
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmerging translational research on magnetic nanoparticles for regenerative medicine
Yu
Gao
,
Jing
Lim
,
Swee-Hin
Teoh
and
Chenjie
Xu
*
Division of Bioengineering, School of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore. E-mail: cjxu@ntu.edu.sg
First published on 12th June 2015
Abstract
Regenerative medicine, which replaces or regenerates human cells, tissues or organs, to restore or establish normal function, is one of the fastest-evolving interdisciplinary fields in healthcare. Over 200 regenerative medicine products, including cell-based therapies, tissue-engineered biomaterials, scaffolds and implantable devices, have been used in clinical development for diseases such as diabetes and inflammatory and immune diseases. To facilitate the translation of regenerative medicine from research to clinic, nanotechnology, especially magnetic nanoparticles have attracted extensive attention due to their unique optical, electrical, and magnetic properties and specific dimensions. In this review paper, we intend to summarize current advances, challenges, and future opportunities of magnetic nanoparticles for regenerative medicine.
 Yu Gao | Yu Gao received his BS and MS in Chemistry from Shandong University (2004) and Central South University (2008), respectively. Then he joined the Research Center for Eco-Environmental Science, Chinese Academy of Sciences to continue his PhD (2008–2012). His project focused on the development of biosensors to investigate the interaction of small molecules with biological targets. He is currently a research fellow under the mentorship of Dr Chenjie Xu at the School of Chemical and Biomedical Engineering, Nanyang Technological University since 2012. His current research focuses on the application of nanotechnology for regenerative medicine, especially cell tracking and controlled drug delivery. |
 Jing Lim | Jing Lim received his BEng (Hons) and MEng (research) from the National University of Singapore in 2010 and 2011, respectively. He was awarded his PhD in Bioengineering from Nanyang Technological University in 2015, under the supervision of Professor Swee-Hin Teoh. His research focused on developing composite biomaterials for the field of bone tissue engineering using solvent-free techniques. He strongly believes that a solvent-free technique for processing biomaterials promotes their translation to clinical use. He is currently working on other innovative composite biomaterials at the interface of nanotechnology and polymer processing. |
 Swee-Hin Teoh | Swee-Hin Teoh received his BEng and PhD from Monash University in 1978 and 1982, respectively. He is Professor of Bioengineering, Chair of the School of Chemical and Biomedical Engineering, and Director of the Renaissance Engineering Program, Nanyang Technological University. He is a Fellow of the Academy of Engineers, Singapore. He has published over 300 papers, filed 8 patents and sat on the board of editors of several journals in the tissue engineering field. He guided Osteopore to obtain FDA approval and a CE mark and implanted it in more than 1500 patients. His main field of research is biomaterials and bone tissue engineering. |
 Chenjie Xu | Chenjie Xu, an Assistant Professor at Nanyang Technological University, received his BS from Nanjing University (2002) and MPhil from Hong Kong University of Science and Technology (2004). After a one-year internship at Stanford University (2005), he continued his PhD under the supervision of Professor Shouheng Sun at Brown University (2005–2009), where he was the recipient of the Vince Wernig Fellowship, Joukowsky Outstanding Dissertation Prize, and Potter Prize for Outstanding Doctoral Thesis. From 2009 to 2012, he was a research fellow under Professor Jeffrey Karp at Brigham and Women's Hospital. His current research focuses on the application of nanotechnology for regenerative medicine. |
1. Introduction
Regenerative medicine, including cell therapy and tissue engineering, replaces or regenerates human cells, tissues or organs, to restore or establish normal function, and it is one of the fastest-evolving interdisciplinary fields in healthcare research.1 It has the potential to tackle many major health problems such as cardiovascular disease, metabolic diseases, cancer, brain and spinal cord injury, as well as organ failure.2–5 So far, over 200 regenerative medicine products, including cell-based therapies, tissue-engineered biomaterials, scaffolds and implantable devices, have been in clinical development for diseases such as diabetes and inflammatory and immune diseases.6 Despite these efforts, unfortunately, few products have negotiated the arduous path to clinical applications, which have been challenged by a range of scientific and engineering questions such as the biomanufacturing of stem cells or tissues (the actual production, packaging, and delivery of a well-defined product), the understanding and control of micro-environmental signals, and the assessment of the efficacy of therapies.7–9The emergence of nanotechnology offers promising perspectives to address these challenges. First of all, the nanoscale of nanomaterials offers comparable sizes to biological molecules and thus improves the interactions between them. Second, nanoscale structures are able to control cellular functions such as adhesion, proliferation, and differentiation. Third, nanomaterials with unique optical and magnetic properties are ideal contrast agents for monitoring cellular behavior in vivo following a transplantation. To substantiate these points, we have seen the application of nanofibrous materials that mimic native extracellular matrix, thereby promoting the adhesion of various cell types. These nanomaterials were subsequently developed as tissue-engineered scaffolds for skin, bone, vasculature, and other tissues.10–14 In addition, nanostructures and nanofibrils have also been shown to control the behavior of primary stem cells such as adhesion, growth and differentiation.15–17 To visualize and track cellular motility, superparamagnetic iron oxide nanoparticles have been developed, which allow non-invasive monitoring of cells in a living organism.18–22
Among these interesting techniques, magnetic nanoparticles (MNPs) are particularly attractive due to their unique magnetic properties and unique dimensions.23 Represented by magnetite nanoparticles (Fe3O4 NPs), MNPs are often made in sizes smaller than 20 nm in diameter and their magnetization directions are subject to thermal fluctuations at room or biological temperatures. Therefore, without an external magnetic field, their overall magnetization value is randomized to zero. Such fluctuations in magnetization direction minimize the magnetic interactions between any two dispersed NP, which makes the dispersion stable in physiological solutions and facilitates coupling of NP with biological agents. On the other hand, once these MNPs are exposed to an external magnetic field, they can align along the field direction, achieving magnetic saturation of a magnitude that far exceeds that from any known biological entity. This unique property of MNPs allows not only the detection of biological samples that contain the MNP, but also the manipulation of these biological samples by an external magnetic field.24–30 In comparison, optically active NPs such as quantum dots, gold NPs, and upconversion NPs are limited to superficial applications due to the penetration depth of related optical methodologies.21 Carbon-based materials have unique optical, electric, and even magnetic properties, but require thorough study of their biocompatibility.31 Gas-filled nano/microbubbles allow remote control and visualization by ultrasound technologies, but suffer from a short halflife in biological systems and technical challenges in functionalization.32
The aim of this paper is to provide an updated, comprehensive and systematic review of MNPs for regenerative medicine. Special emphasis will be placed on the fundamental background of regenerative medicine and its unmet challenges and how the innovation and utilization of MNPs can help address these needs and thus facilitate the translation of MNPs to regenerative medicine. In general, successful translation to regenerative medicine needs collaboration between researchers from different disciplines, including materials, nanotechnology, and molecular biology.
2. The unmet challenges in regenerative medicine
Major improvements in healthcare all over the world in terms of sterilization techniques, the availability of off-the-shelf replacements for body parts, and advances in drug delivery techniques have contributed to an increase in lifespan, while evident changes in lifestyle (to being more physically active) also contribute to a high demand for tissue replacements. Collectively, the need for tissue replacements and drug delivery devices provided the motivation for the development of regenerative medicine (including tissue engineering). In fact, the beginning of regenerative medicine could be traced back as early as 1902 (Alexis Carrel) in the field of cardiovascular and transplant surgery.33 In the use of materials as grafts, gold plates were probably one of the first biocompatible materials that were used (as bone replacements) back in the 16th century.34 Since then, the field of regenerative medicine has seen significant progress, owing to our improved understanding of tissue and organ epidemiology, as well as disease etiology and traumatology.35–37 Although the gold standard of replacement remains the autograft, the lack of appropriate autografts, combined with the increasing demand for tissue replacements, fuels the need for the development and clinical translation of biomaterials.The approach to regenerative medicine varies according to the requirements of the target tissue and organ, as most people would expect. Therefore, there are distinct principles from which regenerative medicine may proceed, for which we have proposed the following five central areas: biomaterials, cells, bioreactors, growth factors, and bioimaging.38 Given the biological complexity and structural make-up of our native tissue, it is not unusual that an applied regenerative technique combines two or more of these areas. For example, Liu et al. employed bioactive polycaprolactone–tricalcium phosphate (PCL–TCP) three-dimensional (3D) scaffolds that were seeded with MSCs and endothelial colony-forming cells (ECFCs) and showed that this combination resulted in higher osteogenic and angiogenic expressions compared to a PCL–TCP MSC-only group. Corresponding studies in the mouse revealed that host neovascularization increased 2.2-fold.39 In another example, Zhang et al. combined PCL–TCP scaffolds with MSCs that had been cultured in a dynamic biaxial rotating bioreactor and demonstrated enhanced osteogenicity in vitro.40 In the approach taken by Khademhosseini, hyperbranched polyesters (HPE) were used to encapsulate hydrophobic drugs, which exhibited sustained release.41 By functionalizing HPE with photocrosslinkable acrylate moieties, the modulus of HPE was tuned to facilitate cell adhesion, spreading, and proliferation. Wulkersdorfer et al. combined polyglycolic acid meshes with growth factors for the generation of tissue-engineered small intestine (TESI) and showed that the type of growth factor incorporated could manipulate the surface area of neomucosa and TESI morphology.42 From the above examples, we can clearly see the importance of the combined use of biomaterials, cells, bioreactors, and growth factors in regenerative medicine research.
Although these approaches are becoming more dynamic and perhaps closer to clinical translation, there remains the need to develop techniques to control the fate and function of engineered tissues to achieve therapeutic effects in vivo. An unfortunate but classic example of our apparent lack of understanding and control would be the use and off-label use of recombinant human bone morphogenetic protein-2 (rhBMP-2), as reported between 2008 and 2010.43–45 Although the Food and Drug Administration (FDA) has approved the clinical use of rhBMP-2 in the lumbar spine together with the lumbar tapered cage, it does not have approval for use in surgical interventions such as anterior cervical fusion applications, posterior lumbar interbody fusion and transforaminal lumbar interbody fusion (TLIF). As a result of off-label use, 4 patients suffered from delayed symptomatic neural compression. Worryingly, 85% of the principal surgical procedures that were conducted in the United States using rhBMP-2 for off-label applications took place without understanding the clinical consequences of its off-label usage. In this light, we feel that more may be done to control the fate and function of engineered tissue in order to achieve therapeutic effects in vivo.
On this note, there are few minimally invasive methods of control that we may apply without post-surgical intervention. To the authors' knowledge, this is the first instance where we comprehensively review how post-surgical control of the fate and function of cells and/or scaffolds may be achieved, specifically via the use of MNPs. As mentioned earlier, MNPs possess interesting characteristics that make them ideal candidates to support and augment current regenerative techniques. More importantly, MNPs are compatible with current imaging techniques available in clinics, such as magnetic resonance imaging (MRI), positron emission tomography (PET), and computed tomography (CT) imaging. Fluorescent probes have evidently been investigated and widely studied in the forms of fluorescent proteins,46,47 molecular fluorophores,48,49 or quantum dots,50,51 but are limited to superficial tissues, due to the low depth of penetration of the imaging tools specific for fluorescent probes. In addition, the decay of fluorescence intensity over time is a concern. Therefore, currently MRI aided by MNPs is still the mainstream effective method in clinics for non-invasively tracking transplanted cells and evaluating the efficacy of regenerative medicine.
In the following section, we will outline each of the five main areas of regenerative medicine and the unmet challenges for their translation into clinics after a brief summary of surface modification strategies for MNPs (Fig. 1).
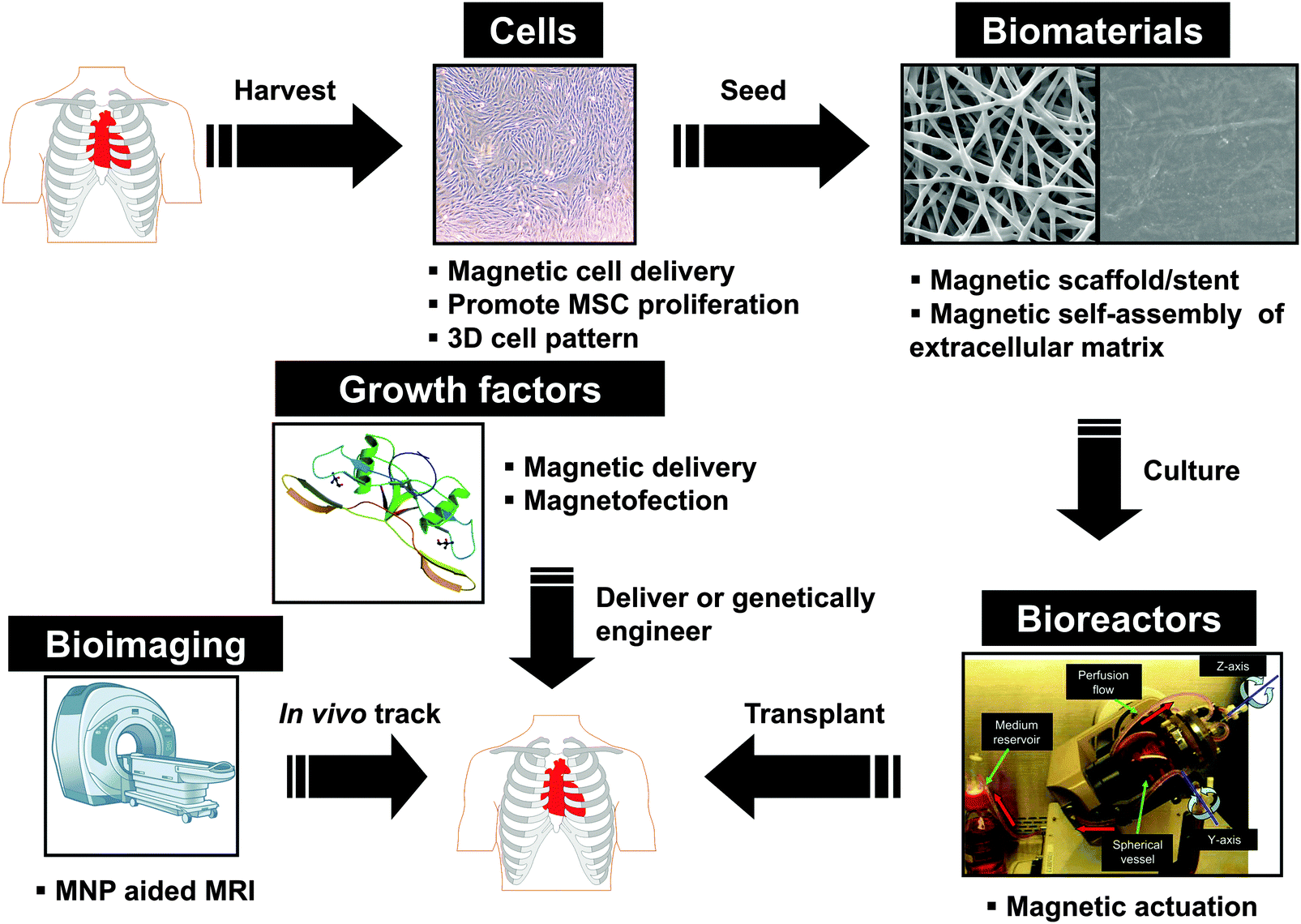 | ||
| Fig. 1 Schematic of the five main areas in regenerative medicine and how magnetic nanoparticles may be useful in each area. The five areas include cells, biomaterials, bioreactors (reproduced from ref. 38 with permission), growth factors, and bioimaging. | ||
3. MNPs to address unmet needs in regenerative medicine
3.1 Surface modification strategies
The physical and chemical characteristics of MNPs are closely related to their size, morphology, and surface chemistry.52,53 As their size and morphology contribute more to the physical properties of MNPs, their surface chemistry determines the stability, biocompatibility, and functions of MNPs in biological systems. Herein, we would like to start from an insight into the significance of surface modification of MNPs for regenerative medicine.If MNPs are to be used in vivo, the requirements for surface chemistry are stricter. It is well known now that any NPs administered through the blood will undergo opsonization, which creates a protein layer on delivered NPs. This protein layer makes NPs identified and cleared by the reticuloendothelial system (RES).53 One idea to weaken this effect is to create a stealth coating. Studies have shown that a neutral, hydrophilic coating such as PEG or dextran could reduce the binding of plasma proteins to the NPs' surface.54 For example, a PEGylated liposome nanocarrier loaded with the anticancer agent doxorubicin, known as Doxil, has been approved for treatment of ovarian cancer and Kaposi's sarcoma since 1995. PEG coatings (5 mol%) effectively reduce the uptake of liposomes by mice peritoneal macrophages to 40% of the value for non-coated forms and significantly increase their plasma circulation time.55
Another factor to consider is the tissue penetration ability of MNPs. Unlike movement in biological fluids, which is driven by magnetic force (speed is inversely proportional to fluid viscosity), movement of MNPs in soft tissue (e.g. in drug delivery) is more complicated due to the complex composition of tissue.57 Therefore, strategies that can control the penetration of particles in tissue are highly desired. Recently, Nance et al. demonstrated that by coating polystyrene NPs (60 or 110 nm) with PEG (5000 Da), the speed of transport of NPs in mouse brain tissue increased by 3 orders of magnitude compared to COOH-coated NPs.58 PEG-coated 60 nm NPs were able to spread to the mouse cerebral cortex as far as 150 μm away from the injection site within 30 min, whereas COOH-coated NPs stayed at the injection site. Another systematic study of the impact of surface charge, shape, and surface modification on the penetration depth of gold NPs in skin was carried out by Fernandes et al.59 In their research, positive charge and rod shape (vs. spherical shape) of gold NPs are favorable factors for penetration of skin. Moreover, when modified with a cell-penetrating peptide, gold NPs penetrated the skin in larger numbers (up to 10 times) in comparison to PEGylated NPs.
Stabilizing and functional molecules on MNPs can be roughly divided into polymers (Mw larger than 10 kDa) and small molecules (Mw less than 10 kDa). Polymers, including chitosan, polyvinylamine (PVA), polyacrylic acid (PAA) and polyethyleneimine (PEI), possess affinity for magnetic materials and can be physisorbed onto an MNP surface via reversible adsorption (e.g. electrostatic adsorption for PEI) without a defined structure.60 Due to the larger size of polymers compared with MNPs, multiple magnetic cores are usually encapsulated by one or multiple polymer chains to form one cluster. Commercial MRI contrast agents (e.g. Feridex and Endorem) are examples in which magnetic cores are encapsulated by dextran. One small problem caused by the use of polymers is an increase in the hydrodynamic diameter of the particles. For example, the hydrodynamic diameters of magnetic NPs with core sizes of 5 nm and 8 nm increased to 45 nm and 90 nm by coating with dextran and PAA, respectively.60,61 A simple way to solve this problem is to choose molecules with low Mw (<10 kDa) that strongly bind to the magnetic core. These molecules, such as dopamine and dimercaptosuccinic acid (DMSA), are usually composed of an anchor part, which strongly binds to the MNPs' surface, a short spacer part, which sterically stabilizes MNPs, and a functional part providing additional functionalities. These small functional molecules are mainly placed on the particle's surface via ligand exchange and they form a monolayer by stretching their hydrophilic heads towards their surroundings.
Besides their size, the presentation and organization of functional molecules are also related to the packing density of surfactants on a NP's surface. Taking 100 nm polylactic acid NPs as an example, the threshold value of the packing density of PEG (2 kDa) for protein resistance would be 0.2 molecules per nm2.62 Due to steric repulsion, a high Mw of PEG will decrease the packing density during grafting. Therefore, PEG with Mw between 1.9 and 5 kDa was found to be optimal for surface modification of MNPs in many biomedical applications.60
The last factor is the architecture of functional molecules on the particle surface. For example, Gillich et al. evaluated the impact of the architecture of the surface coating on the solubility, stability, hydrodynamic diameter, and thermoresponsive behavior of MNPs by grafting linear or hyperbranched PEG by covalent binding of nitrocatechol (anchor group) to the surfaces of iron oxide NPs.63 Although linear and hyperbranched PEG with similar Mw (2737 and 2477 Da, respectively) had similar packing densities on the surface of MNPs (0.69 and 0.73 nm−2, respectively), hyperbranched PEG formed a thinner polymeric shell with lower hydration and higher stiffness, which provided higher colloidal stability compared to linear PEG.63 Besides colloidal stability, the architecture of functional groups on the NP surface would influence their interaction with cells, because an ordered surface conformation can lower the energy barrier during translocation across the membrane.64,65
3.2 Cell and growth factor delivery: current techniques and unmet needs
Cell and growth factor delivery are important elements of regenerative medicine. Successful regeneration relies on the survival, proliferation, and differentiation of transplanted cells at the correct locations following adequate delivery. During regeneration, growth factors, which are naturally occurring substances, are important for regulating a variety of cellular processes such as cellular growth, proliferation, healing and differentiation. For example, growth factors such as BMP have been utilized widely for the stimulation of bone regeneration,66,67 whereas vascular endothelial growth factor (VEGF) has been used for stimulating angiogenic and vasculogenic events.68,69 Localized delivery of platelet-derived growth factor (PDGF) was achieved using tricalcium phosphate matrices and resulted in the promotion of long-term, stable clinical improvements to periodontal defects.70 The use of growth factors in regeneration requires cellular targets, which may either already be present within the site or are transplanted together with the respective growth factors. For example, ex vivo bone tissue engineering strategies now place emphasis on co-culture systems of MSCs and endothelial progenitor cells (EPCs) as strategies for generating highly osteogenic and well-vascularized scaffolds,39,71,72 which stresses the importance of cellular components in regenerative medicine. With this aim, we have recently reviewed some of the key strategies for targeting vascularization and discussed how various vasculogenic/angiogenic cells may be employed in co-culture systems or as part of pre-vascularization strategies to create vascular networks that may quickly anastomose with existing blood vessels and capillaries.73 The incorporation of proteins and growth factors that have the potential to stimulate cellular responses has also been investigated. For example, recombinant human bone morphogenetic proteins (rhBMPs) were found to induce osteogenesis32 and have since been successfully translated into clinical use.74 Combinatorial approaches incorporating BMPs like BMP-2 or BMP-7 have since been developed75–78 and we have also shown that discontinuous triphasic release of BMP from polycaprolactone (PCL) scaffolds led to a higher bone volume fraction and trabecular thickness.79 To provide an alternative to or complement BMPs, You et al. intensively studied heparan sulfates (HS), which are glycosaminoglycan sugars.80 They showed in many studies that HS possess potential osteogenic effects, which may be of interest to the clinical community.81–85 In short, we have seen the creation of novel scaffolds with the incorporation of relevant cell types and growth factors.Although this approach that combines cellular therapeutics and growth factors is promising, key challenges such as distribution and retention after delivery impede the success of this technique. For example, retention of peripheral blood mononuclear cells (PBMCs) after various routes of administration to the heart was notably poor.86 Moreover, the dosage and release profile of growth factors remain a hotly debated topic,87 while chronic inflammation with the use of transplanted scaffolds reduces the efficacy of regenerative medicine.88 To address these issues, a potential solution lies in the use of NPs, specifically MNPs, for controlling targeted cell/drug (including growth factor) delivery,89,90 nucleic acid delivery and transfection.91,92 Construction of cell patterns and 3D tissue by magnetic manipulation also holds great promise for scaffold-free tissue regeneration, which will hopefully reduce the inflammatory response caused by scaffold implantation. Finally, techniques that promote the adhesion and proliferation of therapeutic cells on a scaffold by magnetically self-assembling biological objects with defined nanostructure to mimic an artificial extracellular microenvironment on a scaffold are also highly needed.
A possible strategy using MNPs for targeted delivery in regenerative medicine could be: (1) generating localized magnetic gradients near magnetic stents/scaffolds under an external magnetic field for in vivo deposition of drug-loaded MNPs; (2) pre-loading MNPs and cargoes into porous scaffolds and remotely controlling the deformation and volume changes of scaffolds by a magnetic field to deliver biological agents on demand; and (3) engineering cells with MNPs in order to enhance the efficiency of delivery by magnetic manipulation.
Magnetic stents have been developed for localized drug delivery in vascular disease with the aim of reducing the incidence of restenosis (i.e. reobstruction) after stenting. For example, by applying a relatively strong uniform external magnetic field (∼1000 G), a 304-grade stainless steel stent wire network was efficiently magnetized and high-level magnetic gradients were generated within the stent (Fig. 2A and B).102 This magnetic stent exhibited nearly superparamagnetic properties (remanent magnetization was around 7% of the saturation magnetization value) and was inert in aqueous environments. The magnetic gradients that were generated could attract MNPs loaded with paclitaxel (200 nm, magnetite, Fig. 2C–E) delivered by a catheter in a rat carotid stenting model, which was hard to target when non-magnetic stents were used, because of the blood flow (Fig. 2F–M). Subsequent release of paclitaxel to the surrounding arterial tissue significantly inhibited in-stent restenosis (Fig. 2N and O) at a relatively low dose of MNP-encapsulated paclitaxel (7.5 μg per stent).102
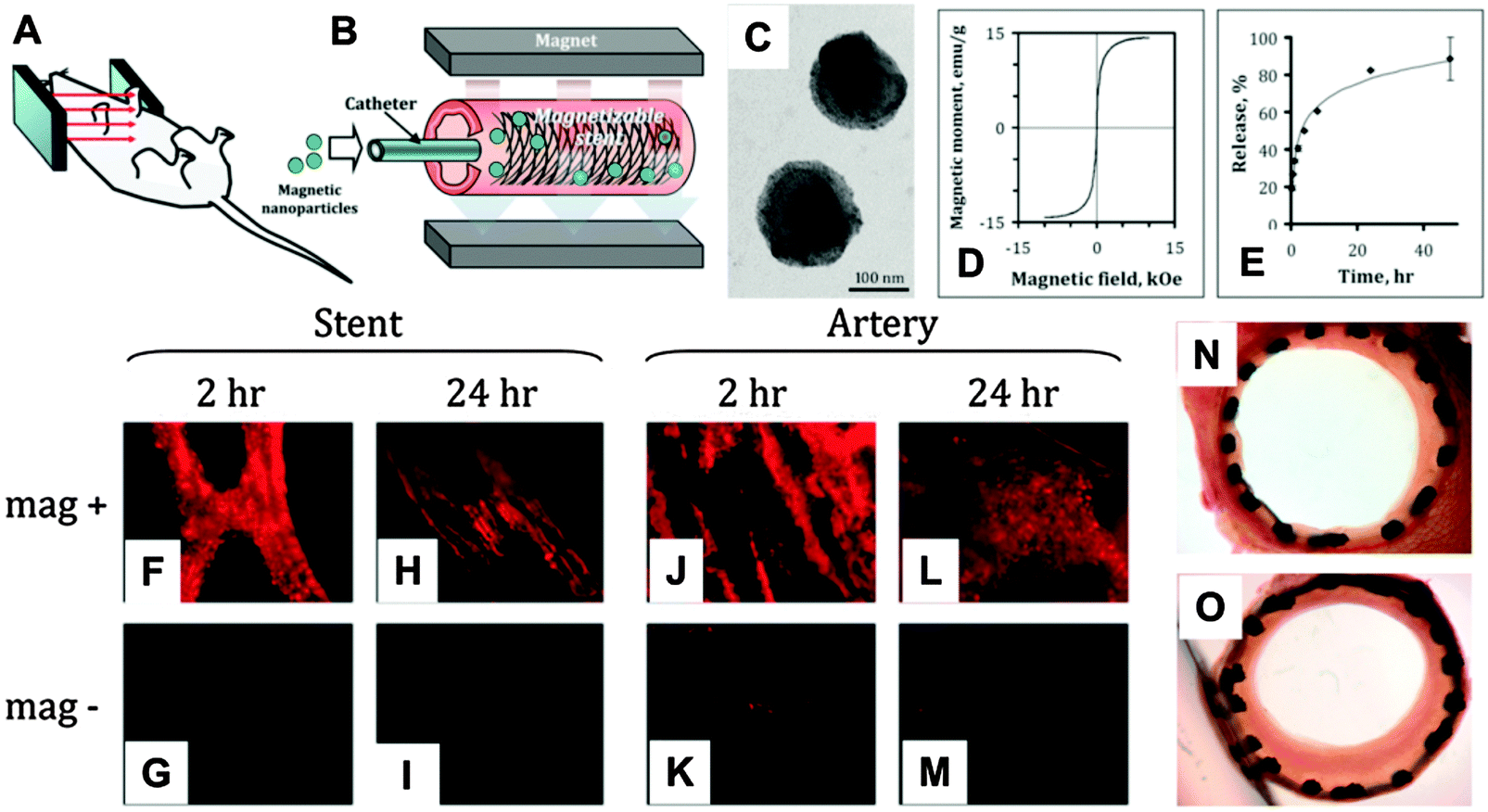 | ||
| Fig. 2 Targeted local delivery of MNPs to a deployed 304-grade stainless steel stent mediated by a uniform-field-induced magnetization effect: (A) schematic figure of the uniform field generated by paired electromagnets on a mouse model. (B) The uniform field that is generated induces high magnetic gradients in the stent and magnetizes drug-loaded MNPs, thus creating a magnetic force that drives MNPs to the stent struts and adjacent arterial tissue. (C) Transmission electron micrograph of MNPs. (D) MNPs exhibit a magnetic moment of 14.3 emu g−1 at saturation. (E) Paclitaxel release kinetics of drug-loaded MNPs. In an in vivo targeting experiment, MNPs covalently labeled with BODIPY 564/570 were delivered over 30 s under magnetic vs. non-magnetic conditions after deployment of a 304-grade stainless steel stent. (F–I) The stent and (J–M) luminal surface of the arteries were examined by fluorescence microscopy 2 h and 24 h after treatment. Original magnification ×200. (N) Representative Verhoeff–van Gieson-stained section of an artery treated with 7.5 μg paclitaxel under magnetic conditions is shown in comparison with (O) a “no treatment” control. Reproduced from ref. 102 with permission. | ||
With the purpose of a magnetic guide, using the same concept as magnetic stents for vascular disease,102 magnetic scaffolds have been fabricated by dip-coating conventional bone scaffolds (i.e. hydroxyapatite and collagen) in ferrofluids containing iron oxide NPs.103 These incorporated MNPs could respond to a magnetic field and provide the scaffolds with magnetization values as high as 15 emu g−1 at 10 kOe. Simulations that were performed for a 1 cm diameter spherical magnetic scaffold showed that the attractive force generated would exceed the weight of a 150 nm MNP within a range of 2–4 mm near the scaffold when the magnetic gradient was more than 13 Oe cm−1 (easily accessible in laboratory and clinical settings). Furthermore, the magnetization of scaffolds did not significantly influence the adhesion, viability, and proliferation of MSCs. Despite still being in the early stage, the authors believed that the magnetic field generated by magnetizing scaffolds could help deliver bioagents for tissue regeneration by deposition of magnetic carriers.
Another strategy is to pre-load cargoes into a magnetic scaffold. After transplantation, the release profile can be controlled by generating mechanical force or heat under external magnetic fields. For example, Hu et al. developed a ferroscaffold using in situ synthesis of iron oxide NPs in the presence of gelatin. In brief, FeCl2 and FeCl3 (molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were added to a gelatin solution at 40 °C until they were completely dissolved and cooled to 4 °C for gelation. Then, the gel was immersed in NH4OH solution to produce iron oxide. After being placed in a freezing bath at −80 °C for 1 day, a ferroscaffold was formed with 50–200 μm pores within the gel, which allowed the release of pre-loaded cargoes (e.g. vitamin B12).104 The content of MNPs in the scaffold could be controlled to be as high as 9.41% with a saturation magnetization of 23.5 emu g−1. When an external magnetic field (an electromagnet of ∼400 Oe) was switched on, the release rate of vitamin B12 was significantly decreased due to deformation of the gel caused by a magnetic inter-particle force, which blocked the pores of the gelatin scaffold. By programming the “on” and “off” of the magnetic field, stepwise release of growth factors could be achieved.104 In contrast to “turn-off” release, a macroporous alginate scaffold embedded with MNPs (10 nm iron oxide NPs) was fabricated (ferrogel), which allowed reversible deformation and volume changes (up to 70%) under a magnetic field, causing the flow of water through interconnected pores.105 Therefore, the release of pre-loaded biological agents could be triggered when a magnetic field was turned on. For example, 50% release of mitoxantrone was achieved within 180 minutes (2 minutes magnetic stimulation in every 30 minutes), in contrast to 5% release without magnetic stimulation. A significant increase in the release profile was also found when delivering plasmid DNA and chemokines such as stromal cell-derived factor-1-alpha (SDF-1α). Interestingly, the cargoes were not only restricted to molecules. When modified with arginine-glycine-aspartic acid (RGD) amino acid before gelation, this magnetic scaffold enabled an increase in cell adhesion via RGD–integrin interaction and achieved the release of a prescribed number of fibroblasts in a Petri dish with viability of 95%. Furthermore, on-demand release of mouse MSCs after being implanted subcutaneously into the back region of nude mice was demonstrated.105
:1) were added to a gelatin solution at 40 °C until they were completely dissolved and cooled to 4 °C for gelation. Then, the gel was immersed in NH4OH solution to produce iron oxide. After being placed in a freezing bath at −80 °C for 1 day, a ferroscaffold was formed with 50–200 μm pores within the gel, which allowed the release of pre-loaded cargoes (e.g. vitamin B12).104 The content of MNPs in the scaffold could be controlled to be as high as 9.41% with a saturation magnetization of 23.5 emu g−1. When an external magnetic field (an electromagnet of ∼400 Oe) was switched on, the release rate of vitamin B12 was significantly decreased due to deformation of the gel caused by a magnetic inter-particle force, which blocked the pores of the gelatin scaffold. By programming the “on” and “off” of the magnetic field, stepwise release of growth factors could be achieved.104 In contrast to “turn-off” release, a macroporous alginate scaffold embedded with MNPs (10 nm iron oxide NPs) was fabricated (ferrogel), which allowed reversible deformation and volume changes (up to 70%) under a magnetic field, causing the flow of water through interconnected pores.105 Therefore, the release of pre-loaded biological agents could be triggered when a magnetic field was turned on. For example, 50% release of mitoxantrone was achieved within 180 minutes (2 minutes magnetic stimulation in every 30 minutes), in contrast to 5% release without magnetic stimulation. A significant increase in the release profile was also found when delivering plasmid DNA and chemokines such as stromal cell-derived factor-1-alpha (SDF-1α). Interestingly, the cargoes were not only restricted to molecules. When modified with arginine-glycine-aspartic acid (RGD) amino acid before gelation, this magnetic scaffold enabled an increase in cell adhesion via RGD–integrin interaction and achieved the release of a prescribed number of fibroblasts in a Petri dish with viability of 95%. Furthermore, on-demand release of mouse MSCs after being implanted subcutaneously into the back region of nude mice was demonstrated.105
Although promising, typical ferrogels have yet to be optimized for applications as implantable scaffolds. This is because the optimal size (e.g. 2 mm thickness) of implantable scaffolds for small animals (e.g. rodents) is smaller than what has been fabricated for in vitro tests (15 mm thickness). The reduced size will cause a decrease in iron oxide concentration and therefore cannot produce enough volume deformation of ferrogels. Elevation of the iron oxide NP concentration in the gel would stiffen the matrix and cause a potential toxicity issue. Recently, Mooney et al. addressed this problem by fabricating a biphasic ferrogel to replace their former monophasic version.106 During polymerization, a magnet was placed under the alginate to attract MNPs (Fig. 3A). Instead of the homogeneous distribution in a monophasic ferrogel, MNPs were only located to one side of the biphasic ferrogel (Fig. 3B and C). Redistribution of MNPs and an increase in macropore size (Fig. 3D and E) resulted in increased deformation in comparison to monophasic ferrogels (Fig. 3F). On-demand release of drugs and viable cells was also demonstrated in the small-sized biphasic ferrogel system. After two weeks following implantation, scaffolds remained at the initial implant site with minimal cell infiltration, which suggests the biocompatibility of this biphasic ferrogel.106
 | ||
| Fig. 3 (A) Schematic of fabrication of monophasic and biphasic ferrogels. (B) SEM/EDS of monophasic and biphasic ferrogels showing iron in yellow and carbon in blue. (C) Photograph (i), micro-computed tomography (micro-CT) (ii), and SEM (iii and iv) images of fully fabricated biphasic ferrogels. (D) Micro-CT images showing the porosity of monophasic and biphasic ferrogels at the locations indicated by red circles in the schematic under each micro-CT image. (E) Quantified open porosity of monophasic (M), biphasic iron-oxide-rich (BF), and biphasic alginate-rich (BA) ferrogel regions. Values represent the mean and standard deviation (n = 4). Data were compared using ANOVA with Bonferroni's post hoc test (*p < 0.05, ***p < 0.001). (F) Photographs of small 7 wt% iron oxide biphasic and monophasic ferrogels in the presence of no magnetic field (field off) and a moderate vertical magnetic field gradient (field on). Reproduced from ref. 106 with permission. | ||
Engineering cells with MNPs allows remote manipulation of labeled cells and directed targeting to locations of interest. For example, in the case of artery regeneration, endothelial progenitor cells (EPCs) were labeled with Endorem, a type of FDA-approved superparamagnetic iron oxide (SPIO) NPs.107 A dose of 5 × 105 labeled EPCs was administered to a common carotid artery injury site in a rat model, while a magnetic actuator was placed adjacent to the neck. Engraftment of EPCs onto the injured vascular surface increased around 4 times with the presence of an external magnetic field.107 Similarly to localized drug delivery in vascular disease, magnetic stents have been developed for targeted delivery of bovine aortic endothelial cells (BAECs) loaded with MNPs into a rat common carotid artery.108 A catheter was introduced via the external carotid into the common carotid near a 304-grade stainless steel stent that was deployed in a common carotid injury rat model. A dose of 0.5 × 106 labeled BAECs was injected through the catheter under a magnetic field of 1000 G and achieved saturated accumulation of 20% of circulating cells near the stent within 50 minutes (10% within the first 6 minutes). However, no detectable cells were captured by the stent without a magnetic field in normal blood flow conditions.108 It should be noted that the saturation magnetization of the stent, MNPs or MNPs-loaded cells occurred in the range of 0.1 to 0.2 T, which might be provided by a typical MRI system (1 to 3 T) in clinical settings. Therefore, this technology takes a step forward to intervention in targeting human vascular injuries.
A similar strategy was used to deliver MSCs to neurological tissues (e.g. retina).109 Specifically, rat MSCs were labeled with SPIO NPs (fluidMAG-D) before intravitreal injection into a rat which was subject to retinal degeneration. By placing a gold-plated neodymium disc magnet outside the eye, researchers increased the amount of deposited MSCs 10 times with minimal collateral damage compared with unlabeled cells. The subsequent increase in the anti-inflammatory molecule interleukin-10 further indicated the therapeutic effect of the delivered MSC in the dystrophic retina.109
Besides cell therapy, MNPs have also been used in tissue engineering. Commercial SPIO NPs (ferucarbotran) have been used to label MSCs for cartilage and bone regeneration. A preclinical study demonstrated the efficacy of regeneration of articular cartilage and osteochondral defects of the patella was enhanced by accumulating magnetically labeled MSCs in both small and large-animal models.110–112 For example, MSCs were labeled with 25 μg Fe per mL ferumoxides overnight using poly-L-lysine as transfection agent.110 These labeled MSCs responded efficiently under a magnetic field generated by a disk-shaped electromagnet (maximum 0.6 T). Accumulation of labeled MSCs in the chondral defects of rabbit and swine models was observed and confirmed by histological analysis after treatment with a magnetic field for 4 hours, whereas the control group (without magnetic field) did not show any notable attachment of MSCs to the defect area.110
Despite these interesting achievements, most MNP-based drug/cell delivery strategies are still at the preclinical stage. The challenge lies in the ability of existing magnetic carriers to penetrate through physiological barriers (e.g. the blood–brain barrier, endothelial luminal layer, or soft tissue). There are two potential solutions to address this challenge. One solution is to increase the mobility of magnetic carriers through the extracellular matrix by functionalizing them with a proteolytic surface.113 Inspired by the process of degradation of the biological barriers of invasive cells, MNPs were immobilized with a microbial protease (e.g. collagenase). When manipulated by an external magnetic field, a significant increase in NP velocity was observed in ECM gel compared to MNPs modified by bovine serum albumin or polyethylene glycol (from 0 to 90 μm h−1). Improved penetration can also be achieved with the help of bubble cavitation. When combined with ultrasound, co-administered microbubbles can generate transient pores on cellular barriers by inertial cavitation, which could also improve the penetration of magnetic carriers in tissues.114 Another solution is to increase the strength of the magnetic field at the tissues of interest. For example, based on a study of delivering therapeutic MNPs into the inner ear of guinea pigs,115 the necessary magnetic field intensity at the working distance required for humans (30–50 cm) would exceed current US FDA safety limits (8 T for adults and 4 T for children).116 Therefore, magnetic implants (e.g. scaffolds or stents) or magnetic carriers with high magnetization could partly address this problem with deep tissue delivery. Besides, delicate clinical standard magnetic devices for specific human treatments are highly needed.117,118
Despite being promising, the translation of genetically engineered stem/progenitor cells for cell therapy has suffered from low transfection efficiency. Lipofection is the simplest and most cost-effective technique among all transfection methods. However, the transfection efficiency for mouse embryonic stem cells (mESCs) was highly variable, ranging from 3 to 30% depending on the size of the DNA.121 Yang et al. developed biodegradable polymeric NPs (e.g. poly(β-amino esters)-DNA NPs) to deliver genes to MSCs with a transfection efficiency of 35%, which was higher than other reported methods using electroporation (16%), poly(L-lysine)–palmitic acid (17%), and commercial transfection reagents (e.g. 3% for FuGene and 5% for DOTAP).122 Viral transfection (e.g. lentiviral transfection) is now the most efficient method for gene transfer but has major limitations to its translation to clinical use for regenerative medicine due to the risk of insertional mutagenesis and oncogene activation.123
Therefore, to enhance the efficiency of transfection, magnetically guided nucleic acid delivery, known as magnetofection, has been developed. In this method, a magnetic field is used to accelerate the sedimentation of magnetic vectors onto a target cell, which increases the internalized dose of DNA within a given period.124 During transfection, a magnetic field has been demonstrated to produce no change in the mechanism of cellular uptake.125 In magnetofection, MNPs are associated with a gene delivery vector (i.e. nucleic acid alone, nucleic acid with a non-viral lipoplex or polyplex, or viral vector) to form a magnetic vector complex. Strategies of fabrication of magnetic vectors include specific ligand–ligand interactions, electrostatic and hydrophobic interactions, and covalent coupling between vectors and MNPs.124
Transplantation of oligodendrocyte precursor cells (OPC) promotes the repair of myelin damage in neurological diseases and spinal cord injury via the production of new myelin by their daughter cells (oligodendrocytes). This idea has been verified in clinical trials, in which OPCs derived from human embryonic stem cells (hESC) successfully repaired spinal cord injury.126 More interestingly, researchers found that genetically modified OPCs that produce therapeutic proteins (e.g. fibroblast growth factor-2) have higher efficacy in promoting cellular repair, axonal outgrowth, and angiogenesis. Currently, OPCs are mainly transfected by viral methods, which encounter significant risks (e.g. safety issues) and challenges (e.g. large-scale production) when being translated into clinical use. The development of large-scale synthesis of MNPs and their minimal safety issues make them ideal vectors for gene transfection. Magnetofection with a 4 Hz oscillating magnetic field was found to increase transfection efficiency (20.6 ± 2.2% for GFP plasmid) by around 3.5-fold compared to the absence of a magnetic field.127 The transplantation potential of these transfected cells was tested in brain slices as the host tissue, where the cells were found to migrate, proliferate, differentiate, and integrate well. To further increase the transfection efficiency, a cationic cell-penetrating peptide (e.g. TAT peptide derived from trans-activating transcriptional activator) could be combined with magnetofection to promote cellular uptake.128 Magnetic vectors were synthesized by mixing polyethyleneimine-coated MNPs with plasmid DNA followed by addition of bis(cysteinyl) histidine-rich TAT peptide. Ternary complexes displayed a fourfold improvement (up to 60%) in transgene expression over binary complexes without TAT peptide. When administered into the spinal cord of rats by lumbar intrathecal injection using a magnet on the back of animals, ternary complexes provided twofold higher transfection efficiency than binary complexes.
The generation of induced pluripotent stem cells (iPS) for specific patients is one of the ultimate goals of modern regenerative medicine. However, this emerging approach has been restricted by the use of potentially harmful viral DNAs. Although the efficiency of the generation of iPS cells from mouse cells using a non-viral system was still very low (at 0.0001–0.0012%), it increased to 0.001–0.003% with the help of MNPs. In this case, PEI-coated MNPs were mixed with an iPS gene to form a magnetic complex.129 These generated iPS cells exhibited embryonic stem cell-like characteristics, which indicated safe non-viral generation of iPS cells.
To date, the main problem with tissue-engineered constructs is their lack of structural complexity and precision. In order to reproduce complex tissues using functional constructs, well-defined spatial organization of cells is required. Although cell patterning methods such as microcontact printing and lithography have been developed, they need specialized surfaces and prolonged procedures. Consequently, using an external force such as an electric field, optical trap, or magnetic field for spatial cell patterning may be an alternative simple approach.
Ito et al. labelled cells with magnetite NPs (10 nm) using cationic liposomes as carriers (MCL). When steel plates that were placed on a magnet were positioned under a cell culture surface, the magnetically labelled cells aligned with the steel plates (Fig. 4A and B) in so-called magnetite tissue engineering (MTE) technology.141 Based on this method, approximately 15-layered cell sheets of ARPE-19 human retinal pigment epithelial cells were formed after 24 hours culture under a magnet (4000 G) for the treatment of choroidal neovascularization.142 Multilayered sheets of human MSCs that were formed by magnetic construction (Fig. 4C) have been demonstrated to be feasible for scaffold-free bone regeneration when transplanted into a bone defect in the crania of nude rats (Fig. 4D–F).143 Fabricated cell sheets can be further constructed into 3D tissue-like structures. For example, when a cylindrical magnet was rolled onto a cell sheet, a tubular structure was formed even after the removal of the magnet. Tubular layers of monotypic urothelial cells and heterotypic layers of endothelial cells, smooth muscle cells and fibroblasts were created, which have the potential to regenerate urinary tissue and vascular tissue, respectively.144 To regenerate skeletal muscle tissue, the myogenic cell line C2C12 labelled with MNPs was linearly patterned on a monolayer of fibroblast cells under a magnetic field. C2C12 cells could differentiate, fuse, and form multinucleated myotubes.145 The expression of muscle-specific markers and an increase in creatine kinase activity during differentiation were further observed. The physiological functionality of artificial skeletal muscle tissue was demonstrated by contraction in response to electric pulses.146
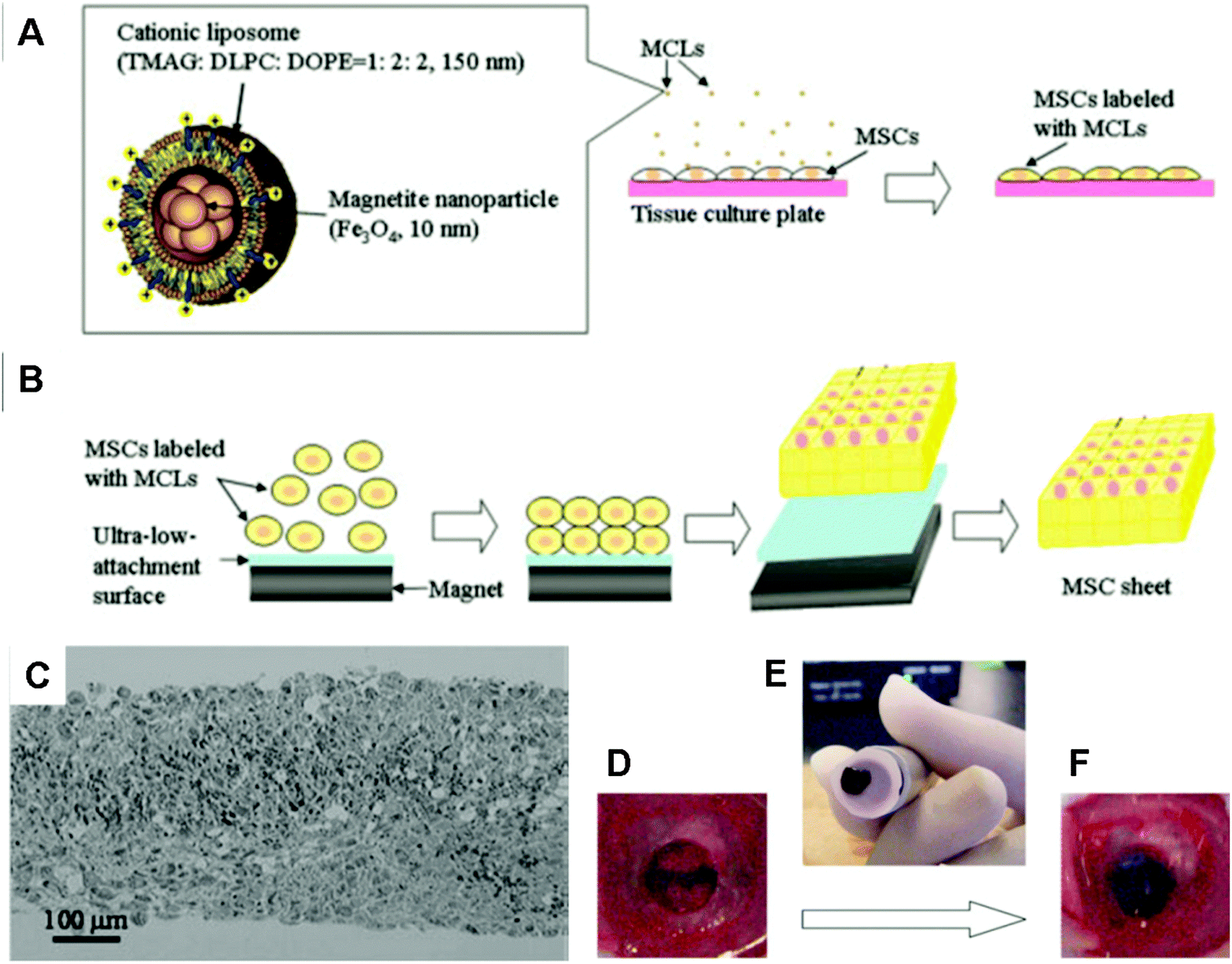 | ||
| Fig. 4 Procedure for construction of MSC sheets by magnetic-force-based tissue engineering (Mag-TE) and fabricated cell structures. (A) Illustration of magnetite cationic liposome (MCL) and scheme for magnetic labelling of MSCs with MCL. (B) Construction of MSC sheets by Mag-TE. Labelled MSCs were seeded onto an ultra-low-attachment plate and a cylindrical magnet was then placed under the plate. Cells that were attracted to the culture surface by magnetic force were cultured to construct a cell sheet. Upon removal of the magnet, cell sheets became detached from the culture surface and could be harvested without enzymatic treatment. (C) Bright-field photograph of hematoxylin and eosin-stained cross-sections of an MSC sheet constructed using MCLs and a magnet. (D–F) Transplantation procedure of MSC sheets using an electromagnet: (D) a 5 mm defect was made in the cranial bone of nude rats. (E) MSC sheets constructed by Mag-TE were harvested and transported to the defect, and (F) transplanted into the bone defect using magnetic force. Reproduced from ref. 143 with permission. | ||
In another study, iPS cell sheets that were created by the MTE method have been demonstrated to accelerate revascularization for reparative angiogenesis.147 Kito et al. created an iPS cell-derived Flk-1+ cell sheet (10–15 layers) by MTE within an extracellular matrix precursor, which reduced the risk of inducing ischemia in inner cell layers. After implantation into a mouse model of hindlimb ischemia, this cell sheet provided a significant increase in limb perfusion 7 to 21 days after surgery compared to control (extracellular matrix only) and Flk-1− groups. Increased expression of VEGF and basic fibroblast growth factor (bFGF) further confirmed the mechanism that therapy with iPS cell-derived Flk-1+ cells mediated angiogenesis via release of angiogenic cytokines rather than direct differentiation into endothelial cells.
Rapid endothelialization of synthetic vascular grafts could address graft failure in small vessels caused by thrombosis and formation of neointima. This strategy mainly depends on the seeding efficiency of endothelial cells on the grafts. Researchers have demonstrated enhanced seeding efficiency of MNPs-labelled endothelial cells using magnetic force.148–150 Uniform cell coverage was found on the magnetized scaffold when transplanted into porcine carotid arteries after one day. However, relatively few labelled cells were seen on the scaffold without a magnetic field because of the blood flow.151
Another interesting example, which provides an alternative to biodegradable porous scaffolds, is 3D cell culture technology that uses magnetic forces to levitate cells while they divide and grow.152,153 Compared with cell cultures grown on flat surfaces, 3D cell cultures tend to form tissues that more closely resemble those inside the body. A hydrogel that encapsulated MNPs and a filamentous bacteriophage was levitated in a medium by placing a coin-sized magnet on top of the lid of the dish. Interestingly, by spatially controlling the magnetic field, the geometry of the cell mass could be manipulated and multicellular clustering of different cell types in co-culture could be achieved.152 Using a 3D levitation tissue culture system, a model of white adipose tissue was reconstructed with closer cell positioning, longer survival time, and more efficient formation of lipid droplets in comparison to a 2D culture system.154
Magnetic assembly of microscale tissues/subunits is currently under the spotlight in tissue engineering and regenerative medicine. Scaling up microtissues into larger tissues using magnetic control provides the possibility of creating well-defined 3D architectures with tissue-specific functions (assembly of different functional microtissues). Using this concept, Liu et al. fabricated microcryogels (i.e. gelatin) loaded with MNPs with the encapsulation of different cells by on-chip cryogelation and micromolding (Fig. 5A).155 These microtissues were controllable in a microfluidic device (Fig. 5B) and had three advanced applications. First, when magnetic microcryogels loaded with hepatic cells (liver functional cells) (Fig. 5C) were co-cultured with non-magnetic microcryogels loaded with stromal fibroblast cells (supporting cells), liver functions, including albumin expression and urea synthesis, were significantly higher compared to hepatic cells in a mono-culture system. Subsequent separation of the hepatic cells from the supporting tissues was easily achieved with a magnet (Fig. 5D), which could be used for drug hepatotoxicity testing. Second, the self-assembly of microtissues into larger tissues could be either random (magnetic field only) or shape-directed (with a magnetic field and the addition of supplementary structural blocks) (Fig. 5E). Third, the engineered tissue could be tracked in vivo by MRI after transplantation (Fig. 5F).
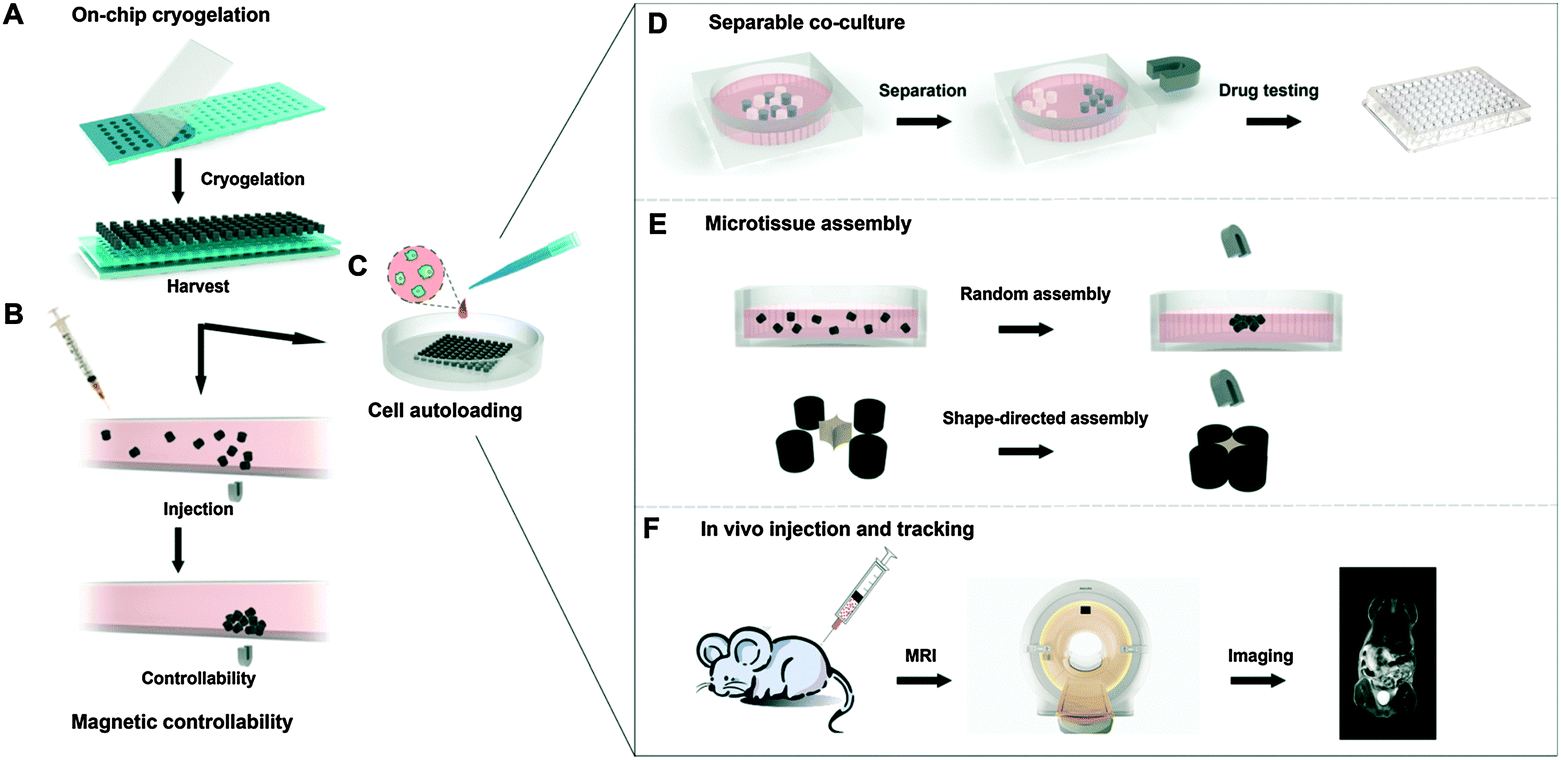 | ||
| Fig. 5 Schematic demonstration of the fabrication, assessment of magnetic controllability and three exemplary applications of magnetically controllable 3D microtissues. (A) Fabrication of magnetic microcryogels from a microstencil array chip; (B) assessment of magnetic controllability under shear stress in vitro; (C) cells autoloaded into magnetic microcryogels; (D) separable co-culture system for functional enhancement of microtissues, which can be used for downstream drug testing; (E) accelerated controllable formation of microtissues by random and shape-directed assembly; (F) in vivo tracking of microtissues by MRI. Reproduced from ref. 155 with permission. | ||
As the field of regenerative medicine has evolved, attention has been focused on recreating the extracellular microenvironment of tissues.156 The hierarchically organized nanocomposite (an intricate interweaving of fibrillar collagen or elastin fibers ranging from 10 to hundreds of nanometers) of extracellular matrix (ECM) regulates essential cellular functions such as morphogenesis, differentiation, proliferation, adhesion and migration.157 This was demonstrated by reconstruction of a rat heart by seeding cardiac and endothelial cells onto decellularized rat hearts that retained the underlying ECM.158 After 8 days, migration and self-organization of cells in the matrix were found in their natural location. The constructs could perform the pump function under physiological load and electrical stimulation.158 Therefore, the ability to develop artificial ECMs with a defined nanostructure on a scaffold may have significant value for tissue engineering and regenerative medicine.
Ingber et al. first developed a method for magnetic self-assembly of fibrin matrices with ordered nanoscale structure.159 Superparamagnetic beads were coated with thrombin, which was able to catalyze in situ the formation of fibrin protein from fibrinogen. Soluble fibrin monomers then self-assembled into fibrin fibers along the bead–bead axis. By magnetically organizing the magnetic beads into periodically ordered arrays (e.g. hexagonal arrays) at the air–liquid interface, oriented nanoscale fibrils were formed within minutes. Human microvascular endothelial cells that were seeded on these fibrin scaffolds could adhere to and spread along the fibers and their actin filaments aligned with the fibrin nanofibers.159
3.3 Magnetic nanoparticles and bioimaging: real-time visualization and tracking stem cells by MRI
Molecular imaging can help researchers and clinicians understand the fate, distribution, and function of therapeutic cells after transplantation. Imaging techniques include optical imaging, MRI, and radionuclide imaging. Typically, contrast agents are required to label cells and generate a signal that is distinguishable from the background of the host tissue. Optical imaging modalities suffer from limitation of their penetration depth (<5 μm) in the human body, which is dependent on the wavelength of the applied light. Radionuclide imaging modalities (e.g. single-photon emission computed tomography, positron emission tomography) possess optimal sensitivity in medical imaging. However, drawbacks exist such as the impossibility of longitudinal cell tracking (due to the low retention time of contrast agents), lack of anatomical information and risks to human health.MRI provides excellent soft tissue contrast with high spatial resolution and can be used for visualization of single cells against a homogeneous background. This technique is based on the principle that magnetization of hydrogen protons in the human body will be aligned with an applied external magnetic field. The presence of MNPs shortens the spin–spin relaxation time and thus produces a negative (decreased) signal in T2 and T2*-weighted MR images.21 Therefore, by labeling the cells/tissues of interest with MNPs, we can visualize them non-invasively. With this aim, our group have been actively involved in the development and use of MNPs for cell tracking.21,160–164 Various MNPs based on Fe, Co, and Ni have been demonstrated to be suitable as cell trackers,162 with the added advantage of being able to be biofunctionalized.165 Lewin et al. developed a cell labeling approach with superparamagnetic NPs (iron oxide core), which demonstrated that intravenous injection into immunodeficient mice allowed detection of single cells by MRI.166 Xie et al. successfully created a triple-functional (PET/near-infrared fluorescence/MRI) MNP and demonstrated in athymic nude mice that accumulation and extravasation rates could be tracked by any of these imaging techniques.167
Zhu et al. demonstrated the feasibility of tracking neural stem cells in patients with brain trauma for at least 7 weeks by MRI.169 Autologous neural stem cells were first collected and cultured during an emergency operation. Then the cells were loaded with Feridex I.V. (SPIO) in the presence of Effectene (a lipofection reagent). After stereotactic implantation of these neural stem cells around the region of brain damage, MRI with a 3.0 T system was performed to image the brain at 24 hours and then every 7 days after transplantation (total 10 weeks). After one week, labeled neural stem cells were found to migrate from the injection site to damaged areas, with accumulation and proliferation around the lesion. The signal increased during the second and third weeks, but could not be observed after 7 weeks due to dilution of the signal caused by cell proliferation.169 Control patients were injected with neural stem cells with no labeling. After implantation, no pronounced change in signal was found around the lesion.
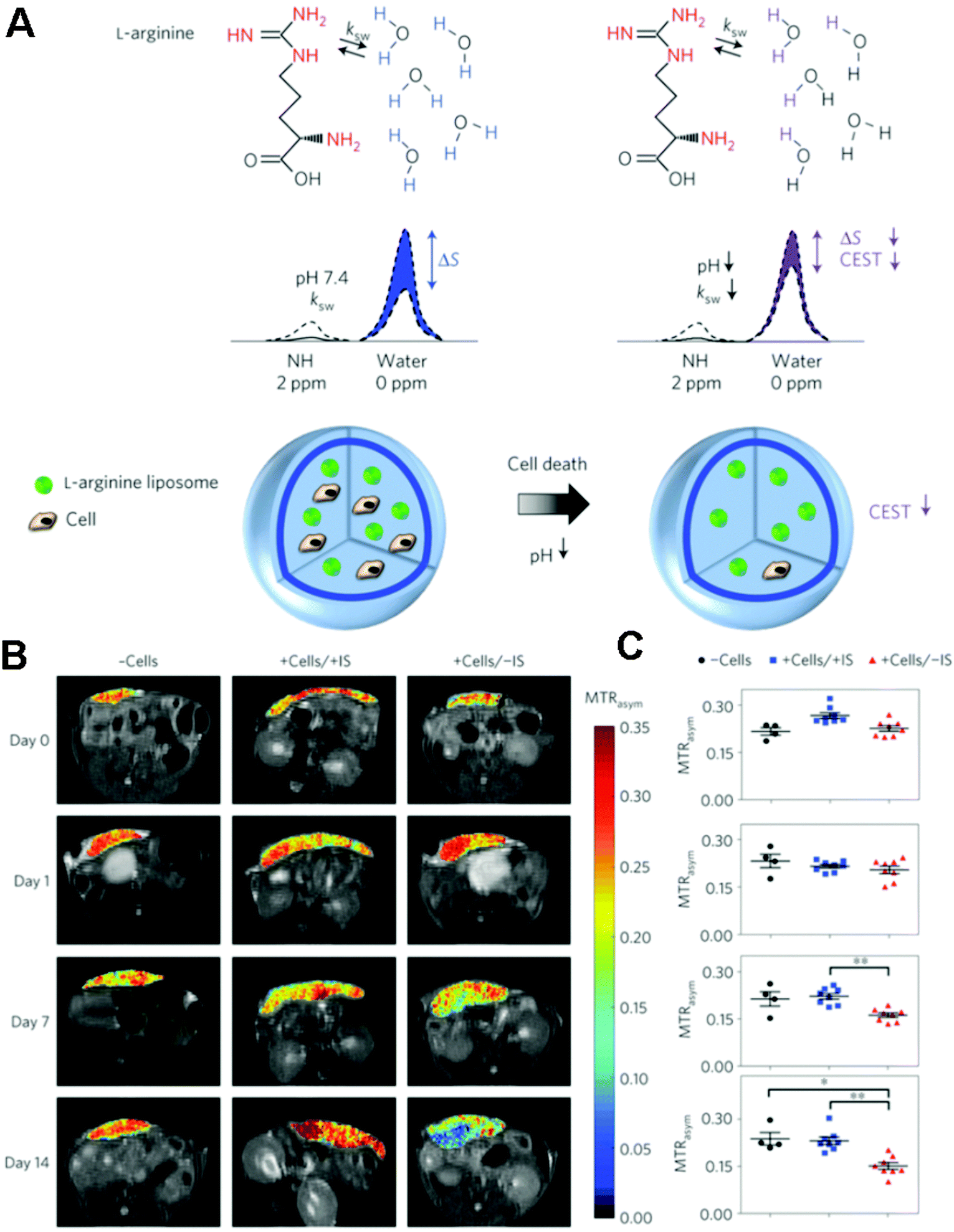 | ||
| Fig. 6 (A) Schematic of the principles of in vivo detection of cell viability using LipoCEST microcapsules as pH nanosensors. (B) In vivo CEST imaging of LipoCEST capsules containing hepatocytes. BALB/c mice were subcutaneously transplanted with 2500 empty LipoCEST capsules (−Cells), with LipoCEST capsules containing hepatocytes while receiving immunosuppression (+Cells/+IS), and with capsules containing cells but no immunosuppression (+Cells/−IS). CEST/MTw (magnetization transfer-weighted) overlays use a frequency offset of 2 ppm at days 0, 1, 7 and 14 after transplantation. (C) Average MTRasym values for −Cells (n = 4, black), +Cells/+IS (n = 8, blue) and +Cells/−IS (n = 8, red). Reproduced from ref. 171 with permission. | ||
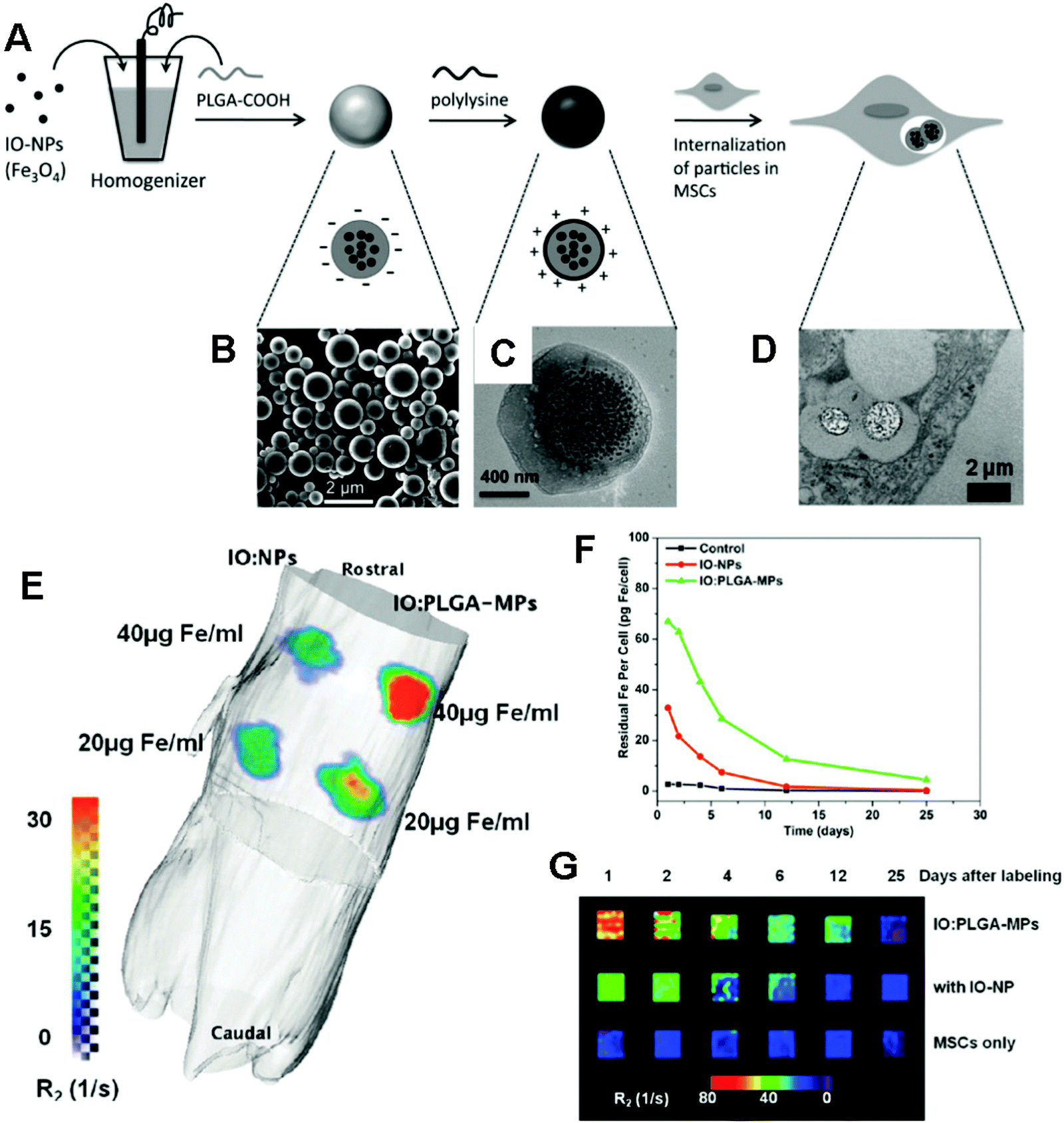 | ||
| Fig. 7 (A) Schematic of the preparation of IO/PLGA-MPs by a single emulsion method. (B) SEM image of IO/PLGA-MPs. (C) TEM image of a representative IO/PLGA-MP. (D) TEM image of IO/PLGA-MPs internalized in a MSC. (E) 3D reconstruction of a mouse with an R2 map collected with a 4.7 T Bruker Pharmascan scanner and calculated within the OsiriX environment. The scale bar indicates the value of R2 (unit: s−1 or Hz). (F) Change in iron content per cell after initial labeling with IO-NPs or IO/PLGA-MPs at an incubation concentration of 50 μg Fe per mL. (G) R2-weighted MR images of 200000 MSCs collected at different time points and suspended in 3% agarose gels (4 × 4 mm □−1). Reproduced from ref. 164 with permission. | ||
For example, our group have synthesized Au–Fe3O4 dumbbell NPs by decomposing iron pentacarbonyl on the surfaces of Au NPs in the presence of oleic acid and oleylamine, with sizes of 8–20 nm (core particle diameter).174 By functionalizing them with epidermal growth factor receptor antibody (EGFRA) on the Fe3O4 surface, these 8–20 nm Au–Fe3O4 NPs can target A431 cells (epidermoid carcinoma) that overexpress EGFR and visualize these cells by both T2-weighted MRI and optical imaging. It is worth noting that the modified nanoparticles exhibit negligible toxicity towards A431 cells at 0.01 mg Fe per mL and 0.004 mg Au per mL. The fact that dumbbell NPs are able to image exactly the same tissue area by both MRI and optical sources implies that they can be used to provide an anatomy scan (MRI) and detection of single cells (optical imaging) in regenerative medicine.174 Other multifunctional imaging platforms such as Fe3O4–CdSe heterodimer NPs18 (CdSe for fluorescence imaging) and gold-coated magnetic silica NPs (gold shell for photoacoustic imaging)28 have demonstrated the ability to integrate fluorescence imaging and photoacoustic imaging with MRI.
3.4 MNPs and therapeutic cells promote cell proliferation
In regenerative therapies that rely on the transplantation of therapeutic cells, one bottleneck is the ex vivo expansion of the cells. To take bone tissue engineering as an example, the use of fresh bone marrow aspirates was the first effective cell-based approach for bone regeneration. However, the quantity of osteoprogenitors in bone marrow was dependent on the age and health of patients. As a result, in some cases cell numbers were not sufficient to produce a therapeutic effect.175 Therefore, to date most research has focused on the isolation, maintenance, and expansion of stem or progenitor cells.7Regarding the labeling of cells with MNPs, most research has focused on MNPs' ability to enhance MRI, as well as their safety issues (e.g. cytotoxicity). Nevertheless, it is interesting to note that some pioneers have been keenly aware of other bioeffects of MNPs, especially on cell growth and differentiation. The ability to increase osteoblast density in the presence of MNPs for bone tissue engineering was first explored by Rajesh et al.176 Firstly, different MNPs with calcium phosphate (CaP) coatings were synthesized and injected into porous bone sites. The CaP-coated MNPs were then directed to and attached to bone tissue under an applied magnetic field. Results showed that γ-Fe2O3 MNPs significantly promoted osteoblast density (cells per well) after 5 and 8 days compared to Fe3O4 MNPs and control groups. When coated with hydroxyapatite (HA, the main inorganic component of bone), γ-Fe2O3 MNPs effectively promoted osteoblast proliferation after 1 day. Although the mechanism was still not well understood, the researchers believed that adsorption of some specific proteins (such as vitronectin and fibronectin) on nanoscale surfaces enhanced the adhesion of osteoblasts, which might be essential for promoting cell functions.176
Recently, Fe3O4 NPs have been reported to possess intrinsic peroxidase-like activity,177 which could catalyze the breakdown of H2O2. Huang et al. demonstrated that when labeled with ferucarbotran (SPIO NPs), the proliferation of MSCs was dramatically increased in a dose-dependent (ferucarbotran concentration) manner (Fig. 8A–D). A decrease in intracellular H2O2, which was able to control proliferation and cell death,178,179 was found in the presence of SPIO NPs and indicates one potential mechanism of stimulating the growth of MSCs. Furthermore, the authors found that leached Fe by lysosomal metabolism of SPIO NPs could play a positive role in cell cycle progression by upregulating cyclins and cyclin-dependent kinases.180 In this way, the other aspect of intracellular SPIO NPs that can promote the growth of MSCs by reducing intracellular H2O2 levels and affect protein regulators of the cell cycle has been demonstrated for the first time (Fig. 8E).
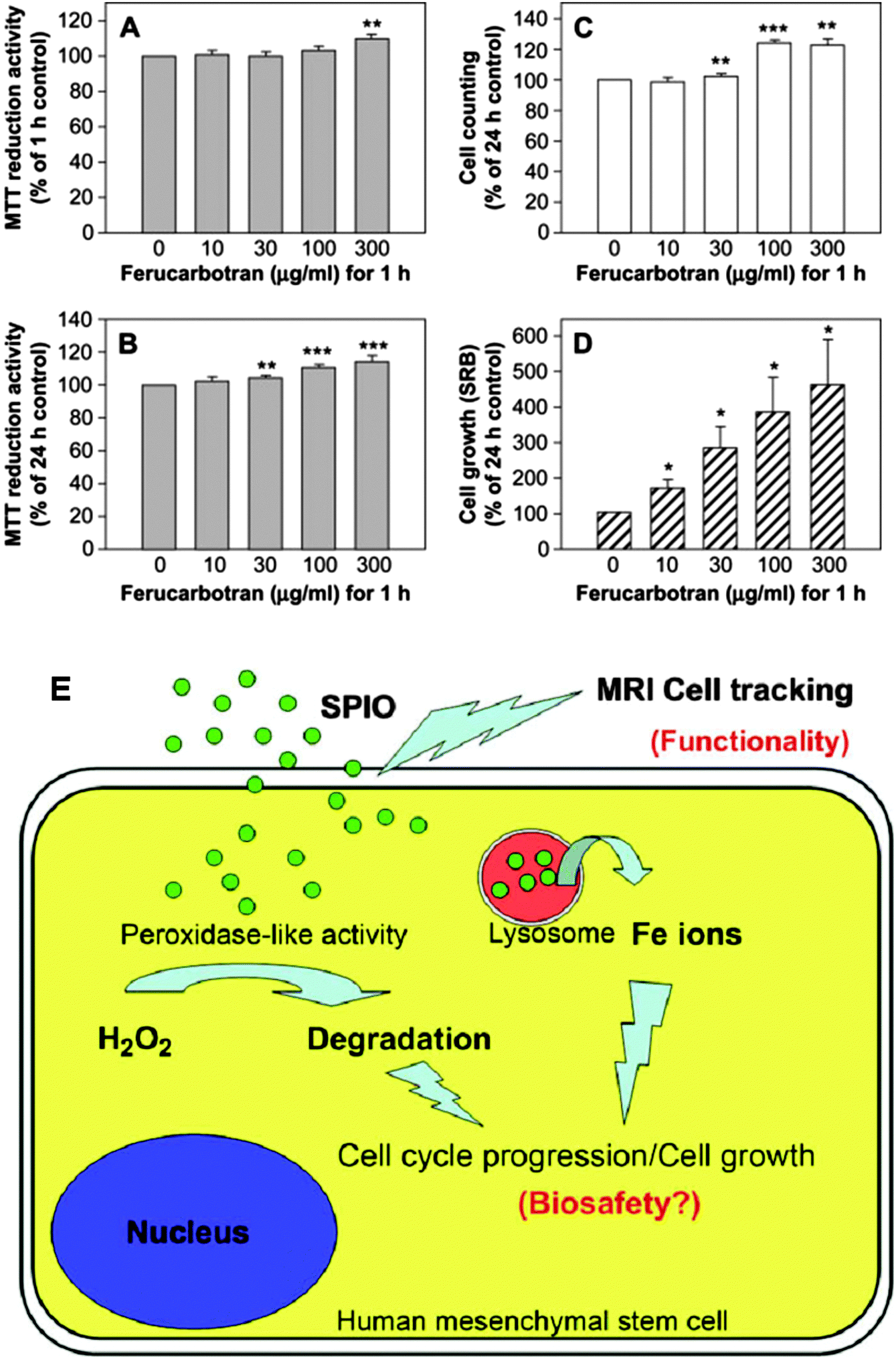 | ||
| Fig. 8 Ferucarbotran (SPIO NPs) dramatically stimulates growth of MSCs. (A) Labeling with ferucarbotran at high concentrations (300 μg mL−1) increases the acute MTT reduction activity of MSCs. (B) Ferucarbotran significantly increases cell viability. (C) Trypan blue exclusion assay shows that ferucarbotran increases the cell number of MSCs. (D) SRB study shows that ferucarbotran dramatically stimulates the proliferation of MSCs. All data are expressed as mean ± standard error of three to eight determinations (each in quadruplicate) for three donors. (*p < 0.05; **p < 0.01; ***p < 0.001). (E) Superparamagnetic iron oxide (SPIO) nanoparticles are capable of labeling human mesenchymal stem cells for magnetic resonance imaging (MRI). After internalization into cells, SPIO can promote cell growth due to its ability to reduce intracellular H2O2 levels via its intrinsic peroxidase-like activity. Moreover, SPIO can accelerate cell cycle progression, which may be mediated by free iron (Fe) released by lysosomal degradation. Reproduced from ref. 180 with permission. | ||
Despite the positive effect on the growth of MSCs, there are potential biosafety concerns. SPIO NPs (ferucarbotran) can cause dose-dependent inhibition of osteogenic differentiation (at concentrations as low as 10 μg mL−1) and prevent differentiation at high concentrations (>100 μg mL−1).181 Potential mechanisms involved promotion of cell migration and activation of signaling molecules (e.g. β-catenin, matrix metalloproteinase-2, etc.) by SPIO NPs, which caused enhanced mobility and detachment of MSCs during osteogenic differentiation. These were attributed to intracellular free Fe, which leached from SPIO NPs, following an iron chelation (by desferrioxamine) experiment, which suppressed all the above actions induced by ferucarbotran.181 A recent work also reported the inhibition of chondrogenic differentiation of MSCs by MNPs (Endorem®). Although MNPs did not exert an influence on cell viability and proliferation at all doses (from 12.5 to 1600 μg mL−1), chondrogenic gene expression (e.g. COL2A2, ACAN, SOX9, COL10, COMP) was significantly suppressed at all doses.182
3.5 MNPs and bioreactors: mechanotransduction-remote control of cellular behaviour
Apart from combining scaffolds, cells, and growth factors to form a regenerative strategy, mechanoinduction also plays an important role in tissue regeneration. For example, Boerckel et al. demonstrated via rat studies that neovascularization is sensitive to mechanical stimulation. They also reported that a delay in the onset of mechanical loading would enhance bone formation and vascular remodeling significantly.183 This study, among others, has prompted research on the bioreactor culture of functional ex vivo tissue, which now forms a tenet of regenerative medicine. According to a review by Martin et al.,184 bioreactors play a few primary roles: (1) enhancing the seeding efficiency and homogeneity of cells on 3D scaffolds; (2) improving mass transport; and (3) providing mechanical cues. Martin et al. have demonstrated the effects of bioreactor culture on engineered cartilage constructs over a period of 6 weeks, demonstrating that the dynamic laminar flow that is generated by a rotating-wall vessel bioreactor resulted in superior biochemical and biomechanical properties compared to static and stirred-flask cultures, with these properties approaching those of native cartilage.185 With specific reference to the generation of ex vivo bone tissue, our group has evaluated various bioreactors and found significant advantages in the biaxial rotating bioreactor,186 with more homogeneous cellular proliferation and differentiation after 28 days of culture. Apart from promoting homogeneous cell distribution and shear forces, bioreactors may also be used to provide compressive loading to alter gene expression and the production of extracellular matrix, as demonstrated in studies by Hunter et al.,187 where chondrocyte metabolism was found to vary according to mechanical stimulation. Terraciano et al. reported that MSCs and human embryoid body-derived (hEBd) cells that were encapsulated in hydrogels responded positively to chondrogenesis when mechanically stimulated in the presence of transforming growth factor-β1.188To control and activate the cellular functions of stem cells after transplantation holds great promise for enhancing the efficacy of regenerative medicine. Mechanical stimulation has been demonstrated to be very important for osteogenic differentiation of MSCs, which triggers the mechanotransduction signaling pathway. As mentioned above, bioreactors not only improve the efficiency of mass transfer but also provide sufficient shear stress that can trigger mechanotransduction signaling pathways, thus upregulating the production of cyclic adenosine monophosphate (cAMP), TGF-beta 1, and nitric oxide.40,189
In addition to biochemical molecules that are involved in cell signaling pathways, mechanical cues also provide significant stimulation to promote the production of functional tissue matrix. Binding MNPs to a cell membrane would allow us to remotely control specific cellular behavior in vitro or even in vivo, due to the response to a magnetic field. There are several ways in which actuation of MNPs controls cellular behavior.190–192 (1) Magnetic twisting: MNPs are coated with RGD peptides and attached to integrin receptors on a cell membrane. A twisting magnetic field following a magnetizing pulse (to provide initial remanent magnetization) will allow twisting of bound MNPs on the cell membrane and thus generate a mechanical force on actin filaments that are linked to the receptors. (2) Activation of a mechanosensitive ion channel: by applying a high-gradient magnetic field, MNPs (bound to integrin receptors) are pulled towards the field direction, causing deformation of a cell membrane and activation of adjacent mechanosensitive ion channels. (3) Targeted activation of an ion channel: an ion channel is forced open by specifically targeting MNPs to the channel via an antibody and applying a high-gradient magnetic field. (4) Receptor clustering: spaced receptors (binding with MNPs) on a cell membrane will form a cluster when a magnetic needle is applied to generate a localized magnetic field due to an interparticle magnetic force.
Instead of mechanical conditioning in a bioreactor, magnetic actuation could activate ion channels. By attaching MNPs (130 nm to 4 μm) to integrin receptors of human primary osteoblasts, mineralization was enhanced by applying a time-varying magnetic field (Bmax ∼ 60 mT) compared to a control group with no MNPs.193 This process has been demonstrated to stimulate intracellular calcium stores, change membrane potential, and upregulate genes related to bone formation. Selective activation of the TREK-1 ion channel, which is a stretch-activated potassium channel, could also be achieved by linking MNPs to 6-His-loop mutant TREK-1 in COS-7 cells by an antibody or nickel and nitrilotriacetic acid (Ni–NTA). Under a 1 Hz magnetic field (∼80 mT), targeted MNPs were manipulated, which led to changes in whole-cell currents as well as TREK-1 activity.194 The significant upregulation of cartilage-related genes in MSCs further demonstrated that this process is able to specifically activate the TREK-1 ion channel in the cartilage differentiation pathway. Based on these results, Dobson et al. summarized this technique, which applies mechanical forces (in the pico/nanonewton range) directly to molecular components of cells by magnetic actuation, as a “magnetic force bioreactor” (MFB). By targeting MNPs to platelet-derived growth factor receptor-α (PDGFRα), they have demonstrated a significant increase in the mineral to matrix ratio of human MSCs after 3 weeks MFB culture and proposed a possible mechanism (Fig. 9). The response profile of stimulation mediated by PDGFRα was strong in the early stage (within 7 days), whereas stimulation mediated by integrin was strong in the medium term (7 to 10 days).195 More recently, this group demonstrated successful expression of smooth muscle α-actin on both protein and gene levels by cyclical mechanical stimulation of PDGFR α and β on MSCs.196
 | ||
| Fig. 9 Schematic illustration of proposed osteogenic differentiation of MSCs induced by magnetomechanical stimulation combined with biochemical signals. MNPs were functionalized with RGD peptide or PDGFRα antibody to target either integrin ανβ3 or PDGFRα on the cell surface membrane of MSCs. When exposed to MFB, induced mechanotransduction coupled with biochemical signals directed the osteogenic differentiation and mineralization of MSCs. Reproduced from ref. 195 with permission. | ||
4. Challenges and the future
As discussed so far, MNPs can have their physicochemical properties tailored or be combined with other types of materials to fit the desired application in regenerative medicine, especially when helping to translate current preclinical research into effective treatments. Despite being promising, several challenges still need to be overcome (Table 1).| Challenges | Proposed solutions |
|---|---|
|
(1) Safety
• Multifactorial197–199 • Release of free iron200–203 |
• Comprehensive preclinical study, case by case
• Utilize surface coating (e.g. PEG)198 and biodegradable microparticles (e.g. PLGA)164 |
| (2) Formation of a protein corona204–208 | • Fingerprint protein coronas and evaluate their biological effects for specific MNPs |
| (3) Transplant rejection209 |
• Magnetic scaffold for controlled and localized release of immunosuppressive drugs
• Magnetic semipermeable microcapsules for MR-visible immunoisolation213 |
|
(4) MRI imaging
• Signal dilution164 • Lack of information about cellular function |
• Utilize biodegradable microparticles (e.g. PLGA)164 or genetically engineer cells to self-synthesize MNPs173
• Multifunctional MNPs174 • New MRI technology (e.g. CEST)171 |
| (5) Lack of effective magnetic field gradient |
• Development of a new type of magnetic control system214–216
• Synthesis of MNPs with high magnetic moment |
| (6) Large-scale fabrication and packaging | • Need collaboration between researchers from different disciplines |
4.1 Safety
Although MNPs have been approved and used in clinical applications, research has demonstrated that the toxicity of MNPs is multifactorial and depends on their composition, properties (such as size, surface characteristics), dose, and route of administration. For example, when incubated with an A549 alveolar epithelial cell line at a high dose (80 μg mL−1), 30 nm Fe3O4 particles caused higher oxidative damage to DNA compared to 0.5 μm particles in a comet assay.197 In terms of surface characteristics, a PEG coating prevents a significant decrease (64%) in cell (primary human fibroblasts) adhesion, cell membrane abnormalities and disruption observed for uncoated iron oxide NPs (50 nm).198 The accumulation and clearance of the same MNPs in different organs or tissues are also different, which causes a variation in LD50 values.199 This indicates a variation in the toxicity of MNPs when adopting different routes of administration.There are concerns about the excessive release of free iron from MNPs in vivo, because this can facilitate the generation of free radicals, leading to oxidative stress and disruption in liver metabolism.200,201 Excess accumulation of MNPs in the brain may also cause oxidative stress by interactions between iron and proteins (e.g. amyloid peptide, metallothionein or neuromelanin) and therefore lead to the risk of neurodegenerative diseases such as Alzheimer's and Parkinson's disease.202,203
As a consequence, it is critical to comprehensively investigate the safety issues of MNPs case by case prior to their clinical use. A number of in vitro toxicity exit investigations have shown no adverse side effects of MNPs in regenerative medicine; however, long-term in vivo studies are highly needed.
4.2 Formation of protein corona
Decades ago, researchers already found that particles that are administered intravenously will be coated with a layer of proteins (i.e. a protein corona) right after injection (within less than 0.5 minutes).204 Recent research has further shown that there are almost 300 kinds of proteins involved.205 The exact amount and type of proteins adsorbed on the surface of NPs depend on the physicochemical properties of NPs (e.g. size, surface charge, etc.) as well as the sources of protein in different biological fluids. These protein coronas modulate the pathobiological effects of NPs. For example, a protein corona coating allowed silica NPs to prevent aggregation of thrombocytes.205 In brief, fresh isolated human thrombocytes were activated rapidly when exposed to silica NPs without a protein corona, which was similar to the natural stimulus by collagen. In contrast, silica NPs re-exposed to human plasma for 0.5 min did not activate thrombocytes. In another study, a protein corona (formed in fetal bovine serum) dramatically decreased the relaxivity of amine-dextran (positive charge)-coated MNPs, slightly increased the relaxivity of carboxyl-dextran (negative charge)-coated MNPs, and had no effect on plain MNPs (dextran coated).206 When MNPs were coated with polyvinyl alcohol instead of dextran, there were more proteins on the surface of MNPs, which prolonged their circulation.207 In another interesting study, 3.5 nm MNPs with different charges were intravenously injected into mice and the mouse brains were imaged by MRI in real time. Both plain and negatively charged MNPs were observed in the brain vessels with strong signals 5 minutes after injection, but positively charged MNPs were not found there.208 This phenomenon was believed to be related to the formation of a unique protein corona (high presence of ApoA-I), which caused uptake of MNP by the brain tissue.In general, the composition, formation dynamics, and functions of protein coronas have not yet been well understood. Unexpected results might emerge when translating MNP to clinical use without a comprehensive understanding of these protein coatings. To fully understand and even potentially make use of protein coronas, fingerprint information about protein coronas for specific MNPs is desired and could be collected by combining quantitative proteomics, state-of-the-art mass spectroscopy, bioinformatics and systems biology.
4.3 Transplant rejection
Currently, the problem with transplanting cells into patients is immune rejection by the body, where the immune system attacks and kills the injected cells. In clinical treatments, immunosuppressive drugs are usually co-administered with the transplanted cells. However, these drugs are coincidentally very toxic to the transplanted cells, which compromises the efficacy of these treatments.209By using a hydrogel scaffold with incorporated MNPs, on-demand localized release of immunosuppressive drugs can be achieved after transplantation simply via an external magnetic field. This strategy not only reduces the side effects of immunosuppressive drugs, but also visualizes the transplanted cells by MRI and lets the doctor know if the cells have been injected correctly and are surviving after transplantation.
Another strategy relies on the immunoisolation of transplanted cells in semipermeable microcapsules that prevent or delay immune rejection following transplantation.210–212 Bulte et al. developed MR-visible alginate microcapsules for immunoisolation and non-invasive imaging of cellular therapeutics.213 These microcapsules are fabricated via three steps: (i) mixing liquid alginate, cells and MNPs to form a micro-sized (average diameter ∼350 μm) solid gel by electrostatic extrusion; (ii) coating gelled spheres with a polycation (e.g. poly-L-lysine) as a crosslinker; and (iii) incubating again in alginate solution to form a secondary alginate layer (semipermeable). This double alginate layer not only allows the diffusion of chemicals and proteins but also prevents the attack of immune cells.213 Therefore, this magnetic microcapsule opens up the possibility of immunoisolating xenogeneic grafts.
4.4 Signal dilution and lack of information about function in MRI imaging
Signal dilution, which is an issue for long-term in vivo tracking of stem cells, is the loss of a signal resulting from cell division and exocytosis. Currently, the most commonly used MRI contrast agents for cell tracking are iron oxide NPs with core size ranging from 4 to 20 nm. However, all of these suffer from a time-dependent decrease in MRI signal. For example, residual Fe in MSCs labeled with iron oxide NPs decreased to 30% at day 5 and undetectable levels at day 12.164 Therefore, concentrating MNPs in biodegradable polymer microparticles or engineering therapeutic cells with the ability to self-synthesize iron oxide NPs has the promise of addressing this problem.As mentioned before, the lack of information about the function and fate of stem cells following engraftment has confined tracking of stem cells by MRI to understanding the biodistribution of transplanted therapeutic cells. Multifunctional MNPs such as Janus NPs that provide dual contrast for both MRI and other imaging methodologies (e.g. fluorescence and photoacoustic imaging) may allow the acquisition of more information compared to MRI tracking alone. New technology such as CEST imaging, which could sense the pH of the microenvironment, is of great significance in future clinical imaging.
4.5 Lack of effective magnetic field gradient
Currently, most magnetic controllers consist of a permanent magnet, which is placed near the target site. As reported, most commercially available magnets can only penetrate a tissue depth of a few millimeters. The development of a new type of magnetic control system, as well as the synthesis of MNPs with high magnetic moments, may solve this problem. Superconducting magnets such as SmBaCuO and YBaCuO can generate strong magnetic gradients with a penetration depth of 20 mm.214 Permanent neodymium–iron–boron magnets (magnetic field strength ranging from 250 to 1000 G) could be combined with MNPs with higher magnetic susceptibility (e.g. a composite of Fe2O3 and Fe3C alloy with a 75:25 Fe:C ratio) to achieve in vivo targeting in a swine model up to a depth of 8–12 cm.215,216 To increase the magnetic moment, other metal ions (Mn2+, Co2+ or Ni2+) are doped into the spinel structure of ferrite, and metallic MNPs (alloys) with a high magnetic moment are also promising. It should be noted that systematic and comprehensive evaluation of the safety of new forms of MNPs is a prerequisite before any clinical application.
4.6 Large-scale fabrication and packaging
Although the large-scale production of commercial MNPs has been achieved, various delicately designed MNPs, MNP-based scaffolds/stents, and specialized magnet systems are still confined to laboratory use. The large-scale fabrication, packaging, storage, and delivery of cellular therapeutics or engineered tissues for regenerative medicine still need to be addressed before being translated to clinical settings.Acknowledgements
The work was supported by the Tier-1 Academic Research Funds by Singapore Ministry of Education (RG64/12 to XCJ; RGT21/13 to TSH).References
- C. Mason and P. Dunnill, Regener. Med., 2007, 3, 1–5 CrossRef PubMed.
- I. Singec, R. Jandial, A. Crain, G. Nikkhah and E. Y. Snyder, Annu. Rev. Med., 2007, 58, 313–328 CrossRef CAS PubMed.
- R. Pardal, M. F. Clarke and S. J. Morrison, Nat. Rev. Cancer, 2003, 3, 895–902 CrossRef CAS PubMed.
- M. F. Pittenger and B. J. Martin, Circ. Res., 2004, 95, 9–20 CrossRef CAS PubMed.
- F. P. Barry and J. M. Murphy, Int. J. Biochem. Cell Biol., 2004, 36, 568–584 CrossRef CAS PubMed.
- http://alliancerm.org/sites/default/files/ARM-Natl-Strat-Apr13.pdf .
- K. E. Healy, T. C. McDevitt, W. L. Murphy and R. M. Nerem, Sci. Transl. Med., 2013, 5, 207ed217 Search PubMed.
- I. Martin, P. J. Simmons and D. F. Williams, Sci. Transl. Med., 2014, 6, 232fs216 Search PubMed.
- I. L. Weissman, Science, 2000, 287, 1442–1446 CrossRef CAS.
- C. Y. Xu, R. Inai, M. Kotaki and S. Ramakrishna, Biomaterials, 2004, 25, 877–886 CrossRef CAS.
- W. J. Li, R. Tuli, C. Okafor, A. Derfoul, K. G. Danielson, D. J. Hall and R. S. Tuan, Biomaterials, 2005, 26, 599–609 CrossRef CAS PubMed.
- C. M. Li, C. Vepari, H. J. Jin, H. J. Kim and D. L. Kaplan, Biomaterials, 2006, 27, 3115–3124 CrossRef CAS PubMed.
- T. J. Sill and H. A. von Recum, Biomaterials, 2008, 29, 1989–2006 CrossRef CAS PubMed.
- Z. Q. Feng, X. H. Chu, N. P. Huang, T. Wang, Y. C. Wang, X. L. Shi, Y. T. Ding and Z. Z. Gu, Biomaterials, 2009, 30, 2753–2763 CrossRef CAS PubMed.
- S. Namgung, K. Y. Baik, J. Park and S. Hong, ACS Nano, 2011, 5, 7383–7390 CrossRef CAS PubMed.
- B. K. K. Teo, S. T. Wong, C. K. Lim, T. Y. S. Kung, C. H. Yap, Y. Ramagopal, L. H. Romer and E. K. F. Yim, ACS Nano, 2013, 7, 4785–4798 CrossRef CAS PubMed.
- S. Lavenus, V. Trichet, S. Le Chevalier, A. Hoornaert, G. Louarn and P. Layrolle, Nanomedicine, 2012, 7, 967–980 CrossRef CAS PubMed.
- J. Gao, W. Zhang, P. Huang, B. Zhang, X. Zhang and B. Xu, J. Am. Chem. Soc., 2008, 130, 3710–3711 CrossRef CAS PubMed.
- N. A. Frey, S. Peng, K. Cheng and S. Sun, Chem. Soc. Rev., 2009, 38, 2532–2542 RSC.
- C. Sun, J. S. H. Lee and M. Q. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS PubMed.
- C. Xu, L. Mu, I. Roes, D. Miranda-Nieves, M. Nahrendorf, J. A. Ankrum, W. Zhao and J. M. Karp, Nanotechnology, 2011, 22, 494001 CrossRef PubMed.
- M. Mahmoudi, H. Hosseinkhani, M. Hosseinkhani, S. Boutry, A. Simchi, W. S. Journeay, K. Subramani and S. Laurent, Chem. Rev., 2010, 111, 253–280 CrossRef PubMed.
- C. Xu and S. Sun, Adv. Drug Delivery Rev., 2013, 65, 732–743 CrossRef CAS PubMed.
- H. Gu, K. Xu, C. Xu and B. Xu, Chem. Commun., 2006, 941–949 RSC.
- C. Xu, K. Xu, H. Gu, R. Zheng, H. Liu, X. Zhang, Z. Guo and B. Xu, J. Am. Chem. Soc., 2004, 126, 9938–9939 CrossRef CAS PubMed.
- C. Xu, K. Xu, H. Gu, X. Zhong, Z. Guo, R. Zheng, X. Zhang and B. Xu, J. Am. Chem. Soc., 2004, 126, 3392–3393 CrossRef CAS PubMed.
- P. Matteini, M. R. Martina, G. Giambastiani, F. Tatini, R. Cascella, F. Ratto, C. Cecchi, G. Caminati, L. Dei and R. Pini, J. Mater. Chem. B, 2013, 1, 1096–1100 RSC.
- Y. Jung, G. Guan, C.-w. Wei, R. Reif, X. Gao, M. O'Donnell and R. K. Wang, J. Biomed. Opt., 2012, 17, 0160151 Search PubMed.
- Y. Pan, X. Du, F. Zhao and B. Xu, Chem. Soc. Rev., 2012, 41, 2912–2942 RSC.
- M. Colombo, S. Carregal-Romero, M. F. Casula, L. Gutierrez, M. P. Morales, I. B. Bohm, J. T. Heverhagen, D. Prosperi and W. J. Parak, Chem. Soc. Rev., 2012, 41, 4306–4334 RSC.
- S. H. Ku, M. Lee and C. B. Park, Adv. Healthcare Mater., 2013, 2, 244–260 CrossRef CAS PubMed.
- A. Alzaraa, G. Gravante, W. Y. Chung, D. Al-Leswas, M. Bruno, A. R. Dennison and D. M. Lloyd, Am. J. Surg., 2012, 204, 355–366 CrossRef PubMed.
- Q. Hu, Z. Tan, Y. Liu, J. Tao, Y. Cai, M. Zhang, H. Pan, X. Xu and R. Tang, J. Mater. Chem., 2007, 17, 4690–4698 RSC.
- F. C. Grant and N. C. Norcross, Ann. Surg., 1939, 110, 488 CrossRef CAS PubMed.
- T. K. Dash and V. B. Konkimalla, J. Controlled Release, 2012, 158, 15–33 CrossRef CAS PubMed.
- V. Sinha, K. Bansal, R. Kaushik, R. Kumria and A. Trehan, Int. J. Pharm., 2004, 278, 1–23 CrossRef CAS PubMed.
- J. H. Lee, N. G. Rim, H. S. Jung and H. Shin, Macromol. Biosci., 2010, 10, 173–182 CrossRef CAS PubMed.
- S. H. Teoh, Engineering Materials for Biomedical Applications, Mainland Press, Singapore, 2004 Search PubMed.
- Y. Liu, S. H. Teoh, M. S. Chong, E. S. Lee, C. N. Mattar, N. s. K. Randhawa, Z. Y. Zhang, R. J. Medina, R. D. Kamm and N. M. Fisk, Stem Cells, 2012, 30, 1911–1924 CrossRef PubMed.
- Z. Y. Zhang, S. H. Teoh, M. S. K. Chong, E. S. M. Lee, L. G. Tan, C. N. Mattar, N. M. Fisk, M. Choolani and J. Chan, Biomaterials, 2010, 31, 608–620 CrossRef CAS PubMed.
- H. Zhang, A. Patel, A. K. Gaharwar, S. M. Mihaila, G. Iviglia, S. Mukundan, H. Bae, H. Yang and A. Khademhosseini, Biomacromolecules, 2013, 14, 1299–1310 CrossRef CAS PubMed.
- B. Wulkersdorfer, K. K. Kao, V. G. Agopian, J. C. Dunn, B. M. Wu and M. Stelzner, J. Surg. Res., 2011, 169, 169–178 CrossRef CAS PubMed.
- D. A. Wong, A. Kumar, S. Jatana, G. Ghiselli and K. Wong, Spine J., 2008, 8, 1011–1018 CrossRef PubMed.
- K. L. Ong, M. L. Villarraga, E. Lau, L. Y. Carreon, S. M. Kurtz and S. D. Glassman, Spine J., 2010, 35, 1794–1800 CrossRef PubMed.
- N.-F. Chen, Z. A. Smith, E. Stiner, S. Armin, H. Sheikh and L. T. Khoo, J. Neurosurg.: Spine, 2010, 12, 40–46 CrossRef PubMed.
- J. Chu, R. D. Haynes, S. Y. Corbel, P. Li, E. González-González, J. S. Burg, A. J. Ataie, A. J. Lam, P. J. Cranfill and M. A. Baird, Nat. Methods, 2014, 11(5), 572–578 CrossRef CAS PubMed.
- K. Nienhaus and G. U. Nienhaus, Chem. Soc. Rev., 2014, 43(4), 1088–1106 RSC.
- H. Keith and F. Padden Jr, J. Appl. Phys., 2004, 35, 1270–1285 CrossRef PubMed.
- Q. Zheng, M. F. Juette, S. Jockusch, M. R. Wasserman, Z. Zhou, R. B. Altman and S. C. Blanchard, Chem. Soc. Rev., 2014, 43(4), 1044–1056 RSC.
- S. J. Kress, P. Richner, S. V. Jayanti, P. Galliker, D. K. Kim, D. Poulikakos and D. J. Norris, Nano Lett., 2014, 14(10), 5827–5833 CrossRef CAS PubMed.
- K. Ding, L. Jing, C. Liu, Y. Hou and M. Gao, Biomaterials, 2014, 35(5), 1608–1617 CrossRef CAS PubMed.
- M. Colombo, S. Carregal-Romero, M. F. Casula, L. Gutierrez, M. P. Morales, I. B. Bohm, J. T. Heverhagen, D. Prosperi and W. J. Parak, Chem. Soc. Rev., 2012, 41, 4306–4334 RSC.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- A. Ito, E. Hibino, K. Shimizu, T. Kobayashi, Y. Yamada, H. Hibi, M. Ueda and H. Honda, J. Biomed. Mater. Res., Part B, 2005, 75B, 320–327 CrossRef CAS PubMed.
- R. Sensenig, Y. Sapir, C. MacDonald, S. Cohen and B. Polyak, Nanomedicine, 2012, 7, 1425–1442 CrossRef CAS PubMed.
- E. Nance, K. Timbie, G. W. Miller, J. Song, C. Louttit, A. L. Klibanov, T.-Y. Shih, G. Swaminathan, R. J. Tamargo, G. F. Woodworth, J. Hanes and R. J. Price, J. Controlled Release, 2014, 189, 123–132 CrossRef CAS PubMed.
- R. Fernandes, N. R. Smyth, O. L. Muskens, S. Nitti, A. Heuer-Jungemann, M. R. Ardern-Jones and A. G. Kanaras, Small, 2015, 11, 713–721 CrossRef CAS PubMed.
- E. Amstad, M. Textor and E. Reimhult, Nanoscale, 2011, 3, 2819 RSC.
- S. Santra, C. Kaittanis, J. Grimm and J. M. Perez, Small, 2009, 5, 1862–1868 CrossRef CAS PubMed.
- M. Vittaz, D. Bazile, G. Spenlehauer, T. Verrecchia, M. Veillard, F. Puisieux and D. Labarre, Biomaterials, 1996, 17, 1575–1581 CrossRef CAS.
- T. Gillich, C. Acikgöz, L. Isa, A. D. Schlüter, N. D. Spencer and M. Textor, ACS Nano, 2013, 7, 316–329 CrossRef CAS PubMed.
- Y. Li, X. Li, Z. Li and H. Gao, Nanoscale, 2012, 4, 3768–3775 RSC.
- A. Verma, O. Uzun, Y. Hu, Y. Hu, H.-S. Han, N. Watson, S. Chen, D. J. Irvine and F. Stellacci, Nat. Mater., 2008, 7, 588–595 CrossRef CAS PubMed.
- S. E. Kim, Y.-P. Yun, Y.-K. Han, D.-W. Lee, J.-Y. Ohe, B.-S. Lee, H.-R. Song, K. Park and B.-J. Choi, Carbohydr. Polym., 2014, 99, 700–709 CrossRef CAS PubMed.
- J. Almodóvar, R. Guillot, C. Monge, J. Vollaire, Š. Selimović, J.-L. Coll, A. Khademhosseini and C. Picart, Biomaterials, 2014, 35, 3975–3985 CrossRef PubMed.
- T.-S. Yeh, Y.-H. Dean Fang, C.-H. Lu, S.-C. Chiu, C.-L. Yeh, T.-C. Yen, Y. Parfyonova and Y.-C. Hu, Biomaterials, 2014, 35, 174–184 CrossRef CAS PubMed.
- A. J. Rufaihah, S. R. Vaibavi, M. Plotkin, J. Shen, V. Nithya, J. Wang, D. Seliktar and T. Kofidis, Biomaterials, 2013, 34, 8195–8202 CrossRef CAS PubMed.
- M. Nevins, R. T. Kao, M. K. McGuire, P. K. McClain, J. E. Hinrichs, B. S. McAllister, M. S. Reddy, M. L. Nevins, R. J. Genco, S. E. Lynch and W. V. Giannobile, J. Periodontol., 2012, 84, 456–464 CrossRef PubMed.
- M. I. Santos and R. L. Reis, Macromol. Biosci., 2010, 10, 12–27 CrossRef CAS PubMed.
- C. J. Kirkpatrick, S. Fuchs and R. E. Unger, Adv. Drug Delivery Rev., 2011, 63, 291–299 CrossRef CAS PubMed.
- J. Lim, M. S. K. Chong, Y. Liu, A. Khademhosseini and T. Swee-Hin, in Engineering Muscuoloskeletal Tissues and Interfaces, ed. N. Syam, C. Laurencin and J. Freeman, Woodhead Publishing, 2014 Search PubMed.
- G. Blair, T. Fannin and D. Gordon, Br. Med. J., 1976, 2, 907–908 CrossRef CAS.
- L. Chiarini, S. Figurelli, G. Pollastri, E. Torcia, F. Ferrari, M. Albanese and P. Francesco Nocini, J. Cranio. Maxill. Surg., 2004, 32, 5–9 CrossRef PubMed.
- P. Scolozzi, A. Martinez and B. Jaques, J. Craniofac. Surg., 2007, 18, 224–228 CrossRef PubMed.
- D. J. Prolo, K. P. Burres, W. T. McLaughlin and A. H. Christensen, Neurosurgery, 1979, 4, 18–29 CrossRef CAS.
- F. Viterbo, A. Palhares and E. Modenese, J. Craniofac. Surg., 1995, 6, 80–82 CrossRef CAS PubMed.
- M. Cabraja, M. Klein and T.-N. Lehmann, Neurosurg. Focus, 2009, 26, E10 CrossRef PubMed.
- L. You, S. Temiyasathit, P. Lee, C. H. Kim, P. Tummala, W. Yao, W. Kingery, A. M. Malone, R. Y. Kwon and C. R. Jacobs, Bone, 2008, 42, 172–179 CrossRef CAS PubMed.
- L. F. Bonewald and M. L. Johnson, Bone, 2008, 42, 606–615 CrossRef CAS PubMed.
- H. Singh, S.-H. Teoh, H. T. Low and D. Hutmacher, J. Biotechnol., 2005, 119, 181–196 CrossRef CAS PubMed.
- Z.-Y. Zhang, S.-H. Teoh, M. S. Chong, E. S. Lee, L.-G. Tan, C. N. Mattar, N. M. Fisk, M. Choolani and J. Chan, Biomaterials, 2010, 31, 608–620 CrossRef CAS PubMed.
- J.-T. Schantz, A. Brandwood, D. W. Hutmacher, H. L. Khor and K. Bittner, J. Mater. Sci.: Mater. Med., 2005, 16, 807–819 CrossRef CAS PubMed.
- S. Aydin, B. Kucukyuruk, B. Abuzayed, S. Aydin and G. Z. Sanus, J. Neurosci. Rural Pract., 2011, 2, 162 CrossRef PubMed.
- D. Hou, E. A.-S. Youssef, T. J. Brinton, P. Zhang, P. Rogers, E. T. Price, A. C. Yeung, B. H. Johnstone, P. G. Yock and K. L. March, Circulation, 2005, 112, I-150–I-156 Search PubMed.
- N. E. Epstein, Surg. Neurol. Int., 2013, 4, S343 CrossRef PubMed.
- G. Chan and D. J. Mooney, Trends Biotechnol., 2008, 26, 382–392 CrossRef CAS PubMed.
- O. Veiseh, J. W. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef CAS PubMed.
- J. Dobson, Drug Dev. Res., 2006, 67, 55–60 CrossRef CAS PubMed.
- Q. A. Pankhurst, J. Connolly, S. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167 CrossRef CAS.
- J. Dobson, Gene Ther., 2006, 13, 283–287 CrossRef CAS PubMed.
- K. Cheng, S. Peng, C. Xu and S. Sun, J. Am. Chem. Soc., 2009, 131, 10637–10644 CrossRef CAS PubMed.
- B. Wang, C. Xu, J. Xie, Z. Yang and S. Sun, J. Am. Chem. Soc., 2008, 130, 14436–14437 CrossRef CAS PubMed.
- M. Arruebo, R. Fernandez-Pacheco, M. R. Ibarra and J. Santamaria, Nano Today, 2007, 2, 22–32 CrossRef.
- O. Veiseh, J. W. Gunn and M. Q. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef CAS PubMed.
- S. C. McBain, H. H. P. Yiu and J. Dobson, Int. J. Nanomed., 2008, 3, 169–180 CAS.
- Y. Liu, J. Lim and S.-H. Teoh, Biotechnol. Adv., 2013, 31, 688–705 CrossRef CAS PubMed.
- F. A. Auger, F. Berthod, V. Moulin, R. Pouliot and L. Germain, Biotechnol. Appl. Biochem., 2004, 39, 263–275 CrossRef CAS PubMed.
- R. M. Nerem, Biomaterials, 2007, 28, 5074–5077 CrossRef CAS PubMed.
- D. J. Mooney and H. Vandenburgh, Cell Stem Cell, 2008, 2, 205–213 CrossRef CAS PubMed.
- M. Chorny, I. Fishbein, B. B. Yellen, I. S. Alferiev, M. Bakay, S. Ganta, R. Adamo, M. Amiji, G. Friedman and R. J. Levy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8346–8351 CrossRef CAS PubMed.
- N. Bock, A. Riminucci, C. Dionigi, A. Russo, A. Tampieri, E. Landi, V. A. Goranov, M. Marcacci and V. Dediu, Acta Biomater., 2010, 6, 786–796 CrossRef CAS PubMed.
- S.-H. Hu, T.-Y. Liu, C.-H. Tsai and S.-Y. Chen, J. Magn. Magn. Mater., 2007, 310, 2871–2873 CrossRef CAS PubMed.
- X. H. Zhao, J. Kim, C. A. Cezar, N. Huebsch, K. Lee, K. Bouhadir and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 67–72 CrossRef CAS PubMed.
- C. A. Cezar, S. M. Kennedy, M. Mehta, J. C. Weaver, L. Gu, H. Vandenburgh and D. J. Mooney, Adv. Healthcare Mater., 2014, 3, 1869–1876 CrossRef CAS PubMed.
- P. G. Kyrtatos, P. Lehtolainen, M. Junemann-Ramirez, A. Garcia-Prieto, A. N. Price, J. F. Martin, D. G. Gadian, Q. A. Pankhurst and M. F. Lythgoe, JACC: Cardiovasc. Interv., 2009, 2, 794–802 CrossRef PubMed.
- B. Polyak, I. Fishbein, M. Chorny, I. Alferiev, D. Williams, B. Yellen, G. Friedman and R. J. Levy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 698–703 CrossRef CAS PubMed.
- A. Yanai, U. O. Häfeli, A. L. Metcalfe, P. Soema, L. Addo, C. Y. Gregory-Evans, K. Po, X. Shan, O. L. Moritz and K. Gregory-Evans, Cell Transplant., 2012, 21, 1137–1148 Search PubMed.
- T. Kobayashi, M. Ochi, S. Yanada, M. Ishikawa, N. Adachi, M. Deie and K. Arihiro, Arthroscopy, 2008, 24, 69–76 CrossRef PubMed.
- A. Kodama, N. Kamei, G. Kamei, W. Kongcharoensombat, S. Ohkawa, A. Nakabayashi and M. Ochi, J. Bone Jt. Surg., Br. Vol., 2012, 94B, 998–1006 CrossRef PubMed.
- T. Kobayashi, M. Ochi, S. Yanada, M. Ishikawa, N. Adachi, M. Deie and K. Arihiro, Arthroscopy, 2009, 25, 1435–1441 CrossRef PubMed.
- S. J. Kuhn, S. K. Finch, D. E. Hallahan and T. D. Giorgio, Nano Lett., 2006, 6, 306–312 CrossRef CAS PubMed.
- J. C. Chappell, J. Song, C. W. Burke, A. L. Klibanov and R. J. Price, Small, 2008, 4, 1769–1777 CrossRef CAS PubMed.
- K. J. Dormer, V. Awasthi, W. Galbraith, R. D. Kopke, K. Chen and R. Wassel, J. Biomed. Nanotechnol., 2008, 4, 174–184 CrossRef CAS PubMed.
- J. F. Schenck, Prog. Biophys. Mol. Biol., 2005, 87, 185–204 CrossRef PubMed.
- B. Shapiro, K. Dormer and I. B. Rutel, in 8th International Conference on the Scientific and Clinical Applications of Magnetic Carriers, ed. U. Hafeli, W. Schutt and M. Zborowski, Amer Inst Physics, Melville, 2010, vol. 1311, pp. 77–88.
- N. Saho, N. Nishijima, H. Tanaka and A. Sasaki, Phys. C, 2009, 469, 1286–1289 CrossRef CAS PubMed.
- J. L. Dragoo, J. Y. Choi, J. R. Lieberman, J. Huang, P. A. Zuk, J. Zhang, M. H. Hedrick and P. Benhaim, J. Orthop. Res., 2003, 21, 622–629 CrossRef CAS.
- D. Sheyn, M. Ruethemann, O. Mizrahi, I. Kallai, Y. Zilberman, W. Tawackoli, L. E. A. Kanim, L. Zhao, H. Bae, G. Pelled, J. G. Snedeker and D. Gazit, Tissue Eng., Part A, 2010, 16, 3679–3686 CrossRef CAS PubMed.
- S. McLenachan, J. P. Sarsero and P. A. Ioannou, Genomics, 2007, 89, 708–720 CrossRef CAS PubMed.
- F. Yang, S. W. Cho, S. M. Son, S. R. Bogatyrev, D. Singh, J. J. Green, Y. Mei, S. Park, S. H. Bhang, B. S. Kim, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3317–3322 CrossRef CAS PubMed.
- S. R. Braam, C. Denning, S. van den Brink, P. Kats, R. Hochstenbach, R. Passier and C. L. Mummery, Nat. Methods, 2008, 5, 389–392 CrossRef CAS PubMed.
- C. Plank, O. Zelphati and O. Mykhaylyk, Adv. Drug Delivery Rev., 2011, 63, 1300–1331 CrossRef CAS PubMed.
- S. Huth, J. Lausier, S. W. Gersting, C. Rudolph, C. Plank, U. Welsch and J. Rosenecker, J. Gene Med., 2004, 6, 923–936 CrossRef CAS PubMed.
- http://clinicaltrials.gov/ct2/show/study/NCT01217008 .
- S. I. Jenkins, M. R. Pickard, N. Granger and D. M. Chari, ACS Nano, 2011, 5, 6527–6538 CrossRef CAS PubMed.
- H. P. Song, J. Y. Yang, S. L. Lo, Y. Wang, W. M. Fan, X. S. Tang, J. M. Xue and S. Wang, Biomaterials, 2010, 31, 769–778 CrossRef CAS PubMed.
- C. H. Lee, J. H. Kim, H. J. Lee, K. Jeon, H. Lim, H. Y. Choi, E. R. Lee, S. H. Park, J. Y. Park, S. Hong, S. Kim and S. G. Cho, Biomaterials, 2011, 32, 6683–6691 CrossRef CAS PubMed.
- D. J. Prolo and S. A. Oklund, Clin. Orthop., 1991, 268, 270–278 Search PubMed.
- W. C. Lara, J. Schweitzer, R. P. Lewis, B. C. Odum, R. F. Edlich and T. J. Gampper, J. Long-Term Eff. Med. Implants, 1997, 8, 43–53 Search PubMed.
- S. Ostrovidov, V. Hosseini, S. Ahadian, T. Fujie, S. P. Parthiban, M. Ramalingam, H. Bae, H. Kaji and A. Khademhosseini, Tissue Eng., Part B, 2014, 20, 403–436 CrossRef PubMed.
- H. B. Gladstone, M. W. McDermott and D. D. Cooke, Otolaryngol. Clin. North Am., 1995, 28, 381–400 CAS.
- A. Smith, I. Jackson and J. Yousefi, Eur. J. Plast. Surg., 1999, 22, 17–21 CrossRef.
- I. T. Jackson and R. Yavuzer, Br. J. Plast. Surg., 2000, 53, 24–29 CrossRef CAS PubMed.
- P. R. Ebeling, D. M. Thomas, B. Erbas, J. L. Hopper, J. Szer and A. P. Grigg, J. Bone Miner. Res., 1999, 14, 342–350 CrossRef CAS PubMed.
- G. Hadad, L. Bassagasteguy, R. L. Carrau, J. C. Mataza, A. Kassam, C. H. Snyderman and A. Mintz, Laryngoscope, 2006, 116, 1882–1886 CrossRef PubMed.
- C. H. Snyderman, I. P. Janecka, L. N. Sekhar, C. N. Sen and D. E. Eibling, Laryngoscope, 1990, 100, 607–614 CrossRef CAS PubMed.
- P. Neligan, S. Mulholland, J. Irish, P. Gullane, J. Boyd, F. Gentili, D. Brown and J. Freeman, Plast. Reconstr. Surg., 1996, 98, 1159–1166 CAS.
- Z. Y. Wang, E. Y. Teo, M. S. Chong, Q. Y. Zhang, J. Lim, Z. Y. Zhang, M. H. Hong, E. S. Thian, J. K. Chan and S. H. Teoh, Tissue Eng., Part C, 2013, 4, 4 Search PubMed.
- A. Ito, Y. Takizawa, H. Honda, K. I. Hata, H. Kagami, M. Ueda and T. Kobayashi, Tissue Eng., 2004, 10, 833–840 CrossRef CAS PubMed.
- A. Ito, E. Hibino, C. Kobayashi, H. Terasaki, H. Kagami, M. Ueda, T. Kobayashi and H. Honda, Tissue Eng., 2005, 11, 489–496 CrossRef CAS PubMed.
- K. Shimizu, A. Ito, T. Yoshida, Y. Yamada, M. Ueda and H. Honda, J. Biomed. Mater. Res., Part B, 2007, 82B, 471–480 CrossRef CAS PubMed.
- A. Ito, K. Ino, M. Hayashida, T. Kobayashi, H. Matsunuma, H. Kagami, M. Ueda and H. Honda, Tissue Eng., 2005, 11, 1553–1561 CrossRef CAS PubMed.
- H. Fujita, K. Shimizu, Y. Yamamoto, A. Ito, M. Kamihira and E. Nagamori, J. Tissue Eng. Regener. Med., 2010, 4, 437–443 CAS.
- Y. Yamamoto, A. Ito, H. Fujita, E. Nagamori, Y. Kawabe and M. Kamihira, Tissue Eng., Part A, 2011, 17, 107–114 CrossRef CAS PubMed.
- T. Kito, R. Shibata, M. Ishii, H. Suzuki, T. Himeno, Y. Kataoka, Y. Yamamura, T. Yamamoto, N. Nishio, S. Ito, Y. Numaguchi, T. Tanigawa, J. K. Yamashita, N. Ouchi, H. Honda, K. Isobe and T. Murohara, Sci. Rep., 2013, 3 Search PubMed.
- H. Perea, J. Aigner, U. Hopfner and E. Wintermantel, Cells Tissues Organs, 2006, 183, 156–165 CrossRef CAS PubMed.
- H. Perea, J. Aigner, J. T. Heverhagen, U. Hopfner and E. Wintermantel, J. Tissue Eng. Regener. Med., 2007, 1, 318–321 CrossRef CAS PubMed.
- K. Shimizu, A. Ito, M. Arinobe, Y. Murase, Y. Iwata, Y. Narita, H. Kagami, M. Ueda and H. Honda, J. Biosci. Bioeng., 2007, 103, 472–478 CrossRef CAS PubMed.
- S. V. Pislaru, A. Harbuzariu, G. Agarwal, T. Witt, R. Gulati, N. P. Sandhu, C. Mueske, M. Kalra, R. D. Simari and G. S. Sandhu, Circulation, 2006, 114, I314–I318 CrossRef PubMed.
- G. R. Souza, J. R. Molina, R. M. Raphael, M. G. Ozawa, D. J. Stark, C. S. Levin, L. F. Bronk, J. S. Ananta, J. Mandelin, M. M. Georgescu, J. A. Bankson, J. G. Gelovani, T. C. Killian, W. Arap and R. Pasqualini, Nat. Nanotechnol., 2010, 5, 291–296 CrossRef CAS PubMed.
- W. L. Haisler, D. M. Timm, J. A. Gage, H. Tseng, T. C. Killian and G. R. Souza, Nat. Protoc., 2013, 8, 1940–1949 CrossRef CAS PubMed.
- A. C. Daquinag, G. R. Souza and M. G. Kolonin, Tissue Eng., Part C, 2013, 19, 336–344 CrossRef CAS PubMed.
- W. Liu, Y. Q. Li, S. Y. Feng, J. Ning, J. Y. Wang, M. L. Gou, H. J. Chen, F. Xu and Y. A. Du, Lab Chip, 2014, 14, 2614–2625 RSC.
- T. Dvir, B. P. Timko, D. S. Kohane and R. Langer, Nat. Nanotechnol., 2011, 6, 13–22 CrossRef CAS PubMed.
- K. Y. Tsang, M. C. H. Cheung, D. Chan and K. S. E. Cheah, Cell Tissue Res., 2010, 339, 93–110 CrossRef CAS PubMed.
- H. C. Ott, T. S. Matthiesen, S. K. Goh, L. D. Black, S. M. Kren, T. I. Netoff and D. A. Taylor, Nat. Med., 2008, 14, 213–221 CrossRef CAS PubMed.
- E. Alsberg, E. Feinstein, M. P. Joy, M. Prentiss and D. E. Ingber, Tissue Eng., 2006, 12, 3247–3256 CrossRef CAS PubMed.
- C. Xu, Z. Yuan, N. Kohler, J. Kim, M. A. Chung and S. Sun, J. Am. Chem. Soc., 2009, 131, 15346–15351 CrossRef CAS PubMed.
- C. Xu, B. Wang and S. Sun, J. Am. Chem. Soc., 2009, 131, 4216–4217 CrossRef CAS PubMed.
- C. Xu and S. Sun, Adv. Drug Delivery Rev., 2013, 65, 732–743 CrossRef CAS PubMed.
- C. Xu and S. Sun, Dalton Trans., 2009, 5583–5591 RSC.
- C. Xu, D. Miranda-Nieves, J. A. Ankrum, M. E. Matthiesen, J. A. Phillips, I. Roes, G. R. Wojtkiewicz, V. Juneja, J. R. Kultima, W. Zhao, P. K. Vemula, C. P. Lin, M. Nahrendorf and J. M. Karp, Nano Lett., 2012, 12, 4131–4139 CrossRef CAS PubMed.
- S. Schlücker, ChemPhysChem, 2009, 10, 1344–1354 CrossRef PubMed.
- M. Lewin, N. Carlesso, C.-H. Tung, X.-W. Tang, D. Cory, D. T. Scadden and R. Weissleder, Nat. Biotechnol., 2000, 18, 410–414 CrossRef CAS PubMed.
- J. Xie, K. Chen, J. Huang, S. Lee, J. Wang, J. Gao, X. Li and X. Chen, Biomaterials, 2010, 31, 3016–3022 CrossRef CAS PubMed.
- I. J. M. de Vries, W. J. Lesterhuis, J. O. Barentsz, P. Verdijk, J. H. van Krieken, O. C. Boerman, W. J. G. Oyen, J. J. Bonenkamp, J. B. Boezeman, G. J. Adema, J. W. M. Bulte, T. W. J. Scheenen, C. J. A. Punt, A. Heerschap and C. G. Figdor, Nat. Biotechnol., 2005, 23, 1407–1413 CrossRef CAS PubMed.
- J. H. Zhu, L. F. Zhou and F. G. XingWu, N. Engl. J. Med., 2006, 355, 2376–2378 CrossRef CAS PubMed.
- Y. Gao, Y. Cui, J. K. Chan and C. Xu, Am. J. Nucl. Med. Mol. Imaging, 2013, 3, 232 CAS.
- K. W. Y. Chan, G. S. Liu, X. L. Song, H. Kim, T. Yu, D. R. Arifin, A. A. Gilad, J. Hanes, P. Walczak, P. C. M. van Zijl, J. W. M. Bulte and M. T. McMahon, Nat. Mater., 2013, 12, 268–275 CrossRef CAS PubMed.
- Y. Gao, Y. Wang, A. Fu, W. Shi, D. Yeo, K. Q. Luo, H. Ow and C. Xu, J. Mater. Chem. B, 2015, 3, 1245–1253 RSC.
- http://www.bellbiosystems.com/ .
- C. Xu, J. Xie, D. Ho, C. Wang, N. Kohler, E. G. Walsh, J. R. Morgan, Y. E. Chin and S. Sun, Angew. Chem., Int. Ed., 2008, 47, 173–176 CrossRef CAS PubMed.
- Y. Liu, J. K. Y. Chan and S.-H. Teoh, J. Tissue Eng. Regener. Med., 2015, 9, 85–105 CrossRef CAS PubMed.
- A. P. Rajesh, T. Erik and J. W. Thomas, Nanotechnology, 2008, 19, 265101 CrossRef PubMed.
- L. Z. Gao, J. Zhuang, L. Nie, J. B. Zhang, Y. Zhang, N. Gu, T. H. Wang, J. Feng, D. L. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed.
- R. H. Burdon, Free Radical Biol. Med., 1995, 18, 775–794 CrossRef CAS.
- C. Nicco, A. Laurent, C. Chereau, B. Weill and F. Batteux, Biomed. Pharmacother., 2005, 59, 169–174 CrossRef CAS PubMed.
- D.-M. Huang, J.-K. Hsiao, Y.-C. Chen, L.-Y. Chien, M. Yao, Y.-K. Chen, B.-S. Ko, S.-C. Hsu, L.-A. Tai, H.-Y. Cheng, S.-W. Wang, C.-S. Yang and Y.-C. Chen, Biomaterials, 2009, 30, 3645–3651 CrossRef CAS PubMed.
- Y.-C. Chen, J.-K. Hsiao, H.-M. Liu, I. Y. Lai, M. Yao, S.-C. Hsu, B.-S. Ko, Y.-C. Chen, C.-S. Yang and D.-M. Huang, Toxicol. Appl. Pharmacol., 2010, 245, 272–279 CrossRef CAS PubMed.
- E. Roeder, C. Henrionnet, J. C. Goebel, N. Gambier, O. Beuf, D. Grenier, B. L. Chen, P. A. Vuissoz, P. Gillet and A. Pinzano, PLoS One, 2014, 9 Search PubMed.
- J. D. Boerckel, B. A. Uhrig, N. J. Willett, N. Huebsch and R. E. Guldberg, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E674–E680 CrossRef CAS PubMed.
- I. Martin, D. Wendt and M. Heberer, Trends Biotechnol., 2004, 22, 80–86 CrossRef CAS PubMed.
- I. Martin, B. Obradovic, S. Treppo, A. Grodzinsky, R. Langer, L. Freed and G. Vunjak-Novakovic, Biorheology, 2000, 37, 141–147 CAS.
- Z.-Y. Zhang, S. H. Teoh, E. Y. Teo, M. S. Khoon Chong, C. W. Shin, F. T. Tien, M. A. Choolani and J. K. Y. Chan, Biomaterials, 2010, 31, 8684–8695 CrossRef CAS PubMed.
- C. J. Hunter, S. M. Imler, P. Malaviya, R. M. Nerem and M. E. Levenston, Biomaterials, 2002, 23, 1249–1259 CrossRef CAS.
- V. Terraciano, N. Hwang, L. Moroni, H. B. Park, Z. Zhang, J. Mizrahi, D. Seliktar and J. Elisseeff, Stem Cells, 2007, 25, 2730–2738 CrossRef CAS PubMed.
- Z. Y. Zhang, S. H. Teoh, W. S. Chong, T. T. Foo, Y. C. Chng, M. Choolani and J. Chan, Biomaterials, 2009, 30, 2694–2704 CrossRef CAS PubMed.
- J. Dobson, Nat. Nanotechnol., 2008, 3, 139–143 CrossRef CAS PubMed.
- N. Wang, J. Butler and D. Ingber, Science, 1993, 260, 1124–1127 CAS.
- F. J. Alenghat, J. D. Tytell, C. K. Thodeti, A. Derrien and D. F. Ingber, J. Cell. Biochem., 2009, 106, 529–538 CrossRef CAS PubMed.
- S. H. Cartmell, J. Dobson, S. B. Verschueren and A. J. El Haj, IEEE Trans. NanoBioscience, 2002, 1, 92–97 CrossRef.
- S. Hughes, S. McBain, J. Dobson and A. J. El Haj, J. R. Soc., Interface, 2008, 5, 855–863 CrossRef CAS PubMed.
- B. Hu, A. J. El Haj and J. Dobson, Int. J. Mol. Sci., 2013, 14, 19276–19293 CrossRef PubMed.
- B. Hu, J. Dobson and A. J. El Haj, Nanomedicine, 2014, 10, 45–55 CrossRef CAS PubMed.
- H. L. Karlsson, J. Gustafsson, P. Cronholm and L. Möller, Toxicol. Lett., 2009, 188, 112–118 CrossRef CAS PubMed.
- A. K. Gupta and A. S. G. Curtis, J. Mater. Sci.: Mater. Med., 2004, 15, 493–496 CrossRef CAS.
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- M. Ahamed, H. A. Alhadlaq, J. Alam, M. Khan, D. Ali and S. Alarafi, Curr. Pharm. Des., 2013, 19, 6681–6690 CrossRef CAS.
- P. Ma, Q. Luo, J. E. Chen, Y. P. Gan, J. Du, S. M. Ding, Z. G. Xi and X. Yang, Int. J. Nanomed., 2012, 7, 4809–4818 CAS.
- Y. Ke and Z. M. Qian, Lancet Neurol., 2003, 2, 246–253 CrossRef CAS.
- P. M. Doraiswamy and A. E. Finefrock, Lancet Neurol., 2004, 3, 431–434 CrossRef CAS.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, U772–U1000 CrossRef PubMed.
- H. Amiri, L. Bordonali, A. Lascialfari, S. Wan, M. P. Monopoli, I. Lynch, S. Laurent and M. Mahmoudi, Nanoscale, 2013, 5, 8656–8665 RSC.
- U. Sakulkhu, M. Mahmoudi, L. Maurizi, J. Salaklang and H. Hofmann, Sci. Rep., 2014, 4 Search PubMed.
- M. Mahmoudi, S. Sheibani, A. S. Milani, F. Rezaee, M. Gauberti, R. Dinarvand and H. Vali, Nanomedicine, 2015, 10, 215–226 CrossRef CAS PubMed.
- L. M. Shaw, B. Kaplan and D. Kaufman, Clin. Chem., 1996, 42, 1316–1321 CAS.
- P. Soon-Shiong, R. E. Heintz, N. Merideth, Q. X. Yao, Z. Yao, T. Zheng, M. Murphy, M. K. Moloney, M. Schmehl, M. Harris, R. Mendez and P. A. Sandford, Lancet, 1994, 343, 950–951 CrossRef CAS.
- T. T. Dang, A. V. Thai, J. Cohen, J. E. Slosberg, K. Siniakowicz, J. C. Doloff, M. Ma, J. Hollister-Lock, K. M. Tang, Z. Gu, H. Cheng, G. C. Weir, R. Langer and D. G. Anderson, Biomaterials, 2013, 34, 5792–5801 CrossRef CAS PubMed.
- S. Schneider, P. J. Feilen, F. Brunnenmeier, T. Minnemann, H. Zimmermann, U. Zimmermann and M. M. Weber, Diabetes, 2005, 54, 687–693 CrossRef CAS.
- B. P. Barnett, A. Arepally, M. Stuber, D. R. Arifin, D. L. Kraitchman and J. W. M. Bulte, Nat. Protoc., 2011, 6, 1142–1151 CrossRef CAS PubMed.
- S. Takeda, F. Mishima, S. Fujimoto, Y. Izumi and S. Nishijima, J. Magn. Magn. Mater., 2007, 311, 367–371 CrossRef CAS PubMed.
- T. Neuberger, B. Schöpf, H. Hofmann, M. Hofmann and B. von Rechenberg, J. Magn. Magn. Mater., 2005, 293, 483–496 CrossRef CAS PubMed.
- S. Goodwin, C. Peterson, C. Hoh and C. Bittner, J. Magn. Magn. Mater., 1999, 194, 132–139 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |