DOI:
10.1039/C4CS00284A
(Tutorial Review)
Chem. Soc. Rev., 2015,
44, 119-135
Supramolecular photochemistry: from molecular crystals to water-soluble capsules
Received
25th August 2014
First published on 15th October 2014
Abstract
Photochemical and photophysical behavior of molecules in supramolecular assemblies are different and more selective than in gas and isotropic solution phases. Knowledge of the inherent electronic and steric properties of the reactant is insufficient to predict the excited state behavior of molecules confined in such assemblies. Weak interactions between the medium and the reactant as well as the free space in a reaction cavity would play a significant role in modulating the excited state properties of molecules when they are included within confined spaces. The concepts of ‘Molecular Photochemistry’ should be modified while applying them to ‘Supramolecular Photochemistry’. In this review we show that the topochemical rules established to understand reactions in crystals could be extended to supramolecular assemblies in general. To make the best use of the medium one needs to understand the features of the medium, the nature of interaction between the medium and the molecule and the rules that govern the behavior of a molecule in that medium. This tutorial provides introduction to these aspects of ‘Supramolecular Photochemistry’.

V. Ramamurthy
| V. Ramamurthy is Professor of Chemistry at University of Miami, Coral Gables, Florida, USA. The author of over 370 publications, editor of fourteen monographs (1991–2011), guest editor of six special journal issues (1987–2014), co-author of a recently (2010) published textbook Modern Molecular Photochemistry of Organic Molecules and Senior Editor of Langmuir, he received his PhD from the University of Hawaii, Honolulu, USA (R. S. H. Liu), and pursued his postdoctoral training at the University of Western Ontario, London, Canada (P. de Mayo) and Columbia University, New York, USA (N. J. Turro). Before joining the University of Miami in 2005, he was Bernard Baus Professor of Chemistry at Tulane University, New Orleans, Louisiana, Senior Scientist at the Du Pont Company in Wilmington, Delaware, and Associate Professor at the Indian Institute of Science, Bangalore, India. His research interests are in the area of supramolecular photochemistry that encompasses photochemistry in crystals, zeolites, cavitands and capsules. |

Shipra Gupta
| Shipra Gupta received her PhD at University of Miami, Coral Gables, Florida, USA (V. Ramamurthy) in 2013 for her thesis titled “Understanding the Influence of Confinement on the Excited State Properties of Small Organic Molecules”. Before joining University of Miami she received her BSc and MSc degrees from Isabella Thoburn College, Lucknow and Indian Institute of Technology, Guwahati respectively. She has interest in supramolecular photochemistry, chemistry of reactive intermediates and industrial and biological applications of photochemistry. She is currently a postdoctoral fellow in the laboratory of Professor J. Saltiel at Florida State University, Tallahassee, Florida, USA. |
Key learning points
• Reaction cavity in crystals and common features of reaction cavities of various supramolecular assemblies.
• Topochemical rules applicable in crystals that can be extended to other supramolecular assemblies.
• Organization of molecules within confined hydrophobic spaces and at hydrophobic–hydrophilic interfaces.
• The various unique features of supramolecular assemblies that can aid manipulation of the excited state behavior of molecules confined in them.
• The choice of the reaction medium for a given situation is specific to the reactant molecule and the goal.
|
1. Introduction
The history of the growth of organic photochemistry from a relatively unknown discipline to a developed one during the last century is fascinating. During this period photochemists discovered new reactions, established mechanisms of photoreactions, and laid out the ground rules for the behavior of molecules in the excited state surfaces.1 Applications of photochemistry in everyday life such as the discovery of photodynamic therapy and Lasik surgery brought out the tremendous potential of light in medical technology.2 The importance of photochemistry in materials science has long been recognized through the discoveries of photography, lithography and xerography.3 The future role of photochemistry in materials science is well placed through recent interests in non-linear optics, two photon spectroscopy, luminescent nanoparticles, and molecular sensors, switches, triggers, and machines. Importantly though, solar energy as an environmentally safe energy source is yet to be fully realized. Given these potentials, explorations in photochemistry are expected to continue for a long time.
Organic photochemistry grew from the important recorded observations of a number of European photochemists, Sestini, Cannizaro, Liberman, Klinger, Ciamician, Silber, Paterno and others during the late 19th and early 20th century.4 Following an initial phase involving the discovery of light induced transformations,5 activity in photochemistry in terms of establishing the ground rules of molecular photochemistry occurred in Japan, Europe and North America during the mid 20th century. Subsequently and continuing till date, the focus has been on controlling the behavior of molecules in the excited state. In this context, of the various approaches, supramolecular photochemistry has been the most promising.6 Thus, broadly, molecular photochemistry developed in three stages: (a) discovery of new reactions; (b) mechanistic pursuit and (c) controlling the outcome of a reaction. This article concerned with the third stage is a personalized tutorial review briefly summarizing the concepts useful in visualizing and planning photoreactions in confined spaces. Photochemical rather than photophysical aspects of supramolecular photochemistry is emphasized. Select examples are included to illustrate the value of this approach. Since it is not intended to be a summary of the activity in the field, recent reviews and monographs should be consulted for extensive coverage of the literature.7–18
The two striking biological phenomena that serve as a source of inspiration to photochemists and illustrate what can be achieved in a confined environment are photosynthesis and visual signal transduction. In the former, the medium (protein) is able to organize a number of active elements so as to predispose them to a desired physical process, while in the latter, the medium (rhodopsin) restricts the rotational mobility on certain parts of a single molecule, retinal. Inspired by, and realizing the complexity of natural systems, chemists have utilized a number of organized media that are less complex than biological systems to study the photochemical behavior of molecules. Examples of organized media which have been investigated include molecular crystals, inclusion complexes (both in the solid and solution states), liquid crystals, micelles and related assemblies such as vesicles, microemulsions and membranes, monolayers, Langmuir–Blodgett (LB) films, surfaces (silica, clay and zeolites), water soluble organic and inorganic hosts (cyclodextrins, cucurbiturils, calixarenes, octa acid, Pd nanocage etc.), polymers and dendrimers and more recently natural systems such as proteins and DNA. These media have been referred to in the literature under various names – organized, confined, constrained, ordered, restricted, microheterogeneous, non-homogeneous and anisotropic media, nanoscopic reactors and molecular containers etc. All these media, despite the different geometries and structures influence the excited state behavior of the reactant and therefore the observed chemistry is ‘supramolecular’ and no longer ‘molecular’. To predict the excited state behavior of a reactant in these media, besides the inherent molecular properties one needs to consider the properties of the medium and the reactant together. The role of these above media, with their different properties and structures on the excited state behavior of included reactants (represented as guest@host) that could be considered under a general model adopted from crystal photochemistry is emphasized in this review. We will focus on what is common with all these seemingly different hosts with different structures.
2. Transitioning from molecular to supramolecular photochemistry
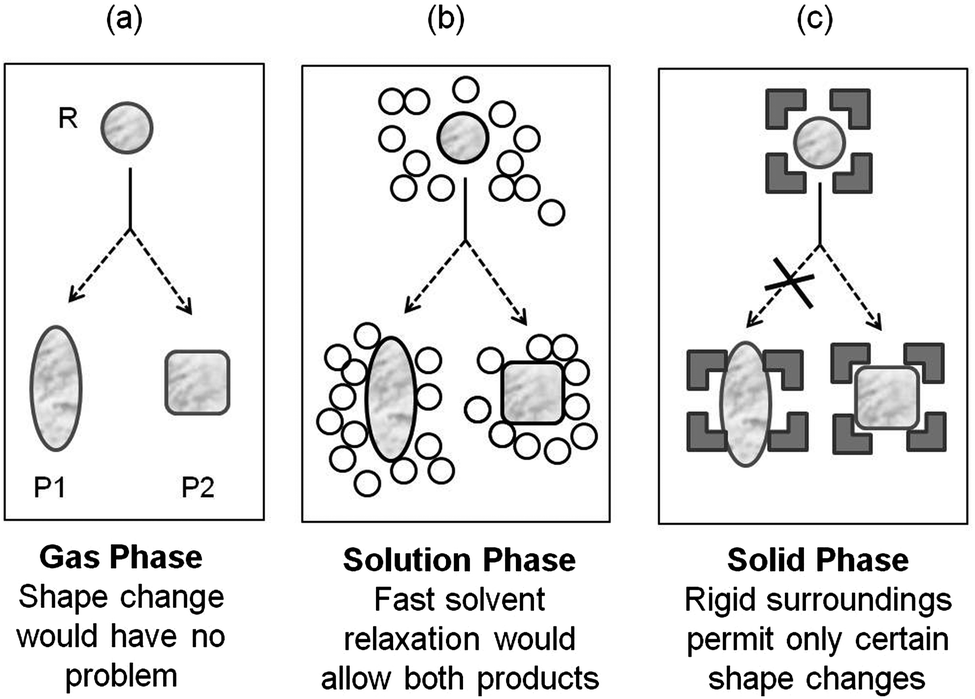
In the gas- and isotropic solution-phases, photoreactions are dictated by the inherent electronic- and steric-features of the reacting molecule. The intermolecular and non-covalent interactions in these media are generally considered to be so weak, non-directional and non-specific, to only play a secondary role in determining the structure and reactivity in systems under study (Fig. 1). The influence of the medium, usually isotropic solvents, is minimal though not completely negligible. Often times the influence of the solvent could be predicted on the basis of the colligative properties of the medium such as dielectric constant, viscosity etc. rather than 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 specific interaction between the solvent and the reactant molecules. Supramolecular chemistry on the other hand is based on the concept that there are situations where introduction of more than one interacting molecular structure is required to understand the chemistry under consideration. An important feature of supramolecular systems is that in general, a supramolecule is held together as an assembly by a number of weak, non-covalent intermolecular bonds whose summation could lead to a strong overall bonding of the components of the supramolecule.19
:1 specific interaction between the solvent and the reactant molecules. Supramolecular chemistry on the other hand is based on the concept that there are situations where introduction of more than one interacting molecular structure is required to understand the chemistry under consideration. An important feature of supramolecular systems is that in general, a supramolecule is held together as an assembly by a number of weak, non-covalent intermolecular bonds whose summation could lead to a strong overall bonding of the components of the supramolecule.19
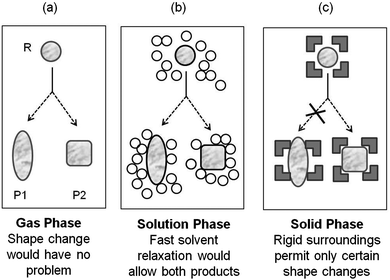 |
| | Fig. 1 Effect of medium on the outcome of a reaction. Influence of the environment on a reactant in (a) gas phase; (b) organic solvent and (c) crystals is illustrated. P1 (oblang) and P2 (square) represent two possible products. | |
During the last five decades beginning with the studies of Schmidt,20,21 Whitten,22 Turro23,24 and de Mayo25,26 supramolecular photochemistry in a variety of organized assemblies capable of organizing and confining reactants and reaction intermediates have been extensively investigated by numerous investigators. Striking examples illustrating the value of these media have appeared in the literature11,13–18 and more examples of ‘show and tell’ variety are being reported even now implying room for the principles of supramolecular photochemistry to fully develop.
3. The earliest supramolecular photochemical assembly: crystals
The first report of supramolecular photochemistry is probably the photodimerization of cinnamic acid derivatives in solid sate reported by Libermann in 1871.27 The enormous interest in solid-state reactions, evident from reports during 1900–1930, waned for lack of experimental techniques to uncover the reaction mechanisms. Schmidt and co-workers' systematic study combining X-ray crystallography and photoreactions in 1960s played an important role in the development of solid-state photochemistry and indirectly in supramolecular photochemistry.20,21 This line of research did not attract the attention of mainstream photochemists till tremendous developments were made in the X-ray diffractometer instrumentation and structure solution methods. Although Schmidt's contributions to solid-state photochemistry should be recognized as the beginnings of supramolecular photochemistry,20,21,28 breakthrough in this area came in 1970s through the pioneering publications of Whitten,22 Turro,23,24 de Mayo25,26 and others in micelle mediated photoreactions in aqueous solution.7 The potential of controlling photoreactions with an organized assembly in solution was immediately recognized and sparked the interests of mainstream organic photochemists leading to the birth of ‘Supramolecular Photochemistry’.8,9
4. Cohen's reaction cavity model extended to supramolecular photochemistry
Following the significant contributions on solid-state photochemistry by Schmidt and his untimely death, Cohen, a senior colleague of Schmidt, formulated a model to understand photoreactions in crystals.29 We believe this reaction cavity model's several interesting features could be adopted for photoreactions in supramolecular assemblies in general.30–32
Definition of a reaction cavity
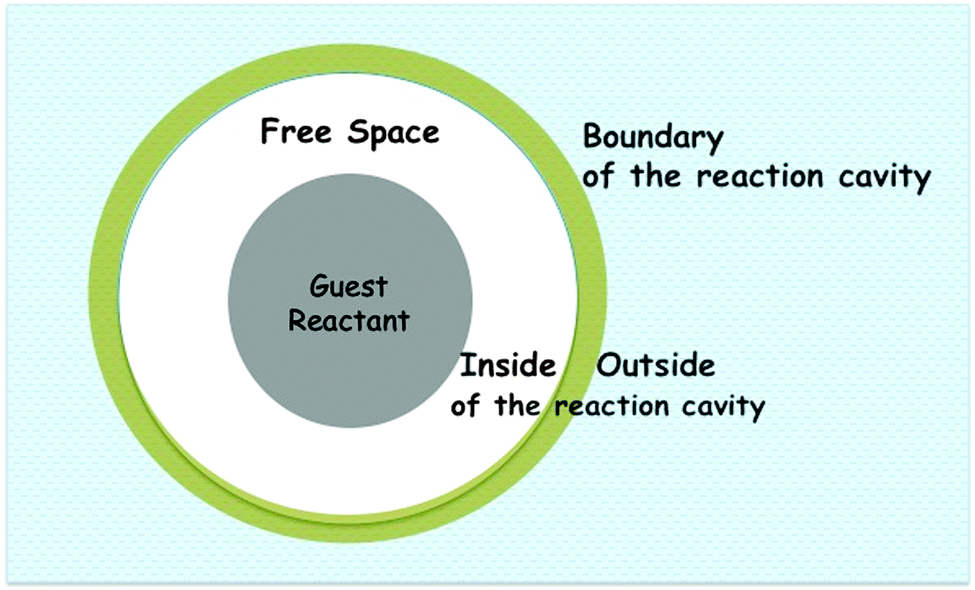
One can envision reactions in an organized medium as occurring within an enclosed space. This space defined as the “reaction cavity” (white and darker space in Fig. 2), is a term originally used by Cohen to describe reactions in crystals. In the larger context of supramolecular photochemistry, one should think of a reaction cavity as the space in which excited state reactant molecules and their pre-product intermediate(s) are confined during their lifetimes. Selectivity seen for reactions in crystals, according to the reaction cavity model, arises due to lattice restraints on the motions of the atoms in reactant molecules within the reaction cavity (Fig. 1c). In other words, severe distortion of the reaction cavity will not be tolerated and only reactions that proceed without much distortion of the cavity are allowed in a crystal. Therefore, the space required to accommodate the displacement of the reactant atoms from their original positions during a chemical reaction must be built largely into the reaction cavity. Packing of polyatomic molecules in crystals leaves some distances between neighboring non-bonded atoms greater than the sum of their van der Waals radii. This creates a certain amount of free volume (white space in Fig. 2), which may be so disposed of to allow the atomic motions required to effect a reaction. We believe the above reaction cavity model could be extended to organized media in general.
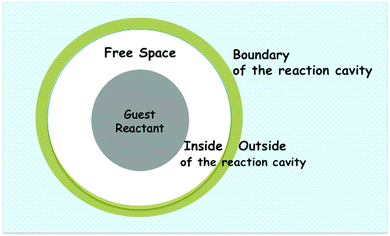 |
| | Fig. 2 A schematic representation of a reaction cavity. Various components of a reaction cavity are indicated. The white area represents the reaction cavity where the guest (darker circle) resides. | |
Boundary, size and shape of a reaction cavity
The boundaries of the reaction cavity in an organized medium are comprised of a network of atoms or molecules. This while common to all condensed media, the size, shape and nature of the reaction cavity differs significantly from one medium to the other. Such factors are responsible in large part for the different influences exerted by different supramolecular structures on the course of reactions of included guest molecules. The size of the reaction cavity may vary from being very small (as in crystals, cyclodextrins etc.) to potentially very large (as in micelles, clay, zeolites and silica). Whether the information regarding the size and shape of the reaction cavity will be registered in the distribution or stereochemistry of the products will depend on the mechanism involved in the product formation.
Free volume within a reaction cavity
Free volume within a reaction cavity is essential to accommodate the shape changes that occur as the reactants transform themselves to products (Fig. 1 and 2). There exists a well-defined free space around the guest in a number of host systems where they are accommodated; in isotropic liquids the free volume is highly mobile; in organized media it may be essentially immobile as in crystals, or have mobility ranging from that of crystals to that of micelles. The free volume needed to accommodate shape changes that occur during the course of a reaction must be present intrinsically within the fixed structure. In flexible structures such as micelles this may become available as the reaction proceeds. In very limited or no free volume situations the reaction may not occur within a confined space, on the other hand, when there is a lot of free volume the reaction may not be selective and the confined space may not influence the reaction as desired. Judicious choice of the supramolecular structure to exert the desired influence on an excited state reaction is thus crucial.
Hard and soft reaction cavities
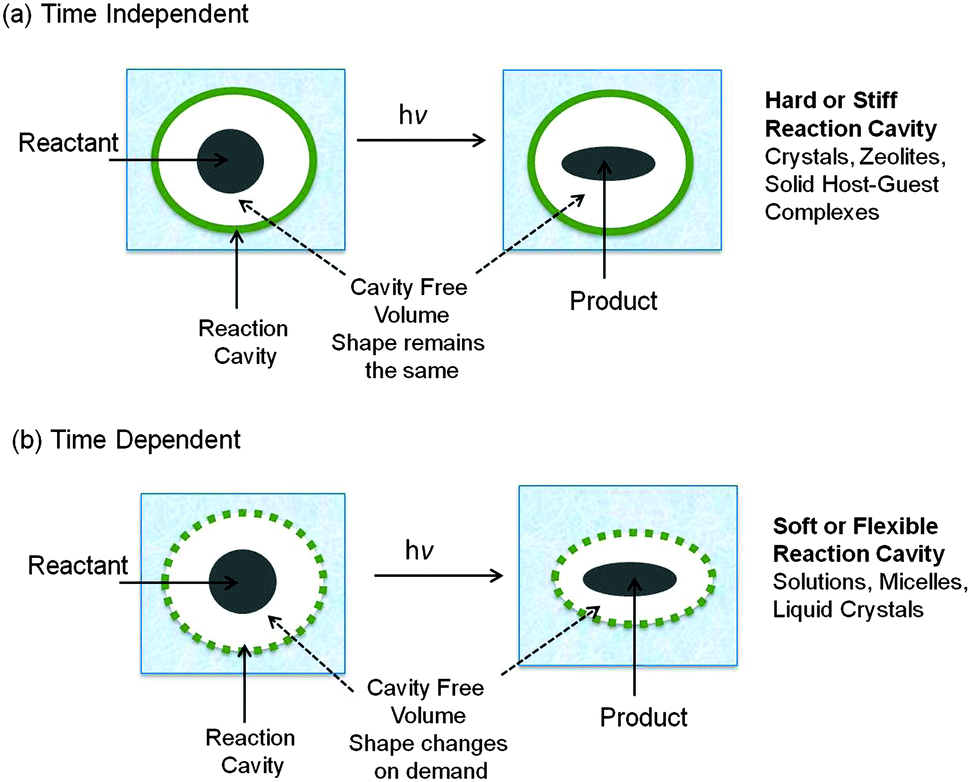
The nature of the reaction cavity distinctly differs between the rigidly organized structures such as crystals and zeolites, and media such as micelles, microemulsions, molecular aggregates, and liquid crystals. In the latter, reaction cavities may contain minimal intrinsic free volume at the time of photoexcitation of guests. However, since the molecules that constitute these organized assemblies are mobile, the reaction cavity can respond to shape changes in the reactant as the reaction occurs. How much and how readily each medium will accommodate shape changes will be an important determinant of the selectivity that will be obtained. Such media are defined to possess “flexible or soft” reaction cavities. In order for a guest molecule to react in the environment provided by a restrictive host, the medium must make necessary adjustments within the time frame determined by the rate limiting spatial decay paths leading to the pre-product transition state(s). Without such a “timely” response of the host medium no reaction will occur as exemplified by media such as crystals, zeolites, clay etc. The reaction cavities in these media are “stiff or hard” (Fig. 3a). Thus, the boundaries of the enclosures in confined media may be of two types: hard (i.e., the walls that are not flexible), as in the case of crystals, or soft (i.e., the walls of the cavity are sufficiently mobile to allow considerable internal motion of the enclosed molecules), as in the case of micelles (Fig. 3b). The required free volume for a reaction in these two extreme situations is either intrinsic (built into the hard reaction cavity) or latent (can be provided on demand by the soft reaction cavity).
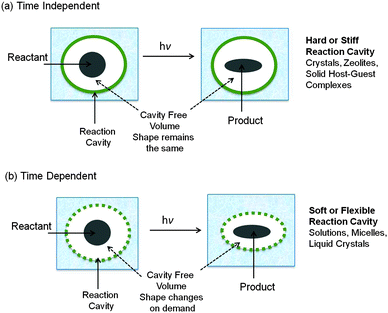 |
| | Fig. 3 Illustration of features of two types of reaction cavities: (a) time dependent soft reaction cavity; (b) time independent hard reaction cavity. Note the shape of the reaction cavity remains constant in (a) and varies with time in (b). | |
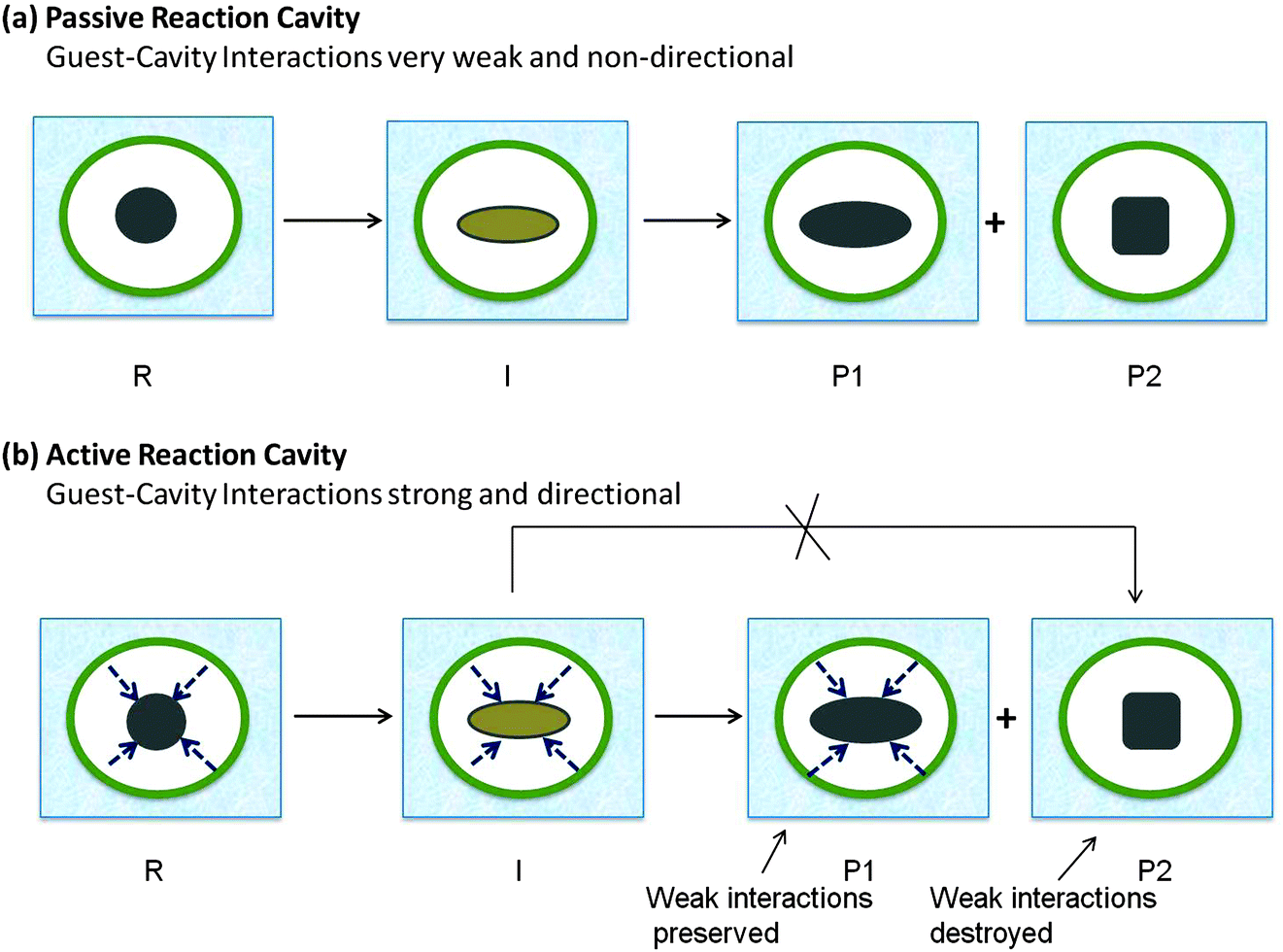
Active and passive reaction cavities
In addition to being soft or hard, reaction cavity walls are characterized as “active” or “passive” (Fig. 4). The cavity wall is considered “active” when there is an interaction between a guest molecule and a wall of the cavity, and “passive” in the absence of any significant interaction. In the latter case where the confinement alone plays a role in the selectivity, predictions on the excited state behavior of guest molecules can be made on the basis of size, shape and flexibility of the reaction cavity. On the other hand, active walls require considerations of the possible wall–guest interactions as well (Fig. 4b). The walls of some active cavities bear functional groups that interact non-covalently with specific functional groups of the guest molecules and their intermediates.19 Such interactions when sufficiently attractive or repulsive will influence the locations or conformations of guest molecules in the cavity. These interactions may vary from weak van der Waals forces, to hydrogen bonds to strong electrostatic forces between charged centers: the functional groups in a number of hosts such as cyclodextrin that form inclusion complexes can hydrogen bond with guest molecules; hydrogen bonds also play an important role in templating photoreactions in the crystalline state; silanol groups of silica surfaces may orient the adsorbed molecules through hydrogen bonding; surfaces of clays and zeolites often carry a number of cations that can interact electrostatically with guests; hydrophobic–hydrophilic interactions orient guest molecules at the interface of micellar, monolayer, and related assemblies and certain cavitand hosts. In active reaction cavities the weak interactions between the reaction cavity and the reactant are likely to influence the product formation (Fig. 4b).
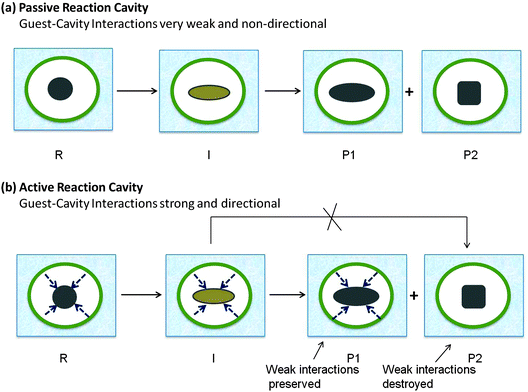 |
| | Fig. 4 Illustration of features of two types of reaction cavities: (a) passive reaction cavity where there is no interaction, (b) active reaction cavity where there is an interaction. Presence of interaction leads to control on the distribution of photoproducts. R is the reactant, I is the intermediate, P1 and P2 are the two possible products. The weak interactions are preserved in R, I and P1 but not in P2. P2 is not expected to be formed in this active reaction cavity. | |
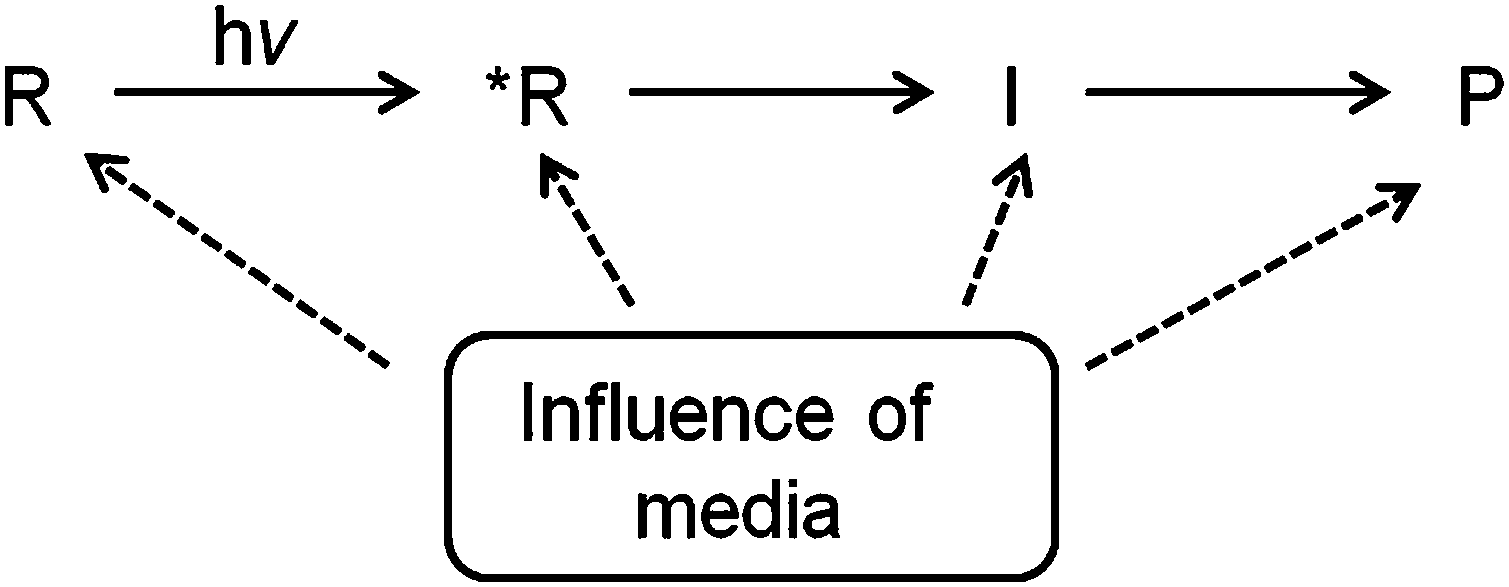
Extending reaction cavity concept to excited state chemistry
It is relatively easy to predict and visualize the geometry and orientation of molecules within reaction cavities based on attractive and repulsive interactions between ground state guest molecules and the host structure. Electronic excitation frequently leads to changes in molecular geometry and polarizability and these differences between the ground and the reactive excited state can affect the interaction between the excited guest molecules and their neighbors. The reaction gets more complex when intermediates intervene between the excited states of reactants and their photoproducts. Clearly, consideration of interactions of the reaction cavity with the ground state, excited states and reaction intermediates of a guest at the atomic level will be required for reliable predictability on the specific influence of anisotropic environments on the course of photoreactions (Fig. 5). Being based on interactions between ground state reactants and the reaction cavity the predictions on the excited state behavior are expected to be qualitative only.
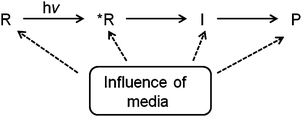 |
| | Fig. 5 The ultimate outcome of a photoreaction in a supramolecular assembly will depend on its influence on reactant, transition state, intermediate and the product. R is the reactant in ground state, R* is the reactant in the excited state, I is the intermediate and P is the product. | |
Differences in reaction cavity in isotropic solvents and in supramolecular assemblies
Comparison of the cavity of a supramolecular host to the solvent cage around a solute in an isotropic solution of a small molecular solvent such as benzene, acetonitrile and water highlights the former's distinction as a reaction cavity (Fig. 1b). The solvent cage is very fluid and flexible and as a result its dimensions, size and shape are fluid and therefore not well defined. Solvent molecules can be easily displaced, so that size matching of the reactant, products and the reaction cavity generally requires very small activation energy. For example, larger molecules can use their thermal energy to just ‘push solvent’ around so that the fluid solvent cage easily adjusts to ‘fit’ around the molecule in the solvent cage. On the other hand, when the reaction cavity of a host possesses a well-defined molecularly rigid boundary, size and shape matching with the guest will become important (Fig. 1c). Thus the reactions in a solvent cage are expected to be controlled only by inherent electronic and steric features of the reactant molecule while in supramolecular assemblies an additional factor, i.e., the influence of the surroundings must be taken into account.
Points to note
The following points on the reaction cavity model emerge from the above discussion:29–32 (1) A reaction cavity is an enclosure reducing the mobility of the reactant molecules with a boundary that the reactant molecules may not cross without overcoming an energy barrier (Fig. 2). (2) The sizes and shapes of the reaction cavities among organized media vary. (3) The free volume within a reaction cavity whose shape, size, location and directionality and the dynamics of their change largely control the extent to which the medium influences a photoreaction (Fig. 2). (4) When the atoms/molecules constituting the walls of the reaction cavity are stationary (i.e., possess time independent positions on the time scale of the guest reaction), the free volume necessary to convert a guest molecule to its photoproduct(s) must be built into the reaction cavity. On the other hand, where the walls are flexible, the free volume may become available during the course of a reaction (Fig. 3). (5) The reaction cavity may contain specific functional groups, which may strongly interact attractively or repulsively with either a guest molecule or/and the transition state or/and the intermediate formed as the guest proceeds to products. Such specific interactions may lead to unique product selectivity and either enhance or decrease the quantum yields for reactions (Fig. 4).
5. Schmidt's topochemical postulates extended to supramolecular photochemistry
Following extensive parallel crystallographic and photochemical investigations on solid-state photodimerizations, Schmidt made a few general important observations that highlighted the differences between photochemical reactions in solution (molecular photochemistry) and crystalline state (supramolecular photochemistry).20,21 These observations generally known as topochemical postulates quoted below, we believe, are also applicable to photoreactions conducted in various organized media listed above. Modifications may be needed depending on the nature of the reaction cavity (hard or soft; active or passive). Important observations made by Schmidt based on his studies on solid-state photoreactions are:20,21,28
• “A given compound reacts differently in the solid state and dispersed phases.
• Reaction in the solid state occurs with a minimum amount of atomic or molecular movement.
• The geometry of the crystal lattice determines the fate of the excited molecule, the course of the chemical transformation and the structure of the product.
• The crystal lattice maintains control over the configuration and conformation of the reacting components, at least until the formation of the transition state of the defined geometry.
• Chemically closely related compounds show significant differences in chemical behavior in the solid state.
• The rule of homology, which has been essential to the development of solution chemistry does not hold in solid-state chemistry.
• The systematic development of solid-state photochemistry will be difficult if not impossible until we understand the intermolecular forces responsible for the stability of crystalline lattice of organic compounds; a theory of the organic solid state is a requirement for the eventual control of molecular packing arrangement.”
The above observations though relating to photodimerization in crystals should be equally applicable to different types of photoreactions and to various confined media. In other words supramolecular photochemistry should be dictated by topochemical postulates with some modifications to suit the nature of the reaction cavity. In this context the prophetic words of Cohen in 1976 are worth noting:33 “As yet topochemistry has dealt with crystals, the epitome of organized systems. But the same principles must be involved, at least in part, in less organized systems. One can think of liquid crystals, membranes, micelles, molecular aggregates, monomolecular films, surfaces, intercalate compounds, macromolecules, and so on, all either synthetic or natural. The range of such systems is vast, and they are of significance not only to chemists, but also biologists, soil and materials scientists. Applications of topochemical ideas in this area are just beginning. It is probable that it is in this direction that topochemistry is destined to make its most significant contribution.”
6. Beginnings of supramolecular solution photochemistry
As indicated earlier extensive studies on solid-state photochemistry of cinnamic acids by Schmidt marked the beginning of supramolecular photochemistry. In spite of its uniqueness such studies remained confined to crystals and mainly to Schmidt's laboratory. In this context, studies in micelles and related assemblies marked the ‘real’ beginning of supramolecular photochemistry. Studies in these assemblies illustrated that the well-known excited state chemistry based on solution behavior could be altered when the reactants were solubilized in micelles and mono and bilayers. Examples from the classic studies of de Mayo, Whitten and Turro are provided below.
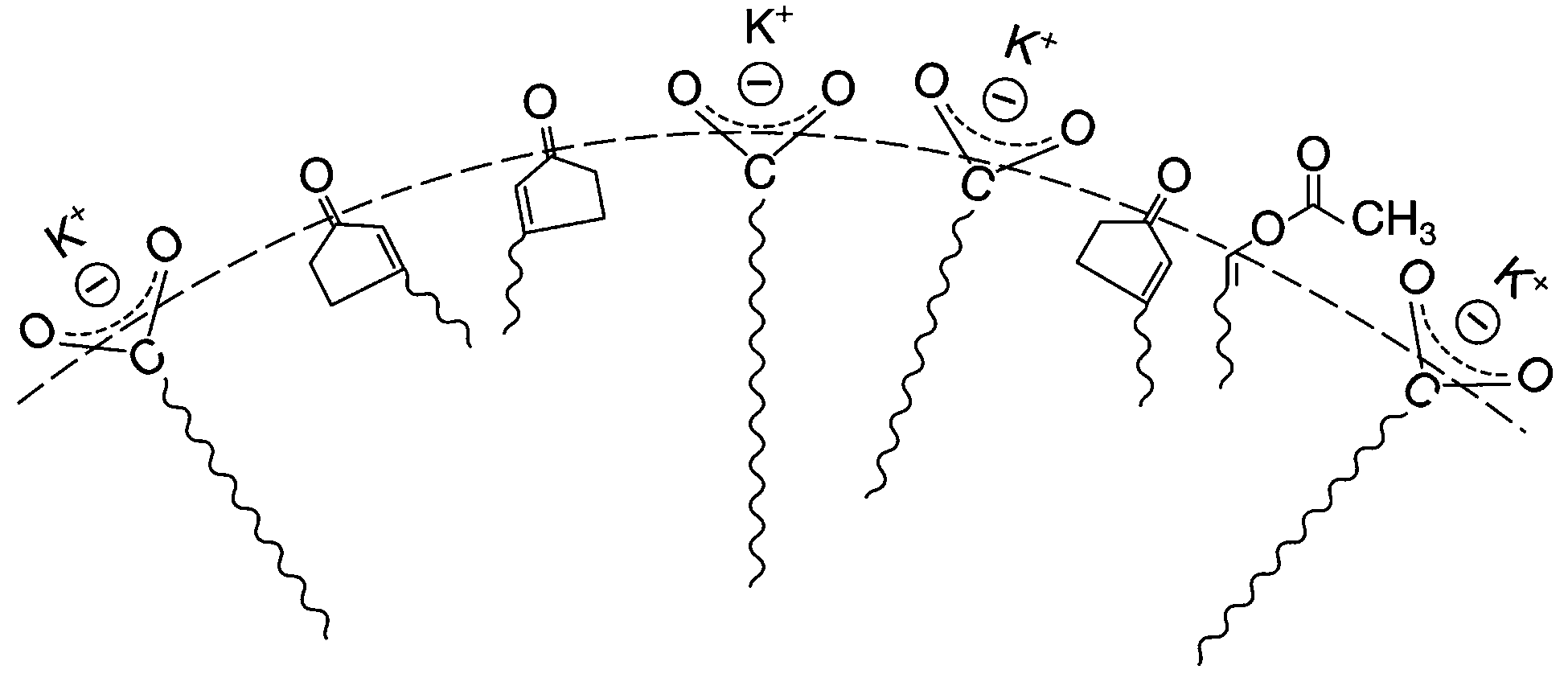
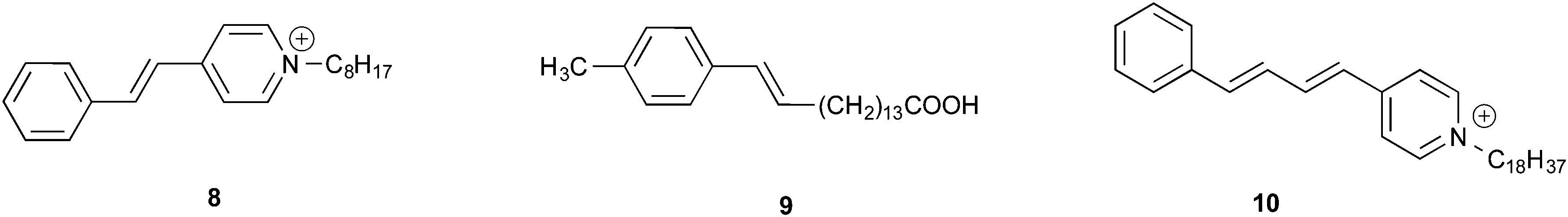
Regioselectivity has been induced by micelles in photocycloaddition reactions of olefins to 3-alkylcyclopentenones. Photocycloaddition of 3-butylcyclopentenone to 1-hexene and 1-octene in organic solvents results in two adducts in roughly equal ratio (slight variations depending on the solvent) (Scheme 1). Preference to one isomer from irradiation of the olefin in potassium dodecanoate micelles is consistent with a model of orientation of cyclopentenone in the micelle with the polar carbonyl functionality at the interface and the hydrophobic butyl chain directed toward the micelle interior (Fig. 6). Regioselectivity increased when the orientation of the olefinic reaction partner was made more pronounced with respect to the micellar interface by the introduction of an acetoxy group (Scheme 2). For example, 1-heptenyl acetate gave 7 exclusively in organic solvents and mostly 6 in micelles. The limited freedom of motion (hard reaction cavities) in LB films affords very well defined interfaces for the alignment of reactant molecules to be readily expressed in the products. In photodimerization of stilbazole derivatives, N-octadecyl-1-(4-pyridyl)-4-(phenyl)-1,3-butadiene, surfactant styrene derivatives 8 and 9, and cinnamic acid derivatives 10 carried out in LB films by Whitten and co-workers,22 single isomeric head–head dimers were obtained (Scheme 3). Geometric isomerization the main reaction in isotropic solvents did not compete with photodimerization in these rigid reaction cavities.
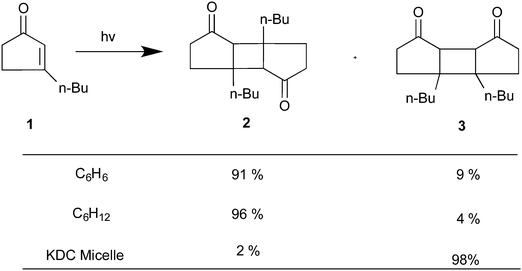 |
| | Scheme 1 Product distribution upon irradiation of 3-n-butylcyclopentenone in organic solvents and micelles. | |
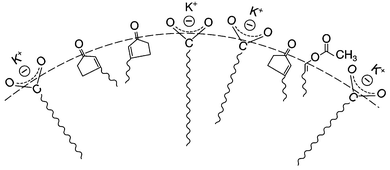 |
| | Fig. 6 Orientation of 3-n-butylcyclopentenone and 1-acetyl heptane at micellar interface. | |
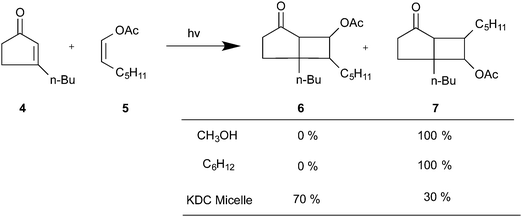 |
| | Scheme 2 Product distribution of irradiated 3-n-butylcyclopentenone and 1-acetyl heptene in organic solvents and micelles. | |
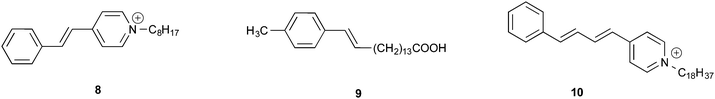 |
| | Scheme 3 Reactants irradiated as LB films. | |
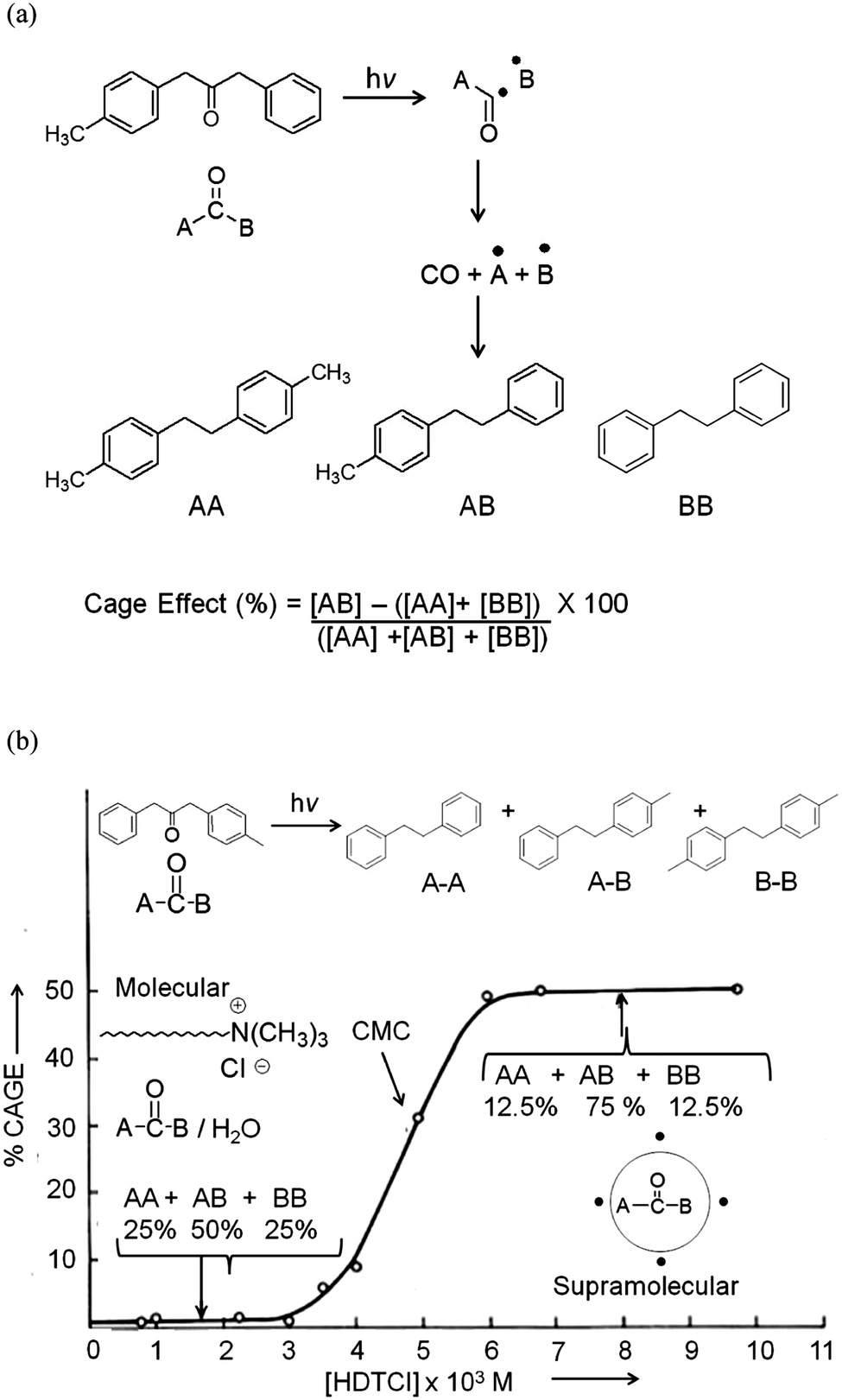
In our opinion the elegant publications on dibenzyl ketones by Turro and his co-workers in micelles brought out the power of confining molecules in a supramolecular assembly such as micelles.23,24 A comparison of the product distribution of reactions conducted in solution and in micelles shown in Fig. 7 unequivocally points out the influence of the media on photoreactions. Photolysis of 3-(4-methylphenyl) 1-phenyl acetone results in a 1:2:1 mixture of three products shown in Fig. 7a; the same molecule when solubilized in hexadecyltrimethylammonium chloride micelle gave a single product (AB). The profile of cage effect versus surfactant concentration clearly showed the importance of micelles in altering the course of photoreaction (Fig. 7b). It must be noted that the change in product distribution is at or above the critical micelle concentration (CMC).
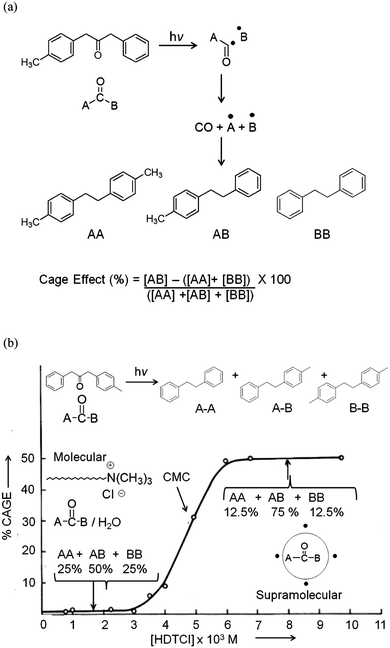 |
| | Fig. 7 (a) Products of photolysis of 3-(4-methylphenyl) 1-phenyl acetone (a dibenzylketone derivative); (b) variation of cage effect with the concentration of hexadecyltrimethyl ammonium chloride in water. The break in cage effect is seen near the critical micelle concentration. | |
7. Supramolecular solution photochemistry extended beyond micelles
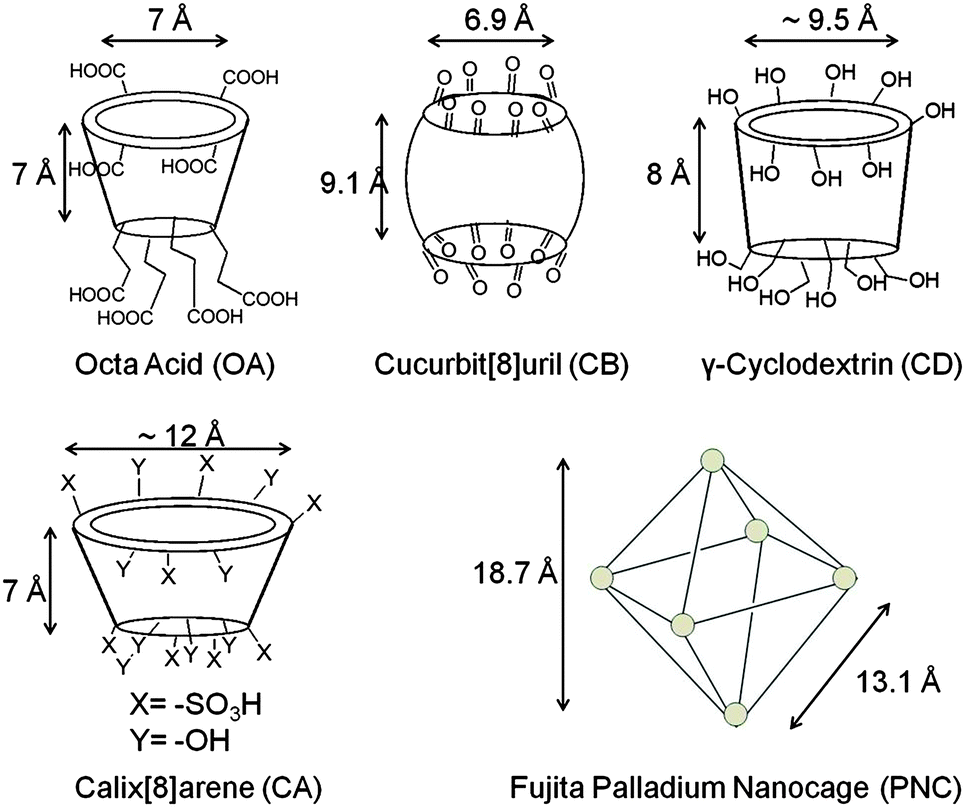
The above concept of orienting molecules within confined spaces has been extended to cavitand type host systems such as cyclodextrins (CD), cucurbiturils (CB), calixarenes (CA), Pd nanocage and cavitand octa acid (Fig. 8). These water soluble host systems are open at more than one end and they in principle, similar to micelle, could expose a guest molecule to hydrophobic–hydrophilic interface. At the same time the reaction cavities offered by these cavitands are rigid and small. These features that amplify intermolecular interactions have been exploited to obtain single dimers from excited olefins.
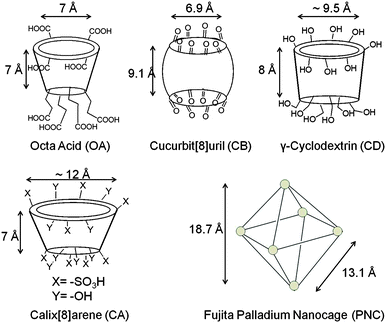 |
| | Fig. 8 Skeletal representation of cavitand type hosts: octa acid, cucurbituril, cyclodextrin, calixarene and palladium nanocage. | |
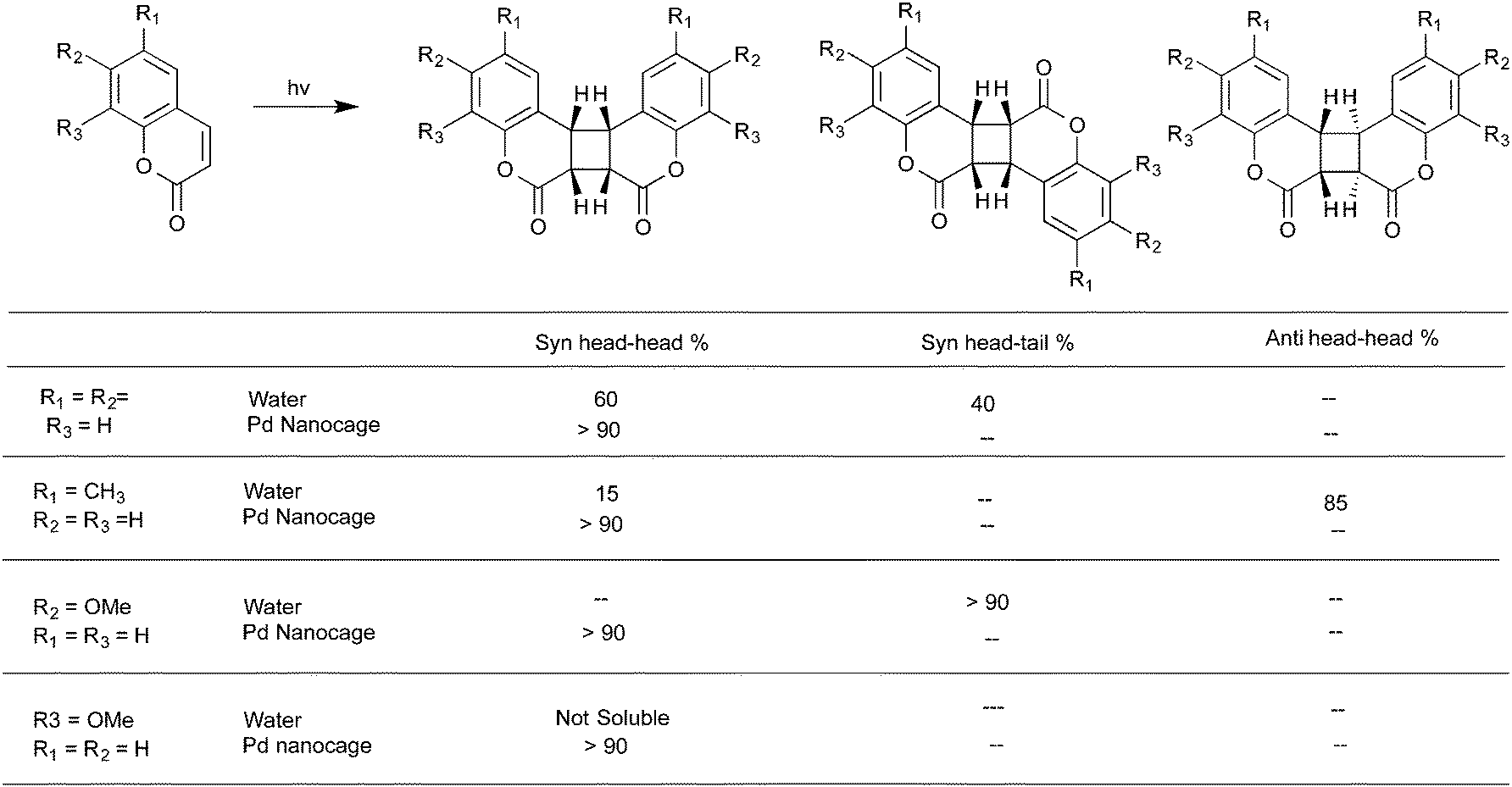
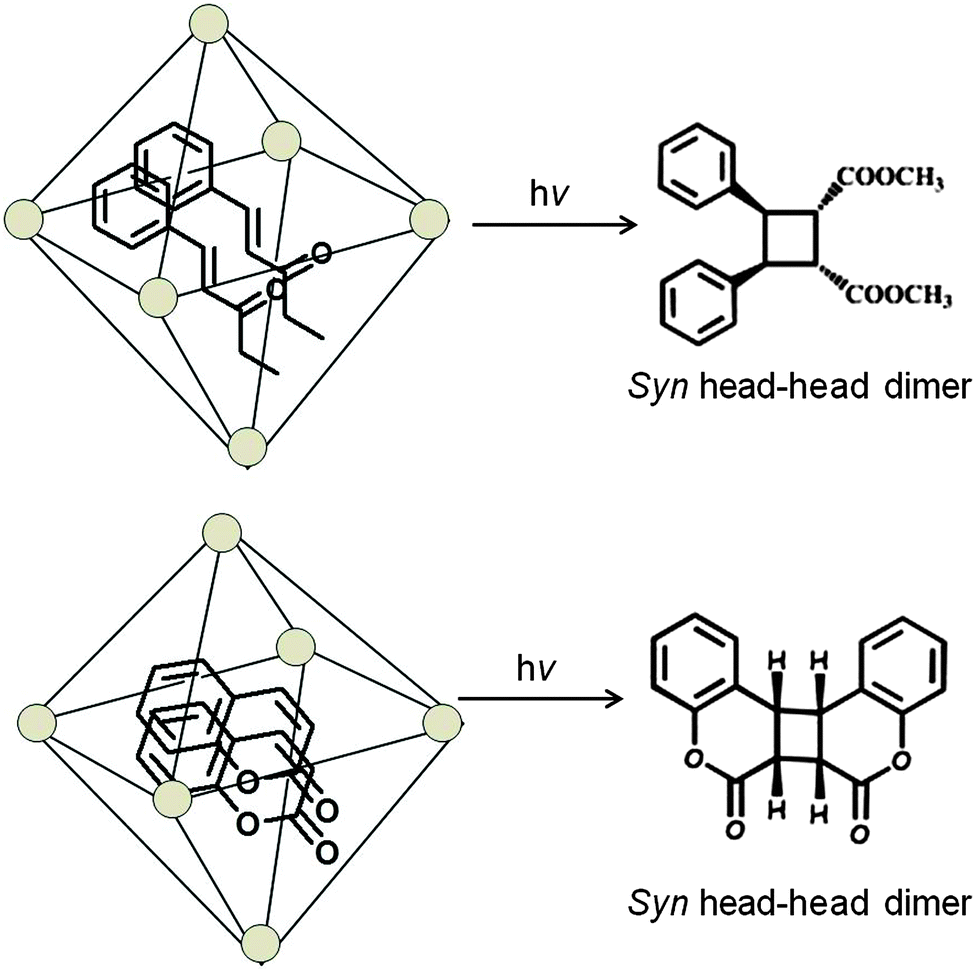
Cucurbiturils with reaction cavities similar to cyclodextrins form water-soluble host–guest complexes and have been employed to pre-align cinnamic acids for photodimerization.13,34 For example, the three cinnamic acids listed in Scheme 4 that do not react in solid state and isomerize in solution upon excitation when included in CB[8] in water readily yielded the syn-head–head dimer exclusively.35 The cinnamic acids listed in Scheme 5 yielding syn-head–tail dimer in the solid state, and the cis isomers in solution resulted in syn-head–head dimers exclusively within CB[8]. While the confined cavity helped bring the two reactive olefin molecules together, the weak intermolecular forces oriented them in a proper geometry for dimerization (Fig. 9). Inclusion of the two olefins, likely prompted by a combination of π–π interaction between phenyl rings, intramolecular hydrogen bonding between COOH groups and hydrophobic interaction, in a head–head fashion, where the two acid groups face water and the two phenyl groups stayed parallel to favor dimerization product, one quite different from that obtained in isotropic solution and solid state.
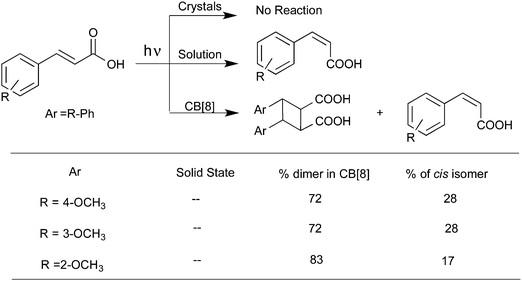 |
| | Scheme 4 Photodimerization of cinnamic acid derivatives in solid phase, solution and in the presence of cucurbituril in water. | |
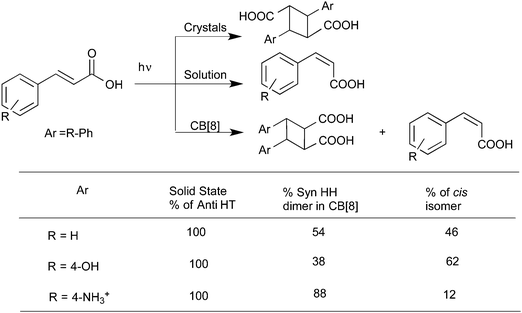 |
| | Scheme 5 Photodimerization of cinnamic acid derivatives in solid phase, in solution and in the presence of cucurbituril in water. | |
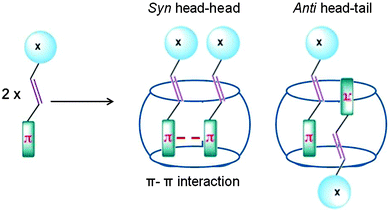 |
| | Fig. 9 Visualized orientation of cinnamic acids included within cucurbituril. X represents the head group, in this case COOH. | |
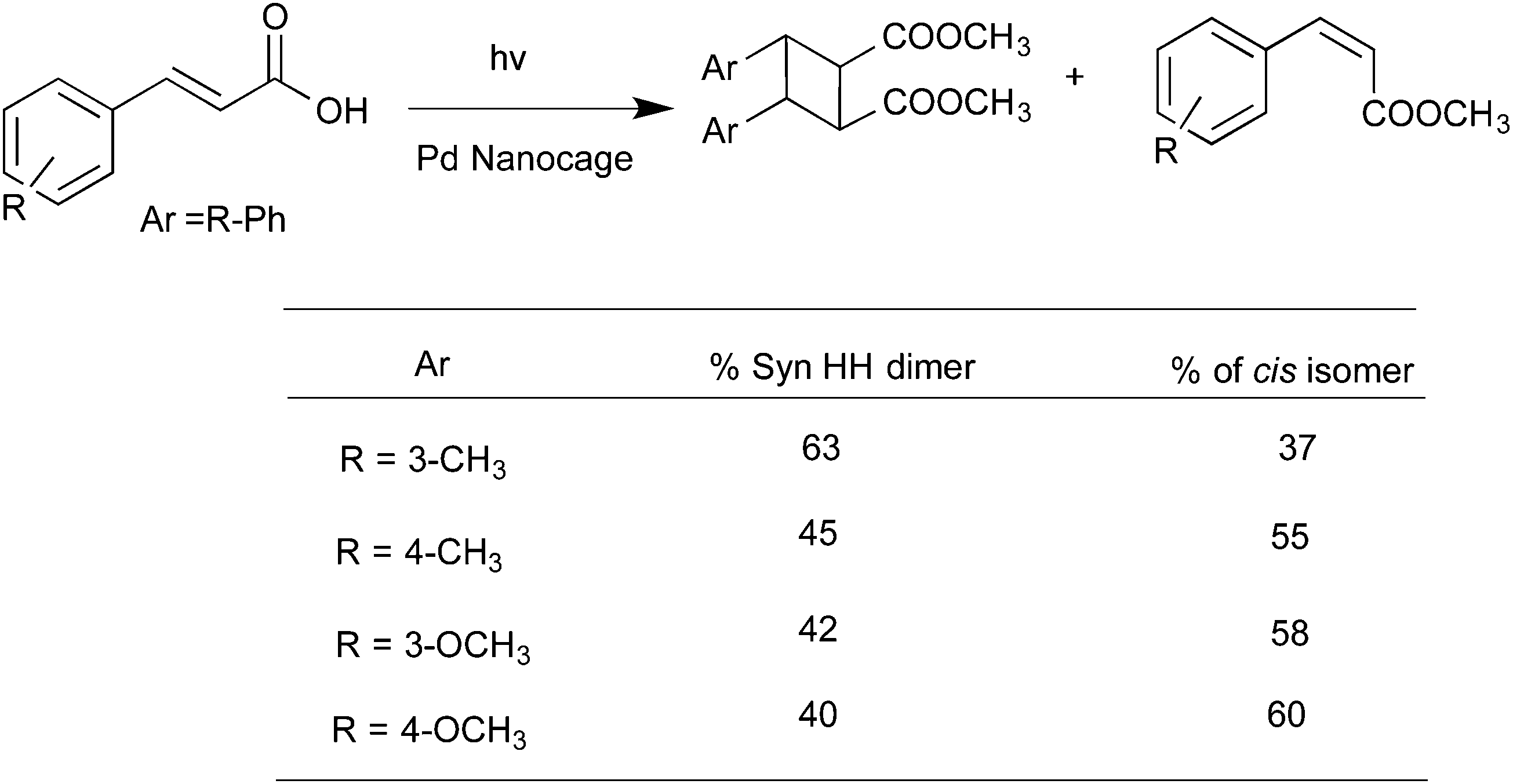
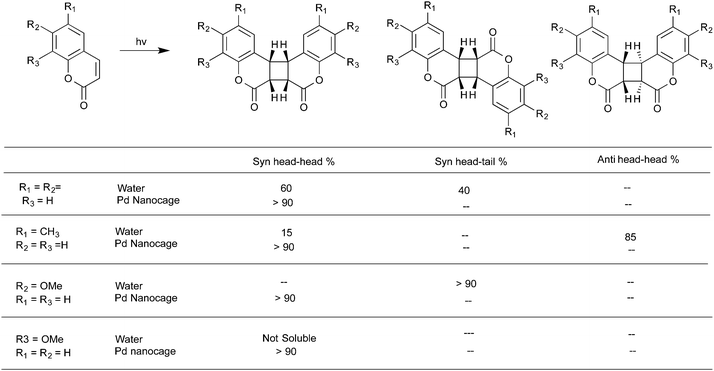
Water-soluble complexes of the Pd nanocage shown in Fig. 8 that readily included the cinnamic acids and coumarins listed in Schemes 6 and 736,37 upon irradiation gave only the respective syn-head–head dimer. The confined hydrophobic reaction cavity of the Pd nanocage clearly served to template the cinnamic acids and coumarins to yield such syn-head–head dimers. The hydrophobic effect forcing cinnamic acid and coumarin molecules to stay within the cage and the likely alignment of the polar groups at the interface between the hydrophobic exterior and the hydrophilic interior toward water favored formation of the syn-head–head dimers (Fig. 10).
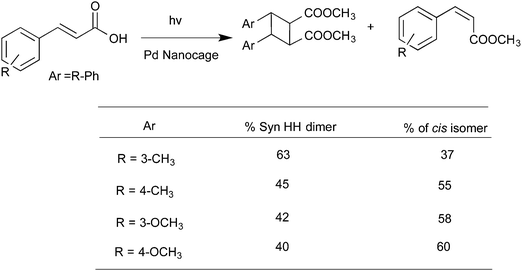 |
| | Scheme 6 Photodimerization of cinnamic acid derivatives in the presence of palladium nanocage. In this example the cis isomer accompanied the dimer. But only one syn HH dimer was formed. | |
 |
| | Scheme 7 Photodimerization of coumarins in the presence of palladium nanocage. | |
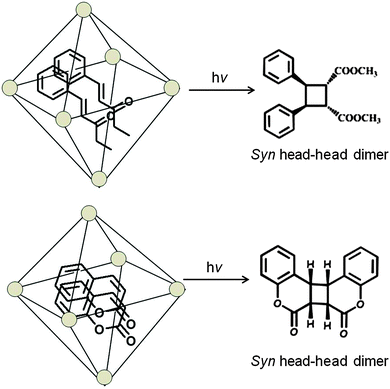 |
| | Fig. 10 Visualized orientation of coumarins included within palladium nanocage. | |
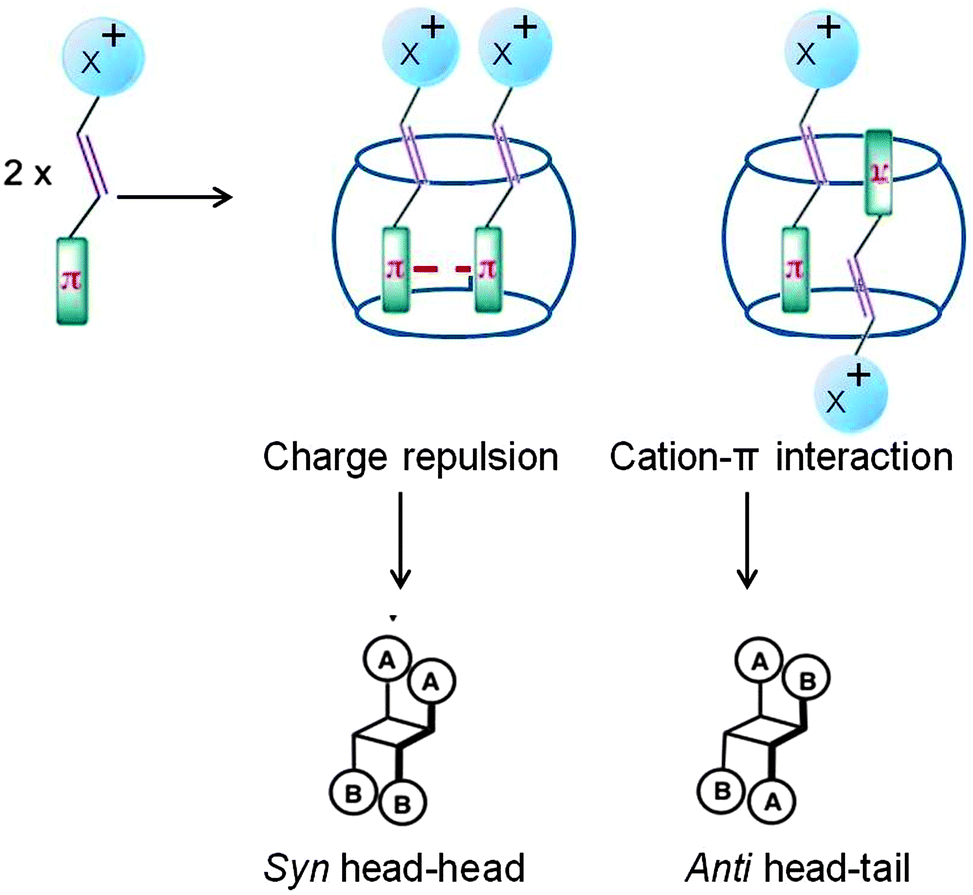
Protonated stilbazoles in water on irradiation yield the following: the corresponding isomerization product, i.e. the cis isomer, the cyclized product and less than 10% dimer. Protonated stilbazoles formed a stable 1:2 (host to guest) inclusion complexes with CB[8] which on irradiation yielded the anti head–tail dimer as the major product (∼90%) while the cis isomer was formed in very small yields (Scheme 8).38–40 Formation of the anti head–tail dimer could be visualized to result from pre-organization of the two olefins as shown in Fig. 11. The charge transfer type of interaction between the protonated pyridyl part of one olefin and phenyl part of the second olefin in these systems, probably, pre-oriented them in a head–tail arrangement. Thus CB[8] localizes the stilbazoles and pre-orients them. It must be noted that in solution such weak interactions are unable to do both these to result in a dimer.
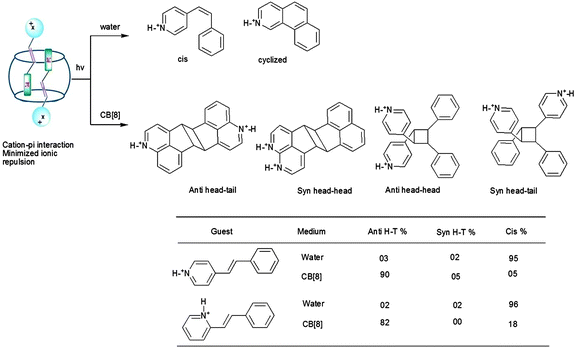 |
| | Scheme 8 Photodimerization of protonated stilbazoles in water and in the presence of cucurbituril in water. Product yields in % are provided in the table. | |
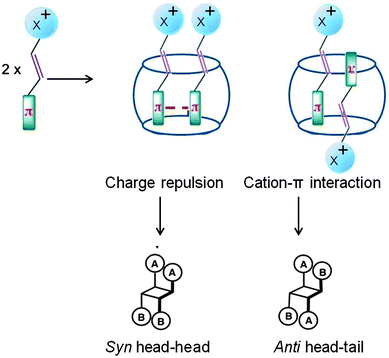 |
| | Fig. 11 Visualized orientation of protonated stilbazoles included within cucurbituril. X represents the head group which is a pyridinium ion. | |
Micelles and open cavitands offer two phases with antithetical properties (hydrophobic interior and hydrophilic exterior) and the interface between these phases orients the guest molecules. The above water-soluble hosts solubilize the water-insoluble organic compounds in the aqueous medium and confine them in the restricted reaction cavity. Based on the above select examples in micelles and open cavitands CB and Pd nanocage we conclude that: (a) a given compound reacts differently in supramolecular assemblies and dispersed phases, (b) the pre-organization within the supramolecular assemblies determines the fate of the excited molecule, the course of the chemical transformation and the structure of the product, (c) the systematic development of supramolecular photochemistry requires an understanding of the intermolecular forces responsible for the stability of host–guest complexes and (d) judicious choice of the host should be made to achieve maximum selectivity and the desired product.
8. Uniqueness of supramolecular photochemistry illustrated with examples
In the following section examples from our recent studies with octa acid (OA) as the host are provided. OA differs from the open containers such as micelles and open cavitands such as cyclodextrins, cucurbiturils and Pd nanocage, in that the OA capsule is a closed container. The eight carboxylic acids on both the top and bottom rims of the cavitand giving the name ‘octa acid’ to the molecule make it water soluble above basic (pH ∼ 9.0) conditions.15 The dimensions of OA cavity are provided in Fig. 12. In presence of a suitable guest (or guests), two molecules of OA assemble in aqueous solution to form a supramolecular, closed capsule. Unlike in micelles and open cavitands the entire guest molecule within OA is enclosed within a hydrophobic container thereby limiting the influence by the capsule entirely to confinement and intermolecular weak interaction between the guest and the capsule. The hydrophobic effect in OA only facilitates the formation of a strong host–guest complex and does not pre-organize the guests. The various aspects of supramolecular photochemistry facilitated by OA capsule are highlighted with examples under the following headings: (a) localization effect, (b) compartmentalization effect, (c) pre-orientational effect, (d) conformational effect, (e) cage effect, and (f) supramolecular steric effect. Note: only a single major effect is highlighted though more than one effect could be operative in a given example. It is important to note that the influence of most supramolecular assemblies can be looked at with these effects in mind.
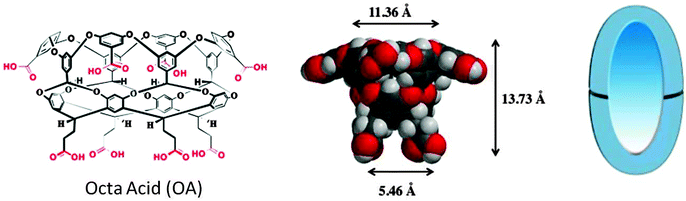 |
| | Fig. 12 Molecular structure, CPK model and cartoon representation of octa acid. | |
Localization effect
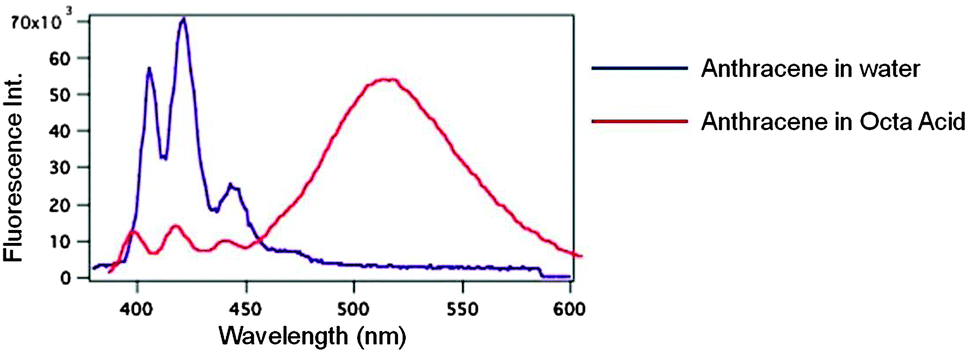
Hydrophobic molecules such as naphthalene and anthracene when accommodated within a larger water-soluble organic host molecule could be dissolved in water. More than a single such molecule can be accommodated within the hydrophobic interior of the water-soluble OA capsule thus bringing them in close proximity. Such a phenomenon of a host localizing more than one molecule within its small cavity providing high local concentration is termed ‘localization effect’. As seen in Fig. 13, only fluorescence from monomer anthracene was recorded in the absence of OA. Generally anthracene dimerizes at high concentrations and does not show excimer emission in any solvent. However as illustrated in Fig. 13 when solubilized in water in presence of OA capsule strong excimer emission was recorded from it41 due to localization effect; dimerization is prevented due to lack of free space. This observation could not be achieved in solution with any other host. Although several substituted anthracenes show excimer emission at higher concentration, at the concentration used in the study none show excimer emission in solution. More importantly, parent anthracene do not exhibit excimer emission in any solvent under any condition.
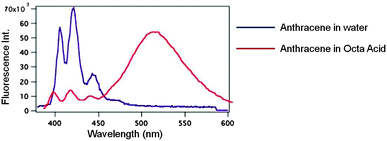 |
| | Fig. 13 Emission spectra of anthracene (10−5 M) in water and included within octa acid. In water the emission is from monomer while in octa acid excimer emission predominates. | |
Compartmentalization effect
The compartmentalization of reactants leading to ‘protection’ of the excited singlet and triplet states of molecules by suppression of bimolecular quenching processes is a feature unique to supramolecular host systems. This property of host OA that results in control of unimolecular photophysical processes is illustrated in the following example: as displayed in Fig. 14, while in perfluoro dimethylcyclohexane solvent the thioketone shows no phosphorescence, when included within OA capsule it does.42 Though there are two molecules within a capsule their restricted mobility prevents self-quenching; the OA capsule protects the thioketone triplet from oxygen as well; the ‘phosphorophore’ is compartmentalized and prevented from interacting with other quencher molecules.
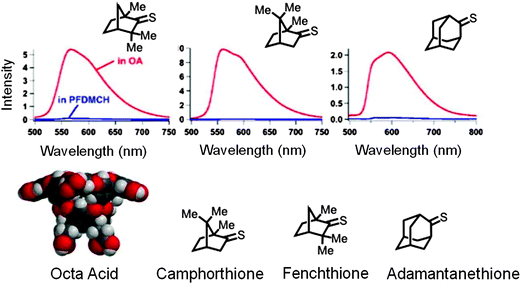 |
| | Fig. 14 Emission spectra of three aliphatic thioketones in perfluorocyclohexane (PFDMCH) and included within octa acid (OA) in water (pH ∼ 9) at room temperature. Phosphorescence is observed only in octa acid. Red curve is in OA and the blue line is in PFDMCH. | |
Pre-orientational effect
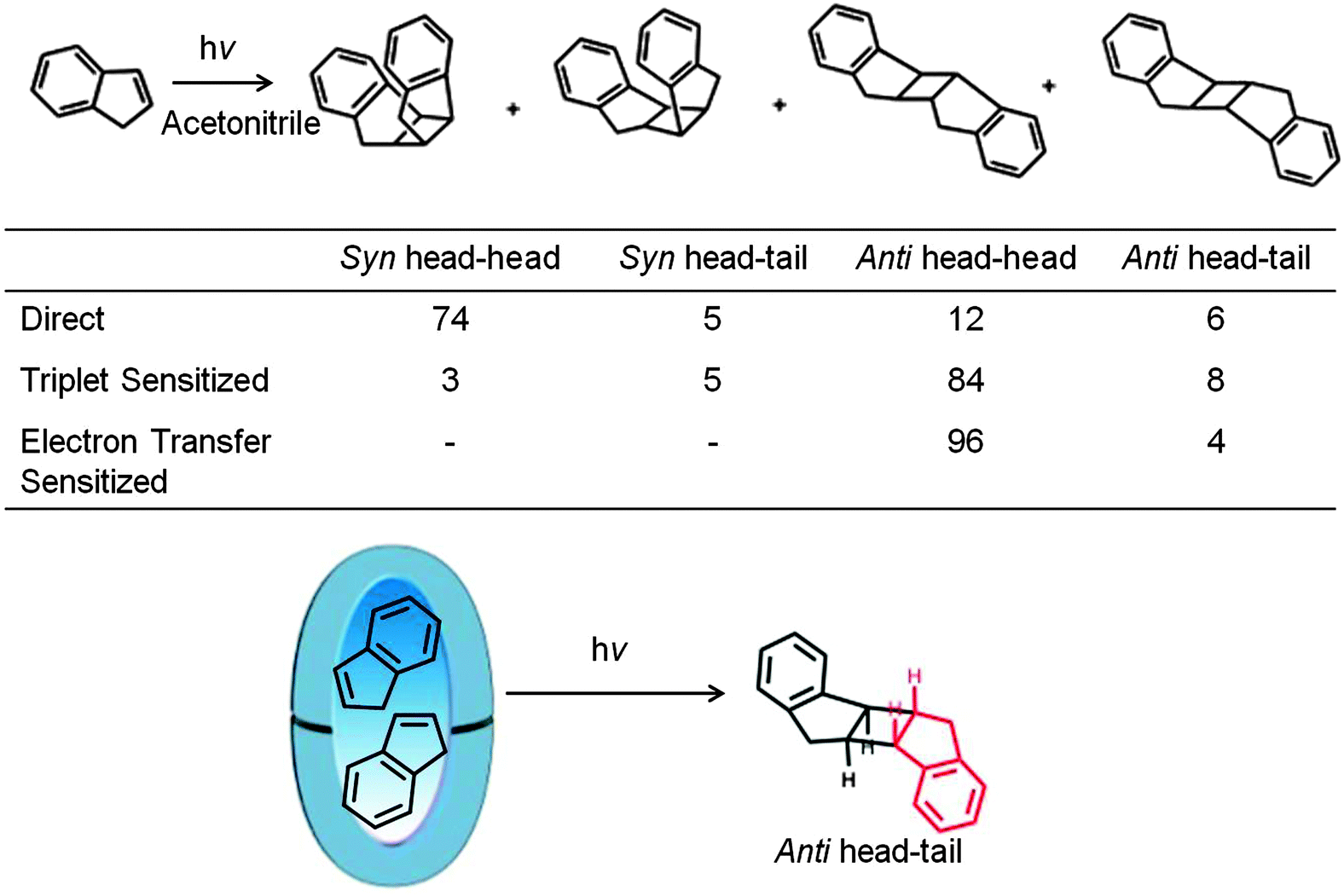
In an aqueous solution, organic compounds with a hydrophilic functional group have been demonstrated to reside with specific orientation, primarily at the hydrophobic–hydrophilic interface (Fig. 6 and 10). This tendency of solubilized molecules to become specifically oriented within a water-soluble reaction container is known as ‘pre-orientational effect’. Within a OA capsule (recall it is a closed container) all parts of the enclosed guest molecules are exposed only to a hydrophobic interior eliminating any orientation from hydrophobic–hydrophilic interfacial feature available in micelles and open cavitands. As shown with an example illustrated in Scheme 9 even in these hydrophobic capsules reactant molecules remain pre-organized to result in a single dimer.43,44 Within the limited space of the hydrophobic capsule the adoption of specific intermolecular orientation by guest molecules to result in a dimer suggest hydrophobic–hydrophilic interface to not be a necessity for pre-orienting molecules.
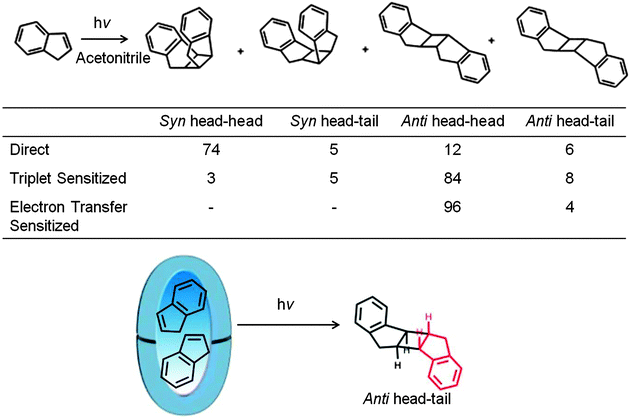 |
| | Scheme 9 Photodimerization of indene under direct, triplet sensitized and electron transfer sensitized conditions in acetonitrile. The photodimerization within octa acid is shown in the equation below. anti-Head–tail dimer is formed only in octa acid. Product yields in % are provided in the table. | |
Conformational effect
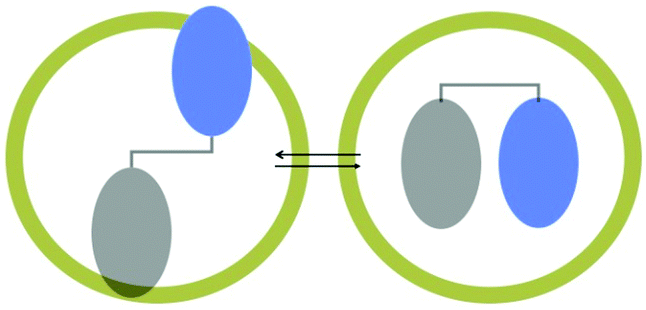
Similar to pre-orientation of two molecules at a hydrophobic–hydrophilic interface, two parts of a single molecule could be oriented at an interface resulting in preference for a single conformer within a supramolecular assembly.45 The ability of a medium to alter the conformer equilibrium is termed ‘conformational effect’, which is illustrated in Fig. 15. The example shown in Fig. 15 has two conformations – extended and folded. While in isotropic solution both conformers would exist, within a confined space, depending on the nature of the end groups one of the two conformer is likely to be preferred. Two examples of conformational control by OA capsule on a guest molecule are provided in Scheme 10 and Fig. 16. Remarkable asymmetric induction in photoproduct from substituted pyridones could be brought forth by this effect (Scheme 10)46 to result in diastereomeric excess as high as 92%. Within the capsule the chiral auxiliary was able to get closer to the reaction center probably due to conformational control by the confined space. In solution, the chiral auxiliary is unable to provide asymmetric induction in product since such a conformation is most likely not preferred.
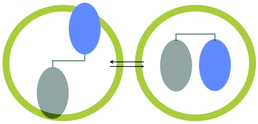 |
| | Fig. 15 A schematic representation of a molecule capable of existing extended and folded conformations. Within octa acid only one could be accommodated. | |
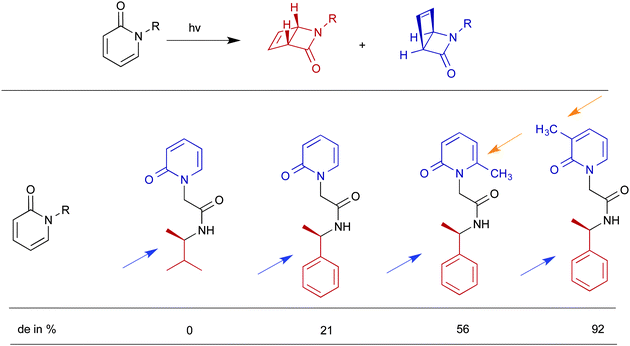 |
| | Scheme 10 Photocylization of pyridones: with chiral R group the diastereomeric products would result. Within octa acid diastereoselectivity of the product varies and in acetonitrile the diastereomeric excess (de) is close to zero. | |
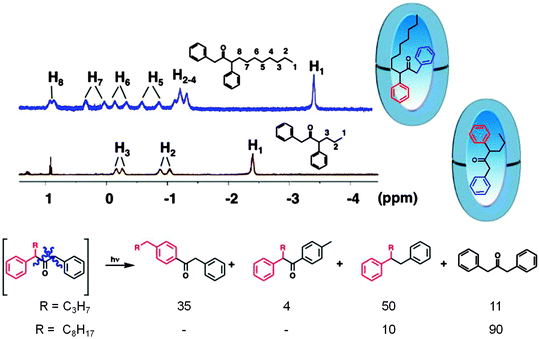 |
| | Fig. 16 Product distribution upon photolysis of α-alkyl ketones within octa acid depends on the chain length of the alkyl group. In solution the product distribution is independent of the chain length. The 1H NMR spectra suggest that the alkyl group resides at the narrow end of the octa acid (note the negative chemical shift of the methyl group). Product yields in % are provided below the products. | |
Interestingly, within OA the photoproduct distribution from α-alkyl dibenzyl ketone depends on the alkyl chain length (butyl vs. octyl) while independent of it in isotropic solution.47 This dependency is attributed to the conformational control by the OA capsule where lack of free space forces the α-octyl dibenzyl ketone to adopt a conformation different from the smaller α-butyl dibenzyl ketone. This variation between the two α-alkyl dibenzyl ketones brings out “The rule of homology does not hold in supramolecular chemistry”. Remember that this was one of the observations made by Schmidt in crystal photochemistry.
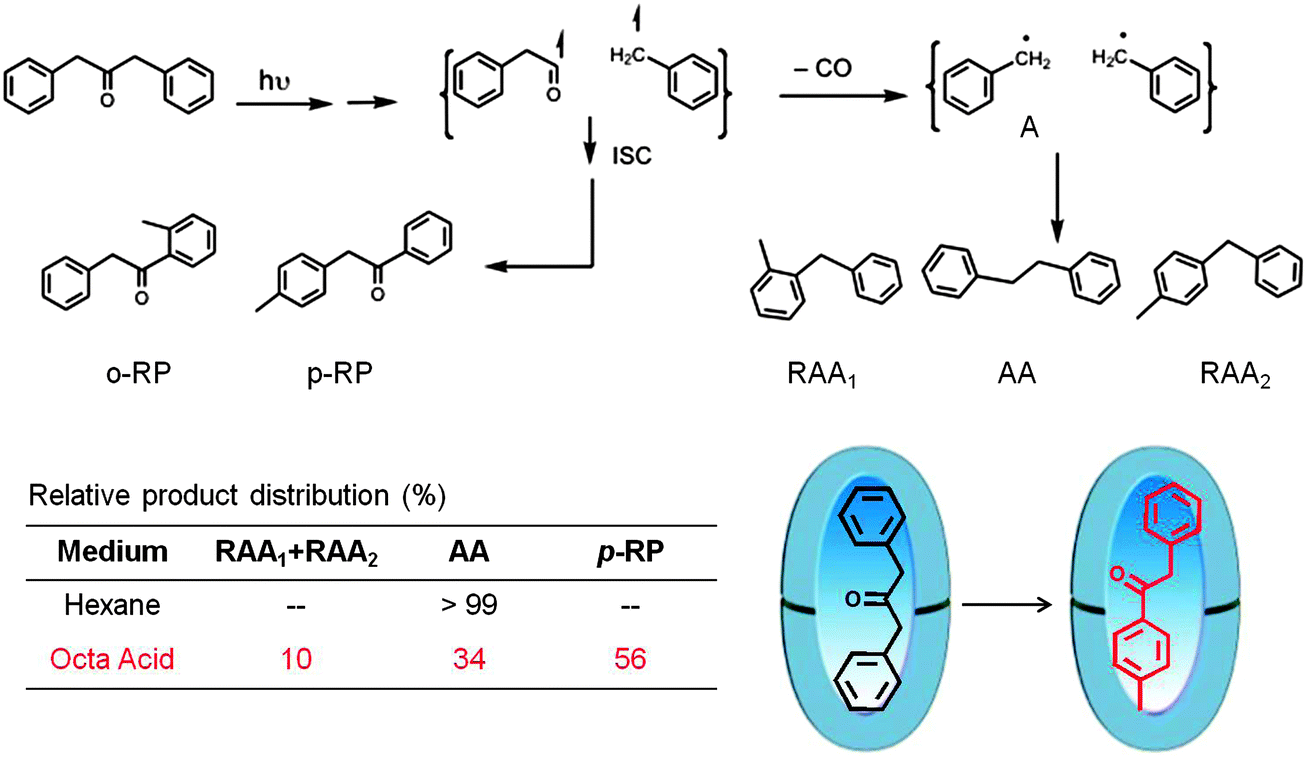
Cage effect
Cage effect arises from the ability of a host to hold two reactive intermediates together, long enough for new reaction to occur. Relative to homogeneous solvents, the magnitude of the large cage effect observed in a supramolecular assembly cannot be explained by the difference in microviscosity between isotropic solution and the above assembly. The main reason for the increased cage effect is that the hydrophobicity of the solvents inhibits diffusion of the intermediates into the aqueous phase, thereby increasing the time spent by the radical intermediates in the restricted space. As mentioned in an earlier section, Turro reported the first remarkable example of cage effect of interest to photochemists in micelles.23 In principle the cage effect does not require a hydrophobic–hydrophilic interface. The example provided below illustrates that the cage effect could be brought forth in a hydrophobic OA capsule.48,49 As shown in Scheme 11 the primary and secondary radical pairs resulting from excited state of dibenzylketone were incarcerated by the OA capsule and allowed to explore new reaction pathways. The dramatic change in the product distribution between the reaction in hexane and within OA capsule (preferential formation of rearranged DBK, p-RP, as the primary product) is the result of cage effect provided by the capsule.
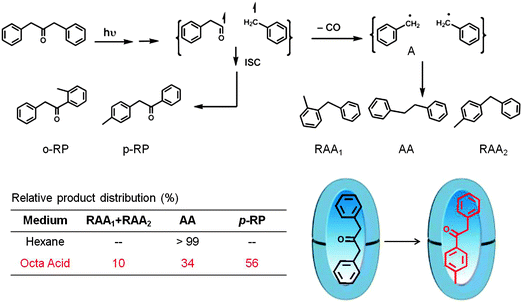 |
| | Scheme 11 Product distribution upon photolysis of dibenzyl ketone in hexane and within octa acid in water (pH ∼ 9). Note that in octa acid the rearrangement product p-RP is predominant and it is not formed in hexane. Product yields in % are provided in the table. | |
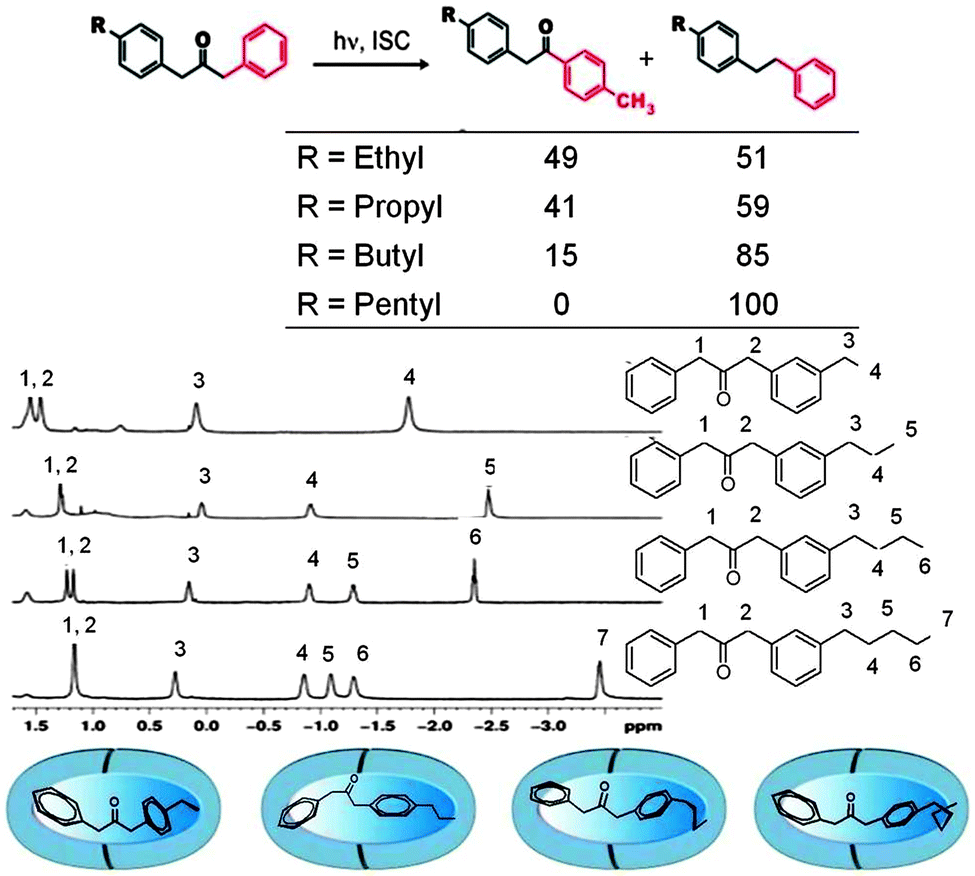
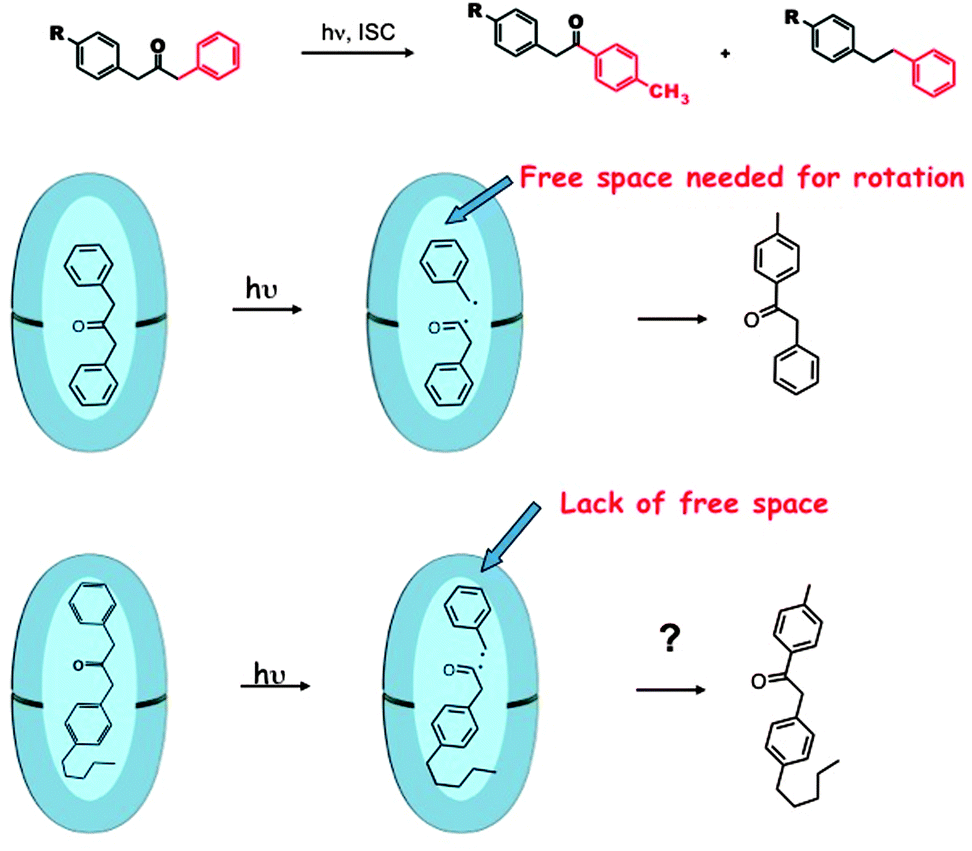
Supramolecular steric effect
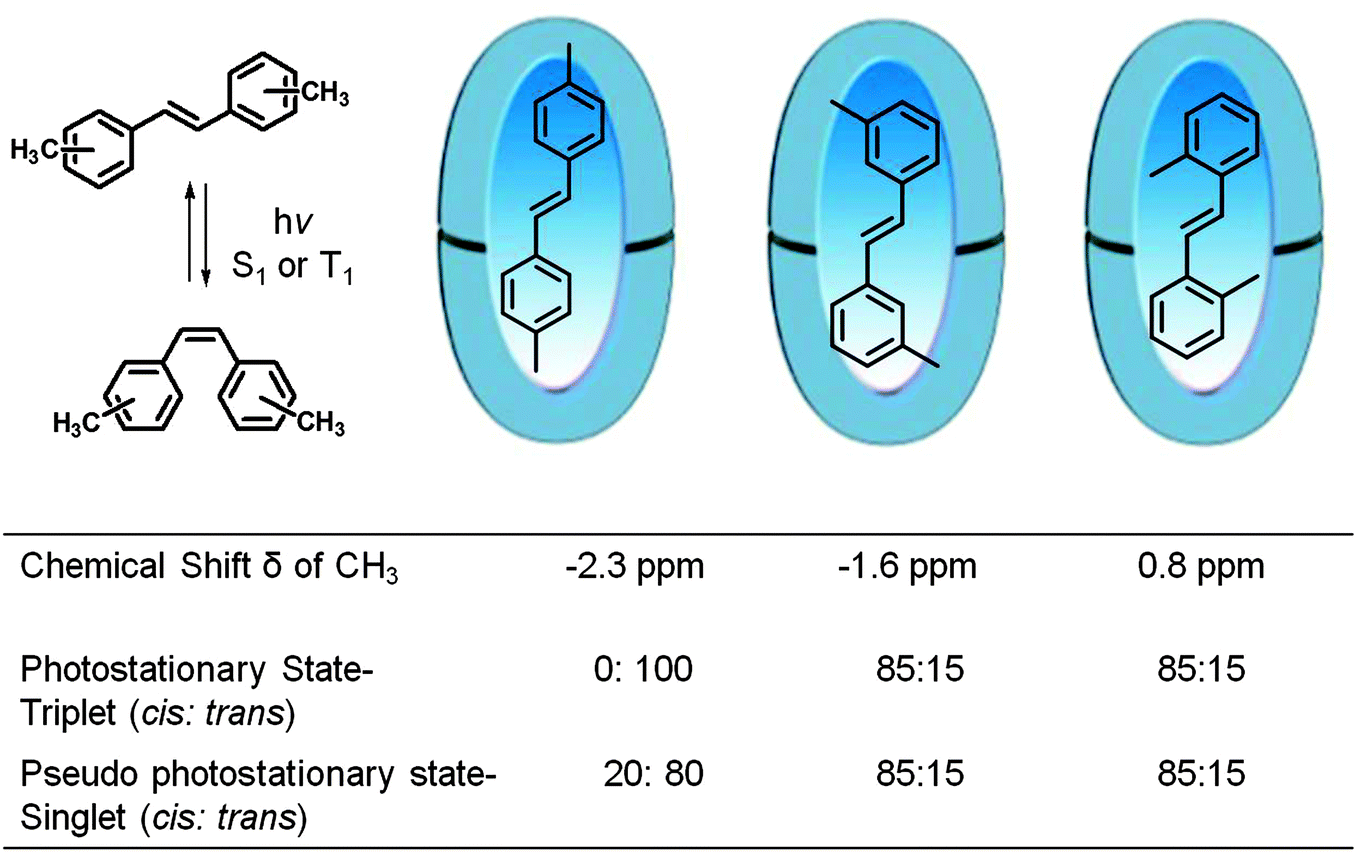
Inherent electronic and steric effects of a reactant molecule that are widely utilized to understand the reactivities of organic molecules may not be the same within a supramolecular container. When the interaction between the guest and the host in a supramolecular environment results in additional steric problems the effect is termed ‘supramolecular steric effect’. Such an effect is more prevalent in ‘hard’ reaction cavities than in ‘soft’ ones. Two examples where the photoproduct distribution is controlled by supramolecular steric effect are presented in Fig. 17 and 19. In the first example the product distribution is controlled by the length of the alkyl chain substituted at the para position of dibenzylketone;49 the longer chain experiences steric problems with the host resulting in restricted rotation of para alkyl phenyl acetyl benzyl radical pair (Fig. 18); this problem is absent with smaller ethyl chain. Thus the different product distribution observed for ethyl and pentyl derivative could be due to the presence of supramolecular steric effect in the latter molecule. In the second example, the para, para′ methyl groups arrest the isomerization of trans-4,4′-dimethyl stilbene.50 Here the steric effect provided by the host OA increases the barrier for rotation in the excited state. The OA capsule's narrow end contains four aromatic rings. C–H⋯π interaction between the methyl hydrogen and the aryl rings at the narrow end probably anchors the two ends of the 4,4′-dimethyl stilbene within the cage. Absence of influence of the OA capsule on the isomerization of trans-2,2′-dimethyl stilbene as well as trans-3,3′-dimethyl stilbene supports this model. This difference clearly brings out the importance of ‘location’ of the alkyl group on the supramolecular steric effect.
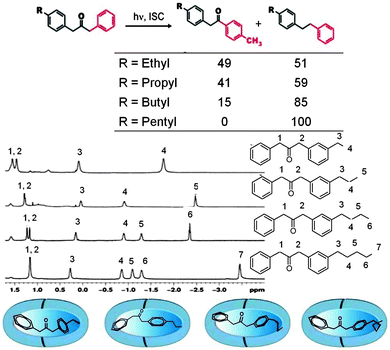 |
| | Fig. 17 Product distribution upon photolysis of para-alkyl ketones within octa acid depends on the chain length of the alkyl group. In solution the product distribution is independent of the chain length. The 1H NMR spectra suggest that the alkyl group resides at the narrow end of the octa acid (see Fig. 16 also). The numbers on the side represent the product yields of the two products in %. | |
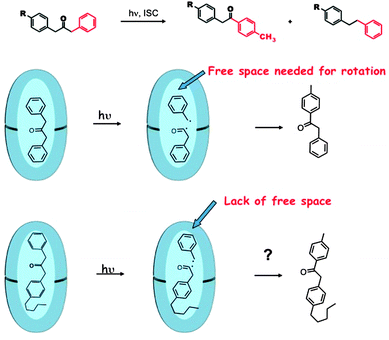 |
| | Fig. 18 Reduced free space by longer alkyl chain alters the product distribution. The reduced steric interaction between the cavity and the alkyl group influences the rotational mobility of the radical pair. | |
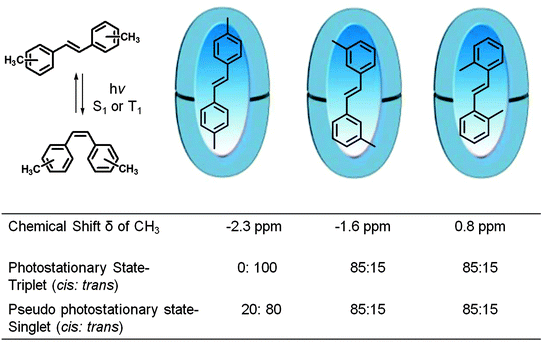 |
| | Fig. 19 Photostationary state trans- and cis-isomer distribution of three alkyl substituted stilbenes. Geometric isomerization of trans to cis of para, para′-substituted stilbene is influenced by the octa acid. The methyl groups anchored at the narrower end of the octa acid capsule are subjected to supramolecular steric effect. | |
9. Summary
During the last five decades extensive explorations of crystals, micelles, cyclodextrins and zeolites have established them as valuable reaction media. The search for better control on photoreactions have led to the synthesis and investigation of numerous synthetic organic and inorganic hosts as reaction vessels amongst which cucurbiturils, Pd nanocage and octa acid have played a predominant role. In this brief review we have demonstrated that the photochemistry based on concepts of physical organic and supramolecular chemistry continues to be a discipline with unlimited potential. Toward providing a common basis for discussion, we have outlined a workable model, which includes the more important identifiable parameters of supramolecular reaction vessels responsible for directing photochemical processes. In spite of the many examples cited in this review it is clear that the model requires a great deal more testing and refinement.
While nature inspired photochemists harken back to the extremely efficient natural systems two aspects remain yet to be fully explored. To our knowledge, catalytic supramolecular reactions are scarce whereas reactions in biological systems are generally so. Recognizing the catalytic value is important and developing catalytic confined media is a challenge. An enzyme, analogous to the supramolecular assemblies discussed above, is often embedded in membranes, yet another supramolecular structure and not free floating. Organizing and investigating supramolecular assemblies on ordered surfaces is thus likely to open a new dimension. Much activity is expected in the coming years in supramolecular surface photochemistry or ‘super-duper photochemistry’ as referred to by Turro.
Acknowledgements
VR is grateful to the National Science Foundation, USA (CHE-1411458) for financial support and dedicates this article to the memory of his mentors Professors P. de Mayo and N. J. Turro who pioneered ‘Supramolecular Photochemistry’.
References
-
N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, CA, 2010 Search PubMed.
-
A. Axel Griesbeck, M. Oelgemöller and F. Ghetti, CRC Handbook of Organic Photochemistry and Photobiology, CRC Press, Boca Raton, London, New York, Washington D.C., 3rd edn, 2012 Search PubMed.
-
W. Gerhartz, Imaging and Information Storage Technology, Verlagsgesellschaft mbH, Weinheim, (Federal Republic of Germany) and VCH Publishers, Inc. New York, NY, U.S.A., Weinheim, Germany and New York U.S.A., 1992 Search PubMed.
- H. D. Roth, Angew. Chem., Int. Ed. Engl., 1989, 28, 1193–1207 CrossRef.
-
A. Schonberg, Preparative Organic Photochemistry, Springer-Verlag New York Inc., New York, 1968 Search PubMed.
-
J.-M. Lehn, Supramolecular Chemistry: Concepts and perspectives, VCH, New York, 1995 Search PubMed.
-
K. Kalyanasundaram, Photochemistry in Microheterogeneous Systems, Academic Press, Inc., New York, 1987 Search PubMed.
-
Photochemistry in Organized and Constrained Media, ed. V. Ramamurthy, Wiley-VCH, New York, 1991 Search PubMed.
-
Supramolecular Photochemistry, ed. V. Ramamurthy and Y. Inoue, John Wiley, Hoboken, 2011 Search PubMed.
- N. J. Turro, J. Org. Chem., 2011, 76, 9863 CrossRef CAS PubMed.
- B. Bibal, C. Mongin and D. M. Bassani, Chem. Soc. Rev., 2014, 43, 4179 RSC.
- C. Bohne, Chem. Soc. Rev., 2014, 43, 4037–4050 RSC.
- N. Vallavojo and J. Sivaguru, Chem. Soc. Rev., 2014, 43, 4084–4101 RSC.
- C. Yang and Y. Inoue, Chem. Soc. Rev., 2014, 43, 4123–4143 RSC.
- J. H. Jordan and B. C. Gibb, Chem. Soc. Rev., 2014 10.1039/c4CS00191e.
- L. S. Shimizu, S. R. Salpage and A. A. Korous, Acc. Chem. Res., 2014, 47, 2116–2127 CrossRef CAS PubMed.
- L. R. MacGillivray, G. S. Papaefstathiou, T. Friscic, T. D. Hamilton, D.-K. Bucar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280–291 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- R. Paulini, K. Muller and F. Diederich, Angew. Chem., Int. Ed., 2005, 44, 1788–1805 CrossRef CAS PubMed.
-
D. Ginsburg and G. M. J. Schmidt, et al., Solid State Photochemistry, Verlag Chemie, GmbH, Weinheim, 1976, (A collection of research publications by G. M. J. Schmidt) Search PubMed.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CrossRef CAS.
- F. H. Quina and D. G. Whitten, J. Am. Chem. Soc., 1977, 99, 877–883 CrossRef CAS.
- N. J. Turro and W. R. Cherry, J. Am. Chem. Soc., 1978, 100, 7431–7432 CrossRef CAS.
- N. J. Turro and B. Kraeutler, J. Am. Chem. Soc., 1978, 100, 7432–7434 CrossRef CAS.
- P. de Mayo and L. K. Sydnes, J. Chem. Soc., Chem. Commun., 1980, 994–995 RSC.
- K. Lee and P. de Mayo, J. Chem. Soc., Chem. Commun., 1979, 493–495 RSC.
- C. T. Libermann, Ann. Chem. Pharm., 1871, 158, 300 Search PubMed.
- M. D. Cohen and G. M. J. Schmidt, J. Chem. Soc., 1964, 1996–2000 RSC.
- M. D. Cohen, Angew. Chem., Int. Ed. Engl., 1975, 14, 386–393 CrossRef.
-
V. Ramamurthy, R. G. Weiss and G. S. Hammond, in Advances in Photochemistry, ed. D. Volman, G. S. Hammond and D. C. Neckers, John Wiley & sons, 1993, vol. 18, pp. 67–234 Search PubMed.
-
N. J. Turro and M. A. Garcia-Garibay, in Photochemistry in Organized & Constrained Media, ed. V. Ramamurthy, VCH Publishers, New York, 1991, pp. 1–38 Search PubMed.
- R. G. Weiss, V. Ramamurthy and G. S. Hammond, Acc. Chem. Res., 1993, 26, 530–536 CrossRef CAS.
-
M. D. Cohen and G. M. J. Schmidt, in Solid State Photochemistry, ed. D. Ginsburg, Verlag Chemie, GmbH, Weinheim, 1976, pp. 233–254 Search PubMed.
- S. Y. Jon, Y. H. Ko, S. H. Park, H.-J. Kim and K. Kim, Chem. Commun., 2001, 1938–1939 RSC.
- M. Pattabiraman, L. S. Kaanumalle, A. Natarajan and V. Ramamurthy, Langmuir, 2006, 22, 7605–7609 CrossRef CAS PubMed.
- S. Karthikeyan and V. Ramamurthy, J. Org. Chem., 2006, 71, 6409–6413 CrossRef CAS PubMed.
- S. Karthikeyan and V. Ramamurthy, J. Org. Chem., 2007, 72, 452–458 CrossRef CAS PubMed.
- M. V. S. N. Maddipatla, L. S. Kaanumalle, A. Natarajan, M. Pattabiraman and V. Ramamurthy, Langmuir, 2007, 23, 7545–7554 CrossRef CAS PubMed.
- R. Kaliappan, L. S. Kaanumalle, A. Natarajan and V. Ramamurthy, Photochem. Photobiol. Sci., 2006, 5, 925–930 CAS.
- R. Kaliappan, M. V. S. N. Maddipatla, L. S. Kaanumalle and V. Ramamurthy, Photochem. Photobiol. Sci., 2007, 6, 737–740 CAS.
- L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2005, 127, 3674–3675 CrossRef CAS PubMed.
- N. Jayaraj, M. V. S. N. Maddipatla, R. Prabhakar, S. Jockusch, N. J. Turro and V. Ramamurthy, J. Phys. Chem. B, 2010, 114, 14320–14328 CrossRef CAS PubMed.
- A. Parthasarathy, S. R. Samantha and V. Ramamurthy, Res. Chem. Intermed., 2013, 39, 73–87 CrossRef CAS.
- V. Ramamurthy and A. Parthasarathy, Isr. J. Chem., 2011, 51, 817–829 CrossRef CAS.
- S. Devanathan and V. Ramamurthy, J. Phys. Org. Chem., 1988, 1, 91–102 CrossRef CAS.
- A. K. Sundaresan, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, Tetrahedron, 2009, 65, 7277–7288 CrossRef CAS PubMed.
- C. L. D. Gibb, A. K. Sundaresan, V. Ramamurthy and B. C. Gibb, J. Am. Chem. Soc., 2008, 130, 4069–4080 CrossRef CAS PubMed.
- L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2004, 126, 14366–14367 CrossRef CAS PubMed.
- A. K. Sundaresan and V. Ramamurthy, Photochem. Photobiol. Sci., 2008, 7, 1555–1564 CAS.
- A. Parthasarathy and V. Ramamurthy, Photochem. Photobiol. Sci., 2011, 10, 1455–1462 CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.