrazone 14e-15b, as expected. Finally, the full DCL consisting of 14a–f and 15a–e was exposed to CDK2 crystals, and 14e-15b was again observed in the ATP-binding pocket, which clearly indicates that X-ray crystallography is an efficient and powerful technique to detect binders form DCLs whilst also providing information on the binding mode. This method also has several advantages over conventional DCC analytical protocols: it is less time-consuming, requires small amounts of protein, circumvents the need for conventional synthesis, purification and analysis of DCL members and provides the binding mode of the ligands identified. Despite these unique advantages, this method has not been followed up on, which is most likely due to the fact that protein X-ray crystallography requires specific expertise and infrastructure that is not readily available in an organic chemistry laboratory.
In 2012, the group of Wanner demonstrated an efficient way to screen DCLs by means of a competitive MS-based binding assay, combining facile library generation with efficient compound screening.34 γ-Aminobutyric acid (GABA) transporter 1 (GAT1), the most important subtype of GABA transporters, which is responsible for neuronal diseases like epilepsy, Parkinson's disease and sleeping disorders, was used to demonstrate the feasibility of this approach.38 A library based on hydrazone chemistry containing compounds that resemble mGAT1 inhibitors 16–18 were chosen as model compounds for the proof-of-principle study (Scheme 6a). Based on these hydrazones, a hydrazine 19 and a set of 36 aldehydes 20 were selected for the generation of DCLs consisting of 36 hydrazones 19-20 (Scheme 6b). The entire library was divided in to nine one-dimensional sub-libraries, each containing four aldehydes and the hydrazine 19 to enable deconvolution. The hydrazine 19 was used in excess to ensure a pseudostatic DCL is formed in presence of the target protein mGAT1. In pseudostatic DCLs, the equilibrium is forced towards products by using a large excess of the single compound with fixed structure. The DCLs was then analyzed by the competitive MS-based binding assay, employing the native marker 21. This is the first report of this highly sensitive assay used for library screening and involves quantification of a native, i.e., unlabeled marker, thereby avoiding all the drawbacks associated with radioisotopes. The assay should be easy to extend to other targets for which only low concentrations can be reached, such as transmembrane proteins, provided that the affinity of the marker is high enough.39
 | ||
| Scheme 6 (a) Structures of GAT1 inhibitors 16–18. (b) Generation of dynamic combinatorial libraries of hydrazones 19-20 using hydrazine 19 and 36 aldehydes 20 for analysis using a competitive MS-based binding assay against the transporter mGAT1.34 | ||
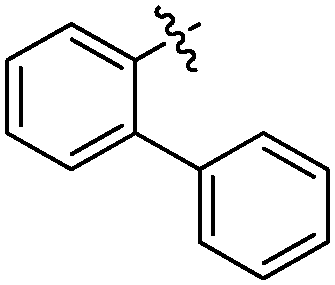
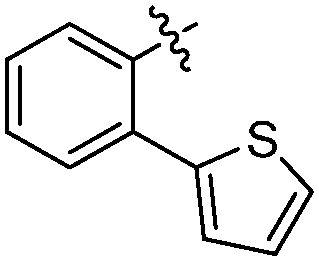
According to deconvolution experiments, the most potent inhibitors are hydrazones 19-20a and 19-20b followed by 19-20c–e that decrease marker binding to around 10% and 30%, respectively. Interestingly, all five hydrazones are derived from ortho-substituted benzaldehyde derivatives. Hydrazone 19-20f was also evaluated as it contains an ortho-biaryl moiety, which decreases marker binding to 65%. To verify the results of the deconvolution experiments, all hydrazones identified were synthesized and evaluated for binding affinity. Ki and IC50 values were determined using the MS-based binding assay for mGAT140 and the [3H]-GABA uptake assay, respectively.41 The MS-based binding assay not only confirms the high affinity of inhibitors 19-20a and 19-20b, but also provides the corresponding inhibitory potencies, enabling the ranking of the six inhibitors according to their inhibitory potency, which is comparable to the results obtained from the deconvolution experiments. The most potent inhibitors 19-20a and 19-20b indeed display the highest potency with pKi values of 6.186 and 6.229, respectively and the medium potent inhibitors 19-20c–f show pKi values of 5.542, 5.577, 5.445 and 4.479, respectively (Table 7).
|
|
||||
|---|---|---|---|---|
| Inhibitors | R | SBa (%) | pKib | pIC50c |
| a Specific binding (SB) of 21 determined in deconvolution experiments. b pKi values were determined by competitive MS-based binding assay with 21. c pIC50 values were determined by [3H]-GABA uptake assay performed in mGAT1-expressing HEK293 cells.34 | ||||
| 19-20a |
|
≤5 | 6.186 ± 0.028 | 5.308 ± 0.096 |
| 19-20b |
|
8.2 ± 0.4 | 6.229 ± 0.039 | 5.542 ± 0.107 |
| 19-20c |
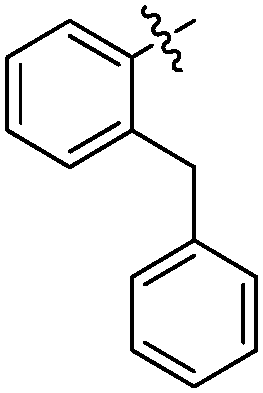
|
27 ± 1 | 5.542 ± 0.042 | 5.186 ± 0.084 |
| 19-20d |
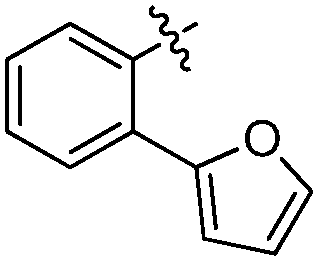
|
37 ± 1 | 5.577 ± 0.037 | 4.895 ± 0.152 |
| 19-20e |
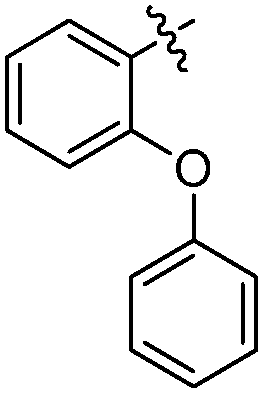
|
24 ± 1 | 5.445 ± 0.075 | 4.879 |
| 19-20f |
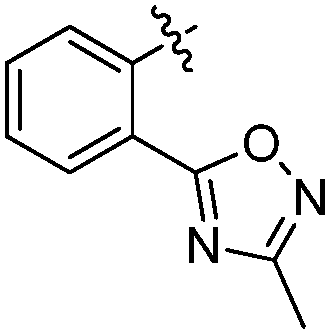
|
65 ± 8 | 4.479 ± 0.064 | 4.022 |
Starting from the two hydrazone-based hits 19-20a and 19-20b with submicromolar activity towards mGAT1, identified using a competitive MS-based binding assay,34 the former 19-20a was used for DCC-based optimization of its affinity for mGAT1 by modification of the biphenyl system attached to the hydrazone linker of both hits (Scheme 7a).35 A library of 36 aldehydes mainly derived from biphenyl-2-carboxyaldehyde (20a) by varying the substituents on the phenyl ring (22) were selected for the formation of a hydrazone library 19-22, which was generated and analyzed as described above using the hydrazine 19 (Scheme 7b).
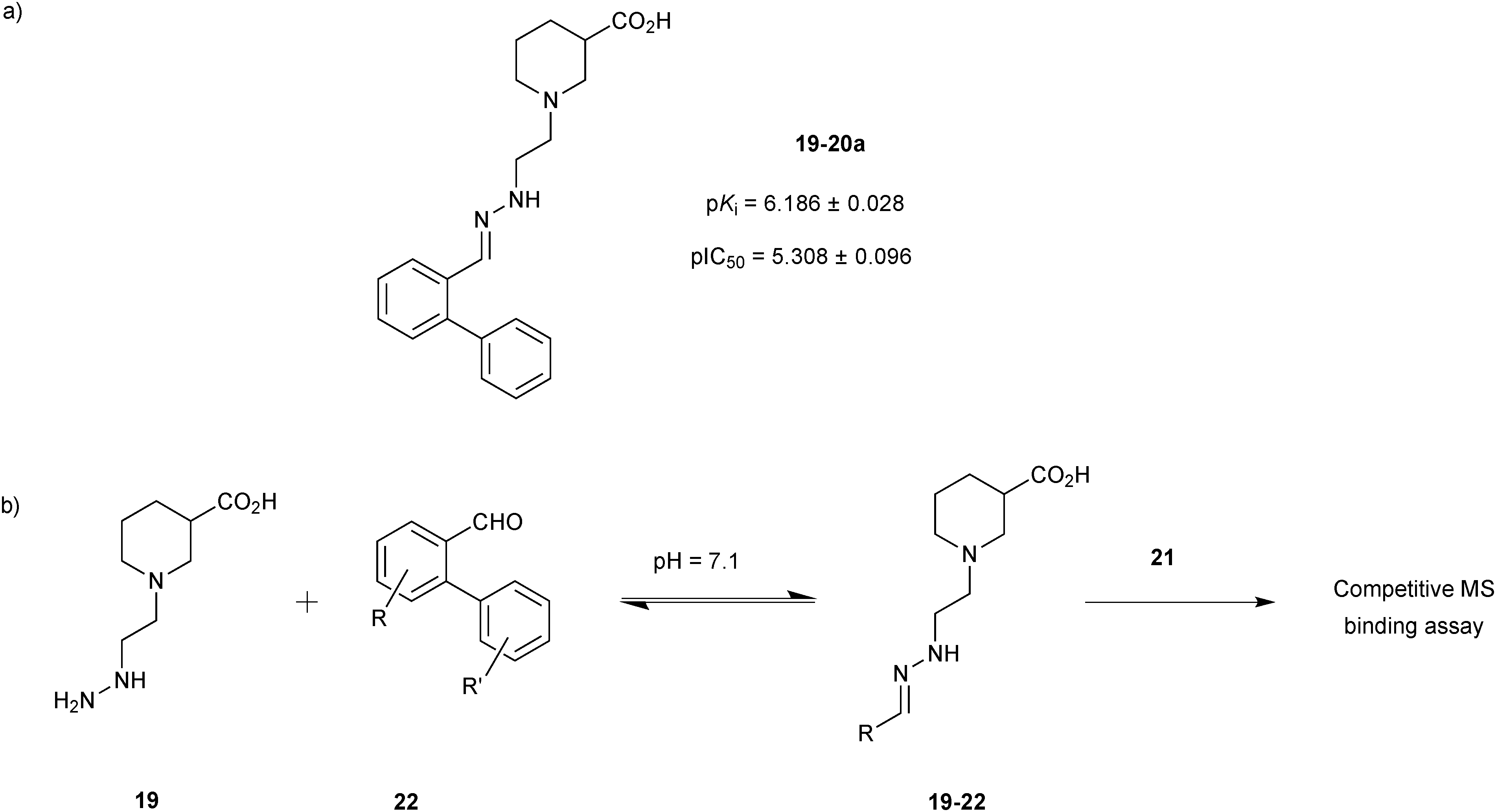 | ||
| Scheme 7 (a) Structures of original hit 19-20a. (b) Generation of dynamic combinatorial libraries of hydrazones 19-22 using hydrazine 19 and 36 aldehydes 22 for analysis by a competitive MS-based binding assay against the transporter mGAT1.35 | ||
In the deconvolution experiments of nine hydrazone libraries, 21 out of the 36 hydrazones showed higher affinity than the original lead compound 19-20a (pKi = 6.186 ± 0.028), reducing the MS marker binding to less than 10%. As previously, hydrazones identified were resynthesized and the binding assay confirmed their higher affinity compared to the original lead compound 19-20a. Hydrazone 19-22a, containing a 2′,4′-dichloro biphenyl moiety emerged as the most potent lead compound with an affinity in the lower nanomolar range, displaying a pKi = 8.094 ± 0.098, which is 2 log units higher than the original lead compound 19-20a (Scheme 8). 2′,4′-Disubstituted biphenyl rings featuring chloride and to a lesser extent fluoride substituents appear to be crucial to improve the affinity for mGAT1. The MS-based binding assay not only confirmed the good potency of all compounds screened but is also in good agreement with the deconvolution studies.
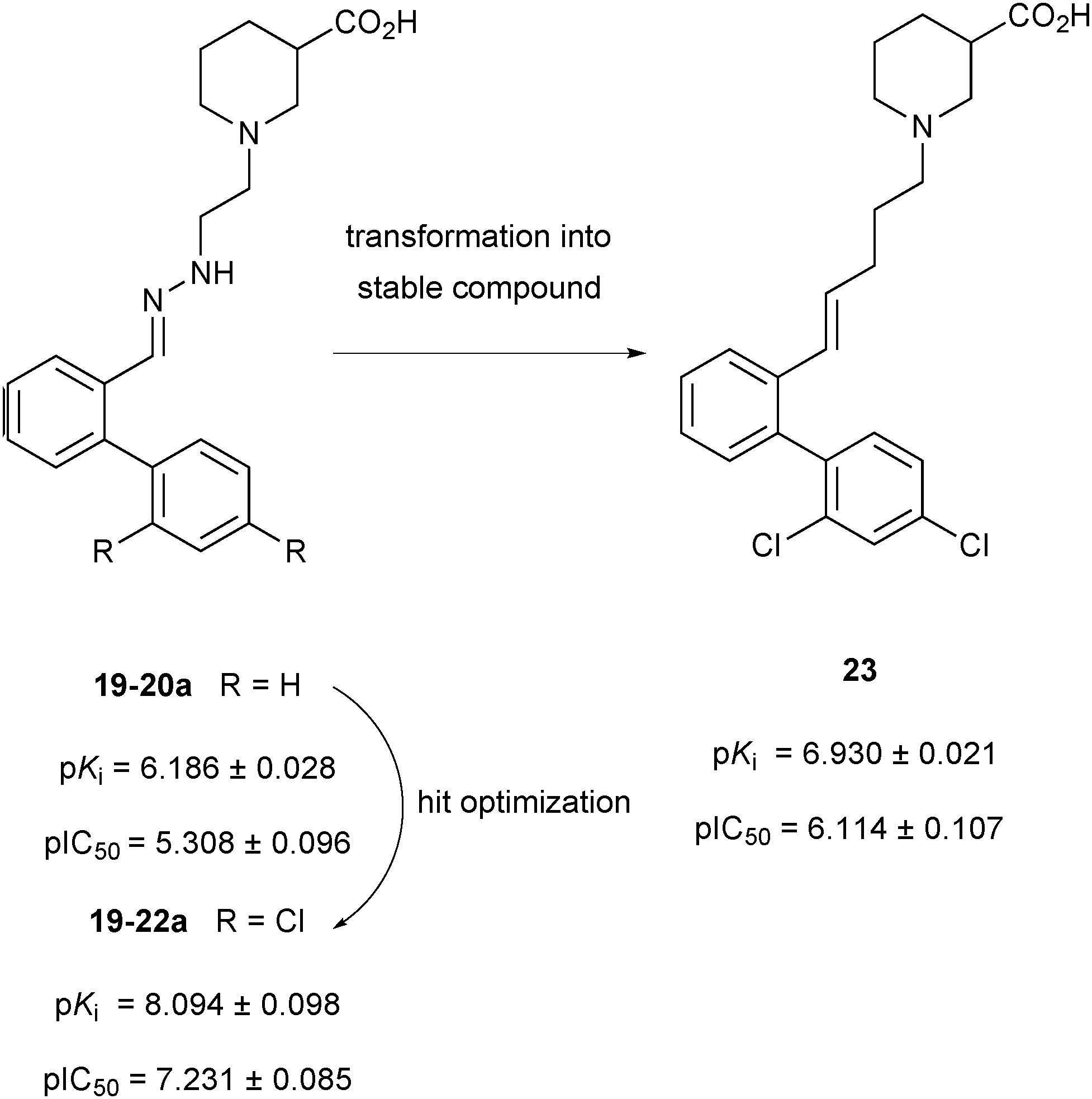 | ||
| Scheme 8 Schematic representation of compounds 19-20a, 19-22a and 23 with their pKi and pIC50 values against the transporter mGAT1.32 | ||
To develop the hydrazone compounds identified into lead compounds for drug development, five stable carba-analogues were synthesized and tested for binding affinity. Like before, the 2′,4′-dichloro biphenyl-substituted analogue 23 emerged to be the most potent mGAT1 binder with a pKi about 1 log unit lower than 19-22a (Scheme 8). This could be the result of the decreased polarity of the carba-analogue compared to the original hydrazone derivative. Taken together, both studies demonstrate that the competitive MS screening assay of pseudostatic hydrazone libraries is a powerful and efficient hit-identification strategy, providing invaluable SAR data, which can be used for the development of stable lead compounds with similar affinity to that of the initial hydrazine hits.
Nucleophilic catalysis of acylhydrazone equilibration was first demonstrated by the group of Greaney in 2010.20 Until this report, acylhydrazone chemistry was considered to be only of limited use for protein-templated DCC given the requirement for acidic pH, which is incompatible with most protein targets. This study has shown that in the presence of target protein, the reversible formation of acylhydrazones can be achieved by using aniline as a nucleophilic catalyst at relatively less acidic pH. In the presence of aniline, DCLs are fully reversible and equilibrate in 6 hours at pH 6.2 instead of 5 days without aniline. The equilibrium of the DCLs can be switched on and off by changing the pH. These DCLs were templated with two isozymes of glutathione S-transferase (GST), which play an important role in cell detoxification and are emerging targets for the treatment of drug resistance in chemotherapy and tropical diseases, and different amplification effects were observed.
DCLs were composed of one aldehyde 24, derived from the known GST substrate chloro-2,4-dinitrobenzene, and a 2.5-fold excess of each of the ten hydrazides 25a–j (eight acyl and two sulfonyl hydrazides) in presence of 10 mM aniline at pH 6.2 (Scheme 9a). The large excess of hydrazides ensures pseudo first-order behavior and also faster equilibration rates with respect to the aldehyde. Two recombinant GST isozymes were exposed to the pre-equilibrated DCLs, and amplification was analyzed. LC-MS analysis showed that in each of the DCLs a different acylhydrazone was amplified: thiophene acylhydrazone 24-25g was amplified in presence of SjGST and t-butylphenyl hydrazone 24-25c in presence of hGST (Scheme 9c). Owing to the poor solubility of the acylhydrazones 24-25g and 24-25c, their accurate IC50 values could not be determined. To overcome this problem and at the same time to anchor the compounds at the active site, the DCLs were regenerated in presence of glutathione (26, GSH)-conjugated aldehyde 27 and the same ten hydrazides 25a–j (Scheme 9a and b). As before, similar amplification was observed for both GST targets: thiophene acylhydrazone 27-25g was selected by SjGST and t-butylphenyl acylhydrazone 27-25c by hGST (Scheme 9c). To confirm that the amplification effects are not due to target-accelerated synthesis, SjGST was added to the pre-equilibrated DCC, and the same equilibrium distribution was achieved. Compounds 27-25g and 27-25c were found to have IC50 values of 22 and 57 μM against SjGST and hGST, respectively.
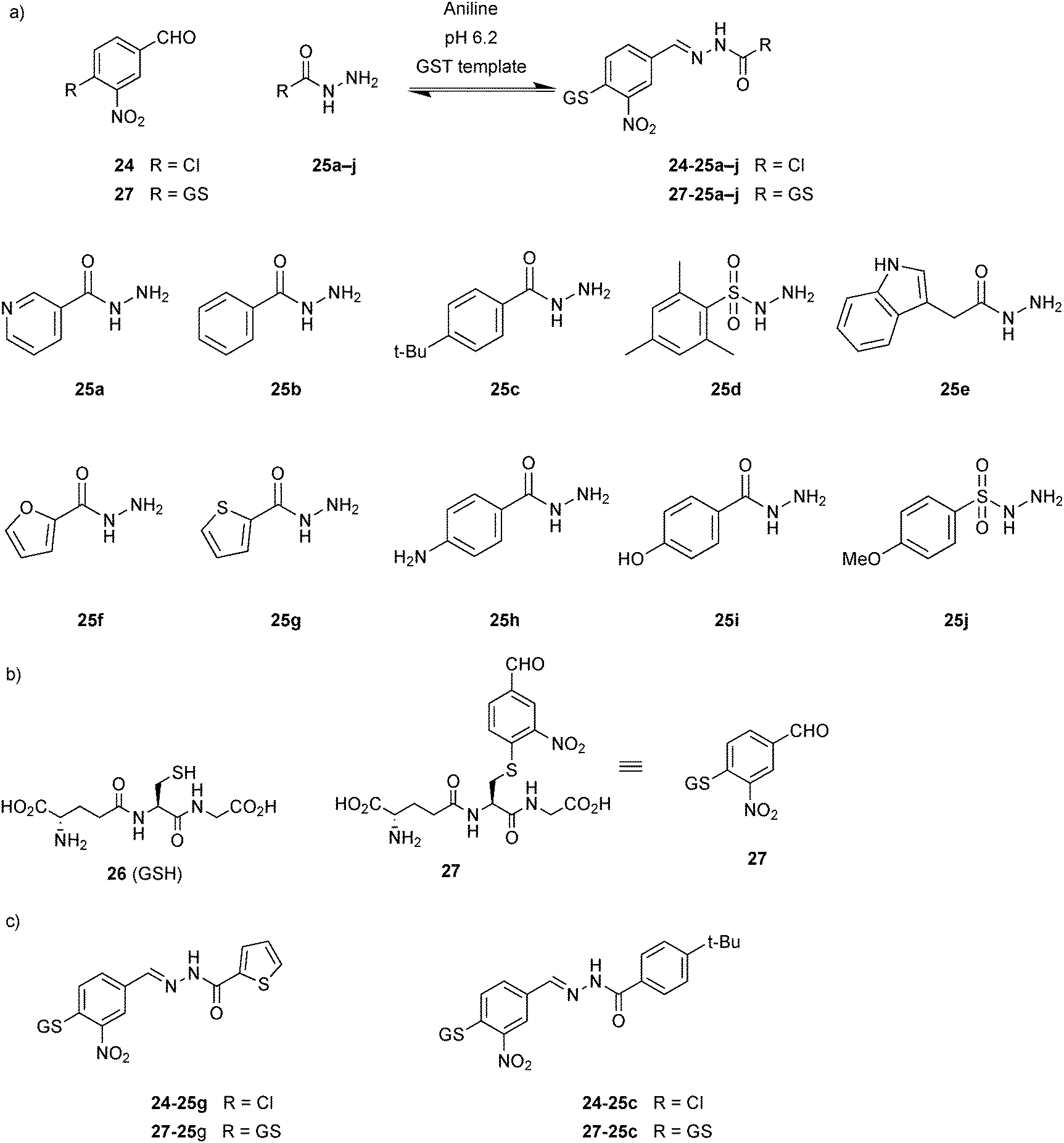 | ||
| Scheme 9 Aniline-catalyzed generation of acylhydrazone-based dynamic combinatorial libraries of (a) aldehydes 24 or 27 and ten hydrazides 25a–j. (b) Structures of glutathione 26 (GSH) and glutathione-conjugated aldehyde 27. (c) Acylhydrazones amplified in the presence of GST isozymes.20 | ||
Building on this initial study, Greaney and co-workers developed stronger, bivalent acylhydrazone inhibitors of GST isozymes, leading to the most potent inhibitor of the M-class of this enzyme to date.46 The majority of GST isozymes exist as homodimers. A bivalent DCC approach has the potential to lead to more (diverse) compounds using the same number of building blocks and more potent and selective inhibitors and represents a novel concept in protein-templated DCC. Potential drawbacks might be the poor solubility of bisacylhydrazones, a more challenging analysis as well as less straightforward synthesis. A bivalent DCC protocol was designed to selectively target the GST isozymes. The DCL was constructed using three nitro-substituted aldehydes 24, 27 and 28, which are derivatives of chloro-2,4-dinitrobenzene, a weakly binding substrate of all GSTs and four bivalent hydrazide linkers 29a–d of varying length and lipophilicity. A DCL of potentially 24 homo- and hetero-bis-acylhydrazones as well as the mono-acylhydrazones, excluding E/Z isomers, was generated in presence of 5 mM aniline at pH 6.2. Four GST isozymes (mGSTM1-1, hGST1-1, SjGST and mGSTA4-4) were exposed to the pre-equilibrated DCL, and the best results were obtained for M-class and Sj isozymes, with the most significant amplification of over 600% by the M-class for homo-bis-acylhydrazone 27-29c-27 (Scheme 10).
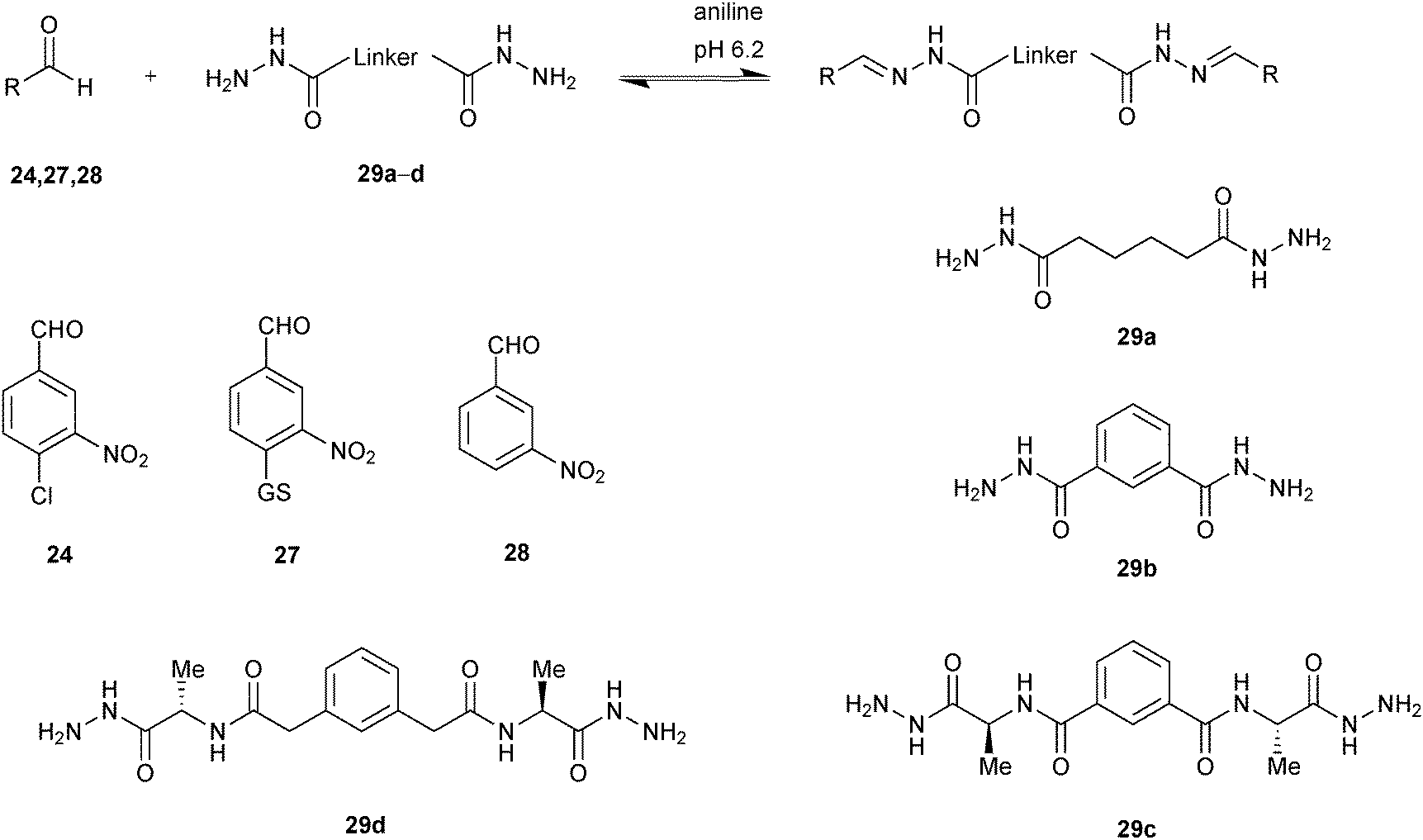 | ||
| Scheme 10 Aniline-catalyzed generation of acylhydrazone-based dynamic combinatorial libraries of aldehydes 24, 27, 28 and bishydrazides 29a–d for templating by GSTs.46 | ||
On the one hand, compounds containing the GSH-tagged aldehyde 27 (27-29c-27, 27-29a-27, 27-29b-27 and 27-29d-27) are strongly responsive to mGSTM1-1 (Fig. 2). On the other hand, the DCL is unresponsive to the mGSTA4-4 isozyme. In contrast, hGSTP-1 appeared to interfere with the DCL equilibrium.
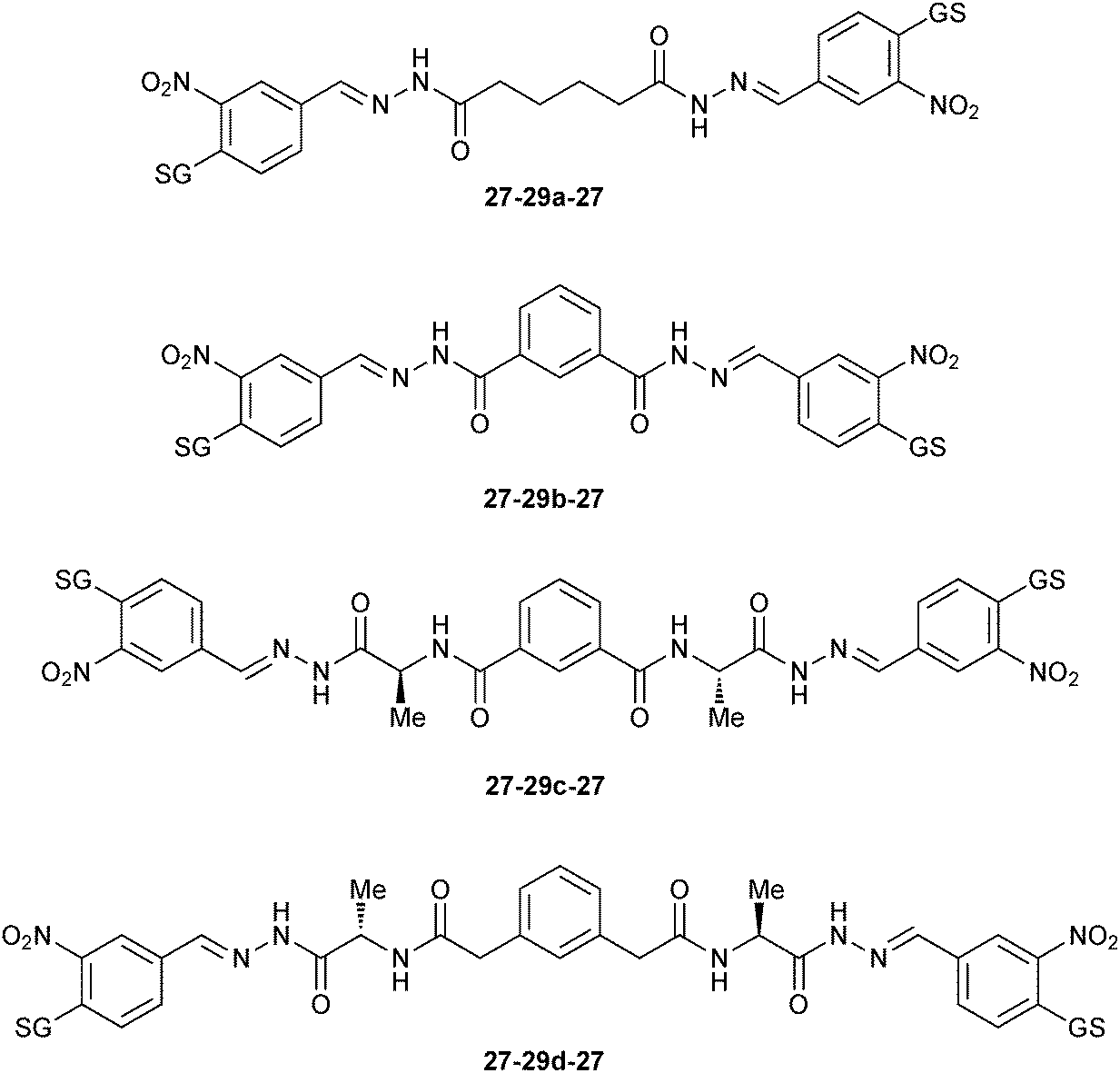 | ||
| Fig. 2 Amplified GSH-tagged homo-bis-acylhydrazones 27-29-27 in presence of mGSTM1-1 isozyme.46 | ||
The biochemical data correlate well with the amplification factors for the mGSTM1-1 isozyme: the inhibitor 27-29c-27 (IC50 = 50 nM) is nearly ten-fold more potent against GST than the other bis-acylhydrazone products. The inhibition values for SjGST correlate less well with the amplification factors. Unfortunately, inhibition data for hGSTP1-1 could not be correlated with amplification factors, presumably owing to the lack of a defined equilibration (Table 8). To elucidate the binding mode of inhibitor 27-29c-27, it was overlaid onto the crystal structure of hGST M1A-1A (PDB code: 1XWK) containing the ligand glutathione S-2,4-dinitrobenzene.47 The model indicates that the linker 29c can span the dimer interface of hGST M1A-1A without introducing any unfavorable interactions.
| Acylhydrazone | IC50 (μM) | |||
|---|---|---|---|---|
| mGSTM1-1 | hGSTP1-1 | SjGST | mGSTA4-4 | |
| 27-29c-27 | 0.050 | 13.45 | 0.989 | >100 |
| 27-29a-27 | 1.207 | 126.5 | 3.471 | >100 |
| 27-29b-27 | 0.337 | 11.81 | 0.252 | >100 |
| 27-29d-27 | 0.413 | 0.356 | 1.8 | >100 |
| Aldehyde 24 | 341.7 | ≥500 | 265.6 | >500 |
| Hydrazide 29c | >500 | >500 | ||
Taken together, both studies by Greaney and co-workers show that use of aniline as a nucleophilic catalyst makes acylhydrazone formation amenable to protein-templated DCC with a large range of protein targets. This reversible reaction has numerous advantages such as easily accessible, versatile building blocks that afford products with the right kinetic and thermodynamic balance enabling facile and direct analysis and, in this case, afforded DCLs displaying isozyme selectivity, leading to the most potent and selective inhibitor of one of the isozymes reported to date. Furthermore, a bivalent DCC protocol should be of particular use for the discovery of inhibitors targeting large protein surface areas.
In 2014, we demonstrated for the first time that the combination of de novo structure-based design (SBD) and DCC is a powerful technique for hit identification and -optimization.11 Whereas SBD is widely used for hit optimization, there are only a few reports of true de novo SBD. We used endothiapepsin as the protein target for this proof-of-concept study. Endothiapepsin belongs to the class of pepsin-like aspartic protease, which play a causative role in numerous diseases such as hypertension or malaria.48 We used two crystal structures of endothiapepsin, representing two alternative binding modes (with and without a crystallographically localized water molecule) to design a library of acylhydrazones, which address the catalytic dyad of the endothiapepsin either directly via hydrogen bonding or hydrogen-bonding interactions via the catalytic water molecule. We used acylhydrazone chemistry as it appeared to be the most suitable central scaffold for efficient anchoring to the active site through a strong hydrogen-bonding network with the catalytic dyad. In addition, the pH optimum of endothiapepsin is 4.5, making it compatible with acylhydrazone chemistry in the absence of aniline as a catalyst. Retrosynthesis of these designed acylhydrazones led to five hydrazides 30a–e and five aldehydes 31a–e, which were used to generate the DCLs (Scheme 11). We used saturation-transfer difference NMR (1H-STD-NMR) spectroscopy to identify which component associations are bound to the enzyme. 1H-STD-NMR spectroscopy enables the direct characterization of protein–ligand interactions in solution and is therefore a powerful technique for the analysis of DCLs. There is only one other report of DCLs analyzed by 1H-STD-NMR spectroscopy by the group of Ramström, which we will discuss in the section on hemithioacetal-based DCC.15 To simplify the analysis of the DCL, we divided the whole library into five sub-libraries, each containing five hydrazides and one aldehyde, potentially forming five acylhydrazones and identified the acylhydrazones bound to the protein by comparing the imine-type and α-carbon proton signals in the 1H-STD-NMR spectra after addition of endothiapepsin to the pre-equilibrated DCLs.
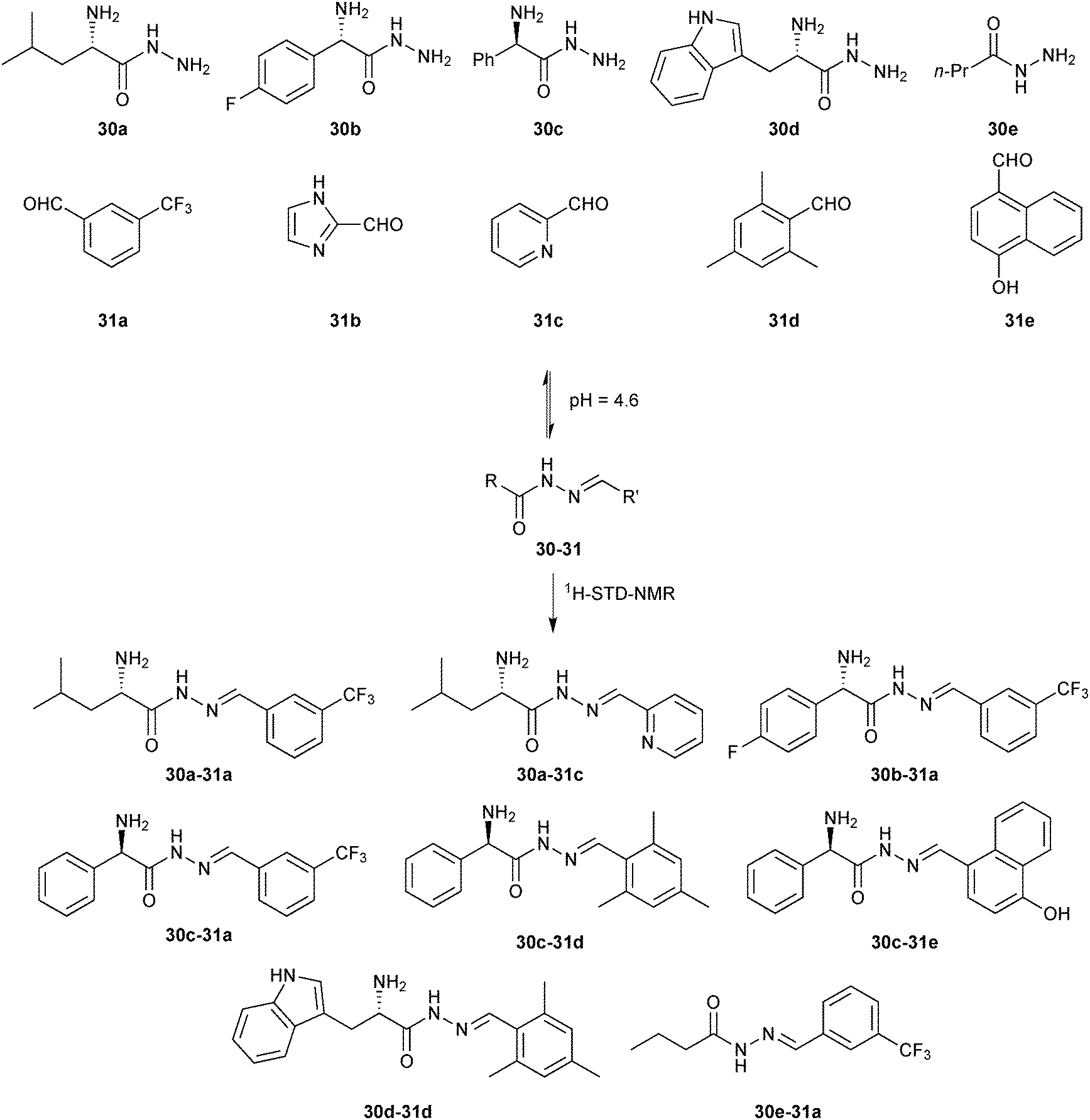 | ||
| Scheme 11 Generation of acylhydrazone-based dynamic combinatorial libraries of hydrazides 30a–e and aldehydes 31a–e for analysis by 1H-STD-NMR analysis of binding to the aspartic protease endothiapepsin.11 | ||
We identified a total of eight acylhydrazones 30-31 from five sub-libraries (Scheme 11). A fluorescence-based enzyme-activity assay confirmed that all eight hits from the 1H-STD-NMR analysis are indeed inhibitors of endothiapepsin with IC50 values in the range of 13–365 μM. The most potent inhibitors 30c-31e and 30d-31d display IC50 values of 14.5 μM and 12.8 μM respectively, which correlate with the calculated free energies of binding (Table 9). Subsequent co-crystal structure determination of 30c-31e (PDB code: 3T7P) and 30d-31d (PDB code: 4KUP) in complex with endothiapepsin at 1.25 Å and 1.31 Å resolution, respectively, confirmed the in silico prediction that either direct or water-mediated hydrogen-bonding interactions with the catalytic dyad can be achieved (Fig. 3).
| Inhibitors | IC50 (μM) | K i (μM) | ΔGEXPTa (kJ mol−1) | ΔGHYDEb (kJ mol−1) |
|---|---|---|---|---|
| a The Gibbs free energy of binding ΔGEXPT was derived from the experimentally determined IC50 values. b The calculated Gibbs free energy of binding ΔGHYDE was calculated using the HYDE scoring function in the LeadIT suite. c Result of the docking run without catalytic water molecule (PDB code: 3PBD). d Result of the docking run with catalytic water molecule (PDB code: 3PI0). | ||||
| 30a-31a | 150 ± 17 | 71 ± 8 | −24 | −28c |
| 30a-31c | 365 ± 95 | 172 ± 45 | −22 | −17d |
| 30b-31a | 177 ± 13 | 83 ± 6 | −23 | −23c |
| 30c-31a | 352 ± 13 | 166 ± 6 | −22 | −26c |
| 30c-31d | 206 ± 19 | 97 ± 9 | −23 | −25c |
| 30c-31e | 14.5 ± 0.5 | 7 ± 0.2 | −30 | −26d |
| 30d-31d | 12.8 ± 0.4 | 6 ± 0.2 | −30 | −32c |
| 30e-31a | Insoluble | — | −23c | |
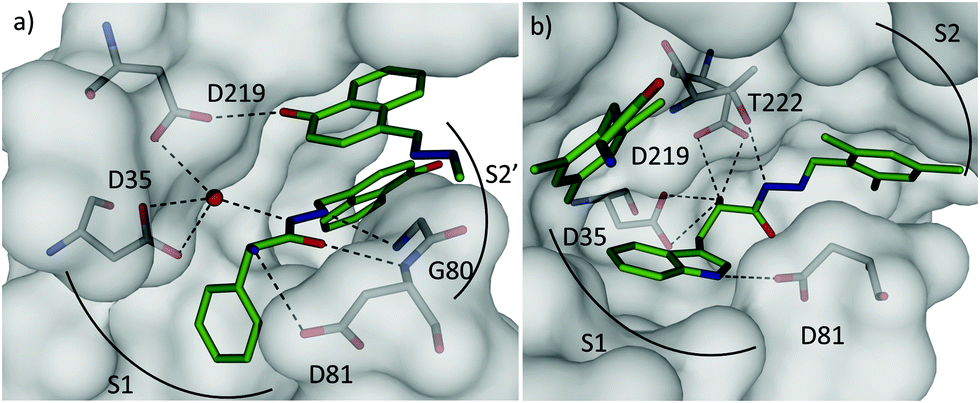 | ||
| Fig. 3 X-ray crystal structures of endothiapepsin co-crystallized with ligands: (a) overview of the two molecules of 30c-31e (C: green, water molecules: red spheres) bound in the active site (PDB code: 3T7P). The central ligand binds to the catalytic dyad (D35 and D219) via catalytic water molecule. (b) Overview of two molecules of 30d-31d (C: green, PDB code: 4KUP). The central molecule binds D35 and D219 directly through its α-amino group.11 | ||
Our report shows for the first time that the combination of DCC and de novo SBD constitutes an efficient and synergistic approach for hit identification and -optimization of the first acylhydrazone-based inhibitors of aspartic proteases but could be extended to potentially any biological target. The binding mode of the most potent hits was validated by protein X-ray crystallography. The combination of techniques should make de novo design less risky given that a certain degree of flexibility can be taken into account during the design stage to account for potentially flexible regions of the protein target. As a result, it should become more widely accessible and find application in a whole range of other classes of drug targets, especially in the early stages of drug discovery.
C–C bond formation: alkene cross metathesis
Cross metathesis (CM) was first introduced by the group of Nicolaou, employing a biphasic reaction medium to connect vancomycin monomers in the presence of dipeptides of the type D-Ala-D-Ala.49In 2006, the group of Poulsen first introduced CM to protein-templated DCC, constituting the first report with a protein target.12 They used an FBDD approach exploiting DCC to identify small-molecule inhibitors for bovine carbonic anhydrase II (bCA II). bCA II is an attractive drug target as it is responsible for glaucoma, epilepsy, gastric ulcers and cancer tumor progression. To facilitate the proof-of-principle studies, an allylic ester benzene sulfonamide 32 was used as an anchor. The bifunctional building block 32 was designed so as to contain both an aromatic sulfonamide moiety and a terminal alkene substituent for recognition of bCA II and to enable CM, respectively. Ten diverse terminal alkenes 33a–j (Fig. 4a) were chosen for CM with the anchor building block 32 to explore additional favorable interactions, which might complement the established binding of an aromatic sulfonamide fragment by bCA II.
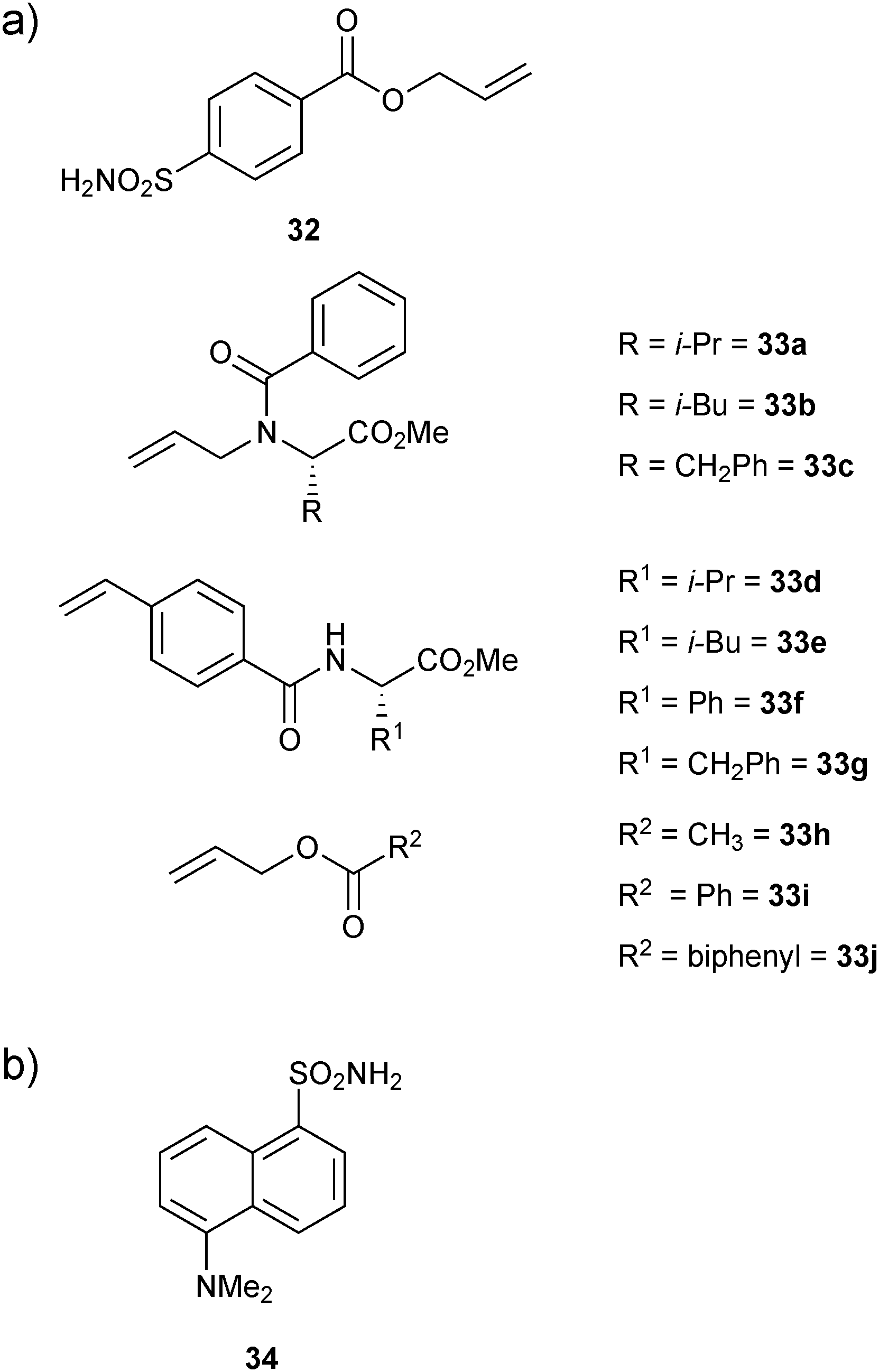 | ||
| Fig. 4 (a) Building blocks 32 and 33a–j used for generation of dynamic combinatorial libraries using alkene cross metathesis for templating by bovine carbonic anhydrase II. (b) Structure of 34 used in competitive fluorescence assay.12 | ||
A total of ten DCLs were designed, each containing scaffold building block 32 and each individual terminal alkene (33a–j), resulting in the formation of three-component DCLs, consisting of homodimers 32-32–33j-33j, etc. individually and heterodimer 32-33a, 32-33b, etc. (Scheme 12). To facilitate the formation of heterodimers, a ten-fold excess of each terminal alkene (33a–j) was used with respect to compound 32 in each DCL. The CM was performed in presence of 20% Grubbs first-generation catalyst immobilized on a polystyrene support in dichloroethane as a solvent. For analysis by ESI-MS and a competitive fluorescence-based assay with the ligand 5-(dimethylamino)-1-naphthalenesulfonamide 34 (dissociation constant Kd (34) = 0.3 μM, Fig. 4b), samples of the pre-equilibrated DCLs21,45 were diluted with acetonitrile and water, respectively.
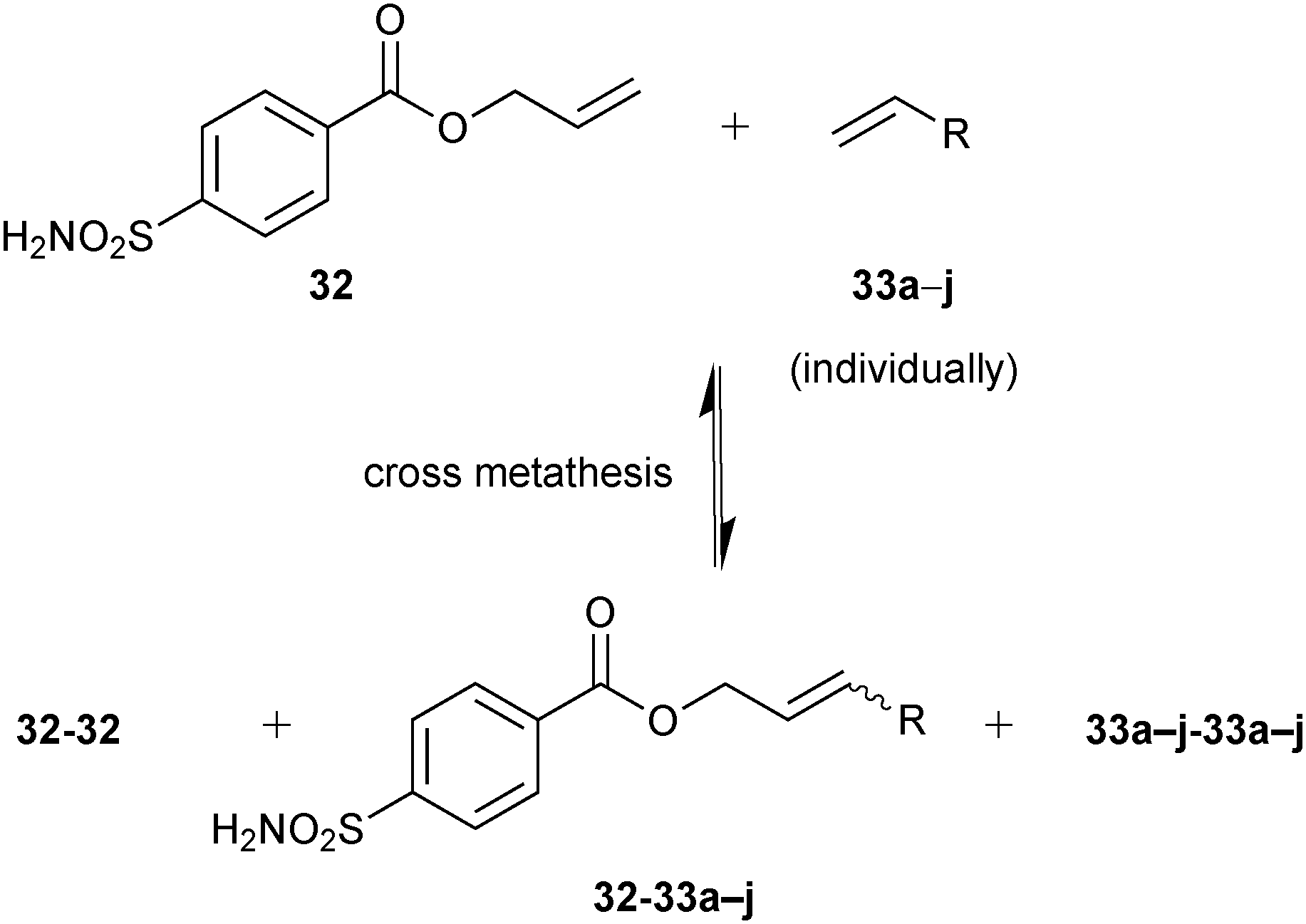 | ||
| Scheme 12 Generation of pre-equilibrated dynamic combinatorial libraries using alkene cross metathesis of building blocks 32 and each of 33a–j individually for templating by bCA II.12 | ||
Screening of the DCLs using the competitive binding assay, revealed that the CM product with the highest affinity is 32-33h featuring a terminal acetate group, which displaces 34 by 77%. Weaker binders 32-33i and 32-33j displace 34 by 45% and 37%, respectively. Next to the heterodimers, only one binding homodimer 32-32 was detected with 88% displacement of 34 (Fig. 4b).
To validate the screening results obtained from crude DCLs, dimers 32-32 and 32-33h–j were synthesized and tested for bCA II binding. They were found to display Ki values of 5.1, 4.9, 6.6 and 8.5 nM, respectively (Table 10). These results are in good agreement with the screening results. This report represents the first application of both heterogeneous catalysis and CM as a reversible reaction in protein-templated DCC. The approach proved to be very efficient in identifying inhibitors of bCA II and has two main advantages: firstly, the good agreement between the affinities for the target obtained from the screen of the DCL without and with prior purification indicates that synthesis and purification of individual CM products can be avoided. Secondly, use of a heterogeneous rather than a homogeneous catalyst enables straightforward control over the reaction by simple filtration and re-addition of the catalyst. Use of a water-soluble Grubb's catalyst should circumvent the requirement for the less elegant pre-equilibrated DCC strategy when using CM as a reversible reaction.50
| Compounds | Structures | K i (nM) |
|---|---|---|
| a bCA II binding data using competitive displacement of 34 from bCA II, Kd (34) = 0.3 μM. | ||
| 32-32 |
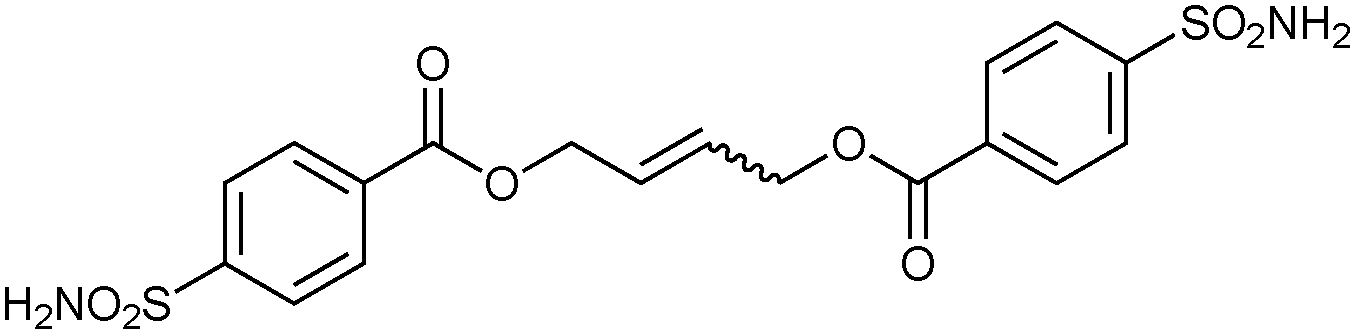
|
5.1 |
| 32-33h |
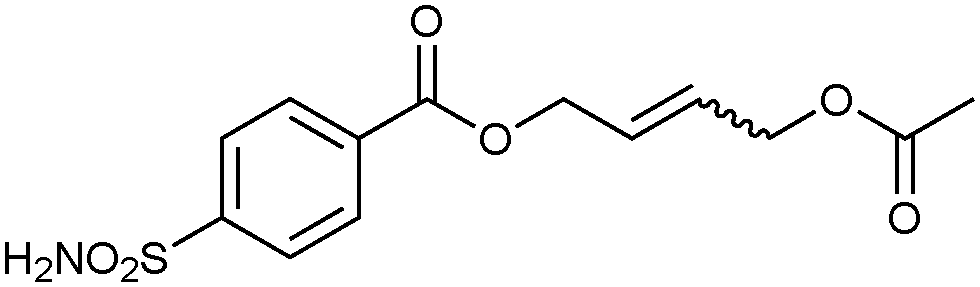
|
4.9 |
| 32-33i |
|
6.6 |
| 32-33j |
|
8.5 |
C–S bond formation
For their proof-of-concept study, Ramström and co-workers used a selection of five thiols 35a–e and two aldehydes 36a and 36b based on the known substrate o-nitrophenyl-β-galactopyranoside (37) and the inhibitor isopropyl-1-thio-β-D-galactopyranoside (38) of β-galactosidase (Scheme 13) to construct a DCL of potentially ten hemithioacetals (20 stereoisomers). Both aldehydes are in equilibrium with their hydrates 39a and 39b (Scheme 13). 1H-STD-NMR spectroscopy was used to identify the binders from the DCL, which we already introduced in the section on acylhydrazone-based DCC. Given that hemithioacetal formation and dissociation are fast with respect to the NMR relaxation time at pH = 7.2, required for protein stability, these conditions lead to virtual DCL formation; only in the presence of enzyme, those hemithioacetal products that also bind to the target are formed, leading to a substantial reduction in the signal-to-noise ratio. 1H-STD-NMR analysis of the DCL supported the hypothesis that only hemithioacetals containing galactose would be selected by the protein: the proton signal from compound 35a and both of the aldehydes 36a and 36b and their hydrates 39a and 39b are clearly visible. Competition experiment showed that only the first compound binds specifically to the target. Comparison of 1H-STD-NMR spectra of the full DCL with subsequent reference experiments suggested that hemithioacetals 35a-36a and 35a-36b, containing galactose, are interacting with β-galactosidase.
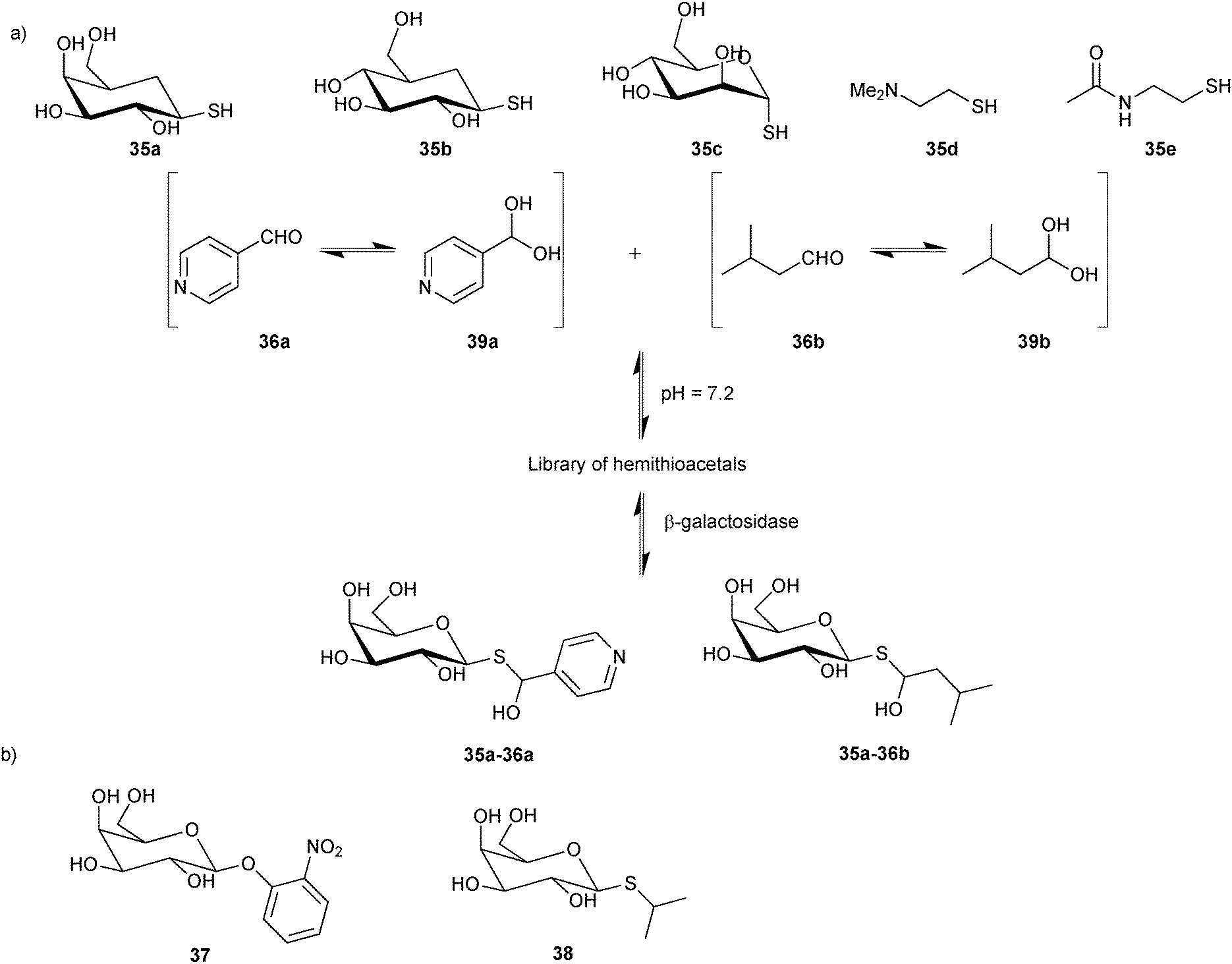 | ||
| Scheme 13 (a) Generation of hemithioacetal-based virtual dynamic combinatorial libraries of thiols 35a–e and aldehydes 36a and 36b for analysis by 1H-STD-NMR spectroscopy for binding to β-galactosidase. (b) Known substrate 37 and inhibitor 38 of β-galactosidase.15 | ||
To confirm the 1H-STD-NMR results, inhibition assays were performed for each combination of thiol and aldehyde using the substrate 37. The hydrolysis of substrate 37 by β-galactosidase to o-nitrophenol was monitored in presence and absence of each dynamic system. The hemithioacetals 35a-36a, 35a-36b and thiol 35a were confirmed as the most potent inhibitors, decreasing the rates of substrate hydrolysis by 12-, 4- and 2-fold, respectively. The combined results of 1H-STD-NMR spectroscopy, enabling in situ identification of the binders, and the inhibition assay confirmed the first successful application of hemithioacetal formation in protein-templated DCC for the discovery of β-galactosidase inhibitors. The rapidly equilibrating system gives rise to virtual DCLs, which enable a rapid and efficient analysis. Given the labile nature of hemithioacetals, however, transformation into stable analogues is an absolute requirement for this reversible system.
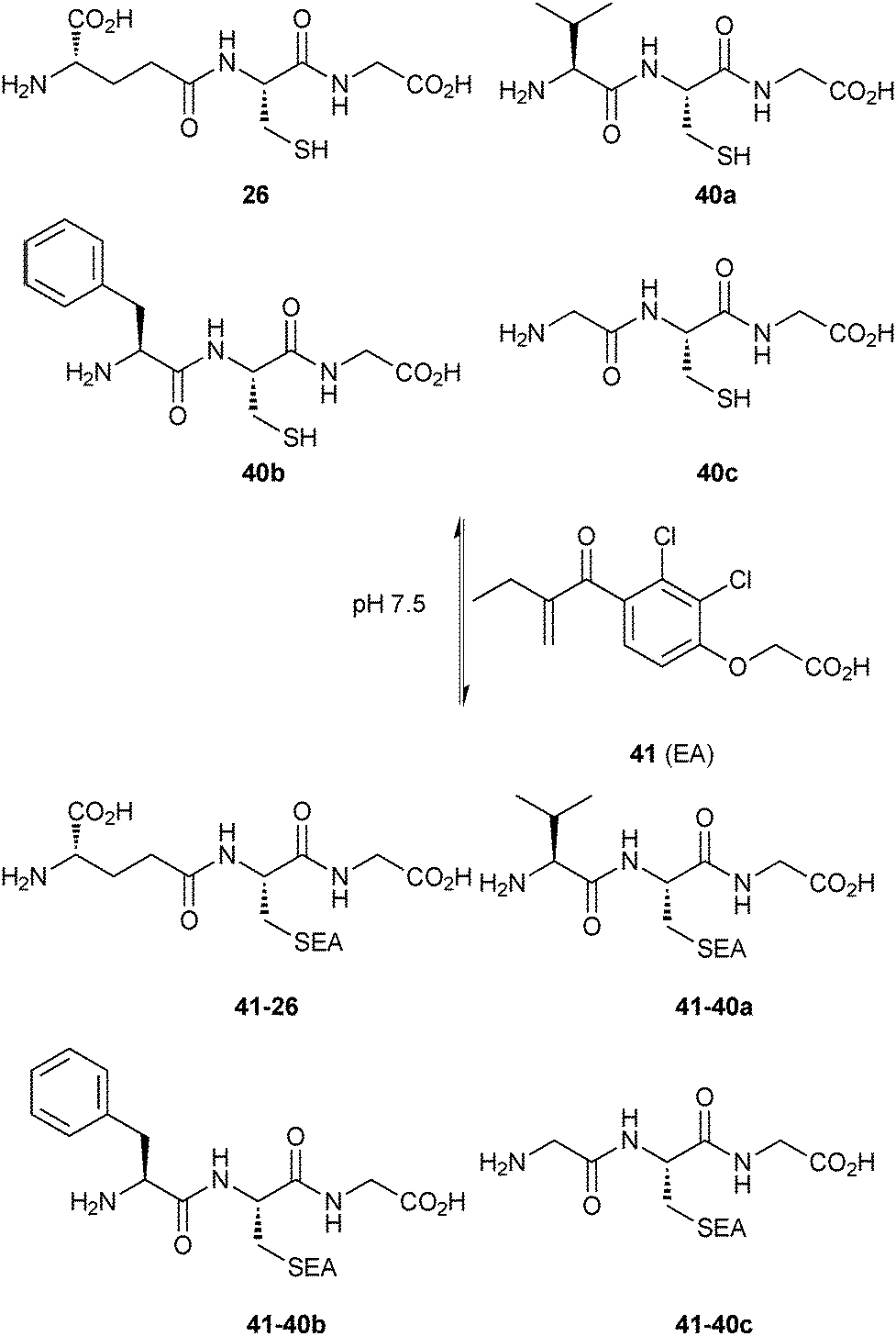 | ||
| Scheme 14 Generation of thioether-based dynamic combinatorial libraries of thiols 26 and 40a–c and enone 41 for templating by GST.18 | ||
The thiols 26, 40a–c and the enone 41 afforded four thioethers 41-26 and 41-40a–c, which reached equilibrium in 1 hour and re-equilibrated upon addition of SjGST, collapsing to only one thioether 41-26 in 10 minutes, indicating that the enzyme accelerates the conjugate addition. Addition of the enzyme to the pre-equilibrated DCL, led to the same result but at a substantially slower rate (6 days), which suggests that the catalytic effect of SjGST on the conjugate addition does not affect the equilibrium distribution of the DCL. These experiments indicate that the amplified product 41-26 is a good binder of SjGST. To confirm that the binding affinity and amplification correlate with each other, the only amplified compound 41-26 and a non-amplified compound 41-40c were synthesized and subjected to the standard GST inhibition assay.53 Inhibition results showed IC50 values of 0.32 μM and 88 μM for 41-26 and 41-40c, respectively, demonstrating that the observed amplification is clearly related to binding affinity.
GSTs contain two binding sites, a GSH-binding site, which is highly conserved and a hydrophobic binding site, called H-site. To explore the H-site of GSTs, 14 enones 42a–n and 26 as a thiol building block were selected to construct a DCL (Scheme 15). The DCL was analyzed in the same way, and three amplified products 26-42a–c emerged. To investigate the binding affinity of these amplified products, compounds 26-42a and 26-42c, a non-amplified product 26-42d and a depleted adduct 26-42e were synthesized and their IC50 values determined to be 0.61 μM and 1.40 μM for 26-42a and 26-42c, respectively. Compounds 26-42d and 26-42e show IC50 values of 8.2 μM and 4.3 μM, which are ten- and seven-fold lower than compound 26-42a (Table 11). These results show that the extent of amplification correlates with the relative binding affinities of the DCL members and that the system is capable of differentiating between compounds differing in IC50 value by one order of magnitude. This study has established a novel reversible system for protein-templated DCC, namely the conjugate addition reaction of thiols to enones.
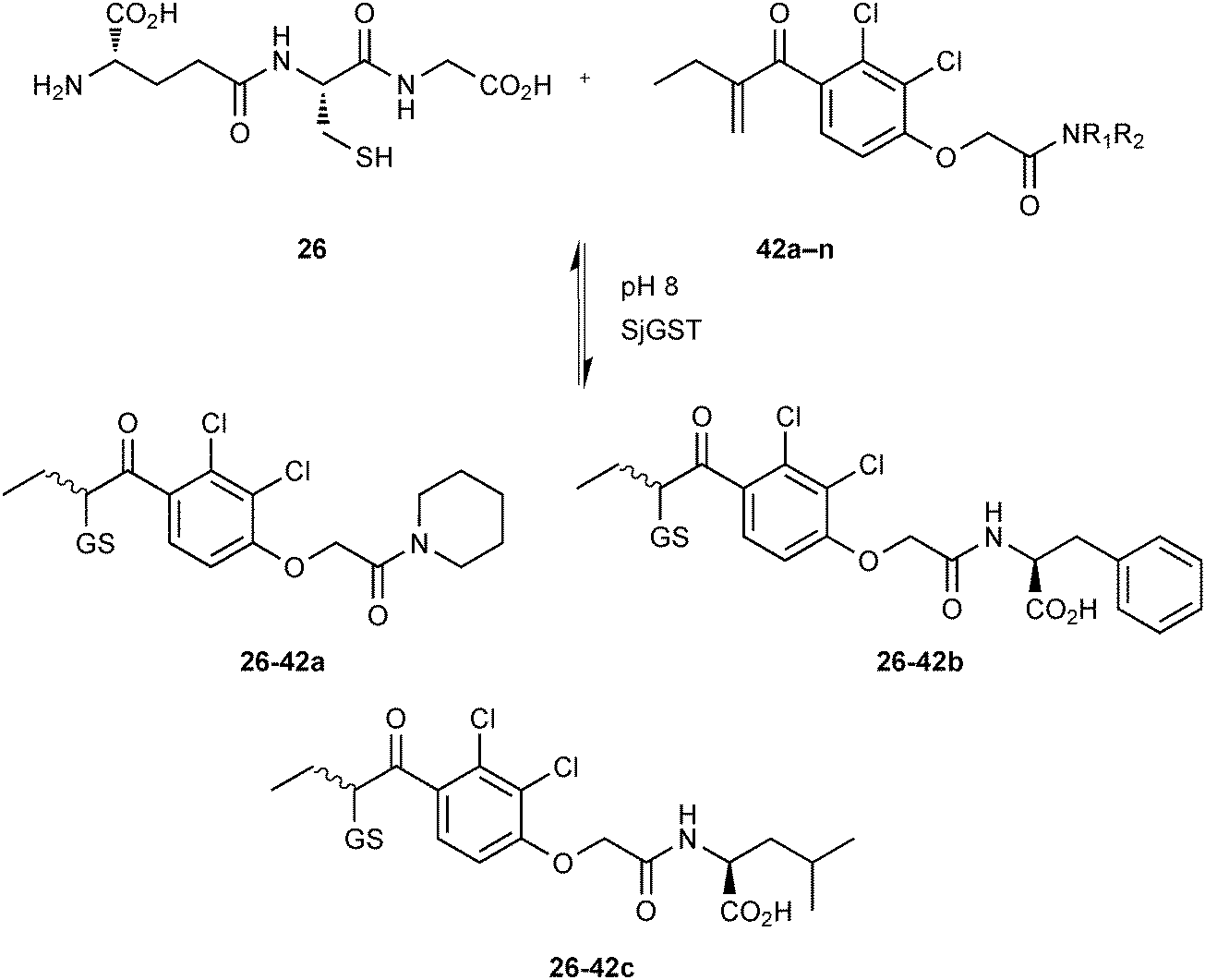 | ||
| Scheme 15 Generation of thioether-based dynamic combinatorial libraries of GSH (26) and fourteen enones 42a–n for templating by GST.18 | ||
| Inhibitor | Structure | IC50 (μM) |
|---|---|---|
| 26-42a |
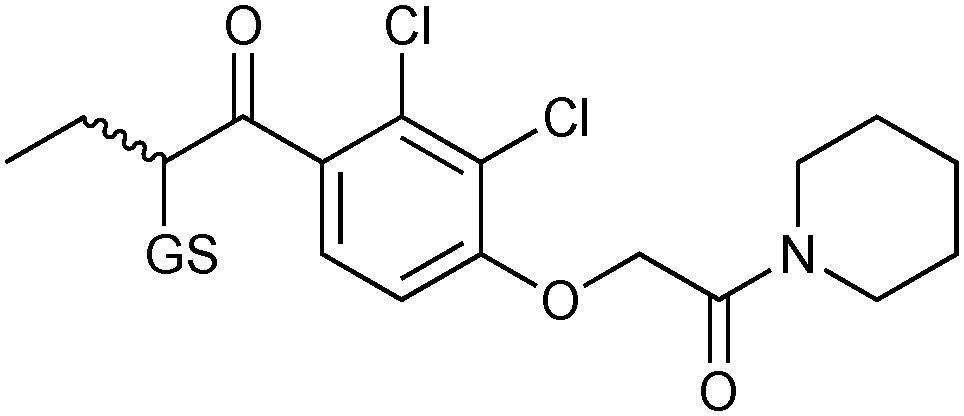
|
0.61 |
| 26-42c |
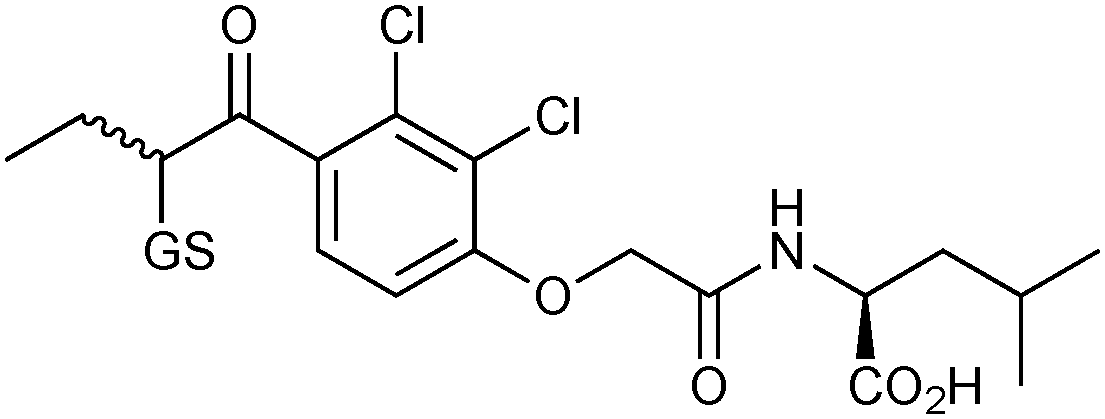
|
1.40 |
| 26-42d |
|
4.30 |
| 26-42e |
|
8.2 |
S–S bond formation
In 2008, the group of Schofield demonstrated that dynamic combinatorial mass spectroscopy (DCMS) analysis can be used to analyze DCLs of thiols/disulfides under non-denaturing conditions.14 DCMS had been introduced by Poulsen for the identification of bCA II inhibitors.19 BcII metallo-β-lactamase (BcII MBL) was used as a target for the proof-of-principle study. The MBL family of enzymes are clinically important as they catalyze the hydrolysis of almost all clinically used β-lactam antibiotics, therefore playing an important role in the development of resistance against this class of antibiotics.61 Dithiol 43, which is an analogue of a known inhibitor of the MBL family,62 was selected as a “support ligand”. In DCMS, the support ligand is anchored reversibly to the protein's active site using one thiol, leaving the second thiol free for disulfide-bond formation with a suitable fragment. In the case of BcII MBL, the support ligand is anchored to the protein through complexation to the active-site zinc cation using one of the thiols. Initially, it was shown, using an ESI-MS binding assay, that compound 43 binds to BcII:Zn2; it is likely that only one thiol chelates the Zn(II) cation, leaving the other available for disulfide-bond formation, which confirms that compound 43 is a suitable support ligand.
The first DCL was generated using a set of 19 thiols 44a–s and the support ligand 43 in presence of BcII (Scheme 16a). Analysis of the library using DCMS, showed the formation of BcII:Zn-disulfide complexes after 30 minutes of exposure to air. Thiophenols 44a–f had reacted with the support ligand 43 to form disulfide complexes BcII:Zn2-43-44a–f. To replace the labile disulfide bond of the compounds identified by a stable linker, a series of cysteine-based analogues were prepared and tested. Despite not being perfect bioisosteres (the amide linker used merely mimics the length and stereochemistry), the stable analogue 45 emerged as the most potent inhibitor with a Ki value of 740 nM, that is a 170-fold increase in inhibitory potency with respect to the anchor 43 (Scheme 16b).
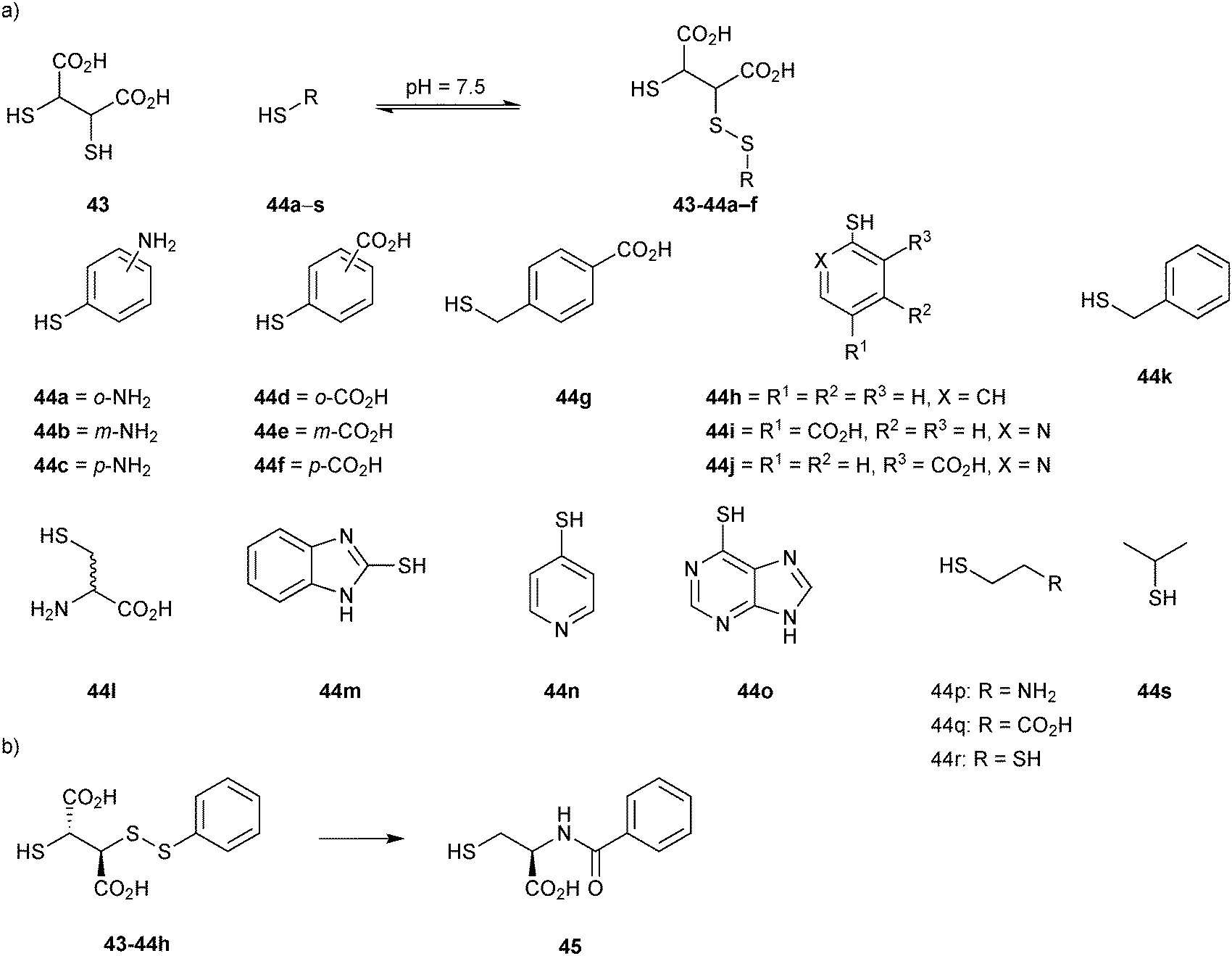 | ||
| Scheme 16 (a) Generation of disulfide-based dynamic combinatorial libraries of support ligand 43 and thiols 44a–s for templating by BcII. (b) Cysteine-based stable analogue 45 of disulfide 43-44h.14 | ||
As the first DCC revealed that the support-ligand 43 specifically forms disulfide bonds with thiophenols 44a–f to afford BcII:Zn-disulfide complexes, a second DCL was generated using a set of thiol-derivatives based on thiophenols 44a–f from the first DCL and the new thiophenols 45a–m as well as the support ligand 43 (Fig. 5). DCMS analysis and deconvolution experiments to distinguish between disulfides of identical/similar mass of the DCL indicated the formation of four disulfide complexes BcII:Zn2-43-44a–c, BcII:Zn2-43-44d–f, BcII:Zn2-43-45i and BcII:Zn2-43-45k. To validate the DCMS results, amide analogues 46 and 47 of disulfides 43-45k and 43-45m (the latter was included as a control given that it was not observed to bind) respectively, were synthesized and tested for inhibition of BcII. Despite remaining relatively potent, compounds 46 (Ki = 13.5 μM) and 47 (Ki = 8.4 μM) did not show any improvement in inhibitory potency, although MS binding affinity of 46 was significantly stronger than 47 for BcII:Zn2, which validated the DCMS results. Overall, this report demonstrates the efficiency of the DCMS technique for rapid identification of inhibitors, which, unlike most other analytical techniques enables direct analysis of the protein–ligand complexes. DCMS provides both the stoichiometry and binding strength of the protein–ligand interaction and can be applied even for protein targets for which the substrate is unknown, a clear advantage over an enzyme-activity assay.
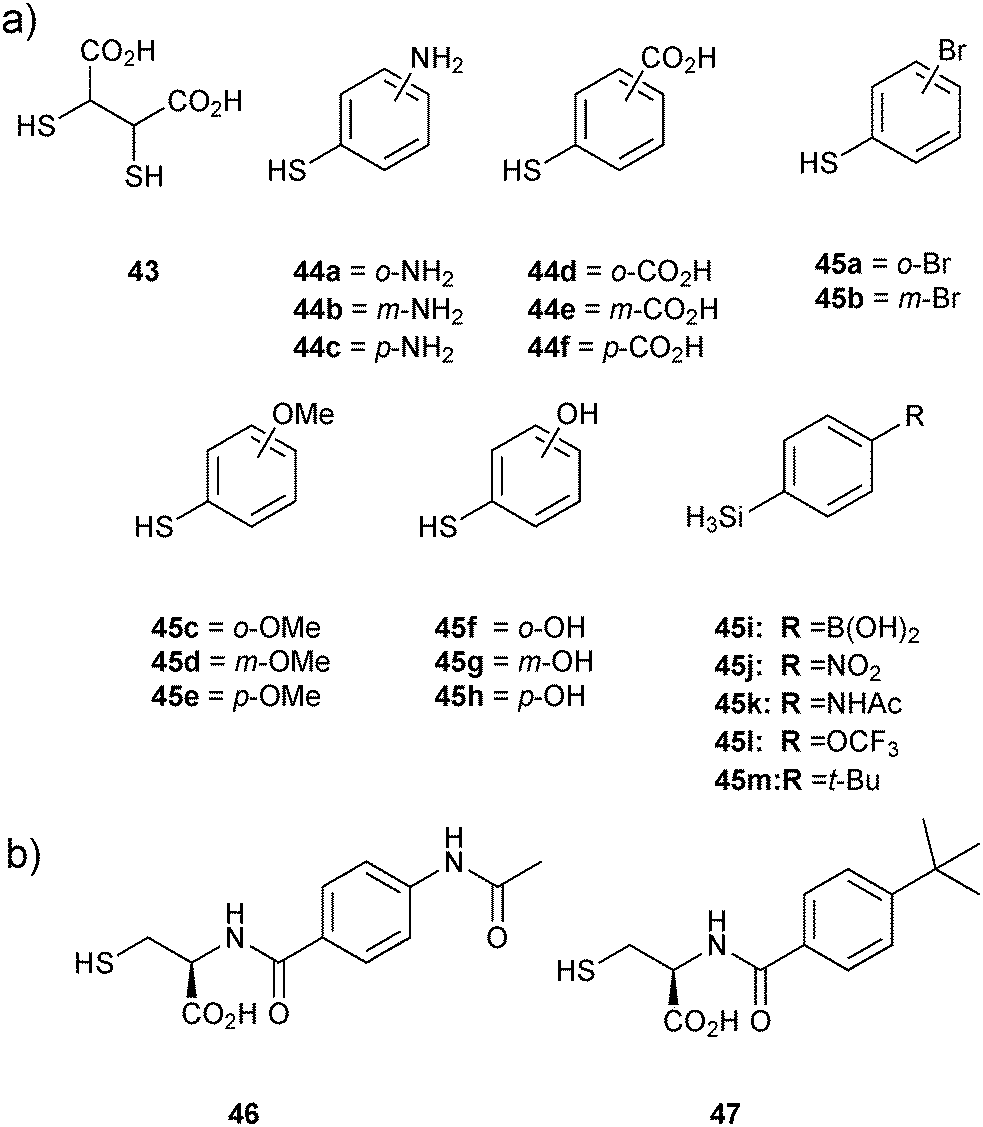 | ||
| Fig. 5 (a) Thiols used for generation of disulfide-based dynamic combinatorial libraries for templating by BcII. (b) Amide analogues 46 and 47 of disulfides 43-45k,m, respectively based on the second DCMS results.14 | ||
The main drawbacks of this method are that not all proteins can be used for non-denaturing MS analysis and that not all of the noncovalent interactions might be translated during the transition from solution to gas phase in the same manner.63 Up until now, multiple reports have shown reasonable agreement between the MS binding results and solution-phase data.14,19,56
Two years later, the established DCMS method14 was used by the same group based on disulfide formation for the ligand identification of JMJD2E, a member of the subfamily of JMJD2 histone demethylases, which plays a causative role in prostate cancer, leukemia, squamous cell carcinoma and mental retardation.56 An initial non-denaturing ESI-MS screen of a library of L- and D-enantiomers of N-oxalyl derivatives of amino acids was carried out based on the structure of 2-oxoglutarate (2OG, co-substrate of JMJD2E). The target was found to have a clear preference for the D-enantiomers. N-oxalyl-D-cysteine (48), which contains a free thiol group, emerged as a binder of JMJD2E, making it a suitable support ligand for DCMS (IC50 = 73 μM). Subsequently, the space available in the substrate-binding pocket of JMJD2E was explored using DCMS. In this study, the support ligand 48 is anchored to the active site through complexation by the N-oxalyl group of the Fe(II) cation at the active site. This binding mode leaves the thiol group free for disulfide-bond formation with thiol building blocks, generating a library of disulfides. The library was then analyzed using non-denaturing ESI-MS to identify which disulfide binds to the protein. A set of five thiols 49a–e and support ligand 48 were used to generate a library of disulfides in presence of JMJD2E (Fig. 6a). The library was then analyzed for binding with JMJD2E by non-denaturing ESI-MS, which revealed the formation of 48-49a·JMJD2E and 48-49b·JMJD2E complexes (Fig. 6b), which were further confirmed by deconvolution experiments.
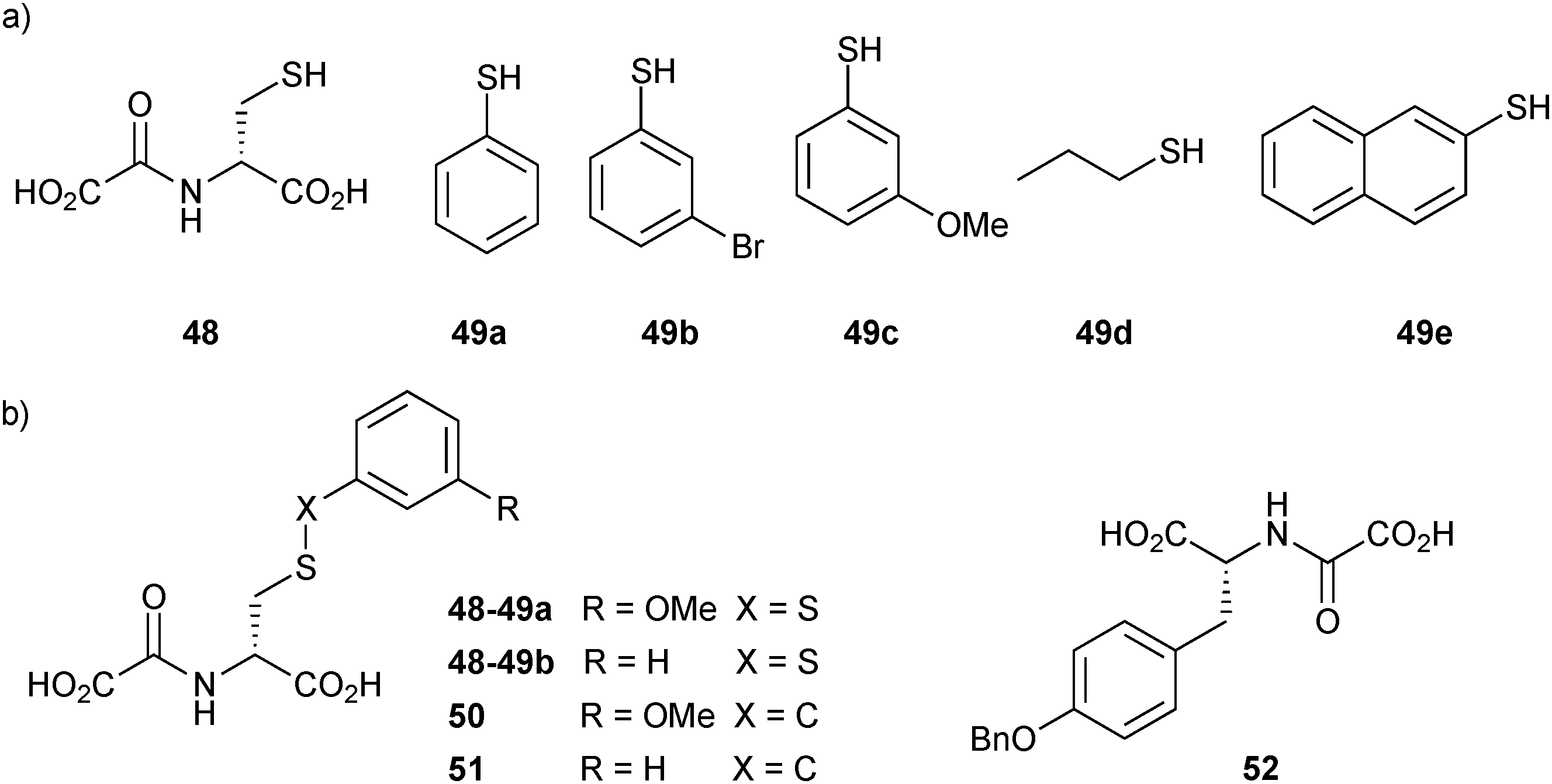 | ||
| Fig. 6 Structures of (a) the support ligand 48 and thiols 49a–e for the generation of disulfide-based dynamic combinatorial libraries for templating by histone demethylase JMJD2E. (b) Inhibitors identified (48-49) and their stable carbon analogues 50–52.56 | ||
Given that disulfides are not stable under the assay conditions, the corresponding carbon analogues 50 and 51, featuring a thioether in place of the disulfide linkage, were synthesized and found to be less potent than the support ligand 48 with IC50 values of 204 μM and 300 μM, respectively. This discrepancy might stem from differences in the interactions in solution and the gas phase. Alternatively, thioethers might simply be a poor bioisostere of disulfide bonds amongst others, due to different conformational preferences of both motifs. The initial DCMS results indicated that a subpocket of JMJD2E's active site is likely to accommodate larger side chains than that of 48. Consequently, compound 50 was optimized to inhibitor 52 (IC50 = 37.1 μM) by screening a series of N-oxalyl-D-tyrosinyl derivatives. The predicted binding mode of inhibitor 52 was validated by solving the co-crystal structure of the complex with JMJD2A (a member of the JMJD2 subfamily) (PDB code: 2WWJ, Fig. 7). These results show that DCMS can be applied for the identification of potent and selective inhibitors of JMJD2E, a member of the JMJD2 histone demethylase subfamily and should be easily extended to fragment growing projects involving other protein targets.
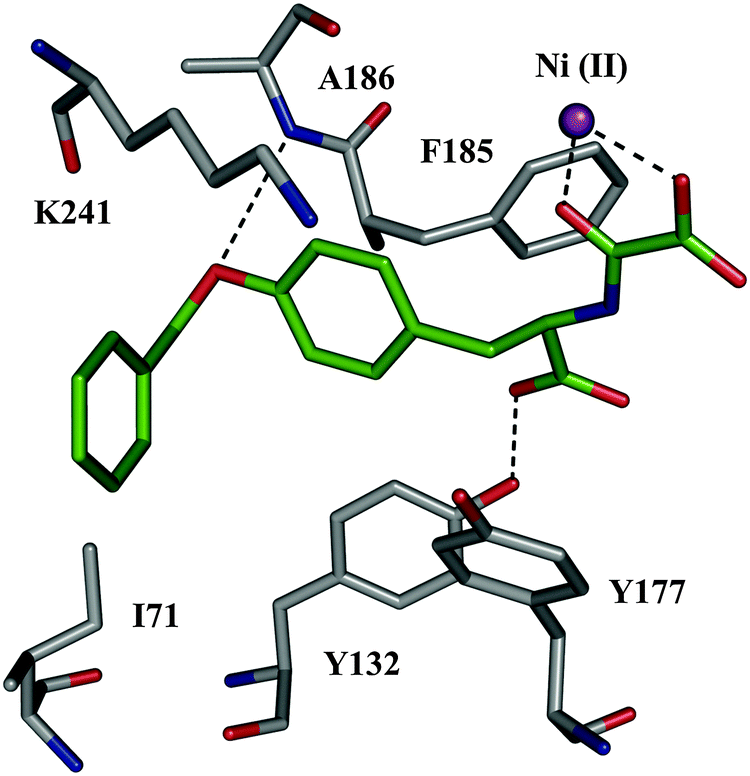 | ||
| Fig. 7 X-ray crystal structure of JMJD2A co-crystallized with inhibitor 52 (color code: protein skeleton: gray; inhibitor skeletons: C: green, N: dark blue, O: red; Ni(II): violet sphere. (PDB code: 2WWJ).56 | ||
Over the next years, the established DCMS method14 was again used by the same group to identify small-molecule probes for the Fe(II)- and 2OG-dependent oxygenase (AlkB), which catalyzes the hydroxylation of methylated N-methyl-modified DNA/RNA, ultimately resulting in demethylation using disulfide-based DCC.57 Small-molecule probes targeting the active site should be invaluable tools to elucidate the exact molecular role of these oxygenases. Initially, a non-denaturing ESI-MS screen of a library of L- and D-enantiomers of N-oxalyl derivatives of amino acids was carried out showing that AlkB has a clear preference for the L-enantiomers, which is in contrast to other 2OG-dependent demethylases. Based on the known, generic inhibitor N-oxalylglycine (53) and by analogy to the previous study, a suitable support ligand N-oxalyl-L-cysteine (54) was identified. To explore the subpocket accessible to the N-oxalyl amino acid series, a library of 37 thiols 55a–u and 56a–p and the support ligand 54 were incubated in presence of AlkB and Fe(II) (Fig. 8a). The DCMS experiments indicated the appearance of two peaks of two different masses, which could correspond to 16 disulfides. Given this ambiguous result, a direct consequence of the choice of building blocks used in the study, ESI-MS analysis of the 16 individual thiols with support ligand 54 was necessary to identify the bound disulfides unambiguously as 54-55g, 54-55h and 54-55q (Fig. 8b). Stable carbon analogues 57–59 were synthesized by replacing the disulfide by a thioether bond and found to be strong binders of AlkB. These results were confirmed with inhibition and thermal shift assays, showing IC50 values of 5.2 μM, 48 μM, 50.4 μM, and melting temperature shifts of 4.0 °C, 1.8 °C 1.3 °C for 57, 58 and 59, respectively (Fig. 8b). The binding mode of the inhibitors was then validated by solving a co-crystal structure of inhibitor 57 and the optimized derivative 60 (IC50 = 0.5 μM) in complex with AlkB; both compounds bind to AlkB in an analogous manner to 2OG. This study illustrates the power of disulfide-based DCC for the efficient identification of potent and selective inhibitors of the 2OG-dependent nucleic acid-modifying oxygenase AlkB, which were further optimized based on a co-crystal structure of the most potent inhibitor identified by DCC. Taken together, the previous two reports further establish the DCMS method as a powerful analytical tool for protein-templated DCC.
 | ||
| Fig. 8 (a) Initial thiol building blocks 55a–u and 56a–p and the support ligand 54 used for the generation of disulfide-based dynamic combinatorial libraries for templating by the 2OG-dependent oxygenase AlkB. (b) Disulfides 54-55 identified and their carbon analogues and 57–60.57 | ||
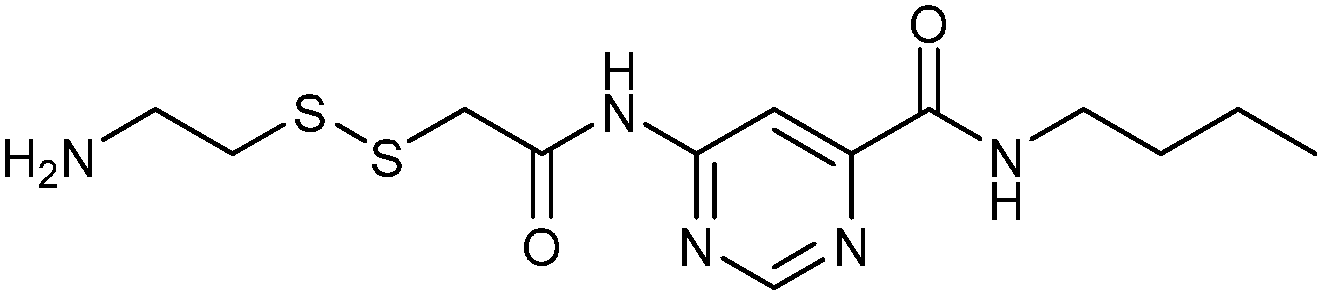
In 2008, the group of Erlanson pioneered site-directed DCC based on disulfide exchange, which enables dynamic combinatorial screening in a specific pocket of the protein.58 The kinase Aurora A, which plays an important role in the regulation of mitosis, was used in this study. Taking inspiration from tethering and tethering with extenders,64,65 which enables identification of suitable fragments for growing lead compounds through irreversible attachment of the tether to a cysteine residue, the concept was extended to a fully reversible system in which the tether is connected to the cysteine residue via a disulfide bond. First, a cysteine residue was introduced in proximity of the ATP-binding pocket through site-directed mutagenesis. The protein was then screened against a library of 4500 disulfide-containing fragments in presence of a doubly dynamic extender 61, which is based on diaminopyrimidine (DAP), known to bind to the ATP-binding site. The extender was designed to feature two disulfides: one disulfide forms a reversible covalent bond to the protein through the modified cysteine residue and the other can react and be extended with the disulfide-bearing fragments. The resulting protein–extender–fragment complexes were then analyzed by ESI-MS. To simplify the screening, several sub-libraries were designed, each of which contained roughly ten fragments of unique mass; an exemplary library containing disulfide fragments 62a–j is shown in Fig. 9.
 | ||
| Fig. 9 Generation of disulfide-based dynamic combinatorial libraries of dynamic extender 61 and exemplary disulfides 62a–j for templating by Aurora A kinase.58 | ||
ESI-MS analysis revealed the formation of a complex between cysteine-modified Aurora A kinase, dynamic extender 61 and the fragment 62. The hit identified (63) was then converted into a stable and soluble inhibitor 64 by replacing one disulfide bond with a flexible alkyl linker and removing the other disulfide from DAP, displaying an IC50 value of 17 μM. Exchanging the DAP moiety for a purine or shortening the linker by one carbon atom afforded more potent inhibitors 65 and 66 with IC50 values of 3.1 and 2.9 μM, respectively. Fragment 62a, which was identified by DCC, was shown to have an important contribution to the binding affinity as illustrated by truncating this substituent, leading to the inactive compound 67 (Table 12). The X-ray crystal structure of 66 in complex with Aurora A validated the predicted binding pose. This study constitutes the first report of site-directed DCC, enabling growing of a kinase inhibitor beyond the ATP-binding pocket into the adaptive region. By exploiting DCC, the requirement for a reactive warhead in “extended tethering” has been circumvented. The drawback of this approach is the requirement for a cysteine residue in proximity to the binding pocket, which needs to be introduced through site-directed mutagenesis unless it is already present.
| Compound | Structure | IC50 (μM) |
|---|---|---|
| 61 |
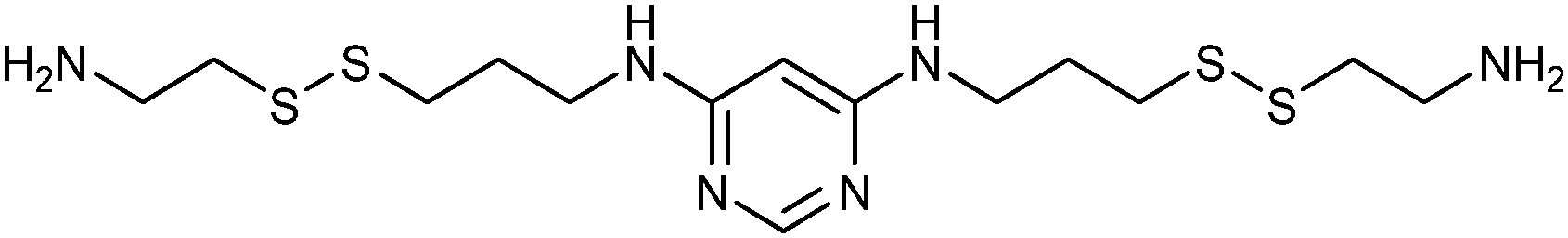
|
— |
| 62a |
|
— |
| 63 |
|
— |
| 64 |
|
17 |
| 65 |
|
3.1 |
| 66 |
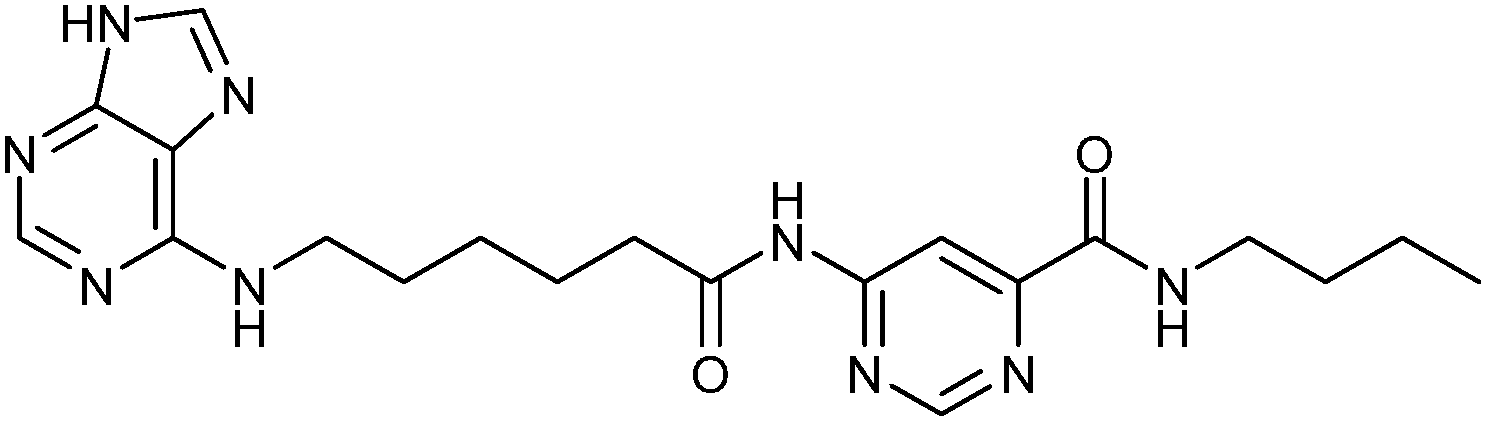
|
2.9 |
| 67 |
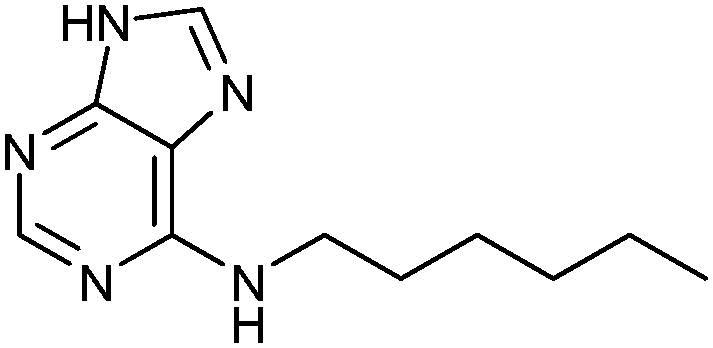
|
>100 |
In 2009, the group of Ciulli employed disulfide-based DCC in combination with SBDD to probe the adenosine-binding site of Mycobacterium tuberculosis pantothenate synthetase, a potential virulence factor for M. tuberculosis that is required for pantothenate (vitamin B5) biosynthesis.59 5′-Deoxy-5′-thioadenosine (68, Scheme 17), derived from the cofactor ATP, was chosen as an anchor having established its binding to the ATP-binding site of the target in an 1H-NMR WaterLOGSY experiment, by isothermal titration calorimetry (ITC) and by solving an X-ray co-crystal structure in complex with the target. A set of hydrophobic thiols 69a–d and hydrophilic thiols 69e–h were selected to grow the anchor into the substrate (pantoate)- and phosphate-binding pockets, respectively (Scheme 17). In a GSH buffer at pH 8.5, the DCLs generated using the anchor 68 and the thiols 69a–h were found to equilibrate within 24 hours.
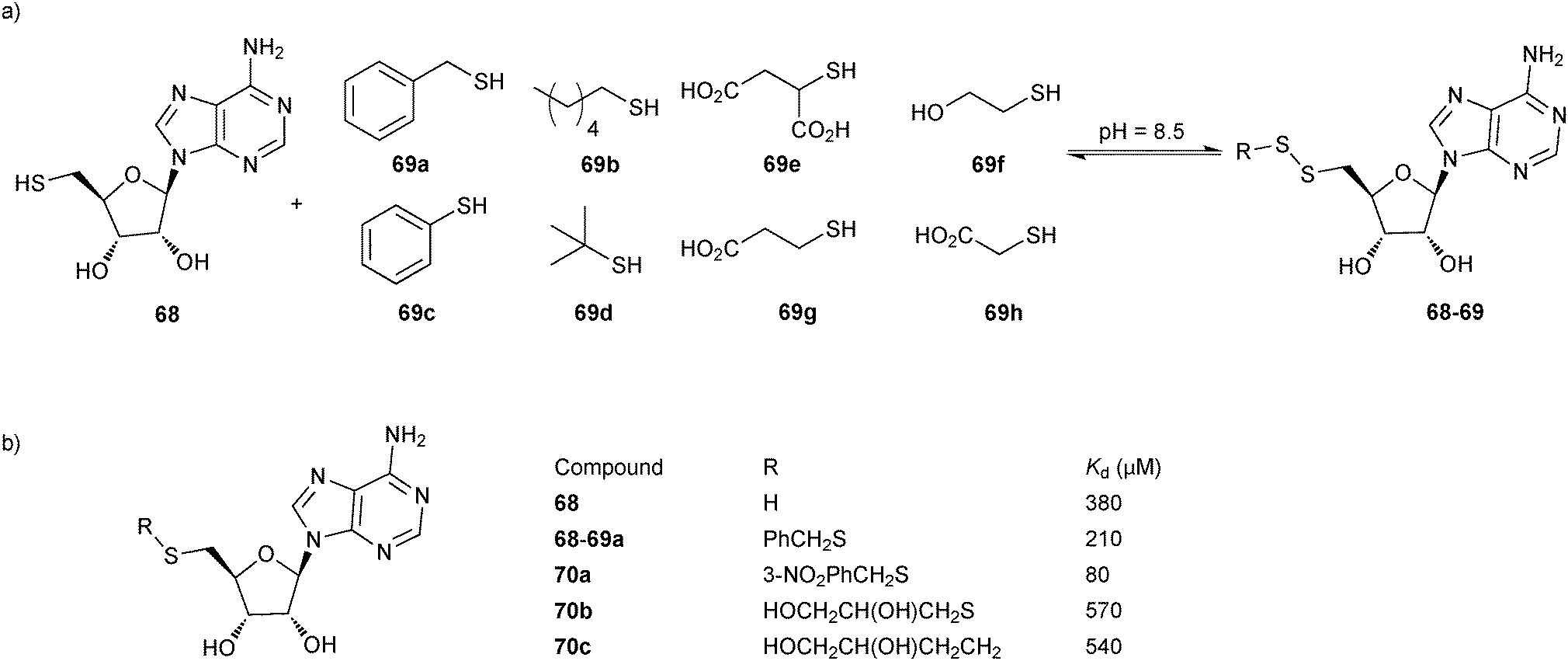 | ||
| Scheme 17 (a) Generation of disulfide-based dynamic combinatorial libraries of thiols 68 and 69a–h for templating by M. tuberculosis pantothenate synthetase. (b) Hit identified and hit optimization to 70a–c.59 | ||
Analysis by HPLC after acidification and ultrafiltration was facilitated by selectively monitoring at the absorption maximum of the anchor 68 (λ = 260 nm) so as to only select disulfides comprising the anchor. The buffer was prepared using a reduced to oxidized GSH ratio of 4:1 to bias the DCL towards the thiols and enable stringent selection of only the strongest disulfide binders. In the absence of protein target, the dominant species in the DCL was the disulfide GSH-68. Upon addition of the protein, a new disulfide 68-69a was amplified as well as the thiol 68 at the expense of the disulfide GSH-68. As predicted by docking studies, the benzyl group is hosted in the hydrophobic substrate-binding pocket. This binding hypothesis was confirmed by solving the X-ray co-crystal structure of the target in complex with the hit 68-69a, which suggested that the benzyl substituent might be decorated with additional functional groups. A glycerol molecule found in the co-crystal structure provided additional ideas for the growing and optimization of the initial hit 68-69a, affording disulfides 70a–b as well as 70c, the stable thioether derivative of 68b. ITC analysis of all derivatives revealed an approximate two-fold improvement in affinity of the initial hit 68-69a (Kd = 210 μM) with respect to 68. meta-Substitution of the benzyl ring with a nitro group in 70a led to a further optimization of the affinity as reflected in the Kd value of 80 μM. 70b and 70c were found to display a decreased affinity, which could be ascribed to the flexible glycerol substituent. Co-crystal structures of 70 and 70c validated the binding hypothesis showing perfect superimposition of the adenosine moieties with that of 68 and providing insight into the observed differences in affinity of the derivatives prepared. Taken together, this study shows the successful identification of a binder of M. tuberculosis pantothenate synthetase, exploiting an anchor to direct DCC to the adenosine-binding pocket of the enzyme. Subsequent optimization based on the co-crystal structure led to a binder with improved affinity. This fragment-based directed approach should be applicable to the whole range of adenosine-binding enzymes, such as kinases.
In 2014, the group of Mahler used DCC based on disulfide formation for the identification of Trx glutathione reductase (TGR) inhibitors.60 TGR is an essential enzyme regulating thiol homeostasis in the flatworm parasite, making it an attractive anti-infective target. A biased DCL was designed based on two anchor thiols, the acid 71 and GSH (26), which bind to the thioredoxin and GSH domains. In addition, four thiol fragments 72a–d were selected for the generation of the DCL at pH 8.8 (Fig. 10), giving rise to potentially 21 disulfides. The DCLs were then analyzed by LC-MS in negative ion mode. The reaction took 24 hours to reach equilibrium in the absence of protein. Protein was added at equilibrium, and the library was left to re-equilibrate in the absence of oxygen for 24 hours. The dynamic equilibrium was frozen by addition of trichloroacetic acid, which also led to denaturation of the protein and concomitant release of the bound inhibitors. The library was then analyzed by LC-MS in negative ion mode and compared to the LC-MS chromatogram of the blank reaction (without protein). LC-MS results revealed the appearance of two new peaks corresponding to the diastereomeric mixture of disulfide heterodimers cis/trans-71-72d. The trans-disulfide 71-72d was then synthesized and its inhibitory potential against TGR was assayed. trans-71-72d displays an IC50 value of 24.6 μM, which was optimized by preparing a series of analogues of 71-72d to afford 73 with an IC50 value 14.0 μM. This study highlights the use of site-directed DCC for the identification of low-affinity fragments, which can be used to grow and optimize known inhibitors and should be directly applicable to other targets.
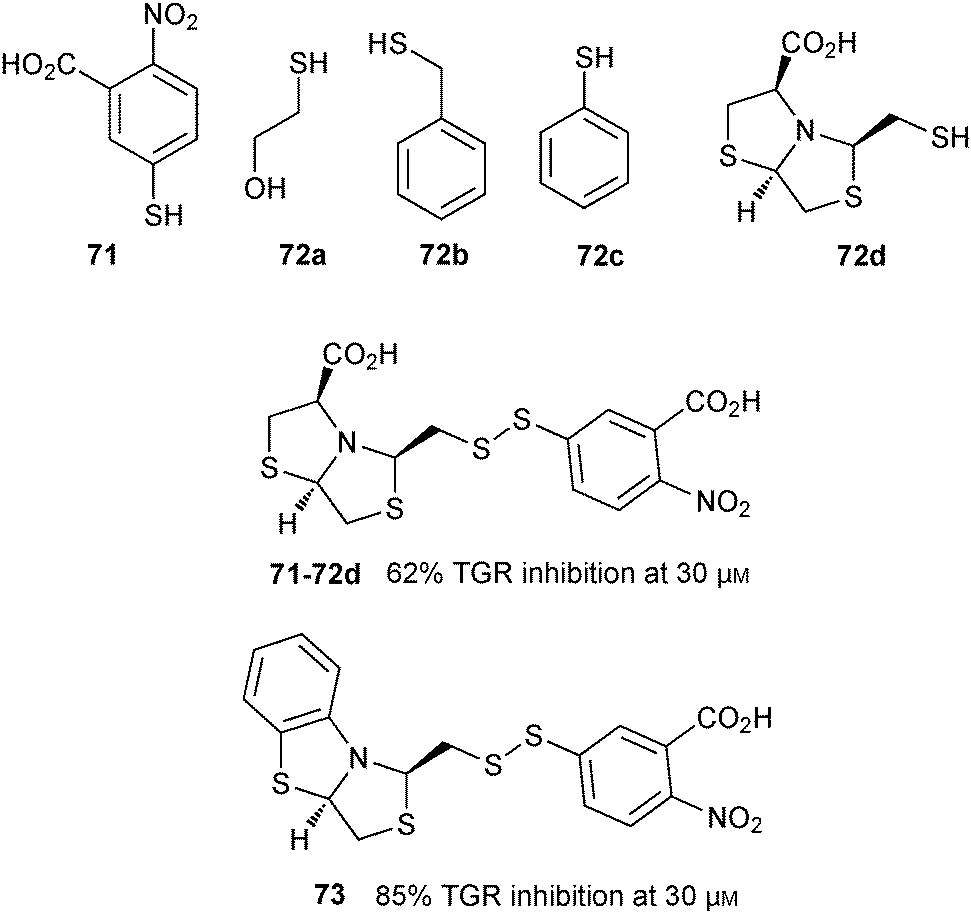 | ||
| Fig. 10 Thiol building blocks 71 and 72a–d used to generate disulfide-based dynamic combinatorial libraries for templating by TGR; disulfide inhibitor 71-72d and its derivative 73.60 | ||
B–O bond formation: boronate ester formation
Boronate ester formation between boronic acids and diols is attractive for DCL formation given that it is reversible in aqueous solution at slightly basic, neutral and slightly acidic pH depending on the pKa of boronic acid and alcohol, making it amenable to a large number of targets for protein-templated DCC.13In 2011, the group of Claridge pioneered reversible boronate ester formation for use in protein-templated DCC. The serine protease α-chymotrypsin (αCT) was chosen as a model enzyme for this proof-of-principle study.1311B-NMR and 1H-waterLOGSY spectroscopy were used to monitor the ternary complexes of enzyme, boronic acids and sugars (diols in a syn-periplanar arrangement such as in furanoses afford more stable complexes) in the DCLs. 11B-NMR spectroscopy has the advantage of not having any protein background signals and is very efficient for differentiating enzyme-bound from free ligands. The quadrupolar nature of the 11B nucleus (I = 3/2) is used to probe the hybridization state of the boron atom: broad or sharper peaks are observed for a 11B nucleus in an sp2-hybridized trigonal planar or highly symmetrical sp3-hybridized tetrahedral environment, respectively.66 Based on known serine-protease inhibitors, 74a–e (IC50 of 5 μM and 14 μM for 74a and 74b, respectively) were selected as model boronic acid fragments, and D-fructose (75a) and L-fructose (75b)67 were selected as the alcohol fragments for use in DCC (Fig. 11). Given that the pKa of most boronic acids is around 7–9, the DCC experiments were conducted at pH 5.8 to ensure the free boronic acid is present in its sp2-hybridized form.
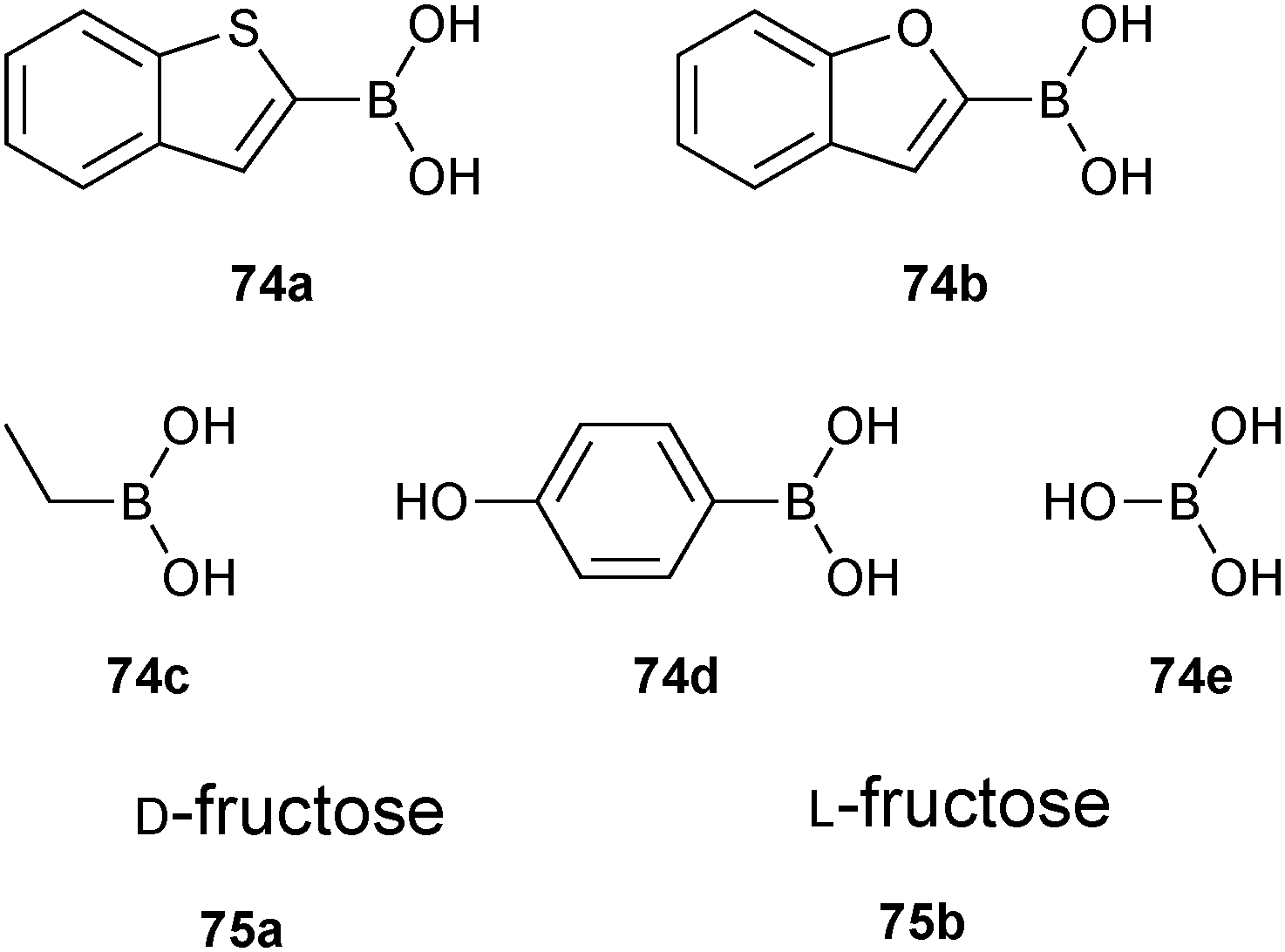 | ||
| Fig. 11 Boronic acids 74a–e and sugars 75a and 75b used to generate boronate ester-based dynamic combinatorial libraries for templating by αCT.13 | ||
Initial experiments were carried out using boronic acids 74a and 74b, 75b and αCT, showing that both 74a and 74b are equally reactive to boronate ester formation in solution. 11B-NMR spectroscopy was used to monitor the interaction of boronic acids 74a and 74b with the enzyme αCT in presence and absence of sugars. Upon addition of 75a, ternary complex αCT-74a-75a formed preferentially over αCT-74b-75a, which demonstrates the subtle active-site selectivity (Scheme 18a). Subsequent experiments using 74a, 75a, 75b and αCT revealed that 75a readily forms a ternary fructose complex αCT-74a-75a whereas 30 times more 75b is required to fully form αCT-74a-75b (Scheme 18b). To demonstrate the pKa dependency of boronate ester formation, boronic acids 74a and 74c–e were selected, and cocktails of boronic acids were treated with αCT in presence of 75a and 75b separately and the formation of enzyme–boronic acid–sugar complexes was monitored by 11B-NMR spectroscopy at pH 5.8. It was found that upon addition of αCT to the boronic acid cocktail, only boronic acid 74a formed a complex with αCT. Addition of D-fructose led to quantitative formation of αCT-74a-75a, whereas addition of 75b only afforded 50% of αCT-74a-75b (Scheme 18c). These results suggest that αCT preferentially forms the complex with 74a out of the cocktail of boronic acids and that 74a and 75a are preferentially bound. 11B-NMR and waterLOGSY spectroscopy have been used for the first time to analyze DCLs, employing high or low protein concentrations, respectively. Even though the latter technique is more sensitive, it is prone to signal overlap for larger DCLs. This study establishes reversible boronate ester formation as a potentially useful reversible reaction for protein-templated DCC, extending the palette of reactions compatible with biomacromolecules, as long as a more suitable analytical technique is found that enables the analysis of the labile ternary complexes.
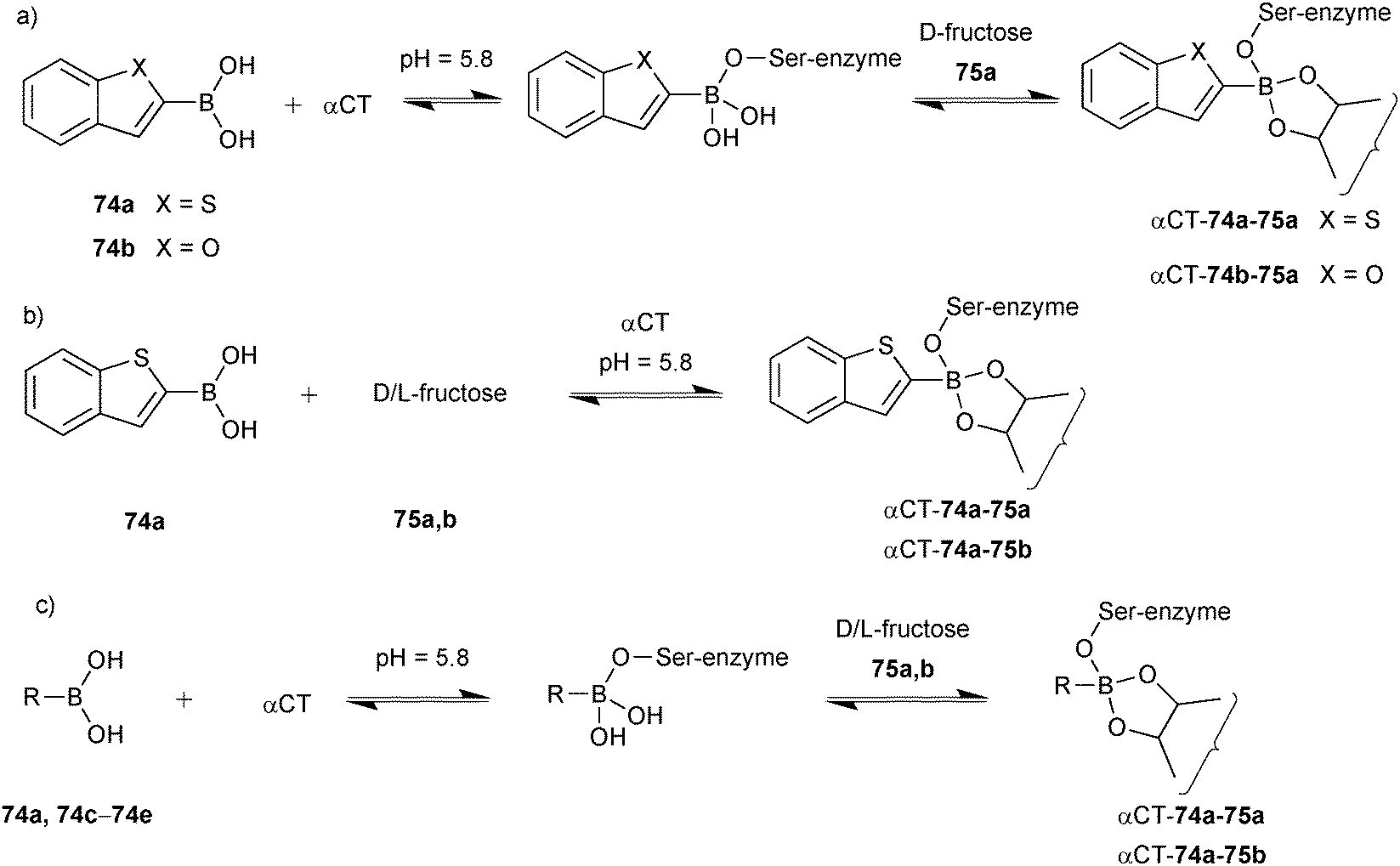 | ||
| Scheme 18 Generation of boronate ester-based dynamic combinatorial libraries for templating by αCT using building blocks (a) 74a, 74b, αCT and 75a; (b) 74a, αCT, 75a and 75b; (c) 74a, 74c–e, αCT, 75a and 75b.13 | ||
Based on this preliminary proof-of-principle study, the Schofield group demonstrated that reversible boronate ester formation in combination with DCMS makes the efficient identification of protein inhibitors possible.69 The DCMS technique had previously been used for the analysis of disulfide- and hydrazone-based DCLS.14,19,57,58 Prolyl hydroxylase domain isoform 2 (PHD2), a Fe(II)- and 2OG-dependent oxygenase that controls the human hypoxic response and is used as a target for the treatment of ischemia-related diseases and anemia, was used as the protein target. The support ligands 76 or 77 (pKa (76) = 7.4 and pKa (77) = 6.9); the latter was included as a control and was predicted not to fit into the active site), which were designed to participate in Fe(II) chelation in the active site and participate in boronate ester formation, were used as boronic acid fragments and forty diols (78) in four sets of ten each were used as alcohol fragments to simplify the analysis for the formation of DCLs at pH 7.5, making this reaction highly compatible for protein-templated DCC (Scheme 19a).
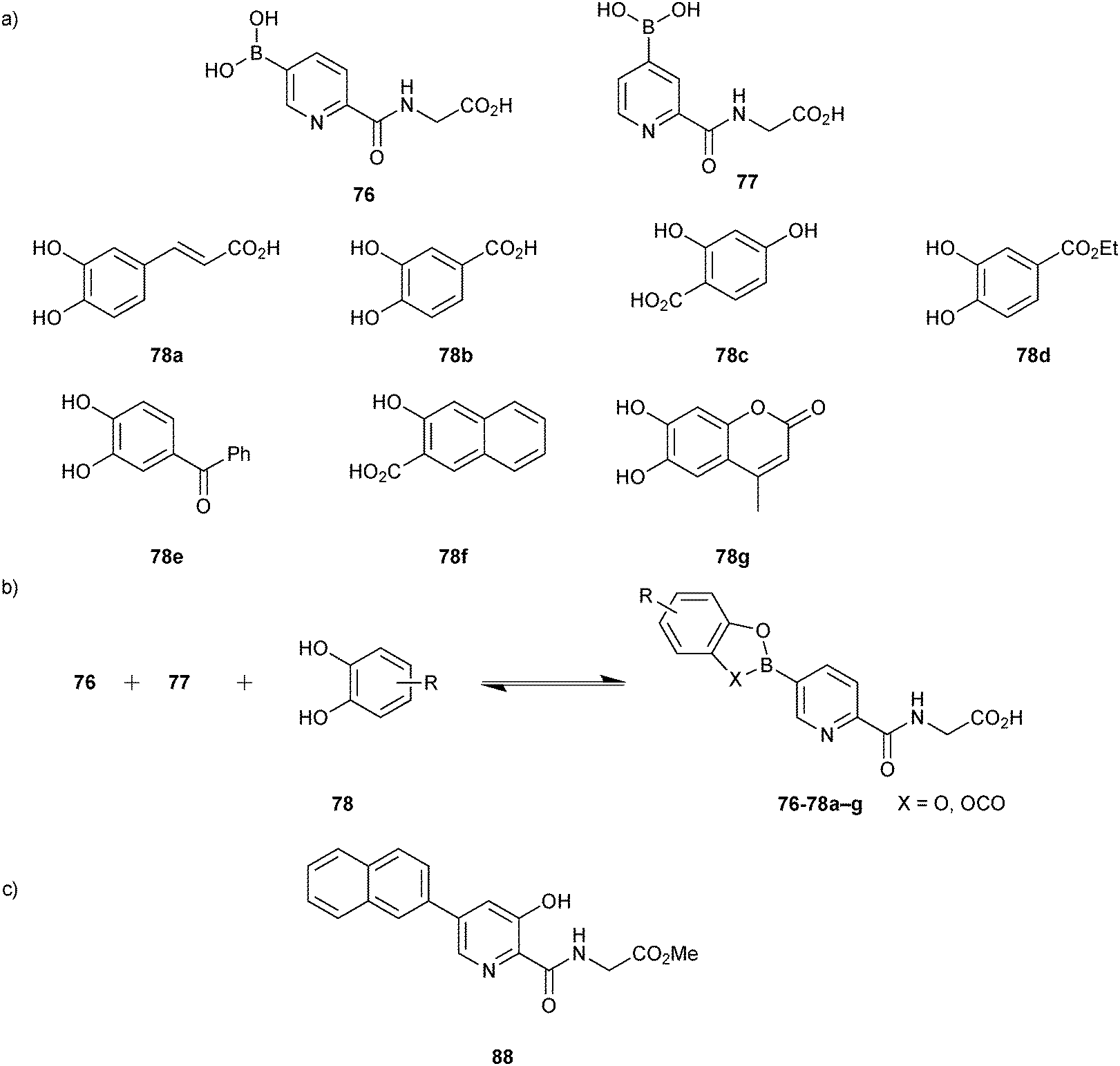 | ||
| Scheme 19 (a) Building blocks used to generate boronate ester-based dynamic combinatorial libraries for templating by PHD2. (b) Reversible formation of boronate esters from boronic acids and diol building blocks. (c) Structure of inhibitor 88.69 | ||
The DCLs were synthesized by treating individual “support ligands” 76 and 77 with forty diols (78) (Scheme 19b). Mild ESI-MS was use to ensure the correct mass shift of different boronate ester complexes of PHD2·Fe(II). DCMS analysis of the DCLs revealed the formation of boronate esters of only boronic acid 76 with seven diols (76-78a–g). ESI-MS analysis of boronic acid 76 with each of the seven individual diols 78a–g validated the DCMS results.
No boronate ester complex formation was observed with support ligand 77 with diols 78a–g and PHD2·Fe(II). In both cases, direct binding of some diols to PHD2·Fe(II) was observed in the absence of 76 and 77. This result also implies that the mass shift observed with 76 represents the boronate ester–protein complexes rather than the simultaneous binding of 77 and diols.
To validate the DCMS results and ensure that they correspond to the solution-state situation, NMR-based water relaxation experiments were performed to measure the binding constants of boronate esters of 76 with PHD2. It was observed that the apparent affinity of 76 for PHD2 (KD = 24.8 μM) increased in presence of the diols, which were identified from DCMS experiments with KD values of 0.6 μM for 76-78a, 1.3 μM for 76-78b and 3 μM for 76-78c. For further verification of the use of the labile boronate esters identified, several stable analogues (79–87) derived from support ligand 76 and diols 78a–g were synthesized, and their binding affinities and IC50 values were determined (Table 13). The stable analogues were found to be stronger binders than the support ligand 76 but weaker than the corresponding boronate esters 76–78. To validate the binding mode of the boronate ester analogues, compound 87 was crystallized in complex with the catalytic domain of PHD2 containing Mn(II), mimicking the Fe(II) cation. The corresponding crystal structure (PDB code: 3HQR) revealed that 87 complexes the metal cation in a bidentate manner. To investigate the efficiency of these compounds in cells, methyl ester derivative 88 (Scheme 19c) of 85 was tested and shown to upregulate H1F1α in cells by selectively inhibiting PHDs but not Factor Inhibiting Hypoxia (FIH). Taken together, this study demonstrates the potential of boronate ester formation as a novel reversible reaction for protein-templated DCC. It enabled identification of boronate esters, which were developed into stable analogues that proved to be selective inhibitors of the hydroxylase PHD2. Taking advantage of DCMS as a convenient analytical tool complemented by NMR-based screening, boronate ester formation should find application to other protein targets.
|
|
||||
|---|---|---|---|---|
| Compound | R | X | K D (μM) | IC50 (μM) |
| 76 |
|
H | 24.8 | 126 |
| 79 | H | H | — | >1000 |
| 80 |
|
H | 9.5 | >500 |
| 81 |
|
H | 7.0 | >500 |
| 82 |
|
H | 1.6 | 107 |
| 83 |
|
H | 8.7 | >100 |
| 84 | H | OH | 3.5 | 409 |
| 85 |
|
OH | 0.5 | 0.017 |
| 86 |
|
OH | 0.8 | 0.013 |
| 87 |
|
OH | 0.9 | 0.004 |
Subsequently, the group of the Schofield and the group of Claridge used reversible boronate ester formation for the identification of inhibitors of 2OG oxygenases.70 Human 2OG oxygenases, such as the hypoxia inducible factor (HIF) hydroxylases71 are validated drug targets for diseases such as cancer, inflammation, ischemia and anemia.72 PHD2 and FIH were used as model systems for this study to introduce a competition-based 1H-NMR method for the identification of 2OG oxygenase binders from the DCL. This constitutes as an important alternative to DCMS, which suffers from the potential limitation that not all non-covalent protein–ligand interactions may be stable during the solution-to-gas-phase transition. The co-substrate 2OG was used as a reporter ligand and the endogenous Fe(II) was substituted by diamagnetic, catalytically inactive Zn(II). This technique depends on the displacement of the reporter ligand 2OG from the binding pocket upon binding of competitive ligands, which is monitored via1H-NMR spectroscopy. An advantage of this technique is that site-specific binding information as well as Kd values both for ligands with high and low affinity are obtained, which is difficult for other ligand-based NMR techniques. Initial screens using known PHD2/2OG inhibitors indicated that the reporter ligand NMR screening is an efficient means to derive relative strengths of binding at a single inhibitor concentration. To demonstrate the potential of the NMR-based method for inhibitor discovery, it was applied to hit validation of the hits discovered in the previous report.69 The ligand 76, which binds to the 2OG-binding pocket and is a weak inhibitor of PHD2, was used as the boronic acid scaffold in combination with the set of diol hits 78a, 78b, 78d and 78e for the DCC experiments (Scheme 20). In this proof-of-principle study, the boronic acid scaffold 76 and the diols were subjected to competitive NMR analysis. The boronic acid caused ∼50% displacement of 2OG, which confirms it as a weak inhibitor of PHD2. Three-fold higher concentrations of diols were used to ensure boronate ester formation in each case. Reporter-ligand displacement was not observed for any diol in the absence of boronic acid, but in presence of boronic acid, ∼75–85% of 2OG displacement was observed, validating the DCMS screening results reported earlier.68 Together with a series of control experiments these results corroborate the hypothesis that boronate esters are responsible for the displacement of 2OG and thus validate the potential of this reporter method for hit identification. This NMR-based method using reporter ligands appears to be a generic method for the analysis of DCLs using members of the 2OG oxygenase superfamily and should be also applicable to other enzyme families. It has the advantage of using substoichiometric amounts of unlabeled protein and provides information on the binding mode as well as the KD values.
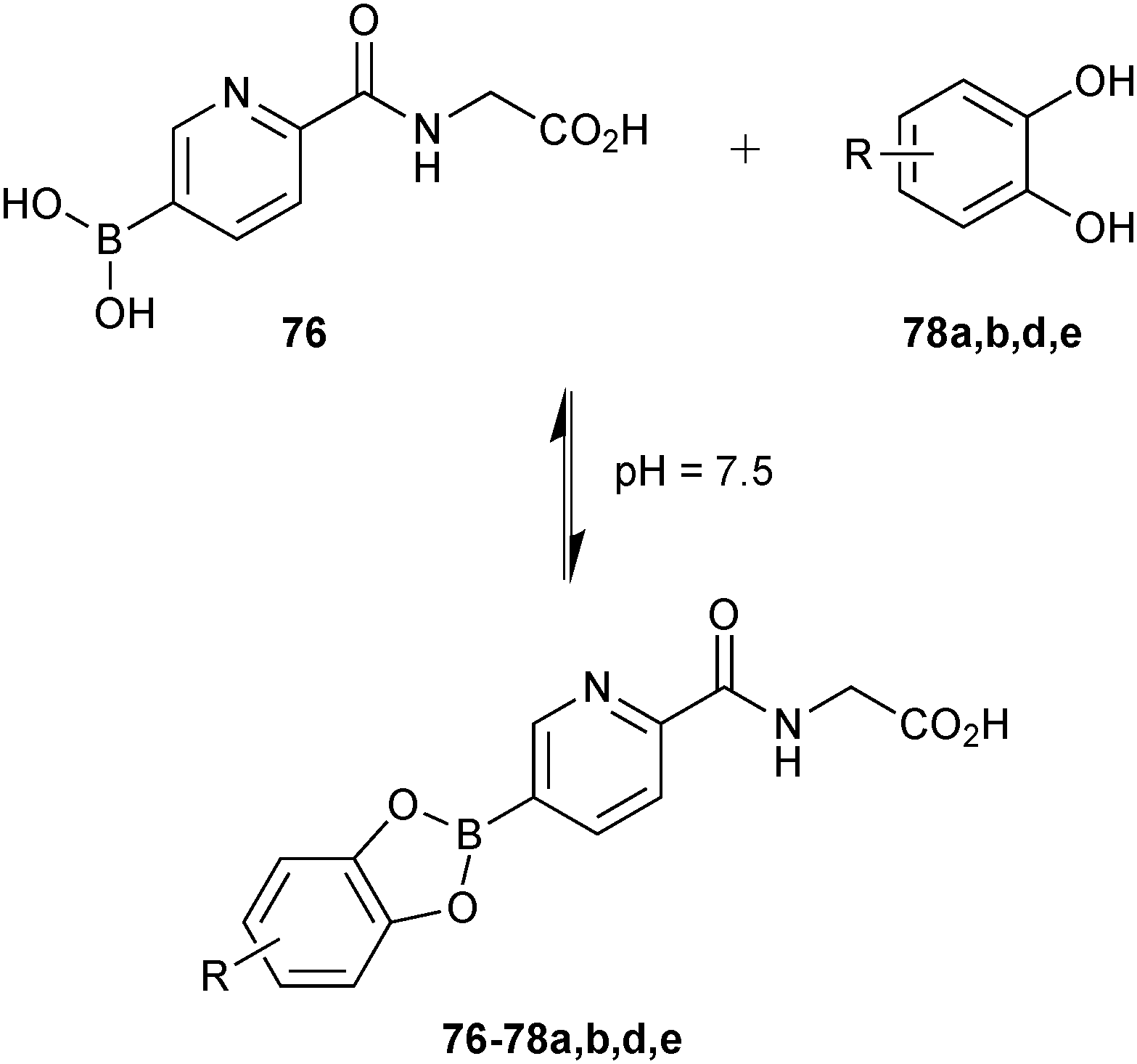 | ||
| Scheme 20 Generation of boronate ester-based dynamic combinatorial libraries using building blocks 76, 78a, 78b, 78d and 78e for analysis by a competition-based NMR method of binding to PHD2.70 | ||
Metal–ligand coordination
The lectin concanavalin A (ConA) was used as the target for the proof-of-principle study. To establish the reversibility of the Fe(II)–bipy complexes, a small DCL was generated by mixing two homoleptic Fe(II)–bipy complexes 89a and 89b in acetonitrile. Due to ligand exchange, a library of four complexes 89a–d was formed and reached equilibrium after five days (Scheme 21a). The library was analyzed using LC-MS.
 | ||
| Scheme 21 (a) Generation of Fe(II)–bipy complex-based dynamic combinatorial libraries of a complexes 89a and 89b. (b) Complexes 89e–g for templating by lectin concanavalin A.73 | ||
Building on this successful result, a bigger library of ten members (89e–n) was constructed by mixing three homoleptic Fe(II)–bipy complexes 89e (Fe-MMM), 89f (Fe-GGG) and 89g (Fe-FFF) (Scheme 21b). Ligands of these Fe(II)–bipy complexes consisted of bipy ligands connected to different sugar moieties such as D-mannoside (M), D-galactoside (G) and L-fucoside (F) via a flexible spacer. These sugars were chosen based on their reported binding affinities towards ConA (M > F, G). A DCL was set up at low temperature (5 °C) to avoid dissociation of Fe(II)–bipy complexes and required 14 days to reach equilibrium. Given that the masses of galactose and mannose are identical, the retention time (Fe-GGG < Fe-MMM < Fe-FFF) was considered alongside the MS result for unequivocal peak assignment. Subsequently, a similar library was set up by mixing three homoleptic FeII–bipy complexes 89e (Fe-MMM), 89f (Fe-GGG) and 89g (Fe-FFF) in presence of sepharose-bound ConA and allowed to equilibrate for 14 days. The bound ligands were separated by filtration and re-suspended in aqueous HCl to release the bound complexes from the protein surface.21 LC-MS analysis of this fraction showed the full mannoside complex 89e (Fe-MMM) as the most abundant species with seven-fold amplification in comparison to the library without protein along with other two mannoside-rich complexes 89h (Fe-GMM) and 89i (Fe-MMF). Taken together, these results show that DCC based on metal–ligand complexation can be used for protein–ligand recognition and can potentially be extended to drug discovery of any target, whilst taking advantage of multivalency to improve binding affinity. The main limitations of this method are the unclear spatial arrangement of the ligands in heterogeneous complexes, highly reversible nature and potential toxicity issues of the metal complexes. Using a target bound to a solid support has the advantage of reducing the analytical challenge simply by removing all unbound library members before analysis.
 | ||
| Fig. 12 Generation of oxorhenium-complex-based dynamic combinatorial libraries of proline analogues 90a–p and thiols 91a–p for templating by human peptidyl-prolyl isomerase hCyp-18.76 | ||
To validate the strategy, a small library of 16 oxorhenium complexes was synthesized by reduction and incubation of disulfides 90a–d and thiols 91a and 91i (Scheme 22a). As expected, all 16 complexes were formed as a mixture of diastereoisomers. Upon addition of hCyp-18, yields of these complexes barely varied except for 90d-91i, which was amplified after addition of the target hCyp-18 (“cylcophilin-enhancing effect”). These complexes bind to hCyp-18 in the submillimolar range except for the complex 90d-91i, which was found to be a strong binder with a Kd value of 11 ± 2 μM (IC50 = 12 μM). Interestingly, upon addition of GSH, all complexes readily dissociated except for 90d-91i, which was stable even at high GSH concentrations. This effect was called “cyclophilin-protecting effect” and results from a protective effect of the protein target against thiol exchange of interacting complexes. The positive “cylcophilin-enhancing effect” and “cyclophilin-protecting effect” from GSH strongly suggest that the oxorhenium complex 90d-91i specifically interacts with the protein hCyp-18 as also indicated by a competition experiment with the known inhibitor cyclosporine.
 | ||
| Scheme 22 (a) Generation of oxorhenium-complex-based dynamic combinatorial libraries of disulfides 90a–d and diols 91a and 91i for templating by human peptidyl-prolyl isomerase hCyp-18; (b) DCLs containing disulfides 90e–p and each of 16 thiols 91a–p.76 | ||
After successful application of the reversible oxorhenium complex formation using a small library of fragments for ligand identification of hCyp-18, a larger library was investigated in the same manner using modules 90e–p and modules 91a–p (Scheme 22b), affording 192 possible complexes. To facilitate analysis, the whole library was divided into 16 sub-libraries, each containing twelve disulfide building blocks 90e–p and one of the 16 thiols 91a–p. LC-MS analysis of these DCLs showed that only complexes 90e-91b, 90i-91b and 90p-91n displayed higher resistance to GSH in the presence of hCyp-18, which were selected as potential cyclophilin inhibitors. Subsequently, these complexes were synthesized as a mixture of diastereoisomers and tested for binding affinity, displaying Kd values of 0.3–2 μM (Table 14). Reversible oxorhenium coordination provides an efficient and straightforward access to protein ligands, affording compounds with submicromolar affinity for hCyp-18. Drawbacks include the sensitivity of the complexes to endogenous thiols such as GSH. Nevertheless, this strategy provides access to the rapid identification of non-peptidic inhibitors, which should be particularly useful to target surface proteins and replacement of the ReO3+ by a TcO3+ core would turn them into molecular imaging agents.
| Compound | Structure | K d (μM) | IC50 (μM) |
|---|---|---|---|
| 90d-91i |
|
11 | 12 |
| 90e-91b |
|
2 | 5.3 |
| 90i-91b |
|
0.3 | 0.3 |
| 90p-91n |
|
0.3 | 0.2 |
Conclusions
Over the past 15 years, DCC has rapidly evolved and our understanding of the underlying concepts has grown tremendously. DCC is an exquisite tool that enables the efficient development of novel compounds with potentially interesting biological activities. Given that it combines in situ synthesis of covalently or non-covalently connected building blocks and screening of the potential binders against the target, it holds the potential of dramatically accelerating the drug-discovery process. Structural diversity can be readily accessed and screened. A variety of protein targets have been used, illustrating that a suitable reversible reaction and analytical method can be selected to enable DCC (Table 15). Mainly covalent reversible reactions but also non-covalent reversible reactions have been used to generate libraries of potential binders at various stages in a medicinal-chemistry or chemical-biology project: it has been used in FBDD to screen for low affinity fragments, hit identification, validation as well as optimization and elucidation of the role or mechanism of a protein of interest. The potential of DCC is certainly largest in the early stages of the drug-discovery process. For targets featuring ill-defined pockets that cannot be addressed easily by SBDD, DCC should represent a particularly elegant alternative.| Target | Reversible system used to form DCL | pH used to generate DCL | Number of compounds generated | Analytical method for identification of strongest binders | Activity of the strongest binder IC50 or Ki [μM] | Ref. |
|---|---|---|---|---|---|---|
| a Imines were generated separately in well-plate. b K m values determined using non-linear regression given that determination of Ki values was impossible due to incomplete inhibition. c pKi was determined by [3H]-GABA uptake assay performed in mGAT1-expressing HEK293 cells. d Library was screened in pools of 10 fragments. e K d values were measured. f Library was screened in pools of 20 compounds. g Library was screened in pools of 12 complexes. | ||||||
| SARS coronavirus SARS-CoV Mpro (cysteine protease) | Imines | 7 | 234 and 110a | Dynamic ligation screening | 50.3 and 2.9, respectively | 17 |
| Human carbonic anhydrase II | Imines | 6.5 | 20 | LC-MS | 0.021 | 25 |
| Human carbonic anhydrase I & II | Imines | 6.5 | 6 | LC-MS | 0.005 | 26 |
| Hen egg-white lysozyme (HEWL) | Imines | 7 | 32 | Size-exclusion chromatography | 0.203 ± 0.028 mg mL−1b |
27 |
| Cyclin-dependent kinase 2 (CKD2) | Hydrazones | 7 | 30 | X-ray crystallography | 0.030 | 16 |
| γ-Aminobutyric acid (GABA) transporter 1 (GAT1) | Hydrazones | 7.1 | 36 | Competitive MS binding assay | 6.229c | 34 |
| γ-Aminobutyric acid (GABA) transporter 1 (GAT1) | Hydrazones | 7.1 | 36 | Competitive MS binding assay | 8.094c | 35 |
| Glutathione S-transferase (GST) | Acylhydrazones | 6.2 | 10 | LC-MS | 22 | 20 |
| Glutathione S-transferase (GST) | Acylhydrazones | 6.2 | 24 | LC-MS | 0.050 | 46 |
| Endothiapepsin (aspartic protease) | Acylhydrazones | 4.6 | 25 | 1H-STD-NMR | 12.8 | 11 |
| Bovine carbonic anhydrase II | Alkene cross metathesis | 7.2 | 10 | Competitive MS binding assay | 4.9 | 12 |
| β-Galactosidase | Hemithioacetals | 7.2 | 10 | 1H-STD-NMR | Not reported | 15 |
| Glutathione S-transferase (GST) | Thioethers | 7.5 and 8 | 4 and 14 | HPLC | 0.32 and 0.61, respectively | 18 |
| BcII metallo-β-lactamase | Disulfides | 7.5 | 19 and 13 | Dynamic combinatorial MS | 0.740 and 8.4, respectively | 14 |
| JMJD2 histone demethylase (JMJD2E) | Disulfides | 7.2 | 5 | ESI-MS | 37.1 | 56 |
| 2OG-dependent oxygenase (AlkB) | Disulfides | 7.5 | 37 | ESI-MS | 0.5 | 57 |
| Aurora A (kinase) | Disulfides | 8 | 4500d | ESI-MS | 2.9 | 58 |
| Mycobacterium tuberculosis pantothenate synthetase | Disulfides | 8.5 | 8 | HPLC | 80e | 59 |
| Trx glutathione reductase (TGR) | Disulfides | 8.8 | 21 | LC-MS | 14 | 60 |
| α-Chymotrypsin (αCT) | Boronate esters | 5.8 | 10 | 11B-NMR | Not reported | 13 |
| Prolyl hydroxylase domain isoform 2 (PHD2) | Boronate esters | 7.5 | 80f | Dynamic combinatorial MS | 0.004 | 69 |
| 2OG oxygenases | Boronate esters | 7.5 | 4 | 1H-NMR | Not reported | 70 |
| Lectin concanavalin A (ConA) | Fe(II)–bipyridine complexes | 7 | 10 | LC-MS | Not reported | 73 |
| Cyclophilin hCyp-18 | Oxorhenium-complexes | 7.8 | 16 and 192g | LC-MS | 12 and 0.2, respectively | 76 |
There are some limitations of protein-templated DCC that will have to be tackled, namely the use of larger and more complex libraries. Other drawbacks can include long equilibration times that are not tolerated by particularly unstable proteins or the pronounced dynamic nature of several reversible reactions, causing the potential inhibitors to hydrolyze or decompose, leading to potentially toxic compounds and hampering straightforward analysis. As a result, protein-templated DCC should be followed by bioisosteric transformation of unstable inhibitors to stable analogues without a concomitant loss in activity.
Most examples reported so far use libraries of small or moderate size and relatively well-established protein targets, which may well be due to the limits of the analytical techniques. On the other hand, some elegant tricks have been developed that should enable use of DCC also with precious protein targets and larger libraries. The time has come for protein-templated DCC to fulfil its full potential and truly revolutionize the drug-discovery process.
Acknowledgements
Funding was granted by the Netherlands Organisation for Scientific Research (NWO-CW, VENI grant to A.K.H.H.) and by the Dutch Ministry of Education, Culture, Science (gravitation program 024.001.035).References
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS PubMed.
- I. Huc and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS.
- O. Ramström and J.-M. Lehn, Nat. Rev. Drug Discovery, 2002, 1, 26–36 CrossRef PubMed.
- S. Otto, R. L. Furlan and J. K. Sanders, Drug Discovery Today, 2002, 7, 117–125 CrossRef CAS.
- C. Karan and B. L. Miller, Drug Discovery Today, 2000, 5, 67–75 CrossRef CAS.
- S. Ladame, Org. Biomol. Chem., 2008, 6, 219–226 CAS.
- A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC.
- M. Mondal, N. Radeva, H. Köster, A. Park, C. Potamitis, M. Zervou, G. Klebe and A. K. H. Hirsch, Angew. Chem., Int. Ed., 2014, 53, 3259–3263 CrossRef CAS PubMed.
- S.-A. Poulsen and L. F. Bornaghi, Bioorg. Med. Chem., 2006, 14, 3275–3284 CrossRef CAS PubMed.
- I. K. H. Leung, T. Brown Jr, C. J. Schofield and T. D. W. Claridge, MedChemComm, 2011, 2, 390–395 RSC.
- B. M. R. Liénard, R. Hüting, P. Lassaux, M. Galleni, J.-M. Frére and C. J. Schofield, J. Med. Chem., 2008, 51, 684–688 CrossRef PubMed.
- R. Caraballo, H. Dong, J. Ribeiro, J. Jiménez-Barbero and O. Ramström, Angew. Chem., 2010, 49, 589–593 CrossRef CAS PubMed.
- M. S. Congreve, D. J. Davis, L. Devine, C. Granata, M. O'Reilly, P. G. Wyatt and H. Jhoti, Angew. Chem., Int. Ed., 2003, 42, 4479–4482 CrossRef CAS PubMed.
- M. F. Schmidt, A. Isidro-Llobet, M. Lisurek, A. El-Dahshan, J. Tan, R. Hilgenfeld and J. Rademann, Angew. Chem., Int. Ed., 2008, 47, 3275–3278 CrossRef CAS PubMed.
- B. Shi, R. Stevenson, D. J. Campopiano and M. F. Greaney, J. Am. Chem. Soc., 2006, 128, 8459–8467 CrossRef CAS PubMed.
- S.-A. Poulsen, J. Am. Soc. Mass Spectrom., 2006, 17, 1074–1080 CrossRef CAS PubMed.
- V. T. Bhat, A. M. Caniard, T. Luksch, R. Brenk, D. J. Campopiano and M. F. Greaney, Nat. Chem., 2010, 2, 490–497 CrossRef CAS PubMed.
- O. Ramström and J.-M. Lehn, ChemBioChem, 2000, 1, 41–48 CrossRef.
- A. Valade, D. Urban and J.-M. Beau, ChemBioChem, 2006, 7, 1023–1027 CrossRef CAS PubMed.
- S. Zameo, B. Vauzeilles and J.-M. Beau, Angew. Chem., Int. Ed., 2005, 44, 965–969 CrossRef CAS PubMed.
- S. Zameo, B. Vauzeilles and J.-M. Beau, Eur. J. Org. Chem., 2006, 5441–5444 CrossRef CAS.
- G. Nasr, E. Petit, C. T. Supuran, J.-Y. Winum and M. Barboiu, Bioorg. Med. Chem. Lett., 2009, 19, 6014–6017 CrossRef CAS PubMed.
- G. Nasr, E. Petit, D. Vullo, J.-Y. Winum, C. T. Supuran and M. Barboiu, J. Med. Chem., 2009, 52, 4853–4859 CrossRef CAS PubMed.
- Z. Fang, W. He, X. Li, Z. Li, B. Chen, P. Ouyang and K. Guo, Bioorg. Med. Chem. Lett., 2013, 23, 5174–5177 CrossRef CAS PubMed.
- I. S. M. Meldal, J. Chem. Soc., Perkin Trans. 1, 1995, 159–1596 Search PubMed.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef CAS PubMed.
- C. Godoy-Alcántar, A. K. Yatsimirsky and J.-M. Lehn, J. Phys. Org. Chem., 2005, 18, 979–985 CrossRef.
- K. Wenck, S. Koch, C. Renner, W. Sun, T. Schrader and V. Uni, J. Am. Chem. Soc., 2007, 129, 16015–16019 CrossRef CAS PubMed.
- J. K. M. Sanders, Pure Appl. Chem., 2000, 72, 2265–2274 CrossRef CAS.
- V. Berl, I. Huc, J.-M. Lehn, A. DeCian and J. Fischer, Eur. J. Org. Chem., 1999, 3089–3094 CrossRef CAS.
- M. Sindelar and K. T. Wanner, ChemMedChem, 2012, 7, 1678–1690 CrossRef CAS PubMed.
- M. Sindelar, T. a. Lutz, M. Petrera and K. T. Wanner, J. Med. Chem., 2013, 56, 1323–1340 CrossRef CAS PubMed.
- P. M. Fischer and D. P. Lane, Curr. Med. Chem., 2000, 7, 1213–1245 CrossRef CAS.
- M. Knockaert, P. Greengard and L. Meijer, Trends Pharmacol. Sci., 2002, 23, 417–425 CrossRef CAS.
- N. O. Dalby, Eur. J. Pharmacol., 2003, 479, 127–137 CrossRef CAS PubMed.
- K. T. W. G. Höfner, C. Zepperitz and K. T. Wanner, in Mass Spectrometry in Medicinal Chemistry, ed. K. T. Wanner and G. Höfner, Wiley-VCH, Weinheim, 2007, pp. 247–283 Search PubMed.
- C. Zepperitz, G. Höfner and K. T. Wanner, ChemMedChem, 2006, 1, 208–217 CrossRef CAS PubMed.
- A. Kragler, G. Höfner and K. T. Wanner, Eur. J. Med. Chem., 2008, 43, 2404–2411 CrossRef CAS PubMed.
- G. R. L. Cousins, S.-A. Poulsen and J. K. M. Sanders, Chem. Commun., 1999, 1575–1576 RSC.
- R. L. E. Furlan, Y. Ng, S. Otto and J. K. M. Sanders, J. Am. Chem. Soc., 2001, 123, 8876–8877 CrossRef CAS.
- J. Liu, K. R. West, C. R. Bondy and J. K. M. Sanders, Org. Biomol. Chem., 2007, 5, 778–786 CAS.
- T. Bunyapaiboonsri, O. Ramström, S. Lohmann, J.-M. Lehn, L. Peng and M. Goeldner, ChemBioChem, 2001, 2, 438–444 CrossRef CAS.
- A. J. Clipson, V. T. Bhat, I. McNae, A. M. Caniard, D. J. Campopiano and M. F. Greaney, Chem. – Eur. J., 2012, 18, 10562–10570 CrossRef CAS PubMed.
- Y. Patskovsky, L. Patskovska, S. C. Almo and I. Listowsky, Biochemistry, 2006, 3852–3862 CrossRef CAS PubMed.
- J. B. Cooper, Curr. Drug Targets, 2002, 2, 155–173 CrossRef.
- K. C. Nicolaou, R. Hughes, S. Y. Cho, N. Winssinger, C. Smethurst, H. Labischinski and R. Endermann, Angew. Chem., Int. Ed., 2000, 39, 3823–3828 CrossRef CAS.
- J. Tomasek and J. Schatz, Green Chem., 2013, 15, 2317–2338 RSC.
- B. Shi and M. F. Greaney, Chem. Commun., 2005, 886–888 RSC.
- C. Anderssonsg, E. Mosialous, A. E. P. Adangllll, G. J. Mulder and A. Van Der, J. Biol. Chem., 1991, 266, 2076–2079 Search PubMed.
- D. Burg, D. V. Filippov, R. Hermanns, G. A. Van Der Marel, J. H. Van Boom and G. J. Mulder, Bioorg. Med. Chem., 2002, 10, 195–205 CrossRef CAS.
- H. Hioki and W. C. Still, J. Org. Chem., 1998, 3375, 904–905 CrossRef.
- S. Otto, R. L. E. Furlan and J. K. M. Sanders, J. Am. Chem. Soc., 2000, 122, 12063–12064 CrossRef CAS.
- N. R. Rose, E. C. Y. Woon, G. L. Kingham, O. N. F. King, J. Mecinović, I. J. Clifton, S. S. Ng, J. Talib-Hardy, U. Oppermann, M. A. McDonough and C. J. Schofield, J. Med. Chem., 2010, 53, 1810–1818 CrossRef CAS PubMed.
- E. C. Y. Woon, M. Demetriades, E. A. L. Bagg, W. Aik, S. M. Krylova, J. H. Y. Ma, M. Chan, L. J. Walport, D. W. Wegman, K. N. Dack, M. a. McDonough, S. N. Krylov and C. J. Schofield, J. Med. Chem., 2012, 55, 2173–2184 CrossRef CAS PubMed.
- M. T. Cancilla, M. M. He, N. Viswanathan, R. L. Simmons, M. Taylor, A. D. Fung, K. Cao and D. A. Erlanson, Bioorg. Med. Chem. Lett., 2008, 18, 3978–3981 CrossRef CAS PubMed.
- D. E. Scott, G. J. Dawes, M. Ando, C. Abell and A. Ciulli, ChemBioChem, 2009, 10, 2772–2779 CrossRef CAS PubMed.
- C. Saiz, V. Castillo, P. Fontán, M. Bonilla, G. Salinas, A. Rodríguez-Haralambides and S. G. Mahler, Mol. Diversity, 2014, 18, 1–12 CrossRef CAS PubMed.
- K. Bush, G. A. Jacoby and A. A. Medeiros, Antimicrob. Agents Chemother., 1995, 39, 1211–1233 CrossRef CAS.
- S. Siemann, A. J. Clarke, T. Viswanatha and G. I. Dmitrienko, Biochemistry, 2003, 42, 1673–1683 CrossRef CAS PubMed.
- J. M. Daniel, S. D. Friess, S. Rajagopalan, S. Wendt and R. Zenobi, Int. J. Mass Spectrom., 2002, 216, 1–27 CrossRef CAS.
- D. A. Erlanson, J. W. Lam, C. Wiesmann, T. N. Luong, R. L. Simmons, W. L. DeLano, I. C. Choong, M. T. Burdett, W. M. Flanagan, D. Lee, E. M. Gordon and T. O'Brien, Nat. Biotechnol., 2003, 33, 308–314 CrossRef PubMed.
- D. A. Erlanson, J. A. Wells and A. C. Braisted, Annu. Rev. Biophys. Biomol. Struct., 2004, 33, 199–223 CrossRef CAS PubMed.
- R. R. Gupta and M. D. Lechner, Chemical Shifts and Coupling Constants for Boron-11 and Phosphorus-31, Springer-Verlag, 1997 Search PubMed.
- H. Suenaga, K. Nakashima and S. Shinkai, J. Chem. Soc., Chem. Commun., 1995, 29–30 RSC.
- H. Suenaga, M. Mikami, H. Yamamoto, T. Harada and S. Shinkai, J. Chem. Soc., Perkin Trans. 1, 1995, 1733–1738 RSC.
- M. Demetriades, I. K. H. Leung, R. Chowdhury, M. C. Chan, M. A. McDonough, K. K. Yeoh, Y.-M. Tian, T. D. W. Claridge, P. J. Ratcliffe, E. C. Y. Woon and C. J. Schofield, Angew. Chem., Int. Ed., 2012, 51, 6672–6675 CrossRef CAS PubMed.
- I. K. H. Leung, M. Demetriades, A. P. Hardy, C. Lejeune, T. J. Smart, A. Szöllössi, A. Kawamura, C. J. Schofield and T. D. W. Claridge, J. Med. Chem., 2013, 56, 547–555 CrossRef CAS PubMed.
- A. C. R. Epstein, J. M. Gleadle, L. A. McNeil, K. S. Hewitson, J. O. Rourke, D. R. Mole, M. Mukherji, E. Metzen, M. I. Wilson, A. Dhanda, Y. Tian, N. Masson, D. L. Hamilton, P. Jaakkola, R. Barstead, J. Hodgkin, P. H. Maxwell, C. W. Pugh, C. J. Schofield, P. J. Ratcliffe, R. Drive and O. Ox, Cell, 2001, 107, 43–54 CrossRef CAS.
- G. L. Semenza, Nat. Rev. Cancer, 2003, 3, 721–732 CrossRef CAS PubMed.
- P. Reeh and J. de Mendoza, Chem. – Eur. J., 2013, 19, 5259–5262 CrossRef CAS PubMed.
- T. Letters, Tetrahedron Lett., 1997, 4039, 8145–8148 Search PubMed.
- V. Goral, M. I. Nelen, A. V. Eliseev and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1347–1352 CrossRef CAS PubMed.
- C. Clavaud, J. Le Gal, R. Thai, M. Moutiez and C. Dugave, ChemBioChem, 2008, 9, 1823–1829 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |