
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–organic framework based mixed matrix membranes: a solution for highly efficient CO2 capture?†
Beatriz Seoane
a,
Joaquin Coronasb,
Ignacio Gascon
c,
Miren Etxeberria Benavidesd,
Oğuz Karvan
d,
Jürgen Caro
e,
Freek Kapteijn
a and
Jorge Gascon
*a
aCatalysis Engineering, Chemical Engineering Department, Delft University of Technology, Julianalaan 131, 2628 BL Delft, The Netherlands. E-mail: j.gascon@tudelft.nl
bChemical and Environmental Engineering Department and Instituto de Nanociencia de Aragón (INA), Universidad de Zaragoza, 50018 Zaragoza, Spain
cPhysical Chemistry Department and Instituto de Nanociencia de Aragón (INA), Universidad de Zaragoza, 50018 Zaragoza, Spain
dTecnalia, Energy and Environmental Division, Parque Tecnológico de San Sebastián, Mikeletegi Pasealekua, 2. E-20009 Donostia-San Sebastian, Spain
eInstitut für Physikalische Chemie und Elektrochemie, Leibniz Universität Hannover, Callinstr. 22, D-30167 Hannover
First published on 18th February 2015
Abstract
The field of metal–organic framework based mixed matrix membranes (M4s) is critically reviewed, with special emphasis on their application in CO2 capture during energy generation. After introducing the most relevant parameters affecting membrane performance, we define targets in terms of selectivity and productivity based on existing literature on process design for pre- and post-combustion CO2 capture. Subsequently, the state of the art in M4s is reviewed against these targets. Because final application of these membranes will only be possible if thin separation layers can be produced, the latest advances in the manufacture of M4 hollow fibers are discussed. Finally, the recent efforts in understanding the separation performance of these complex composite materials and future research directions are outlined.
 Beatriz Seoane | Beatriz Seoane (1985) received her MSc in 2009 in Chemical Engineering at the University of Zaragoza. The same year she started her PhD at the Chemical and Environmental Engineering Department in Zaragoza on the development of new strategies for MOF synthesis and their application as filler in polymeric membranes for gas separation. After finishing her PhD cum laude in 2014, she moved to The Netherlands to start as a post-doc in the Catalysis Engineering section of the Delft University of Technology. Her research interest focuses on the synthesis of engineered microporous compounds and the development of MOF-based Mixed-Matrix Membranes for gas separation. |
 Ignacio Gascon | Ignacio Gascon (1975) obtained his MSc (1998) and PhD (2002) in Chemistry at the University of Zaragoza, Spain. After post-doctoral stays at the University of Sheffield and Paul Sabatier (Toulouse) he returned to the University of Zaragoza where he became an Associate Professor of Physical Chemistry in 2010. His research interests include the fabrication and characterization of ultrathin films of molecular, polymeric and nano-structured materials and the study of thermophysical properties of liquid systems. He has co-authored more than 70 publications in peer-reviewed journals. |
 Oğuz Karvan | Oğuz Karvan (1972) received his PhD in Materials Science and Engineering from Istanbul Technical University, Turkey (2007). He took part in Dr W. J. Koros Membrane Research Group at Georgia Institute of Technology, Atlanta, USA between 2008–2011 as postdoctoral fellow and research engineer. He joined TECNALIA on 2012 as a researcher in the “Materials for Energy Business Area” at the Energy and Environment Division of TECNALIA. He acts as a project manager in Membrane Technology Research Department. He focuses on polymeric, hybrid and carbon membranes for gas separation applications. He has experience in developing, synthesis and characterization of porous materials, adsorbents and membranes. |
 Jürgen Caro | Jüergen Caro (1951) received his MSc in Chemistry in 1974 and his PhD in Physical Chemistry in 1977, both at Leipzig University. He joint the Academy of Sciences in Berlin, where he was from 1988–1889 the Deputy Director of the Institute of Physical Chemistry. In 1989 he got Dr sc. Nat., in 1992 Dr rer nat habil. Caro was one of the founders of the Institute of Applied Chemistry in Berlin-Adlershof where he was until 2001 the Head of the Department “Inorganic materials and chemical engineering science”. In 2001 Caro became C4 professor for physical chemistry at the University Hannover. Caro is author of 300 publications and 38 patents. He got in 2013 the Breck Award of the International Zeolite association and in 2013 the Ostwald medal of the Saxonian Academy of Sciences. |
 Freek Kapteijn | Freek Kapteijn (1952), MSc in Chemistry and Mathematics, received his PhD on ‘Metathesis of alkenes’ in 1980 at the University of Amsterdam. After post-doc positions (Coal Science) in Amsterdam and Nancy (ENSIC), he became Associate professor in Amsterdam. Moved to Delft University of Technology in 1992, became ‘Anthonie van Leeuwenhoek professor’ in 1999, and is since 2008 chair of Catalysis Engineering, with visiting professorships at ETH Zürich, Tianjin and Zhejiang Normal University. Research interest focuses on the interplay of catalysis and engineering, comprising structured and multifunctional catalysts, adsorption, separation and (catalytic) membranes. Co-authored over 500 publications in peer-reviewed journals and as book chapters. |
 Jorge Gascon | Jorge Gascon (1977) received his MSc. in Chemistry in 2002 and his PhD in Chemical Engineering in 2006, both at the University of Zaragoza (Spain). Starting as post-doc at TUDelft he is since 2014 ‘Anthonie van Leeuwenhoek Professor’ of Catalysis Engineering. Research interests include fundamental aspects and applications of new nano-structured materials and composites. He has co-authored over 110 publications, several patents and has edited the book: “Metal–Organic Frameworks as Heterogeneous Catalysts”. He has been the recipient of the prestigious VENI (2010), VIDI (2013) and ERC Starting (2013) personal grants. Recently he received the 2013 ExxonMobil Chemical European Science and Engineering Award. |
A Introduction – setting the scene
The urgent need for strategies to reduce global atmospheric concentrations of greenhouse gases has prompted international action from governments and industries, and a number of collaborative programs have been established including the European Strategic Energy Technology Plan (SET-Plan), the European Technology Platform for Zero Emission Fossil Fuel Power Plants (ZEP), the Intergovernmental Panel on Climate Change (IPCC), the United Nations Framework Commission on Climate Change, and the Global Climate Change Initiative.1–4 In addition to the continuous development of non CO2 emitting generation of energy from wind, solar or hydro and geothermal sources, the capture and sequestration of carbon dioxide, the predominant greenhouse gas, is a central strategy in these initiatives, as it offers the opportunity to meet increasing demands for fossil fuel energy on the short- to medium-term, whilst reducing the associated greenhouse gas emissions.5 In this spirit the EU, through the SET-Plan and the CCS Technology Roadmap, has agreed to enable the cost competitive deployment of CCS after 2020 and to further develop the technologies to allow application in all carbon intensive industrial sectors, with an objective of 90% CO2 capture with less than 8 percentage point efficiency losses.2,3,6Broadly, three lines of capturing technologies exist to reduce CO2 emissions in combustion processes: post-combustion, pre-combustion, and oxyfuel combustion (Fig. 1).
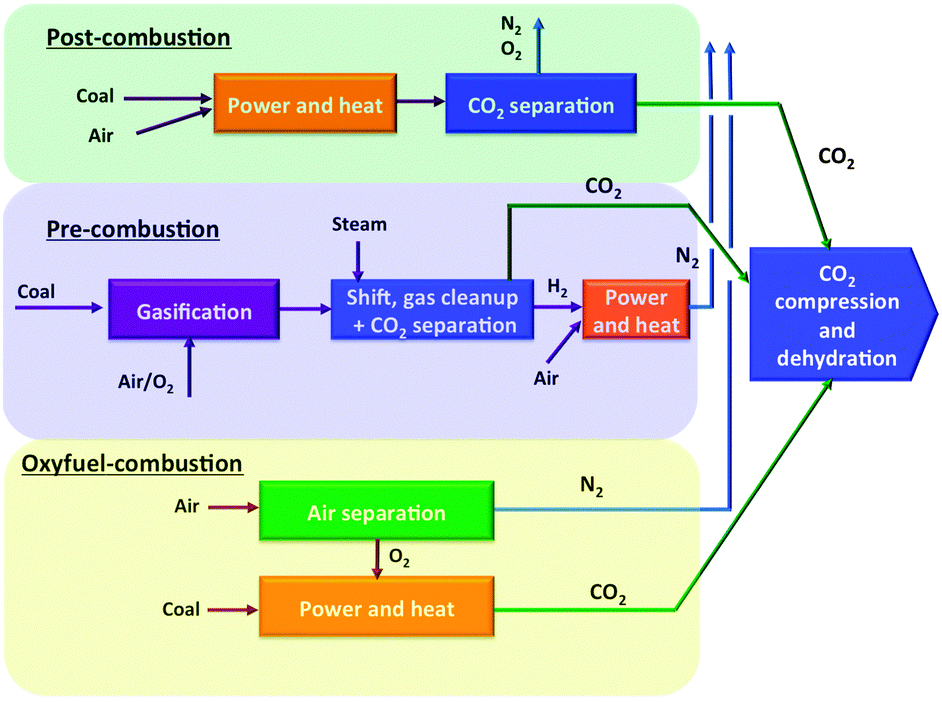 | ||
| Fig. 1 Technical options for CO2 capture from coal-power plants.1 | ||
Post-combustion CO2 capture comprises of capturing CO2 from the flue gases produced after fossil fuels or other carbonaceous materials (such as coal or biomass) are burned. Combustion-based power plants provide most of the world's electricity today. In modern natural gas and coal-fired power plants, the combustible is mixed with air and burned. The heat released by combustion generates steam, which drives a turbine-generator. The hot combustion gases exiting the boiler consist mainly of nitrogen (from air) plus lower concentrations of water vapour and CO2, with the concentration of the latter depending on the combustible used. Additional products formed during coal combustion from impurities in coal include sulphur dioxide, nitrogen oxides and particulate matter (fly ash). These regulated air pollutants, as well as other trace species such as mercury, must be removed to meet applicable emission standards. In some cases, additional removal of pollutants (especially SO2) is required to provide a sufficiently clean gas stream for subsequent CO2 capture. The absence of impurities in natural gas results in a clean flue gas stream, so that no additional clean-up is needed for effective CO2 capture.7
With current technology, the most effective method of CO2 capture from flue gases is chemical absorption in an aqueous solution of an amine based organic, such as mono- or diethanolamine (MEA, DEA). In the absorber, the flue gas is countercurrently “scrubbed” with an amine solution, typically capturing 85 to 90 percent of the CO2. The CO2-laden solvent is then pumped to a second vessel (stripper), where heat is supplied in the form of steam to release the CO2. The resulting stream of concentrated CO2 is then compressed and piped to a storage site, while the depleted solvent is recycled back to the absorber. The regeneration requires considerable energy, as not only the captured CO2 has to be released at higher temperatures, also the evaporation losses of water are considerable.1
To remove carbon from fuel prior to combustion it must first be converted into a form amenable to capture. For a coal-fuelled plant this is accomplished by reacting coal with steam and oxygen at high temperature and pressure, a process called partial oxidation, or gasification. The result is a gaseous fuel consisting mainly of carbon monoxide and hydrogen mixture known as synthesis gas (syngas), which can be burned to generate electricity in a combined cycle power plant. This approach is known as integrated gasification combined cycle (IGCC) power generation. After particulate impurities are removed from the syngas, a two-stage shift reactor converts the carbon monoxide to CO2via a reaction with steam (H2O). The result is a mixture of CO2 and hydrogen (and water). A solvent, such as the widely used commercial Selexol® (which employs a glycol-based solvent) and Rectisol® (using refrigerated methanol), then captures the CO2, leaving a stream of nearly-pure hydrogen that is burned in a combined cycle power plant to generate electricity.8 Although the fuel conversion steps of an IGCC plant are more elaborate and costly than traditional coal combustion plants, CO2 separation is much easier and cheaper because of the high operating pressure and high CO2 concentration of this design. Thus rather than requiring a chemical reaction to capture CO2 (as with amine systems in post-combustion capture), the mechanism employed in pre-combustion capture involves physical absorption into a solvent (although pressures above ∼20 bar are required), followed by release of the CO2 when the pressure is reduced, typically in several stages. Nonetheless, there is still a significant energy penalty associated with CO2 capture due to the need for a shift reactor and other processes. In oxyfuel processes pure oxygen is used for the combustion, resulting in a flue gas containing mainly water vapour and carbon dioxide. Condensation of the water results in a nearly pure carbon dioxide stream. The major energy penalty here is the production of pure oxygen by air separation.
In general, the higher the power plant efficiency, the smaller the energy penalty and associated impact for CO2 separation. For this reason, replacing or repowering an old, inefficient plant by a new, more efficient unit with CO2 capture can still yield a net efficiency gain that decreases all plant emissions and resource consumption. Thus, the net impact of the CO2 capture energy penalty must be assessed in the context of a particular situation or strategy for reducing CO2 emissions and developing sustainable processes.9
The energy requirements of current CO2 capture systems are roughly ten to a hundred times greater than those of other environmental control systems (e.g. de-NOx, SOx capture, fly ash removal) employed at a modern electric power plant. This energy “penalty” lowers the overall (net) plant efficiency globally by 20–30% and significantly increases the net costs of CO2 capture, as indicated in Table 1.
| Power plant and capture system type | Net plant efficiency (%) w/o CCS | Net plant efficiency (%) with CCS | CCS energy penalty | |
|---|---|---|---|---|
| Additional energy input (%) per net kW h outputa | Reduction in net kW h output (%) for a fixed energy input | |||
| a This is the definition of the incremental primary energy needed to supply one unit of electric power (e.g., 1 kW h) to the grid. b PC stands for pulverized coal. | ||||
| Existing subcritical PC,b post-combustion capture | 33 | 23 | 43 | 30 |
| New supercritical PC, post-combustion capture | 40 | 31 | 29 | 23 |
| New supercritical PC, oxy-combustion capture | 40 | 32 | 25 | 20 |
| New IGCC (bituminous), pre-combustion capture | 40 | 33 | 21 | 18 |
| New Natural Gas comb. cycle, pre-combustion capture | 50 | 43 | 16 | 14 |
A recent analysis has shown that the thermodynamic minimum energy demand for capturing 90% of the CO2 from the flue gas of a typical coal-fired power plant is approximately 3.5% (assuming a flue gas containing 12–15% CO2 at 40 °C).11 By comparison with data presented in Table 1, column 5, it is clear that current technology is far from ideal. In addition, although absorber–stripper units represent a proven, well-accepted technology in the gas processing industry, the high-pressure absorber tower in particular is an expensive, large, thick-walled, heavy vessel. The need to heat and cool the recirculating fluids requires careful, well-monitored, expensive operating procedures.12,13 Furthermore, the degradation of the amine absorbent leads to corrosive mixtures. Although the use of inhibitors reduces degradation (e.g. FLUOR's Econamine FGplus technology14), the need for regular maintenance hinders the use of amine absorber–strippers in remote locations and in small electricity plants.15 On the other hand, the use of amines and solvents is environmentally unfriendly due to the contamination of the gas with solvent vapours and the likely degradation of amines due to the high temperature treatments required to regenerate the absorbent. To prevent this, task specific ionic liquids, which exhibit extremely low partial pressures up to 300 °C, have been designed as solvents or active absorbents. However, the frequently used ionic liquids with phosphine anions have the tendency to decompose via Beckman rearrangement at moderate temperatures16 which limits their use.
Fig. 2 displays the Levelized Cost Of Energy (LCOE) of new power plants with and without CCS, considering different Emissions Trading System (EU ETS) values as reported in recent studies based on current commercial post-combustion and pre-combustion capture processes.1
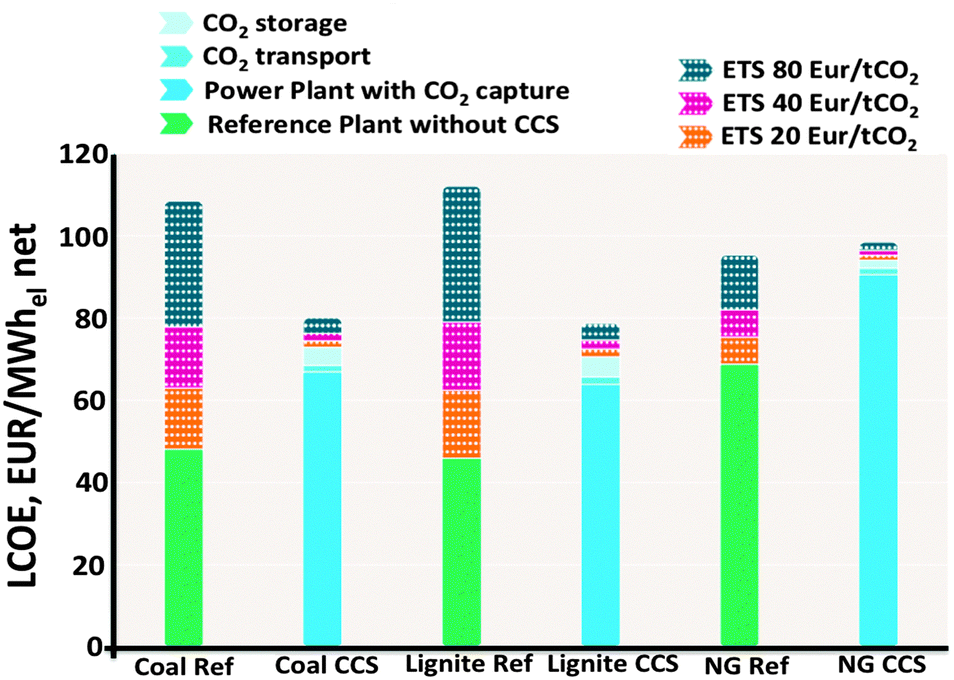 | ||
| Fig. 2 The levelized cost of energy (LCOE) of integrated CCS projects (blue bars) compared to the reference plants without CCS (green bars).1 | ||
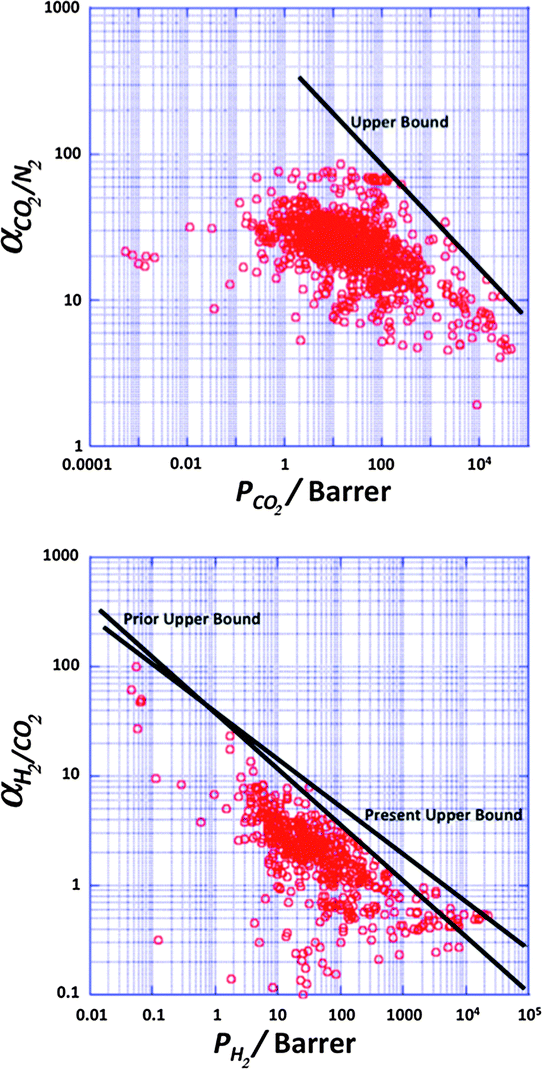 | ||
| Fig. 3 Robeson plots for the separation of CO2 from N2 (top) and H2 from CO2 (bottom). This plot shows the selectivity obtained from the ratio of pure-gas permeabilities plotted against permeability of one component for different polymeric membranes. No commercial polymeric membranes currently operate above the upper bound.23 | ||
The LCOE (in € per MW h) is shown for power plants burning bituminous coal, lignite or natural gas. The LCOE includes the costs of CCS, from which 80 to 90 percent is for capture (including compression), the rest for transport and storage.11Fig. 2 can be used to calculate the cost per MW h of electricity produced: for a coal reference plant 45 € per MW h without CCS vs. 70 € per MW h using CCS. This cost difference is equivalent to the “carbon price”, for new supercritical coal plants this is currently about €24–35/ton CO2 (one should note that the amount of tons of CO2 released per MW h of electricity generated depend on the efficiency of the plant on the nature of the coal).
These economic, energetic, operational and environmental evaluations underscore the immense opportunities and incentives that exist for improved CO2 capture processes and materials.
Alternative processes, still in different stages of development, comprise chemical looping combustion (CLC) using metal oxides, carbon capture during water gas shift (clay type materials), and adsorption–regeneration processes using solid adsorbents (zeolites, activated carbons, metal–organic frameworks). These are all cyclic uptake–regeneration processes based on solid materials, that are either recirculated from one reactor system to another, or used in fixed beds in swing operation.17–19 These processes operate at quite different temperature levels, with the adsorption–desorption processes at the lowest since these are mainly based on (exothermal) physical adsorption.
Gas separation membranes offer a number of benefits over other gas separation technologies.20 Conventional technologies such as the cryogenic distillation, adsorption, condensation and amine absorption require a gas–liquid phase change. This phase change adds a significant energy cost to the separation cost. Membrane gas separation, on the other hand, does not require a phase change. In addition, gas separation membrane units are smaller than other types of plants, such as amine stripping plants, and therefore have relatively small footprints. The lack of mechanical complexity in membrane systems is another advantage. Membrane devices for gas or vapour separation usually operate under continuous, steady-state conditions. The feed stream passes along one side of the membrane. The non-permeating molecules that are retained at the feed-stream side exit the membrane as the retentate stream. A pressure difference across the membrane drives the permeation process. The mechanism of permeation (sorption of molecules and diffusion) depends on the membrane material. In case of membranes with well-defined pores (i.e. zeolites, metal–organic frameworks, carbon molecular sieves (CMS)) adsorption, diffusion and eventually molecular sieving dominate membrane performance, whereas in case of polymeric membranes permeation takes place mostly through a solution–diffusion mechanism.
In 1980, Permea (now a division of Air Products) launched its hydrogen-separating Prism membrane.21 This was the first large industrial application of gas separation membranes. Since then, membrane-based gas separation has grown almost exponentially.22,23 Membranes were known to have the potential to separate important gas mixtures long before 1980, but the technology to fabricate economically high-performance membranes and modules was lacking. The development of high-flux anisotropic membranes and large surface area membrane modules for reverse osmosis applications occurred in the late 1960s and early 1970s. Permea then adapted this technology to membrane gas separation.24 Its polysulfone hollow-fiber membrane was an immediate success, particularly for the separation and recovery of hydrogen from the purge gas streams of ammonia plants. Within a few years, Permea systems were installed in many such plants. This success encouraged other companies to advance their own technologies.
The first membrane systems (anisotropic cellulose acetate) to separate carbon dioxide from natural gas were introduced in the mid 1980s by Cynara (now part of Natco), Grace Membrane Systems Separex (now part of UOP), and GMS (now part of Kvaerner).25 In the last decade, cellulose acetate has begun to be challenged by newer membranes, such as polyimide (Air Liquide) and perfluoropolymer membranes (ABB/MTR).21 At about the same time, Generon (now part of MG) introduced a membrane system based on poly(4-methyl-1-pentene) (TPX) to separate nitrogen from air. These membranes were only competitive in a few niche areas requiring 95% nitrogen, but by 1990, Generon, Praxair, and Medal all had produced custom polymers with higher oxygen selectivities.25 This application has grown to represent about one-third of the new nitrogen production capacity; to date more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nitrogen systems have been installed worldwide.22 Finally, membranes are also being used for a variety of small but growing applications, such as the dehydration of compressed air and the separation of hydrocarbons from nitrogen or air.25
000 nitrogen systems have been installed worldwide.22 Finally, membranes are also being used for a variety of small but growing applications, such as the dehydration of compressed air and the separation of hydrocarbons from nitrogen or air.25
As observed above, to date only polymeric membranes have found their way towards large-scale industrial implementation in gas separation. This is to a large extent due to their easy processing and mechanical strength.26 However, a poor resistance to contaminants, low chemical and thermal stability and a limit in the trade-off between permeability and selectivity, the so called Robeson upper bound limit,22,23 are among their main disadvantages.
In parallel to the development of polymeric membrane materials, much research effort has been devoted to develop pure inorganic membranes, among others by several of our authors. Inorganic membranes refer to membranes made of materials such as ceramics,27 carbon,28 zeolite,29 various oxides (alumina, titania, zirconia),30 metal–organic frameworks,31 and metals such as palladium, silver and their alloys.32 Inorganic membranes can be classified into two major categories based on their structure: porous inorganic membranes and dense (non-porous) inorganic membranes. Microporous inorganic membranes include both amorphous and crystalline membranes. Although inorganic membranes offer unique properties for gas separation (i.e. excellent thermal and chemical stability, good erosion resistance and high gas flux and selectivity), certain aspects still require further attention such as mechanical resistance, reproducibility, long term stability, scaling up and, more importantly, fabrication costs. Other types of hybrid membranes such as organosilica based33,34 share the same fabrication prize issue.
The cost of inorganic membranes is dominated by that of the support on which the selective layer is deposited, with Pd membranes as exception.31,35 Only zeolite A membranes are deployed commercially for alcohol dehydration by vapour permeation.
In order to overcome the limitations of both polymeric and inorganic membranes, the so-called Mixed Matrix Membranes (MMMs, consisting of a blend of filler particles in a polymeric matrix) have been identified to provide a solution to go beyond the upper-bound trade-off limit of the polymeric membranes as well as the inherent obstacles of brittleness and lack of reproducibility associated with inorganic membranes. MMMs potentially combine the advantages in separation performances of both inorganic and polymeric membranes and overcome their drawbacks, although it introduces the issue of compatibility between the constituents. A good adhesion is essential to avoid non-selective voids in such membranes.
Indeed, during the last few decades, several solutions have been proposed to boost the performance of polymeric membranes. Various polymers have been modified with inorganic fillers such as zeolites, mesoporous silicas, activated carbons, carbon nanotubes and even non-porous solids to produce Mixed Matrix Membranes (MMMs).36–40 A mixed matrix membrane is a composite of filler particles in a polymeric matrix. As it will unveiled in this review, both polymer as well as filler properties affect MMM morphology and separation performance.
Recent advances have shifted towards the addition of new fillers, namely carbon nanotubes, layered silicates (sometimes after delamination) and MOFs as potential fillers in the polymer matrix.41,42 MOFs are among the most sophisticated nanostructured materials.43 In addition to a high surface area and pore volume, their chemical nature can be fine-tuned by selecting the appropriate building blocks and/or by post-synthetic modification, thus leading to tailored porous materials with great promise for the selective adsorption of strategic gases. More importantly, the porosity of MOFs is, in general, much higher than that of their inorganic counterpart, zeolites, justifying the designation ‘framework’ and challenging the scientific community to make an effective use of such empty space. In addition to the facile functionalization, many MOFs are known to undergo structural changes upon adsorption of different molecules (‘breathing’),44 facilitating the design of, for instance, dynamic composites.45–47 When it comes to MMMs, the use of MOFs as fillers might result in a breakthrough in the MMM field, since compatibility issues can eventually be overcome by optimizing the MOF linker–polymer interactions.46 Since the first report in 2004,48 research into M4s has experienced an unprecedented explosion. Certainly, as highlighted in recent reviews,49–52 MOF based mixed matrix membranes (M4s) have the potential to overcome current challenges in membrane separation, both in terms of membrane synthesis and performance. Because of these reasons, we believe that it is now the right moment to critically evaluate the recent advances in the field. In this review, after introducing the most relevant parameters affecting membrane performance, we will define targets in terms of selectivity and productivity based on existing literature on process design for pre- and post-combustion CO2 capture. Subsequently, the state of the art in M4s is reviewed against the previously defined targets. Because final application of these membranes will only be possible if thin separating layers can be produced, we will then review the latest advances in the manufacture of M4s hollow fibers. Last but not least, the recent efforts in understanding the separation performance of these complex composite materials will be discussed. This article is finally wrapped up with our personal opinion and possible future directions in the development of new generations of M4s.
B Describing transport in mixed matrix membranes
The lab-scale manufacture of M4s is similar to the one applied for the synthesis of other MMMs. In the general procedure, the first step is the dispersion of the filler in the solvent in an ultrasonic bath. Polymer is then added, usually maintaining a ratio 90/10 wt% solvent/filler–polymer mixture. The whole mixture is stirred overnight. Before the casting, different intervals of sonication and stirring take place to ensure a well dispersion, provided that sonication does not result in deterioration of the polymer (something very important when high flux polymers such as PIM-1 are used in the manufacture of the composite). Subsequently, the membranes are cast on a flat surface, either Petri-type dishes or Doctor Blade system, and then left overnight for evaporation of solvent at room temperature. Once dried, the films are placed in a vacuum oven for 24 h at a specific temperature (depending on the polymer glass transition temperature) high enough to remove the remaining solvent.Permeability and separation factor are the two key parameters generally used to characterize polymeric membranes. Permeability, officially called permeability coefficient, Pi, a normalized productivity of a specific gas component by the membrane, is defined (eqn (1)) as the diffusive Flux of gas i through the membrane (flow per unit membrane area A) normalized by the partial pressure difference of that component across the membrane per unit thickness of the membrane (l).
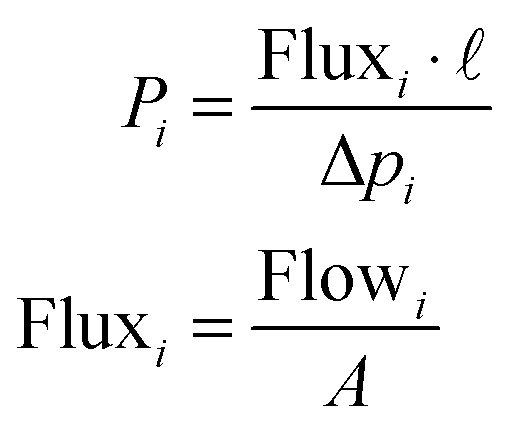 | (1) |
However, permeability values can only be given when the thickness of the separating layer is well known, something not possible in case of very thin membranes or advanced membrane configurations such as hollow fibers and asymmetric films. In this case, Permeance (pressure normalized flux) is used, with Gas Permeance Units (GPU) being the most widely applied units in polymer membrane separations: 1 GPU = 10−6 cm3(STP) cm−2 cmHg−1 = 0.344 × 10−10 mol m−2 s−1 Pa−1.
The separation factor or permselectivity reflects the capability of a membrane to separate one gas from another. If the permeabilities of two individual components are known, the ideal selectivity, Sij (eqn (2)), is given by the ratio of the two pure gas permeabilities:
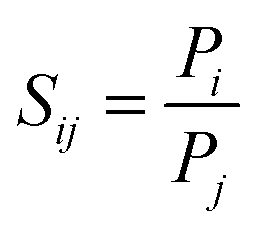 | (2) |
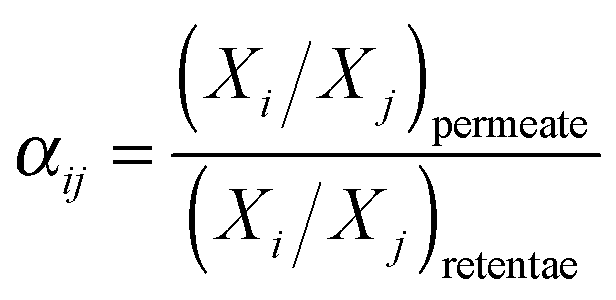 | (3) |
In order to be able to understand composite performance, the development of appropriate models that describe transport is crucial. For an extensive overview on modeling of MMMs we strongly recommend the recent review by Vinh-Thang and Kaliaguine.55 In short, during the last few decades different models have been proposed to estimate the permeation performance of MMMs by developing different theoretical expressions depending on MMM morphology. These models capture to different levels of complexity the presence of filler in a continuous polymer matrix, and the effect of voids and rigidified polymer regions. Barrer and Petropoulos were the first proposing a model for the performance of polymer–filler blends.56 Their formulation assumes concentration-independent diffusivities and Henry's law for adsorption, and it deals with the inherent two-dimensionality of the situation through the introduction of several unknown correction factors. The reliance on such correction factors is one good reason to seek more satisfying treatments; the restrictive assumption of Henry's law adsorption is another. Cussler has proposed perhaps the most sophisticated model by reducing the three-dimensional diffusion problem to an essentially one-dimensional problem through a series of approximations.57 Aside from the limitations of these approximations, Cussler's model employs Fickian diffusivity with a constant diffusivity and an equilibrium condition between phases that requires identical adsorption isotherms in both materials. These are both serious limitations, particularly when trying to describe the performance of a composite containing a zeolite phase. The Maxwell formulation can be also extended to MMMs by combining the flux through the polymer and filler in parallel and series pathways, in a clear analogy to electrical circuits. This model is however only applicable for low filler loadings since it assumes that the streamlines associated with diffusive mass transport around filler particles are not affected by the presence of nearby particles. The Bruggeman model,58 which can be considered to be an improved version of the Maxwell model, accounts for these effects and extends the applicability to highly loaded MMMs.59 Sheffel and Tsapatsis60,61 introduced later a more extended model for diffusive transport in microporous MMMs utilizing the Maxwell–Stefan formulation and different models to account for multi-component mixtures: Henry's law, extended Langmuir model, and ideal adsorbed solution theory (IAST).
Any model attempting to describe diffusion within a membrane containing a microporous filler phase must include a realistic treatment of diffusion in both the porous filler and the continuous phase. Diffusion in the gas phase or in relative large pores (>100 nm (ref. 62)) is dominated by inter-molecular collisions and the flux of component i can be described by the Maxwell–Stefan (MS) approach,63 in which forces acting on molecules (in diffusional processes the gradient in thermodynamic potential) are balanced by the friction between molecules and, in case of porous materials, with a solid. In the latter case this model was named the ‘Dusty Gas Model’. The often used Fick's law is a simplification of the generalized MS equations for thermodynamically ideal systems.63
While Fickian diffusion can be used to describe transport through polymers, in the case of a porous material a correction needs to be made to account for the porosity (ε) and tortuosity (τ) of the material, leading to an ‘effective’ diffusivity. In this way, molar flux (Ni, mol m−2 s−1) can be defined as:
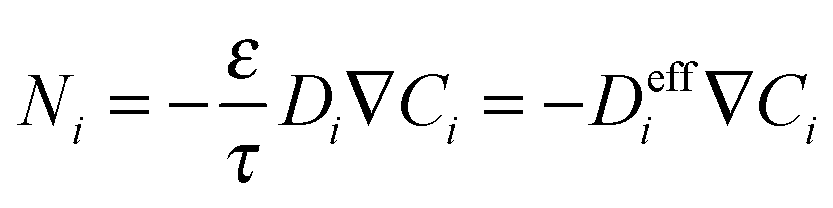 | (4) |
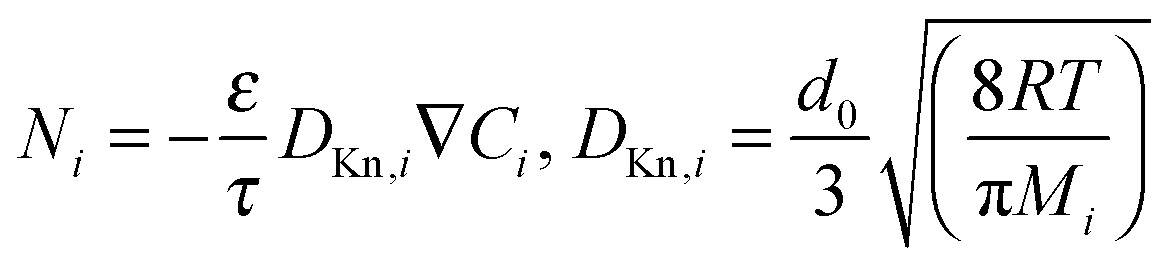 | (5) |
| Ni = −ρDi∇qi | (6) |
The adsorbed phase (qi) in eqn (6) is related to the gas phase fugacity through an adsorption isotherm of which the classical example is the Langmuir isotherm:
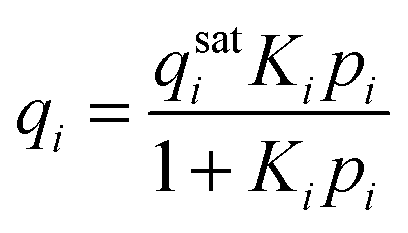 | (7) |
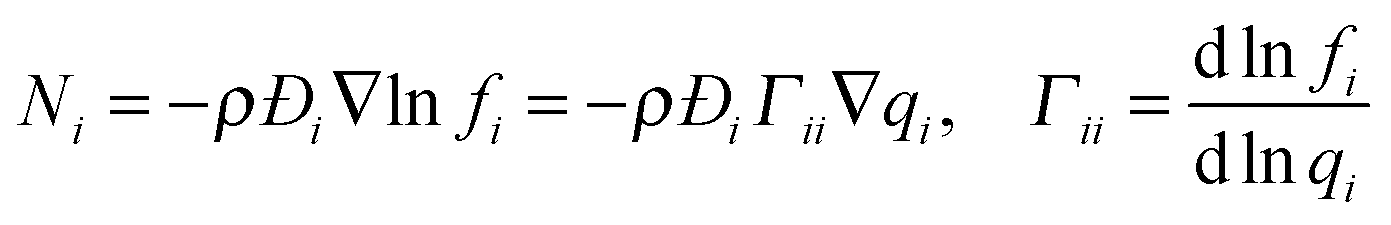 | (8) |
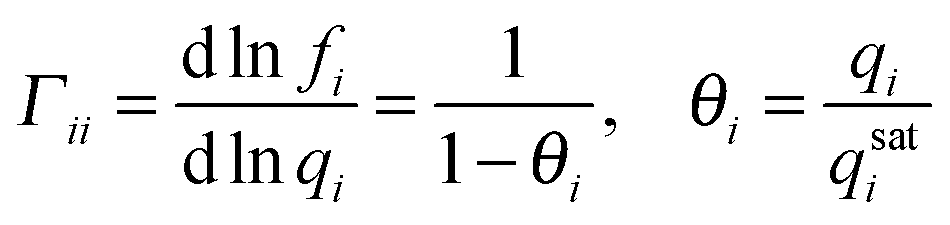 | (9) |
When multicomponent adsorption needs to be considered (i.e. separation through membranes), due to the relatively high concentrations of adsorbates, interactions between molecules can play a significant role in terms of ‘speeding up’ or ‘slowing down’ other components. In the Maxwell–Stefan approach besides the interaction (or ‘friction’) of the individual molecule with pore walls, also the interaction between the different diffusing molecules is accounted for and balanced with the driving force for mass transport:
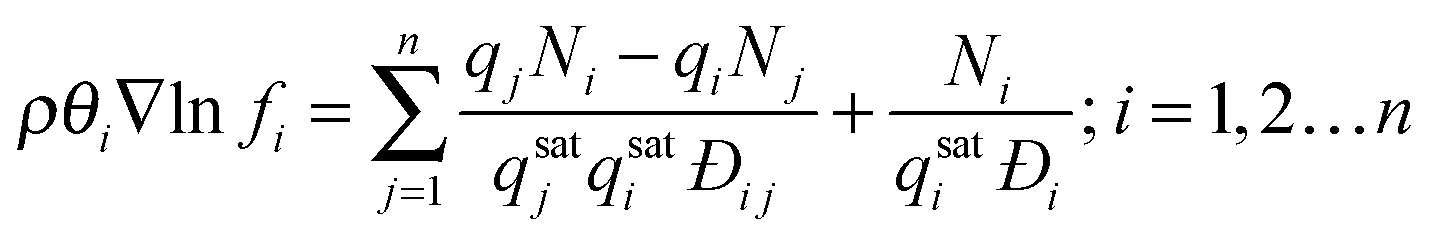 | (10) |
For a single component system of tagged and untagged species the saturation capacities are equal and one can show67 that the single component exchange coefficient is related to the self diffusivity and MS diffusivity as
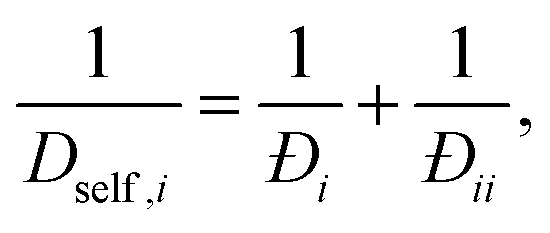 | (11) |
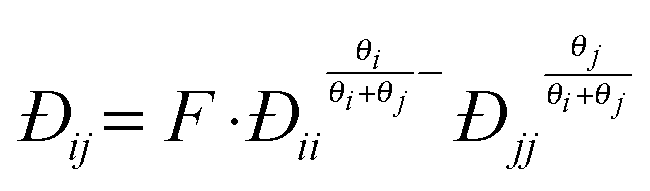 | (12) |
It is evident that in the case of mixture diffusion an accurate estimation of the individual component loading and the driving force is required to satisfactory model such a system. For zeolitic and MOF systems, IAST68 provides an acceptable mixture prediction based on the single component isotherms,62,69,70 but when adsorption heterogeneity becomes manifest IAST also tends to fail.71
At significant loading the molecular interaction can play an important role, strongly influencing the reactant and product concentration profiles. When the loading is relatively low the cross-correlation effects can often be ignored, i.e. the system can be modelled as single component system (eqn (8)).
Once diffusion and adsorption for both components have been defined, a model able to describe transport through the composite can be established. For instance, the Maxwell model can be used to describe the effective molar flux (Neff) of a gas species in a MMM for a suspension of spherical filler particles in a continuous polymer matrix as:59,72,73
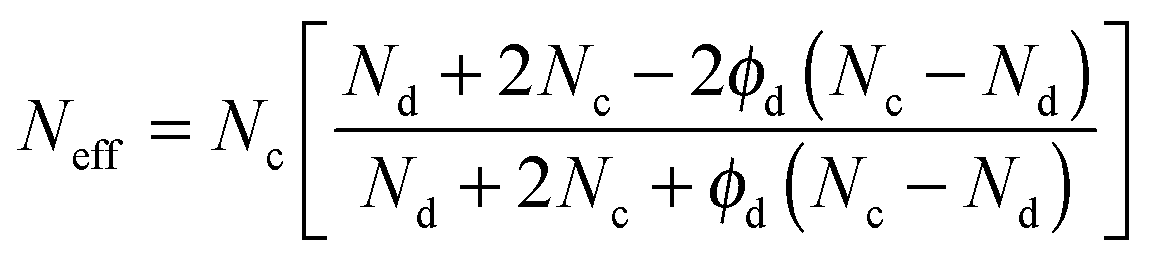 | (13) |
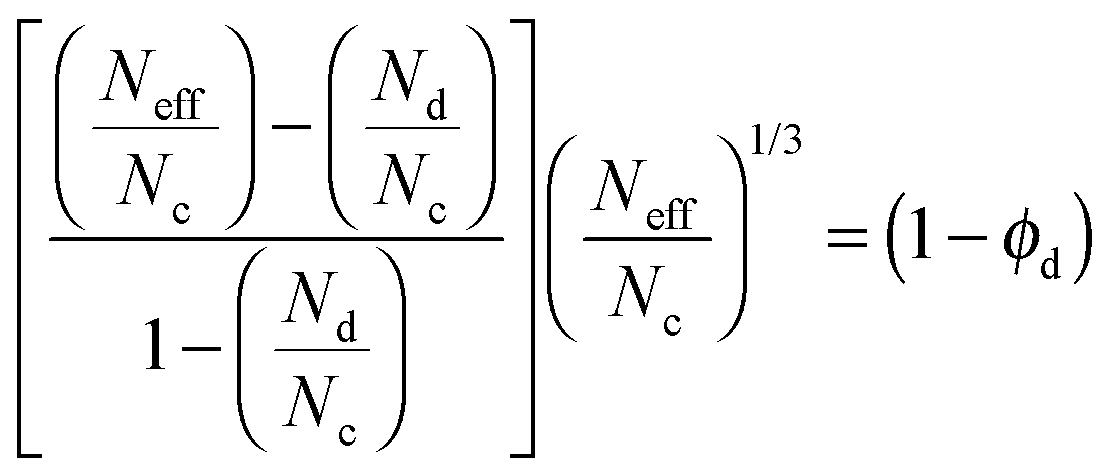 | (14) |
Modeling mixture permeation through MMMs is more complicated than describing pure gas permeation since the gas permeabilities of each species can be affected by competition effects between the two species. The most widely applied method for calculating mixture permeation is the so-called dual mode/partial immobilization model.59,74 The model proposes that sorption can occur in either the Langmuir or the Henry's Law regime (i.e. dual mode sorption) and that the diffusion through these regimes can also be different (partial immobilization). This approach is only based on parameters supplied by pure gas measurements. In this case, the generalized expressions for the permeability coefficients of species i and j in a binary gas mixture with a vacuum downstream are:
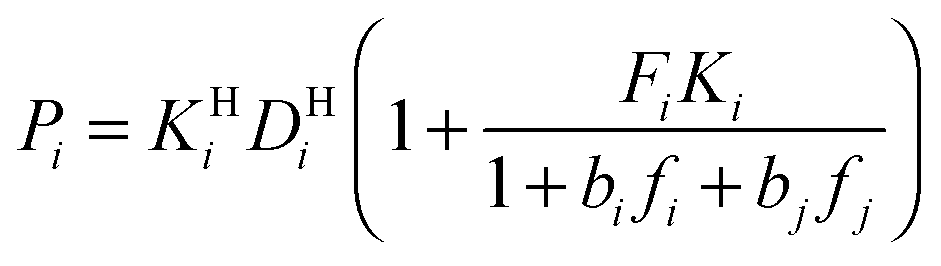 | (15) |
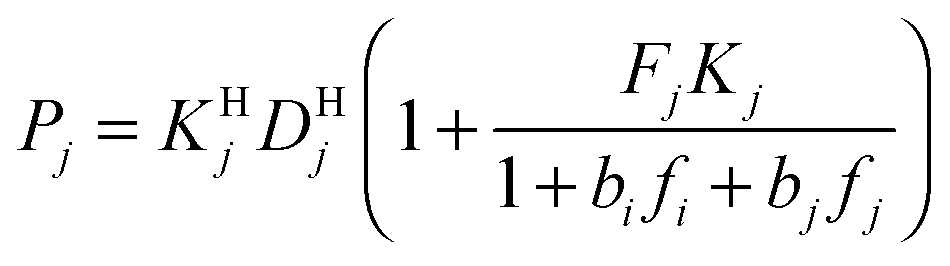 | (16) |
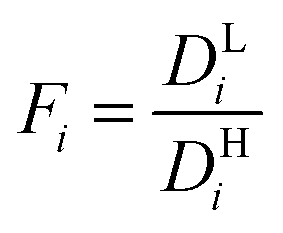 ,
, 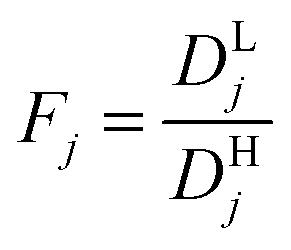 ,
, 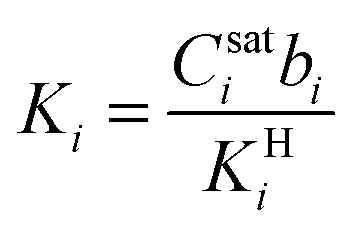 ,
, 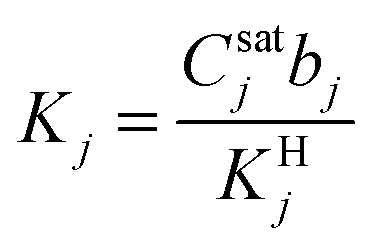 , and fi and fj correspond to the upstream fugacities of components i and j. In these expressions, DHi and DLi are the diffusivities of species i in the Henry and Langmuir environments, respectively; KHi is the Henry adsorption coefficient of species i, and Csatiand bi are the Langmuir capacity constant and affinity constant for species i, respectively.
, and fi and fj correspond to the upstream fugacities of components i and j. In these expressions, DHi and DLi are the diffusivities of species i in the Henry and Langmuir environments, respectively; KHi is the Henry adsorption coefficient of species i, and Csatiand bi are the Langmuir capacity constant and affinity constant for species i, respectively.
When it comes to MOF-MMMs, performance modeling has hardly been explored, mostly a post analysis was performed. Keskin and Sholl studied MMMs consisting of Matrimid® and MOF-5 by using Maxwell and Bruggeman permeation models to predict single gas permeabilities for low and high filler loadings respectively. To calculate mixture permeation, the authors applied a dual mode/partial immobilization method to describe gas transport through MMMs containing IRMOF-1 in Matrimid®. The performance of Cu(hfipbb)(H2hfipbb)0.5 MMMs was predicted using Maxwell and Bruggeman models.
They illustrated that 20 wt% Cu(hfipbb)(H2hfipbb)0.5 was enough to bring the MMM above the Robeson's upper bound with a CO2/CH4 selectivity of 72 and CO2 permeability of 15.7 Barrer. Keskin and co-workers have further expanded their modelling activities by combining molecular simulations to predict MOF and polymer properties and Maxwell, extended Maxwell and modified Maxwell models to predict M4 performance for a variety of systems comprising ZIFs and most popular MOFs.72,76–79
More recently, Nair and co-workers developed a completely new approach for the simulation of MMMs by constructing detailed and large-scale 3D mixed-matrix membrane (MMM) models, which were then solved by finite-element methods (see Fig. 4).75 Such models explicitly account for the effects of matrix-filler interfacial equilibrium in addition to the differences in Fickian diffusivity between the two phases. By doing so, they demonstrated that the individual values of the interfacial equilibrium constant or partition coefficient, K, the equilibrium ratio between the concentration in the filler and the polymer, and the diffusivity ratio of the filler and the matrix, Df/Dm, and not the combined permeability ratio Pf/Pm, determine the MMM permeability. This is in contrast to most commonly applied analytical equations (e.g., Maxwell model) that can only predict the MMM permeability under an implicit assumption that K and Df/Dm can be lumped into a single parameter, the permeability ratio Pf/Pm = KDf/Dm. This approach certainly looks like the way to go for modeling of these complex composites.
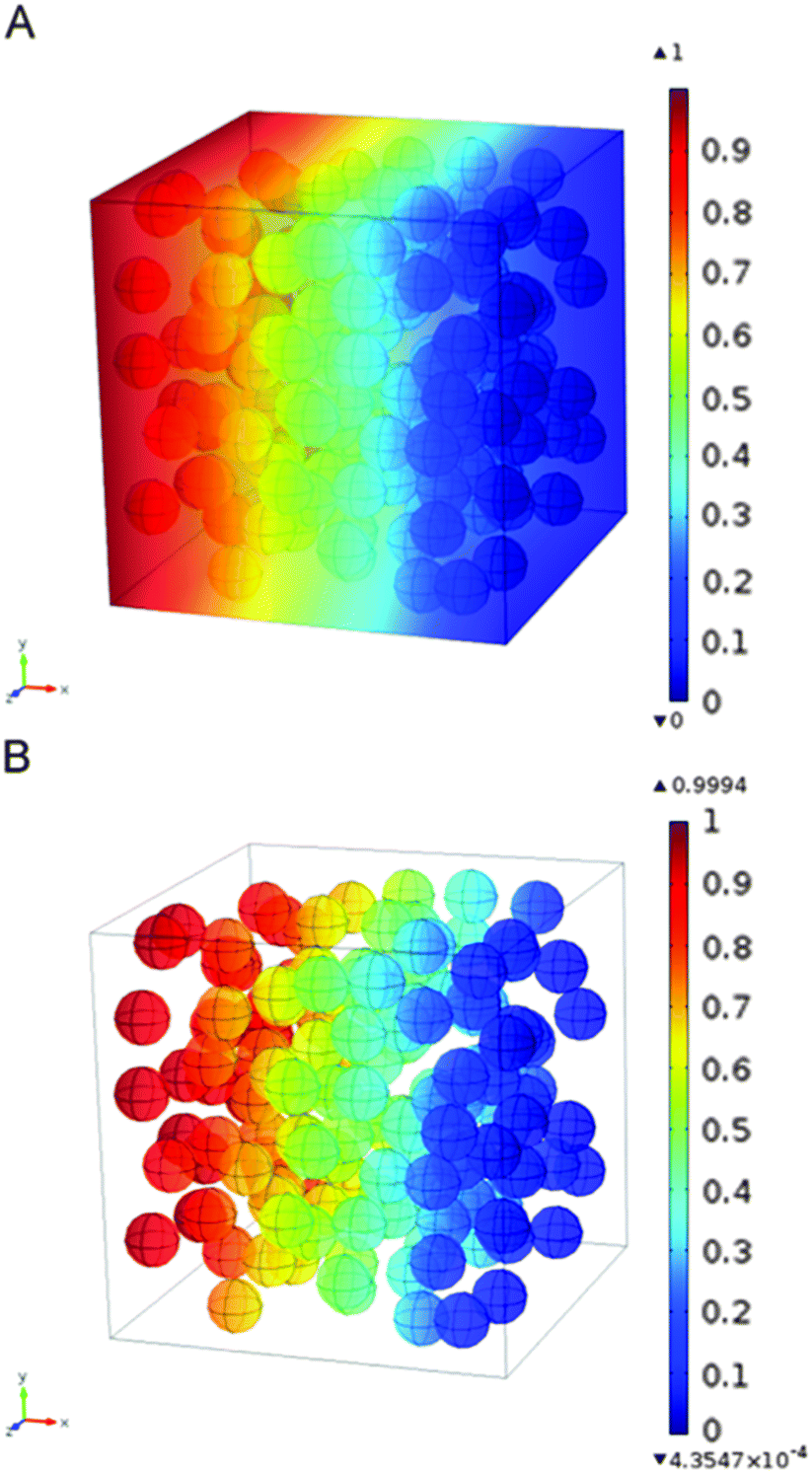 | ||
| Fig. 4 Concentration profiles of (a) both matrix and filler domains, and (b) filler domain only, of a membrane model with randomly distributed filler. Length units are in μm and concentration in mol m−1.75 Reproduced with permission from Elsevier. | ||
C Challenges and targets in developing membranes for post- and pre-combustion CO2 capture
The main challenge in post-combustion CO2 capture is the low partial pressure of CO2 and the huge amount of the flue gas to process. The CO2 content (volume basis) can be as low as 4% in a gas turbine plant, around 15% for coal power plants, and more concentrated (∼20–30%) for cement and steel production plants. This low CO2 partial pressure represents an enormous challenge for any CO2 capture technology: in the case of adsorption and absorption based processes, the use of adsorbents and absorbents with very high affinities is necessary, making regeneration very energy intensive.80 In the case of membranes, the driving force (ratio of feed to permeate partial pressure) becomes the limiting parameter, while no regeneration is needed. The routes to increase the driving force of the process are: (i) pressurizing the feed stream, (ii) applying partial vacuum on the permeate side of the membrane or (iii) using a sweep gas on the permeate side of the membrane module.Favre et al.9,81 have shown that the energy penalty for carbon capture is reduced if membranes with higher selectivities are used, especially when flue gases with high CO2 concentrations are involved (i.e. biogas combustion). Indeed, several researchers have demonstrated that a single stage membrane process can fulfil the targets for a lower energy penalty if the CO2 concentration is higher than 50%. In contrast, when it comes to lower CO2 concentrations, multi-stage membrane configurations are needed: Merkel et al.82,83 compared different multi stage membrane configurations (cross-flow, counter-flow and sweep flow) using a pressure ratio of 5, gas composition data from a 600 MWe coal-fired power plant (11 vol% CO2) and the MTR's membrane Polaris® as the base case (Permeance 1000 GPU, CO2 selectivity α = 50). For the optimal configuration (two-step counter-flow/sweep membrane process, see Fig. 5), a 90% CO2 recovery can be achieved at a price of 18 € per ton CO2 (including compression). This analysis stresses the importance of advanced engineering analysis in parallel to membrane development: a four-fold reduction in membrane area could be achieved by proper process design.82 Similar conclusions were reached by Ramasubramanian and coworkers84 using a cheaper, more permeable (3000 GPU) and more selective membrane than Polaris®. In the latter case, it is possible to reach a similar separation target at feed pressures close to 1 bar with multi stage air sweep process, in good agreement with previous results.85 A similar approach was followed by Koros et al.86 employing asymmetric hollow fiber modules instead of the spiral wound modules considered by Merkel and Ramasubramania.82,84 Koros and coworkers developed asymmetric hollow fibers from a highly permeable glassy polymer and investigated the performance of modules of these fibers for the same process configuration.86 The results show that although hollow fiber modules can be more expensive than spiral wound ones, in terms of CO2 capture cost both membrane configurations are comparable: the authors concluded that if hollow fibers (HFbs) can be produced with a permeance higher than 1000 GPU and a moderate selectivity (∼20), their modules can reduce the CO2 capture cost to less than 18 € per ton (including compression). As a rough calculation, 200 Barrer would be equivalent to ∼1300 GPU for membranes with a selective layer of 150 nm. This means that membranes with PCO2 > 500 Barrer (equivalent to 3300 GPU for 150 nm thick HFbs) and selectivities in the range of 30–40 will certainly achieve the SET objective of 90% CO2 capture at a cost below 25 € per MW h: such membranes will result in CO2 capture costs below 15 € per ton CO2 which, depending on the energy plant, would lower the CO2 capture cost to less than 10 € per MW h.
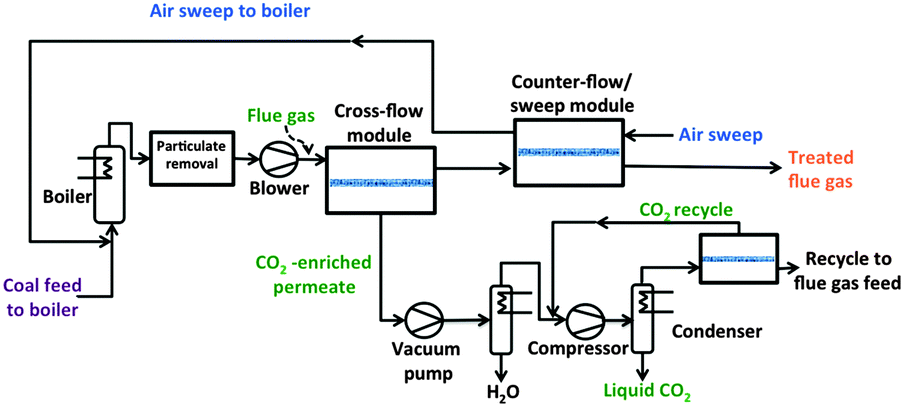 | ||
| Fig. 5 Simplified flow diagram of a two-step counter-flow/sweep membrane process to capture and sequester CO2 in flue gas from a coal-fired power plant.82 | ||
In the case of pre-combustion CO2 capture, the application of membranes offers several advantages: (i) the mixture of CO2 and H2 following the shift reactor is already at high pressure, unlike post-combustion applications and (ii) the application of selective H2 permeable membranes can deliver CO2 at high pressure, greatly reducing compression costs, while subsequent combustion of H2 to produce electricity does not require high pressures. However, in terms of membrane performance (see Fig. 3) separation is very challenging, since achieving H2/CO2 selectivities higher than 10 implies very low permeances, unsuitable for the treatment of large effluent amounts. For pre-combustion CCS, the gas transport performance of the polymers at elevated temperatures (150–250 °C) is more important than the ambient temperature data used on the Robeson plot. Separation at high temperatures is preferred in order to increase membrane selectivity towards hydrogen. However, only a few systematic gas permeability studies have been performed with polymer containing membranes at higher temperatures and only a couple of studies involved M4s (vide infra). Currently, the metal supported polybenzimidazole (PBI) membrane under development at DOE's Los Alamos National Laboratory (LANL) represents the state of the art polymeric H2 separating membrane for pre-combustion CO2 capture.87,88 PBI has attracted attention due to its thermal stability and good processability but it has poor performance at 30 °C (PH2 = 1 Barrer; α(H2/CO2) = 15), although this improves markedly at 250 °C (PH2 = 100 Barrer; α(H2/CO2) = 22). The LANL study shows that more permeable PBI derivatives also display much improved performance at elevated temperatures. For example, the permeability and selectivity of 6F-PBI at 30 °C (PH2 = 250 Barrer; α(H2/CO2) = 1.5) is greatly improved at 250 °C (PH2 = 1000 Barrer; α(H2/CO2) = 6). In a separate programme of research, MTR's proprietary polymer membrane, Proteus™, displays very promising performance at 150 °C (PH2 = 600 Barrer; α(H2/CO2) = 15). Spiral-wound membranes based on this polymer have been the focus of a successful pilot-scale CCS trial that demonstrated good performance over several weeks of operation. Recently Ku et al.89 published a detailed study on membrane performance requirements for pre-combustion CO2 capture applying a single step high temperature membrane process. In electricity generation applications, the permeate stream is combusted to produce power. Gas turbines capable of accepting feed streams with up to 45 vol% hydrogen have been in operation for over 10 years, with more than 80000 h of operation of the fleet leader. This can be used to advantage in membrane systems, by using the N2 as a sweep gas to increase the separation driving force. For membranes displaying higher permeances than 1000 GPU, the estimated H2/CO2 membrane selectivity requirement for IGCC with 90% CO2 capture ranged from about 20 to 60, considerably lower than for industrial H2 production. This is not surprising given the ultra-high purity requirement (99.999%) for the latter case. Ideally, membranes displaying such permeances and separation factors larger than 60 would allow high H2 recovery rate (>90%). At a 90% overall CO2 capture rate, the required H2/CO2 selectivity drops from about 60 to about 20 as the H2 recovery is reduced from 90% to 70%. In summary, high performance membranes will support pre-combustion CCS at an efficiency that matches the targets of 90% carbon capture with only 10% extra cost.90 Hence, the development of membranes with target properties of PH2 > 500 Barrer and α(H2/CO2) > 30 at >150 °C will allow the SET targets to be reached or exceeded. A permeability of 500 Barrer would be equivalent to a permeance of 1600 GPU for membranes with a selective layer of 300 nm. In case of pre-combustion capture, in view of the envisaged higher temperature and pressure operation, slightly thicker membranes than for post-combustion capture should be used.
D MOF based mixed matrix membranes for gas separation
As already discussed above, the use of MOFs as fillers offers potential advantages over other porous materials mostly due to: (i) the better affinity of the polymer chains for MOFs in comparison to other inorganic fillers due to their partially organic nature, helping to avoid the so-called “sieve-in-a-cage morphology”,38,91,92 the most common MMM deficiency; and (ii) their easily adjustable cavities in terms of size, shape and chemical functionalities that can be tuned by choosing the appropriate ligands in the synthesis93 or by post-synthetic functionalization.94 Furthermore, when comparing MOFs with other fillers, it needs to be considered that MOFs commonly have a higher pore volume and a lower density than zeolites, meaning that their effect on the membrane properties can be larger for a given weight percentage of the filler. Table 2 summarizes most of the publications on the topic, while in Fig. 6 the reported results have been plotted in the shape of Robeson plots for the most relevant CO2 capture related gas pairs.| M4 | wt% loading (best MMM performance)b | Example (best performance)c | Graph coded | Type of analysis | Operation conditions (optimal value)e | Published year and ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MOFa | Polymera | P CO2 (Barrer) | CO2/CH4 selectivity (—) | P CO2 (Barrer) | CO2/N2 selectivity (—) | P H2 (Barrer) | H2/CO2 selectivity (—) | T (°C) | ΔP (bar) | ||||
| a Abbreviations: μ-BPA: 1,2-bis(4-pyridyl) ethane; μ-BPP: 1,3-bis(4-pyridyl)propane; 1,4-BDC: 1,4-benzenedicarboxylate; 2,6-NDC: 2,6-napthalenedicarboxylate; 2-amBzIM: 2-aminobenzimidazole; 4,4′-BPDC: 4,4′-biphenyl dicarboxylate; 4,4′-BPY: 4,4′-bipyridine; 4,4′-SDA: bis(4-aminophenyl) sulphide; 4MPD (or durene): 2,3,5,6-tetramethyl-1,4-phenylenediamine; 6FDA: 4,4′-(hexafluoroisopropylidene)diphthalic anhydride; APTMDS: bis(3-aminopropyl)tetramethyldisiloxane; b: bulk; BuI: 5-tert-butylisophthalic acid; BzIM: benzimidazole; DABA: 3,5-diaminobenzoic acid; DABCO: 1,4-diazabicyclo[2.2.2]octane; DAM (or TMPDA): 2,4,6-trimethyl-m-phenylenediamine; DBzPBI: PBI after N-substitution reaction with 4-tert-butylbenzyl bromide; DMPBI: PBI after N-substitution reaction with methyl iodide; DSDA: 3,3′,4,4′-diphenylsulfone tetracarboxylic dianydride; Glu: glutarate; HFS: hexafluorosilicate; MSS: mesoporous silica spheres; nc: nanoparticle crystals; ns: nanosheets; ODA: 4,4′-oxydianiline; ODPA: 4,4′-oxydiphthalicanhydride; PAET: poly(3-acetoxyethylthiophene); PAI: polyamide-imide; PBI: polybenzimidazole; PDMS: polydimethylsiloxane; PEI: polyetherimide; PES: polyestersulfone; PLLA: poly(L-lactic acid); PMDA: pyromellitic dianhydride; PMMA: poly(methyl methacrylate); PMP: poly(4-methyl-1-pentyne); POZ: polyoxazoline; PPEES: poly(1,4-phenylene ether-ether-sulfone); PPO: poly(2,6-dimethyl-1,4-phenylene oxide); PSF: polysulfone; PVAc: poly(vinyl acetate); TED: triethylenediamine; XLPEO: cross-linked polyethylene oxide. b Maximum MOF loading in terms of MMM permselectivity performance. Values are given in brackets. c CO2 and H2 permeabilities and CO2/CH4, CO2/N2 and H2/CO2 selectivities of the pure polymer and the MMMs with the optimal MOF loading. A permeance of 1 GPU corresponds to a membrane exhibiting an intrinsic permeability of 1 Barrer and having a selective layer thickness of 1 μm. Results with * are calculated from graphs. d Code of the different publications represented in Fig. 6. e Optimum operation conditions that maximized gas separation performance of the MMMs. Values are given in brackets. | |||||||||||||
| Cu 4,4′-BPDC-TED | PAET | 10–30 (30) | 1.4– (0.7) | 18.0– (3.2) | — | — | — | — | [1] | Single gas CO2, O2, N2, CH4 | 25 | 2 | 200448 |
| — | — | — | — | — | — | — | Gas mixture CH4/CO2 (10:90) |
||||||
| [Cu2(PF6)(NO3)(4,4′-bpy)4]2PF6·2H2O | PSF | 2.5–5 (5) | — | — | — | — | — | — | — | Single gas He, H2, O2, N2, CH4 | 35 | 1 | 200595 |
| [Zn2(1,4-bdc)2 (dabco)]·4DMF·0.5H2O | PAI | 30 | 46.7– (109) | 49.7– (40.4) | 46.7– (109) | 28.3– (24.6) | 79.2– (191) | 1.7– (1.8) | [2A] | Single gas | — | — | 2006110 |
| 6FDA-4MPD | 1000– (3330) | 23.0– (19.6) | 1000– (3330) | 21.4– (19.1) | 743– (1890) | 0.7– (0.6) | [2B] | ||||||
| PDMS | 2830– (4010) | 3.4– (3.7) | 2830– (4010) | 10.5– (10.0) | 673– (955) | 0.2– (0.2) | [2C] | ||||||
| HKUST-1 | PDMS | 10–40 (30, 10, 40) | 2500– (2900)* | 3.1– (3.6)* | 2500– (3050)* | 7.0– (8.9)* | 550– (900)* | 0.2– (0.4)* | [3A] | Single gas H2, CO2, O2, N2, CH4 | — | — | 200696 |
| PSF | 5–10 (5, 10) | 6.5– (7.5)* | 18.0– (21.5)* | 6.5– (7.5)* | 20.0– (25.0)* | 9.8– (15.0)* | 1.5– (1.9)* | [3B] | |||||
| Mn(HCOO)2 | PSF | 5–10 (10, 5) | 6.5– (7.0)* | 18.0– (9.5)* | 6.5– (7.0)* | 20.0– (25.5)* | 9.5– (10.5)* | 1.5– (1.6)* | [3C] | ||||
| Cu-4,4′-BPY-HFS | Matrimid® | 10–40 (20, 30) | 7.3– (9.9) | 34.7– (27.6) | 7.3– (9.9) | 33.1– (31.9) | 17.5– (20.3) | 2.4– (2.0) | [4] | Single gas H2, CO2, O2, N2, CH4 | 35 | 2 | 2008101 |
| 20 | — | 36.3– (20.5) | — | — | — | 2.6– (2.6) | — | Gas mixture H2/CO2 (50 CO2/CH4 (50 CH4/N2 (94 |
|||||
| IRMOF-1 | Matrimid® | 20 | 10.0– (38.8) | 28.2– (29.2) | — | — | 33.1– (114.9) | 3.3– (3.0) | [5A] | Single gas H2, CO2, CH4 | 50 | 7 | 2009111 |
| Ultem® | 10, 20 (20) | 2.0– (3.0) | 30.3– (26.3) | — | — | 11.2– (16.9) | 5.7– (5.7) | [5B] | |||||
| HKUST-1 | Matrimid® | 30 | 10.0– (22.1) | 28.2– (29.8) | — | — | 33.1– (66.9) | 3.3– (3.0) | [5C] | ||||
| MOF-5 | Matrimid® | 10–30 (30) | 9.0– (20.2) | 41.7– (44.7) | 9.0– (20.2) | 36.0– (38.8) | 24.4– (53.8) | 2.7– (2.7) | [6] | Single gas H2, CO2, O2, N2, CH4 | 35 | 2 | 2009102 |
| 30 | — | 38.0– (29.0) | — | — | — | 2.3– (2.3) | — | Gas mixture H2/CO2 (75 CH4/N2 (94 CO2/CH4 (10 |
|||||
| ZIF-8 | PPEES | 10–30 (30) | 6– (25) | — | 6– (25) | — | — | — | — | — | 30 | 1, 2, 3, 5, 7, 10 | 2010112 |
| Cu 1,4-BDC | PVAc | 15 | 2.4– (3.3) | 34.9– (40.4) | 2.4– (3.3) | 32.1– (35.4) | — | — | [7] | Single gas He, CO2, O2, N2, CH4 | 35 | 4.5 (0.1 for CO2) | 2010100 |
| ZIF-8 | Matrimid® | 20–60 (50) | 9.5– (4.7) | 39.7– (124.9) | 9.5– (4.7) | 30.6– (26.2) | 28.9– (18.1) | 3.0– (3.8) | [8] | Single gas H2, CO2, O2, N2, CH4, C3H8 | 25 | 2.7 | 2010103 |
| 50–60 (50, 60) | — | 42.1– (89.2) | — | — | — | 2.6– (7.0) | — | Gas mixture H2/CO2 (50 CO2/CH4 (10 |
|||||
| HKUST-1 | Matrimid® | 10–30 (30) | 10.0– (17.5)* (GPU) | 18.0– (24.0)* | 11.0– (18.5)* (GPU) | 23.5– (24.5)* | — | — | [9] | Gas mixture CO2/CH4 (10 CO2/N2 (10 |
35 | 10 | 2010113 |
| HKUST-1 | PMDA-ODA | 3–6 (3, 6) | 306.6– (227.2) | 12.0– (7.0)* | 306.6– (227.2) | 8.0– (5.5)* | 3066– (4445) | 10.0– (27.8) | [10] | Single gas H2, CO2, O2, N2, CH4 | 25 | 10 | 2010114 |
| ZIF-90 | Ultem® | 15 | 1.4– (2.9)* | 38– (39)* | — | — | — | — | [11A] | Single gas CO2, CH4 | 35 | 4.5 | 201040 |
| Matrimid® | 7.5– (10.5)* | 34– (35)* | — | — | — | — | [11B] | ||||||
| 6FDA-DAM | 390– (720) | 24– (37) | — | — | — | — | [11C] | Gas mixture CO2/CH4 (50:50) |
25 | 2 | |||
| ZIF-20 | PSF | 8 | — | — | — | — | — | — | — | Gas mixture O2/N2 (50:50) |
35 | 2 | 201198 |
| NH2-MIL-53(Al) | PSF | 8, 16, 25, 40 (25) | 2.0– (2.4) | 45– (117) | — | — | — | — | [12] | Gas mixture CO2/CH4 (50:50) |
−10, 35 | 1, 3, 5, 7, 10, 13 | 201145 |
| ZIF-7 | PBI | 10, 25, 50 (50) | — | — | — | — | 3.7– (26.2) | 8.7– (14.9) | — | Single gas H2, CO2 | 35 | 3.5 | 2011115 |
| 75– (440)* | 8.5– (7.2)* | [13] | Gas mixture H2/CO2 (50:50) |
35, 60, 80, 120, 150, 180 | 7 | ||||||||
| ZIF-8 + S1C | PSF | 16 + 0, 8 + 8 (16 + 0) | 4.6– (12.1) | 24.3– (19.8) | 5.9– (12.3) | 24.6– (19.5) | — | — | [14A] | Gas mixture CO2/CH4 (50 CO2/N2 (50 O2/N2 (50 H2/CH4 (50 |
35 | 2 | 201197 |
| HKUST-1 + S1C | 16 + 0, 8 + 8 (8 + 8) | 4.6– (4.9) | 24.3– (22.4) | 5.9– (8.4) | 24.6– (38.0) | — | — | [14B] | |||||
| HKUST-1 | Matrimid® | 10, 20, 30 (30) | 10.0– (17.5)* (GPU) | 18.5– (23.0)* | 11.5– (19.5)* (GPU) | 18.0– (23.5)* | — | — | [15A] | Gas mixture CO2/CH4 (10 CO2/N2 (10 |
35 | 10 | 2011116 |
| ZIF-8 | 10.0– (22.5)* (GPU) | 18.5– (19.5)* | 11.5– (20.0)* (GPU) | 18.0– (19.5)* | — | — | [15B] | ||||||
| MIL-53(Al) | 10.0– (20.0)* (GPU) | 18.5– (22.5)* | 11.5– (20.0)* (GPU) | 18.0– (23.0)* | — | — | [15C] | ||||||
| ZIF-8 | PPEEs | 10, 20, 30 (30) | 5.4– (50.0) | 22.9– (20.8) | 5.4– (50.0) | 30.1– (24.5) | 6.7– (92.3) | 1.3– (1.8) | [16] | Single gas H2, CO2, O2, N2, CH4, C2H4, C2H6 |
10, 20, 30, 40 | 1 | 201199 |
| ZIF-8 | 6FDA-DAM | 16.4, 28.7, 48 (48) | — | — | — | — | — | — | — | Single gas C3H6, C3H8 | 35 | 2 | 2012117 |
| Gas mixture C3H6/C3H8 (50:50) |
1.4, 2.8, 4.1, 5.5 | ||||||||||||
| MIL-101(Cr) | PSF | 8, 16, 24 | — | — | — | — | — | — | — | Single gas O2, N2 | 30 | 3 | 2012118 |
| MOF-508a(Zn) | 8 | — | — | — | — | — | — | — | |||||
| MIL-53(Al) | — | — | — | — | — | — | — | ||||||
| MIL-100(Fe) | — | — | — | — | — | — | — | ||||||
| MIL-53(Al) | 6FDA-ODA | 25 | 14.5– (21.0)* | 48.0– (44.0)* | — | — | — | — | — | Single gas CO2, CH4 | 35 | 10 | 2012122 |
| 14.5– (21.0)* | 42.0– (42.5)* | — | — | — | — | [17A] | Gas mixture CO2/CH4 (50:50) |
||||||
| NH2-MIL-53(Al) | 10, 15, 20, 25, 30, 32, 35 (32) | 14.5– (14.7)* | 48.0– (76.0)* | — | — | — | — | — | Single gas CO2, CH4 | ||||
| 14.5– (14.7)* | 42.0– (53.0)* | — | — | — | — | [17B] | Gas mixture CO2/CH4 (50:50) |
||||||
| ZIF-8 | Ultem® | 10, 13 (13) | — | — | 14.0– (26.0) (GPU) | 30.0– (36.0) | — | — | — | Single gas CO2, N2 | 25, 30, 35, 45 | 6.7 | 2012119 |
| — | — | (26.0)* (GPU) | (32.0) | — | — | [18] | Gas mixture CO2/N2 (20:80) |
25, 35, 45 | 1.4, 2.1, 2.8, 3.4 | ||||
| MIL-53(Al) | PMDA-ODA | 5 | 0.30– (0.21) (GPU) | 72.1– (50.5) | 0.30– (0.21) (GPU) | 34.8– (27.5) | 0.35– (0.42) (GPU) | 1.1– (2.0) | [19A] | Single gas He, H2, CO2, N2, CH4 | 25 | 6 | 2012120 |
| MOF-5 | 0.30– (0.27) (GPU) | 72.1– (56.8) | 0.30– (0.27) (GPU) | 34.8– (14.1) | 0.35– (0.24) (GPU) | 1.1– (0.9) | [19B] | ||||||
| HKUST-1 | 0.30– (0.32) (GPU) | 72.1– (73.6) | 0.30– (0.32) (GPU) | 34.8– (38.1) | 0.35– (0.44) (GPU) | 1.1– (1.3) | [19C] | ||||||
| ZIF-8 | Matrimid® | 10, 25 (25) | 10.7– (23.2) | 34– (39) | — | — | — | — | [20] | Single gas CO2, CH4 | 35 | 4.5 | 2012121 |
| ZIF-8 | Matrimid® | 5, 10, 20, 30, 40 (20, 30) | 8.1– (16.6) | 35.2– (35.8) | 8.1– (16.6) | 22.4– (19.0) | 32.7– (112.1) | 4.0– (3.9) | [21] | Single gas H2, CO2, O2, N2, CH4 | 22 | 4 | 2012122 |
| UiO-66 | 6FDA-ODA | 25 | 14.4– (50.4) | 44.1– (46.1) | — | — | — | — | [22A] | Single gas CO2, CH4 | 35 | 10 | 2012123 |
| — | 41.7– (42.3) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| NH2-UiO-66 | 14.4– (13.7) | 44.1– (51.6) | — | — | — | — | [22B] | Single gas CO2, CH4 | |||||
| — | 41.7– (44.7) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| HKUST-1 | 14.4– (21.8) | 44.1– (51.2) | — | — | — | — | [22C] | Single gas CO2, CH4 | |||||
| — | 41.7– (50.7) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| NH2-HKUST-1 | 14.4– (26.6) | 44.1– (59.6) | — | — | — | — | [22D] | Single gas CO2, CH4 | |||||
| — | 41.7– (52.4) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| UiO-67 | 14.4– (20.8) | 44.1– (15.0) | — | — | — | — | [22E] | Single gas CO2, CH4 | |||||
| — | 41.7– (15.0) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| ZIF-8 | PBI | 18, 20, 29, 34, 59 (29) | — | — | — | — | 3.7– (105.4) | 8.6– (12.3) | [23A] | Single gas H2, CO2 | 25 | 3.5 | 2012124 |
| PBI/Matrimid® | 10, 20, 33 (10) | — | — | — | — | 2.1– (8.9) (GPU) | 6.2– (9.5) (GPU) | — | |||||
| 10, 20, 33 (10) | — | — | — | — | (65.4) (GPU) | (12.3) (GPU) | [23B] | Gas mixture H2/CO2 (50:50) |
25, 35, 50, 80, 120, 150, 180 | 7 | |||
| ZIF-8 | 6FDA-DAM:DABA 4:1 |
20 | — | — | 211.4– (553) | 21.3– (19.3) | — | — | [24] | Single gas CO2, N2 | 30 | 1.4 | 201286 |
| ZIF-8 | PBI | 30, 60 (30) | — | — | — | — | 4.1– (82.5) | 8.9– (12.0) | [25A] | Single gas H2, CO2 | 35 | 3.5 | 2013107 |
| — | — | — | — | (470) | (26.3) | [25B] | Gas mixture H2/CO2 (50 H2/CO2/CO (49.5 |
35, 60, 120, 180, 230 | 2 | ||||
| ZIF-7 | Pebax® | 8, 22, 34 (22) | 72– (111) | 14– (30) | 72– (111) | 34– (97) | — | — | [26] | Single gas CO2, N2, CH4 | 20 | 6.5 (2.75 for CO2) | 2013125 |
| ZIF-8 | PIM-1 | 13.8, 24.2, 32.4, 39.0 (39.0) | 4390– (6300) | 14.2– (14.7) | 4390– (6300) | 24.4– (18.0) | 1630– (6680) | 0.4– (1.1) | [27] | Single gas He, H2, CO2, O2, N2, CH4 | 20–22 | 1 | 2013126 |
| HKUST-1 | P84 | 20 | — | — | — | — | — | — | — | Gas mixture C2H4/C2H6 (80:20) |
— | 5, 10, 15 | 2013127 |
| FeBTC | — | — | — | — | — | — | — | ||||||
| MIL-53(Al) | — | — | — | — | — | — | — | ||||||
| HKUST-1 | P84 | 10, 20, 40 (20) | — | — | — | — | — | — | — | Gas mixture C2H4/C2H6 (80:20) |
— | 5, 10, 15 | 2013127 |
| ZIF-90 | PBI | 10, 25, 45 (45) | — | — | — | — | 4.1– (24.5) | 8.9– (25) | — | Single gas H2, CO2 | 35 | 3.5 | 2013128 |
| 45 | — | — | — | — | (226.9) | (13.3) | [28] | Gas mixture H2/CO2 (50:50) |
35, 60, 80, 120, 180 | 7 | |||
| HKUST-1 | PPO | 10, 20, 30, 40, 50 (40) | 68.7– (115)* | 16.4– (34)* | 68.7– (115)* | 16.0– (26)* | 75.0– (119)* | 1.1– (1.0)* | [29] | Single gas H2, CO2, N2, CH4 | 30 | — | 2013129 |
| ZIF-8 | 6FDA-durene | 33.3 | 468.5– (1552.9) | 15.6– (11.0) | 468.5– (1552.9) | 13.4– (11.3) | 518.5– (2136.6) | 1.1– (1.4) | [30A] | Single gas H2, CO2, O2, N2, CH4 | 35 | 3.5 | 2013130 |
| 6FDA-durene (cross-linked) | 0.4– (23.7) | (16.9) | 0.4– (23.7) | (11.9) | 52.1– (283.5) | 130.3– (12.0) | [30B] | ||||||
| NH2-MIL-53(Al) | 6FDA:DSDA-4MPD:4,4′-SDA 1:1 |
0, 5, 10, 15 (15) | 57.9– (66.5) | 35.1– (36.9) | — | — | 90.1– (100) | 1.6– (1.8) | [31A] | Single gas H2, CO2, CH4 | 35 | 3 | 2013131 |
| 6FDA-4MPD:4,4′-SDA 1:1 |
10 | 134– (137) | 30.2– (27.2) | — | — | 169– (175) | 1.3– (1.3) | [31B] | |||||
| NH2-MIL-101(Al) | 6FDA:DSDA-4MPD:4,4′-SDA 1:1 |
0, 5, 10 (10) | 57.9– (70.9) | 35.1– (41.6) | — | — | 90.1– (114) | 1.6– (1.6) | [31C] | ||||
| 6FDA-4MPD:4,4′-SDA 1:1 |
10 | 134– (151) | 30.2– (29.6) | — | — | 169– (191) | 1.3– (1.3) | [31D] | |||||
| NH2-CAU-1 | PMMA | 5, 10, 15, 20, 25 (15) | — | — | — | — | 5000– (11000) |
3– (13) | [32] | Single gas H2, CO2 | RT | 3 | 2013132 |
| — | — | — | — | — | 2– (10)* | — | Gas mixture H2, CO2 (—) | ||||||
| MIL-68(Al) | PSF | 4, 8 (8) | 5.4– (4.7) | 31.1– (36.5) | — | — | — | — | [33] | Gas mixture CO2/CH4 (50:50) |
35 | 2 | 2013131 |
| HKUST-1 | PLLA | 5 | — | — | — | — | — | — | — | –CO2, O2 | 23 | — | 2013133 |
| ZIF-8 | 6FDA-durene (400 °C) | 20 | 541– (1090) | 13.1– (13.0) | — | — | — | — | [34A] | Single gas CO2, CH4, C3H6, C3H8 | 35 | 10 (3.5 for C3H6 and C3H8) | 2013134 |
| 6FDA-durene:DABA 9:1 (200 °C) |
5, 10, 15, 20, 30, 40 (40) | 256– (779) | 19.5– (20.9) | — | — | — | — | [34B] | |||||
| 6FDA-durene:DABA 7:3 (400 °C) |
20 | 429– (698) | 26.0– (25.8) | — | — | — | — | [34C] | |||||
| 6FDA-durene:DABA 9:1 (400 °C) |
20, 40 (20) | 305– (728) | 13.8– (19.6) | — | — | — | — | [34D] | Gas mixture CO2/CH4 (50:50) |
35 | 20 | ||
| NH2-MIL-53(Al) | Matrimid® | 15 | 6.2– (9.2) | 31.0– (2.1) | — | — | — | — | [35A] | Single gas CO2, CH4 | 35 | 10 | 2013135 |
| — | 28.5– (2.1) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| Ultem® | 15 | 1.5– (3.0) | 39.5– (36.2) | — | — | — | — | [35B] | Single gas CO2, CH4 | ||||
| — | 31.6– (36.1) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| 6FDA-ODA:DAM 1:1 |
15, 20, 22 (10) | 54.1– (51.2) | 23.5– (34.1) | — | — | — | — | [35C] | Single gas CO2, CH4 | ||||
| — | 23.6– (31.8) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| 6FDA-ODA:DAM 1:4 |
10, 15, 20 (15) | 130.0– (113) | 23.2– (28.2) | — | — | — | — | [35D] | Single gas CO2, CH4 | ||||
| — | 23.6– (28.5) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| 6FDA-ODA:DAM 1:1 (APTMDS) |
15, 20, 25, 30, 32, 35 (30) | 32.2– (58.5) | 18.9– (36.6) | — | — | — | — | [35E] | Single gas CO2, CH4 | ||||
| — | 20.2– (33.9) | — | — | — | — | — | Gas mixture CO2/CH4 (10 CO2/CH4 (35:65) CO2/CH4 (50 CO2/CH4 (60 CO2/CH4 (80 CO2/CH4 (85 |
30, 35, 45, 60, 75 | 10 | ||||
| MIL-53 | Matrimid® | 15 | 6.2– (6.7) | 31.0– (9.4) | — | — | — | — | [35F] | Single gas CO2, CH4 | 35 | 10 | 2013135 |
| — | 28.5– (8.5) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| Ultem® | 15 | 1.5– (1.8) | 39.5– (43.1) | — | — | — | — | [35G] | Single gas CO2, CH4 | ||||
| — | 31.6– (42.8) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| 6FDA-ODA:DAM 1:1 |
20 | 54.1– (61.5) | 23.5– (12.5) | — | — | — | — | [35H] | Single gas CO2, CH4 | ||||
| — | 23.6– (13.0) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| 6FDA-ODA:DAM 1:4 |
25 | 130.0– (123) | 23.2– (18.1) | — | — | — | — | [35I] | Single gas CO2, CH4 | ||||
| — | 23.6– (19.1) | — | Gas mixture CO2/CH4 (50:50) |
||||||||||
| 6FDA-ODA:DAM 1:1 (APTMDS) |
25 | 32.2– (76.4) | 18.9– (8.9) | [35J] | Single gas CO2, CH4 | ||||||||
| — | 20.2– (8.8) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
||||||
| TKL-107 | Matrimid® | 5, 10, 20, 30 (20) | 7– (17)* | 36– (64.6) | — | — | — | — | — | Single gas CO2, CH4 | 25 | 2 | 2013136 |
| 6– (15)* | 24– (50.3) | — | — | — | — | [36] | Gas mixture CO2/CH4 (20:80) |
||||||
| CPO-27(Mg) | XLPEO | 10 | — | — | 380– (250) | 22– (25) | — | — | [37A] | Single gas CO2, N2 | 25 | 2 | 2013137 |
| 6FDA-TMPDA | 10 | — | — | 650– (850) | 14– (23) | — | — | [37B] | |||||
| PDMS | 20 | — | — | 3100– (2100) | 9.5– (12) | — | — | [37C] | |||||
| Silica-(ZIF-8) core–shell | PSF | 8, 12, 16, 20, 32 (32) | 11.8– (73.1)* | 10.2– (5.5)* | — | — | 35.0– (224.1)* | 3.4– (3.9)* | [38] | Gas mixture H2/CO2 (50 CO2/CH4 (10 |
35, 60, 90,120, 150 | 2 | 2014138 |
| 35, 60, 90, 120 | |||||||||||||
| ZIF-8 | Pebax® | 5, 10, 15, 20, 25, 30, 35 (35) | 351– (1287) | 8.3– (9.0) | 351– (1287) | 33.8– (32.3) | — | — | [39] | Single gas CO2, O2, N2, CH4 |
RT | 2, 6 | 2014139 |
| 5, 10, 15, 20, 25, 30, 35, 40, 50 (25) | — | — | 200– (900)* | 66– (53)* | — | — | — | Gas mixture CO2/N2 (10:90) |
25 | 1.5 | |||
| MIL-53 | Matrimid® | 5, 10, 15, 20 (15) | 6.4– (12.4) | 28.2– (51.8) | — | — | — | — | [40] | Single gas CO2, CH4 | 35 | 3 | 2014140 |
| ZIF-8 | PSF | 8 | — | — | — | — | — | — | — | Gas mixture H2/CH4 (50 O2/N2 (50 |
35 | 2 | 2014141 |
| NH2-MIL-53(Al) | — | — | — | — | — | — | — | ||||||
| [Cd26FDA(H2O)]25H2O | 6FDA-ODA | 10 | 20.6– (37.8) | 33.1– (44.8) | 20.6– (37.8) | 26.4– (35.1) | — | — | [41] | Single gas CO2, N2, CH4 | 25 | 2 | 2014142 |
| MIL-53(Al) | PMP | 5, 10, 15, 20, 25, 30, 35, 40 (30) | — | — | — | — | 100– (365)* | 0.11– (0.04) | — | Single gas H2, CO2 | 30 | 2, 4, 6, 8 | 2014143 |
| ZIF-8 | 6FDA-durene | 3, 5, 7, 10, 15, 20, 30 (30) | 1468.3– (2185.5) | 22.6– (17.1) | 1468.3– (2185.5) | 25.4– (17.0) | — | — | [42] | Single gas CO2, O2, N2, CH4 | RT | 2, 6 | 2014144 |
| — | — | — | — | — | — | — | Gas mixture CO2/N2 (10:90) |
25 | 1.5 | ||||
| NH2-MIL-53(Al) + MSSs | PSF | 16 + 0, 12 + 4, 8 + 8, 4 + 12 (4 + 12) | — | — | — | — | — | — | — | Gas mixture O2/N2 (50 H2/CH4 (50 |
35 | 2 | 2014145 |
| NH2-MIL-53(Al) + MSSs | Matrimid® | 8 + 8, 4 + 12 (4 + 12) | — | — | — | — | — | — | — | ||||
| NH2-MIL-53(Al) | Matrimid® | 15, 20, 25 (25) | 4.8– (3.9) | 100– (107) | — | — | — | — | [43A] | Gas mixture CO2/CH4 (50:50) |
0, 25, 35 | 3, 5, 9, 12 | 201447 |
| PSF | 15, 20, 25 (25) | 5.2– (5.4) | 23.0– (27.5) | — | — | — | — | [43B] | 35 | 3 | |||
| NH2-MIL-101(Al) | Matrimid® | 8, 15, 25 (25) | 4.8– (3.0) | 100– (98) | — | — | — | — | [43C] | 0, 25, 35 | 3, 5, 9, 12 | ||
| PSF | 8, 15, 25 (25) | 5.2– (8.4) | 23.0– (28.5) | — | — | — | — | [43D] | 35 | 3 | |||
| ZIF-8 | 6FDA-DAM | 17, 30 (17) | — | — | — | — | — | — | — | Single gas O2, N2 | 35 | 2 | 2014146 |
| Gas mixture C3H6, C3H8 (50:50) |
1.4 | ||||||||||||
| ZIF-8 | Matrimid® | 15 | 9– (26)* | 34.5– (35)* | — | — | — | — | — | Single gas CO2, CH4 | 35 | 3.45 | 2014147 |
| ZIF-7-8-(20) | 9– (20)* | 34.5– (35.5)* | — | — | — | — | — | ||||||
| ZIF-8-ambz-(15) | 9– (12)* | 34.5– (36)* | — | — | — | — | — | ||||||
| ZIF-8-ambz-(30) | 9– (11)* | 34.5– (38.5)* | — | — | — | — | — | ||||||
| ZIF-7-8-(20) | 8– (19)* | 43– (41)* | — | — | — | — | [44A] | Gas mixture CO2/CH4 (50:50) |
— | 6.9, 13.8, 27.6, 41.4 | |||
| ZIF-8-ambz-(15) | 8– (14)* | 43– (40)* | — | — | — | — | [44B] | ||||||
| ZIF-8-ambz-(30) | 8– (11)* | 43– (42.5)* | — | — | — | — | [44C] | ||||||
| FeBTC | Matrimid® | 10, 20, 30 | 14– (14)* | 55– (35)* | — | — | — | — | — | Single gas CO2, CH4 | 35 | 2– 40 (40) | 2014148 |
| 14– (8.2)* | 22– (28)* | — | — | — | — | [45] | Gas mixture CO2/CH4 (50:50) |
— | 5 | ||||
| ZIF-8 | PBI-BuI | 10, 20, 30 (30) | 2.3– (5.2) | 57.0– (43.6) | 2.3– (5.2) | 26.8– (16.0) | 6.2– (22.1) | 2.7– (4.2) | [46A] | Single gas He, H2, CO2, N2, CH4 | 35 | 20 | 2014149 |
| DMPBI-BuI | 10, 20, 30 (30) | 3.8– (53.9) | 47.2– (15.7) | 3.8– (53.9) | 21.7– (11.3) | 12.8– (127.5) | 3.4– (2.4) | [46B] | |||||
| DBzPBI-BuI | 10, 20 (20) | 25.8– (89.8) | 15.9– (11.6) | 25.8– (89.8) | 12.9– (14.3) | 61.4– (180.3) | 2.4– (2.0) | [46C] | |||||
| NH2-MIL-53(Al) | PMP | 5, 10, 15, 20, 25, 30, 35, 40 (30) | 96.5– (358.2) | 8.8– (24.4) | — | — | — | — | — | Single gas CO2, CH4 | 30 | 2, 4, 6, 8 | 2014150 |
| 80.1– (339.5) | 8.1– (22.9) | — | — | — | — | [47] | Gas mixture CO2/CH4 (10:90) |
||||||
| MIL-53(Al)-ht | Matrimid® | 33.3, 37.5 (37.5) | 8.4– (51) | 39.4– (47.0) | 8.4– (51) | 33.6– (28.3) | 25.7– (103) | 3.1– (2.0) | — | Single gas H2, CO2, O2, N2, CH4 | 35 | 2 | 2014151 |
| MIL-53(Al)-as | 37.5 | 8.4– (40) | 39.4– (90.1) | 8.4– (40) | 33.6– (95.2) | 25.7– (66.0) | 3.1– (1.7) | [48] | |||||
| c-MOF-5 | PEI | 5, 15, 25 (25) | 1.7– (5.4) | 18.7– (23.4) | 1.7– (5.4) | 16.8– (28.4) | 10.1– (28.3) | 6.0– (5.3) | [49] | Single gas H2, CO2, N2, CH4 | 25 | 6 | 2014152 |
| HKUST-1 | Ultem® | 10, 20, 30, 35, 40 (35) | 1.1– (4.1) | 36.8– (34.0) | 1.1– (4.1)* | 28.0– (28.0)* | — | — | [50] | Single gas CO2, O2, N2, CH4 | 35 | 3.5 | 2014153 |
| HKUST-1 | ODPA-DAM (annealed 200 °C 24 h) | 10, 15, 20, 30, 40, 50 (40) | 47.7– (260.7) | 29– (28)* | — | — | — | — | [51A] | Single gas CO2, O2, N2, CH4 | 35 | 2 | 2014154 |
| Matrimid® (annealed 200 °C 24 h) | 20 | 7.6– (24.8) | 37.5– (37.8) | — | — | — | — | [51B] | |||||
| ZIF-8 | ODPA-DAM (annealed 200 °C 24 h) | 47.7– (134)* | 29– (26)* | — | — | — | — | [51C] | |||||
| ZIF-71 | 6FDA-durene | 10, 20, 30 (20) | 959– (4006) | 16.4– (12.8) | 959– (4006) | 14.7– (12.9) | 756– (2310) | 0.8– (0.6) | [52A] | Single gas H2, CO2, O2, N2, CH4, C2H4, C2H6, C3H6, C3H8 | 35 | 3.5 (2 for C2H4, C2H6, C3H6 and C3H8) | 2014155 |
| 10, 20, 30 (20) | 917– (3435) | 21.8– (16.0) | — | — | — | — | [52B] | Gas mixture CO2/CH4 (50:50) |
35 | 7 | |||
| [Cu2(Glu)2(μ-bpa)]·(CH3CN) | POZ | 5, 10, 15, 20 (15) | — | — | 28– (11.6)* | 1– (55)* | — | — | [53A] | Single gas CO2, N2 | — | 3.1 | 2014156 |
| [Cu2(Glu)2(μ-bpp)]·(C3H6O) | — | — | 28– (16.0)* | 1– (7)* | — | — | [53B] | 0.4 | |||||
| MIL-53(Al) | Matrimid® | 10, 20, 30 (30) | 14– (24)* | 55– (66)* | — | — | — | — | — | Single gas CO2, CH4 | 35 | 2.5, 5, 7.5, 10, 12.5, 15, 20, 25, 30, 40 | 2014157 |
| ZIF-8 | 14– (24)* | 55– (72)* | — | — | — | — | — | ||||||
| HKUST-1 | 14– (18)* | 55– (52)* | — | — | — | — | — | ||||||
| MIL-53(Al) | 9– (18)* | 5– (40)* | — | — | — | — | [54A] | Gas mixture CO2/CH4 (50:50) |
2.5, 5, 7.5, 10, 15, 20 | ||||
| ZIF-8 | 9– (20)* | 5– (37)* | — | — | — | — | [54B] | ||||||
| HKUST-1 | 9– (14)* | 5– (46)* | — | — | — | — | [54C] | ||||||
| ZIF-11 | PSF | 4.7 | — | — | — | — | — | — | — | — | — | — | 2014158 |
| PES | 4.7 | — | — | — | — | — | — | — | |||||
| PBI | 16.1, 29.7, 39.5 (39.5) | — | — | — | — | 17.2– (464.7) | 5.0– (3.6) | [55] | Single gas H2, CO2 | RT | — | ||
| b-Cu 1,4-BDC | Matrimid® | 8 | 5.8– (5.2) | 59.8– (45) | — | — | — | — | — | Gas mixture CO2/CH4 (50:50) |
25 | 3, 4.5, 6, 7.5 | 2014159 |
| nc-Cu 1,4-BDC | 8 | 5.8– (5.0) | 59.8– (49.4) | — | — | — | — | — | |||||
| ns-Cu 1,4-BDC | 2, 4, 8 (8) | 5.9– (2.8) | 47.7– (88.2) | — | — | — | — | — | 3, 4.5, 6, 7.5 | ||||
| ns-Cu 2,6-NDC | 8 | 5.8– (6.3) | 59.8– (43.5) | — | — | — | — | [56] | 3 | ||||
| ZIF-8 100 nm | PSF | 5 | 25.7– (15.6) (GPU) | 19.4– (28.5) | — | — | — | — | — | Single gas CO2, CH4 | 27 | 4 | 2014160 |
| ZIF-8 300 nm | 25.7– (25.9) (GPU) | 19.4– (5.8) | — | — | — | — | — | ||||||
| ZIF-8 500 nm | 25.7– (28.1) (GPU) | 19.4– (5.8) | — | — | — | — | — | ||||||
| ZIF-8 | PDMS | 2.5, 5, 10, 15, 20 | — | — | — | — | — | — | — | Single gas C3H8, N2 | — | 1.5 | 2015161 |
| — | — | — | — | — | — | — | Gas mixture C3H8/N2 (10:90, 20:80, 30:70, 40:60) |
20, 27, 35, 45, 55 | 0.5, 1.5, 2.5, 3.5, 4.5 | ||||
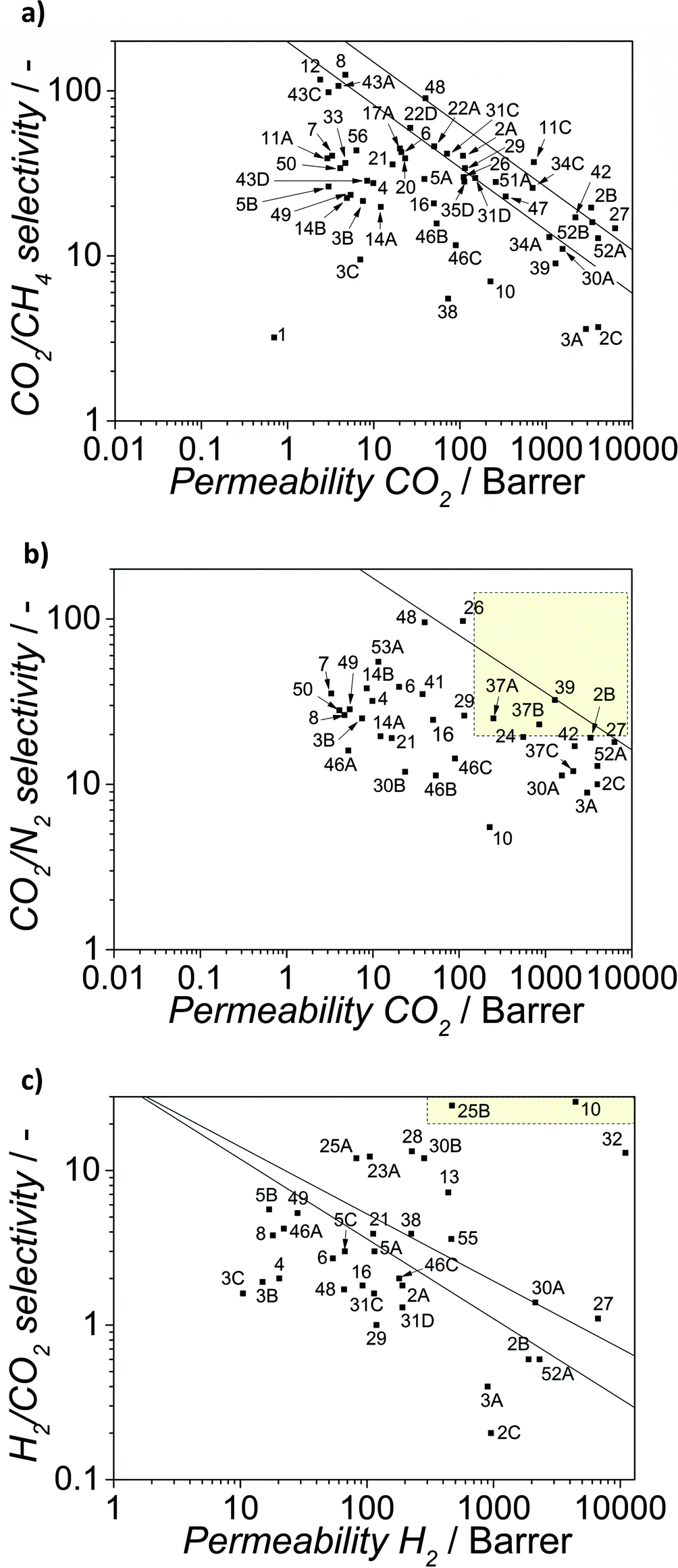 | ||
| Fig. 6 Robeson plots for the separation of CO2 from CH4 (a) CO2 from N2 (b) and H2 from CO2 (c). The graphs contain the most relevant results reported in literature for M4s. See Table 2 for references. | ||
The first M4 reported48 comprised a three dimensional copper(II) biphenyl dicarboxylate-triethylenediamine MOF embedded in PAET (poly(3-acetoxyethylthiophene)) and was applied in gas separation. The authors claimed that the increase in hydrophobicity of the MMMs resulted in preferential adsorption of methane, leading to an increase in CH4 permeability at 20 and 30 wt% of MOF loading together with a decrease in CO2 permeability; thus, giving rise to a reduction of the CO2/CH4 selectivity. Since this pioneering work, the field of research of MOF based MMMs has experience an exponential growth and a large number of different membranes have been reported in literature. Regarding the fillers, HKUST-1, ZIF-8 and MIL-53(Al) with and without amino group have been the most studied MOFs. As for the polymers, the organic phase used as continuous phase can be classified into low flux glassy polymers (i.e. PSF,95–98 PPEES,99 PVAc,100 Ultem®, Matrimid®101–104 or PBI105–107) and more interesting high flux polymers: rubbery, such as PDMS96 and PMPS (polymethylphenylsiloxane),108 and glassy, such as 6FDA-DAM.40,109
As a general trend, in a large percentage of the reported results, improvements in flux at constant selectivities with respect to the bare polymer have been reported and only in circa 10% of the cases improvements in both flux and selectivity were achieved. Furthermore, for all the membranes tested at high pressures it was observed that upon MOF addition, the plasticization of the membrane at high CO2 pressures was partially suppressed, maintaining large separation factors over a wider pressure range than that observed for the pure polymer135,157 or even increasing the selectivity at high pressures.45,143
This behaviour has very important consequences in applications in which the retentate has to be kept pressurized. These improvements in permeability and/or selectivity upon filler addition demonstrate the tremendous potential of MOF-based MMMs for efficient CO2 capture, as can be seen when results are put in perspective using the customary Robeson plots with the latest upper limits (see Fig. 6).23
For CO2/CH4 separation, membranes comprising high permeable 6FDA-containing polyimides (e.g. 6FDA-4MPD,40,109 6FDA-ODA123,162 and 6FDA-DSDA163) have generally a performance beyond the Robeson limit of 1991,164 surpassing the Robeson limit of 200823 when ZIF-9040 and [Zn2(1,4-bdc)2(dabco)]·4DMF·0.5H2O are used (graph codes 11C and 2B, respectively). Furthermore, for this gas mixture M4 based on a microporous polymer PIM-1, also exhibits a behaviour above the latest Robeson limit, reaching permeabilities up to 6300 Barrer together with selectivities of 14.2 for ZIF-8 loadings of 39 wt%.126 However, as expected, low permeable polymers, such as PSF, Ultem®, PPEES or Matrimid® lead to composites with separation properties well below the state of the art, with permeabilities typically in the range 2 to 70 Barrer and selectivities between 8 and 135. In the case of CO2/N2 separation, the best results have been obtained for Pebax® and PIM-1 whose permselectivities have been improved up to above the latest Robeson limit upon ZIF-7 and ZIF-8 addition. Interestingly, M4 comprising Pebax® and 35 wt% ZIF-8139 and 6FDA-DAM with 10 wt% CPO-27(Mg)137 have attractive separation properties for the separation of CO2 from flue gas on a large scale when cross-flow modules with membranes with selective layers thinner than 300 nm operated at pressure ratios of 5–10 are considered.82
Moreover, membranes comprising ZIF-8 and 6FDA-DAM:DABA, PIM-1 or 6FDA-durene (graph codes 24, 27 and 42 respectively),126 CPO-27 and XLPEO (graph code 37A),137 and [Zn2(1,4-bdc)2(dabco)]·4DMF·0.5H2O and 6FDA-4MPD (graph code 2B) exhibit permselectivities very close to those required for an attractive membrane-based post-combustion CO2/N2 separation.82
However, it must be highlighted that the optimal membrane performance calculated by Merkel et al.82 concerns flue gases consisting of low CO2 concentrations in N2 at 40–50 °C saturated in water. In this sense, more realistic measurements of these membranes, including water vapour, should be performed to assess their viability. Finally, for H2/CO2 separation, membranes comprising PBI,105 PIM-1126 and 6FDA114 containing polyimides and ZIFs (namely, ZIF-8,107 ZIF-90,128 ZIF-7105 and ZIF-11158) exhibit the best performance. The most outstanding results were obtained for asymmetric membranes prepared with HKUST-1 and PMDA-ODA30 and dense membranes containing ZIF-8 and PBI107 for which the commercial attractive region89 is reached with MOF loadings of 6 wt% and 30 wt%, respectively. Interestingly, the membranes were tested up to 230 °C in the latter case, under conditions relevant for pre-combustion CCS.
E Towards high productivity M4s: progress in hollow fiber and thin layer membranes
A membrane module with a proper permselectivity for a real industrial application should also have a meaningful productivity. Even membrane materials displaying excellent separation performance would be useless if productivity is low. By controlling the morphology it is possible to create asymmetric (anisotropic) membranes with very thin selective layers that decrease mass transfer resistance and increase productivity.Generally the target is to have a selective layer with a thickness lower than a micrometer. However, such a thin layer of polymeric or mixed matrix material needs a support. This is the basic definition of asymmetric membranes: a thin selective dense layer on a non-selective porous support providing the strength. Several methods to manufacture asymmetric membranes are available, such as phase separation, interfacial polymerization, solution-coating, plasma polymerization, etc.165
A fundamental question when it comes to the application of thin separating layers regards their geometry. Basically, two different membrane modules can be envisaged: (i) spiral wound flat sheets, (ii) supported composites and (iii) hollow fiber (HFb, see Fig. 7 membrane modules). Although spiral wound modules were the first commercialized, HFb modules offer significant advantages; the most important being their high packing density (over 10000 m2 m−3),166–168 about ten times higher than for flat sheet (plate and frame) membranes. In addition, HFb membranes can handle very high transmembrane pressure differences (up to 70 bar) and their fabrication costs are 5 to 20 times lower than that of equivalent membranes for spiral wound modules (5–20 US $ per m2versus 5–100 US $ per m2).158 Although already some reports exist on M4 based asymmetric flat membranes,113,116,120,125,169,170 with promising results and providing important insight, because of the above reasons we focus here on asymmetric hollow fiber membranes.
 | ||
| Fig. 7 SEM images of the cross section of a polysulfone hollow fiber membrane, (a) overall, (b) outer edge. | ||
The preparation of hollow fiber membranes relies on the phase separation technique developed by Loeb and Sourirajan,165,171 most specifically, phase inversion spinning (also called dry jet-wet quench spinning or wet spinning).167,168 In this process, a dense layer is integrally formed over a porous layer. The outer dense thin layer (selective skin layer) is the selective part of the structure while the inner porous layer is only a support without any important transport resistance. In the process, a polymer solution and a bore fluid are coextruded through a nozzle (spinneret) and precipitated in a non-solvent bath to create the asymmetric structure. Thickness of the selective layer and the morphology of the substructure determine the efficiency of the membrane.
Besides the parameters to control for the fabrication of polymer-only fibers, spinning mixed matrix membranes in asymmetric hollow fiber geometry (MM-HFbM) brings a few more issues to address, namely the compatibility of the polymer and filler particles and the distribution of the particles within the fiber wall. Zhang and co-workers146 defined the preparation of MM-HFbM as the development of an asymmetric structure from compatible components (polymer and filler) with a very thin selective layer where the filler particles are well dispersed without any major defect at an economically attractive cost.
The first key parameter to achieve this objective is the preparation of a homogenous dispersion of the particles within the spinning solution, since stability of the dope during the spinning process is crucial to avoid sedimentation or cluster formation. Studies with polymer–zeolite dense films and MM-HFbMs showed that surface interactions between filler particles and other components of the polymer solution (‘dope’) is the parameter to tune in order to increase the stability and the performance of the membranes. This is mostly discussed as polymer–filler interaction or compatibility and becomes even more important in the case of sub-micron particles.146,167 The controlling parameters are: (a) the surface properties of filler particles, such as the degree of hydrophobicity, (b) surface chemistry, (c) surface charge, (d) geometry and (e) the size of the particles. It is also known that the nature and degree of interaction of the (non)-solvents with filler particles are important parameters that control the phase separation kinetics.73
The distribution of filler particles within the polymer matrix in the thin separating layer is the other key issue that affects performance of a MM-HFbM. There are different methods for dispersing particles during the dope preparation process.146,166,167 Mostly, particles are wetted and dispersed in a small portion of the solvent of the dope formulation. Then this dispersion is added to the dope solution or vice versa. The energy required for dispersing is introduced by mechanical agitation or sonication. Mechanical agitation is generally performed by high speed mixers. Ultrasound baths (indirect sonication) or horns (direct sonication) are used when mechanical agitation is not enough. Indeed, indirect sonication seems to be the most efficient method to avoid the formation of agglomerates.121
Even if a good dispersion is obtained, agglomeration and settling of the particles before spinning could be a problem. This issue may be controlled by tuning the flow behaviour of the dope. It has also been observed that agglomeration of particles during the phase inversion is possible depending on the concentration of the particles within the dope.166 Agglomeration of particles can cause serious defects within the outer dense selective layer and ends with performance loss. Distribution, compatibility and agglomeration issues should be considered together. Goh et al. showed that the agglomeration of filler particles and gap formation at the filler–polymer interface are strongly related.172 They observed that suppressing the agglomeration yields fewer defects at the selective surface. Also they claim that the formation of large voids with tear-drop shape in the sub-structure is strongly related to agglomeration phenomenon. Spinning parameters, related to the flow behaviour – momentum and velocity profiles at the outlet of the spinneret nozzle – have a great influence on the orientation of the polymer chains and the redistribution of particles along the fiber wall.173–178 There are two sources of stress induced on dope and/or nascent fiber during the spinning process; the shear stress induced within the spinneret and the stress on the free falling (or pulled by a take-up drum) nascent fiber from the spinneret nozzle. These stresses change the alignment and the orientation of polymer chains and filler particles. On the contrary, they also may have a negative effect on the adhesion of the polymer on the surface of the fillers.167,179
Last but not least, selectivity of an asymmetric MM-HFbM is very sensitive to the state of the selective layer. There is a strong relation between the degree of defectiveness and thickness of the selective layer.168 In order to overcome the formation of defects during preparation, different approaches can be followed: (i) addition of non-solvents into the dope formulation, (ii) performing the spinning at high temperatures (>50 °C) or (iii) optimizing the shear and elongational force to manipulate the polymer chain orientation and at the same time orienting the MOF particles with large aspect ratios. In addition, high filler loadings cause high viscosity that makes handling and spinning process more difficult and also can cause defects on the surface.146 Also, high particle loading, especially nano-size particles, is more prone to cause agglomeration. A careful attention has to be paid as well as intensive laboratory work is necessary to optimize the dope, not only to achieve good dispersion and defect free fibers, but also for handling the dope and to achieve an excellent utilization of the filler, allowing the use of lower concentrations.
Even when the issues above have been solved, almost in all cases, industrial fiber spinning yields defective membranes. The most common healing technique to allow application of these fibers consist of a thin coating with a secondary polymer (i.e. PDMS or Polyaramid) to clog the possible pin holes or scratches.180,181 This layer must be more permeable than the original selective skin to maintain productivity. The nature and the selectivity of this coating may also affect the separation characteristics of the membrane.146 Annealing above Tg (glass transition temperature) has also been reported as another post-treatment to heal the defects in the skin layer. Coatings by plasma polymerization and/or plasma treatment of the membrane surface are recently proposed defect healing techniques.116
The dual layer asymmetric hollow fiber concept represents a step forward to overcome all these issues and it offers important advantages, since this configuration allows using different polymers or different dope formulations within the same fiber. Dual layer hollow fibers are fabricated by co-extrusion of two dopes and a bore fluid, allowing the use of different polymers for support and separating layers167–169,182 Although this method adds further complexity, it allows the use of cheaper and unselective polymers -for the support layer. Moreover, different dope formulations can be used for the inner and outer layers.40,168,183,184 Another advantage is that this configuration allows the use of highly selective but not spinnable polymers (as a single layer HFb) for the skin layer. Populating the selective filler particles within the dense selective layer; dual layer fiber spinning with mixed matrix dope on the sheath and polymer only dope for the bore is an excellent alternative to control the distribution of particles (Fig. 8) and to minimize filler consumption.168,177 In order to avoid the formation of defects on the surface, it is necessary to use filler particles much smaller than the thickness of the selective layer.
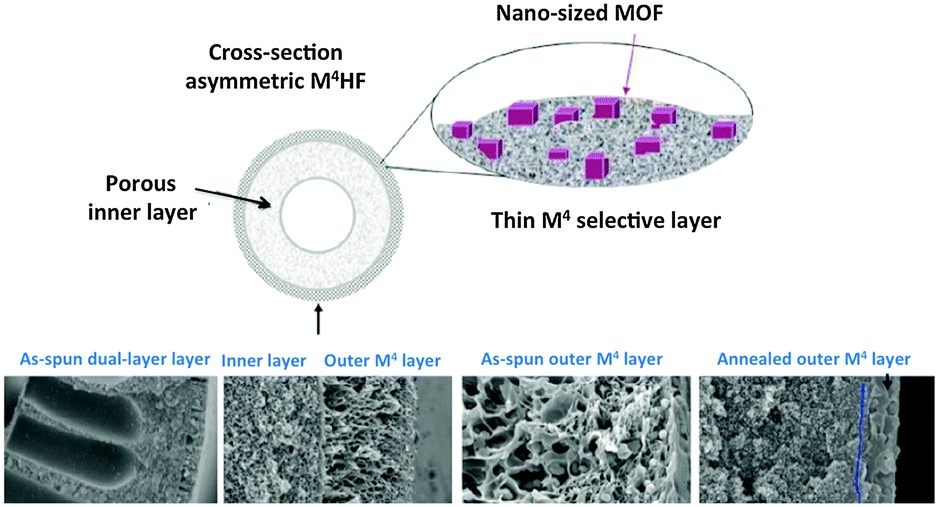 | ||
| Fig. 8 Schematic cross-section morphology of the dual-layer hollow fiber with a polymer–particle mixed matrix skin.168 Reproduced with permission from Elsevier. | ||
Although most of the current experience on MM-HFbMs originates from studies on polymer–zeolite MMMs, already the first reports on M4-HFbs have been published. Hu and co-workers spun polyimide (PI)–Cu3(BTC)2 blends.114 Their results proved that particles are homogenously dispersed within the polymer matrix with only a few small agglomerates and no serious interface voids. Authors claim that the pore blocking of MOF with PI chains and the rigidification of PI chains within the interface negatively affect the diffusion of all gases, reaching a maximum permeance for H2 of 1270 GPU with a selectivity over CO2 of 27 (already within the desired range for pre-combustion CO2 capture). In a more recent study, Dai et al. prepared dual layer M4-HFbs with a commercial polyetherimide (Ultem®100) and ZIF-8 fillers for CO2/N2 separation.119 Morphological characterization studies showed a homogeneous dispersion of the filler material (size ∼ 200 nm) and a very good adhesion between the core and the sheath regions. Gas permeation measurements demonstrated that both permeance and permselectivity of dual-layer MM-HFbMs are higher than that of polymer only HFb membranes. At similar test conditions, Ultem®-ZIF-8 MM-HFbMs yielded a CO2/N2 selectivity (= 36) with relatively low permeance for CO2 (= 26 GPU). The actual challenge is to decrease the skin thickness to achieve higher fluxes. In the latest report on the subject, researchers reported the successful formation of high-loading mixed matrix hollow fibers containing ZIF-8 (up to 30% wt).146 Although the study targeted hydrocarbon separations, it proves that it is feasible to transfer the knowledge generated from dense film studies on MOF containing mixed matrix membranes to industrial scale highly productive asymmetric hollow fiber membranes.
F Structure performance relationships in M4s
As thoroughly discussed above, polymer as well as filler properties affect MMM morphology and separation performance. Regarding the filler, chemical structure, surface chemistry, particle size distribution and aspect ratio are the most important variables. Indeed, poor filler–polymer adhesion and filler segregation or blocking of its porosity by the polymer are the main reasons why traditional MMM fillers like zeolites, silicas or activated carbons have not made the final steps towards industrial implementation. Due to these limitations, in general only low filler loadings can be achieved without compromising the separation performance unless laborious filler post-treatments are applied.157 As to the polymer, it is very important to match its properties with those of the MOF filler.4For every MOF–polymer couple, the MOF loading should be maximized. Loadings lower than a certain value do not alter in a significant way the transport properties of the polymer membrane, while rigidity and mechanical strength of the composite are increased, as determined by differential scanning calorimetry and dynamic mechanical analysis.37,101,115,185,186 At a certain loading, a good dispersion of the filler with an excellent interfacial contact with the polymer chains (composite interface) results in an optimum MMM performance. However, at higher loadings polymer chains are not completely able to enwrap the particles, so that the latter may agglomerate, reducing their dispersion in the polymer matrix and forming undesirable transport channels.96,103 High permeabilities are also attributed to the disruption of the polymer chain packing and linking due to the presence of the molecular sieves which implies also an increase in polymer free volume.187 In this section, we first discuss how to assess the exact structure of these composites, then the different synthetic approaches utilized to improve MOF–polymer performance. Finally, we will introduce the use of Hansen solubility parameters to predict chemical interactions between MOF and polymer.
Structural features of MMMs such as the spatial distribution of the filler crystals and the existence of voids at the filler-matrix boundary are essential in determining the mechanical properties and gas separation performance. However, these parameters are also particularly difficult to assess experimentally. Generally, the membranes are fractured after immersion in liquid N2, often referred to as cryo-fracturing, in order to gain access to the cross-section of the membrane with imaging techniques such as SEM. This provides only 2D, local information, while furthermore the cryo-fracturing approach often results in rough membrane cross-section surfaces. Therefore, a number of surface motifs derived from the fracturing process are imaged, leading to an incomplete or deceptive picture of the structural features of the membrane, most particularly the fraction of voids in the membrane.
As an interesting alternative, the use of FIB-SEM tomography188 in the characterization of M4s was recently introduced by Rodenas et al.46,189 In FIB-SEM imaging the Ga+ primary focused ion beam is used to controllably sputter a selected area of the specimen, precisely removing thin slices of material and enabling a series of consecutive cross-sections to be studied individually. Here FIB-SEM was used to study the spatial distribution of the MOF filler in the MMMs as well as a mean to quantify the contact between the filler and the matrix phases, as depicted in Fig. 9. After alignment of the stack of SEM micrographs, the 3D structure of the analysed volume could be reconstructed in 3D and depicted along three orthogonal cross-sections. Segmentation of the individual phases, i.e. PI matrix, MOF filler and voids, was performed by image thresholding. Despite the relatively high filler loading (25 wt%), a homogeneous distribution of the MOF crystals within the polymer matrix is observed, indicating adequacy of the procedure employed to cast the MMMs. In addition, quantification of the segmented volume allowed determining the mass based MOF loading, obtaining a remarkably good agreement with the bulk MOF loading (as derived from TGA), which validates the image analysis results. With this new technique, it is now possible to have a much more realistic picture of the internal structure of the membrane. In combination with rigorous mathematical modelling, as the one developed by Nair and co-workers, a unique tool for the understanding of MMMs should be available for future studies.
 | ||
| Fig. 9 3D reconstructed volume of the portion of a NH2-MIL-53(Al)/PI_25% membrane studied with FIB-SEM (a); the corresponding surface-rendered view of the volume corresponding to the MOF crystals (b). Panel (c) shows a detail of the volume indicated in the yellow frame in panel (a). In panel (d), half of the material in (c) has been removed to improve visualization. And (e) shows the surface-rendered view of the volume corresponding to the voids. Box size (a, b, e): 14.3 μm × 10.7 μm × 7.5 μm.46 Reproduced with permission from WILEY-VCH Verlag GmbH & Co. | ||
One of the most critical concerns for the development of MMMs is the lack of compatibility between filler and polymer matrix, which produces a decrease in membrane performance. To overcome this issue, different strategies have been developed in the last years. An elegant solution is the use of MOFs that contain organic linkers similar to elements of polymer units. In particular, ZIFs have been regarded as promising fillers for MMMs because the linkers in ZIF structure are expected to allow a better affinity and interaction with certain polymeric materials such as polybenzimidazole (PBI). Yang et al.105 showed that it is possible to prepare M4s containing up to 50 wt% of ZIF-7 nanoparticles by mixing the as-synthesized ZIF-7 nanoparticles without the traditional drying process with PBI. In these M4s the permeability of H2 was significantly higher than in pure PBI membranes, although H2/CO2 selectivity was not improved. In a similar way, M4s containing PBI and high loadings of ZIF-8 or ZIF-90 nanocrystals106,128,190 have been fabricated. As expected, all these composite membranes showed improved H2 permeability together with an enhanced H2/CO2 separation selectivity.128 Recently, Yao et al.158 fabricated M4s incorporating ZIF-11 crystals into PBI. Gas permeabilities of H2 and CO2 increased upon incorporation of ZIF-11. Additionally, the H2/CO2 ideal selectivity was also slightly improved in the composite membrane containing a 16 wt.% of MOF.
MOFs containing amino groups are also considered good candidates to optimize polymer matrix-filler interactions. In particular, the flexible NH2-MIL-53(Al) has shown excellent adhesion with different polymers such as polysulfone or polyimide.45,162 Nik et al.123 reported that fillers containing amino groups may lead to the rigidification of the polymer at the MOF–filler interface, thus decreasing gas permeability and increasing selectivity. However, almost no systematic study about the influence of functional groups on the MOF–polymer interactions has been performed. Seoane et al.163 synthesized thioether- and sulfone-containing copolyimides, 6FDA:DSDA/4MPD:4,4′-SDA (polymer 1) and 6FDA/4MPD:4,4′-SDA (polymer 2), with the aim of studying the effect of polymer functional groups in the preparation and performance of M4s containing NH2-MIL-53(Al) or NH2-MIL-101(Al). The main difference between both polymers was that in polymer 1 a part of the 6FDA monomer was substituted by the more flexible DSDA unit. This modification increased the interaction between the amino-functionalized MOFs and polymer 1, showing that the flexibility of the polymer had an influence on the filler–polymer matrix interaction and, consequently, on the overall performance of the membranes.
MOF–polymer interactions can also be improved using the one-pot synthesis methodology developed by Seoane et al.131 A common solvent for the MOF synthesis and membrane casting is necessary in this approach.
Other methodologies have been used to improve the MOF distribution in the polymer matrix. Interfacial polymerization of thin film nanocomposite membranes was first developed by Jeong et al.,191 and allows embedding the filler nanoparticles into a thin polymer film during monomer polymerization. Sorribas et al.192 have used this procedure to obtain MOF [ZIF-8, MIL-53(Al), NH2-MIL-53(Al) and MIL-101(Cr)]-polyamide (PA) thin layers on top of cross-linked polyimide porous supports. The use of fillers of different nature within the same composite is another interesting approach towards improved separation performance, as introduced by Zornoza et al.97 in a study where polysulfone based MMMs containing one MOF (HKUST-1 or ZIF-8) and zeolite silicalite-1 were manufactured. Later Valero et al.145 obtained MMMs by a combination of silica MCM-41 and MOF NH2-MIL-53(Al) in glassy commercial polymers (polysulfone Udel® or polyimide Matrimid®) following a similar approach.
The so-called Hansen solubility parameters (HSP) have classically been applied to the evaluation of solvent-polymer chemical interactions,193 but also to study barrier properties and chemical resistance of protective clothing,193 prediction of cytotoxic drug interactions with DNA,194 optimization of the extraction of bioactive compounds from biomass with subcritical water,195 identification of an alternative, less toxic solvent used in a microencapsulation process,196 and preparation of stable dispersions of TiO2 and hydroxyapatite nanoparticles in organic solvents,197 among other examples. In the case of polymeric membranes, these parameters have been used to evaluate possible solvents and non-solvents to prepare membranes by phase inversion,198,199 to study membrane fouling,200 for the analysis of compatibility between polymers and IPA in IPA/water distillation201 and to explain the acetone, butanol and ethanol interaction with silicalite-1/PDMS membranes.202
Regarding the application of HSP to metal–organic framework (MOF) materials, there are two recent reports; one dealing with the formation of composites between MOF HKUST-1 (with 5 wt% loading) and poly(L-lactic acid) (PLLA),203 and other discussing the encapsulation of caffeine into MOFs ZIF-8 and NH2-MIL-88B.204 Thanks to HSP the basic principle of “like dissolves like”, i.e. the qualitative idea behind most of the previous examples, is expressed in numbers easy to handle and compare. However, a limitation of HSP application is related to insufficient availability of HSP data for systems of interest (in particular, MMM polymers and mainly MOFs).
The chemical interactions established between MOF and solvent or ligand can be of different nature, i.e. dispersion, polar or hydrogen bonds. In case of solvents these interactions can be discussed in terms of Hansen solubility parameters.193 These parameters (δD, δP and δH for dispersion or London interaction, polar interaction and hydrogen bonds, respectively) are given in Table 1 for some selected membrane polymers, MOF ligands and MOF HKUST-1 (the only available MOF to date with HSP203). The MOF ligands included in this table are commonly used for the synthesis of some of the most typical and studied MOFs: 2-methylimidazolate (2MI, for ZIF-8205), benzenedicarboxylate (BDC, used for MOF-5,206 MIL-53,207 MIL-101,208 UiO-66,209etc.), NH2-benzenedicarboxylate (NH2-BDC, used for NH2-MIL-53,210 NH2-MIL-88B,211etc.) and benzenetricarboxylate (BTC, for HKUST-1,212 MIL-96,213etc.). In terms of HSP, the interaction between two substances 1 and 2 can be obtained calculating the parameter Ra193 with the following eqn (1):
| Ra2 = 4(δD1 − δD2)2 + (δP1 − δP2)2 + (δH1 − δH2)2 | (17) |
R a values in Table 3 suggest that common linkers are not totally compatible with the selected membrane polymers (Ra above 7.5); however, 2MI (in special) and BDC are compatible with four out of five polymers (excluding PDMS). This is in agreement with the great availability of MMMs with MOFs obtained from 2MI and BDC (in particular the above mentioned ZIF-8 and MIL-53). When Ra values for the same polymers are compared for HKUST-1 and BTC (the linker in HKUST-1), the analysis always favours the MOF over its linker. This relative discrepancy between BTC-HKUST-1 pair (Ra in Table 1 for HKUST-1 is as good as the values commented for 2MI and BDC), is basically due to the fact that the available value for the BTC δH (17.0) is higher than expected because most probably all the three acid –COOH groups in BTC would not be available for H-bonding, as the experimental HKUST-1 δH (10.7) obtained by Auras et al.203 suggests, and as already indicated by Paseta et al.204 Furthermore, it is obvious from this discussion that the availability of HSP for MOFs of interest (so that the speculations made here for linkers would make more sense) would facilitate the selection of MOF–polymer couples without the necessity of synthesizing and testing their corresponding composite membranes. Indeed, HSP for polymers and MOFs would help selecting the best membrane polymer material for every desired MOF. Finally, it is worth emphasizing that most likely the same MOF material would exhibit different HSP values depending on the particle size (when nano- and micro-sized particles of the same MOF phase would be considered) and perhaps on the form of the MOF in those cases in which flexibility is a key issue.207 Indeed, in addition to the chemical compatibility of polymer and filler, another important aspect in MMM performance is the morphology of the filler.
| HSP [MPa0.5] | R a [MPa0.5] | |||||||
|---|---|---|---|---|---|---|---|---|
| δ D | δ P | δ H | 2MI | BDC | NH2-BDC | BTC | CuBTC | |
| PES | 19.6 | 10.8 | 9.2 | 1.7 | 5.2 | 7.9 | 8.1 | 3.8 |
| PEI Ultem 1000 | 19.6 | 7.6 | 9.0 | 3.6 | 3.9 | 7.8 | 8.3 | 4.4 |
| PI Matrimid | 18.7 | 9.6 | 6.7 | 3.2 | 7.1 | 10.6 | 10.8 | 4.3 |
| PDMS | 15.9 | 0.1 | 4.7 | 13.1 | 13.5 | 17.5 | 17.7 | 12.2 |
| PSF Udel P-1700 | 19.0 | 5.9 | 6.1 | 6.0 | 7.1 | 11.2 | 11.7 | 6.5 |
| 2MI | 18.8 | 10.7 | 9.7 | |||||
| BDC | 20.0 | 7.2 | 12.8 | |||||
| NH2-BDC | 20.8 | 8.6 | 16.4 | |||||
| BTC | 20.3 | 9.3 | 17.0 | |||||
| CUBTC | 17.9 | 9.9 | 10.7 | |||||
Particle morphology is crucial for many applications and allows the properties of a certain solid to be tuned without changing the material composition. For instance, the improved performance of gold or silver nanoparticles for surface plasmon resonance,217 semi-conductor nanodots for quantum confinement,218 and metal or metal oxides for catalysis219,220 is strongly dependent on crystal morphology. In case of MMMs, it is easy to envisage that the performance of membranes containing exactly the same material but with a different particle configuration will result in different separation performance. For instance, most of the outstanding examples in terms of separation reported so far in literature made use of MOF nanoparticles.40,117 The improved performance of composites incorporating nanoparticles is usually ascribed to the larger external to internal surface ratio in these nanoparticles, that allows a much better interaction with the polymer. Moreover, lower filler loadings can be used for an improved separation performance, preserving to a large extent the mechanical properties of the polymer. However, although microwave,221 electrochemical222 protocols and the use of chemical additives223 are powerful synthesis tools for the manufacture of homogeneous MOF nano-crystals, conventional MOF synthesis procedures render agglomerated powders consisting of isotropic micron-sized crystals or barely dispersible nano-particles. In this spirit, the availability of high-aspect-ratio, ideally ultrathin, MOF nanostructures represents an advanced solution to improve the integration between both components in the composite materials, thereby circumventing the aforementioned hurdles. The synthesis of MOF nanosheets and their application in M4s has now been reported by Rodenas et al.159 In this article, a bottom-up synthesis strategy leading to highly crystalline, intact MOF nanosheets that could be readily dispersed into a polymer matrix is reported. The synthesis strategy to produce MOF nanosheets relies on the diffusion-mediated modulation of the MOF growth kinetics, with the synthesis medium consisting of three liquid layers composed of mixtures of DMF and a suitable miscible co-solvent in appropriate ratios, that are vertically arranged according to their different densities. By applying this method, the authors were able to prepare free standing nanosheets of CuBDC homologs. MOF–polymer composites were prepared by incorporating CuBDC nanosheets within a polyimide (PI) matrix at different filler loadings (2–12 wt%). The same procedure was employed to prepare comparative composites incorporating either bulk-type or sub-micron sized (nanoparticle) isotropic CuBDC MOF crystals as fillers. The internal structure of the composite membranes was again studied with FIB-SEM, as illustrated in Fig. 10. Despite the identical filler content, striking differences in the nanostructure were immediately evident. Whereas the regular MOF crystals leave a significant fraction of the composite volume unoccupied in b-CuBDC(8)@PI, due to their bulky nature, the MOF lamellae are uniformly distributed over the inspected volume for ns-CuBDC(8)@PI. Image analysis of the FIB-SEM tomograms allowed quantification of a number of structural parameters of the composite membranes (Fig. 10). The MOF nanosheets in ns-CuBDC(8)@PI exposed ca. one order of magnitude larger surface area than the bulk-type crystals incorporated to b-CuBDC(8)@PI (2.2 × 10−3vs. 2.9 × 10−4 nm2 nm−3 MOF), enormously increasing their interaction with gas molecules. As a result, at every studied trans-membrane pressure difference, the separation selectivity for the nanosheet-CuBDC(8)@PI membranes is 30–80% higher than for the polymeric membrane and 75% to 8-fold higher than for the bulk-CuBDC(8)@PI counterpart in the range of operation conditions investigated. The similar intrinsic sorption properties of bulk-type and nanosheet CuBDC crystals cannot account for such remarkable differences in separation performance, which were therefore attributed to the different MOF crystal morphology, which turned out key for the filler–polymer integration and the occupation of the gas permeation pathways by the molecular sieve. Most remarkably, the selectivity achieved with ns-CuBDC(8)@PI was retained or even increased slightly upon increasing the upstream pressure. This significant finding is completely opposite to the general observation for both polymeric and conventional MOF–polymer membranes,55 that the separation selectivity decreases with increasing partial pressure of CO2. These results confirm the relevance for the separation performance of the extent to which the MOF filler occupies the membrane cross-section perpendicular to the gas flux and the importance of crystal engineering in the development of efficient composites for gas separation. Moreover, while based on the Maxwell model for MMMs with homogeneously dispersed fillers an optimal performance requires similar permeabilities of both components, using fillers with large aspect ratios this requirement is strongly relaxed.60,224
 | ||
| Fig. 10 Surface-rendered views of the segmented FIB-SEM tomograms for composite membranes containing bulk-type (a) and nanosheet (b) CuBDC metal–organic-framework embedded in polyimide. Full projections along the y-direction of the reconstructed volumes (c, d). Angular histogram showing the orientation of MOF lamellae with respect to the gas flux direction (y axis) for a composite material containing MOF nanosheets embedded in polyimide (e). Histogram of the efficiency with which the individual MOF nanosheets cover the membrane cross-section, defined as the ratio between the area of the MOF lamellae (Alam) and that projected on the plane perpendicular to the gas flux (Aproj), as schematically depicted in the inset to the panel (f). In the same inset figure, α represents the angle of inclination of each MOF lamellae with respect to the y-axis. Green bars correspond to experimental data while the red line shows the exponential fit. Reproduced with permission from Nature publishing group.159 | ||
G Summary and future perspectives
Metal–organic framework–polymer composites hold great promise for application as gas selective membranes. In this review we have shown that already reported M4s, if applied in pre and post-combustion CO2 capture during energy generation, may be able to facilitate the development of efficient and economically affordable capture technologies. The success of these new membranes lies in the rich chemistry behind MOF formation, both in terms of chemical composition and of particle morphology. The combination of these features with highly permeable polymers can, in principle, deliver membranes that meet the most important requirements for the capture of CO2 under relevant post-combustion conditions and of H2 under relevant pre-combustion conditions. Having said this, one should also realize that the final success of these membranes will depend on several critical issues, viz. the manufacture of thin membranes and the closely related structure–performance relationship of M4s, as discussed below.One of the most important aspects for the final application of M4s will be the development of efficient methods for the synthesis of thin separating layers, preferably with hollow fiber architecture. Although already some work has been devoted to this important field, much more is necessary. One of the main barriers to optimize HFb manufacture is the scaling up of state of the art MOF fillers and polymers, since large amounts of both materials are needed in regular spinning setups. In this sense, alternative thin film preparation techniques may facilitate the development of membranes with this morphology on a more controllable manner. The Langmuir–Blodgett (LB) method is a well-known technique for the fabrication of monolayer films at the air–water interface. Moreover, these films can be transferred onto almost any desired substrate to obtain films with a controlled thickness by consecutive depositions.225 Therefore, it is a powerful tool for the fabrication of very thin selective layers onto porous supports for the development of asymmetric membranes such as hollow fibers. Many studies dealing with the use of LB films for gas separations resulted in disappointing results, since the selectivities obtained were lower than the values predicted from Graham's law. This indicated that gas transport occurred mainly through film defects.226 To improve the selectivity of LB films in gas separations, the group of Regen started in the early 1990's a systematic study to obtain defect-free LB films making use of different surfactants based on the calix[6]arene framework.227 In a recent publication,228 the authors have shown that an asymmetric membrane formed by a 7 nm thick bilayer composed by a quaternary ammonium derivative of poly(maleic anhydride-alt-1-octadecene) ionically cross-linked with poly(acrylic acid) deposited onto a cast film made from poly[1-(trimethylsilyl)-1-propyne] (PTMSP) exhibits a remarkable H2/CO2 selectivity of 200 (with a H2 permeability of about 9 GPUs). These contributions have shown that it is possible to obtain very thin dense layers that are suitable for the development of gas separation membranes by the LB technique. Furthermore, Tsolatas et al.229 obtained two-dimensional MOF LB films using three kinds of MOF particles with different sizes and morphologies, Cu3(BTC)2, Cu2(BDC)2(BPY) and Al12O(OH)18(H2O)3(Al2(OH)4)(btc)6. These authors showed that the morphology of the MOF crystals determines the particle orientation on the substrates; moreover, crystal density on the films could be controlled with the LB method. Additionally, Lu et al.230 have obtained monolayers of polyvinylpyrrolidone-modified UiO-66 microcrystals at the air-liquid interface using sodium dodecyl sulfate to consolidate the films. Hybrid films MOF–polymer obtained by the LB method would allow a deeper understanding of the filler–polymer interactions and also a more detailed characterization of the membrane structure and properties and of the subsequent influence on the MMM performance. Different strategies may be used to obtain mixed MOF–polymer LB films. Xu and Goedel231 produced polymer–silica hybrid LB films spreading a chloroform solution containing hydrophobized silica colloids (140 nm of diameter) and polyisoprene amphiphiles (47/53 wt%) onto a water surface. After compression, the hybrid monolayer was cross-linked by UV light and transferred onto different solid supports to obtain a freestanding cross-linked hybrid membrane. The fabrication of hybrid monolayers polymer/nanospheres at the air–water interface has been reported by Hu et al.232 The authors spread polystyrene spheres with diameters ranging from 100 nm to 1 μm onto the water (containing 1–3 ppm of polyethylene oxide) surface. In a few hours, the polymer was adsorbed onto the surface of the nanospheres obtaining a closed packed hybrid monolayer that could be transferred onto a solid support. Recently, Martin-Garcia and Velazquez233 obtained hybrid LB films composed by CdSe quantum dots (3.5 nm) and poly(styrene-co-maleic anhydride) partial 2-butoxyethyl ester cumene terminated (PS-MA-BEE) by successive compression–expansion cycles234 of the monolayer at the air–water interface. This study has shown that the morphology of the hybrid films can be modulated by shear stress. In addition, the LB method can be used to fabricate alternate LB films by successive deposition of monolayers of different materials, what would allow obtaining polymer–MOF–polymer sandwich-like structures with a controlled density of MOF particles. All these strategies and many others that can be proposed for the fabrication of mixed MOF–polymer LB films open an attractive field of research for the next years. On a different type of approach, the preparation of pure MOF coatings on polymeric HFb supports by interfacial microfluidic processing has also attracted considerable attention in the last few years, with very promising results.235–239
In spite of the preparation method, it is very important to gain much more insight into the relationship between composite structure, components and membrane performance. The development of accurate mathematical models to describe transport among M4s, the use of adequate techniques for the characterization of these membranes and the appropriate selection of components are of the utmost importance. Preparation and testing of M4s should not rely on serendipity but on the judicious choice of components. In this sense, the development and use of indicators like the Hansen solubility parameters may offer great advantages in the future and should help researchers make a first selection of components rather than trying every MOF and polymer “from the self”.
In this review we have highlighted the importance of crystal engineering as a powerful tool to further enhance membrane performance. Fillers with large aspect ratios and/or in nanoparticulate form seem to be the most feasible ones to achieve improved separation performance and thin films, the latter being a must to achieve the necessary productivities for pre- and post-combustion CO2 capture.
Last but not least, separation performance under conditions relevant for practice (e.g. long-term operation at high temperature and/or pressure with realistic multicomponent mixtures) will be necessary to convince industry about the applicability of M4s. MOFs are still seen as unstable materials by a large part of the scientific community and by most industry. Although M4 literature points at a very beneficial effect of the polymer matrix into MOF stability, more proofs and long term testing under real separation conditions (both in terms of gas composition and temperature) will be required.
In summary, M4s are at the forefront of MOF and membrane research and the next few years will be crucial in the future of these thrilling composites. We are however confident that even more exciting results will be achieved by the scientific community and these should pave the way to the, perhaps, first large scale application of metal–organic frameworks.
Acknowledgements
The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 608490, project M4CO2 and (FP/2007–2013)/ERC Grant Agreement no. 335746, CrystEng-MOF-MMM (J. Gascon).Notes and references
- The Global Status of CCS: 2011 The Global CCS Institute, Canberra, Australia, 2011 Search PubMed.
- 2011 Technology Map of the European Strategic Energy Technology Plan (SET-Plan), European Commission, Joint Research Centre: Institute for Energy and Transport, © European Union, 3rd edn, 2011 Search PubMed.
- F. Princiotta, Global Climate Change – The Technology Challenge, Springer, 2011 Search PubMed.
- CCS EII Implementation Plan 2010-2012, Zero Emissions Platform, http://www.zeroemissionsplatform.eu/library.html, 2010.
- A. L. Kohl and R. Nielsen, Gas Purification, Gulf Publishing, 5th edn, 1997 Search PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- E. S. Rubin, H. Mantripragada, A. Marks, P. Versteeg, J. Kitchin, H. Mantripragada, A. Marks, P. Versteeg and J. Kitchin, Prog. Energy Combust. Sci., 2012, 38, 630–671 CrossRef CAS.
- J. Franz, P. Maas and V. Scherer, Appl. Energy, 2014, 130, 532–542 CrossRef CAS.
- B. Belaissaoui, D. Willson and E. Favre, Chem. Eng. J., 2012, 211–212, 122–132 CrossRef CAS.
- E. S. Rubin, C. Chen and A. B. Rao, Energy Policy, 2007, 35, 4444–4454 CrossRef.
- E. P. R. Institute, Program on Technology Innovation: Post-combustion CO2 Capture Technology Development, Electric Power Research Institute, Palo Alto, 2008 Search PubMed.
- L. Nurrokhmah, T. Mezher and M. R. M. Abu-Zahra, Environ. Sci. Technol., 2013, 47, 13644–13651 CrossRef CAS PubMed.
- M. Wilson, P. Tontiwachwuthikul, A. Chakma, R. Idem, A. Veawab, A. Aroonwilas, D. Gelowitz, J. Barrie and C. Mariz, Energy, 2004, 29, 1259–1267 CrossRef CAS.
- M. T. Sander and C. L. Mariz, Energy Convers. Manage., 1992, 33, 341–348 CrossRef CAS.
- Y. Li, Q. Wang and P. Wang, in Resources and Sustainable Development, Pts 1-4, ed. J. Wu, X. Lu, H. Xu and N. Nakagoshi, 2013, vol. 734–737, pp. 1881–1886 Search PubMed.
- E. V. Nikitin, A. S. Romakhin, V. A. Zagumennov and Y. A. Babkin, Electrochim. Acta, 1997, 42, 2217–2224 CrossRef CAS.
- V. Jiménez, A. Ramírez-Lucas, J. A. Díaz, P. Sánchez and A. Romero, Environ. Sci. Technol., 2012, 46, 7407–7414 CrossRef PubMed.
- J. Liu, P. K. Thallapally, B. P. Mcgrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- M. Tagliabue, D. Farrusseng, S. Valencia, S. Aguado, U. Ravon, C. Rizzo, A. Corma and C. Mirodatos, Chem. Eng. J., 2009, 155, 553–566 CrossRef CAS.
- P. Bernardo, E. Drioli and G. Golemme, Ind. Eng. Chem. Res., 2009, 48, 4638–4663 CrossRef CAS.
- R. W. Baker and K. Lokhandwala, Ind. Eng. Chem. Res., 2008, 47, 2109–2121 CrossRef CAS.
- L. M. Robeson, Curr. Opin. Solid State Mater. Sci., 1999, 4, 549–552 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- M. M. Qiu, S. T. Hwang and Y. K. Kao, Ind. Eng. Chem. Res., 1989, 28, 1670–1677 CrossRef CAS.
- R. W. Baker, Ind. Eng. Chem. Res., 2002, 41, 1393–1411 CrossRef CAS.
- M. A. Aroon, A. F. Ismail, T. Matsuura and M. M. Montazer-Rahmati, Sep. Purif. Technol., 2010, 75, 229–242 CrossRef CAS.
- S. Smart, C. X. C. Lin, L. Ding, K. Thambimuthu and J. C. Diniz da Costa, Energy Environ. Sci., 2010, 3, 268 CAS.
- A. F. Ismail and L. David, J. Membr. Sci., 2001, 193, 1–18 CrossRef CAS.
- J. Caro and M. Noack, Microporous Mesoporous Mater., 2008, 115, 215–233 CrossRef CAS.
- S. Basu, A. L. Khan, A. Cano-Odena, C. Liu and I. F. J. Vankelecom, Chem. Soc. Rev., 2010, 39, 750 RSC.
- J. Gascon, F. Kapteijn, B. Zornoza, V. Sebastian, C. Casado and J. Coronas, Chem. Mater., 2012, 24, 2829–2844 CrossRef CAS.
- N. W. Ockwig and T. M. Nenoff, Chem. Rev., 2007, 107, 4078–4110 CrossRef CAS PubMed.
- I. Agirre, P. L. Arias, H. L. Castricum, M. Creatore, J. E. ten Elshof, G. G. Paradis, P. H. T. Ngamou, H. M. van Veen and J. F. Vente, Sep. Purif. Technol., 2014, 121, 2–12 CrossRef CAS.
- C. Guizard, A. Bac, M. Barboiu and N. Hovnanian, Sep. Purif. Technol., 2001, 25, 167–180 CrossRef CAS.
- J. Coronas, Chem. Eng. J., 2010, 156, 236–242 CrossRef CAS.
- B. Zornoza, C. Tellez, J. Coronas, J. Gascon and F. Kapteijn, Microporous Mesoporous Mater., 2013, 166, 67–78 CrossRef CAS.
- B. Zornoza, S. Irusta, C. Tellez and J. Coronas, Langmuir, 2009, 25, 5903–5909 CrossRef CAS PubMed.
- C. M. Zimmerman, R. Mahajan and W. J. Koros, Abstr. Pap. Am. Chem. Soc., 1997, 214, 270 Search PubMed.
- B. Zornoza, O. Esekhile, W. J. Koros, C. Téllez and J. Coronas, Sep. Purif. Technol., 2011, 77, 137–145 CrossRef CAS.
- T.-H. Bae, J. S. Lee, W. Qiu, W. J. Koros, C. W. Jones and S. Nair, Angew. Chem., Int. Ed. Engl., 2010, 49, 9863–9866 CrossRef CAS PubMed.
- A. Galve, D. Sieffert, E. Vispe, C. Tellez, J. Coronas and C. Staudt, J. Membr. Sci., 2011, 370, 131–140 CrossRef CAS.
- T. Rodenas, in Encyclopedia of Membranes, ed. E. Drioli and L. Giorno, Springer, 2014 Search PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- G. Ferey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
- B. Zornoza, A. Martinez-Joaristi, P. Serra-Crespo, C. Tellez, J. Coronas, J. Gascon and F. Kapteijn, Chem. Commun., 2011, 47, 9522 RSC.
- T. Rodenas, M. van Dalen, E. Garcia-Perez, P. Serra-Crespo, B. Zornoza, F. Kapteijn and J. Gascon, Adv. Funct. Mater., 2014, 24, 249–256 CrossRef CAS.
- T. Rodenas, M. van Dalen, P. Serra-Crespo, F. Kapteijn and J. Gascon, Microporous Mesoporous Mater., 2014, 192, 35–42 CrossRef CAS.
- H. Yehia, T. J. Pisklak, J. P. Ferraris, K. J. Balkus and I. H. Musselman, Abstr. Pap. Am. Chem. Soc., 2004, 227, U351 Search PubMed.
- I. Erucar, G. Yilmaz and S. Keskin, Chem. – Asian J., 2013, 8, 1692–1704 CrossRef CAS PubMed.
- M. Rezakazemi, A. Ebadi Amooghin, M. M. Montazer-Rahmati, A. F. Ismail and T. Matsuura, Prog. Polym. Sci., 2014, 39, 817–861 CrossRef CAS.
- J. P. Ferraris, I. H. Musselman and K. J. Balkus Jr, in Advanced Materials for Membrane Preparation, ed. M. G. Buonomenna, Betham Science Publishers, Richardson, TX, USA, 2012, pp. 83–93 DOI:10.2174/97816080530871120101.
- H. B. Tanh Jeazet, C. Staudt and C. Janiak, Dalton Trans., 2012, 41, 14003–14027 RSC.
- W. Koros, Y. H. Ma and T. Shimidzu, Pure Appl. Chem., 1996, 68, 1479–1489 CrossRef CAS.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- H. Vinh-Thang and S. Kaliaguine, Chem. Rev., 2013, 113, 4980–5028 CrossRef CAS PubMed.
- R. M. Barrer and J. H. Petropoulos, Br. J. Appl. Phys., 1961, 12, 691 CrossRef CAS.
- E. L. Cussler, J. Membr. Sci., 1990, 52, 275–288 CrossRef CAS.
- R. H. B. Bouma, A. Checchetti, G. Chidichimo and E. Drioli, J. Membr. Sci., 1997, 128, 141–149 CrossRef CAS.
- D. Q. Vu, W. J. Koros and S. J. Miller, J. Membr. Sci., 2003, 211, 335–348 CrossRef CAS.
- J. A. Sheffel and M. Tsapatsis, J. Membr. Sci., 2007, 295, 50–70 CrossRef CAS.
- J. A. Sheffel and M. Tsapatsis, J. Membr. Sci., 2009, 326, 595–607 CrossRef CAS.
- F. Kapteijn, W. Zhu, J. A. Moulijn and T. Q. Gardner, in Structured catalysts and reactors, ed. A. Cybulski and J. A. Moulijn, CRC Taylor & Francis, Boca Raton, USA, 2nd edn, 2006, pp. 700–746 Search PubMed.
- R. Krishna and J. A. Wesselingh, Chem. Eng. Sci., 1997, 52, 861–911 CrossRef CAS.
- J. van den Bergh, J. Gascon and F. Kapteijn, in Zeolites and Catalysis: Synthesis, Reactions and Applications, ed. J. Cejka, A. Corma and S. I. Zones, 2010, vol. 1, pp. 361–386 Search PubMed.
- J. M. van de Graaf, F. Kapteijn and J. A. Moulijn, J. Membr. Sci., 1998, 144, 87–104 CrossRef CAS.
- J. M. van de Graaf, F. Kapteijn and J. A. Moulijn, AIChE J., 1999, 45, 497–511 CrossRef CAS.
- R. Krishna and J. M. van Baten, Chem. Eng. Sci., 2009, 64, 3159–3178 CrossRef CAS.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- F. Kapteijn, J. A. Moulijn and R. Krishna, Chem. Eng. Sci., 2000, 55, 2923–2930 CrossRef CAS.
- R. Baur and R. Krishna, Catal. Today, 2005, 105, 173–179 CrossRef CAS.
- M. Murthi and R. Q. Snurr, Langmuir, 2004, 20, 2489–2497 CrossRef CAS PubMed.
- S. Keskin and D. S. Sholl, Energy Environ. Sci., 2010, 3, 343–351 CAS.
- R. Mahajan and W. J. Koros, Ind. Eng. Chem. Res., 2000, 39, 2692–2696 CrossRef CAS.
- R. T. Chern, W. J. Koros, B. Yui, H. B. Hopfenberg and V. T. Stannett, J. Polym. Sci., Polym. Phys. Ed., 1984, 22, 1061–1084 CrossRef CAS.
- T. Singh, D.-Y. Kang and S. Nair, J. Membr. Sci., 2013, 448, 160–169 CrossRef CAS.
- I. Erucar and S. Keskin, Ind. Eng. Chem. Res., 2011, 50, 12606–12616 CrossRef CAS.
- I. Erucar and S. Keskin, J. Membr. Sci., 2012, 407, 221–230 CrossRef.
- G. Yilmaz and S. Keskin, Ind. Eng. Chem. Res., 2012, 51, 14218–14228 CrossRef CAS.
- G. Yilmaz and S. Keskin, J. Membr. Sci., 2014, 454, 407–417 CrossRef CAS.
- S. Cavenati, C. A. Grande and A. E. Rodrigues, Ind. Eng. Chem. Res., 2008, 47, 6333–6335 CrossRef CAS.
- E. Favre, Chem. Eng. J., 2011, 171, 782–793 CrossRef CAS.
- T. C. Merkel, H. Lin, X. Wei and R. Baker, J. Membr. Sci., 2010, 359, 126–139 CrossRef CAS.
- T. C. Merkel, R. P. Gupta, B. S. Turk and B. D. Freeman, J. Membr. Sci., 2001, 191, 85–94 CrossRef CAS.
- K. Ramasubramanian, H. Verweij and W. S. W. Ho, J. Membr. Sci., 2012, 421–422, 299–310 CrossRef CAS.
- L. Zhao, E. Riensche, R. Menzer, L. Blum and D. Stolten, J. Membr. Sci., 2008, 325, 284–294 CrossRef CAS.
- R. P. Lively, M. E. Dose, L. Xu, J. T. Vaughn, J. R. Johnson, J. A. Thompson, K. Zhang, M. E. Lydon, J. S. Lee, L. Liu, Z. Hu, O. Karvan, M. J. Realff and W. J. Koros, J. Membr. Sci., 2012, 423–424, 302–313 CrossRef CAS.
- J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried and R. D. Srivastava, Int. J. Greenhouse Gas Control, 2008, 2, 9–20 CrossRef CAS.
- S. H. Han, H. J. Kwon, K. Y. Kim, J. G. Seong, C. H. Park, S. Kim, C. M. Doherty, A. W. Thornton, A. J. Hill, Á. E. Lozano, K. A. Berchtold and Y. M. Lee, Phys. Chem. Chem. Phys., 2012, 14, 4365 RSC.
- A. Y. Ku, P. Kulkarni, R. Shisler and W. Wei, J. Membr. Sci., 2011, 367, 233–239 CrossRef CAS.
- L. Schmid, M. S. Schneider, D. Engel and A. Baiker, Catal. Lett., 2003, 88, 105–113 CrossRef CAS.
- R. Mahajan, R. Burns, M. Schaeffer and W. J. Koros, J. Appl. Polym. Sci., 2002, 86, 881–890 CrossRef CAS.
- R. Mahajan, D. Q. Vu and W. J. Koros, J. Chin. Inst. Chem. Eng., 2002, 33, 77–86 CAS.
- J. Gascon, U. Aktay, M. D. Hernandez-Alonso, G. P. M. van Klink and F. Kapteijn, J. Catal., 2009, 261, 75–87 CrossRef CAS.
- Z. Q. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- J. G. Won, J. S. Seo, J. H. Kim, H. S. Kim, Y. S. Kang, S. J. Kim, Y. M. Kim and J. G. Jegal, Adv. Mater., 2005, 17, 80–84 CrossRef CAS.
- A. Car, C. Stropnik and K. V. Peinemann, Desalination, 2006, 200, 424–426 CrossRef CAS.
- B. Zornoza, B. Seoane, J. M. Zamaro, C. Tellez and J. Coronas, ChemPhysChem, 2011, 12, 2781–2785 CrossRef CAS PubMed.
- B. Seoane, J. M. Zamaro, C. Tellez and J. Coronas, RSC Adv., 2011, 1, 917–922 RSC.
- K. Diaz, M. Lopez-Gonzalez, L. F. del Castillo and E. Riande, J. Membr. Sci., 2011, 383, 206–213 CrossRef CAS.
- R. Adams, C. Carson, J. Ward, R. Tannenbaum and W. Koros, Microporous Mesoporous Mater., 2010, 131, 13–20 CrossRef CAS.
- Y. F. Zhang, I. H. Musseman, J. P. Ferraris and K. J. Balkus, J. Membr. Sci., 2008, 313, 170–181 CrossRef CAS.
- E. V. Perez, K. J. Balkus, J. P. Ferraris and I. H. Musselman, J. Membr. Sci., 2009, 328, 165–173 CrossRef CAS.
- M. J. C. Ordonez, K. J. Balkus, Jr., J. P. Ferraris and I. H. Musselman, J. Membr. Sci., 2010, 361, 28–37 CrossRef CAS.
- S. Basu, M. Maes, A. Cano-Odena, L. Alaerts, D. E. De Vos and I. F. J. Vankelecom, J. Membr. Sci., 2009, 344, 190–198 CrossRef CAS.
- T. X. Yang, Y. C. Xiao and T. S. Chung, Energy Environ. Sci., 2011, 4, 4171–4180 CAS.
- T. X. Yang, G. M. Shi and T. S. Chung, Adv. Energy Mater., 2012, 2, 1358–1367 CrossRef CAS.
- T. Yang and T.-S. Chung, Int. J. Hydrogen Energy, 2013, 38, 229–239 CrossRef CAS.
- X. L. Liu, Y. S. Li, G. Q. Zhu, Y. J. Ban, L. Y. Xu and W. S. Yang, Angew. Chem., Int. Ed., 2011, 50, 10636–10639 CrossRef CAS PubMed.
- C. Zhang, Y. Dai, J. R. Johnson, O. Karvan and W. J. Koros, J. Membr. Sci., 2011 Search PubMed.
- D. Fritsch, K.-V. Peinemann and D. D. F. Gomes, US Pat., 7,658,784. 9, 2010 Search PubMed.
- C. Liu, B. McCulloch, S. T. Wilson, A. I. Benin and M. E. Schott, US Pat., 7637983, 2009 Search PubMed.
- K. Diaz, L. Garrido, M. Lopez-Gonzalez, L. F. del Castillo and E. Riande, Macromolecules, 2010, 43, 316–325 CrossRef CAS.
- S. Basu, A. Cano-Odena and I. F. J. Vankelecom, J. Membr. Sci., 2010, 362, 478–487 CrossRef CAS.
- J. Hu, H. P. Cai, H. Q. Ren, Y. M. Wei, Z. L. Xu, H. L. Liu and Y. Hu, Ind. Eng. Chem. Res., 2010, 49, 12605–12612 CrossRef CAS.
- T. Yang, Y. Xiao and T.-S. Chung, Energy Environ. Sci., 2011, 4, 4171–4180 CAS.
- S. Basu, A. Cano-Odena and I. F. J. Vankelecom, Sep. Purif. Technol., 2011, 81, 31–40 CrossRef CAS.
- C. Zhang, Y. Dai, J. R. Johnson, O. Karvan and W. J. Koros, J. Membr. Sci., 2012, 389, 34–42 CrossRef CAS.
- H. B. T. Jeazet, C. Staudt and C. Janiak, Chem. Commun., 2012, 48, 2140–2142 RSC.
- Y. Dai, J. R. Johnson, O. Karvan, D. S. Sholl and W. J. Koros, J. Membr. Sci., 2012, 401–402, 76–82 CrossRef CAS.
- H. Ren, J. Jin, J. Hu and H. Liu, Ind. Eng. Chem. Res., 2012, 51, 10156–10164 CrossRef CAS.
- J. A. Thompson, K. W. Chapman, W. J. Koros, C. W. Jones and S. Nair, Microporous Mesoporous Mater., 2012, 158, 292–299 CrossRef CAS.
- Q. Song, S. K. Nataraj, M. V. Roussenova, J. C. Tan, D. J. Hughes, W. Li, P. Bourgoin, M. A. Alam, A. K. Cheetham, S. A. Al-Muhtaseb and E. Sivaniah, Energy Environ. Sci., 2012, 5, 8359–8369 CAS.
- O. G. Nik, X. Y. Chen and S. Kaliaguine, J. Membr. Sci., 2012, 413, 48–61 CrossRef.
- T. Yang, G. M. Shi and T.-S. Chung, Adv. Energy Mater., 2012, 2, 1358–1367 CrossRef CAS.
- T. Li, Y. Pan, K.-V. Peinemann and Z. Lai, J. Membr. Sci., 2013, 425, 235–242 CrossRef.
- A. F. Bushell, M. P. Attfield, C. R. Mason, P. M. Budd, Y. Yampolskii, L. Starannikova, A. Rebrov, F. Bazzarelli, P. Bernardo, J. C. Jansen, M. Lanc, K. Friess, V. Shantarovich, V. Gustov and V. Isaeva, J. Membr. Sci., 2013, 427, 48–62 CrossRef CAS.
- J. Ploegmakers, S. Japip and K. Nijmeijer, J. Membr. Sci., 2013, 428, 445–453 CrossRef CAS.
- T. X. Yang and T. S. Chung, J. Mater. Chem. A, 2013, 1, 6081–6090 CAS.
- L. Ge, W. Zhou, V. Rudolph and Z. H. Zhu, J. Mater. Chem. A, 2013, 1, 6350–6358 CAS.
- S. N. Wijenayake, N. P. Panapitiya, S. H. Versteeg, C. N. Nguyen, S. Goel, K. J. Balkus, I. H. Musselman and J. P. Ferraris, Ind. Eng. Chem. Res., 2013, 52, 6991–7001 CrossRef CAS.
- B. Seoane, V. Sebastian, C. Tellez and J. Coronas, CrystEngComm, 2013, 15, 9483–9490 RSC.
- L. J. Cao, K. Tao, A. S. Huang, C. L. Kong and L. Chen, Chem. Commun., 2013, 49, 8513–8515 RSC.
- A. Kathuria, M. G. Abiad and R. Auras, Polym. Int., 2013, 62, 1144–1151 CAS.
- M. Askari and T. S. Chung, J. Membr. Sci., 2013, 444, 173–183 CrossRef CAS.
- X. Y. Chen, V. T. Hoang, D. Rodrigue and S. Kaliaguine, RSC Adv., 2013, 3, 24266–24279 RSC.
- D. S. Zhang, Z. Chang, Y. F. Li, Z. Y. Jiang, Z. H. Xuan, Y. H. Zhang, J. R. Li, Q. Chen, T. L. Hu and X. H. Bu, Sci. Rep., 2013, 3, 3312 Search PubMed.
- T. H. Bae and J. R. Long, Energy Environ. Sci., 2013, 6, 3565–3569 CAS.
- S. Sorribas, B. Zornoza, C. Tellez and J. Coronas, J. Membr. Sci., 2014, 452, 184–192 CrossRef CAS.
- V. Nafisi and M. B. Hagg, J. Membr. Sci., 2014, 459, 244–255 CrossRef CAS.
- F. Dorosti, M. Omidkhah and R. Abedini, Chem. Eng. Res. Des., 2014, 92, 2439–2448 CrossRef CAS.
- P. Burmann, B. Zornoza, C. Tellez and J. Coronas, Chem. Eng. Sci., 2014, 107, 66–75 CrossRef CAS.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- R. Abedini, M. Omidkhah and F. Dorosti, Int. J. Hydrogen Energy, 2014, 39, 7897–7909 CrossRef CAS.
- V. Nafisi and M. B. Hagg, Sep. Purif. Technol., 2014, 128, 31–38 CrossRef CAS.
- M. Valero, B. Zornoza, C. Tellez and J. Coronas, Microporous Mesoporous Mater., 2014, 192, 23–28 CrossRef CAS.
- C. Zhang, K. Zhang, L. Xu, Y. Labreche, B. Kraftschik and W. J. Koros, AIChE J., 2014, 60, 2625–2635 CrossRef CAS.
- J. A. Thompson, J. T. Vaughn, N. A. Brunelli, W. J. Koros, C. W. Jones and S. Nair, Microporous Mesoporous Mater., 2014, 192, 43–51 CrossRef CAS.
- S. Shahid and K. Nijmeijer, J. Membr. Sci., 2014, 459, 33–44 CrossRef CAS.
- A. Bhaskar, R. Banerjee and U. Kharul, J. Mater. Chem. A, 2014, 2, 12962–12967 CAS.
- R. Abedini, M. Omidkhah and F. Dorosti, RSC Adv., 2014, 4, 36522–36537 RSC.
- J. O. Hsieh, K. J. Balkus, J. P. Ferraris and I. H. Musselman, Microporous Mesoporous Mater., 2014, 196, 165–174 CrossRef CAS.
- M. Arjmandi and M. Pakizeh, J. Ind. Eng. Chem., 2014, 20, 3857–3868 CrossRef CAS.
- C. J. Duan, G. D. Kang, D. D. Liu, L. N. Wang, C. Jiang, Y. M. Cao and Q. Yuan, J. Appl. Polym. Sci., 2014, 131, 40719 Search PubMed.
- C. J. Duan, X. M. Jie, D. D. Liu, Y. M. Cao and Q. Yuan, J. Membr. Sci., 2014, 466, 92–102 CrossRef CAS.
- S. Japip, H. Wang, Y. C. Xiao and T. S. Chung, J. Membr. Sci., 2014, 467, 162–174 CrossRef CAS.
- S. Y. Lim, J. Choi, H. Y. Kim, Y. Kim, S. J. Kim, Y. S. Kang and J. Won, J. Membr. Sci., 2014, 467, 67–72 CrossRef CAS.
- S. Shahid and K. Nijmeijer, J. Membr. Sci., 2014, 470, 166–177 CrossRef CAS.
- L. X. Li, J. F. Yao, X. J. Wang, Y. B. Cheng and H. T. Wang, J. Appl. Polym. Sci., 2014, 131, 41056 Search PubMed.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. Llabrés i Xamena and J. Gascon, Nat. Mater., 2015, 14, 48–55 CrossRef CAS PubMed.
- N. A. H. M. Nordin, A. F. Ismail, A. Mustafa, R. S. Murali and T. Matsuura, RSC Adv., 2014, 4, 52530–52541 RSC.
- M. Fang, C. Wu, Z. Yang, T. Wang, Y. Xia and J. Li, J. Membr. Sci., 2015, 474, 103–113 CrossRef CAS.
- X. Y. Chen, H. Vinh-Thang, D. Rodrigue and S. Kaliaguine, Ind. Eng. Chem. Res., 2012, 51, 6895–6906 CrossRef CAS.
- B. Seoane, C. Tellez, J. Coronas and C. Staudt, Sep. Purif. Technol., 2013, 111, 72–81 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- R. W. Baker, Membrane Technology and Applications, John Wiley & Sons, England, 2nd edn, 2004 Search PubMed.
- M. F. A. Wahab, A. F. Ismail and S. J. Shilton, Sep. Purif. Technol., 2012, 86, 41–48 CrossRef CAS.
- S. Husain and W. J. Koros, J. Membr. Sci., 2007, 288, 195–207 CrossRef CAS.
- N. Peng, N. Widjojo, P. Sukitpaneenit, M. M. Teoh, G. G. Lipscomb, T.-S. Chung and J.-Y. Lai, Prog. Polym. Sci., 2012, 37, 1401–1424 CrossRef CAS.
- T.-S. Chung, L. Y. Jiang, Y. Li and S. Kulprathipanja, Prog. Polym. Sci., 2007, 32, 483–507 CrossRef CAS.
- N. Nordin, A. Ismail and A. Mustafa, Jurnal Teknologi, 2014, 69, 73–76 CrossRef.
- S. Loeb and S. Sourirajan, Saline Water Conversion-II, ACS, Washington, DC, USA, 1963, ch. 9, vol. 38, pp. 117–132 Search PubMed.
- P. S. Goh, B. C. Ng, A. F. Ismail, M. Aziz and Y. Hayashi, J. Colloid Interface Sci., 2012, 386, 80–87 CrossRef CAS PubMed.
- C. Cao, T. S. Chung, S. B. Chen and Z. J. Dong, Chem. Eng. Sci., 2004, 59, 1053–1062 CrossRef CAS.
- L. Y. Jiang, T. S. Chung, C. Cao, Z. Huang and S. Kulprathipanja, J. Membr. Sci., 2005, 252, 89–100 CrossRef CAS.
- J. J. Qin, R. Wang and T. S. Chung, J. Membr. Sci., 2000, 175, 197–213 CrossRef CAS.
- N. Widjojo, T. S. Chung and S. Kulprathipanja, J. Membr. Sci., 2008, 325, 326–335 CrossRef CAS.
- N. Widjojo, S. D. Zhang, T.-S. Chung and Y. Liu, J. Membr. Sci., 2007, 306, 147–158 CrossRef CAS.
- Y. Xiao, K. Y. Wang, T.-S. Chung and J. Tan, Chem. Eng. Sci., 2006, 61, 6228–6233 CrossRef CAS.
- O. M. Ekiner and G. Vassilatos, J. Membr. Sci., 1990, 53, 259–273 CrossRef CAS.
- I. Pinnau, J. G. Wijmans, I. Blume, T. Kuroda and K. V. Peinemann, J. Membr. Sci., 1988, 37, 81–88 CrossRef CAS.
- K. Kimmerle, T. Hofmann and H. Strathmann, J. Membr. Sci., 1991, 61, 1–17 CrossRef CAS.
- Y. Li, T. S. Chung, Z. Huang and S. Kulprathipanja, J. Membr. Sci., 2006, 277, 28–37 CrossRef CAS.
- O. M. Ekiner and S. S. Kulkarni, US, 6,663,805, 2003 Search PubMed.
- D. Bastani, N. Esmaeili and M. Asadollahi, J. Ind. Eng. Chem., 2013, 19, 375–393 CrossRef CAS.
- G. S. Sur, H. L. Sun, S. G. Lyu and J. E. Mark, Polymer, 2001, 42, 9783–9789 CrossRef CAS.
- Y. Zhang, K. J. Balkus Jr, I. H. Musselman and J. P. Ferraris, J. Membr. Sci., 2008, 325, 28–39 CrossRef CAS.
- M. Moaddeb and W. J. Koros, J. Membr. Sci., 1997, 125, 143–163 CrossRef CAS.
- S. Mitchell, N.-L. Michels, K. Kunze and J. Perez-Ramirez, Nat. Chem., 2012, 4, 825–831 CrossRef CAS PubMed.
- G. Prieto, in Encyclopedia of Membranes, ed. E. Drioli and L. Giorno, Springer, 2014 Search PubMed.
- G. M. Shi, T. X. Yang and T. S. Chung, J. Membr. Sci., 2012, 415, 577–586 CrossRef.
- B. H. Jeong, E. M. V. Hoek, Y. S. Yan, A. Subramani, X. F. Huang, G. Hurwitz, A. K. Ghosh and A. Jawor, J. Membr. Sci., 2007, 294, 1–7 CrossRef CAS.
- S. Sorribas, P. Gorgojo, C. Tellez, J. Coronas and A. G. Livingston, J. Am. Chem. Soc., 2013, 135, 15201–15208 CrossRef CAS PubMed.
- C. M. Hansen, Prog. Org. Coat., 2004, 51, 77–84 CrossRef CAS.
- C. M. Hansen, Eur. Polym. J., 2008, 44, 2741–2748 CrossRef CAS.
- K. Srinivas, J. W. King, J. K. Monrad, L. R. Howard and C. M. Hansen, J. Food Sci., 2009, 74, E342–E354 CrossRef CAS PubMed.
- C. Bordes, V. Freville, E. Ruffin, P. Marote, J. Y. Gauvrit, S. Briancon and P. Lanteri, Int. J. Pharm., 2010, 383, 236–243 CrossRef CAS PubMed.
- J. U. Wieneke, B. Kommoss, O. Gaer, I. Prykhodko and M. Ulbricht, Ind. Eng. Chem. Res., 2012, 51, 327–334 CrossRef CAS.
- P. Vandezande, X. F. Li, L. E. M. Gevers and I. F. J. Vankelecom, J. Membr. Sci., 2009, 330, 307–318 CrossRef CAS.
- G. X. Dong, H. Y. Li and V. Chen, J. Membr. Sci., 2010, 353, 17–27 CrossRef CAS.
- P. S. Yune, J. E. Kilduff and G. Belfort, J. Membr. Sci., 2011, 370, 166–174 CrossRef CAS.
- Z. H. Yang, G. L. Zhang, F. N. Liu and Q. Meng, Ind. Eng. Chem. Res., 2010, 49, 11594–11601 CrossRef CAS.
- H. L. Zhou, Y. Su, X. R. Chen and Y. H. Wan, Sep. Purif. Technol., 2011, 79, 375–384 CrossRef CAS.
- D. Elangovan, U. Nidoni, I. E. Yuzay, S. E. M. Selke and R. Auras, Ind. Eng. Chem. Res., 2011, 50, 11136–11142 CrossRef CAS.
- L. Paseta, G. Potier, S. Abbot and J. Coronas, Org. Biomol. Chem., 2015, 13, 1724–1731 CAS.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. D. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- C. Serre, F. Millange, C. Thouvenot, M. Nogues, G. Marsolier, D. Louer and G. Ferey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- B. Arstad, H. Fjellvag, K. O. Kongshaug, O. Swang and R. Blom, Adsorption, 2008, 14, 755–762 CrossRef CAS.
- S. Bauer, C. Serre, T. Devic, P. Horcajada, J. Marrot, G. Ferey and N. Stock, Inorg. Chem., 2008, 47, 7568–7576 CrossRef CAS PubMed.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- C. Volkringer and T. Loiseau, Mater. Res. Bull., 2006, 41, 948–954 CrossRef CAS.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, CRC Press, Boca Raton, 2007 Search PubMed.
- H. W. Milliman, D. Boris and D. A. Schiraldi, Macromolecules, 2012, 45, 1931–1936 CrossRef CAS.
- S. Abbot, C. M. Hansen and H. Yamamoto, Available from http://www.hansen-solubility.com, 4th edn, accessed October 2014.
- L. M. Liz-Marzan, Langmuir, 2006, 22, 32–41 CrossRef CAS PubMed.
- G. Q. Ding, W. Z. Shen, M. J. Zheng, W. L. Xu, Y. L. He and Q. X. Guo, J. Cryst. Growth, 2005, 283, 339–345 CrossRef CAS.
- E. V. Ramos-Fernandez, C. Pieters, B. van der Linden, J. Juan-Alcañiz, P. Serra-Crespo, M. W. G. M. Verhoeven, H. Niemantsverdriet, J. Gascon and F. Kapteijn, J. Catal., 2012, 289, 42–52 CrossRef CAS.
- A. Quintanilla, M. Valvo, U. Lafont, E. M. Kelder, M. T. Kreutzer and F. Kapteijn, Chem. Mater., 2010, 22, 1656–1663 CrossRef CAS.
- S. H. Jhung, J. H. Lee, J. W. Yoon, C. Serre, G. Férey and J. S. Chang, Adv. Mater., 2007, 19, 121–124 CrossRef CAS.
- A. Martinez Joaristi, J. Juan-Alcañiz, P. Serra-Crespo, F. Kapteijn and J. Gascon, Cryst. Growth Des., 2012, 12, 3489–3498 CAS.
- A. Umemura, S. Diring, S. Furukawa, H. Uehara, T. Tsuruoka and S. Kitagawa, J. Am. Chem. Soc., 2011, 133, 15506–15513 CrossRef CAS PubMed.
- H. K. Jeong, W. Krych, H. Ramanan, S. Nair, E. Marand and M. Tsapatsis, Chem. Mater., 2004, 16, 3838–3845 CrossRef CAS.
- G. Roberts, Langmuir-Blodgett films, Plenum Press, New York, 1990 Search PubMed.
- M. H. Wang, V. Janout and S. L. Regen, Acc. Chem. Res., 2013, 46, 2743–2754 CrossRef CAS PubMed.
- M. D. Conner, V. Janout, I. Kudelka, P. Dedek, J. Y. Zhu and S. L. Regen, Langmuir, 1993, 9, 2389–2397 CrossRef CAS.
- M. H. Wang, S. Yi, V. Janout and S. L. Regen, Chem. Mater., 2013, 25, 3785–3787 CrossRef CAS.
- M. Tsotsalas, A. Umemura, F. Kim, Y. Sakata, J. Reboul, S. Kitagawa and S. Furukawa, J. Mater. Chem., 2012, 22, 10159–10165 RSC.
- G. Lu, C. L. Cui, W. N. Zhang, Y. Y. Liu and F. W. Huo, Chem. – Asian J., 2013, 8, 69–72 CrossRef CAS PubMed.
- H. Xu and W. A. Goedel, Langmuir, 2002, 18, 2363–2367 CrossRef CAS.
- C. C. Ho, P. Y. Chen, K. H. Lin, W. T. Juan and W. L. Lee, ACS Appl. Mater. Interfaces, 2011, 3, 204–208 CAS.
- B. Martin-Garcia and M. M. Velazquez, Langmuir, 2014, 30, 509–516 CrossRef CAS PubMed.
- J. Y. Kim, S. Raja and F. Stellacci, Small, 2011, 7, 2526–2532 CAS.
- Y. C. Pan, B. Wang and Z. P. Lai, J. Membr. Sci., 2012, 421, 292–298 CrossRef.
- A. J. Brown, N. A. Brunelli, K. Eum, F. Rashidi, J. R. Johnson, W. J. Koros, C. W. Jones and S. Nair, Science, 2014, 345, 72–75 CrossRef CAS PubMed.
- A. J. Brown, J. R. Johnson, M. E. Lydon, W. J. Koros, C. W. Jones and S. Nair, Angew. Chem., Int. Ed., 2012, 51, 10615–10618 CrossRef CAS PubMed.
- M. Pera-Titus, R. Mallada, J. Llorens, F. Cunill and J. Santamaria, J. Membr. Sci., 2006, 278, 401–409 CrossRef CAS.
- F. Cacho-Bailo, S. Catalán-Aguirre, M. Etxeberría-Benavides, O. Karvan, V. Sebastian, C. Téllez and J. Coronas, J. Membr. Sci., 2015, 476, 277–285 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cs00437j |
| This journal is © The Royal Society of Chemistry 2015 |