
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoparticle colloidal stability in cell culture media and impact on cellular interactions†
Thomas L.
Moore
*a,
Laura
Rodriguez-Lorenzo
a,
Vera
Hirsch
a,
Sandor
Balog
a,
Dominic
Urban
a,
Corinne
Jud‡
a,
Barbara
Rothen-Rutishauser
a,
Marco
Lattuada§
a and
Alke
Petri-Fink§
*ab
aAdolphe Merkle Institute, University of Fribourg, 1700 Fribourg, Switzerland. E-mail: thomaslee.moore@unifr.ch; Tel: +41 26 300 9516
bChemistry Department, University of Fribourg, 1700 Fribourg, Switzerland. E-mail: alke.fink@unifr.ch; Tel: +41 26 300 9501
First published on 9th June 2015
Abstract
Nanomaterials are finding increasing use for biomedical applications such as imaging, diagnostics, and drug delivery. While it is well understood that nanoparticle (NP) physico-chemical properties can dictate biological responses and interactions, it has been difficult to outline a unifying framework to directly link NP properties to expected in vitro and in vivo outcomes. When introduced to complex biological media containing electrolytes, proteins, lipids, etc., nanoparticles (NPs) are subjected to a range of forces which determine their behavior in this environment. One aspect of NP behavior in biological systems that is often understated or overlooked is aggregation. NP aggregation will significantly alter in vitro behavior (dosimetry, NP uptake, cytotoxicity), as well as in vivo fate (pharmacokinetics, toxicity, biodistribution). Thus, understanding the factors driving NP colloidal stability and aggregation is paramount. Furthermore, studying biological interactions with NPs at the nanoscale level requires an interdisciplinary effort with a robust understanding of multiple characterization techniques. This review examines the factors that determine NP colloidal stability, the various efforts to stabilize NP in biological media, the methods to characterize NP colloidal stability in situ, and provides a discussion regarding NP interactions with cells.
 Thomas L. Moore | Thomas Moore studied Bioengineering at Clemson University in South Carolina, USA and received his BSc in 2009. He continued at Clemson University in Bioengineering in the Nanomedicine laboratory under the direction of Prof. Frank Alexis, and received his PhD in 2013. His dissertation was related to theranostic nanoparticles to measure drug delivery to cancer. He joined the BioNanomaterials group in the Adolphe Merkle Institute at the University of Fribourg, Switzerland in 2014 as a postdoctoral researcher. His research interests are nanomedicine, studying the interactions of nanomaterials with biological systems, and studying the colloidal stability of nanoparticles in complex biological media. |
 Laura Rodriguez-Lorenzo | Laura Rodriguez-Lorenzo studied Chemistry at the University of Vigo in Spain. She received her PhD from the University of Vigo in 2012, under the direction of Professors Luis M. Liz-Marzan and Ramon A. Alvarez-Puebla. Her thesis was related to plasmonics, surface-enhanced Raman scattering (SERS) and their application in sensing. Since 2012, she has been a postdoctoral researcher in the BioNanomaterials group at the Adolphe Merkle Institute in Fribourg, Switzerland. Her present research interests mainly focus on the synthesis of metallic nanostructures and hybrid composites, self-assembly, sensing application, plasmonics and surface-enhanced spectroscopies. |
 Sandor Balog | Sandor Balog studied physics at the University of Debrecen, Hungary, and worked at the European Organization for Nuclear Research (CERN) before receiving his MSc in 2003. In 2004 he began at the University of Fribourg, Switzerland, under the direction of Prof. Frank Scheffold, and received his PhD in 2007. He continued at the École Polytechnique Fédérale de Lausanne (EPFL) and the Paul Scherrer Institute, and joined the Adolphe Merkle Institute, Switzerland in 2012 as a senior scientist. He is dedicated to explore and understand diverse aspects of physical phenomena regarding the interactions of man-made and engineered nanomaterials with biological structures. |
 Barbara Rothen-Rutishauser | Prof. Barbara Rothen-Rutishauser received her PhD in Cell Biology in 1996 from the Swiss Federal Institute of Technology in Zurich (ETHZ), Switzerland. Afterwards, she worked for ten years in the research group of Prof. Gehr at the Institute of Anatomy, the University of Bern, Switzerland. Since 2011, she became the new chair in BioNanomaterials at the Adolphe Merkle Institute, the University of Fribourg, Switzerland, a position shared equally with Prof. Alke Petri-Fink. B. Rothen-Rutishauser is an expert in the field of cell–nanoparticle interactions in the lung, with a special focus on 3D lung cell models. |
 Marco Lattuada | Prof. Marco Lattuada received his MS degree in 1998 from the Politecnico di Milano, Italy and his PhD in Chemical Engineering at the Swiss Federal Institute of Technology in Zurich (ETHZ). After two years of post-doctoral work at MIT, he came back to ETHZ as a senior scientist in 2006. In 2012, he moved to the Adolphe Merkle Institute, the University of Fribourg, Switzerland, as an Associate Swiss National Science Foundation Professor. His research is dedicated to the preparation and engineering of novel nanoparticles and to understanding their self-assembly behavior, with the final goal of designing novel materials. |
 Alke Petri-Fink | Prof. Alke Petri-Fink received her PhD in Chemistry from the University of Ulm, Germany in 1999. After a post-doctoral stay at the University of Gainesville, Florida, she joined the Institute of Materials Science at the École Polytechnique Fédérale de Lausanne (EPFL), first as a post-doctoral researcher, then as a senior scientist. She became an Associate Swiss National Science Foundation Professor in the Department of Chemistry at the University of Fribourg in 2009, and a Full Professor in 2011 at the Adolphe Merkle Institute, Switzerland. Her research focuses on inorganic nanoparticles, their synthesis, surfaces, and interactions with biological cells. |
1. Introduction
The turn of the century saw a dramatic rise in the amount of research reporting the use of nanotechnology for biomedical purposes such as implant coatings, imaging agents, diagnostics, drug delivery systems, and other therapies.1,2 Nanoparticles (NPs) used in medicine, nanomedicine, are appealing not only because the nano-size results in unique physico-chemical properties, which can mediate biological response (e.g. the enhanced permeability and retention (EPR) effect can improve uptake of drug delivery NPs into tumors, nano-textured surfaces can improve implant performance, superparamagnetic NPs can enhance MRI contrast), but NPs also offer distinct advantages for technology commercialization (e.g. enabling new indications for drugs, rediscovering drugs that may have previously failed clinically, providing stronger intellectual property protection).1,3,4 The development of the field nanomedicine also saw a concurrent growth in new analytical techniques and instruments for characterization. Thus, the excitement surrounding nanotechnology and nanomedicine resulted in large global investments into the field. The U.S. are predicted to invest $1.5 billion to various agencies through the National Nanotechnology Initiative (NNI) in 2015, and the NNI annual budget grew from $0.5 billion in 2001 to almost $2.0 billion in 2010. Under the Seventh Framework Programme funding initiative (FP7), the European Commission invested €2.5 billion (approximately $3.4 billion) from 2007–2011 and investment into nanotechnologies is explicitly outlined in the European Union's largest ever research endeavor (€80 billion over 7 years), the Horizon 2020 program. However despite the surge in funding and the significant investigation into NPs used for biomedical applications, their translation into clinical use has encountered variable success.5,6 There still exists a wide gap in the number of investigational reports, and those formulations that enter clinical trials. This gap is of course a complex matter influenced by many factors. However, it may in part be due to difficulty in understanding and controlling the most fundamental nanoparticle (NP) interactions with the biological environment. As more nanomedicines enter clinical trials, regulatory agencies require more physico-chemical characterization and fundamental understanding in order to determine how the physico-chemical properties influence biological behavior.7 To this point, it is critical to fully understand how NP properties and more importantly colloidal behavior in biological solutions such as cell culture medium or body fluids impact cellular interactions and cell responses.Upon contact with physiological fluids, NPs will interface with a wide range of biomacromolecules.8 In the past many studies have focused on the interplay between the intrinsic properties of the NPs and the components of the physiological environment, and the subsequent cellular responses towards the NPs in vitro.9–12 One factor that continues to be understated is the colloidal stability of NPs in biological media. In this complex environment NP aggregation is a common phenomenon.13 Aggregation refers to the usually irreversible inter-particular adherence, which leads to the formation of large and irregularly shaped clusters.14,15 NP aggregation can lead to misrepresentative results and impedes experimental reproducibility by changing the cellular uptake and the toxicity profile of the particles.11,16,17 Despite the increasing use of in vitro systems to evaluate cellular responses towards NPs, the possible change of their colloidal properties upon suspension in physiological fluids is rarely taken into consideration.16,18–20 This is even more surprising as aggregation of NPs has been shown to directly (i.e. due to their increased size) and indirectly (i.e. due to their altered diffusion and sedimentation velocities) influence the in vitro cellular response.
In a broader context, understanding the factors that influence NP colloidal stability and aggregation when introduced into biological media is important for the development of safe and effective nano-therapeutics for clinical use. NP colloidal stability, or lack thereof, will determine their biodistribution, pharmacokinetics, and systemic toxicity in vivo.21,22 For example Aoki et al.23 found in an in vivo toxicity study of nano-hydroxyapatite administered at high doses intravenously to Wistar rats that the cause of morbidity was likely NP aggregation and subsequent capillary blockage in the lungs. Similar studies have shown that NP aggregation and pulmonary capillary blockage can lead to morbidity in vivo.24–26 Furthermore, studies have shown that NPs which are stable in blood have a prolonged circulation half-life, and are less likely to be rapidly cleared by the body's reticuloendothelial system (RES).27–29 In most cases the in situ formation of aggregates for intravenously administered nano-therapeutics is undesirable as this will lead to rapid clearance in the liver and RES, thereby limiting the probability that the NPs reach their therapeutic targets. Thus, a fundamental understanding of NP aggregation is important to rationally design NP whereby it can be understood how they may behave at molecular, cellular, and systemic levels within biological systems.
The purpose of this review is to draw the attention of the nano-community to the impact of colloidal interactions of NPs in physiological media used for cell cultures: What are the factors affecting NP colloidal stability, which methodologies are used for characterization, and what are key challenges when undertaking any in vitro studies?
2. Colloids – theoretical considerations
Colloidal materials are composed of at least two phases, where one solid phase is dispersed in the second, frequently liquid, phase. The dimensions corresponding to the dispersed phase are considered to be in the sub-microscopic region, yet well above the atomic size range. At these sizes NPs are able to interact with biological systems on a sub-cellular level.2.1 Colloidal behavior in water
Aqueous solutions, containing simple electrolytes, are the only systems where colloidal stability of NPs can be effectively quantified. Colloidal stability is determined by inter-particle behavior resulting from intermolecular and surface forces (e.g. van der Waals (vdW) forces, the repulsive electrostatic double layer (EDL), and structural forces such as depletion attraction). The balance of these forces determines the colloidal stability of NPs in an aqueous suspension. The attractive vdW forces result from the interaction of induced, instantaneous, or permanent dipoles in the interatomic bonds of NPs, which destabilize a colloidal system (Fig. 1a).30–32 The attractive vdW forces are repulsed by the EDL of the NPs, which stabilizes the dispersion. Most NPs carry some surface charge in aqueous environments due to the ionization/dissociation of surface groups, or due to the adsorption of charged molecules or ions to the particle surface. The net charge of the system is balanced by the formation of a cloud of counterions around the particle. This cloud is composed of the Stern layer, which consists of counterions adsorbed on the charged surface of the NP, and the diffuse layer, an atmosphere of ions of opposite net charge surrounding the NP. Characterization of nanoparticle surface charge is commonly done in terms of the particle zeta potential, which is the electrostatic potential of the particles measured at the so-called shear plane, i.e. at the distance from the surface where ions are not bound to the particle. Overlapping EDLs of two like-particles induces a repulsive force caused by the osmotic interactions between counterions (Fig. 1b).32–37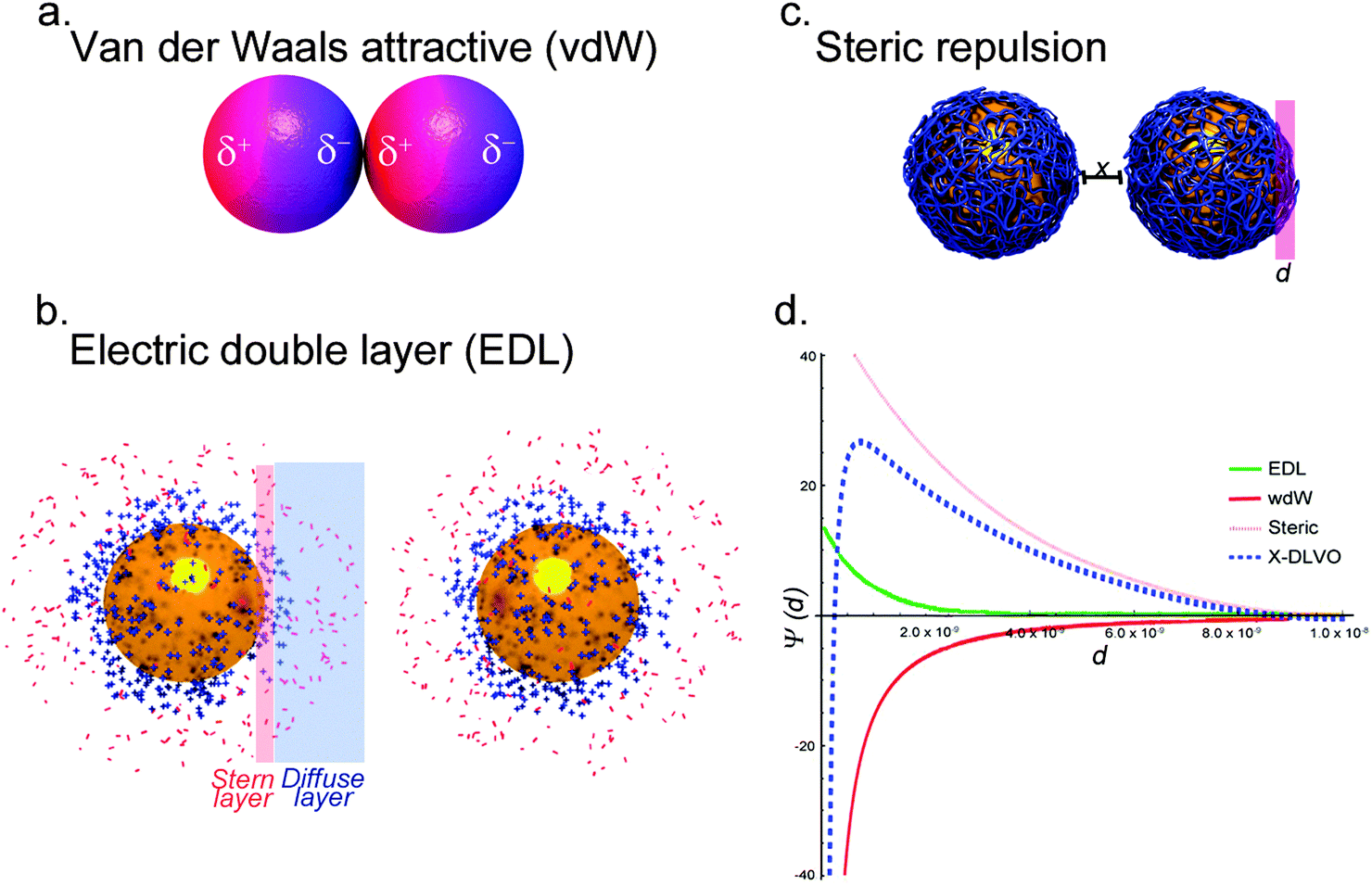 | ||
| Fig. 1 Colloidal interactions. (a) At the most basic level, NP aggregation is governed by van der Waals interactions. Permanent or induced dipoles within the NP can result in net attractive forces between NPs and subsequent aggregation. (b) Macromolecules, such as proteins or polymers, can physically stabilize NP. Hydrophilic macromolecules can shield the NP from interaction, and steric stabilization can be combined with charge stabilization for electro-steric stabilization. (c) The inherent surface charge of NPs, caused by surface ions or functional groups, results in the formation of the stern layer, oppositely charged ions adsorbed to the NP surface. Ions with the opposite charge from the stern layer form the diffuse layer, and all together this is referred to as the electric double layer (EDL). The EDL forms a net charge, and when two like-particles are in close proximity the EDL repels the two. (d) Illustration of potential energy distance curves. They can be used to predict the contribution of the different stabilization approaches, and model the stability of NP in solution. | ||
A classical theoretical approach to determine the state of a colloid can be found in the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory. The theory uses the superposition of attractive vdW and repulsive forces caused by the EDL to predict the stability of a colloidal system.33,38 According to the classical DLVO theory, colloidal stability increases with increasing net surface charge. For particles with amphoteric surfaces, e.g. metal oxides, the surface charge of a particle is dependent on the pH of the suspension. At pH values near the isoelectric point or the point of zero charge of NPs the overall surface charge of the particles tends to be neutral. As a consequence the repulsion of the NPs, caused by the EDL, decreases in favor of attractive vdW forces, promoting aggregation.39 The EDL can also be influenced by the ionic strength of the suspension. High ion concentrations result in a compression of the EDL, which may further induce the collapse of colloidal systems.40
In addition to the aforementioned forces, depending on the composition of the colloidal material, other contributions to the stability of the system have to be included. The adsorption of macromolecules, such as synthetic polymers or biopolymers, onto the surface of NPs can lead to a steric stabilization of the colloid. Steric stabilization provides a powerful tool to enhance the dispersion state of NPs under otherwise harsh conditions by preventing two particles from forming attractive vdW interactions due to osmotic pressure and elastic recoil effects (Fig. 1c). Osmotic pressure acts at longer distances, whereas elastic recoil effects dominate at shorter distances. The magnitude of these interactions further depends on the grafting density of the macromolecules to the surface of the particle and its interaction with the dispersion media.41–43 It has been shown that by using charged macromolecules or proteins, the stabilization of colloidal systems could be improved due to the combined electrosteric repulsion.44–46 However, steric stabilization is only effective if a uniform polymer coating is present on the particle surface. In the case of a patchy polymer layer, steric stabilization is not effective. In fact, it is worth noting that macromolecules can also introduce aggregation of NPs due to bridging effects, where a polymer chain of high molecular weight interacts with more than one NP and therefore leads to the formation of aggregates. Bridging flocculation is based on the strong interaction of, for example, a polymer with the NP surface, and occurs in the presence of very high molecular weight and not too high concentrations of macromolecules in the suspension where the amount of polymer chains is not sufficient to completely cover the surface of each particle.47
Besides electrosteric stabilization, other repulsive forces (e.g. hydration forces), as well as specific attractive forces (e.g. depletion, hydrophobic or magnetic forces) contribute to the stabilization or destabilization of a colloidal system.48 The resulting extended DLVO (X-DLVO) theory is the addition of these individual intermolecular and surface forces assuming that each of these interactions is totally independent of the other. By superimposing these various influences into an energy–distance curve, it is possible to understand and predict the behavior of colloids. An example of such an energy–distance plot is shown in Fig. 1d. Depending on the influencing forces and their magnitude, primary minima, secondary minima as well as energy barriers (maxima) can be predicted. Minima depict particle distances where flocculation or aggregation will occur, whereas maxima represent energy barriers, which prevent particles from coming into contact.49,50
The current theories might fail to describe all systems in general, be it due to the incomplete description of the colloidal systems, the difficulty in obtaining the correct parameters required for the calculations, or due to the fact that the models neglect certain physical phenomena.51 However, the DLVO theory and its extended versions have significantly advanced our understanding of colloidal systems, and have been applied in a number of studies of different research fields, such as ceramic processing,52 industrial separation processes, microbial adhesion,53 virus adsorption to surfaces,54,55 transport of colloids in groundwater,56 and aquatic environments.57,58
When it comes to evaluating NP interactions with cellular systems the colloidal behavior of the respective materials in idealized, simplified aqueous solutions does not fit these complex physiological environments. Due to the high ionic strength and presence of macromolecules (e.g. proteins), understanding the effect of physiological fluid composition on colloidal stability is paramount.59,60
2.2 Colloidal interactions in cell culture medium
The influence of physico-chemical properties of NPs on cellular interaction is routinely assessed using in vitro systems, where NPs interact with the components of the present cell culture medium prior to any cellular contact.61,62 The complexity of the environment a NP is confronted with once suspended in the protein-supplemented and electrolyte rich cell culture medium (CCM) is illustrated in Fig. 2.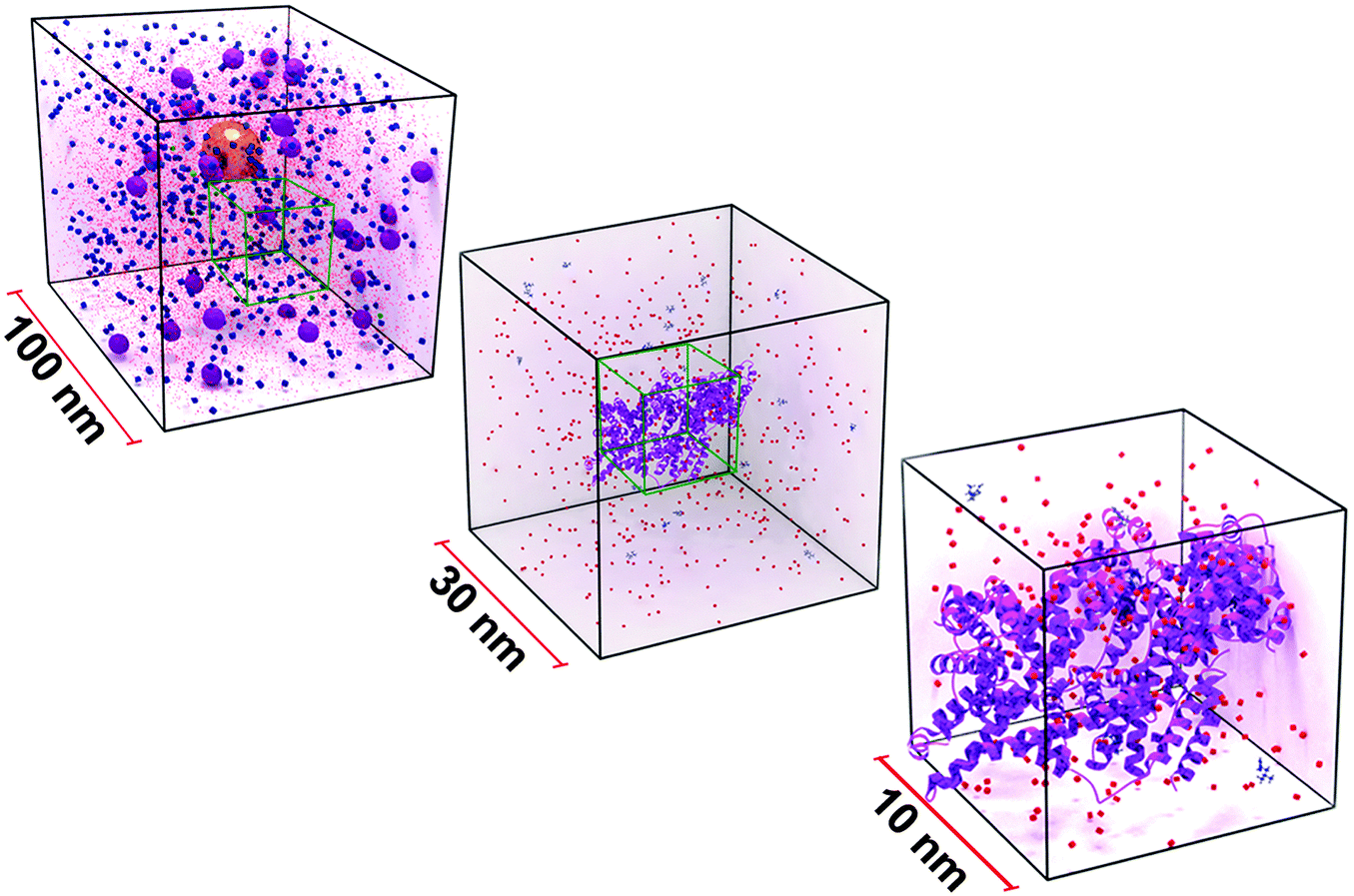 | ||
| Fig. 2 Scaled illustration of a gold NP suspended in 10% serum-supplemented cell culture medium. The boxes represent the volume of medium at different size scales. The gold NP can be seen in the 100 nm3, enveloped by serum proteins (violet spheres). | ||
The environment that the NPs, represented as a 40 nm gold sphere, are found in when delivered into cell culture media is shown. Medium is a buffered solution that is principally comprised of proteins such as serum albumin or globulins (violet spheres), and a number of biomolecules including amino acids (blue spheres), and ionic salts (red dots). These constituents influence the hydrodynamic behavior of NPs. Stabilized NPs can also become unstable through molecule/protein adsorption or loss of surface functionality, resulting in the formation of aggregates. These processes can further influence the in vitro behavior and significantly alter NP mobility.
The impact of this complex biological fluid on the colloidal behavior of the NPs is, however, rarely or insufficiently taken into consideration despite the fact that changes in the dispersion state of NPs have been shown to influence the ensuing cellular interaction and response.11,16,18,20,63 Admittedly, experimental assessment of the colloidal stability in the biological environment is challenging and the multitude of different CCM with a range of different components further contributes to the complexity. When NPs are delivered into cell culture media, their colloidal and chemical properties are not only altered due to the presence of proteins but also because of the high ion content (Fig. 2). Studies addressing the influence of electrolyte ionic strength on colloidal stability recognized quite early that stability can collapse due to screening of the electrostatic interactions.64 This screening can result in aggregation, and multivalent electrolytes were found to be more efficient than monovalent ions at suppressing the stabilizing effect of the electric double layer (EDL).65 Furthermore, the ionic strength and nature of electrolytes were reported to define the morphology and the rate of aggregate formation.66 In the case of polyelectrolytes adsorbed onto nanoparticles, the pH-induced conformational change was found to be significant for colloidal stability.67 Considering chemical changes, it has been shown that the surface charge of carboxyl and amine terminated NPs depends on pH due to the equilibrium of protonation (COOH or NH3+)–deprotonation (COO– or NH2) of these functional groups.68 Alternatively, several studies have demonstrated that NPs, such as Ag, quantum dots or ZnO NPs, can release ions and that the rate of ion release is influenced by particle size, particle functionalization, and the local environment.69–72 Moreover this degradation has been correlated with their cytotoxicity (e.g., the ability to generate reactive oxygen species).73
While this review focuses on NP colloidal stability in CCM, the effect of the intracellular environment cannot be discounted. The majority of NPs are taken up via endocytotic mechanisms (for reviews see ref. 74–76) during which they are exposed to varying conditions, such as pH changes from 7.4 (in the extracellular medium), to 5.5 (in late endosomes), and finally to 4.5 (in lysosomes).77 This altered environment can have several consequences, for example it was shown that bound or adsorbed proteins can be rapidly degraded.78 Ag NPs, some of the most widely studied engineered NPs, were shown to dissolve in endosomes and lysosomes.79 In addition to their acidic pH, lysosomes also contain high levels of hydrolytic enzymes that can result in the degradation of the entire NPs or their coating.80 Thus, the dynamic nature of the cellular environment, with electrolytes, proteins, and enzymes affecting NP stability and dissolution, must be kept in mind.
A brief analysis of the composition of different CCM, as well as comparison to human plasma, shows the difference in biological media composition (Table 1). This can help explain the effect these differences have on the colloidal stability of NPs.
| Classification | Component-details | DMEM + 10% FBS | MEM + 10% FBS | RPMI-1640 + 10% FBS | Human plasma | |
|---|---|---|---|---|---|---|
| a Recently, our understanding of the composition of human plasma and serum has been greatly enlarged by the Human Plasma Proteome Project (HPPP).83,87 | ||||||
| Amino acids | Total | (mM) | 10.65 | 5.43 | 6.44 | 2.32–4.05 |
| Vitamins | Total | (mM) | 0.15 | 0.04 | 0.24 | <0.07 |
| Cations | Sodium, Na+ | (mM) | 155.31 | 144.44 | 124.27 | 142.00 |
| Potassium, K+ | (mM) | 5.33 | 5.33 | 5.33 | 4.00 | |
| Calcium, Ca2+ | (mM) | 1.80 | 1.80 | 0.42 | 2.50 | |
| Magnesium, Mg2+ | (mM) | 0.81 | 0.81 | 0.41 | 1.50 | |
| Iron, Fe3+ | (mM) | 0.25 | n/a | n/a | 10.00–27.00 | |
| Anions | Chloride, Cl− | (mM) | 117.47 | 124.37 | 100.16 | 103.00 |
| Bicarbonate, HCO3− | (mM) | 44.05 | 26.19 | 23.81 | 27.00 | |
| Sulfate, SO4− | (mM) | 0.81 | 0.81 | 0.41 | 0.50 | |
| Nitrate, NO3− | (mM) | 0.74 | n/a | 0.85 | 20.00 | |
| Phosphate, PO43− | (mM) | 0.92 | 1.01 | 5.63 | 1.00 | |
| Proteins | Total | (g L−1) | 3.00–4.50 | 3.00–4.50 | 3.00–4.50 | 65.00–80.00 |
| Serum albumin | (mM) | 0.05 | 0.05 | 0.05 | 0.58 | |
| α-Globulins | (g L−1) | 0.30 | 0.30 | 0.30 | 8.10 | |
| β-Globulins | (g L−1) | 0.27 | 0.27 | 0.27 | 11.50 | |
| γ-Globulins | (g L−1) | 0.07 | 0.07 | 0.07 | 15.60 | |
| IgG | (mM) | 3.56 × 10−0.05 | 3.56 × 10−0.05 | 3.56 × 10−0.05 | 0.08 | |
| Parameters | pH range | 7.00–7.40 | 7.00–7.40 | 7.00–7.40 | 7.34–7.42 | |
| Osmolality | (mOsm kg−1) | 320–360 | 280–320 | 270–310 | 276–295 | |
For example, RPMI contains approximately 5-fold more phosphate ions (5.63 mM) compared to DMEM (0.92 mM), MEM (1.01 mM), and human plasma (1.00 mM). PBS has twice as many phosphate ions (10 mM) as RPMI, and therefore contains approximately 10-fold more phosphate compared to DMEM, MEM, and human plasma. RPMI also contains half the concentration of magnesium ions (0.41 mM) compared to both DMEM (0.81 mM) and MEM (0.81 mM), and approximately a quarter of the magnesium ions in human plasma (1.5 mM). Aside from the complexity of ions within CCM, the presence of macromolecules (e.g. proteins, lipids) further complicates the interaction of NPs with their in vitro environment.
Notably, the total protein content of a CCM, which is commonly supplemented with 10% fetal bovine serum (FBS), is near 3.0–4.5 g L−1. This is approximately 20 times lower than the protein levels in human plasma (65–80 g L−1). Considering that numerous studies have shown that protein adsorption plays a significant, albeit conflicted, role in NP stability and interaction with biological systems, the presence of proteins in CCM is a non-trivial issue.88–90 Depending on the species of the serum source, sera further differ in the levels of fatty acids. Human serum for example is 7.5 times richer in lipids (especially polyunsaturated fatty acids) than FBS.91 Furthermore, since FBS suppliers only analyze certain components of the sera due to financial restraints, the lipid content is generally not assessed. Neither is the amount of many small molecules (e.g. growth factors) or specific proteins. CCM can therefore be characterized as a heterogeneous environment, and considering the variable composition of supplemented CCM, results obtained by characterizing NPs in water or buffered solutions do not represent the colloidal system under in vitro conditions.
2.3 Approaches to stabilize NPs in cell culture media
Surface modification of NPs is one of the most widely accepted methods to improve colloidal stability.11,92 Therefore, it is important to develop a robust understanding of the mechanisms governing NP stability, and to apply these methodologies in biological applications. Table 2 summarizes different types of NPs, organized by the fundamental approach used to stabilize them in cell culture media. Approaches designed to avoid the formation of aggregates in complex biological media include: electrostatic, steric, or electrosteric stabilization.61,93| Stabilization approach | Nanostructure | Stabilizing molecule | Medium | Result | ||||
|---|---|---|---|---|---|---|---|---|
| Electrostatic | Al2O318 | Bare oxide surface | RPMI-1640 with 10% FBS | Unstable | ||||
| Au110 | Bare Au surface | Lysoyzme solution | ||||||
| Au137 | Carboxylic-acid functionalized hydrocarbon | DMEM with 10% FBS | ||||||
| CeO216 | Bare oxide surface | RPMI-1640 with Glutomax and 10% FBS | ||||||
| CeO2138 | Bare oxide surface | DMEM + 10% FBS | ||||||
| CeO2139 | Tetramethylammonium hydroxide | DMEM + 10% FBS | ||||||
| CoO139 | Tetramethylammonium hydroxide | DMEM + 10% FBS | ||||||
| γ-Fe2O3140 | Citrate coating | RPMI | ||||||
| γ-Fe2O3140 | Citrate coating | RPMI + 10% FBS | ||||||
| γ-Fe2O3113 | Citrate coating | DMEM with 10% FBS | ||||||
| Fe3O4139 | Tetramethylammonium hydroxide | DMEM + 10% FBS | ||||||
| Iron/graphite141 | Bare graphite surface | RPMI-1640 with 10% FBS | ||||||
| SiO2-coated magnetite141 | Bare SiO2 surface | RPMI-1640 with 10% FBS | ||||||
| SiO2141 | Bare SiO2 surface | RPMI-1640 with 10% FBS | ||||||
| SiO289,142 | Bare SiO2 surface | MEM with 10% FBS | ||||||
| SiO2143 | (3-Aminopropyl)-trimethoxysilane | PBS | ||||||
| TiO296 | Bare oxide surface | DMEM | ||||||
| TiO2144 | Bare oxide surface | Keratinocyte media | ||||||
| TiO2144 | Bare oxide surface | Keratinocyte media + 10% FBS | ||||||
| Au60 | Tris(3-sulphophenyl) phosphine trisodium salt | RPMI-1640 | ||||||
| SiO295 | L-Lysine | DMEM + 10% FBS | ||||||
| SiO295 | L-Arginine | DMEM + 10% FBS | ||||||
| SiO295 | (3-Aminopropyl) trimethoxysilane | DMEM + 10% FBS | ||||||
| Ag139 | Citrate coating | DMEM + 10% FBS | Stable | |||||
| Au137 | Carboxylic-acid functionalized hydrocarbon | Deionized H2O | ||||||
| Au139 | Citrate coating | DMEM + 10% FBS | ||||||
| Au60 | Tris(3-sulphophenyl) phosphine trisodium salt | RPMI-1640 + 10% FBS | ||||||
| Au100 | Citrate coating | Human plasma | ||||||
| γ-Fe2O3140 | Citrate coating | PBS | ||||||
| SiO297 | Bare SiO2 surface | RPMI-1640 | ||||||
| SiO297 | Bare SiO2 surface | RPMI-1640 with 10% FBS | ||||||
| SiO295 | N-(6-Aminohexyl)-aminopropyltrimethoxysilane | DMEM + 10% FBS | ||||||
| TiO2145 | Bare TiO2 surface | RPMI-1640 with 10% FBS | ||||||
|
• EDL repulsion is not great enough to stabilize particles.
• Destabilization occurs due to: ○The difference in ionic strength when changing from water to CCM. ○Desorption of the stabilizing molecule through the competitive exchange with other CCM components. ○Inter-particle attractive forces, e.g. magnetic attraction from IONP, hydrogen bonding between silica NP surface hydroxyl groups. • In general, serum decreases aggregation because proteins adsorb onto NP surfaces and provide electrosteric repulsion. |
||||||||
| Steric | Surface-adsorbed molecules | Fe2O3114 | Polyvinyl alcohol | DMEM; RPMI | Unstable | |||
| CeO2113 | Phosphonate terminated poly(ethylene glycol) | DMEM with 10% FBS | ||||||
| Ag59 | Polyvinylpyrrolidone | RPMI | ||||||
| Au60 | Polyvinylpyrrolidone | RPMI | ||||||
| Polystyrene107 | Lutensol AT50 | DMEM with 10% FBS | ||||||
| Fe2O363 | Polyvinyl alcohol | DMEM with 10% FBS | ||||||
| Au60 | Polyvinylpyrrolidone | RPMI-1640 with 10% FBS | Stable | |||||
| Ag59 | Polyvinylpyrrolidone | RPMI-1640 with 10% FBS | ||||||
| Fe2O3114 | Polyvinyl alcohol | DMEM with 10% FBS or RPMI-1640 with 10% FBS | ||||||
| SiO2141 | Poly(ethylene glycol) | RPMI-1640 with 10% FBS | ||||||
| Covalently bound surface molecules | Magnetite117 | Poly(ethylene oxide)-DOPA | PBS, PBS with 10% FBS | Unstable | ||||
| Au90 | Thiolated poly(ethylene glycol) | DMEM with 5% FBS | ||||||
| PLGA146 | Chitosan modified poly(lactide-co-glycolide) | RPMI-1640 | ||||||
| Au112 | Thiolated poly(ethylene glycol) | Phosphate buffer with lysozyme | Stable | |||||
| Fe2O3147 | Poly(lactide)-poly(ethylene glycol) micelles | Human blood plasma | ||||||
| Au90 | Thiolated-mercaptododecanoic acid-poly(ethylene glycol) | DMEM with 5% FBS | ||||||
| Magnetite117 | Poly(ethylene oxide)-triNDOPA | PBS, PBS with 10% FBS | ||||||
|
Molecules adsorbed onto NP surface (neutral polymers, oligomers, non-ionic surfactant):
• Steric repulsion forces improve colloidal stability. • NP stability is independent of the solution's ionic strength. • Stabilizing molecule may be desorbed through competitive exchange with other components in media. • Proteins either stabilize NPs through adsorption onto particle surface, or destabilize through competitive exchange of stabilizing molecules. Molecules covalently bound to NP surface (e.g. chitosan, PEG, PLA-PEG): • Generally enhanced colloidal stability. • Coating can be improved through multipoint attachments between polymer and NP. • Robust polymer coating reduces protein adsorption. |
||||||||
| Electrosteric | Au148 | Phosphonate amphiphilic diblock-copolymer coating | DMEM, PBS + 1% L-Glu + 1% P/S and DMEM + 1% L-Glu + 1% P/S | Unstable | ||||
| Fe2O363 | Vinylalcohol/vinylamine copolymer coating | DMEM with 10% FBS | ||||||
| Fe2O3114 | Vinylalcohol/vinylamine copolymer coating | DMEM | ||||||
| Fe2O3114 | Vinylalcohol/vinylamine copolymer coating | RPMI-1640 | ||||||
| Fe2O3114 | Vinylalcohol/vinylamine copolymer coating | RPMI-1640 with 10% FCS | ||||||
| Fe2O3114 | Polyethylenimine | DMEM | ||||||
| Fe2O3114 | Polyethylenimine | DMEM with 10% FCS | ||||||
| Fe2O3114 | Polyethylenimine | RPMI-1640 with 10% FCS | ||||||
| Poly(L-lactide) cationic107 | Cetyltrimethylammonium chloride | DMEM with 10% FCS | ||||||
| Polystyrene cationic107 | Cetyltrimethylammonium chloride | DMEM with 10% FCS | ||||||
| Polystyrene-carboxyl63 | Native anionic polymer charge | DMEM with 10% FBS | ||||||
| Polystyrene-amine63 | Native cationic polymer charge | DMEM with 10% FBS | ||||||
| SiOx93 | HSA coating | PBS | ||||||
| SiOx93 | Tween 80 coating | PBS | ||||||
| TiO293 | Tween 80 coating | PBS | ||||||
| TiO296 | BSA coating (2 mg mL−1) | Bronchial epithelial growth medium, DMEM, Luria-Bertani Broth, Tryptic Soy Broth | ||||||
| ZnO93 | HSA coating | PBS | ||||||
| ZnO93 | Tween 80 coating | PBS | ||||||
| Ag93 | HSA coating | PBS | Stable | |||||
| Ag93 | Tween 80 coating | PBS | ||||||
| Ag93 | Mouse serum coating | PBS | ||||||
| Au148 | Phosphonate amphiphilic diblock-copolymer coating | PBS, PBS + BSA, DMEM + BSA and DMEM + 10% FBS | ||||||
| Au148 | Trimethylammonium amphiphilic diblock-copolymer coating | PBS, DMEM, PBS + 1% L-Glu + 1% P/S, DMEM + 1% L-Glu + 1% P/S, PBS + BSA, DMEM + BSA, and DMEM + 10% FBS | ||||||
| Au149 | Cysteine-functionalized alginate-derived polymer covalent bonding | PBS | ||||||
| Au150 | SH-C11alkyl-PEG-NH2 covalent bonding | DMEM | ||||||
| Au150 | SH-C11alkyl-PEG-COOH covalent bonding | DMEM | ||||||
| Au150 | SH-PEG-NH2 covalent bonding | DMEM | ||||||
| Au150 | SH-PEG-COOH covalent bonding | DMEM | ||||||
| Au68 | Vinylalcohol/vinylamine copolymer coating | RPMI-1640 with 10% FBS | ||||||
| Au68 | SH-PEG-NH2 covalent bonding | RPMI-1640 with 10% FBS | ||||||
| Au68 | Mixture of vinylalcohol/vinylamine copolymer coating and SH-PEG-NH2 coating | RPMI-1640 with 10% FBS | ||||||
| Au68 | Carboxylated polyvinyl alcohol coating | RPMI-1640 with 10% FBS | ||||||
| Au68 | SH-PEG-COOH covalent bonding | RPMI-1640 with 10% FBS | ||||||
| Au68 | Mixture of carboxylated polyvinyl alcohol and SH-PEG-COOH coating | RPMI-1640 with 10% FBS | ||||||
| CeO2113 | Poly(acrylic acid) | DMEM with 10% FBS | ||||||
| Fe2O3113 | Poly(acrylic acid) | DMEM with 10% FBS | ||||||
| Fe2O363 | Carboxylated polyvinyl alcohol coating | DMEM with 10% FBS | ||||||
| Fe2O3114 | Vinylalcohol/vinylamine copolymer coating | DMEM with 10% FBS | ||||||
| Fe2O3114 | Polyethylenimine | RPMI-1640 | ||||||
| Fe2O3140 | Poly(acrylic acid) coating | RPMI-1640 | ||||||
| Fe2O3140 | Poly(acrylic acid) coating | RPMI-1640 with 10% FBS | ||||||
| Fe2O3151 | FBS coating | RPMI-1640 with 4% FBS | ||||||
| N-Acetylated-chitosan nanocapsules115 | Native hydrophilic acetylated aminopolysaccharide surface | MEM with 10% FBS | ||||||
| N-Acetylated-chitosan nanocapsules115 | Native hydrophilic acetylated aminopolysaccharide surface | RPMI-1640 with 6% FBS | ||||||
| N-Acetylated-chitosan nanocapsules115 | Native hydrophilic acetylated aminopolysaccharide surface | Endothelial cell growth medium (ECGM) with 5% FCS | ||||||
| NaYF4116 | Cross-linked poly(maleic anhydride-alt-1-octadecene)-bis(hexamethylene)triamine coating | RPMI-1640 with 10% FBS | ||||||
| Poly(L-lactide) anionic107 | Sodium dodecylsulfate | DMEM with 10% FBS | ||||||
| Polystyrene anionic107 | Sodium dodecylsulfate | DMEM with 10% FBS | ||||||
| Polystyrene-amine93 | Native charged polymeric nature | PBS | ||||||
| Polystyrene-carboxylated152 | Transferrin coating | Human blood plasma | ||||||
| Polystyrene-carboxylated152 | Transferrin covalent bonding | Human blood plasma | ||||||
| Polystyrene-carboxylated93 | Native charged polymeric nature | PBS | ||||||
| Polystyrene-sulfonated152 | Transferrin coating | Human blood plasma | ||||||
| SiOx93 | Mouse serum coating | PBS | ||||||
| TiO293 | Human serum albumin (HSA) coating | PBS | ||||||
| TiO293 | Mouse serum coating | PBS, and RPMI-1640 with 10% FBS | ||||||
| TiO296 | FBS coating (1–5%) | Bronchial epithelial growth medium, DMEM, Luria-Bertani Broth, Tryptic Soy Broth, synthetic defined medium, yeast extract peptone dextrose medium | ||||||
| TiO296 | BSA coating (2 mg mL−1) | DMEM, synthetic defined medium, yeast extract peptone dextrose medium | ||||||
| ZnO93 | Mouse serum coating | PBS | ||||||
|
• Efficacy varies between different media due to different solution chemistries.
• Strongly adsorbed/bound macromolecules act as a physical barrier against aggregation. Charged macromolecules provide extra electrosteric repulsion. • Strong binding of macromolecule reduces protein adsorption. • In general, positively charged NPs/coatings aggregate more due to bridging flocculation between negatively charged proteins and positively charged NPs. • Negatively charged NPs are generally stable in serum media. • Hydration forces provide additional stability. |
||||||||
Mahl et al.60 reported gold NPs electrostatically stabilized with a tris(sodium-m-sulfonatophenyl)phosphine (TPPTS) coating. The hydrodynamic diameter (dH) of TPPTS-stabilized gold NPs in H2O was 25 nm but dispersion in RPMI-1640 cell culture medium without serum caused aggregation to a dH of 750 nm. Dispersion of these NPs in RPMI-1640 supplemented with either fetal calf serum (FCS), or bovine serum albumin (BSA), resulted in dH of 50 nm. Similar results have been reported for other nanomaterials. Chen et al.94 demonstrated that 2,3-dimercaptosuccinnic acid (DMSA)-modified iron oxide (Fe2O3) NPs aggregated in media without FCS, but were stable in RPMI with 10% v/v FCS. Graf et al.95 studied the influence of surface modifying silica particles (SiO2) and their colloidal stability on CCM. They demonstrated that particles stabilized by L-arginine, L-lysine, and amino silane with short alkyl chains undergo aggregation in un-supplemented, and FCS-supplemented cellular media.
Ji et al.96 reported that titanium dioxide (TiO2) NPs increased their dH in the presence of CCM without serum. However, the addition of BSA or fetal bovine serum (FBS) was shown to somewhat stabilize NPs, as there was little change in size following the addition of proteins. Similar findings were reported with a library of NPs including SiO2, ceria (CeO2), Fe2O3, and TiO2.97 NPs were characterized in H2O, RPMI without serum, RPMI supplemented with 10% FBS, and RPMI supplemented with 1% BSA. In general, NPs which had a negative zeta potential in H2O tended to form larger aggregates in media with 10% FBS compared to un-supplemented RPMI or RPMI with 1% BSA. Conversely, NPs which had a positive zeta potential in H2O tended to form large aggregates in un-supplemented RPMI, and then were stabilized by the presence of FBS or BSA. Here the addition of serum proteins in media was shown to decrease aggregation in certain situations, likely due to protein adsorption. The adsorption of proteins on the NP surface starts as soon as the NPs are dispersed in medium, which then provides electrosteric repulsion that occurs faster than NP agglomeration. These trends were not observed across comparable studies, and these differences may be attributed to NP material, experimental method, or characterization. However, this clearly demonstrates that electrostatic stabilization is not sufficient to avoid aggregation in biological media. More advanced methods to stabilize NPs go beyond electrostatic interactions at the particle surface.
PEG is a well investigated and frequently used polymer in the field since it has been shown to decrease protein adsorption, and in vivo pharmacokinetic studies have revealed that PEGylation of NPs can result in a prolonged circulation half-life. This is generally explained by reduced adsorption of opsonizing/serum proteins and increased stability of NPs in blood/serum.27,28,104 Gref et al.27 showed that the adsorption of proteins onto NP surfaces was dependent on PEG coating molecular weight, and PEG has also been shown to improve colloidal stability.105,106 Thus, functionalization of the NP surface with a non-ionic surfactant,107 or neutral polymers can improve colloidal stability.108–111 Many studies have also reported the formation of aggregates when sterically stabilized NPs were introduced to CCM. Mason et al.108 and Hirsch et al.111 demonstrated the formation of aggregates in cellular media of PEG-coated gold NPs and PVA-coated superparamagnetic iron oxide NPs, respectively. Stebounova et al.109 also reported PVP-coated silver NPs that were not stable under in vitro conditions. This poor stability is often explained by polymer–medium interactions. In some systems the polymer coating can become unstable and dissociate from the NP surface in the high ionic strength fluids. Zhang et al.112 showed that gold NPs functionalized by thiol terminated-PEG (PEG-SH) could be de-stabilized via ligand exchange by the small molecule dithiothreitol (DTT), whereby DTT dissociates the Au–S bond of PEG-SH, and releases the PEG coating. However, gold NPs functionalized with PEG-thioctic acid (PEG-TA), which contained multiple thiol groups that act as anchoring ligands, were shown to be more stable. Larson et al.90 reported similar results where PEG-SH can be displaced by cysteine and cystine, two amino acids present in physiological media, resulting in gold NPs being subsequently destabilized by serum proteins. To mitigate this issue, gold NPs were functionalized with PEG terminated by a thiolated alkyl group. This end group provides an alkyl linker that acts a hydrophobic shield between the PEG molecule and the NP surface, thus reducing competitive displacement of PEG by other small, thiol-containing molecules.
Steric stabilization has proven to be an effective method to improve NP colloidal stability. It can reduce protein adsorption, and polymeric coatings can act as a physical barrier to prevent NP aggregation.
Interestingly, exceptional stability of chitosan-based nanocapsules in cellular media has been reported.115 Here, it was demonstrated that colloidal stability cannot solely be explained by electrosteric repulsion. The stability was attributed to the hydrophilic nature of chitosan nanocapsules which demonstrates another stabilization mechanism based on short-range repulsive hydration forces.
Thus, additional mechanisms of stabilization are being explored to improve colloidal stability in biological media. In order to design new generations of NPs, the investigation is being extended to not only particle–media interactions, but also particle–stabilizer interactions.116–118 However, these previously discussed studies show that surface functionalization alone may not be sufficient to totally stabilize particles in CCM.
2.4 The role of proteins
The presence of proteins in CCM results in complex and highly variable effects on NP stability. Proteins have been shown to sterically stabilize or de-stabilize NPs upon their suspension in CCM. For example, citrate capped gold NPs aggregate in PBS, but are stable in the presence of proteins.60 The ability of proteins to stabilize colloidal systems has been shown to strongly depend on both the protein concentration and the composition of the NPs. SiO2 NPs, for example, aggregate in PBS supplemented with low protein concentrations, but are well-dispersed in PBS containing high protein concentrations.95,119 Polystyrene NPs, on the other hand, form large aggregates upon suspension in plasma independent of the protein concentration, which was explained by the slow protein adsorption on these NPs hindering steric stabilization.120,121 Hondow et al.122 concluded that streptavidin-coated quantum dots undergo aggregation in both pure cellular medium and cellular medium plus serum, but the number of QDs per aggregate was less in serum.When NPs are added to CCM, they enter in contact first with albumin and other serum proteins, forming the so-called protein corona,19,88 and BSA has been widely explored for dispersing NPs.60,93,96 Mahl et al.60 reported that the presence of BSA in the cellular medium efficiently prevented gold NP aggregation. They also observed that the diameter of gold NPs was 2-fold larger for tris(sodium-m-sulfonatophenyl)phosphine(TPPTS)–gold NPs and 1.4-fold larger for PVP–gold NPs in the presence of the proteins, indicating a surface layer of adsorbed protein which offered additional stability. By applying fluorescence correlation spectroscopy it has been demonstrated that the human serum albumin (HSA) corona on polymer-coated FePt and CdSe/ZnS NPs consisted of a single protein monolayer.123 Several reports have demonstrated that FBS at a typical concentration of 10% can improve NP colloidal stability in various cellular media.59,60,94,96 Chen et al.94 reported that surface functionalized magnetic NPs dispersed in cellular medium (RPMI-1640) with FCS exhibited increased stability due to extra electrosteric repulsion provided by proteins adsorbed on the particle surface. However, many reports have demonstrated that the formation of a protein layer destabilizes the NPs. When NP dispersions are added to serum, protein adsorption strongly occurs, which in turn affects the surface charge distribution.121
Thus, the effect of protein adsorption on aggregation has not been totally established. It is clear that proteins and molecules present in cellular media can significantly alter the aggregation behavior of NPs. The reasons why NP–protein interactions have been shown to cause the collapse of a colloidal system is, however, still under discussion. For example, strongly positive charged chitosan latex particles underwent charge neutralization and electrostatic screening upon interaction with BSA, resulting in low colloidal stability.124 On the other hand it was shown that the zeta potential of NPs is shifted to comparable negative values after incubation with supplemented CCM, independent of their intrinsic surface charge.16,63,125 The low colloidal stability of positively charged latex NPs could therefore also be explained by bridging flocculation caused by the favorable interaction of the reactive amine groups on the particle surface with the negative surface charge of the protein.124,126
Whether NP–protein interaction leads to the collapse or stabilization of colloidal systems has been shown to depend on an array of different parameters, both with the components of the media and the physico-chemical NP properties. It therefore remains difficult to predict how protein adsorption affects the colloidal stability of NPs. The difference in protein concentration of supplemented CCM and human plasma further impedes the direct extrapolation of data obtained in in vitro experiments to colloidal behavior of NPs in vivo. However, probing and taking possible aggregation of NPs under physiological conditions into consideration is a first step towards accurate interpretation of cellular responses in vitro, which in turn helps to better predict subsequent biological effects in vivo.
2.5 Fundamental interactions between proteins and surfaces
Due to the complexity of NP interactions in complex sera, some groups have addressed surface-protein interactions in a much more idealized situation. As is well known that proteins have a strong tendency to adsorb on surfaces, researchers have investigated the interactions between surfaces coated with proteins.127,128 Most part of the published studies have used flat surfaces coated by different types of proteins and have used either a surface force apparatus or an atomic force microscope (AFM) to measure the force between surfaces. The interactions have generally not only been measured between highly flat surfaces, such as mica,129–131 silicon wafers,132 or silica,133–135 but also polystyrene136 coated by uniform layers of different proteins under different pH conditions and ionic strengths. The proteins investigated include human apolipoprotein AII,131 lactoferrin,132 protein A,130 b-casein,129 BSA,133,136 and apoferritin.134Some general conclusion can be drawn from these investigations. For not too high ionic strengths and not too small separation distances, DLVO theory seems to provide a reasonably good quantitative description of the interactions.127,131,132,135 At short separation distances, instead, a strong repulsive interaction is typically observed.129,130,132,134–136 This is usually a result of steric interactions, and/or hydration interactions.127–129,135 The results are difficult to model since short range interactions are strongly dependent on the detailed structure of the adsorbed proteins. There is also a debate on whether hydration forces rather than steric interactions should be held responsible for the strong repulsive potential.127,135 In some cases, it was verified that proteins were not adsorbed in a monolayer, but rather in a multilayer, thus making it even more difficult to precisely model what happens at short separation distances. At higher ionic strengths, when electrostatic repulsions are strongly shielded, DLVO theory is no longer able to describe the interactions.131 Once again, the behavior is strongly protein dependent, and difficult to rationalize. In an interesting work, Dickinson et al.128 used a more advanced self-consistent field modeling approach, originally developed for polymers, to describe the structure of adsorbed casein proteins on hydrophobic proteins. Their findings were able to explain the different stabilization ability of as1-casein and b-casein, based on the different amino acid sequences of the proteins. b-casein was shown to be able to provide strong electrosteric repulsion; while as1-casein leads to bridging flocculation among oppositely charged patches. This shows that strong differences are observed even among proteins belonging to the same family.
Most of the aforementioned studies treat the ideal case of uniformly coated surfaces. Some work has also addressed the effect of partial surface coverage. Ramos et al.,131 for example, have also measured the forces between mica surfaces not fully covered by apolipoprotein AII. Their results indicate that the force is fully repulsive only in the case of fully covered surfaces. When only partially covered surfaces approach one another, attractive interactions are measured. This phenomenon is likely driven by bridging and intercalation. The experiments on flat surfaces covered by proteins certainly provide insightful information, which however cannot always be immediately applied to particles. More realistic experiments involve the use of colloidal probe techniques, where a colloidal particle is attached to the tip of an AFM, and used to probe interactions with either a flat surface or other particles positioned on a surface.132,133 Due to the large size of the particles used in these systems, no significant differences are observed in comparison with measurements done between flat surfaces.
The effect of protein adsorption on the stability of particles has also been probed directly by a few groups using scattering techniques.153–161 Two major findings have emerged from all these investigations. Contrary to what was expected for charge-stabilized particles from traditional DLVO theory, which foresees a progressive destabilization of particles as the ionic strength increases, protein coated particles show a restabilization at high ionic strengths.157,159 At low ionic strengths a progressive destabilization is measured with an increase in ion concentration, at high ionic strength the particles regain stability. The effect has been primarily observed in the case of particles coated by various antibodies and antigens, and is referred to as anomalous colloidal stability. It has been explained in terms of hydration forces generated by counterion adsorption on the proteins.160 Very often this effect is accompanied by a change in overall surface charge of the particles, as electrophoretic mobility measurements indicate. Attempts to provide more physically sound explanation using the extension of DLVO theory introduced by Ruckenstein et al. have proved to be quite unsuccessful.155,162 The second consequence of the presence of a protein layer on the surface of the NPs is the peculiar mechanical weakness of the aggregates formed.159 This has been explained by looking at the estimated interaction energy profiles of particles. The presence of short range repulsion at short separation distances promotes aggregation of protein coated particles in secondary energy minimum. Secondary energy minima are much shallower than primary energy minima, thus leading to much weaker bonds than those formed when particles aggregate in a primary energy minimum. This leads to difficulties in characterization of the structure of these aggregates, which tend to be very prone to restructuring.
An important consideration can be drawn from all these investigations. When particles are exposed to complex media, the kinetics of adsorption of proteins and other biomolecules is likely to play a crucial role. Even though full coverage of surfaces can provide additional stabilization, at least under certain circumstances, the presence of only a few patches of organic molecules is most likely to induce destabilization and promote bridging flocculation effects, as some investigations indicate. Furthermore, the modeling of colloidal stability using traditional DLVO theory is only expected to provide reasonably accurate results under ideal conditions, which are typically very far from those encountered in physiological solutions. These idealized studies provide controlled examination of NP protein interactions. Conversely, a more developed understanding of NP aggregation can emphasize the relevance of this phenomenon when considering NP behavior in vitro.
3. Aggregation
Until this point, our discussion has crudely treated NPs as “dispersed” or “aggregated.” However, NP aggregation behavior can be elegantly modeled and described in certain cases. Heterogeneity (e.g. high ionic strength, salt content, or the presence of biomacromolecules) is an innate feature of CCM, and forces defining inter-particle interactions will determine the fate of the colloidal suspensions. Briefly, a repulsive net force ensures stability, while an unbalanced attractive net force will cause the stability to quickly vanish. Mixing otherwise stable NP colloids with CCM can unbalance the inter-particle interactions. Consequently, the destabilized suspension will rapidly collapse and the NPs will aggregate.13,18,20 Upon aggregation, NP transport dynamics, such as diffusion and sedimentation, will drastically alter compared to stable, dispersed NPs. The large size will result in a larger hydrodynamic radius, and therefore aggregates may be rendered susceptible to sedimentation, whereas single small NPs will typically exhibit only Brownian motion. However, the aggregates will not behave the same as a larger single particle of an equally large hydrodynamic radius, as aggregates will have a variable packing density which will further impact particokinetics.163 The specific surface area of aggregates will also drastically change compared to monodispersed NPs, which may heavily influence the interface between NP aggregates and cells. Thus understanding the aggregation formation is crucial to understand the impact of NP aggregation on biological systems.In the absence of external forces, aggregates of NPs are irregular and exhibit an arbitrary random shape and a disordered structure. Such aggregates have a so-called mass-fractal structure and exhibit a striking feature called self-similarity, where the characteristic features of randomness and disorder can remain invariant over a large range of length scales (Fig. 3).164
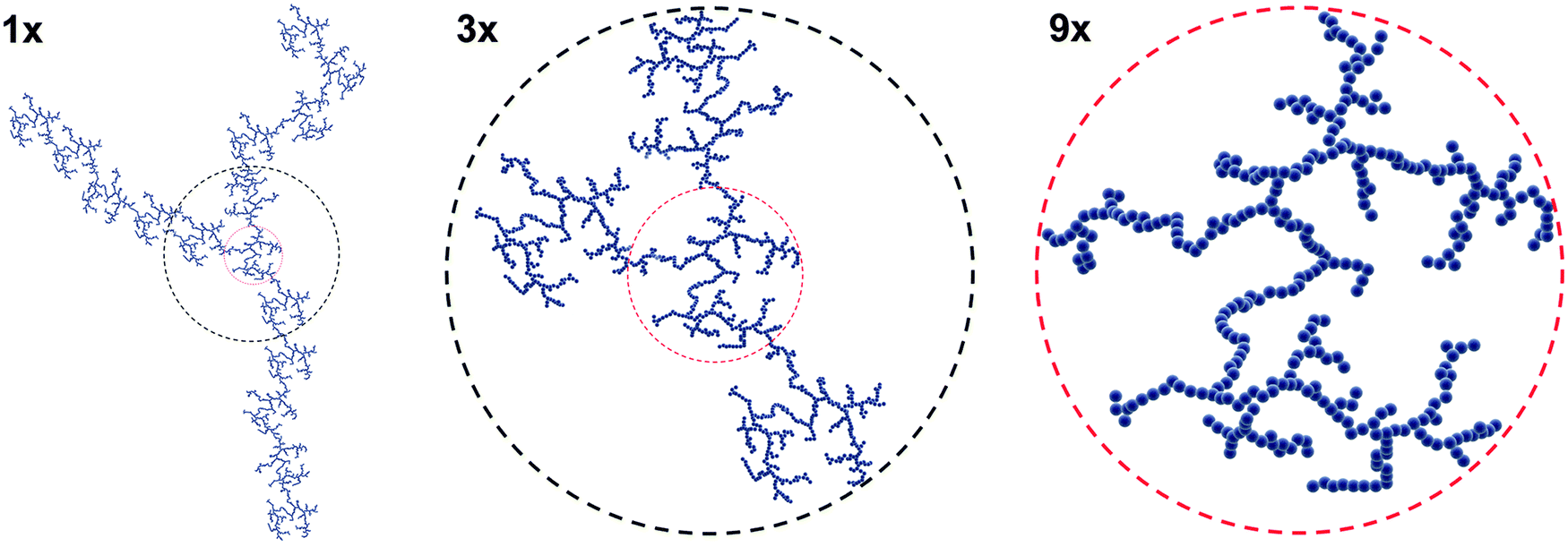 | ||
| Fig. 3 Illustration of self-similarity adapted from ref. 165. Certain characteristic features of the fractal are independent of the level of magnification. | ||
Fractal clusters are composed of loosely aggregated NPs and also empty voids, which are clearly visible on TEM micrographs.165,166 A fundamental concept to describe the structure of mass fractals is the fractal dimension, df, which can be related to the typical packing density of particles within an aggregate.167 Owing to the arbitrary shape and the random structure, aggregates are ill-defined in nature; yet, one may still quantify properties relevant for the study of NP–cell interactions via the fractal dimension. In fluid, the relevant characteristic size is the hydrodynamic radius, R, whose value is close to the cluster radius of gyration.168 Both the hydrodynamic radius and the effective mass density are functions of df, and aggregation number, N. These values therefore influence the transport of aggregates via diffusion and sedimentation.169 In solution the motion of aggregated and clustered NPs is generally different from that of the primary NPs, and this results in differences in the amount of NP which will reach a cell's surface in vitro.163,170
The df therefore accounts for the nonlinear relationship between the number of particles, and the corresponding size and mass of the aggregation cluster.169 Such power-law relationship is very characteristic for self-similar structures. The formation of aggregates and clusters can be reversible or irreversible. The rate and nature of the aggregate formation are complex functions of the now-imbalanced inter-particle forces and NPs dynamics. The fractal dimension is a function of the probability that NPs collide and bind together. The rate of collisions is proportional to the particle concentration and the Brownian dynamics, and the chance of binding upon colliding is a function of the inter-particle forces. Recently a novel experimental approach has been presented which accurately measures the effective density of agglomerates in suspension based on the volume of the pellet obtained by centrifugation of nanomaterials in suspension.171
When the inter-particle interactions are dominated by attraction, the rate and structure of aggregate formation is influenced solely by the Brownian motion because the NPs bind as soon as they collide. This limiting case is diffusion-limited cluster aggregation (DLCA), and the fractal dimension of DLCA clusters is close to 1.8.166,172–174 Interestingly, the fractal dimension of diffusion-limited clusters is universal and independent of the NP size as long as the polydispersity of the NPs is moderate.173 The opposite case is when the NPs must collide more than once before becoming bound. The kinetics of this process, reaction-limited cluster aggregation (RLCA), may be very slow, and the fractal dimension of RLCA clusters is near to 2.1.14,173 A higher fractal dimension indicates a more closely, densely packed structure. As a function of time, the average cluster size has been observed to grow exponentially for RLCA, while a power-law behavior has been observed for DLCA. However, most colloidal systems display a complex behavior including the features of both DLCA and RLCA. For example, Limbach et al.16 demonstrated that small variations in the surface chemistry of the particle may have crucial impact on the stability, and therefore, may significantly slow down the aggregation rate. Similarly, electrolyte concentration, pH, temperature, shear rate, and particle concentration all influence the rate and type of aggregation.175,176 Furthermore, if the bonds among the particles are either very weak or partially reversible it allows for movement of the particles in the aggregates and restructuring, thus resulting in a higher fractal dimension value. Understanding how these numerous factors interact can provide a more clear idea of how NPs aggregate, and understanding aggregation provides more predictive abilities for how the NP will behave in vitro. To this point, thorough NP characterization provides a means for understanding NP properties, aggregation, and ultimately behavior and interaction with cells.
4. Nanoparticle characterization
The assessment of the colloidal stability in physiological fluids is non-trivial. The variety of physical and chemical forces involved, the complexity of analytical methods and theories upon which these analytical methods are based all compound the difficulty in accurately evaluating the dispersion state of NPs in biological environments. These aspects are further complicated by the multitude of different, highly complex physiological fluids NPs interact with. However, there are a number of techniques that are commonly used to evaluate NP physico-chemical properties and the dispersion state, as well as a growing number of state-of-the-art methods. Here we discuss the most commonly used characterization approaches, and comment on considerations that must be made when characterizing NP colloidal stability.Particle size, stability, the onset of aggregation and its evolution in time should ideally always be probed in situ, since ex situ methods require deposition onto a substrate, which may bring in uncontrollable, and hence, unpredictable artifacts. Three major categories dominate the currently applied experimental approaches: scattering, optical spectroscopy, and microscopy. Light scattering techniques are by far the most frequently used in situ methods when describing size and polydispersity of suspended particles. Dynamic light scattering (DLS) is a technique where particle size and polydispersity are determined by monitoring the time-dependent fluctuations in the intensity of the scattered light due to particle motion, which, in dilute samples, can be directly related to the particle's diffusion coefficient. In suspension, due to the Brownian motion of NPs, the scattering intensity exhibits temporal fluctuations from which particle size can be recovered from the diffusion coefficient, which is inversely proportional to the size according to the Stokes–Einstein equation.177 DLS is also relatively straightforward to perform and highly quantitative. Commercial cutting-edge instruments are widely available and affordable. However, the subtleties of the theory behind DLS can be obscured by the experimental simplicity, especially when dealing with polydispersity. An inherent characteristic feature of scattering is that the measured size is intensity weighed, i.e. the light scattered by a particle is proportional to the sixth power of its size. In the obvious case of polydisperse samples, this implies that large particles or aggregates produce very strong signals that can dominate over those signals produced by small particles. Unfortunately, the deconvolution of DLS data is mathematically an ill-posed problem and prone to generate artifacts, even more so because the intensity-weighed statistical distribution should be transformed into number-weighted statistical distribution in order to diminish the effect of aggregated particles and better report the size of smaller, dispersed NP. Furthermore, these techniques generally require a dilute suspension to ensure single scattering events and to mitigate inter-particle interactions. Depolarized DLS (DDLS) extends the technique for particles possessing optical or shape anisotropy, e.g. nanorods.178 The experimental procedure of static light scattering (SLS) is very similar, however, instead of analyzing the time-dependent fluctuation of the scattered intensity, the angle-dependence of the scattered intensity is analyzed. The scattering intensity as a function of momentum transfer, q, relates directly to the size and form of the particles, via Fourier transformation. The scattered intensity of a fractal system follows a power law as a function of momentum transfer, which renders scattering to be essential in the assessment of fractal properties.168
Compared to static scattering, optical spectroscopy is a rapid technique to monitor the colloidal stability of noble metal NPs (i.e. gold, silver). These particles display unique and strongly enhanced optical properties known as localized surface plasmon resonances (LSPRs). The basis of LSPRs is the coherent collective oscillations of conduction electrons upon interaction with the incident light (electromagnetic radiation).179 The LSPR gives rise to a pronounced extinction band in the visible-near infrared (NIR) region of the electromagnetic spectrum, and the position and width of the LSPR band are highly affected by particle aggregation. When plasmonic NPs are close, individual plasmon oscillation can couple with each other via near-field interactions, resulting in coupled LSPR modes,180 which impact the distribution of the electric field around the nanostructure.181 As a consequence, particle aggregation results in a spectral red shift and broadening of the peak.182 These changes indicate a decrease in the concentration of single NPs, which is confirmed by a color shift of the colloid from deep red to purple for gold NPs or from yellow to black for silver NPs.11,183
In certain cases LSPR is suitable for monitoring particle–protein interaction in biological media, as LSPR is sensitive to the local dielectric environment.179 Metal NPs incubated in biological media exhibit a red shift in their LSPR spectra and an increase in band intensity over time due to the formation of a stable protein corona on the NP metal surface.139,184 This approach may be valid for “bare” particles, e.g. citrate-coated gold or silver NPs, but should be carefully revised when particles are coated, with for example a polymer shell. The polymer may act as a dielectric spacer and insulator, thus decreasing the sensitivity to refractive index changes.185 As a consequence UV-Vis can become practically insensitive, and monitoring the protein adsorption of surface-functionalized NPs in biological media cannot be conclusive. Therefore complementary techniques should be used.
In contrast with scattering and optical spectroscopy, NPs can be directly visualized by microscopic imaging. These methods of choice allowing resolving NPs in the range of Ångstrom to nanometer include electron microscopy such as transmission (TEM) or scanning electron microscopy (SEM), and atomic force microscopy (AFM). TEM is a microscopy technique whereby a beam of coherent electrons (the primary beam) is transmitted through a sufficiently thin specimen (<500 nm) thereby offering a convenient method for measuring the diameter of electron dense NP, and resolving structures at the Ångstrom level.186 TEM is however limited because samples must be dried onto a grid for imaging, as TEM is performed under vacuum. This can result in drying artifacts (e.g. aggregation of NPs), and it is therefore difficult to gain information about the NP dispersion state in situ. SEM works by scanning an electron beam over the sample, which scatters secondary electrons from the sample to detectors.186 This method provides detailed information about the NP morphology, topology, and composition, and can resolve structures at the nanometer level. Conventional SEM is also performed under vacuum, and therefore is limited by drying artifacts of samples. Furthermore, depending on the NP material, some samples need to be prepared with an ultrathin electrically conducting material (e.g. gold, palladium, platinum). However, environmental SEM (ESEM) provides a means of imaging samples in a hydrated, non-desiccated state. This allows for samples to be imaged in a somewhat aqueous environment and can be done without coating with an electrically conducting material.187,188
AFM is a technique whereby a fine tip is mounted on a highly sensitive cantilever and scanned across a surface. Measuring the deflection of the cantilever provides a quantitative image of the surface's topography. Thus, AFM can be used to quantitatively measure the size and morphology of NPs, and can resolve structures on the angstrom scale.189 AFM has the advantage of operating in an aqueous environment. However can still be limited by either aggregation artifacts based on NP interactions on the surface, or NP–AFM tip interactions.39
Characterization of NPs is critical for not only obtaining physico-chemical information regarding the NP, but also to fully understand how NP behavior is tied to these properties. To that end, it is of utmost importance to not only robustly characterize NPs with multiple techniques, but to also understand the limitations of each characterization method.
5. Conclusions
The above discussion attempts to summarize the factors which dictate NP stability in physiological fluids such as cell culture media. However, it can be seen that NP aggregation is highly system dependent and one must consider a number of factors: materials, NP physico-chemical properties, surface coatings, solution properties and components, the presence of macromolecules, etc. Thus providing a comprehensive framework to predict NP stability in media or physiological solutions (based on experimental or theoretical data) is exceedingly difficult. General, albeit well-established, trends indicate that the presence of a stabilizing coating (e.g. PEG) improves NP stability and reduces the adsorption of proteins to the NP surface. Likewise the strength or stability of the interaction between the stabilizing coating and the NP is also important to maintain colloidal stability, as constituents of the solution have been shown to displace stabilizing molecules and initiate aggregation. One must also consider the inter-molecular interactions of coatings, and inter-particle forces. Conversely, the composition of the physiological solution must be considered. Thus, the complicated question of NP aggregation depends on the system presented.It is apparent that NP stability and the propensity of NPs to aggregate in complex physiological fluids merit further investigation. In order to more easily comprehend NP behavior in biological experiments, it is necessary for nanomedicine research to (1) heavily rely on interdisciplinary research between physicists, materials scientists, biologists, nanotoxicologists, medicine, etc. (2) supply robust and universal characterization methods with regard to physico-chemical properties and be aware of their limitations, (3) use, if possible, more than one technique to study the materials, and (4) characterize NPs in an environment, which is as close to the experimental environment as possible. Currently the issue of NP aggregation is understated in the literature, which may in part lead to the difficulty in translating nanomedicines from the bench top to the clinic. However, with a concentrated interdisciplinary effort with a heavy focus on both analytics and understanding fundamental behavior of NPs in solution, it may be possible to more successfully advance the use NPs for biomedical applications.
As of today, theoretical models such as the XDLVO theory cannot be used to provide reliable predictions of the stability of NPs in complex biological media. This is partially due to the inability of theory to correctly describe the behavior of the interface between particles and media, but also to the intrinsic experimental inaccuracy in the values of parameters, such as surface charge density, ligand density, adsorption of biomolecules, etc. which need to be supplied to the theory. While much research effort on the theoretical side is certainly necessary, researchers currently have to primarily rely on multiple characterization techniques to probe colloidal stability of NPs in complex media and to engineer better NPs. Characterization includes non-invasive scattering techniques, which are the most reliable tools to spot aggregation, but also analytical techniques, such as titration, calorimetry, spectroscopy, etc. to gain a deeper understanding of how the biological environment impacts NP colloidal stability.
Acknowledgements
Financial support from the Swiss National Science Foundation (Grants #PP00P2126483/1 and 133597), the Adolphe Merkle Foundation and the Swiss National Science Foundation through the National Centre of Competence in Research Bio-Inspired Materials is gratefully acknowledged. L.R.-L. acknowledges the financial support from the L'Oreal Switzerland and UNESCO's fellowship program “For Women in Science 2013”. We also gratefully acknowledge Dr B. Michen's input and discussion in the early stages of this project.References
- V. Wagner, A. Dullaart, A.-K. Bock and A. Zweck, Nat. Biotechnol., 2006, 24, 1211–1217 CrossRef CAS PubMed.
- M. L. Etheridge, S. A. Campbell, A. G. Erdman, C. L. Haynes, S. M. Wolf and J. McCullough, Nanomedicine, 2013, 9, 1–14 CrossRef CAS PubMed.
- P. Burgess, P. B. Hutt, O. C. Farokhzad, R. Langer, S. Minick and S. Zale, Nat. Biotechnol., 2010, 28, 1267–1270 CrossRef CAS PubMed.
- L. Y. Rizzo, B. Theek, G. Storm, F. Kiessling and T. Lammers, Curr. Opin. Biotechnol., 2013, 24, 1159–1166 CrossRef CAS PubMed.
- S. Svenson, Curr. Opin. Solid State Mater. Sci., 2012, 16, 287–294 CrossRef CAS PubMed.
- V. J. Venditto and F. C. Szoka Jr, Adv. Drug Delivery Rev., 2013, 65, 80–88 CrossRef CAS PubMed.
- S. Tinkle, S. E. McNeil, S. Mühlebach, R. Bawa, G. Borchard, Y. Barenholz, L. Tamarkin and N. Desai, Ann. N. Y. Acad. Sci., 2014, 1313, 35–56 CrossRef CAS PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- I. Lynch, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2009, 4, 546–547 CrossRef CAS PubMed.
- A. Albanese and W. C. W. Chan, ACS Nano, 2011, 5, 5478–5489 CrossRef CAS PubMed.
- E. Mahon, A. Salvati, F. Baldelli Bombelli, I. Lynch and K. A. Dawson, J. Controlled Release, 2012, 161, 164–174 CrossRef CAS PubMed.
- K. Rausch, A. Reuter, K. Fischer and M. Schmidt, Biomacromolecules, 2010, 11, 2836–2839 CrossRef CAS PubMed.
- P. Meakin, J. Colloid Interface Sci., 1984, 102, 491–504 CrossRef CAS.
- G. Nichols, S. Byard, M. J. Bloxham, J. Botterill, N. J. Dawson, A. Dennis, V. Diart, N. C. North and J. D. Sherwood, J. Pharm. Sci., 2002, 91, 2103–2109 CrossRef CAS PubMed.
- L. K. Limbach, Y. Li, R. N. Grass, T. J. Brunner, M. A. Hintermann, M. Muller, D. Gunther and W. J. Stark, Environ. Sci. Technol., 2005, 39, 9370–9376 CrossRef CAS.
- P. Wick, P. Manser, L. K. Limbach, U. Dettlaff-Weglikowska, F. Krumeich, S. Roth, W. J. Stark and A. Bruinink, Toxicol. Lett., 2007, 168, 121–131 CrossRef CAS PubMed.
- R. C. Murdock, L. Braydich-Stolle, A. M. Schrand, J. J. Schlager and S. M. Hussain, Toxicol. Sci., 2008, 101, 239–253 CrossRef CAS PubMed.
- J. S. Gebauer, M. Malissek, S. Simon, S. K. Knauer, M. Maskos, R. H. Stauber, W. Peukert and L. Treuel, Langmuir, 2012, 28, 9673–9679 CrossRef CAS PubMed.
- D. B. Warheit, C. M. Sayes, K. L. Reed and K. A. Swain, Pharmacol. Ther., 2008, 120, 35–42 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- K. L. Aillon, Y. Xie, N. El-Gendy, C. J. Berkland and M. L. Forrest, Adv. Drug Delivery Rev., 2009, 61, 457–466 CrossRef CAS PubMed.
- H. Aoki, H. Aoki, T. Kutsuno, W. Li and M. Niwa, J. Mater. Sci.: Mater. Med., 2000, 11, 67–72 CrossRef CAS.
- G. Xie, J. Sun, G. Zhong, L. Shi and D. Zhang, Arch. Toxicol., 2010, 84, 183–190 CrossRef CAS PubMed.
- T. L. Moore, J. E. Pitzer, R. Podila, X. Wang, R. L. Lewis, S. W. Grimes, J. R. Wilson, E. Skjervold, J. M. Brown, A. Rao and F. Alexis, Part. Part. Syst. Charact., 2013, 30, 365–373 CrossRef CAS PubMed.
- T. L. Moore, A. S. Schreurs, R. A. Morrison, E. K. Jelen, J. Loo, R. K. Globus and F. Alexis, J. Nanomed. Nanotechnol., 2014, 5, 1000237 Search PubMed.
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. H. Müller, Colloids Surf., B, 2000, 18, 301–313 CrossRef CAS.
- D. E. Owens III and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef PubMed.
- Z. Poon, J. B. Lee, S. W. Morton and P. T. Hammond, Nano Lett., 2011, 11, 2096–2103 CrossRef CAS PubMed.
- J. Gregory, J. Colloid Interface Sci., 1981, 83, 138–145 CrossRef CAS.
- M. A. Butkus and D. Grasso, J. Colloid Interface Sci., 1998, 200, 172–181 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic press, revised 3rd edn, 2011 Search PubMed.
- B. Derjaguin, Kolloid-Z., 1934, 69, 155–164 CrossRef.
- R. Hogg, T. W. Healy and D. W. Fuerstenau, Trans. Faraday Soc., 1966, 62, 1638–1651 RSC.
- L. N. McCartney and S. Levine, J. Colloid Interface Sci., 1969, 30, 345–354 CrossRef.
- J. E. Sader, S. L. Carnie and D. Y. C. Chan, J. Colloid Interface Sci., 1995, 171, 46–54 CrossRef CAS.
- J.-P. Hsu, Interfacial Forces and Fields: Theory and Applications, CRC Press, 1999 Search PubMed.
- E. J. W. Verwey, J. T. G. Overbeek and J. T. G. Overbeek, Theory of the Stability of Lyophobic Colloids, Courier Dover Publications, 1999 Search PubMed.
- M. Baalousha, Sci. Total Environ., 2009, 407, 2093–2101 CrossRef CAS PubMed.
- S. A. Edwards and D. R. M. Williams, Phys. Rev. Lett., 2004, 92, 248303 CrossRef CAS.
- D. H. Napper, Polymeric Stabilization of Colloidal Dispersions, Academic Press, London, 1983 Search PubMed.
- P. G. De Gennes, Adv. Colloid Interface Sci., 1987, 27, 189–209 CrossRef CAS.
- A. Lozsán, M. García-Sucre and G. Urbina-Villalba, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 061405 CrossRef.
- D. J. McClements, Langmuir, 2005, 21, 9777–9785 CrossRef CAS PubMed.
- A. J. Pelley and N. Tufenkji, J. Colloid Interface Sci., 2008, 321, 74–83 CrossRef CAS PubMed.
- R. F. Domingos, N. Tufenkji and K. J. Wilkinson, Environ. Sci. Technol., 2009, 43, 1282–1286 CrossRef CAS.
- S. Biggs, M. Habgood, G. J. Jameson and Y.-d. Yan, Chem. Eng. J., 2000, 80, 13–22 CrossRef CAS.
- M. Kobayashi, F. Juillerat, P. Galletto, P. Bowen and M. Borkovec, Langmuir, 2005, 21, 5761–5769 CrossRef CAS PubMed.
- W. R. Schowalter and A. B. Eidsath, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3644–3651 CrossRef CAS PubMed.
- C.-J. Chin, S. Yiacoumi and C. Tsouris, Langmuir, 2001, 17, 6065–6071 CrossRef CAS.
- M. Boström, D. R. M. Williams and B. W. Ninham, Phys. Rev. Lett., 2001, 87, 168103 CrossRef.
- J. A. Lewis, J. Am. Ceram. Soc., 2000, 83, 2341–2359 CrossRef CAS PubMed.
- M. Hermansson, Colloids Surf., B, 1999, 14, 105–119 CrossRef CAS.
- B. Michen, A. Diatta, J. Fritsch, C. Aneziris and T. Graule, Sep. Purif. Technol., 2011, 81, 77–87 CAS.
- B. Michen, J. Fritsch, C. Aneziris and T. Graule, Environ. Sci. Technol., 2013, 47, 1526–1533 CAS.
- J. N. Ryan and M. Elimelech, Colloids Surf., A, 1996, 107, 1–56 CrossRef CAS.
- A. R. Petosa, D. P. Jaisi, I. R. Quevedo, M. Elimelech and N. Tufenkji, Environ. Sci. Technol., 2010, 44, 6532–6549 CrossRef CAS PubMed.
- A. R. Petosa, S. J. Brennan, F. Rajput and N. Tufenkji, Water Res., 2012, 46, 1273–1285 CrossRef CAS PubMed.
- S. Kittler, C. Greulich, J. S. Gebauer, J. Diendorf, L. Treuel, L. Ruiz, J. M. Gonzalez-Calbet, M. Vallet-Regi, R. Zellner and M. Köller, J. Mater. Chem., 2010, 20, 512–518 RSC.
- D. Mahl, C. Greulich, W. Meyer-Zaika, M. Koller and M. Epple, J. Mater. Chem., 2010, 20, 6176–6181 RSC.
- X. Jiang, S. Weise, M. Hafner, C. Röcker, F. Zhang, W. J. Parak and G. U. Nienhaus, J. R. Soc., Interface, 2009, 7, S5–S13 CrossRef PubMed.
- E. C. Cho, Q. Zhang and Y. Xia, Nat. Nanotechnol., 2011, 6, 385–391 CrossRef CAS PubMed.
- V. Hirsch, J. Salaklang, B. Rothen-Rutishauser and A. Petri-Fink, IEEE Trans. Magn., 2013, 49, 402–407 CrossRef.
- L. H. Allen and E. Matijević, J. Colloid Interface Sci., 1969, 31, 287–296 CrossRef CAS.
- L. Zha, J. Hu, C. Wang, S. Fu and M. Luo, Colloid Polym. Sci., 2002, 280, 1116–1121 CAS.
- R. A. French, A. R. Jacobson, B. Kim, S. L. Isley, R. L. Penn and P. C. Baveye, Environ. Sci. Technol., 2009, 43, 1354–1359 CrossRef CAS.
- S. Ghosh, W. Jiang, J. D. McClements and B. Xing, Langmuir, 2011, 27, 8036–8043 CrossRef CAS PubMed.
- L. Rodriguez-Lorenzo, K. Fytianos, F. Blank, C. von Garnier, B. Rothen-Rutishauser and A. Petri-Fink, Small, 2014, 10, 1341–1350 CrossRef CAS PubMed.
- S. Kittler, C. Greulich, J. Diendorf, M. Köller and M. Epple, Chem. Mater., 2010, 22, 4548–4554 CrossRef CAS.
- T. S. Peretyazhko, Q. Zhang and V. L. Colvin, Environ. Sci. Technol., 2014, 48, 11954–11961 CrossRef CAS PubMed.
- S. J. Soenen, J.-M. Montenegro, A. M. Abdelmonem, B. B. Manshian, S. H. Doak, W. J. Parak, S. C. De Smedt and K. Braeckmans, Acta Biomater., 2014, 10, 732–741 CrossRef CAS PubMed.
- H. Ma, P. L. Williams and S. A. Diamond, Environ. Pollut., 2013, 172, 76–85 CrossRef CAS PubMed.
- P. Rivera-Gil, D. Jimenez De Aberasturi, V. Wulf, B. Pelaz, P. Del Pino, Y. Zhao, J. M. De La Fuente, I. Ruiz De Larramendi, T. Rojo, X.-J. Liang and W. J. Parak, Acc. Chem. Res., 2013, 46, 743–749 CrossRef CAS PubMed.
- C. Muhlfeld, P. Gehr and B. Rothen-Rutishauser, Swiss Med. Wkly., 2008, 138, 387–391 Search PubMed.
- T.-G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS PubMed.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718–2739 RSC.
- I. Hamer, G. Van Beersel, T. Arnould and M. Jadot, Curr. Drug Metab., 2012, 13, 1371–1387 CrossRef CAS.
- M. Chanana, P. Rivera_Gil, M. A. Correa-Duarte, L. M. Liz-Marzán and W. J. Parak, Angew. Chem., Int. Ed., 2013, 52, 4179–4183 CrossRef CAS PubMed.
- R. Foldbjerg, X. Jiang, T. Miclaus, C. Chen, H. Autrup and C. Beer, Toxicol. Res., 2015, 4, 563–575 RSC.
- S. J. Soenen, W. J. Parak, J. Rejman and B. Manshian, Chem. Rev., 2015, 115, 2109–2135 CrossRef CAS PubMed.
- H. Eagle, Science, 1955, 122, 501–504 CAS.
- W. J. Bettger and W. L. McKeehan, Physiol. Rev., 1986, 66, 1–35 CAS.
- N. Psychogios, D. D. Hau, J. Peng, A. C. Guo, R. Mandal, S. Bouatra, I. Sinelnikov, R. Krishnamurthy, R. Eisner, B. Gautam, N. Young, J. Xia, C. Knox, E. Dong, P. Huang, Z. Hollander, T. L. Pedersen, S. R. Smith, F. Bamforth, R. Greiner, B. McManus, J. W. Newman, T. Goodfriend and D. S. Wishart, PLoS One, 2011, 6, e16957 CAS.
- C. Lazzari, A. Spreafico, A. Bachi, H. Roder, I. Floriani, D. Garavaglia, A. Cattaneo, J. Grigorieva, M. G. Vigano and C. Sorlini, J. Thorac. Oncol., 2012, 7, 40–48 CrossRef CAS PubMed.
- R. Dulbecco and G. Freeman, Virology, 1959, 8, 396–397 CrossRef CAS.
- G. E. Moore, R. E. Gerner and H. Franklin, JAMA, J. Am. Med. Assoc., 1967, 199, 519–524 CrossRef CAS.
- W. T. Geigy, Aufl. Ciba-Geigy: Basel, 1985, 1–252 Search PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- T. A. Larson, P. P. Joshi and K. Sokolov, ACS Nano, 2012, 6, 9182–9190 CrossRef CAS PubMed.
- M. Lagarde, B. Sicard, M. Guichardant, O. Felisi and M. Dechavanne, In Vitro, 1984, 20, 33–37 CrossRef CAS.
- M. Iijima and H. Kamiya, KONA Powder Part. J., 2009, 27, 119–129 CrossRef CAS.
- P. Bihari, M. Vippola, S. Schultes, M. Praetner, A. Khandoga, C. Reichel, C. Coester, T. Tuomi, M. Rehberg and F. Krombach, Part. Fibre Toxicol., 2008, 5, 14 CrossRef PubMed.
- Z. P. Chen, Y. Zhang, S. Zhang, J. G. Xia, J. W. Liu, K. Xu and N. Gu, Colloids Surf., A, 2008, 316, 210–216 CrossRef CAS PubMed.
- C. Graf, Q. Gao, I. Schütz, C. N. Noufele, W. Ruan, U. Posselt, E. Korotianskiy, D. Nordmeyer, F. Rancan, S. Hadam, A. Vogt, J. Lademann, V. Haucke and E. Rühl, Langmuir, 2012, 28, 7598–7613 CrossRef CAS PubMed.
- Z. Ji, X. Jin, S. George, T. Xia, H. Meng, X. Wang, E. Suarez, H. Zhang, E. M. V. Hoek, H. Godwin, A. E. Nel and J. I. Zink, Environ. Sci. Technol., 2010, 44, 7309–7314 CrossRef CAS PubMed.
- J. Cohen, J. Teeguarden and P. Demokritou, Part. Fibre Toxicol., 2014, 11, 1–12 CrossRef PubMed.
- V. Ayala, A. P. Herrera, M. Latorre-Esteves, M. Torres-Lugo and C. Rinaldi, J. Nanopart. Res., 2013, 15, 1–14 CrossRef PubMed.
- S. G. Jang, D. J. Audus, D. Klinger, D. V. Krogstad, B. J. Kim, A. Cameron, S.-W. Kim, K. T. Delaney, S.-M. Hur, K. L. Killops, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, J. Am. Chem. Soc., 2013, 135, 6649–6657 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, A. K. Patri, J. Zheng, J. D. Clogston, N. Ayub, P. Aggarwal, B. W. Neun, J. B. Hall and S. E. McNeil, Nanotechnol. Biol. Med., 2009, 5, 106–117 CrossRef CAS PubMed.
- S. M. Moghimi, I. S. Muir, L. Illum, S. S. Davis and V. Kolb-Bachofen, Biochim. Biophys. Acta, Mol. Cell Res., 1993, 1179, 157–165 CrossRef CAS.
- D. Dutta, S. K. Sundaram, J. G. Teeguarden, B. J. Riley, L. S. Fifield, J. M. Jacobs, S. R. Addleman, G. A. Kaysen, B. M. Moudgil and T. J. Weber, Toxicol. Sci., 2007, 100, 303–315 CrossRef CAS PubMed.
- J. L. Perry, K. G. Reuter, M. P. Kai, K. P. Herlihy, S. W. Jones, J. C. Luft, M. Napier, J. E. Bear and J. M. DeSimone, Nano Lett., 2012, 12, 5304–5310 CrossRef CAS PubMed.
- M. M. Frank and L. F. Fries, Immunol. Today, 1991, 12, 322–326 CrossRef CAS.
- K. Avgoustakis, A. Beletsi, Z. Panagi, P. Klepetsanis, E. Livaniou, G. Evangelatos and D. S. Ithakissios, Int. J. Pharm., 2003, 259, 115–127 CrossRef CAS.
- S. Louguet, A. C. Kumar, N. Guidolin, G. Sigaud, E. Duguet, S. Lecommandoux and C. Schatz, Langmuir, 2011, 27, 12891–12901 CrossRef CAS PubMed.
- A. Musyanovych, J. Dausend, M. Dass, P. Walther, V. Mailänder and K. Landfester, Acta Biomater., 2011, 7, 4160–4168 CrossRef CAS PubMed.
- J. Manson, D. Kumar, B. Meenan and D. Dixon, Gold Bull., 2011, 44, 99–105 CrossRef CAS PubMed.
- L. Stebounova, E. Guio and V. Grassian, J. Nanopart. Res., 2011, 13, 233–244 CrossRef CAS.
- Y. L. Zhang, Y. Yang, J. H. Zhao, R. Q. Tan, P. Cui and W. J. Song, J. Sol-Gel Sci. Technol., 2009, 51, 198–203 CrossRef CAS PubMed.
- V. Hirsch, C. Kinnear, M. Moniatte, B. Rothen-Rutishauser, M. J. D. Clift and A. Fink, Nanoscale, 2013, 5, 3723–3732 RSC.
- G. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D. Liang and C. Li, Biomaterials, 2009, 30, 1928–1936 CrossRef CAS PubMed.
- B. Chanteau, J. Fresnais and J. F. Berret, Langmuir, 2009, 25, 9064–9070 CrossRef CAS PubMed.
- A. Petri-Fink, B. Steitz, A. Finka, J. Salaklang and H. Hofmann, Eur. J. Pharm. Biopharm., 2008, 68, 129–137 CrossRef CAS PubMed.
- F. Goycoolea, A. Valle-Gallego, R. Stefani, B. Menchicchi, L. David, C. Rochas, M. Santander-Ortega and M. Alonso, Colloid Polym. Sci., 2012, 290, 1423–1434 CAS.
- G. Jiang, J. Pichaandi, N. J. J. Johnson, R. D. Burke and F. C. J. M. van Veggel, Langmuir, 2012, 28, 3239–3247 CrossRef CAS PubMed.
- S. L. Saville, R. C. Stone, B. Qi and O. T. Mefford, J. Mater. Chem., 2012, 22, 24909–24917 RSC.
- S. Wang, H. Song, W. Y. Ong, M. Y. Han and D. Huang, Nanotechnology, 2009, 20, 425102 CrossRef PubMed.
- E. Izak-Nau, M. Voetz, S. Eiden, A. Duschl and V. F. Puntes, Part. Fibre Toxicol., 2013, 10, 56 CrossRef PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- N. Hondow, R. Brydson, P. Wang, M. Holton, M. R. Brown, P. Rees, H. Summers and A. Brown, J. Nanopart. Res., 2012, 14, 1–15 CrossRef.
- C. Rocker, M. Potzl, F. Zhang, W. J. Parak and G. U. Nienhaus, Nat. Nanotechnol., 2009, 4, 577–580 CrossRef PubMed.
- C. S. Chern, C. K. Lee and C. C. Ho, Colloid Polym. Sci., 1999, 277, 979–985 CAS.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS PubMed.
- P. P. Karmali and D. Simberg, Expert Opin. Drug Delivery, 2011, 8, 343–357 CrossRef CAS PubMed.
- P. M. Claesson, E. Blomberg, J. C. Froberg, T. Nylander and T. Arnebrant, Adv. Colloid Interface Sci., 1995, 57, 161–227 CrossRef CAS.
- E. Dickinson, V. J. Pinfield, D. S. Horne and F. A. M. Leermakers, J. Chem. Soc., Faraday Trans., 1997, 93, 1785–1790 RSC.
- T. Nylander and N. M. Wahlgren, Langmuir, 1997, 13, 6219–6225 CrossRef CAS.
- S. Ohnishi, M. Murata and M. Hato, Biophys. J., 1998, 74, 455–465 CrossRef CAS.
- S. Ramos, J. Campos-Teran, J. Mas-Oliva, T. Nylander and R. Castillo, Langmuir, 2008, 24, 8568–8575 CrossRef CAS PubMed.
- L. Meagher and H. J. Griesser, Colloids Surf., B, 2002, 23, 125–140 CrossRef CAS.
- W. R. Bowen, N. Hilal, R. W. Lovitt and C. J. Wright, J. Colloid Interface Sci., 1998, 197, 348–352 CrossRef CAS.
- J. J. Valle-Delgado, J. A. Molina-Bolivar, F. Galisteo-Gonzalez, M. J. Galvez-Ruiz, A. Feiler and M. W. Rutland, Langmuir, 2005, 21, 9544–9554 CrossRef CAS PubMed.
- J. J. Valle-Delgado, J. A. Molina-Bolivar, F. Galisteo-Gonzalez and M. J. Galvez-Ruiz, Curr. Opin. Colloid Interface Sci., 2011, 16, 572–578 CrossRef CAS PubMed.
- J. J. Valle-Delgado, J. A. Molina-Bolivar, F. Galisteo-Gonzalez, M. J. Galvez-Ruiz, A. Feiler and M. Rutland, Phys. Chem. Chem. Phys., 2004, 6, 1482–1486 RSC.
- A. C. Sabuncu, J. Grubbs, S. Qian, T. M. Abdel-Fattah, M. W. Stacey and A. Beskok, Colloids Surf., B, 2012, 95, 96–102 CrossRef CAS PubMed.
- H. Kato, K. Fujita, M. Horie, M. Suzuki, A. Nakamura, S. Endoh, Y. Yoshida, H. Iwahashi, K. Takahashi and S. Kinugasa, Toxicol. In Vitro, 2010, 24, 1009–1018 CrossRef CAS PubMed.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. F. Puntes, Small, 2011, 7, 3479–3486 CrossRef CAS PubMed.
- M. Safi, J. Courtois, M. Seigneuret, H. Conjeaud and J. F. Berret, Biomaterials, 2011, 32, 9353–9363 CrossRef CAS PubMed.
- B. Díaz, C. Sánchez-Espinel, M. Arruebo, J. Faro, E. de Miguel, S. Magadán, C. Yagüe, R. Fernández-Pacheco, M. R. Ibarra, J. Santamaría and Á. González-Fernández, Small, 2008, 4, 2025–2034 CrossRef PubMed.
- K. Shapero, F. Fenaroli, I. Lynch, D. C. Cottell, A. Salvati and K. A. Dawson, Mol. BioSyst., 2011, 7, 371–378 RSC.
- F. Rancan, Q. Gao, C. Graf, S. Troppens, S. Hadam, S. Hackbarth, C. Kembuan, U. Blume-Peytavi, E. Rühl, J. Lademann and A. Vogt, ACS Nano, 2012, 6, 6829–6842 CrossRef CAS PubMed.
- R. Y. Prasad, K. Wallace, K. M. Daniel, A. H. Tennant, R. M. Zucker, J. Strickland, K. Dreher, A. D. Kligerman, C. F. Blackman and D. M. DeMarini, ACS Nano, 2013, 7, 1929–1942 CrossRef CAS PubMed.
- Z. E. Allouni, M. R. Cimpan, P. J. Høl, T. Skodvin and N. R. Gjerdet, Colloids Surf., B, 2009, 68, 83–87 CrossRef CAS PubMed.
- N. Nafee, M. Schneider, U. F. Schaefer and C.-M. Lehr, Int. J. Appl. Pharm., 2009, 381, 130–139 CrossRef CAS PubMed.
- A. Bakandritsos, G. Mattheolabakis, R. Zboril, N. Bouropoulos, J. Tucek, D. G. Fatouros and K. Avgoustakis, Nanoscale, 2010, 2, 564–572 RSC.
- D. Hühn, K. Kantner, C. Geidel, S. Brandholt, I. De Cock, S. J. H. Soenen, P. Rivera_Gils, J.-M. Montenegro, K. Braeckmans, K. Müllen, G. U. Nienhaus, M. Klapper and W. J. Parak, ACS Nano, 2013, 7, 3253–3263 CrossRef PubMed.
- A. Kodiyan, E. A. Silva, J. Kim, M. Aizenberg and D. J. Mooney, ACS Nano, 2012, 6, 4796–4805 CrossRef CAS PubMed.
- L. Maus, O. Dick, H. Bading, J. P. Spatz and R. Fiammengo, ACS Nano, 2010, 4, 6617–6628 CrossRef CAS PubMed.
- H. T. R. Wiogo, M. Lim, V. Bulmus, L. Gutiérrez, R. C. Woodward and R. Amal, Langmuir, 2012, 28, 4346–4356 CrossRef CAS PubMed.
- A. S. Pitek, D. O'Connell, E. Mahon, M. P. Monopoli, F. Baldelli Bombelli and K. A. Dawson, PLoS One, 2012, 7, e40685 CAS.
- H. Casanova, J. S. Chen, E. Dickinson, B. S. Murray, P. V. Nelson and M. Whittle, Phys. Chem. Chem. Phys., 2000, 2, 3861–3869 RSC.
- L. Davalos-Pantoja, J. L. Ortega-Vinuesa, D. Bastos-Gonzalez and R. Hidalgo-Alvarez, Colloids Surf., B, 2001, 20, 165–175 CrossRef CAS.
- T. Lopez-Leon, P. M. Gea-Jodar, D. Bastos-Gonzalez and J. L. Ortega-Vinuesa, Langmuir, 2005, 21, 87–93 CrossRef CAS PubMed.
- T. Lopez-Leon, A. B. Jodar-Reyes, J. L. Ortega-Vinuesa and D. Bastos-Gonzalez, J. Colloid Interface Sci., 2005, 284, 139–148 CrossRef CAS PubMed.
- J. A. Molina-Bolivar, F. Galisteo-Gonzalez and R. Hidalgo-Alvarez, J. Colloid Interface Sci., 1998, 206, 518–526 CrossRef CAS.
- J. A. Molina-Bolivar, F. Galisteo-Gonzalez and R. Hidalgo-Alvarez, Colloids Surf., B, 1999, 14, 3–17 CrossRef CAS.
- J. A. Molina-Bolivar, F. Galisteo-Gonzalez and R. Hidalgo-Alvarez, J. Chem. Phys., 1999, 110, 5412–5420 CrossRef CAS PubMed.
- J. A. Molina-Bolivar and J. L. Ortega-Vinuesa, Langmuir, 1999, 15, 2644–2653 CrossRef CAS.
- J. A. Molina-Bolivar, F. Galisteo-Gonzalez and R. Hidalgo-Alvarez, Colloids Surf., B, 2001, 21, 125–135 CrossRef CAS.
- E. Ruckenstein and M. Manciu, Langmuir, 2002, 18, 7584–7593 CrossRef CAS.
- J. G. Teeguarden, P. M. Hinderliter, G. Orr, B. D. Thrall and J. G. Pounds, Toxicol. Sci., 2007, 95, 300–312 CrossRef CAS PubMed.
- C. P. Johnson, X. Li and B. E. Logan, Environ. Sci. Technol., 1996, 30, 1911–1918 CrossRef CAS.
- R. Jullien, Contemp. Phys., 1987, 28, 477–493 CrossRef PubMed.
- P. C. Hiemenz and R. Rajagopalan, Principles Colloid Surf. Chem., revised and expanded, CRC Press, 1997 Search PubMed.
- A. Y. Kim and J. C. Berg, Langmuir, 2000, 16, 2101–2104 CrossRef CAS.
- M. Lattuada, H. Wu and M. Morbidelli, J. Colloid Interface Sci., 2003, 268, 96–105 CrossRef CAS PubMed.
- M. C. Sterling Jr, J. S. Bonner, A. N. S. Ernest, C. A. Page and R. L. Autenrieth, Water Res., 2005, 39, 1818–1830 CrossRef PubMed.
- P. Hinderliter, K. Minard, G. Orr, W. Chrisler, B. Thrall, J. Pounds and J. Teeguarden, Part. Fibre Toxicol., 2010, 7, 36 CrossRef CAS PubMed.
- G. DeLoid, J. M. Cohen, T. Darrah, R. Derk, L. Rojanasakul, G. Pyrgiotakis, W. Wohlleben and P. Demokritou, Nat. Commun., 2014, 5, 3514 Search PubMed.
- T. A. Witten Jr and L. M. Sander, Phys. Rev. Lett., 1981, 47, 1400 CrossRef.
- M. Y. Lin, H. M. Lindsay, D. A. Weitz, R. Klein, R. C. Ball and P. Meakin, J. Phys.: Condens. Matter, 1990, 2, 3093 CrossRef.
- P. Tandon and D. E. Rosner, J. Colloid Interface Sci., 1999, 213, 273–286 CrossRef CAS PubMed.
- J. L. Burns, Y.-d. Yan, G. J. Jameson and S. Biggs, Langmuir, 1997, 13, 6413–6420 CrossRef CAS.
- Z. Zhou and B. Chu, J. Colloid Interface Sci., 1991, 143, 356–365 CrossRef CAS.
- B. J. Berne and R. Pecora, Dynamic Light Scattering: with appl. chem., biol., phys., Courier Dover Publications, 2000 Search PubMed.
- S. Balog, L. Rodriguez-Lorenzo, C. A. Monnier, B. Michen, M. Obiols-Rabasa, L. Casal-Dujat, B. Rothen-Rutishauser, A. Petri-Fink and P. Schurtenberger, J. Phys. Chem. C, 2014, 118, 17968–17974 CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- P. K. Jain and M. A. El-Sayed, Chem. Phys. Lett., 2010, 487, 153–164 CrossRef CAS PubMed.
- E. Hao, S. Li, R. C. Bailey, S. Zou, G. C. Schatz and J. T. Hupp, J. Phys. Chem. B, 2004, 108, 1224–1229 CrossRef CAS.
- D. Aili, P. Gryko, B. Sepulveda, J. A. G. Dick, N. Kirby, R. Heenan, L. Baltzer, B. Liedberg, M. P. Ryan and M. M. Stevens, Nano Lett., 2011, 11, 5564–5573 CrossRef CAS PubMed.
- H. Deng, Y. Xu, Y. Liu, Z. Che, H. Guo, S. Shan, Y. Sun, X. Liu, K. Huang and X. Ma, Anal. Chem., 2012, 84, 1253–1258 CrossRef CAS PubMed.
- G. Maiorano, S. Sabella, B. Sorce, V. Brunetti, M. A. Malvindi, R. Cingolani and P. P. Pompa, ACS Nano, 2010, 4, 7481–7491 CrossRef CAS PubMed.
- A. Vanderkooy, Y. Chen, F. Gonzaga and M. A. Brook, ACS Appl. Mater. Interfaces, 2011, 3, 3942–3947 CAS.
- D. B. Williams and C. B. Carter, Transmission Electron Microscopy, Springer, 2009, pp. 3–22 Search PubMed.
- S. A. Wight and C. J. Zeissler, Microsc. Res. Tech., 1993, 25, 393–397 CrossRef CAS PubMed.
- G. D. Danilatos, Microchim. Acta, 1994, 114, 143–155 CrossRef.
- D. G. de Oteyza, P. Gorman, Y.-C. Chen, S. Wickenburg, A. Riss, D. J. Mowbray, G. Etkin, Z. Pedramrazi, H.-Z. Tsai and A. Rubio, Science, 2013, 340, 1434–1437 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cs00487f |
| ‡ Current address: Agroscope, Institute for Livestock Sciences ILS, 1725 Posieux, Switzerland. |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |