
Peptide stapling techniques based on different macrocyclisation chemistries
Yu Heng
Lau
,
Peterson
de Andrade
,
Yuteng
Wu
and
David R.
Spring
*
University Chemical Laboratory, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
First published on 8th September 2014
Abstract
Peptide stapling is a strategy for constraining short peptides typically in an alpha-helical conformation. Stapling is carried out by covalently linking the side-chains of two amino acids, thereby forming a peptide macrocycle. There is an expanding repertoire of stapling techniques based on different macrocyclisation chemistries. In this tutorial review, we categorise and analyse key examples of peptide stapling in terms of their synthesis and applicability to biological systems.
 Yu Heng Lau | Yu Heng Lau obtained his BSc and MSc in organic chemistry at the University of Sydney, receiving first class Honours with the University Medal for research on azamacrocyclic sensors under the supervision of Dr Matthew H. Todd and Dr Peter J. Rutledge. In 2010, he was awarded a Bragg Cambridge Australia scholarship to study at the University of Cambridge, where he is currently completing his PhD in chemical biology on new peptide stapling techniques with Prof. David Spring. |
 Peterson de Andrade | Dr Peterson de Andrade obtained his degree as a pharmacist in 2005 at the School of Pharmaceutical Sciences – University of São Paulo. From the same institution, he achieved his Masters (2008) and PhD (2012) in organic synthesis (carbohydrate chemistry) under the supervision of Prof. Ivone Carvalho. Recently (2012–2014), he was funded by the Brazilian government (CNPq) to work as a postdoctoral research associate with Prof. David Spring at the University of Cambridge (Department of Chemistry), where he has been working on new approaches to synthesise stabilised alpha-helix mimetic inhibitors for protein–protein interactions. |
 Yuteng Wu | Yuteng Wu was born in China and grew up in Auckland, New Zealand. He received his MSci from Imperial College London in 2013 under the supervision of Prof. Alan Armstrong. During that time he undertook an industrial placement with GlaxoSmithKline. Yuteng moved to Cambridge to undertake his PhD in the group of Prof. David Spring, studying double-click peptide stapling strategies. |
 David R. Spring | David Spring is currently a Professor at the University of Cambridge within the Chemistry Department. He received his DPhil (1998) at Oxford University under Sir Jack Baldwin. He then worked as a Wellcome Trust Postdoctoral Fellow at Harvard University with Stuart Schreiber (1999–2001), after which he joined the faculty at the University of Cambridge. His research programme is focused on synthetic chemistry and chemical biology. |
Key learning points(1) A growing number of stapling techniques are being developed, each utilising different macrocyclisation chemistry.(2) Stapling techniques can be broadly categorised as one- or two-component reactions. (3) Different stapling chemistries have strengths and weaknesses in terms of both their synthetic ease and potential utility for addressing biological questions. (4) Two-component reactions enable efficient modular construction of peptides bearing different staple linkages, orthogonal to changes in the peptide sequence. (5) There are many opportunities to try alternative stapling techniques for a given PPI, as most studies to date only utilise one type of stapling chemistry. |
1. Introduction
The alpha-helix is a common secondary structure motif which plays an important functional role in many proteins. There is great interest in developing molecules which can mimic alpha-helices found at the binding interface between two proteins, as such molecules can act as competitive inhibitors of protein–protein interactions (PPIs).1 The ability to selectively inhibit PPIs may enable therapeutics to be developed against a wide range of new protein targets previously thought to be ‘undruggable’.Whilst the native alpha-helical peptide is a clear starting point for designing a mimetic, the peptide itself is a poor therapeutic candidate. In isolation, short peptides usually do not retain their native conformation and binding capability, as they lack the structural reinforcement provided by the remainder of the protein. Peptides are also susceptible to proteolytic degradation and typically cannot cross the cell membrane. For these reasons, many research groups are investigating synthetic methods for modifying peptides, with the aim of reinforcing their native alpha-helical conformation and thereby restoring binding affinity towards their protein targets. At the same time, these peptide modifications may also lead to improvements in proteolytic stability and cell permeability.
One of the most established methods for generating alpha-helices is peptide stapling. Starting from the native peptide sequence, two amino acids are chosen which lie on the same face of the helix (Fig. 1). These two residues are then substituted for non-native amino acids which have side-chains that can be covalently joined or ‘stapled’ together. If the length and position of the resulting covalent staple are optimal, this macrocyclisation process can impart structural rigidity to the peptide and reinforce the desired alpha-helical conformation.
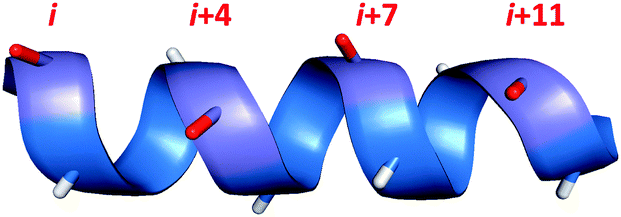 | ||
| Fig. 1 Amino acid residues which lie on the same face of an alpha helix are shown in red, with i,i + 4/7/11 spacing. | ||
Early methods of peptide stapling utilised the chemistry of proteogenic amino acids, forming staples via lactam formation between Lys and Glu/Asp residues.2 However, the stapling technique which has made the greatest impact on this field so far is hydrocarbon stapling, which involves the ring-closing metathesis of olefin-bearing amino acids.3 Indeed, the term ‘stapled peptides’ was first introduced by Verdine and co-workers,4 who have expanded on seminal work by Blackwell and Grubbs5 and led the development of hydrocarbon stapled peptides over the past 15 years. Along with hydrocarbon stapling, there is a growing number of alternative stapling techniques, each utilising a different form of macrocyclisation chemistry and giving rise to stapled peptides with different bioactive properties. By expanding and diversifying the array of stapling techniques available, the chances of developing effective inhibitors against helix-mediated PPIs are maximised.
In this tutorial review, we compare and evaluate the various side-chain macrocyclisation techniques used for peptide stapling in terms of their synthetic accessibility, potential to address biological questions, and current stage of development. Furthermore, we categorise stapling techniques as either one- or two-component reactions (Fig. 2), with the aim of highlighting the broad similarities and differences between the various techniques. We define one-component stapling as a direct bond-forming reaction between the side-chains of the two non-native amino acids, whilst two-component stapling involves a separate bifunctional linker to bridge the two non-native side-chains together.
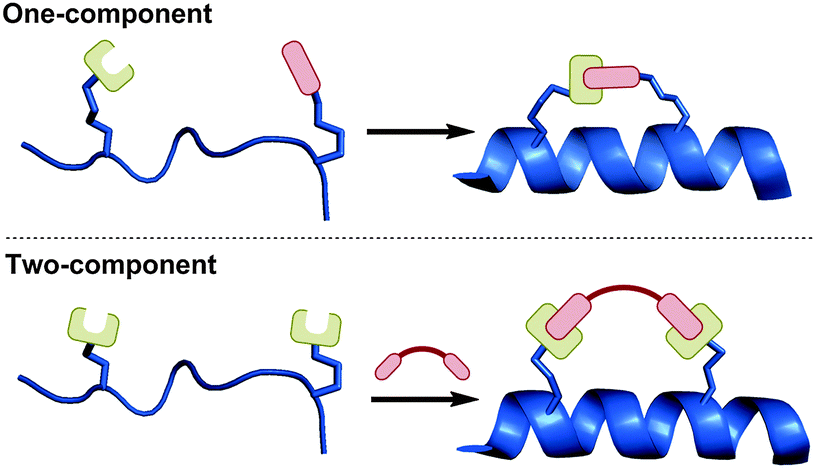 | ||
| Fig. 2 One- vs. two-component stapling. One-component stapling involves direct cyclisation between two side-chains, whilst two-component stapling utilises a separate bifunctional linker to bridge the two side-chains together. | ||
Besides peptide stapling, there are other promising alpha-helix mimetic strategies based on different peptide modifications (e.g. hydrogen-bond surrogates, beta-peptides, metal-chelating clips), as well as non-peptidic mimics (e.g. terphenyl scaffolds), and the reader is directed to other excellent reviews which cover the progress achieved in these fields.6,7 There is also a wealth of literature which addresses general peptide macrocyclisation techniques not specific to alpha-helices.8
2. One-component stapling techniques
One-component stapling techniques use non-native amino acids bearing complimentary side-chain functional groups that can be directly coupled together. The length, structure and chemical functionality of the resulting staple linkage are all determined by the choice of non-native amino acids during solid-phase peptide synthesis (SPPS). Most early examples of peptide stapling techniques are one-component reactions. Whilst the most established techniques utilise either ring-closing metathesis or lactamisation, there are other ligation reactions which have also shown promise for stapling peptides. Here we present an overview of one-component stapled peptides categorised by stapling chemistry.2.1 Ring-closing metathesis
Alkene ring-closing metathesis was first employed as a peptide stapling reaction by Blackwell and Grubbs,5 who reported the solution-phase metathesis and subsequent hydrogenation of model hydrophobic heptapeptides containing either O-allyl serine or homoserine residues with i,i + 4 spacing (Fig. 3A). Although the parent model peptide was already helical, and stapling did not significantly affect peptide conformation, this preliminary study demonstrated the feasibility of conducting metathesis on helical peptide side-chains.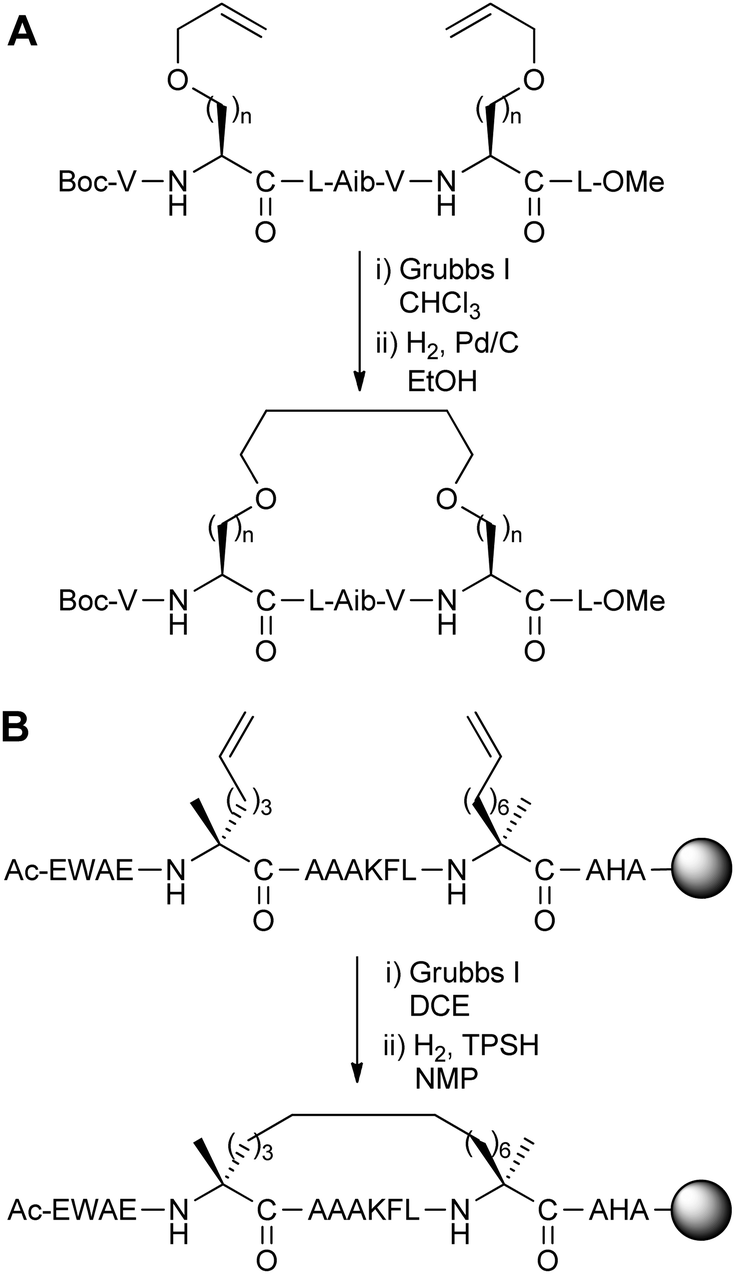 | ||
| Fig. 3 (A) Ring-closing metathesis of peptides containing O-allyl serine and homoserine (n = 1, 2), (B) synthesis of the all-hydrocarbon stapled peptide reported by Schafmeister et al. with the greatest helicity.3 TPSH: 2,4,6-triisopropylbenzenesulphonyl hydrazide, DCE: 1,2-dichloroethane, NMP: N-methylpyrrolidinone. | ||
Following this work, Schafmeister et al. reported a variant of metathesis stapling using α,α-disubstituted amino acids bearing olefinic side-chains of different lengths and stereochemistry (Fig. 3B).3 α,α-Disubstituted amino acids were chosen for their increased helix propensity, especially over D-amino acids. Unlike the previous work by Grubbs, ring-closing metathesis and hydrogenation were carried out on solid-phase prior to peptide cleavage from the resin. Upon stapling a collection of i,i + 4/7 peptides based on the C-peptide sequence of RNAse A, the resulting ‘all-hydrocarbon’ stapled peptides were found to have variable levels of helicity by circular dichroism. The optimal i,i + 7 stapled peptide displayed increased helicity over the native and unstapled peptides, as well as increased stability to proteolysis by trypsin.
With this initial success in hand, the Verdine lab (in collaboration with Walensky, Korsmeyer and co-workers) and other research groups have extensively developed this hydrocarbon stapling technique to address a range of PPI targets as well as creating chemical probes and tool compounds. An early example of their work is the stapling of BID BH3 peptides for binding proteins in the BCL-2 family.4 In addition to increased helicity and in vivo stability, Walensky et al. showed that their optimised stapled peptide could bind BCL-2 with greater affinity than the native peptide, induce apoptosis in leukaemia cells, and inhibit the growth of human leukaemia xenografts in mice.
A recent notable example was reported by Sawyer and co-workers,9 who have developed a hydrocarbon stapled peptide dual inhibitor of the oncogenic p53-MDM2/MDMX interaction which has shown promising results in terms of in vitro binding affinity, cellular activity and suppression of human xenograft tumours in animal models. These findings are now leading to the progression of a p53 stapled peptide into clinical trials.
From a synthetic perspective, the route for preparing hydrocarbon stapled peptides is now established and optimised for i,i + 3/4/7 spacings. Furthermore, the ‘all-hydrocarbon’ linkage is appealing in terms of staple design, as it is a simple and structurally minimal motif. X-ray crystallographic data and molecular dynamics simulations have shown that the hydrocarbon linkage can bind hydrophobic pockets on the surface of target proteins.10 However, it still remains to be seen if these binding interactions involving the staple are a general phenomenon, and whether alternative staple motifs are also able to form such binding contacts.
Despite many literature examples of PPI inhibition by hydrocarbon stapled peptides, there is still no guarantee that hydrocarbon stapling will improve the binding and pharmacokinetic properties of a peptide.11 Extensive optimisation may be needed to find a stapled peptide with the desired characteristics. In some cases, the in vitro binding affinity of peptides may be weakened upon stapling, such as the hydrocarbon stapled p53 peptides reported by Lane and co-workers.12 Despite a drop in affinity, the stapled peptides in this example showed superior activity over their unstapled counterparts in a range of cellular assays. In other cases, cell permeability cannot be achieved by stapling alone. There are examples in the literature, such as the stapled p53 peptides reported by Bernal et al.,13 where modifications in the peptide sequence are necessary to achieve cellular uptake. Although these issues are not confined to any single stapling technique, they have primarily been reported in the context of hydrocarbon stapling, due to the popularity of this stapling technique in the literature. Regarding hydrocarbon stapling in particular, questions have been raised about the necessity of α,α-disubstituted amino acids. Wilson and co-workers have recently shown that for BID BH3 stapled peptides, the equivalent monosubstituted olefinic amino acids behave similarly in terms of helicity, proteolytic stability and binding affinity towards BCL-xL (Fig. 4).14
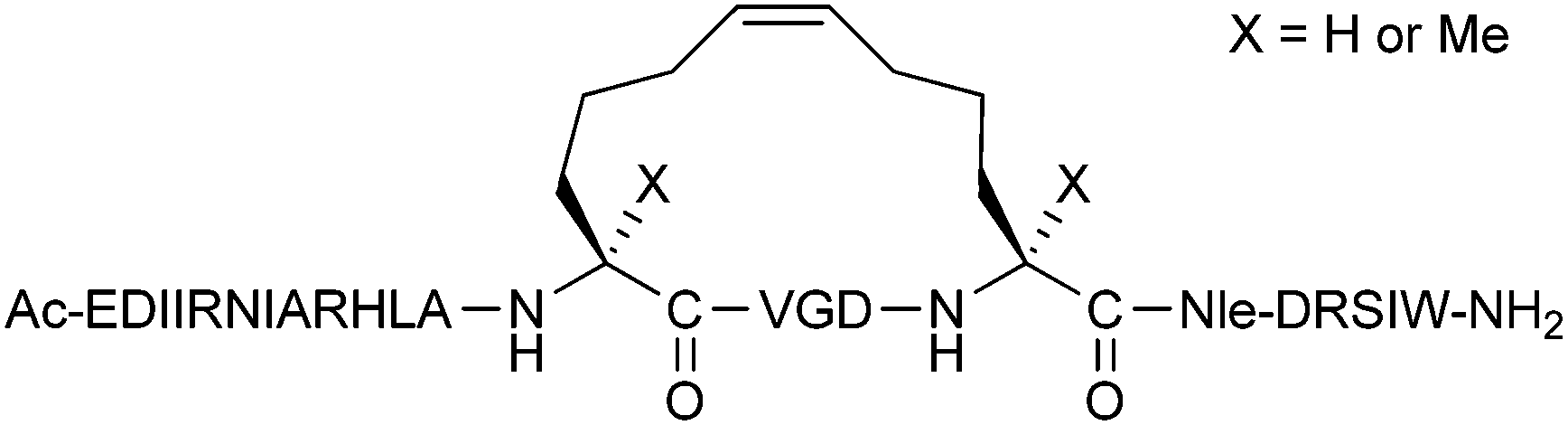 | ||
| Fig. 4 Monosubstituted vs. disubstituted olefinic amino acids for the stapling of BID BH3 peptides. | ||
Nevertheless, hydrocarbon stapling is one of the most thoroughly investigated and successful peptide stapling techniques to date. Readers interested in a comprehensive overview of hydrocarbon stapled peptides can refer to a recent perspective published by Walensky and Bird, and references therein.15
2.2 Lactamisation
The earliest reports of alpha-helix stabilisation via side-chain stapling are based on intramolecular amide-bond formation between i,i + 4 spaced amino acids. Felix et al. were the first to investigate side-chain lactamisation, coupling the side-chains of Lys and Asp residues in shortened analogues of growth hormone releasing factor.2 A combination of NMR studies and circular dichroism were used to show that helicity and growth hormone releasing bioactivity was preserved upon macrocyclisation.Since this initial report, many studies featuring side-chain lactamisation have explored amide linkages with different chain lengths and positioning, with the aim of optimising helix stabilisation for different peptide systems. In terms of biological applications, early studies use side-chain lactamisation to synthesise hormone mimetics or to investigate protein folding. An excellent review covering early examples of lactam stapled peptides and their optimisation has been compiled by Taylor.16 Here, we will present a selection of more recent developments in this field. We note that there are also bis-lactam stapling strategies, and these two-component reactions will be covered in Section 3.3.
Fairlie and co-workers carried out a study of lactamisation between Orn/Lys and Asp/Glu residues on minimal model pentapeptides and hexapeptides.17 Although this was not the first study to optimise the amino acids for lactamisation stapling in short peptides, the authors were able to systematically and quantitatively compare the various possible combinations by circular dichroism to find the shortest possible peptides which are alpha-helical in water (Fig. 5). Remarkably, the most helical pentapeptide remained structurally stable when treated with 8 M guanidinium chloride, and showed superior stability to trypsin proteolysis compared to the uncyclised peptide. Furthermore, the addition of helix-promoting solvent trifluoroethanol did not affect the circular dichroism spectrum, suggesting that a maximum level of helicity was obtained.
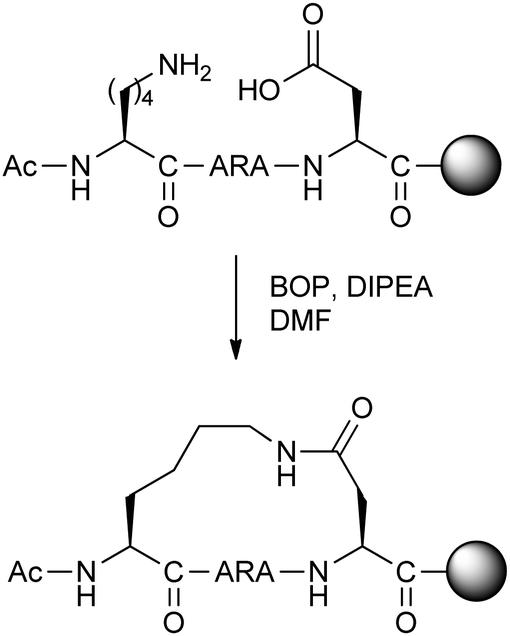 | ||
| Fig. 5 Synthesis of optimal lactam stapled pentapeptide studied by Fairlie and co-workers.17 BOP: (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate, DIPEA: N,N-diisopropylethylamine. | ||
With this minimal i,i + 4 stapling motif in hand, Fairlie and co-workers have since applied this stapling strategy to a number of biological targets. These include the potent inhibition of respiratory syncytial virus fusion by a doubly lactam-stapled peptide, improved antibacterial activity by stapling quorum sensing peptide CSP-1, and enhanced in vitro RNA binding of a lactam-stapled peptide derived from mRNA transport protein HIV Rev.18 Lactam-stapled versions of peptide hormone nociceptin were found to induce greater levels of ERK phosphorylation in cells and thermal analgesia in mice.19 In all the examples mentioned, there was an improvement in helicity as well as biological activity upon lactamisation.
Norton and co-workers have examined various Asp/Lys i,i + 4 combinations for lactam-stapling the helical region of μ-conotoxin KIIIA, a peptide natural product which acts as a potent analgesic in mice by targeting voltage-gated sodium channels (VGSCs).20 The stapled peptides showed different levels of helix stabilisation (not necessarily always the canonical alpha-helix), as well as varied inhibitory activity over different VGSC subtypes in Xenopus laevis oocytes. The optimal stapled peptides showed micromolar activity, although further work is still needed to optimise selectivity for the desired VGSC subtypes.
In terms of synthesis, the proteogenic amino acids for lactam stapling are easier to obtain and incorporate when compared to stapling techniques which require non-proteogenic modified amino acids. However, one counterpoint is that an extra orthogonal protecting group strategy is needed to allow for selective deprotection of the amine and acid functionalities prior to lactamisation. Another caveat is that stapling Lys/Asp residues is only compatible with i,i + 4 spacings, with longer linkages requiring modified amino acids with longer side-chains.
From a biological standpoint, the large body of literature on peptide lactamisation demonstrates that this stapling technique can generate peptide therapeutics with superior bioactivity. Along with alpha-helicity, proteolytic stability is often enhanced, despite the lactam linkage being less biologically inert than the C–C bonds of hydrocarbon stapled peptides for example (although see Section 4 for a comparative study). However, we note that in all the cases discussed above where success was achieved beyond in vitro characterisation, the biological targets are either extracellular or membrane-bound. Compared to hydrocarbon stapling, there is much less evidence in the literature to suggest that lactamisation can improve cell permeability. Without extensive literature directly addressing this issue, it is difficult to judge whether lactam stapled peptides have inherently poorer uptake characteristics, or rather if less attention has been directed towards lactamisation for intracellular targets so far.
In summary, lactamisation is a stapling technique which is at a relatively advanced stage of development. There has been extensive optimisation and application of this technique in many different model systems and biological contexts, although its general applicability for intracellular targets is still to be determined.
2.3 Cycloadditions
The Cu(I)-catalysed azide–alkyne cycloaddition (CuAAC) ‘click’ reaction is a clear candidate for peptide stapling, given its widespread popularity as a biocompatible ligation technique.21 Chorev, D'Ursi and co-workers reported the first instance of generating helical structures using the CuAAC reaction between i,i + 4 residues of a model peptide based on parathyroid hormone-related peptide.22 The stapling reaction was carried out in solution-phase on the fully cleaved and deprotected peptide. Circular dichroism and NMR experiments indicated conformational similarities to an analogous lactam stapled peptide in the literature. Optimisation of side-chain lengths showed that five or six methylene units in the staple loop were able to stabilise alpha-helicity, whilst other lengths led to non-canonical conformations.Applying this methodology to inhibiting the oncogenic interaction of BCL9 with beta-catenin, Wang and co-workers also screened different linker lengths, finding that five methylene units was optimal (Fig. 6).23 Further screening of the triazole position along the staple loop as well as overall staple position on the peptide gave a library of triazole stapled peptides with different helicity and in vitro binding affinity towards beta-catenin. Peptides bearing two triazole staples were also successfully synthesised. For the optimised peptides, there was both a modest improvement in binding affinity in vitro, together with an increased proteolytic stability when incubated in cell media.
 | ||
| Fig. 6 Optimised CuAAC-stapling of BCL9 peptides developed by Wang and co-workers.23 | ||
An alternative cycloaddition, the UV-induced reaction between tetrazoles and alkenes, has been used by Madden et al. to staple peptides for inhibition of the p53-MDM2/MDMX interaction.24 The i,i + 4 stapling reaction was carried out by simply exposing the unprotected linear peptides in solution to UV irradiation (Fig. 7). Upon stapling, the resulting peptides displayed stronger binding towards MDM2 and MDMX in a fluorescence polarisation assay, but were not cell permeable. By modifying various residues to Arg, cellular uptake and modest levels of cellular p53 activation could be achieved.
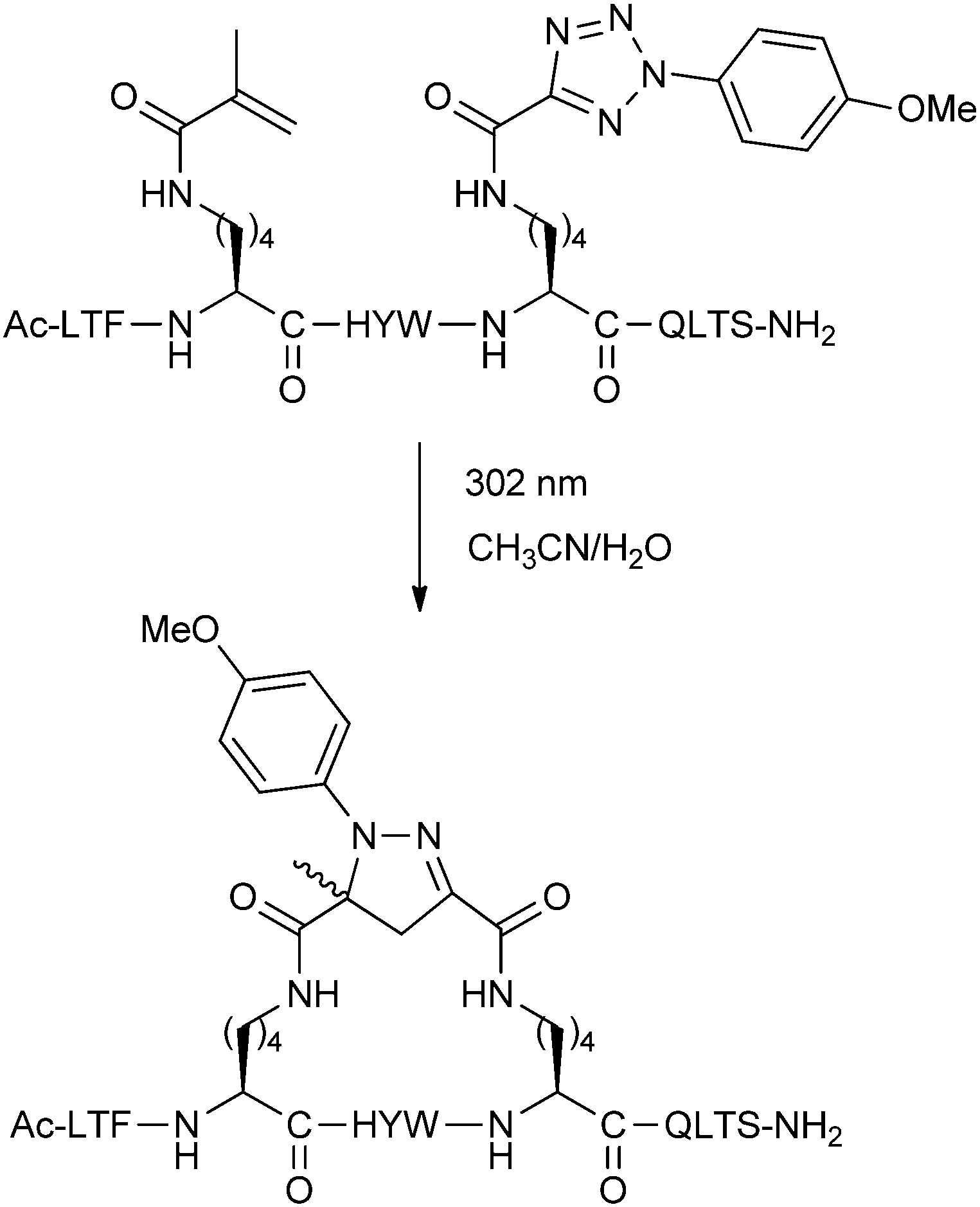 | ||
| Fig. 7 UV-promoted cycloaddition for stapling of p53 peptides. | ||
Overall, stapling peptides with cycloaddition chemistry is an emerging technique which shows great promise. The amino acids required for triazole stapling are readily accessible,25 and the conditions for the intramolecular CuAAC are now established. For the UV stapling protocol, the stapling reaction appears to be simple to execute, although the lysine-derived amino acids required are non-standard. Further studies using these techniques may reveal the extent of their applicability in other biological contexts.
2.4 Reversible reactions
Disulphide bridges between pairs of Cys residues play an important structural role in many proteins. Schultz and co-workers reported an i,i + 7 stapling methodology using disulphide formation between D and L amino acids with thiol-bearing side-chains.26 The amino acids were installed with acetamidomethyl (Acm) protecting groups, and then simultaneously deprotected and oxidised with iodine to give the disulphide stapled peptide (Fig. 8). Circular dichroism spectra of disulphide stapled peptides displayed a high level of alpha-helicity, whilst the Acm-protected precursors were significantly less helical.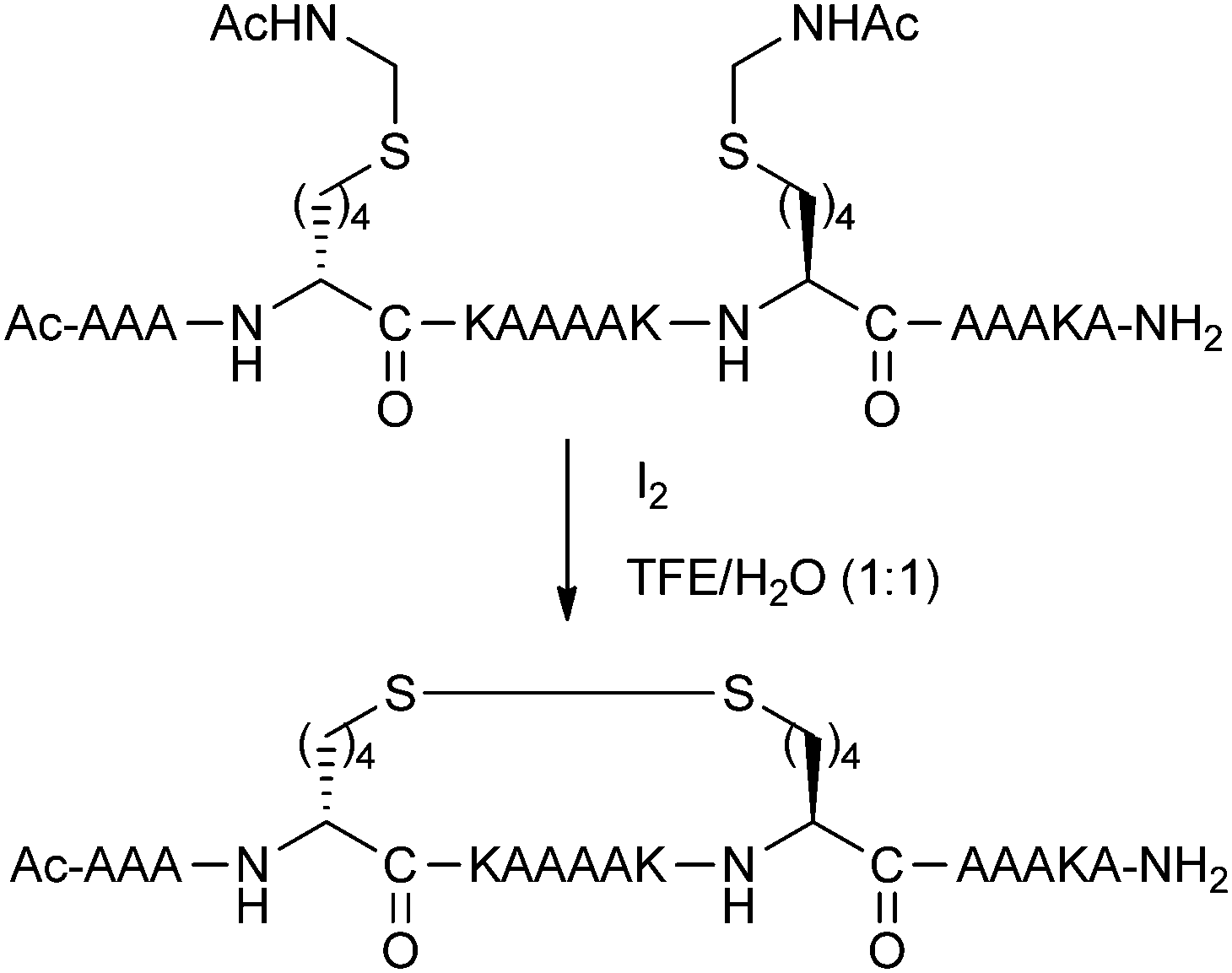 | ||
| Fig. 8 Optimised i,i + 7 disulphide stapling of a model peptide. TFE: 2,2,2-trifluoroethanol. | ||
Although disulphide bridging was one of the first stapling techniques to be reported, there have been relatively few examples reported in the literature since. The major limitation associated with disulphide stapled peptides is their instability in reducing environments, restricting their applicability towards intracellular targets.
A more recent reversible reaction which has featured in peptide stapling is the formation of oxime linkages. Horne and co-workers have developed amino acids bearing aminooxy and 1,2-aminoalcohol groups.27 Upon treatment with periodate, the 1,2-aminoalcohol is oxidatively cleaved to the aldehyde, which can then couple with the aminooxy functionality to give the desired oxime linkage as a mixture of E/Z regioisomers (Fig. 9). These oxime stapled peptides were found to stabilise helicity when placed at i,i + 4 positions. Addition of O-methylhydroxylamine could cause slow exchange between the intramolecular cyclic oxime and the intermolecular linear oxime products. Whilst still in early development, this reversible stapling system may have potential uses in biologically-templated dynamic covalent chemistry. A recent two-component version of this stapling methodology is covered in Section 3.3.
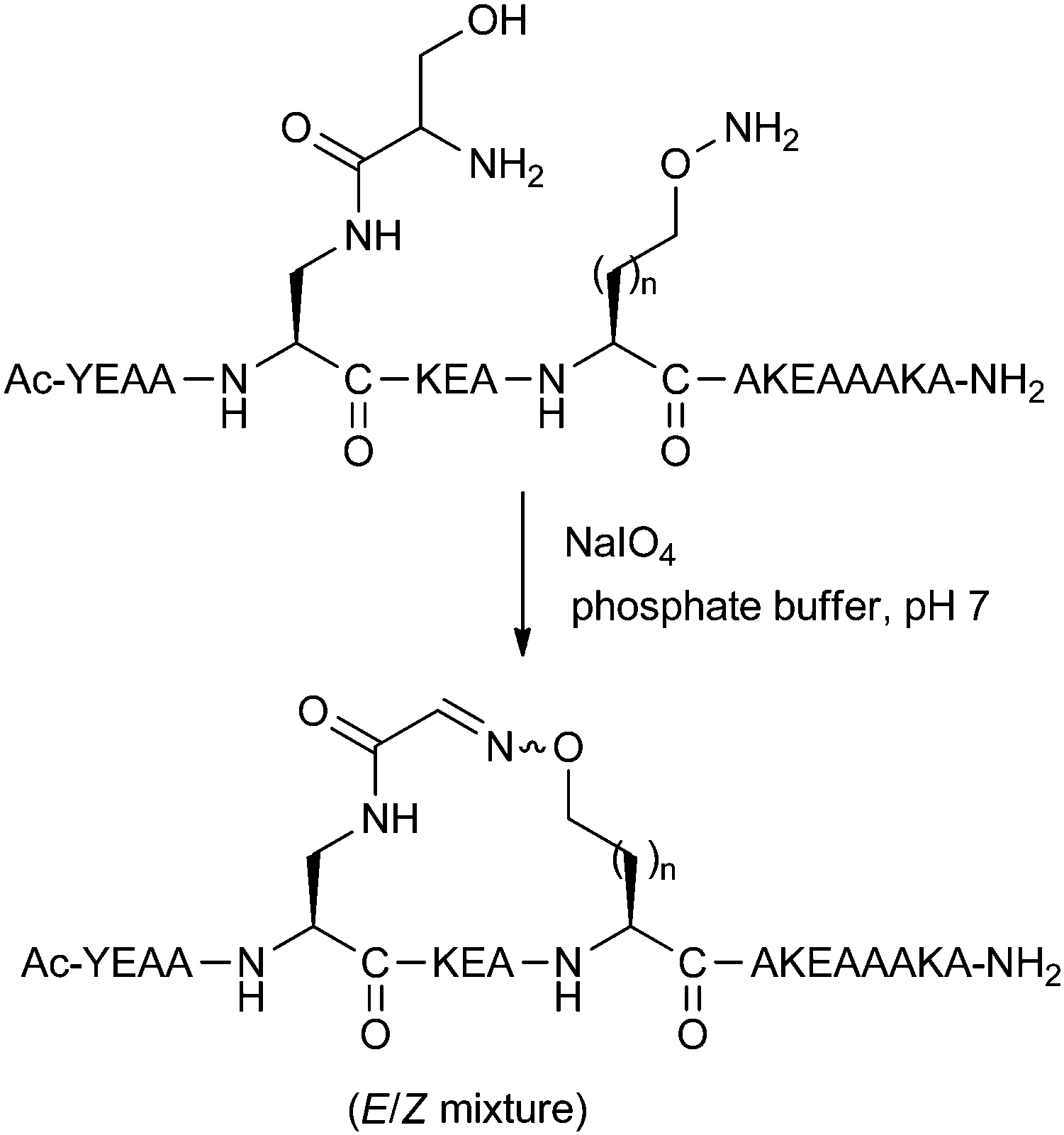 | ||
| Fig. 9 Oxime-stapling of an i,i + 4 model peptide (n = 1, 2). | ||
2.5 Thioether formation
Brunel and Dawson have developed a thioether stapling protocol which uses the reaction between Cys thiol and alpha-bromo amide groups (Fig. 10).28 The stapling reaction was found to proceed in buffered 6 M guanidinium chloride. Designed to mimic the ring size of the previously reported lactam staples, thioether linkages were introduced into peptide epitopes from gp41, as a potential method for developing vaccines against HIV fusion. Both i,i + 3 and i,i + 4 linkages were successfully formed, although circular dichroism data was only shown for the i,i + 3 stapled peptide. This peptide showed increased helicity over its unstapled counterpart, as well as the analogous lactam stapled peptide. After optimisation of the peptide sequence, related thioether stapled peptides were found to bind the gp41-specific antibody 4E10 more effectively than corresponding uncyclised peptides.29 This data suggests that thioether stapling may be more suited than lactam stapling for this shorter i,i + 3 distance in this instance, whilst lactam staples are more appropriate for i,i + 4 stapling. However, thioether stapling is at an early stage of development, and its general applicability still remains to be seen. For further discussion about comparisons between different stapling techniques, we refer the reader to Section 4 of this review.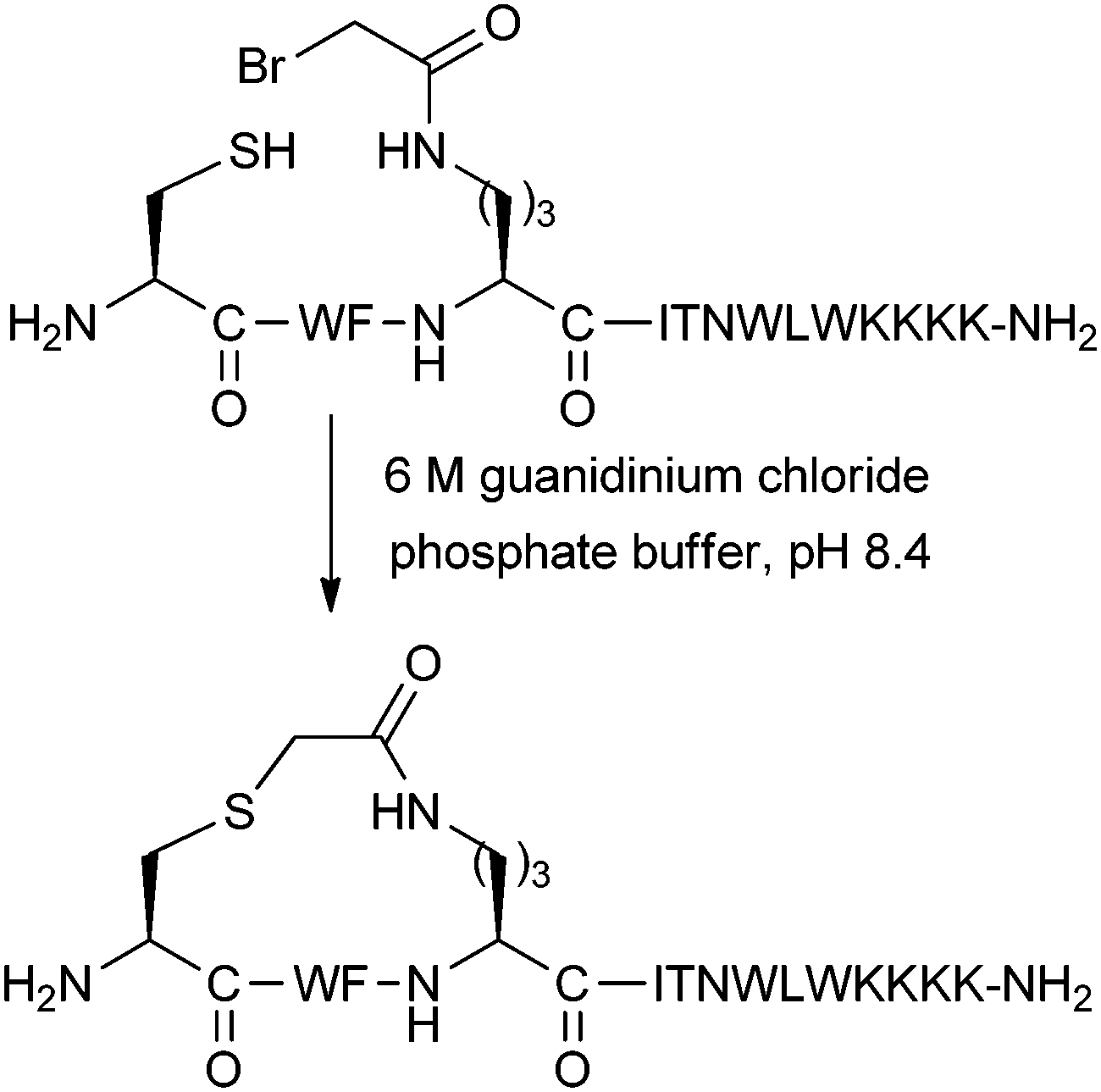 | ||
| Fig. 10 Thioether stapling of an i,i + 3 peptide for inhibiting HIV fusion. | ||
3. Two-component stapling techniques
Two-component stapling involves a bifunctional linker compound which forms a staple by reacting with two complementary non-native amino acids in the peptide of interest. Most two-component reactions are carried out in solution phase due to potential issues of site-isolation on resin. In comparison to one-component stapling, more by-products can potentially be generated in two-component stapling, as the reaction pathway is more complex (Fig. 11).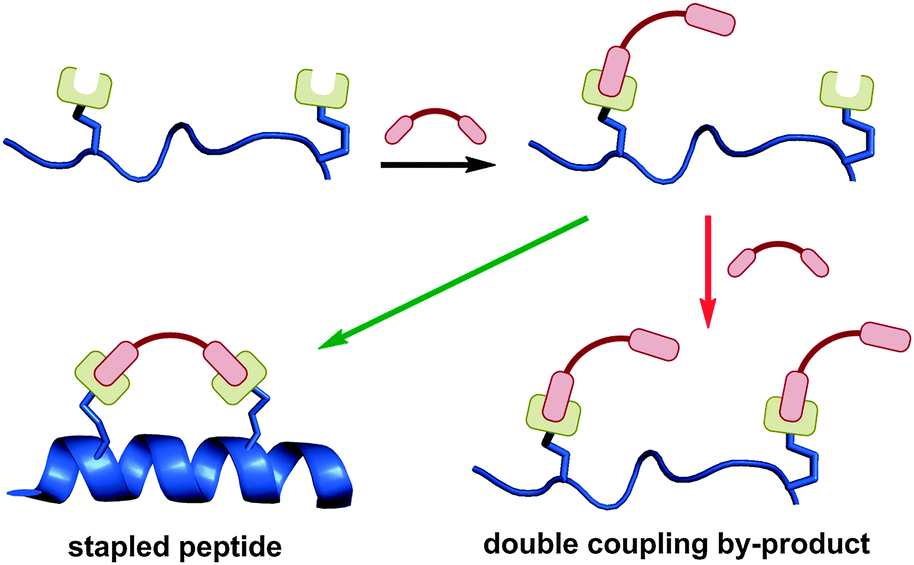 | ||
| Fig. 11 The reaction pathway for two-component stapling involves initial coupling of linker and peptide, followed by intramolecular cyclisation. Introduction of two linker units can lead to a double coupled linear by-product. | ||
The first step is the intermolecular coupling of the linker and peptide to give a linear coupled precursor, followed by a second intramolecular coupling to give the final macrocyclised peptide. A competing pathway is coupling of two linker moieties to a single peptide, one at each non-native amino acid, giving a double coupled linear peptide by-product. This side reaction is not possible in one-component stapling, and exists in addition to other possible oligomerisation side reactions. Despite the theoretical synthetic complexities, there are now numerous effective two-component stapling techniques in the literature. The intramolecular cyclisation may be favoured by conformational pre-organisation of the peptide, positioning the unreacted end of the linker close to the complementary amino acid side-chain.
The major advantage of two-component stapling is the ability to introduce more diverse staple linkages, without needing to synthesise complex non-native amino acids and optimise their incorporation via SPPS. In contrast to the previous section where we categorised peptides in terms of their stapling chemistry, here we have grouped the following examples by their functional features, to highlight the different applications which are possible using two-component stapling chemistry.
3.1 Photoswitchable linkers
The ability to control peptide helicity using UV or visible light is ideal for studying the effects of conformational changes in biological systems. The helicity of stapled peptides can be made switchable by incorporating a photoisomerisable group into a staple linker. Kumita et al. reported the first photoswitchable stapled peptide using an azobenzene-based i,i + 7 linker with 2-iodoacetamide groups for attachment to cysteine residues (Fig. 12).30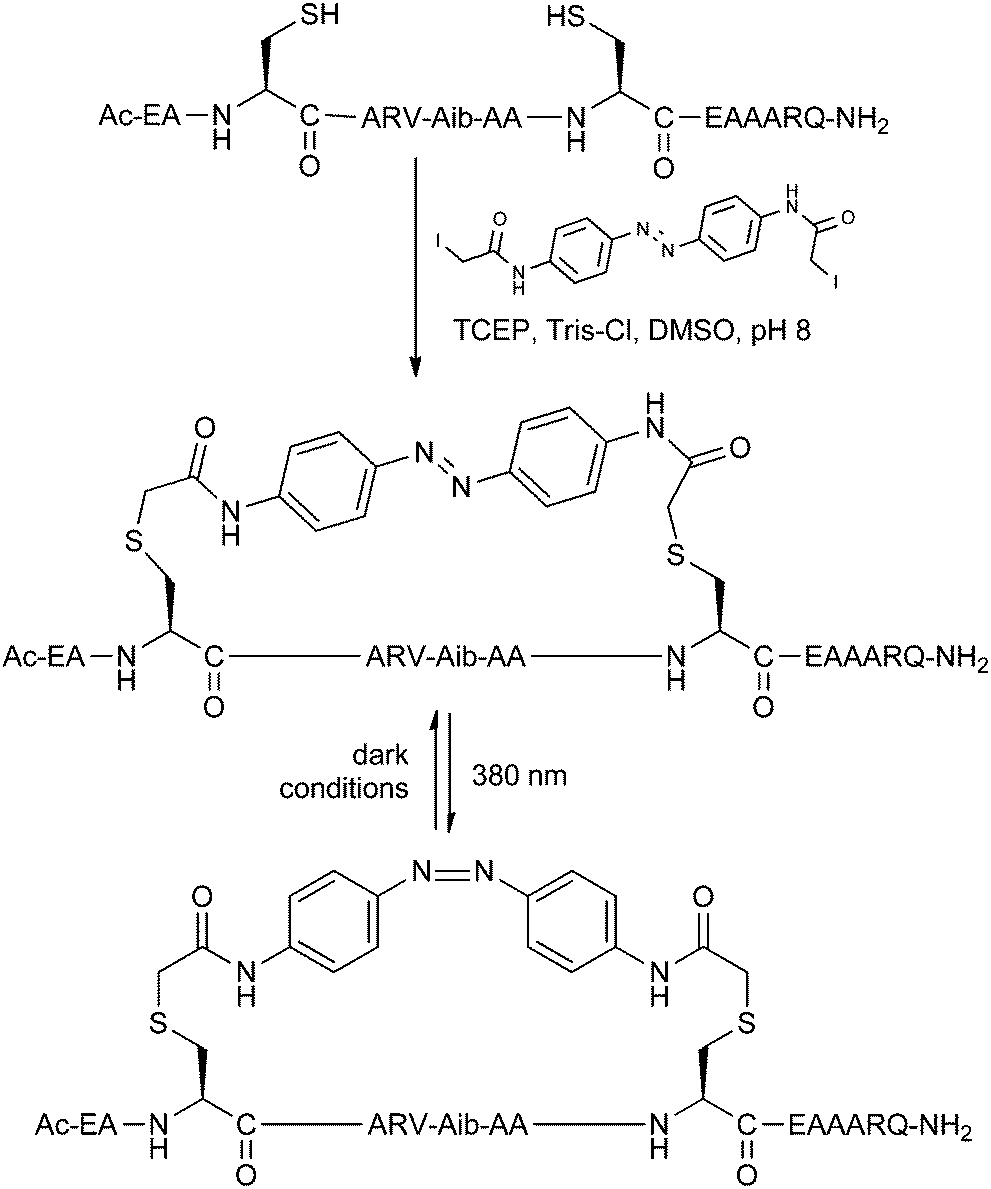 | ||
| Fig. 12 Benzophenone stapling and photoisomerisation of a model peptide. TCEP: tris(2-carboxyethyl)phosphine, Tris: tris(hydroxymethyl)aminomethane. | ||
Starting from a model non-helical trans-azobenzene stapled peptide, the helical cis-azobenzene stapled peptide could be obtained after irradiation at 380 nm for 5 minutes, as determined by circular dichroism. Gradual reversion to the trans form was observed when keeping the peptide in dark conditions over several hours. In follow-up studies, Woolley and co-workers have showed that i,i + 4 stapling also promotes helicity whilst i,i + 11 causes destabilisation with the azobenzene in its cis form.31
Allemann and co-workers have applied i,i + 4/7/11 azobenzene stapling to peptides which bind the oncogenic protein Bcl-xL,32 finding that switching of helicity correlates with the ability of the peptide to bind its protein target in vitro. The biological application of photoswitchable linkers has been further advanced by Nevola et al.,33 who have created stapled peptides for regulating clathrin-mediated endocytosis (CME) by targeting the beta-adaptin subunit of the AP2 complex, a key player in the CME process. The optimised stapled peptides were shown to bind their protein target in vitro using a fluorescence polarisation assay, with the expected reversible changes in affinity upon photoswitching. Furthermore, fluorescently-labelled versions of these peptides were found to be cell-permeable. Using the stapled peptides in cellular studies, endocytosis of transferrin receptor protein could be controlled by switching of the azobenzene linker, as determined by measuring the cellular uptake of labelled transferrin. Finally, microscopy on labelled clathrin-coated pits provided direct evidence of switchable control over the rate of CME.
It is notable that the two-component synthetic approach allows for facile introduction of the azobenzene linkage onto the peptide. Using a one-component strategy, one could envisage synthetic difficulties associated with introducing the azobenzene during SPPS.
With azobenzene stapling proving useful in several biological settings, there is now potential to apply this switchable system to other biological targets. At the same time, other photoisomerisable groups may be used to create new peptide linkers with different optical switching properties. One such example is the spyropyran linkers developed by Fujimoto et al.,34 although this system could benefit from further development to achieve a greater window of helix switchability and biological applicability.
3.2 Functionalised double-click linkers
Whilst one-component triazole stapled peptides were described in Section 2.3, the CuAAC ‘click’ reaction has also been used in two-component stapling systems. Bong and co-workers incorporated azidoalanine residues at i,i + 4 positions in a peptide derived from the GCN4 leucine zipper.35 The peptide was chosen with two glycine residues in the middle of the sequence to impair the native helix dimer structure of the native peptide. After trying several dialkynes, reaction of the peptide with 1,5-hexadiyne either on-resin or in solution phase gave the bis-triazole stapled peptide, restoring helicity and dimerisation as determined by circular dichroism and analytical ultracentrifugation.In the Spring group, we developed an i,i + 7 double-click stapling technique, applied to the inhibition of the p53-MDM2 interaction.36,37 The aim of our investigations was to exploit the two-component nature of the double-click reaction by stapling a single peptide with different functionalised dialkynyl linkers (Fig. 13). This approach enables efficient synthetic access to a library of peptides bearing staples with different properties.
 | ||
| Fig. 13 Double-click stapling of a p53 peptide with differently functionalised linkers (unfunctionalised and cationic arginine-functionalised staple shown). THPTA: tris(3-hydroxypropyltriazolylmethyl)amine. | ||
After introducing two azidoornithine residues into a p53-based peptide, the solution-phase double-click reaction was initially carried out with 3,5-diethynylbenzene, giving peak-to-peak conversion to the desired stapled peptide without any protecting groups. This peptide was found to bind with nanomolar affinity to MDM2 by fluorescence polarisation and isothermal calorimetry, representing an improvement over the corresponding wild-type and unstapled peptides. However, the stapled peptide showed poor uptake and was not active in a cellular p53 reporter assay. Harnessing the two-component nature of this stapling technique, modified dialkynyl linkers bearing cationic arginine groups were stapled onto the same p53 peptide. By increasing the net positive charge via the staple linker, cellular uptake and p53 activation was achieved.
The success of this double-click stapling methodology relies on the ability to introduce many staples to a single peptide in a two-component reaction. Furthermore, the bioorthogonal nature of the CuAAC reaction allows staples to be introduced in a single step, without need for synthetic optimisation and use of protecting groups. There are theoretically many other potential staples and applications for double-click stapling which may expand the utility of this technique, with the caveat that in each case, the bis-triazole motif cannot be avoided. The impact of this motif on biological activity, and the question of whether triazoles can participate in target binding still require further investigation. In the following section, we discuss related stapling techniques which utilise alternative chemistry to also access to different staples in a divergent manner.
3.3 Staple screening systems
The large number of two-component stapling techniques utilise alkylation or arylation chemistry on Cys/Lys residues (such as azobenzene stapling in Section 3.1), or acylation chemistry on Asp/Glu residues. Here we first discuss two notable examples, one early and one recent, to showcase advances in this field (Fig. 14). We then focus on studies which take full advantage of the two-component setup by screening different stapling linkers.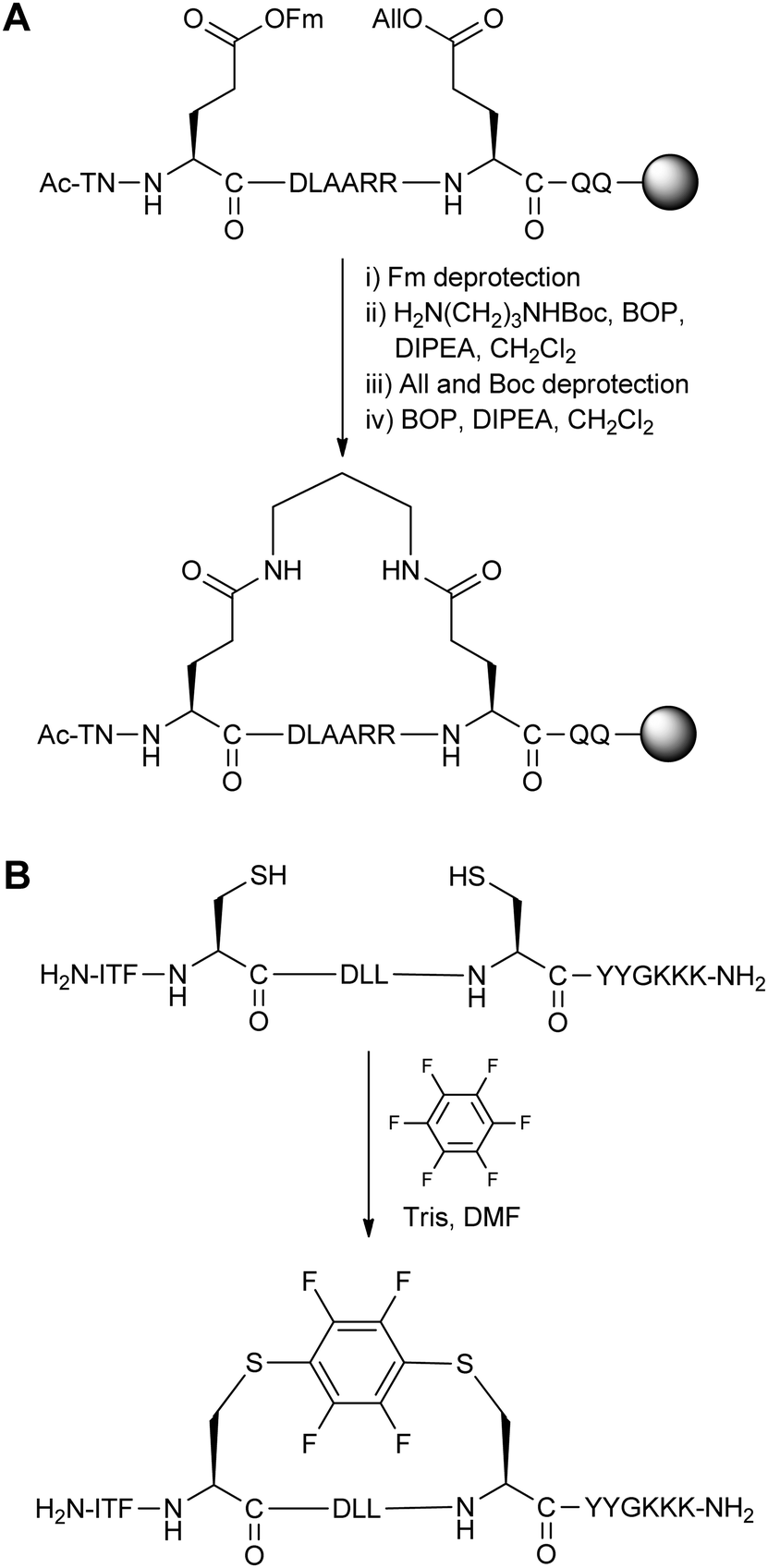 | ||
| Fig. 14 A comparison of early and recent two-component stapling techniques. (A) Bis-lactam stapled peptide developed by Phelan and co-workers.38 (B) Bis-arylation stapling developed by Pentelute and co-workers.39 | ||
An early two-component bis-lactam stapling technique was reported by Phelan and co-workers.38 The stapling reaction involved two i,i + 7 Glu residues coupling with a diaminoalkane. As direct bis-lactamisation resulted in poor yields and purity, the authors chose to use 9-fluorenylmethyl and allyl ester protecting groups on the Glu residues and a mono-Boc-protected diaminoalkane to install the staple linkage in a stepwise manner (Fig. 14A). NMR and circular dichroism analysis revealed that four or five methylene units in the diaminoalkane linkage were optimal when stapling two different peptide sequences (derived from apamin and the C-peptide of RNAse A). This bis-lactam stapling methodology was found to be superior to the analogous disulphide stapled peptides (as described in Section 2.4) in terms of helix stabilisation.
Pentelute and co-workers recently developed an SNAr-based stapling technique for the bis-arylation of i,i + 4 Cys residues.39 Peptide stapling was carried out by combining hexafluorobenzene and unprotected peptide in the presence of Tris base and DMF (Fig. 14B). When applied to a peptide binder of the HIV-1 capsid assembly polyprotein, there were improvements in helicity, proteolytic stability and in vitro binding affinity, as well as cellular uptake by confocal microscopy. Compared to the previous bis-lactam example, this recent work demonstrates the improvements over the last 15 years in both the synthesis and the biological analysis of stapled peptides.
Woolley and co-workers have used a two-component strategy to study the effects of linker flexibility on stapled peptide helicity.40 Three linkers were chosen for i,i + 11 Cys stapling, each with similar extended length but different degrees of flexibility as determined by Langevin dynamics simulations. When stapled to a model peptide, there was a correlation between linker flexibility and peptide helicity by circular dichroism.
A larger scale screen of different stapling linkers was done by Fujimoto et al.,41 investigating twelve different rigid disuccinimidyl esters for coupling to amino acids bearing alkylamine side-chains (Fig. 15A). Model systems for i,i + 4/7/11 stapling gave a large library of peptides, all of which were analysed by circular dichroism to find the optimal linker-peptide combination for maximal helicity.
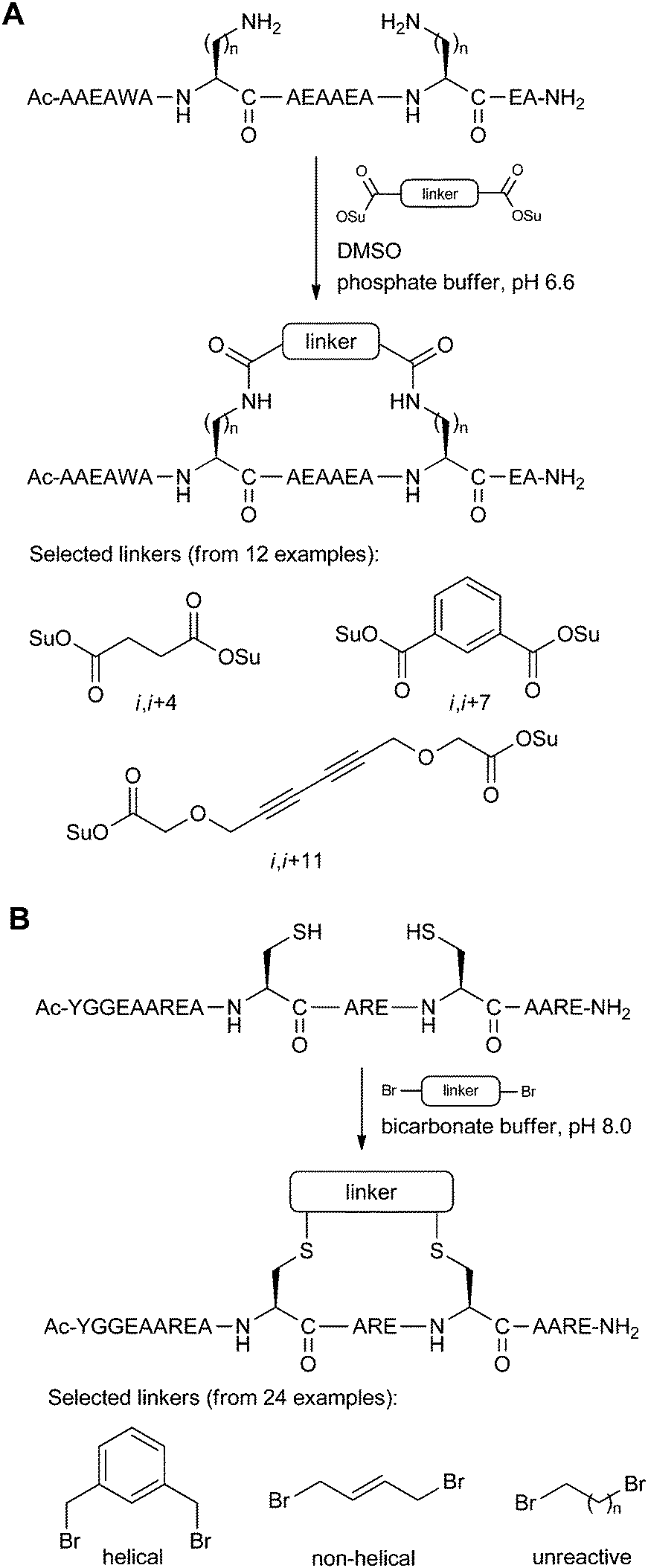 | ||
| Fig. 15 Staple screening systems using a library of linkers. (A) Model peptides (n = 1–4) by Fujimoto et al. which were screened with 12 linkers. Only the i,i + 7 peptide is shown here.41 (B) Model peptide by Jo et al. which was screened with 24 linkers.42 | ||
A similar staple screen was reported by Jo et al.,42 in this case using a variety of bis-electrophilic systems to react with i,i + 4 Cys residues on a model peptide (Fig. 15B). The logic behind the screen was to determine which linkers would react efficiently to give the cleanest macrocyclisation reactions, as this indicates the peptide-linker combinations which were the most compatible for low-strain intramolecular cyclisation. The optimised stapling system was then applied to the inhibition of calpain, a calcium-dependent cysteine protease, as well as the development of an activity-based probe using the stapled peptide motif.
Extending the oxime stapling technique described in Section 2.4, Haney and Horne have developed a two-component version of their methodology.43 Peptides bearing either two aminooxy or hydrazide amino acids with i,i + 4 or 11 spacing were reacted with a small collection of dialdehydes to give the bis-oxime and bis-hydrazone stapled peptides respectively. At this stage, only circular dichroism studies have been carried out, although this technique is closer to realising a dynamic covalent approach for selecting helical peptide inhibitors.
In all the examples of staple screening mentioned in this section so far, the goal of the screening exercise has been to obtain optimal linkers for helicity, using the staple as a distance matching scaffold. Recent work by Lin and co-workers extends staple screening to studying cell permeability, but in a different manner to that previously described for double-click stapled peptides in Section 3.2.44 This work follows an earlier report describing the synthesis and optimisation of inhibitors for Mcl-1, a BCL-2 family protein.45 The stapling technique involves alkylation of i,i + 7 Cys residues using a bis-bromide linker. Whilst modifications in the peptide sequence were carried out to optimise cellular uptake in the earlier work, this work describes eight different linkers for stapling a single dicysteinyl peptide. The authors found no correlation between in vitro binding affinity and cell permeability, with uptake appearing to correlate with hydrophobicity instead. This study highlights the importance of considering factors other than helicity or binding affinity when optimising stapled peptides.
The two-component stapling techniques discussed in this section exemplify the diversity of synthetic approaches available. Standard proteogenic amino acids are utilised in most cases, increasing the synthetic accessibility of these techniques. At the same time, this also increases the chances of cross-reactivity with other unprotected functional groups, either elsewhere on the peptide or on the staple linkage itself. Indeed, most of the staple linkages described in this section do not have any complex functionality (apart from the group involved in the stapling reaction itself), in contrast with the double-click stapling technique covered in Section 3.2. Nevertheless, the growing number of two-component staple screening techniques is giving rise to a rich collection of different staple linkages, each with potentially different physicochemical properties.
4. Comparative studies
A striking feature of the stapled peptide literature to date is how few studies utilise different stapling techniques when developing inhibitors for a given PPI target. This fact may arise from the synthetic challenge of optimising several different stapling chemistries for a particular peptide. The following examples represent the current state of comparative studies on different stapling techniques.Leduc et al. have investigated peptides based on the LXXLL nuclear receptor box motif as selective oestrogen receptor α binders.46 The stapling techniques compared in this study were i,i + 4 lactamisation between Glu/Lys and i,i + 3 disulphide stapling between D/L-Cys. Whilst neither technique gave helical peptides in aqueous solution by circular dichroism, the addition of trifluoroethanol induced helicity for the disulphide stapled peptide. The disulphide stapled peptide also displayed higher affinity binding to oestrogen receptor α, with a quasihelical conformation upon binding as determined by X-ray crystallography.
A comparison of i,i + 4 lactam and hydrocarbon stapled peptides was reported by Giordanetto and co-workers, working on agonists of vasoactive intestinal peptide receptor 2 (VPAC2).47 Using both techniques at a variety of different staple positions, the stapled peptides all showed superior helicity over the wild type peptide. The lactam stapled peptides also showed an improvement over the unstapled versions, whilst both the unstapled and stapled hydrocarbon peptides were significantly helical, indicating the importance of the α,α-disubstituted amino acids in this case. The stapled peptides were also more potent agonists of VPAC2 than the unstapled peptides, with the most potent candidates having a higher affinity than the wild type peptide. Interestingly, there was a wide spread of EC50 values, despite the helicity being similar in every case. The most potent lactam and hydrocarbon stapled peptides also were superior in an insulin secretion assay in pancreatic rat islets. Despite these promising results, the stapled peptides were found to have poor resistance to trypsin proteolysis over 15 minutes, although shorter times were not investigated. Furthermore, the peptides were not cell-permeable by passive diffusion as determined by a Caco-2 monolayer assay. This result however is not unprecedented, as it is thought that stapled peptides generally enter cells by active uptake through endocytosis.
Finally, two independent studies have recently explored direct comparisons over a broader range of stapling techniques, although only assessed in terms of their alpha-helicity in model systems. Frankiewicz et al. used a model peptide derived from vasoactive intestinal peptide to compare lactamisation of Lys/Asp residues, metathesis of O-allylserine residues (before and after hydrogenation) and one-component CuAAC stapling, all with an i,i + 4 spacing.48 Only the triazole stapled peptide was found to be helical by circular dichroism, and in addition, helicity was only observed upon addition of trifluoroethanol.
A more comprehensive study was conducted by Fairlie and co-workers (Fig. 16), using their minimal pentapeptide system previously described in Section 2.2.49 This study compared Lys/Asp lactamisation, hydrocarbon stapling, CuAAC stapling, bis-thioether stapling (seen in the calpain inhibitors in Section 3.3), perfluorobenzene stapling (also in Section 3.3) and regular thioether stapling (Section 2.4). Circular dichroism indicated the greatest amount of helicity was induced by lactamisation, CuAAC and hydrocarbon stapling. NMR studies then revealed that lactamisation gave the greatest alpha-helicity, whilst the peptides stapled using the other two techniques had some degree of 310-helicity.
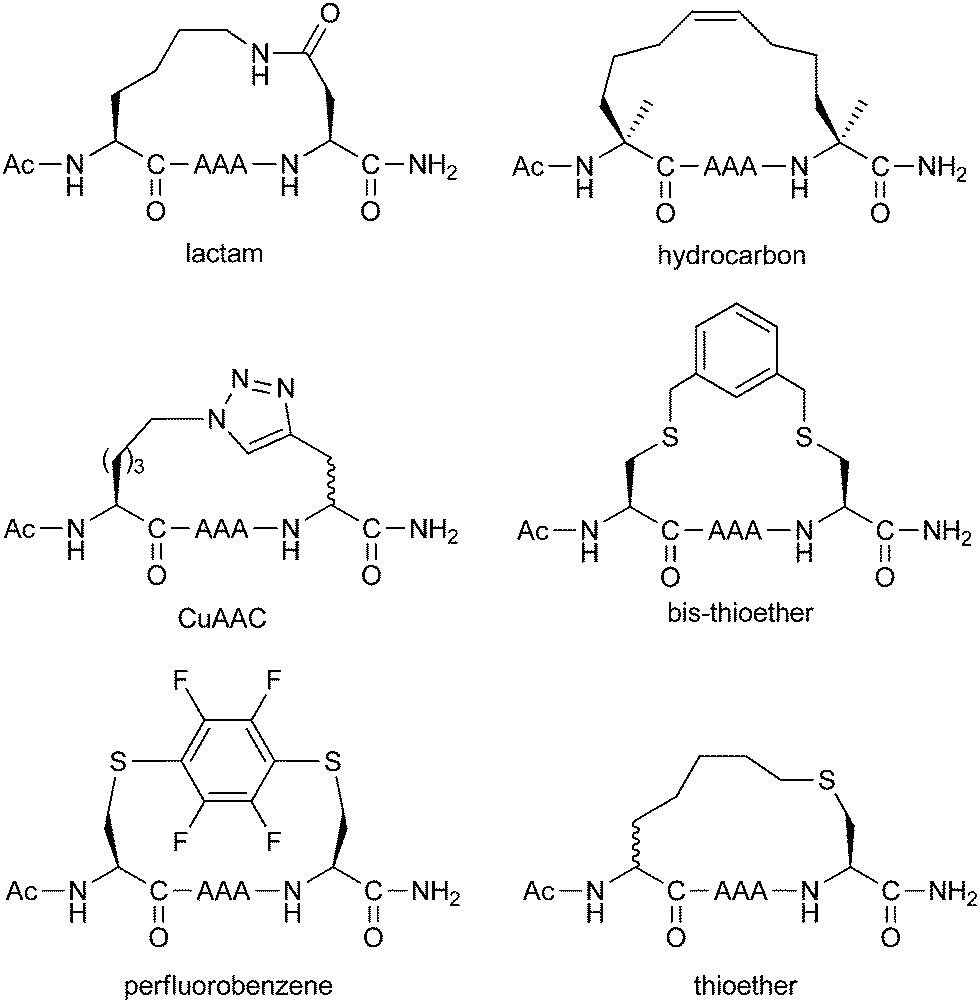 | ||
| Fig. 16 Comparison of different stapling chemistries on model pentapeptides. | ||
5. Conclusions and future directions
From the examples discussed in this review, it is clear that there is no single optimal stapling strategy for all cases, and that choice of stapling technique depends both on the biological questions being investigated and the individual nature of the PPI being studied. Hydrocarbon stapled peptides are currently leading the way for intracellular PPI targets, whilst there is significant literature precedence for lactam stapled peptides against extracellular and membrane-bound targets. At the same time, alternative stapling techniques are being developed and have yet to realise their full potential.A major challenge for the future is to engineer stapled peptides in favour of in vivo efficacy, rather than simply helicity or binding affinity. In particular, the factors governing cell permeability is a vital issue which has yet to be resolved. Another important consideration is the multifaceted role of the staple linkage itself. The staple can no longer be considered an inert structural motif, as it is known to play a critical role in the overall physicochemical properties of stapled peptides. Development of two-component stapling techniques has helped to draw attention to the staple as a customisable element in the overall peptide design.
As this field matures and stapling techniques become more well-documented, we expect to see more studies taking advantage of different stapling techniques in the near future. This diverse approach will maximise the chances of establishing effective peptide-based therapeutics for a variety of PPI targets.
Acknowledgements
This work was supported by the European Union, Engineering and Physical Sciences Research Council, Biotechnology and Biological Sciences Research Council, Medical Research Council and Wellcome Trust. YHL acknowledges a scholarship from the Cambridge Trusts. PDA thanks the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).Notes and references
- B. N. Bullock, A. L. Jochim and P. S. Arora, J. Am. Chem. Soc., 2011, 133, 14220–14223 CrossRef CAS PubMed.
- A. M. Felix, E. P. Heimer, C.-T. Wang, T. J. Lambros, A. Fournier, T. F. Mowles, S. Maines, R. M. Campbell, B. B. Wegrzynski, V. Toome, D. Fry and V. S. Madison, Int. J. Pept. Protein Res., 1988, 32, 441–454 CrossRef CAS PubMed.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281–3284 CrossRef CAS.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- V. Haridas, Eur. J. Org. Chem., 2009, 5112–5128 CrossRef CAS.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K.-H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445–E3454 CrossRef CAS PubMed.
- T. L. Joseph, D. Lane and C. S. Verma, Cell Cycle, 2010, 9, 4560–4568 CrossRef CAS.
- T. Okamoto, K. Zobel, A. Fedorova, C. Quan, H. Yang, W. J. Fairbrother, D. C. S. Huang, B. J. Smith, K. Deshayes and P. E. Czabotar, ACS Chem. Biol., 2012, 8, 297–302 CrossRef PubMed.
- C. J. Brown, S. T. Quah, J. Jong, A. M. Goh, P. C. Chiam, K. H. Khoo, M. L. Choong, M. A. Lee, L. Yurlova, K. Zolghadr, T. L. Joseph, C. S. Verma and D. P. Lane, ACS Chem. Biol., 2012, 8, 506–512 CrossRef PubMed.
- F. Bernal, A. F. Tyler, S. J. Korsmeyer, L. D. Walensky and G. L. Verdine, J. Am. Chem. Soc., 2007, 129, 2456–2457 CrossRef CAS PubMed.
- D. J. Yeo, S. L. Warriner and A. J. Wilson, Chem. Commun., 2013, 49, 9131–9133 RSC.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- J. W. Taylor, Pept. Sci., 2002, 66, 49–75 CrossRef CAS PubMed.
- N. E. Shepherd, H. N. Hoang, G. Abbenante and D. P. Fairlie, J. Am. Chem. Soc., 2005, 127, 2974–2983 CrossRef CAS PubMed.
- R. S. Harrison, N. E. Shepherd, H. N. Hoang, G. Ruiz-Gómez, T. A. Hill, R. W. Driver, V. S. Desai, P. R. Young, G. Abbenante and D. P. Fairlie, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11686–11691 CrossRef CAS PubMed.
- R. S. Harrison, G. Ruiz-Gómez, T. A. Hill, S. Y. Chow, N. E. Shepherd, R.-J. Lohman, G. Abbenante, H. N. Hoang and D. P. Fairlie, J. Med. Chem., 2010, 53, 8400–8408 CrossRef CAS PubMed.
- K. K. Khoo, M. J. Wilson, B. J. Smith, M.-M. Zhang, J. Gulyas, D. Yoshikami, J. E. Rivier, G. Bulaj and R. S. Norton, J. Med. Chem., 2011, 54, 7558–7566 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- M. Scrima, A. Le Chevalier-Isaad, P. Rovero, A. M. Papini, M. Chorev and A. M. D'Ursi, Eur. J. Org. Chem., 2010, 446–457 CrossRef CAS.
- S. A. Kawamoto, A. Coleska, X. Ran, H. Yi, C.-Y. Yang and S. Wang, J. Med. Chem., 2011, 55, 1137–1146 CrossRef PubMed.
- M. M. Madden, A. Muppidi, Z. Li, X. Li, J. Chen and Q. Lin, Bioorg. Med. Chem. Lett., 2011, 21, 1472–1475 CrossRef CAS PubMed.
- Y. H. Lau and D. R. Spring, Synlett, 2011, 1917–1919 CAS.
- D. Y. Jackson, D. S. King, J. Chmielewski, S. Singh and P. G. Schultz, J. Am. Chem. Soc., 1991, 113, 9391–9392 CrossRef CAS.
- C. M. Haney, M. T. Loch and W. S. Horne, Chem. Commun., 2011, 47, 10915–10917 RSC.
- F. M. Brunel and P. E. Dawson, Chem. Commun., 2005, 2552–2554 RSC.
- F. M. Brunel, M. B. Zwick, R. M. F. Cardoso, J. D. Nelson, I. A. Wilson, D. R. Burton and P. E. Dawson, J. Virol., 2006, 80, 1680–1687 CrossRef CAS PubMed.
- J. R. Kumita, O. S. Smart and G. A. Woolley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3803–3808 CrossRef CAS.
- D. G. Flint, J. R. Kumita, O. S. Smart and G. A. Woolley, Chem. Biol., 2002, 9, 391–397 CrossRef CAS.
- S. Kneissl, E. J. Loveridge, C. Williams, M. P. Crump and R. K. Allemann, ChemBioChem, 2008, 9, 3046–3054 CrossRef CAS PubMed.
- L. Nevola, A. Martín-Quirós, K. Eckelt, N. Camarero, S. Tosi, A. Llobet, E. Giralt and P. Gorostiza, Angew. Chem., Int. Ed., 2013, 52, 7704–7708 CrossRef CAS PubMed.
- K. Fujimoto, M. Amano, Y. Horibe and M. Inouye, Org. Lett., 2006, 8, 285–287 CrossRef CAS PubMed.
- O. Torres, D. Yüksel, M. Bernardina, K. Kumar and D. Bong, ChemBioChem, 2008, 9, 1701–1705 CAS.
- Y. H. Lau, P. de Andrade, S.-T. Quah, M. Rossmann, L. Laraia, N. Skold, T. J. Sum, P. J. E. Rowling, T. L. Joseph, C. Verma, M. Hyvonen, L. S. Itzhaki, A. R. Venkitaraman, C. J. Brown, D. P. Lane and D. R. Spring, Chem. Sci., 2014, 5, 1804–1809 RSC.
- Y. H. Lau, P. de Andrade, N. Skold, G. J. McKenzie, A. R. Venkitaraman, C. Verma, D. P. Lane and D. R. Spring, Org. Biomol. Chem., 2014, 12, 4074–4077 CAS.
- J. C. Phelan, N. J. Skelton, A. C. Braisted and R. S. McDowell, J. Am. Chem. Soc., 1997, 119, 455–460 CrossRef CAS.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- F. Zhang, O. Sadovski, S. J. Xin and G. A. Woolley, J. Am. Chem. Soc., 2007, 129, 14154–14155 CrossRef CAS PubMed.
- K. Fujimoto, M. Kajino and M. Inouye, Chem. – Eur. J., 2008, 14, 857–863 CrossRef CAS PubMed.
- H. Jo, N. Meinhardt, Y. Wu, S. Kulkarni, X. Hu, K. E. Low, P. L. Davies, W. F. DeGrado and D. C. Greenbaum, J. Am. Chem. Soc., 2012, 134, 17704–17713 CrossRef CAS PubMed.
- C. M. Haney and W. S. Horne, J. Pept. Sci., 2014, 20, 108–114 CrossRef CAS PubMed.
- A. Muppidi, K. Doi, C. P. Ramil, H.-G. Wang and Q. Lin, Tetrahedron, 2014 DOI:10.1016/j.tet.2014.05.104.
- A. Muppidi, K. Doi, S. Edwardraja, E. J. Drake, A. M. Gulick, H.-G. Wang and Q. Lin, J. Am. Chem. Soc., 2012, 134, 14734–14737 CrossRef CAS PubMed.
- A.-M. Leduc, J. O. Trent, J. L. Wittliff, K. S. Bramlett, S. L. Briggs, N. Y. Chirgadze, Y. Wang, T. P. Burris and A. F. Spatola, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11273–11278 CrossRef CAS PubMed.
- F. Giordanetto, J. D. Revell, L. Knerr, M. Hostettler, A. Paunovic, C. Priest, A. Janefeldt and A. Gill, ACS Med. Chem. Lett., 2013, 4, 1163–1168 CrossRef CAS PubMed.
- L. Frankiewicz, C. Betti, K. Guillemyn, D. Tourwé, Y. Jacquot and S. Ballet, J. Pept. Sci., 2013, 19, 423–432 CrossRef CAS PubMed.
- A. D. de Araujo, H. N. Hoang, W. M. Kok, F. Diness, P. Gupta, T. A. Hill, R. W. Driver, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 6965–6969 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |