
Strategies for interfacing inorganic nanocrystals with biological systems based on polymer-coating
Goutam
Palui†
,
Fadi
Aldeek†
,
Wentao
Wang
and
Hedi
Mattoussi
Florida State University, Department of Chemistry and Biochemistry, 95 Chieftan Way, Tallahassee, Florida 32306, USA
First published on 16th July 2014
Abstract
Interfacing inorganic nanoparticles and biological systems with the aim of developing novel imaging and sensing platforms has generated great interest and much activity. However, the effectiveness of this approach hinges on the ability of the surface ligands to promote water-dispersion of the nanoparticles with long term colloidal stability in buffer media. These surface ligands protect the nanostructures from the harsh biological environment, while allowing coupling to target molecules, which can be biological in nature (e.g., proteins and peptides) or exhibit specific photo-physical characteristics (e.g., a dye or a redox-active molecule). Amphiphilic block polymers have provided researchers with versatile molecular platforms with tunable size, composition and chemical properties. Hence, several groups have developed a wide range of polymers as ligands or micelle capsules to promote the transfer of a variety of inorganic nanomaterials to buffer media (including magnetic nanoparticles and semiconductor nanocrystals) and render them biocompatible. In this review, we first summarize the established synthetic routes to grow high quality nanocrystals of semiconductors, metals and metal oxides. We then provide a critical evaluation of the recent developments in the design, optimization and use of various amphiphilic copolymers to surface functionalize the above nanocrystals, along with the strategies used to conjugate them to target biomolecules. We finally conclude by providing a summary of the most promising applications of these polymer-coated inorganic platforms in sensor design, and imaging of cells and tissues.
 Goutam Palui | Dr Goutam Palui received his BSc and MSc degrees in Chemistry from Jadavpur University before completing his PhD from the Indian Association for the Cultivation of Science (IACS) in 2008 under the supervision of Dr Arindam Banerjee, both in Kolkata, India. During his doctoral research he worked on materials self-assemblies using synthetic short peptides and pseudo-peptides. Currently he is a postdoctoral research fellow at Florida State University, working with Professor Mattoussi, on the design and development of functional water-soluble nanomaterials (metallic and semiconductor QDs) and their applications in imaging and sensing. |
 Fadi Aldeek | Dr Fadi Aldeek is an organic/inorganic chemist. He received his bachelor's and master's degrees in Molecular and Supramolecular Chemistry from Louis Pasteur University, Strasbourg, France. He then pursued his postgraduate study at Henri Poincare University, Nancy, France, earning a PhD in Materials Science in 2010. He is currently working as a Postdoctoral Associate at the Florida State University. His research interest focuses on developing the technology of chemically synthesized inorganic nanocrystals. This includes challenges in making new compositions of nanocrystals and multifunctional ligands, with the ultimate goal of incorporating the nanocrystals into hybrid organic–inorganic devices and biological systems. |
 Wentao Wang | Wentao Wang received a bachelor's degree in Physical/Organic Chemistry from Jilin University, China, in 2009. He then joined the Florida State University in the fall of 2011. He has been a PhD candidate under the supervision of Prof. Hedi Mattoussi, in the Department of Chemistry and Biochemistry since January 2012. His research focuses on the design of multidentate and multifunctional polymer ligands readily adaptable to various metal-rich surfaces, including QDs, metal and metal oxide nanocrystals. He is also developing novel approaches for surface modification of inorganic nanocrystals and the development of biological sensing and imaging using these optimally designed inorganic nanoprobes. |
 Hedi Mattoussi | Hedi Mattoussi has been a professor at the Florida State University since August 2009. Prior to that, he spent 12 years working as a senior Scientist at the Naval Research Laboratory (Washington). He received a bachelor's degree in Physics from the Faculty of Sciences in Tunis and a PhD in Condensed Matter Physics from the University of Pierre & Marie Curie in 1987. In 1994, he received a Habilitation to direct Research, Materials Physics, also from Paris. He presently focuses on interfacing inorganic nanoparticles with biological systems using chemical and photochemical means, to develop novel tools for imaging, sensing and diagnostics. |
1. Introduction
Due to their unique physical, chemical, electronic, and optical properties nanostructures made of metals, metal oxides and metal chalcogenides have attracted a great deal of interest and much activity in the past two decades.1–5 This has been motivated by the great promise they offer for use in numerous applications, ranging from developing optical and electronic devices to cellular imaging and biological sensing.6–9 For example, semiconductor nanocrystals (quantum dots, QDs) exhibit size- and composition-tunable broad absorption along with narrow and symmetric emission spectra; they also exhibit a remarkable photo- and chemical-stability compared to organic dyes and fluorescent proteins.3,10 Similarly, gold and silver nanoparticles (AuNPs and AgNPs) exhibit size- and shape-dependent Surface Plasmon Resonance (SPR) bands ranging from the UV to the near infrared (NIR) region of the optical spectrum.9,11,12 Nanostructures made of other transition metals, such as Fe3O4, Mn-doped Fe3O4, FePt, Ni, and Co show strong size- and composition-dependent coercivity.13–16 These unique features made them very promising to design platforms that can be applied in biology, including imaging, sensing and as diagnostic tools.17–20Aqueous phase synthesis is in principle the simplest method to prepare water dispersible nanocrystals.21 For instance, one of the earlier methods to use this route to grow QDs, including CdTe, CdSe and CdS, involved mixing of cadmium precursors in the presence of a suitable stabilizer (e.g., thioalkyl acids or amines) in aqueous solutions followed by injection of tellurium, selenium or sulfur precursors. This method provides nanocrystals that are capped with small thioalkyl acids (e.g., mercaptoacetic acid, 3-mercaptopropionic acid, or cystamine).22–24 Similarly, there are several water-based chemical routes for growing Fe3O4 and other magnetic nanoparticles, based on the reduction of precursors such as FeCl3·6H2O and FeCl2·4H2O.25,26 However, these water-based routes tend to provide nanocrystals with rather large size distribution, and water dispersions of such materials often exhibit limited colloidal stability to pH changes and to added electrolytes and/or redox-active agents. Furthermore, this route does not allow easy, straightforward and controllable functionalization of the nanocrystals, a necessary property for further coupling to target biomolecules.
High temperature reduction of organometallic precursors in the presence of hydrophobic coordinating molecules (ligands) has thus far provided the highest quality nanocrystals, with low size dispersity and good control over size, morphology and core crystallinity.1,4,27 Commonly used ligands in this “hot injection” reaction include trioctyl-phosphine (TOP), trioctyl-phosphine oxide (TOPO), alkylamine and alkylcarboxy for luminescent QDs, didodecyldimethylammonium bromide (DDAB) for AuNPs, and oleic acid or oleylamine for iron oxide nanoparticles.1,27 The resulting materials are capped with hydrophobic ligands, which make them dispersible only in organic solvents. Thus, use of these materials in biomedical applications requires surface-modification with hydrophilic and biocompatible molecules. In the last two decades, this has been widely done by several groups using various chemical approaches, which can essentially be grouped into two main strategies:3,8,28–30 (1) removal of the native capping molecules and replacing them with bifunctional hydrophilic ligands (cap or ligand exchange), or (2) encapsulation of the native hydrophobic nanocrystals within micelle structures made of amphiphilic polymers or phospholipids.
Ligand exchange requires the presence of strong anchoring groups to replace the native cap and drive the metal–ligand coordination onto the inorganic surface of the nanocrystals, along with hydrophilic segments to promote affinity to water. This strategy relies on small molecules as well as polymers. In comparison, the encapsulation strategy preserves the native cap, as these interdigitate with the hydrophobic block of the amphiphilic polymer or phospholipid (usually made of aliphatic chains), via entropy-driven hydrophobic interactions, while the hydrophilic moieties promote affinity to water media. Interdigitation between the native cap of the nanoparticles and the hydrophobic block of the amphiphilic polymers is stable enough to preserve the nanocrystal coating and impart colloidal stability in aqueous media.
Polymers, whether synthetic or biological, have provided researchers with a great platform for designing a variety of ligands capable of functionalizing various nanocrystals via either of the strategies introduced above (ligand exchange or encapsulation). The wealth of knowledge and expertise gained over the past few decades for designing novel polymerization techniques allow remarkable control over the chemical make-up, architecture and molecular weight of the polymer materials. This can be fully exploited to develop effective surface functionalization strategies applicable to a wide range of nanoparticles based on encapsulation or ligand exchange.31,32 For instance, simple or more complex chemical transformations have allowed groups to design and test several amphiphilic polymers with control over the hydrophobic and hydrophilic blocks as well as the overall polymer size. Similarly, simple chemical coupling allowed the development of several polymer ligands, where the ability to insert several anchoring groups along a single polymer chain can enhance the ligand-to-nanoparticle interactions and provide materials with great colloidal stability.
We will start by summarizing the synthetic strategies developed so far to prepare inorganic nanoparticles, including water-based routes as well as those relying on the high temperature reduction of metal precursors (also referred to as hot injection routes). We then summarize the recent advances in the synthesis of several amphiphilic block copolymers and their use to promote water solubilization via either ligand exchange or an encapsulation process. We then conclude by providing a summary of a few representative biological applications using those polymer-functionalized nanoparticles.
2. Growth of inorganic (metal, semiconducting and magnetic) nanocrystals
Reduction of metal salts in water media (e.g., growth in inverse micelles or via arrested precipitation) was one of the initial routes developed to grow several metal, metal oxide and semiconductor nanocrystals.33–35 This route is easy to implement, often carried out using slight heating, and has the advantage of providing materials already dispersed in aqueous media. It requires water-soluble precursors and suitable capping ligands for stabilizing the nanoparticles. In comparison, the growth of nanocrystals at high temperature (or via hot injection method) relies on the reduction of organometallic precursors at high temperatures; it is primarily carried out in organic hydrophobic solutions. This growth route has been applied to an array of materials, including magnetic nanocrystals, semiconductor quantum dots as well as gold and other metal nanoparticles; it has been shown to reproducibly provide homogeneous materials with crystalline cores and, more importantly, low size dispersity. This involves a temporally discrete nucleation event driven by saturation in the precursor concentrations, followed by slower controlled growth and ripening with further annealing. Rapid injection of precursors into the reaction vessel increases their concentrations above the nucleation threshold. A short nucleation burst partially relieves this saturation, and subsequent annealing at high temperatures promotes the growth of more homogeneous and uniform nanocrystals.The ability of this growth route to provide homogeneous crystalline nanocrystals with reduced size dispersity has allowed researchers to carry out sophisticated characterization studies and probe the fundamental photophysical, spectroscopic and chemical properties of such nanoparticles. The collected results have been tested against proposed conceptual models,36–45 which have permitted scientists to develop a much better understanding of these systems. This experimental success has also brought these nano-structured materials closer to the realm of targeted applications, including integration into electronic, optical and biological systems.
2.1. Luminescent quantum dots (QDs)
In the last two decades, a variety of colloidal semiconductor nanostructures have been prepared; they range from spherical, cubic, rod-like, branched, tetrapod-like and platelet materials.27,46–50 The first reports describing the effects of carrier confinement within nanometer size nanocrystals of semiconductors were published in 1981–1982. In those studies, the authors reported measurement of size-dependent optical spectra of CuCl, CdS or CdSe nanocrystals embedded into silicate glasses.51,52 Efros and Efros showed that glass matrices containing precipitated crystallites of CdSxSe1−x can be used to build tunable optical filters where variations in the size and/or stoichiometry of the crystallites allow tuning of the corresponding absorption band.53 During the same period similar results detailing the growth of CdSe nanoparticles precipitated in glasses were reported by Borrelli and co-workers at Corning Inc.54,55 The growth of colloidal QDs using reverse micelles reported by Brus and co-workers, and Henglein and co-workers in the early 1980s introduced another highly important dimension into the field, as nanocrystals with size-tunable optical features that can be studied and processed from solution conditions became available.33,34,56,57A major breakthrough in the growth of high quality colloidal QDs was developed in 1993 by Bawendi and co-workers. The authors showed that high temperature reduction of dimethyl cadmium (CdMe2) and tri-n-octylphosphine–selenium (TOP:Se) in a hot coordinating solution (at 280–350 °C) made of trioctylphosphine and trioctylphosphine oxide (TOP/TOPO) can provide high quality CdSe QDs, with homogeneous core crystallinity, low size dispersity and high room temperature photoluminescence quantum yields.27 In particular, they prepared CdSe nanocrystals that exhibit narrow and size-tunable symmetric photoluminescence (PL) spectra, a high molar extinction coefficient and high chemical stability. In subsequent studies, Peng and co-workers further refined the synthetic rationales underlying the effectiveness of this synthetic route by showing the importance of introducing additional alkylphosphonic acid ligands into the growth reaction. They also introduced less volatile cadmium precursors (e.g., cadmium oxide, CdO and cadmium acetylacetonate, Cd(acac)2), which are also easy to store under ambient conditions.58,59 A flurry of reaction modifications and adjustments followed those studies where several groups further optimized the reaction conditions and expanded those chemical rationales to grow other nanocrystals.4,60–62 In some of those reports, researchers substituted the TOP/TOPO and hexadecyl amine (HDA) coordinating solution with other non-coordinating materials made of long alkane or alkene chains such as 1-octadecene (ODE) or even olive oil.63,64 Those materials do not play any major role in the stabilization of the nanoparticles during the growth process. These adjustments produce a smaller number of nuclei than the “conventional” route where TOP–Se is used as a precursor, and this is attributed to the fact that TOP provides better solubility to selenium compared to 1-octadecene or other non-coordinating solvents.
By using different combinations of core materials (e.g., CdTeSe, CdHgTe), one can expand the photoemission of the nanocrystals from the red to near infrared (NIR) region of the optical spectrum compared to those made of CdSe cores.65–67 More recently, a few groups expanded the high temperature reduction route to grow Cd-free QDs (namely, CuInS2 and CuIn5Se8 nanocrystals) with emission extending into the NIR.62,68 However, those dots still exhibit broad emission with absorption spectra reflecting less defined crystalline structure. Further refinements will undoubtedly improve those properties.
Solution phase grown nanocrystals have a large fraction of their atoms arrayed at their surfaces that are poorly passivated by the ligands. This creates surface defects which affect the rate of exciton radiative recombination, and reduce the overall photoluminescence quantum yields (PL QYs).69,70 Borrowing from the ideas developed for band gap engineering in semiconductor physics, several groups overcoated the native core with a thin layer (a few atomic monolayers) of wider band gap semiconducting materials to enhance the quantum yield and photochemical stability of the resulting core–shell QDs. Examples include the overcoating of CdSe QDs with a thin layer of ZnS, CdS, ZnSe, or ZnSSe.71–74 More recently a few groups have shown that a very thick layer of CdS on a CdSe core can bring the PL quantum yield (QY) close to one.75,76 Overcoating is usually carried out using high temperature reduction of the desired precursors, but at lower values (120–190 °C) than those used for the core growth. A variety of precursors such as diethylzinc (ZnEt2), zinc acetate (Zn(OAc)2), zinc acetylacetonate (Zn(acac)2), zinc diethyldithiocarbamate (Zn(S2CNEt2)2), hexamethyldisilathiane (TMS2S), and elemental sulfur have been used for overcoating CdSe with ZnS shells.4,71–73,77–79 We should note that overcoating with ZnS, ZnSe, CdS, CdSe, etc. has also been applied to other QD materials such as those made of PbSe, CuInS2 and AgInS2 cores.80–82 We would like to note that the exact nature of the surface capping ligands on QDs prepared using these various high temperature strategies is still unclear. Even though the commonly accepted premise has been for a long time that TOP and TOPO constitute the majority of surface ligands along with smaller fractions of alkylamines and phosphonic acids, recent studies have indicated that TOP/TOPO may not be the dominant ligands on the nanocrystal surface.83
2.2. Iron oxide nanoparticles
The growth of iron-based magnetic nanoparticles initially relied on the precipitation of Fe salts, namely FeCl3 in aqueous media, and materials prepared via this route have been used in various studies and applications.84–87 This route provided an easy synthetic route to prepare “ready to use” hydrophilic nanoparticles. However, control over size, core crystallinity and size dispersity of the nanoparticles was only marginally achieved. Several iron-based magnetic nanoparticles (e.g., Fe5HO8, Fe5(O4H3)3, Fe2O3, Fe3O4, and FeOOH) have been grown using various methods, including chemical precipitation,85 sol–gel and forced hydrolysis,84 hydrothermal techniques,86 surfactant-mediated template synthesis,87 microemulsion,88 biomimetic mineralization,89 flow injection synthesis,90 electrochemical methods,91 sonochemical techniques,92 and high-temperature decomposition. Following the success of the hot injection method reported for growing QDs,27 several groups expanded this route to grow various magnetic nanocrystals.16,93–101 For instance, high quality iron oxide nanocrystals with homogeneous crystalline cores and low size distribution have been prepared via decomposition of iron precursors, such as Fe(Cup)3 (Cup = N-nitrophenylhydroxylamine), Fe(CO)5, FeCl3 and Fe(acac)3 at high temperature in reaction media made of organic solvents and coordinating surfactants. In one of the earlier growth strategies published by Hyeon and co-workers, the authors started by developing an organometallic iron complex, iron–oleate, prepared by reacting iron chloride (FeCl3·6H2O) and sodium oleate in a mixture of ethanol, water and hexane at ∼70 °C for four hours.1,16 Washing the above mixture with distilled water followed by evaporation of the organic solvent(s) yields a waxy solid compound, which could be stored for further use. Briefly, in a typical reaction to grow 12 nm (diameter) NPs, the desired amount of iron–oleate complex is dissolved in 1-octadecene (a non-coordinating organic solvent) and the mixture is heated and annealed at ∼320 °C. After 30 min, the colorless solution turns brownish black, indicating the formation of iron oxide nanoparticles. The size of the nanoparticles can be controlled by varying the solvent used (e.g., 1-hexadecene (b.p. 287 °C), 1-octadecene (b.p. 317 °C), trioctylamine (b.p. 365 °C), octyl ether (b.p. 286 °C), and 1-eicosene (b.p. 330 °C)) and the annealing temperature; larger sizes are usually prepared when using solvents having higher boiling points. Also, the size of the iron oxide nanocrystals can be tuned by varying the concentration of oleic acid in the reaction mixture. For example, 11, 12, and 14 nm sized iron oxide nanoparticles were produced using solutions containing 1.5, 3, and 4.5 mM of oleic acid, respectively. Peng and co-workers further expanded this approach and showed that other metal oxide nanocrystals such as Cr2O3, MnO, and Co3O4 can be grown using high temperature reaction starting with various metal fatty acid salts as precursors.100The high temperature growth strategy has further been expanded to prepare metal-doped nanoparticles with enhanced coercivity, because the spin contribution from the dopants can alter the final magnetic moment per crystal unit and increases the magnetic susceptibility of the resulting nanoparticles. Indeed, the magnetic properties of iron oxide nanoparticles can be controlled by doping the core with magnetically susceptible elements, such as Mn, Ni and Co ions. The resulting transition metal-doped iron oxide nanoparticles exhibit mass magnetization values that can vary from one system to another, with the highest value measured for MnFe2O4 nanoparticles (110 emu per gram Mn, Fe), as demonstrated by Cheon and co-workers.97,102 These magnetism-engineered iron oxide (MEIO) nanoparticles can induce significant MR contrast-enhancement effects, and the resulting nanoparticles were applied for visualizing (via magnetic resonance imaging, MRI) a few specific biological events.14,103,104 Similarly, high temperature growth has been applied to prepare magnetic nanocrystals made of metal alloys, such as FeCo and FePt.105
2.3. Gold nanoparticles (AuNPs)
Chemical reduction of gold precursors at room temperature in either the aqueous phase or a biphasic water–organic mixture has been effectively used by several groups to grow Au nanoparticles.9,106 In one of the early pioneering studies, Turkevich and co-workers were the first to report the growth of ∼10–20 nm AuNPs using water-based reduction of chloroauric acid (HAuCl4) in the presence of trisodium citrate.107 Frens and co-workers used this synthesis route to grow several size AuNPs with the diameter ranging from 16 to 140 nm, by varying the molar ratio of citrate-to-gold precursors used.108 There have also been instances where polymers such as poly(N-vinylpyrrolidone) (PVP), poly(4-vinylpyridine), poly(vinyl alcohol) (PVA), polyethyleneimine (PEI), poly(diallyl dimethylammonium chloride) (PDDA) have been used to grow and stabilize Au nanoparticles.109,110 We should stress that the as-prepared citrate-stabilized AuNPs exhibit very limited colloidal stability to pH changes and added salts. Aggregation is often observed in even slightly acidic buffers or in the presence of low concentration of added electrolytes. They have recently been shown to strongly and nonspecifically interact with serum proteins, producing what has commonly been referred to as corona on inorganic nanoparticles in biological media.111–113 A major development was the synthesis of hydrophobic AuNPs functionalized with thioalkyl ligands using two-phase (toluene–water) reaction reported by Brust and Schiffrin.106 In this method, HAuCl4 was transferred from water to toluene (organic phase) using the surfactant tetraoctylammonium bromide (TOAB), and then reduced by sodium borohydride (NaBH4) in the presence of dodecanethiol. Recently, our group has developed a one-phase aqueous growth method of AuNPs stabilized with dithiol-terminated hydrophilic molecules (i.e., PEG- or zwitterion-appended lipoic acid, LA–PEG or LA–ZW ligands). This route permitted control over the NP diameter in the range of 2–20 nm.114 It has more recently been expanded to grow Ag nanoparticles over a broad size range as well as fluorescent clusters of Au and Ag.12,115 Another approach for synthesizing and controlling the size and shape of AuNPs is the seed-mediated growth. Here, small metal nanoparticles are prepared first and then used as seeds (nucleation centers) along with dissolved Au precursors to grow larger size AuNPs and Au nanorods (AuNRs).116–118 Thus far, most of the water-based growth methods used thiol-containing compounds to provide monolayer-protected AuNPs, a choice motivated by the strong metal-coordination of sulfur onto gold surfaces.119 Peng and co-workers developed a single-phase (organic) reaction to grow AuNPs with low size dispersity. Here, tetrabutylammonium borohydride (TBAB) mixed with hydrazine (in toluene) was used as a reducing reagent and fatty acids or aliphatic amines were used as ligands.48 Briefly, fatty acid ligands were first dissolved in toluene, followed by the addition of TBAB dissolved in didodecyldimethylammonium bromide (DDAB). Then, a gold precursor dissolved in DDAB was injected into the above solution at room temperature. Finally, thiol ligands were added to the reaction mixture to stop the growth of the nanoparticles. Improvement of the nanoparticle quality while reducing size distribution was achieved by thermal annealing at 120 °C. The size of the particles was controlled from 1.5 to 15 nm by varying the nature of the reducing agent and capping ligands, the TBAB-to-gold molar ratio, and growth temperature.Despite the great success of the room temperature reduction route for growing AuNPs and AuNRs, it has been recently shown that the hot injection method provides better quality and more homogeneous AuNPs, as was demonstrated for QDs and magnetic nanocrystals above. For example, Williams and co-workers applied the reduction of Au(acac)PPh3 at ∼105 °C in a solution containing a mixture of TOPO and HDA.120 They reported control over the nanoparticle diameter from 10 to 50 nm by varying the precursor concentration, the nature of the coordinating solvent(s) and the reaction time used. In a parallel study, Osterloh and co-workers used oleylamine as a reducing agent and stabilizer to prepare alkylamine-stabilized gold nanoparticles with low dispersity over the size range of 6–21 nm.121,122 The Au precursor was rapidly injected into a solution containing oleylamine and toluene at 110 °C, and the reaction mixture was left stirring for two hours before cooling to room temperature. They controlled the size of the AuNPs by varying the precursor concentrations and reaction time. Recently, Swihart and co-workers reported the synthesis of homogeneous 10 nm AuNPs using a solution containing pure oleylamine. Here, the oleylamine was used as a solvent, reducing agent and stabilizer for the nanoparticles.123
3. Water dispersion strategies
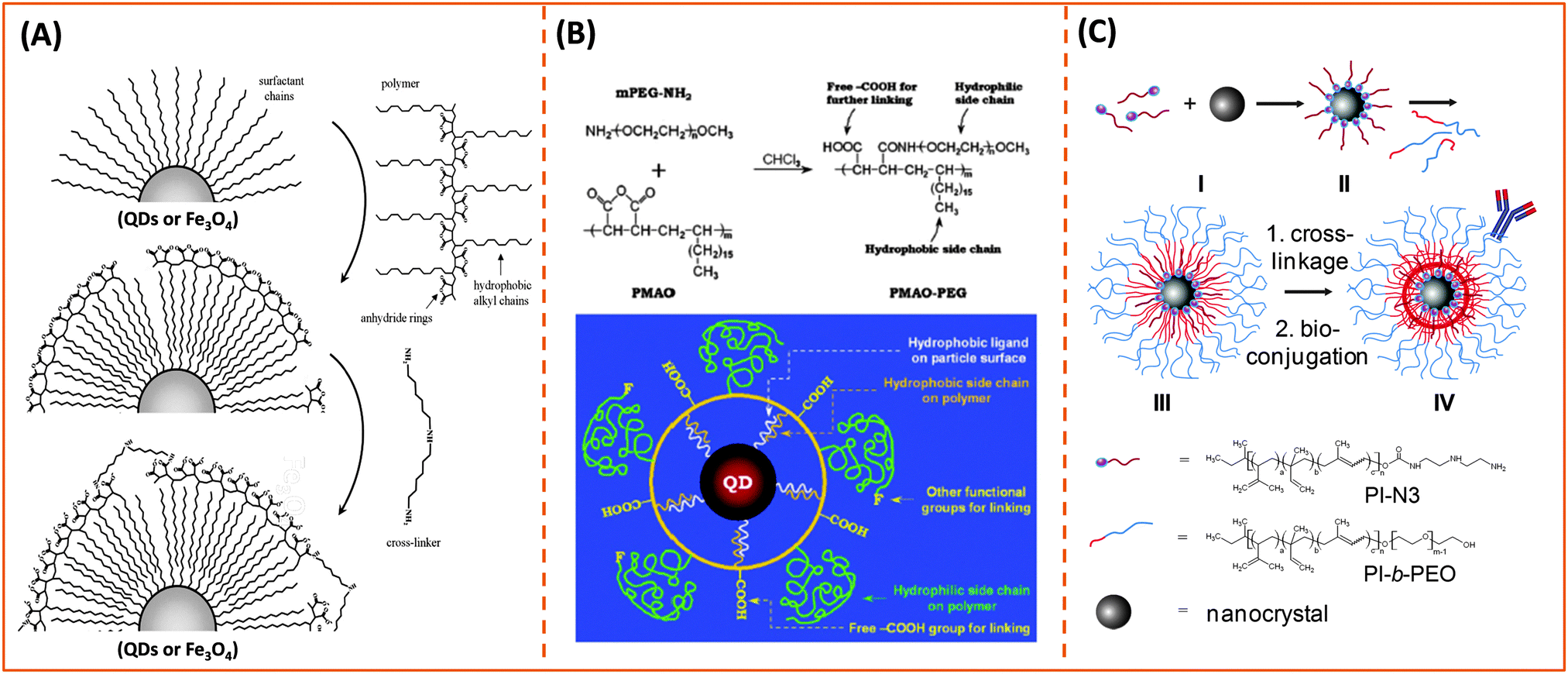
One key requirement for the successful integration of these inorganic nanocrystals/nanoparticles into biology is the implementation of an effective surface-modification strategy that renders those materials hydrophilic and compatible with commonly used bio-conjugation techniques to target biomolecules. This requirement is valid regardless of the initial growth method or the nature of the inorganic nanocrystals used. For example, citrate-stabilized AuNPs and cetyltrimethylammonium bromide (CTAB)-coated AuNRs, even though prepared in water they exhibit limited colloidal stability to added electrolytes and pH changes. This limits one's ability to integrate them with biomolecules, or introduce them into live cells. Additional surface-functionalization with appropriate hydrophilic ligands has been used to expand their colloidal stability and impart target specific biological activities to these materials. However, nanocrystals prepared via a high temperature reduction route are hydrophobic, and a judicious surface functionalization strategy is critically important to promote water-solubility and bio-functionality to these systems.Overall, the strategies developed thus far for achieving surface-functionalization and biocompatibility of inorganic nanostructures can be grouped into two main types. The first involves the removal of the native hydrophobic organic coating and replacing it with bifunctional hydrophilic molecules, i.e. ligand exchange (see Fig. 1).124–129 The second route relies on encapsulation of the native hydrophobic nanocrystals with amphiphilic block copolymers or phospholipid micelles.10,130–132 Because the ligand exchange process requires the removal of the native organic capping shell, the bifunctional molecules used for phase transfer must present one or multiple metal-coordinating groups to anchor onto the inorganic surface, along with reactive functions for attaching the NPs to biomolecules. Conversely, encapsulation relies on the entropy-driven interdigitation between the hydrophobic segments of the amphiphilic molecules and the native cap on the nanocrystals, leaving the hydrophilic blocks (segments) laterally free to interact with the surrounding buffer and promote affinity to water (Fig. 1). In either strategy, polymers have provided researchers with a tremendous wealth of chemical and physical knowledge, along with a wide variety of structures to work with and build on. For example, there are several chemical routes that can be utilized to introduce new functional and/or coordinating groups within the polymer macromolecules (block co-polymer) for optimal functionalization of the nanocrystals.31,32 In addition, several block-copolymers have an extremely low critical micelle concentration (CMC), which makes them stable under a wide range of physiologically-relevant conditions and thus suitable for therapeutic applications.133 A summary of the various polymer designs for either strategy is provided in Table 1.
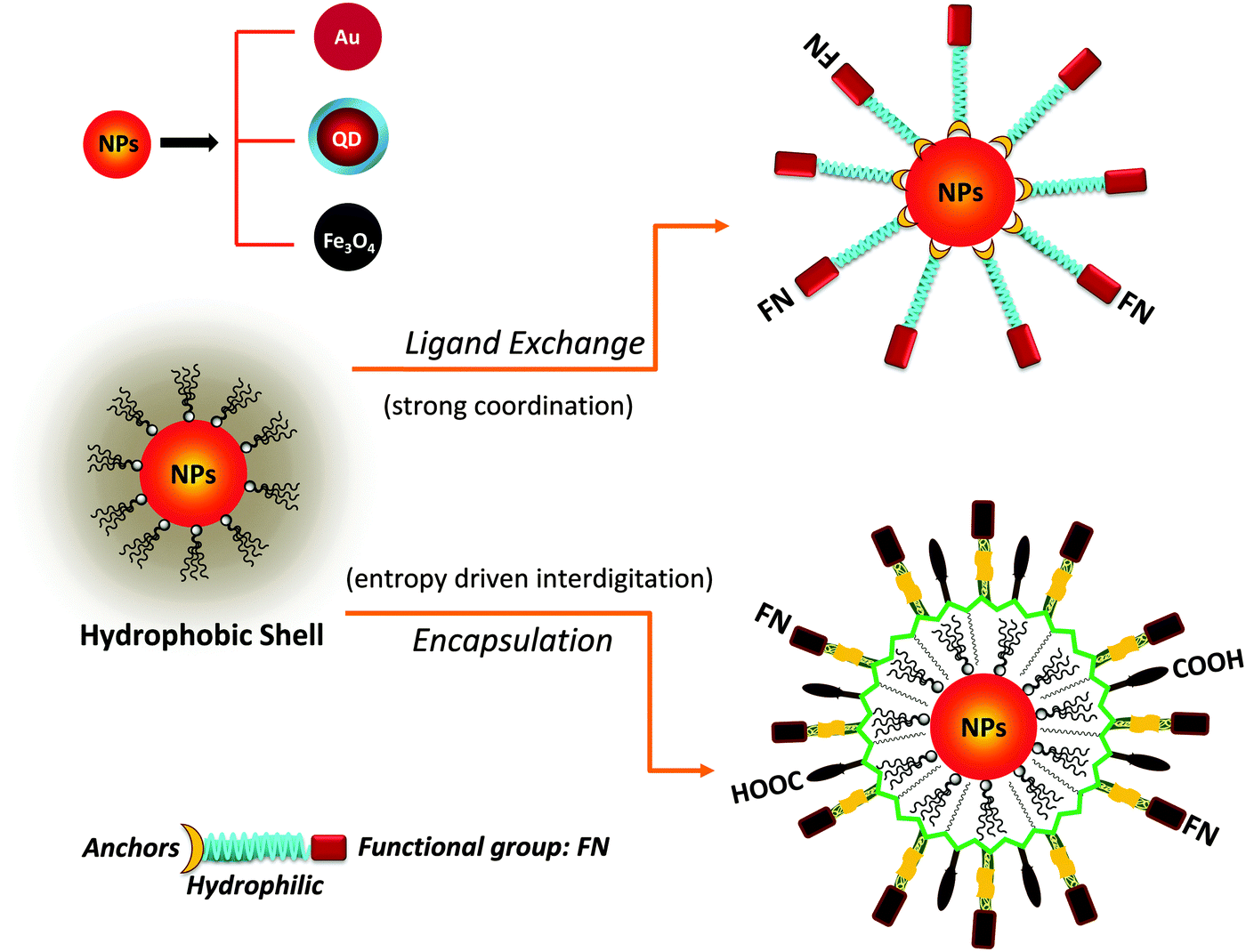 | ||
| Fig. 1 Schematic representation of phase transfer via: (top) ligand exchange which relies on the presence of strong anchoring groups on the nanoparticle surface; (bottom) encapsulation of the hydrophobic nanoparticles (NPs) within an amphiphilic block-copolymer. Encapsulation involves the entropy driven-interdigitation between the native ligands and the hydrophobic blocks of the copolymer. Semiconducting nanocrystals (QDs), metal (Au) and metal oxide (Fe3O4) nanoparticles are shown. | ||
| Surface-modification strategy | Polymer platform used | Coordinating groups/interdigitating blocks | Nanocrystals | References |
|---|---|---|---|---|
| Ligand exchange | Thiolated poly(L-lysine)-graft-poly(ethylene glycol) (PLL-g-[PEG:SH]) | –Thiols: (–SH)n | AuNPs | 149 |
| Poly(acrylic acid)-graft-mercaptoethylamine (PAA-g-MEA) | –Thiols: (–SH)n | CdSe–ZnS | 153 | |
| Multi dihydrolipic acid-graft-poly(methacrylate) | –Thiols: (–DHLA)n | CdSe–ZnS | 150 and 151 | |
| Lipoic acid and poly(ethylene glycol) modified poly(acrylic acid) (PAA-g-[PEG:LA]) | –Thiols: (–LA)n or (–DHLA)n | AuNPs and CdSe–ZnS | 152 | |
| Sulfobetaine and lipoic acid modified poly(acrylic acid)(PAA-g-[LA:ZW]) | –Thiols: (–DHLA)n | CdSe–ZnS | 155 | |
| Methacrylate modified sulfobetaine and lipoic acid (LA:ZW) | –Thiols: (–DHLA)n | CdSe–ZnS | 154 | |
| Poly(methacryloyloxyethyl phosphorylcholine (MPC))-co-poly(dihydro lipoic acid) | –Thiols: (–DHLA)n | AuNRs | 158 | |
| Methacrylate modified poly(ethylene glycol) and imidazole (polyimidazole ligands, PILs) | –Imidazoles | CdSe–CdS–ZnS | 124 | |
| Poly(maleic anhydride)-graft-imidazole (PMAH-g-IL) | –Imidazoles | CdSe–ZnS | 145 | |
| Methacrylate modified sulfobetaine modified-graft and imidazole (SBPILs) | –Imidazoles | CdSe–Cd/ZnS | 163 | |
| Dopamine modified poly(acrylic acid)-graft-poly(ethylene glycol) (OligoPEG-Dopa) | –Dopamines: (–DOPA)n | Fe3O4 | 143 | |
| Dopamine-modified poly(isobutylene-alt-maleic anhydride)-graft-poly(ethylene glycol) (PEG:DOPA) | –Dopamines: (–DOPA)n | Fe3O4 | 172 | |
| Poly(L-3,4-dihydroxyphenylalanine)-graft-poly(ethylene glycol) | –Dopamines: (–DOPA)n | Fe3O4 | 173 | |
| Encapsulation | Polystyrene-block-poly(acrylic acid), PS-b-PAA | –Hydrophobic interactions (polystyrene and alkyl chains) | AuNPs | 178 |
| Poly(methyl methacrylate)-block-poly(acrylic acid), PMMA-b-PAA | –Hydrophobic interactions (polystyrene and alkyl chains) | AuNPs | 178 | |
| Polystyrene-block-poly(acrylic acid), PS-b-PAA block-copolymer: PS100-b-PAA13, PS160-b-PAA13 and PS250-b-PAA13 | –Hydrophobic interactions (polystyrene and alkyl chains) followed by chemical cross linking | AuNPs | 179 | |
| [Polystyrene-co-poly(4-vinyl benzophenone)]-block-poly(acrylic acid) [(PS-co-PVBP)-b-PAA] | –Hydrophobic interactions (polystyrene and alkyl chains) followed by photo-induced cross linking | AuNPs | 180 | |
| [Poly(styrene)-co-poly(4-vinyl benzophenone)]-block-poly-(ethylene oxide) [(PS-co-PVBP)-b-PEO] | –Hydrophobic interactions (polystyrene and alkyl chains) followed by photo-induced cross linking | AuNPs | 180 | |
| Poly(acrylic acid)-graft-dodecylamine | –Hydrophobic interactions (polydodecyl and alkyl chains) | AuNPs | 182 | |
| Poly(ethylene oxide)–poly(n-butyl acrylate), PEO–PnBA | –Micelle assembly through PnBA | AuNRs | 183 | |
| Poly(styrene sulfonate), PSS | –Electrostatic adsorption | AuNRs | 190 | |
| Poly(acrylic acid)-graft-octylamine | –Hydrophobic interactions (Poly alkylamine and alkyl chains) | CdSe–ZnS | 10 and 200 | |
| Poly(maleic anhydride alt-1-tetradecene)-graft-alkyl amine and/or poly ethylene glycol | –Hydrophobic interactions (poly alkyl and TOP/TOPO, oleylamine or alkane chains) | CdSe–ZnS, Fe3O4, AuNPs | 130, 132 and 204 | |
| Poly(styrene-co-maleic anhydride)-graft-poly(ethylene glycol) | –Hydrophobic interactions (poly styrene and TOP/TOPO) | CdSe–ZnS | 205 | |
| Poly(ethylene glycol-b-2-N,N-dimethylaminoethyl methacrylate) (PEG-b-PDMA) | –Hydrophobic interactions (poly styrene and TOP/TOPO) | CdSe, CdSe–ZnS | 206 and 207 | |
| Polyisoprene-block-poly(ethylene oxide) diblock (PI-b-PEO or PI-b-(PEO)2 star) | –Hydrophobic interactions | CdSe–CdS/ZnS, Fe3O4, AuNPs | 209, 211 and 212 | |
3.1. Exchanging the native cap with hydrophilic ligands
Ligand exchange as a strategy involves the removal of the native coating (e.g., CTAB, oleic acid or TOP/TOPO) from the surface of nanoparticles and its replacement with multifunctional hydrophilic ligands. Thus to be effective, this strategy requires a judicious choice of the polymer ligand. The latter must combine high affinity anchoring groups, hydrophilic blocks and reactive groups (Fig. 2 and 3). The first two components (i.e., anchoring groups and hydrophilic moieties) control the stability of the nanocrystal-to-ligand binding and thus the colloidal stability of the resulting dispersions, while the reactive groups allow one to apply the optimal coupling strategy for attaching the desired number and type of target molecules (e.g., peptides and proteins) to the inorganic platform of interest. The selection of the anchoring group(s) depends on the nature of the inorganic surface of the nanocrystals (Fig. 2).134–139 For example, thiol groups exhibit much higher affinity to AuNPs and to Zn- and Cd-rich QD surfaces, though coordination onto AuNPs is much stronger. Au-to-thiol (or Au–sulfur) interaction has been described in several instances as covalent binding,119 and thiol-modified ligands are believed to be the most effective for functionalizing AuNPs and AuNRs.140–142 In comparison, dopamine has been shown to provide strong coordination onto the surface of iron oxide NPs, but its ability to coordinate onto Au and semiconductor surfaces is rather weak. Carboxyl- and amine-appended alkyls such as oleylamine and oleic acid have been used in the high temperature growth of QDs and iron oxide nanocrystals; they provide good anchoring groups for the metal surfaces in organic solutions.1,16,35,100,143,144 These groups have also been proposed and utilized as anchoring groups to promote the dispersion of AuNPs, iron oxide NPs and QDs in buffer media. Their effectiveness as coordinating groups in aqueous media is rather weak, nonetheless, as often nanoparticles prepared using this strategy exhibit limited colloidal stability to pH changes and in the presence of soluble electrolytes.143 More recently, a few studies have shown that the amino acid histidine, if judiciously inserted into a polymer structure (organic or biological), can promote strong affinity to AuNPs and core–shell QDs.124,136,145 In those studies, the authors have exploited the known metal-coordinating capacity of the imidazole group of histidine and designed a few polymer ligands laterally appended with histidine derivatives (e.g., histamines). They have shown that such polyhistamine-modified polymers can coordinate onto semiconductor nanocrystals and promote their dispersion in biological media.124,145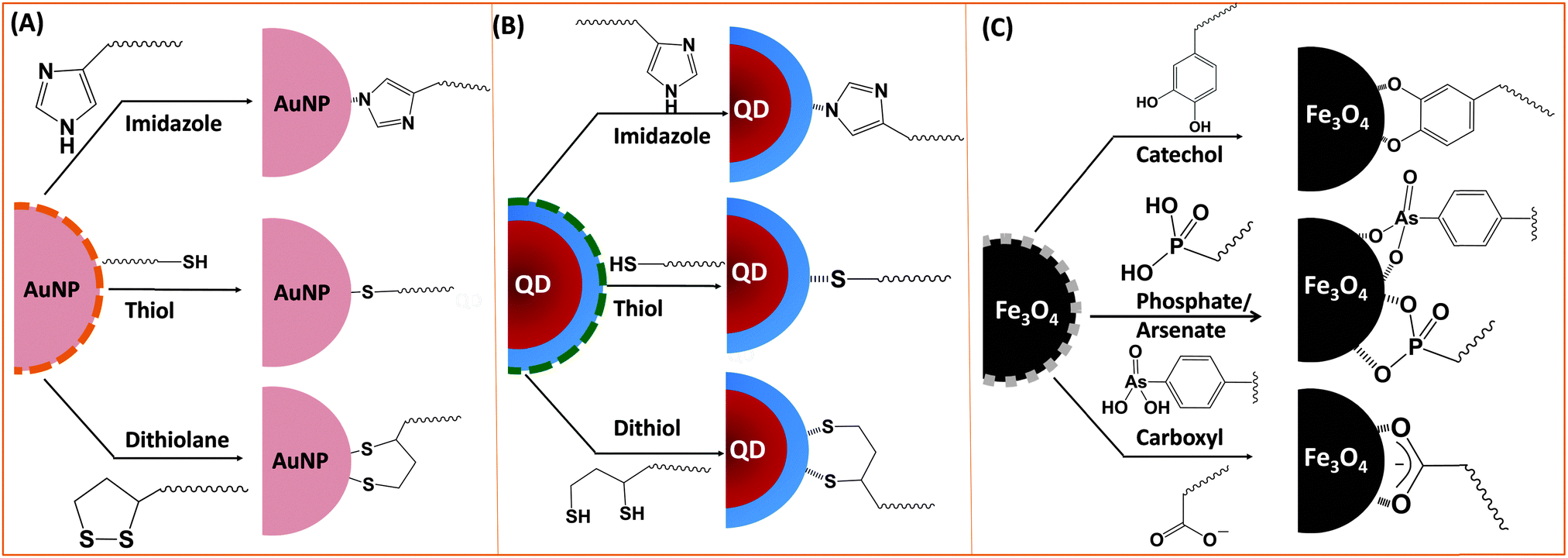 | ||
| Fig. 2 Schematic representation of various metal-anchoring groups often employed using the ligand exchange strategy: (A) metallic (AuNPs), (B) semiconductor (QDs), and (C) magnetic (iron oxide) nanocrystals are shown. | ||
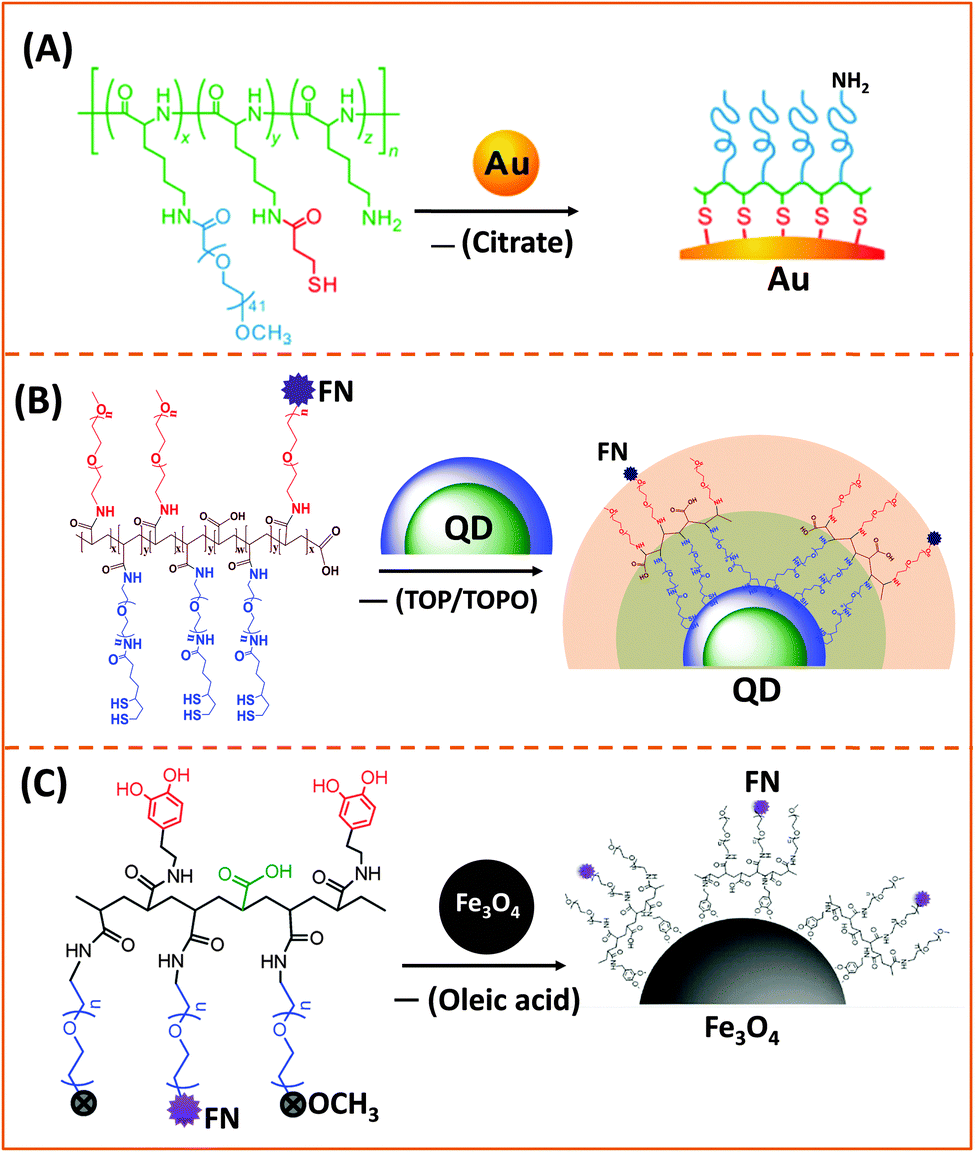 | ||
| Fig. 3 Representative examples of designing biocompatible nanoparticles via cap-exchange applied to: (A) citrate-stabilized gold nanoparticles, (B) TOP/TOPO-capped QDs, and (C) oleic acid-capped magnetic nanocrystals.143,149,152 (Figures are reproduced from the above references with permission from the American Chemical Society.) | ||
AuNPs used for cap exchange are generally stabilized by weakly binding ligands such as citrate or CTAB, and a few groups have recently explored the use of multi-coordinating functional block-copolymers to install stronger binding cap and/or introduce hydrophilic and reactive groups for interfacing with biological entities (antibodies and DNA). In one of their recent reports Taton and co-workers tested the effectiveness of poly(L-lysine)-graft-poly(ethylene glycol) (PLL-g-PEG) copolymers to passivate and disperse AuNPs in buffer media. They incorporated several thiol groups and PEG chains (via amide bond formation) into the poly(lysine) backbone, by sequential addition of NHS-ester-terminated PEG-(mPEG-SCM) and a thiol linker (N-succinimidyl-3-(2-pyridyldithio)-propionate).149 In addition, by leaving a few of the amine groups in the lysine residues intact, this opens up the possibility for coupling the NPs to carboxyl-terminated biomolecules (Fig. 3A).149
Instead of monothiol anchors, a few groups grafted lipoic acid (dithiolane) or lipoic acid-modified with a short PEG segment onto the polymer backbones. In one of the early studies, Raymo and co-workers designed a polymer construct made of the polymethacrylate backbone presenting several lateral LA groups along with few PEG segments to transfer hydrophobic QDs to buffer media.150,151 Their synthetic strategy was based on the radical copolymerization of methacrylate monomers pre-functionalized with lipoic acid, and PEG moieties with varying chain lengths, or PEG moieties presenting lateral amine or carboxyl groups (Fig. 4A). They showed that following borohydride reduction of the LA groups the resulting polymer ligands provided QDs with enhanced long term stability compared to small mono-thiol ligands. Here the larger PEG chain tended to increase the effective hydrodynamic size of the water-dispersible QDs. To potentially reduce the hydrodynamic size of the hydrophilic nanocrystals, a few groups used poly(acrylic acid) oligomers (with a molecular weight of ∼1800).152,153 For example, Liu and co-workers designed a multidentate polymer ligand made of polyacrylic acid (PAA) coupled with mercaptoethylamine (MEA) via carbodiimide chemistry using dicyclohexyl carbodiimide (DCC). The produced multi-thiol polymer was used to transfer QDs to buffer media.153 The resulting PAA-g-MEA capped water-soluble QDs have relatively small hydrodynamic diameters (around 13 nm) and exhibit colloidal stability over a broad pH range (3–14) and added salt (up to saturated NaCl solution).
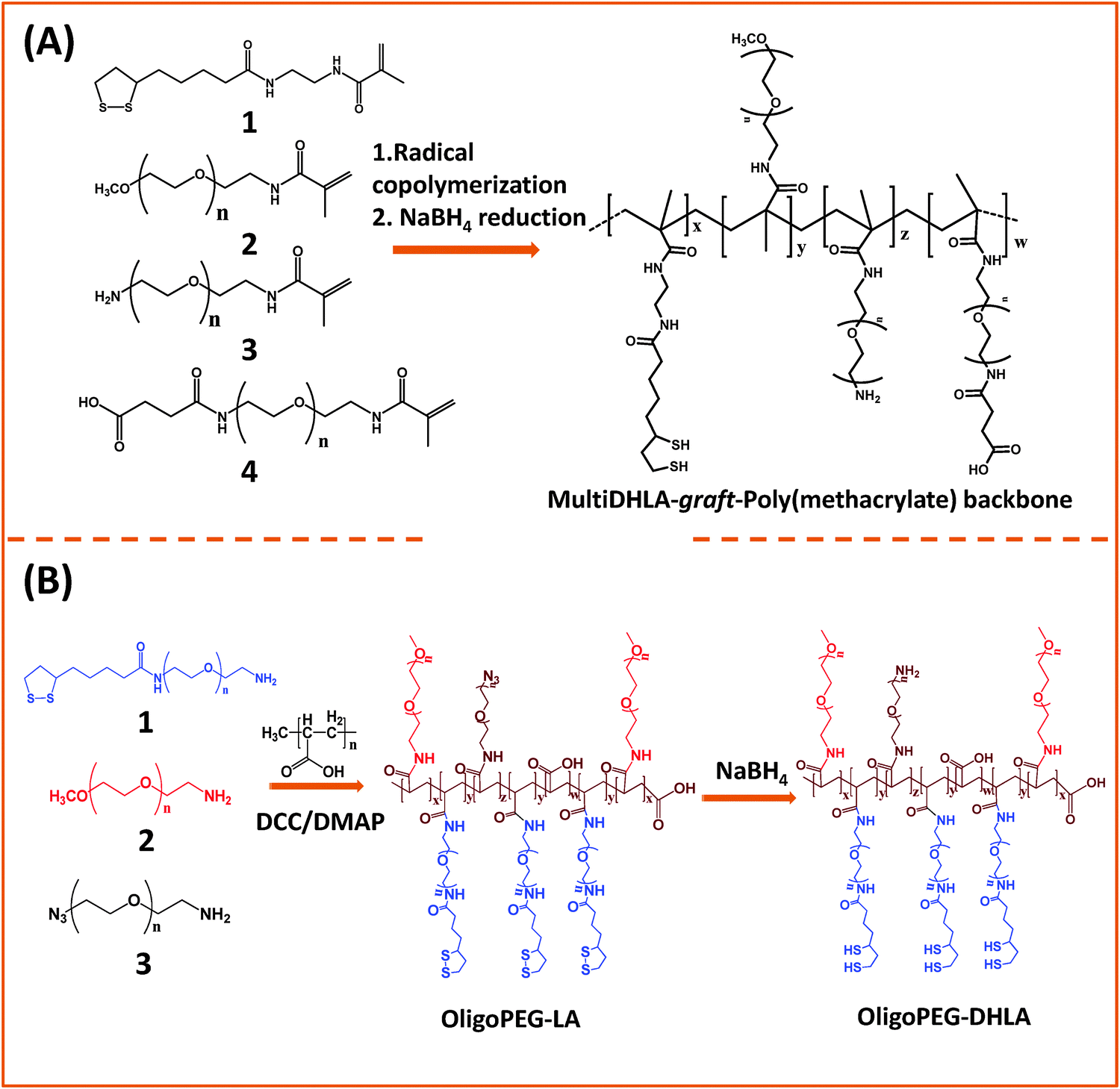 | ||
| Fig. 4 Synthesis of two representative polymer ligands: (A) a block-copolymer prepared via radical polymerization of reactive methacrylate groups; (B) OligoPEG polymer prepared via carbodiimide chemistry starting from a poly(acrylic acid) backbone. Both polymers present multiple lipoic acid moieties per polymer chain.150,152 (Figures are adapted from the above references with permission from the American Chemical Society.) | ||
Our group used this PAA short chain to prepare a series of PEG- and LA-modified oligomer ligands having a central backbone laterally appended with combinations of LA–PEG, methoxy–PEG, amine–PEG, and azide–PEG moieties (OligoPEG ligands).152 The use of smaller PEG moieties (Mw ∼ 600 or 750) eventually reduces the overall extension of the polymer coating on the nanocrystals (Fig. 3B and 4B). These LA-modified OligoPEG ligands were applied either to cap AuNPs, or after borohydride reduction to functionalize QDs. This route provided colloidal dispersions of QDs and AuNPs that remained stable over a broad range of conditions and over extended periods of storage time. With the same aim of reducing the hydrodynamic size of the polymer-capping, Zweit and co-workers, and Giovanelli and co-workers, took slightly different approaches for achieving the synthesis of multi-coordinating zwitterionic co-polymers (Fig. 5A and C).154,155 In particular, Giovanelli and co-workers synthesized a polymer containing molecular lipoic acid anchors and a sulfobetaine containing zwitterionic groups via a two-step process. They first modified the lipoic acid and zwitterion with methacrylamides and then performed the polymerization reaction to obtain a randomly grafted copolymer.154 In order to functionalize the polymer with reactive groups for further conjugation with biomolecules, they introduced a methacrylamide monomer bearing a reactive amine function during the polymerization step (Fig. 5C). This functionalization has been confirmed by coupling the cap exchanged NPs with a dye (fluorescein) via carbodiimide coupling. Finally, the multi-LA–zwitterion appended polymer exhibited strong affinity to QD surfaces, with reduced desorption rates compared to their lower coordination ligand counterparts and increased colloidal and intracellular stability.
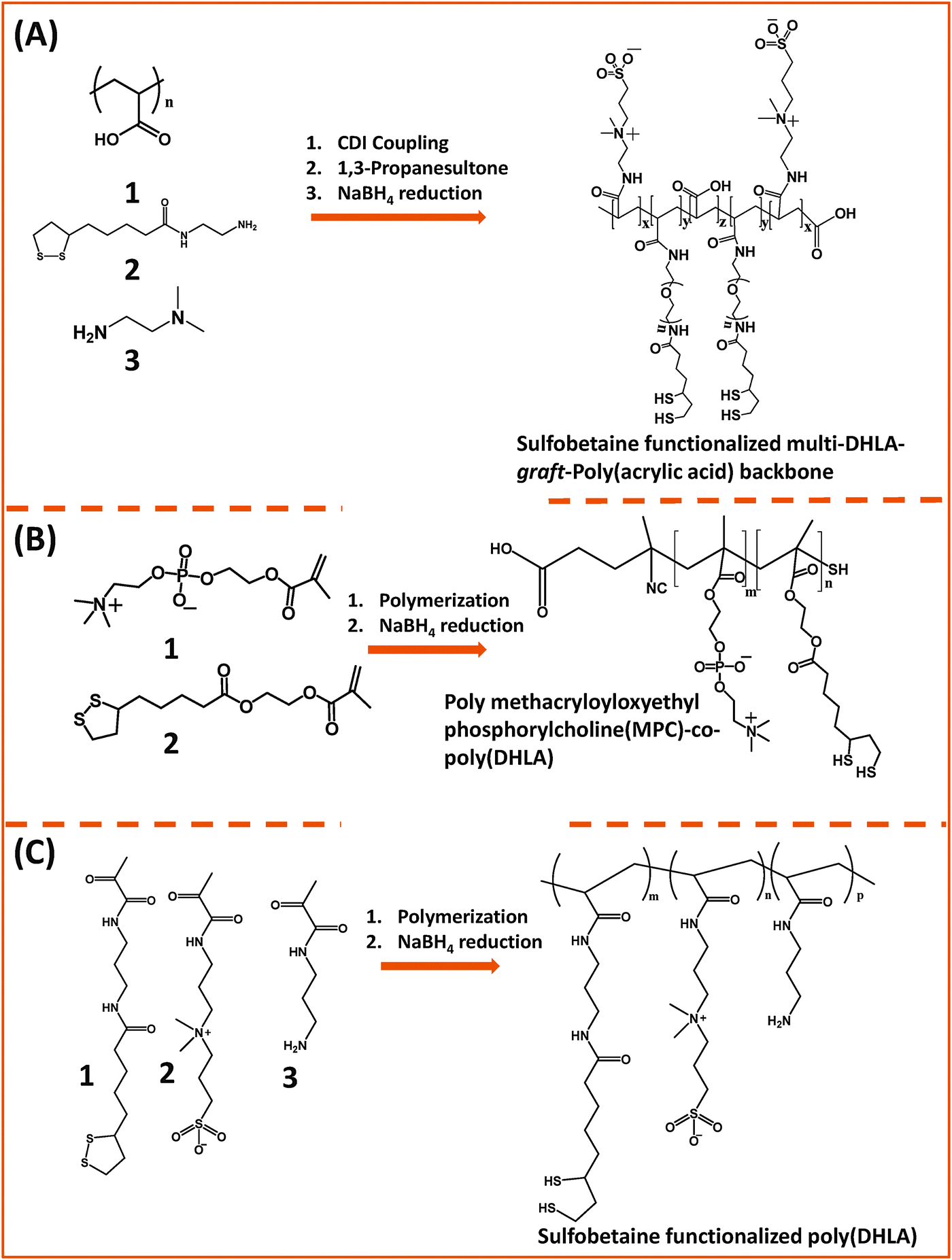 | ||
| Fig. 5 Synthesis of the poly(DHLA)-zwitterionic block-copolymer using: (A) carbodiimide chemistry starting from a poly(acrylic acid) precursor; (B) and (C) radical polymerization starting with LA and ZW moieties pre-modified with reactive methacrylate groups. Sodium borohydride has been used to reduce the dithiolane ring of the lipoic acid, a process required for cap exchange on QDs.154,155,158 (The figures are adapted from the above references with permission from the American Chemical Society.) | ||
To perform cap exchange on CdSe and ZnS-overcoated QDs, reduction of the 1,2-dithiolane to dithiol is required, as only the thiolated form of the ligand can coordinate onto the surface of QDs; the oxidized ligands do not cap these QDs.126,129 This chemical reduction is routinely carried out using NaBH4 as a reducing agent. Though effective, chemical reduction of the dithiolane ring using NaBH4 is not suitable for certain sensitive functional groups (e.g., azide and aldehyde) often introduced into the ligand structure for further modification of the resulting nanocrystals. For polymeric molecules the purification process is even more tedious, and after purification the DHLA-based ligands need to be stored under inert conditions. In order to address these problems, our group has recently introduced a new strategy to transfer QDs to polar and buffer media using lipoic acid-based ligands.156 In this strategy, the ligand exchange is promoted photo-chemically, and involves the in situ reduction of lipoic acid in the presence of QDs. This idea was motivated by a previous study by Sander and co-workers reporting that a well-defined absorption at ∼350 nm originating from the cyclic disulfide ring of the lipoic acid can be altered under UV irradiation.156,157 Indeed, we found that the photoligation and cap exchange on QDs can be easily applied with our molecular scale LA–PEG and LA-zwitterion ligands. Furthermore, the resulting materials exhibit great colloidal stability over a wide range of conditions. This idea should be easily applicable to polymers bearing multiple LA groups.
Emrick and co-workers developed a multi-coordinating zwitterion polymer by appending several LA groups onto a phosphorylchlorine (PC) block co-polymer. They first prepared a hydroxyethyl methacrylate (HEMA)-terminated lipoic acid (LA) monomer via EDC coupling. The HEMA–LA compound was then mixed with a methacrylamide phosphorylcholine (MPC) along with 4-cyanopentanoic acid dithiobenzoate (CTP) as the chain transfer agent and 4,4′-azobis(4-cyanovaleric acid) (ACVA) as the initiator for radical addition-fragmentation chain transfer (RAFT) polymerization (Fig. 5B).158 The authors showed that following chemical reduction of the LA groups the resulting DHLA-rich methacrylamide phosphorylcholine zwitterion polymer can be effectively applied to cap exchange CTAB-capped AuNRs, and the resulting dispersions exhibited great colloidal stability.
There is also growing interest in developing ligands that incorporate metal anchors other than the ubiquitous thiols, carboxy or amines. This idea was inspired by earlier demonstrations showing conjugation of hydrophilic QDs to polyhistidine (Hisn)-tagged proteins and peptides, promoted by metal-affinity interactions. Indeed, several groups have explored this conjugation method, due to the ease of implementation and the fact that His-tagged biomolecules are ubiquitous. For instance, we have demonstrated the conjugation of CdSe–ZnS QDs with His-tagged proteins and peptides, and showed that such interactions require direct access of the histidine tag to the Zn-rich QD surfaces.159,160 Learning from these developments, a few groups recently explored the ability of imidazole-modified ligands, or polyhistidine-appended peptides and proteins to effectively interact with core–shell QDs and AuNPs.124,161,162
In one of those developments, Bawendi and co-workers used RAFT polymerization to design a random brush co-polymer having both PEG and imidazole as side groups along with an aliphatic backbone (Fig. 6).124 They started by preparing monomer precursors bearing the necessary acrylate group to endow the final copolymer with the desired multi-functionality; those precursors present an imidazole anchoring group, a hydrophilic PEG segment, and a reactive amine group. The first monomer was prepared by DCC and NHS coupling between acrylic acid and histamine dihydrochloride, followed by imidazole nitrogen protection using di-tert-butyl dicarbonate (BOC2O). The second monomer consisting of a OMe–PEG11-terminated with acrylate function was synthesized via two modification steps: (1) transformation of the hydroxyl group of poly(ethylene glycol) methylether to amine; (2) reaction of the amine–PEG–OMe with the NHS-ester of acrylic acid. The third monomer was synthesized by coupling amine–(PEG)3/11–NH(Boc) with the NHS ester of acrylic acid. The removal of the Boc-protecting group was carried out using trifluoroacetic acid, TFA. These monomers were chosen to control three parameters: binding affinity of the polymer onto the nanocrystal, colloidal stability and functionality of surface. The stoichiometric ratio of those monomers was varied during the polymerization reaction to eventually control the relative ratio of anchors, hydrophilic and reactive groups. In addition, to minimize the potential for polymer cross-linking and aggregation of QDs after ligand exchange, small molecular weight polymers were used (a degree of polymerization smaller than 30). They showed that this imidazole-rich polymer can effectively displace the native TOP/TOPO cap and coordinate onto QD surfaces, providing water-dispersible relatively compact QDs with long term stability at pH > 5.124 In subsequent studies, they extended that design and substituted the PEG moieties with zwitterionic groups.163 They designed two sulfobetaine-functionalized poly(imidazole) ligands (BPILs) using the above methodology: sulfobetaine poly(imidazole), SBPILs, and carboxybetaine-functionalized poly(imidazole), CBPILs (Fig. 6B). These zwitterion-co-polymer ligands were successfully used for capping various types of QDs emitting from the near infrared to the visible region (e.g., InAs–CdZnS, CdSe–CdS, and CdSe–CdZnS).
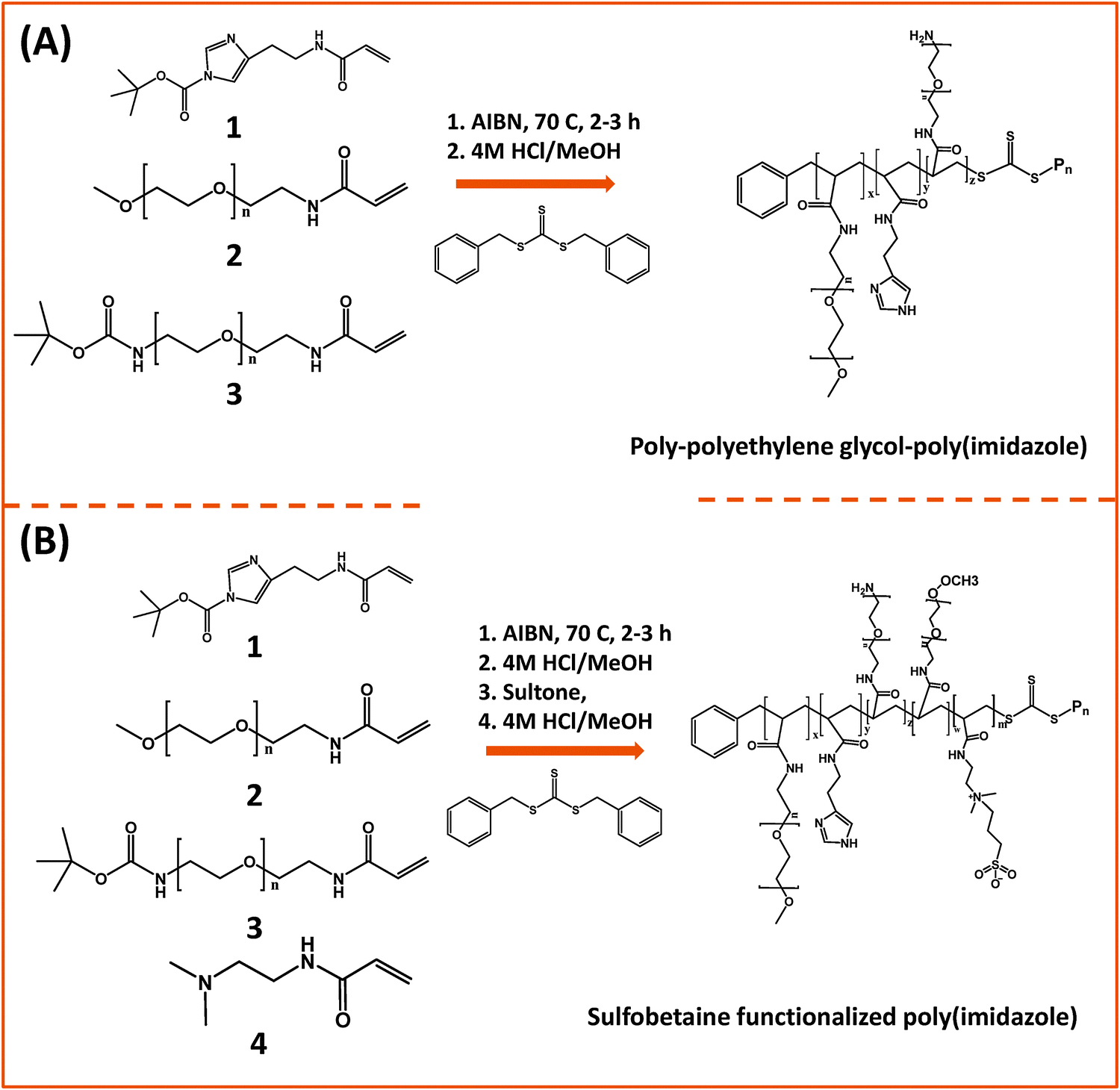 | ||
| Fig. 6 (A) Synthesis of poly(imidazole) block-copolymer prepared via RAFT; polymerization was carried out using methacrylate groups pre-modified with imidazole or PEG moieties. (B) Synthesis of a sulfobetaine functionalized poly(imidazole) block-copolymer using a similar approach.124,163 (Figures are adapted from the above references with permission from the American Chemical Society and from Wiley.) | ||
Another example using an imidazole-modified polymer as a ligand for QDs was reported by Cai and co-workers.145 This multidentate polymer was prepared by reacting poly(maleic anhydride) (PMAH) with either pure histamine or a mixture of histamine and N3–PEG–NH2 to obtain azide-functionalized QDs. They examined the effects of PMAH coating on the hydrodynamic size and optical properties of CdSe–ZnS QDs with varying core size emitting at 525 nm, 605 nm, and 705 nm. They found that this ligand design produced nanocrystals with high quantum yields along with minimal increase in the hydrodynamic size (∼2 nm after cap exchange). They also reported that PMAH-His-capped QDs were stable in the presence of H2O2 and under UV irradiation.
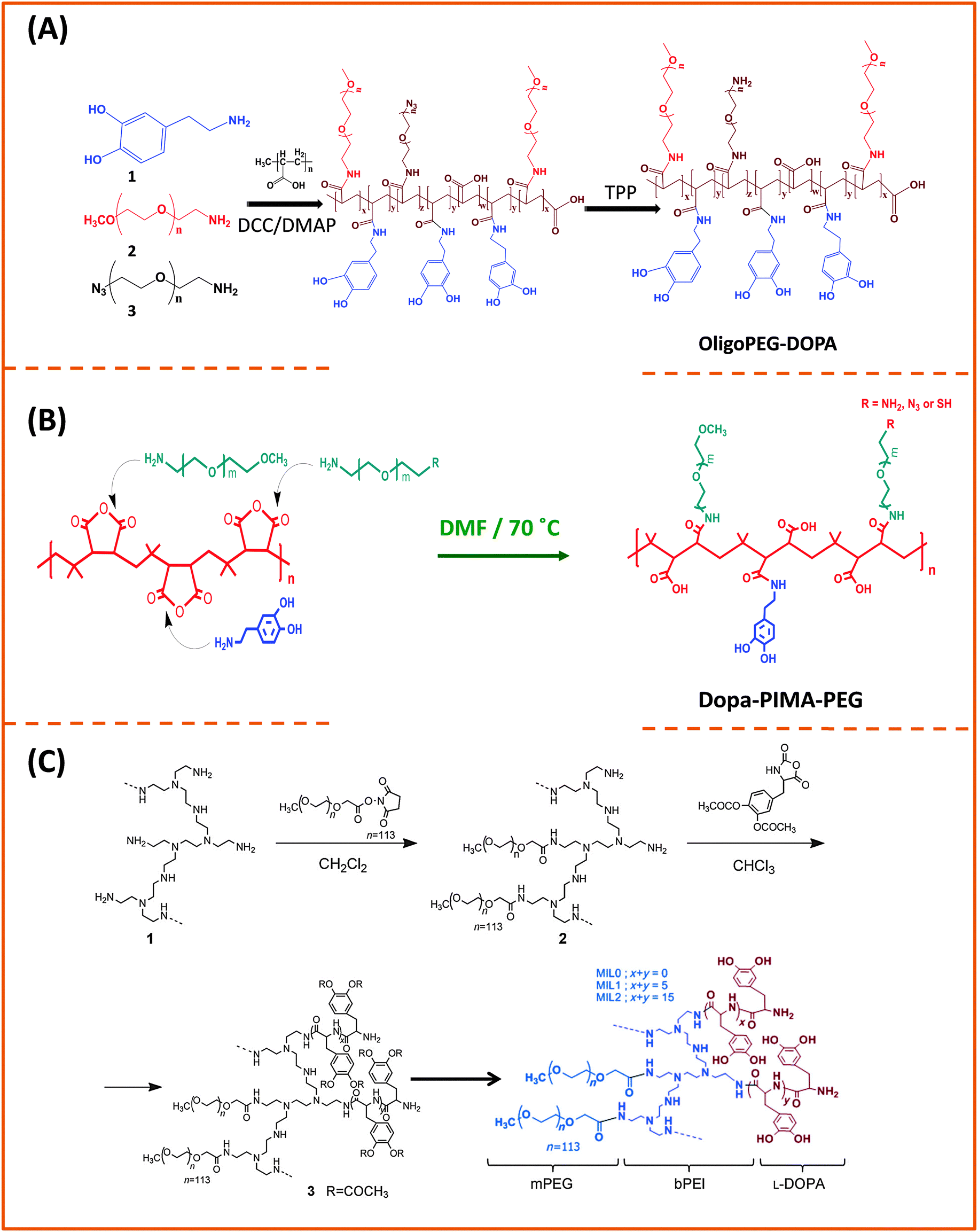 | ||
| Fig. 7 Synthesis of three multi-anchoring poly(dopamine) polymers, starting with: (A) poly(acrylic)acid and DCC coupling; (B) poly(isobutylene-alt-maleic anhydride) and nucleophilic addition reaction; (C) poly-amine as the precursor polymer and NHS along with amine–anhydride coupling.143,172,173 (Figures are adapted from the above references with permission from the American Chemical Society and from Wiley.) | ||
There is an alternative route for designing ligands with strong coordination onto iron oxide nanoparticles, which relies on the use of phosphonic acid groups (instead of carboxyl or dopamine) as anchors.35,174,175 In this approach, phosphonic acid groups are inserted along a polymer chain (with or without polyethylene glycol moieties) to yield multi-phosphonic acid ligands. Such phosphonic acid based polymer ligands were tested by a few groups and were shown to exhibit a higher coordinating affinity towards metal oxide surfaces than their carboxylic acid counterparts, especially under acidic conditions.35,175 Polysaccharides have also been used for coating iron oxide nanoparticles, due to their biocompatibility.176,177 The chelation is driven by the interaction between the hydroxyl groups and the iron oxide surface of the nanoparticles. A ubiquitous polysaccharide that has often been used in these studies is dextran; it is composed exclusively of glucopyranosyl units with varying degrees of branching and chain length. A drawback of this polymer-coating is the relatively high rate of desorption from the nanoparticle surfaces, due to the naturally weak hydrogen bonding; desorption from the nanoparticle surfaces can occur at high dilution or upon heating. However, cross-linking can be used to enhance dextran polymer coating and provide higher colloidal stability in aqueous media.
3.2. Phase transfer via encapsulation within amphiphilic block copolymers
The use of amphiphilic block copolymers for encapsulating various nanocrystals has been widely reported, since this strategy is believed to preserve the photo-physical properties of the native (hydrophobic) nanoparticles. The polymers must contain two distinct blocks with drastically different solubilties, and a balance between the hydrophilic and hydrophobic blocks is crucial for the effectiveness of the encapsulation strategy. We will describe a few established examples where this strategy has been applied for the encapsulation of metal, metal oxide and semiconductor nanoparticles.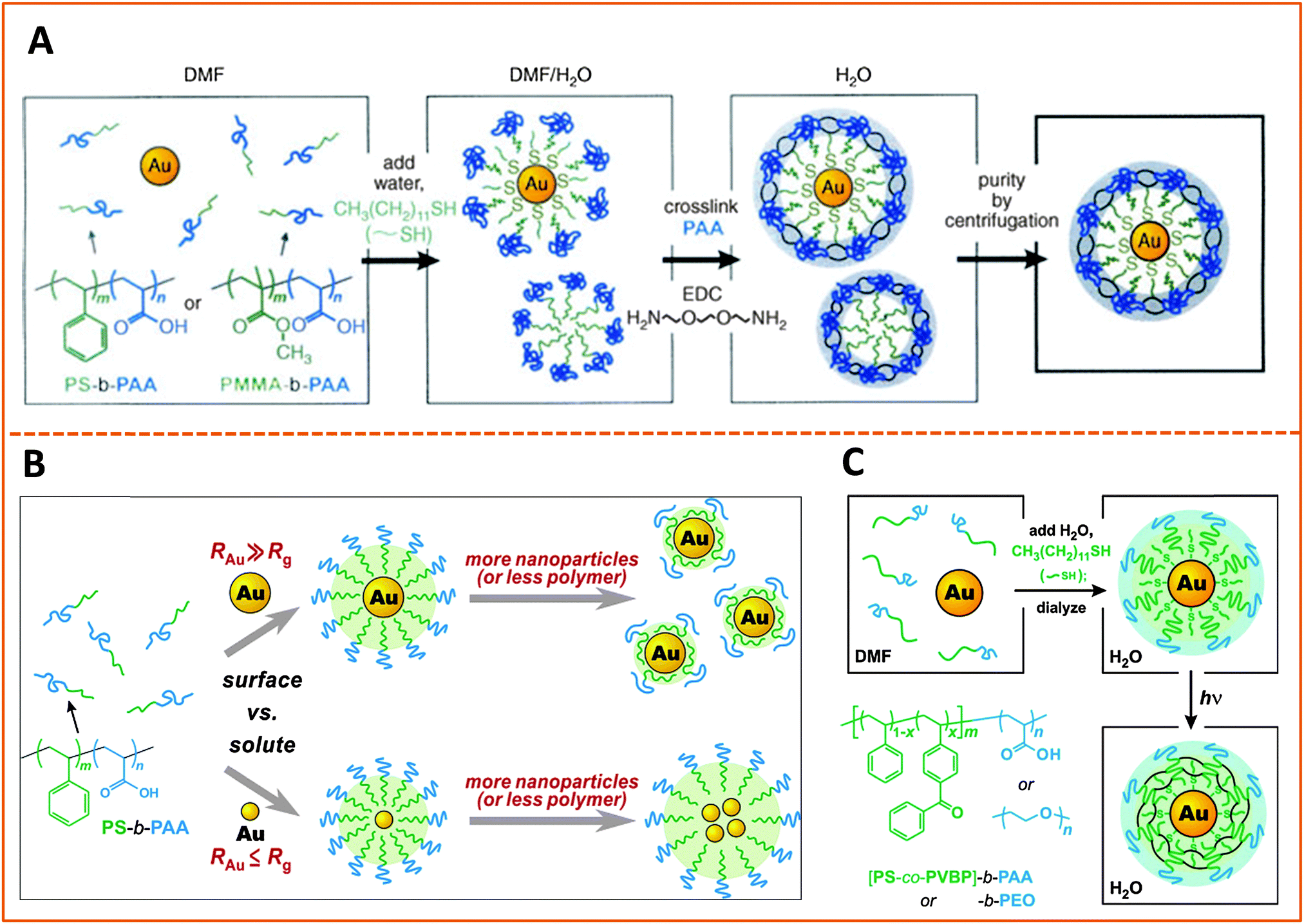 | ||
| Fig. 8 Strategies for encapsulating AuNPs within amphiphilic block-copolymers. (A) Encapsulation using PS-b-PAA and PMMA-b-PAA block-copolymers, followed by EDC cross-linking. (B) Effects of varying the size of the hydrophobic block in the copolymer and/or the size of the AuNP on the structure of the polymer-templated AuNP capsules. Shown are instances where capsules containing single AuNPs vs. a few AuNPs are controlled by changing the ratio between the NP and polymer dimensions, RAu/Rg. (C) Use of photochemically-active benzophenone as a cross linking agent to prepare AuNPs encapsulated within cross-linked PS-b-PAA or PMMA-b-PAA.178–180 (Figures are reproduced from the above references with permission from the American Chemical Society and Wiley.) | ||
In another example Nie and co-workers designed an amphiphilic polymer by chemically substituting 40% of the carboxyl groups of the PAA chain with the 12-carbon aliphatic chain (dodecylamine) via carbodiimide chemistry, and used this co-polymer for the growth of gold nanoparticles. The authors suggested that the amphiphilic polymer forms a three-layer coating on the nanoparticles, with one hydrophobic layer resulting from the self-assembly of two polymer chains, intercalating between two carboxyl-rich lateral layers; one of the carboxyl-rich layers coordinates onto the metal surface while the other one interacts with water, promoting dispersion of the nanoparticles in alkaline solutions.182 With this growth (and coating) route they found that the polymer capsules exhibit pH-dependent conformation, and shedding of the polymer outer layer in acidic pH alters the nanoparticle solubility (NPs become compatible with nonpolar media) along with a decrease in the hydrodynamic radius. They reported that the size of the AuNPs can be controlled from 2 to 15 nm (in situ during the growth phase) by varying the Au-to-polymer molar ratios; lower gold-to-polymer ratios provide small NPs, and vice versa.
The encapsulation strategy has also been applied to surface functionalize AuNRs, albeit with less frequency. In one study, Kim and co-workers utilized a poly(ethylene oxide)–poly(n-butyl acrylate), PEO–PnBA diblock copolymer to encapsulate CTAB-capped AuNRs.183 In this design, the hydrophobic PnBA chains exhibited strong affinity to the gold–water interface, which resulted in the formation of dense micelle assemblies on the Au surface, while the poly(ethylene oxide) block allowed dispersion of nanorods in water media. We should note that the PEO–PnBA polymer used in this study is different from other more commonly used amphiphilic polymers often based on the PS–PAA motif. Here, switching the hydrophilic block from PAA (an ionic system) to PEO (nonionic) provided polymer-coated NPs that are insensitive to changes in the ionic strength of the medium.
Finally, we would like to stress that another form of surface-functionalization using charged polymers (i.e., polyelectrolytes) has been applied by a few groups.184–187 This approach may also be treated as another form of encapsulation within polymeric materials. Here, adsorption of polyelectrolytes, either via direct interaction with the metal-rich surface of the nanostructures, or via layer-by-layer (LBL) self-assembly, has been used to functionalize citrate-stabilized Au nanoparticles or CTAB-coated nanorods.186,188,189 For instance, starting with CTAB-capped AuNRs El-Sayed and co-workers applied LBL to prepare NRs with negatively charged surfaces. For this they simply mixed CTAB-nanorods (positively-charged) with a solution of poly(styrene sulfonate), PSS, which promoted the electrostatic adsorption of the polymer onto the NR, producing a dispersion of PSS-modified and negatively charged AuNRs.190 The materials were further coupled to an antibody and tested for use as photo-thermal therapy platforms (see below). Layer-by-layer self-assembly of polyelectrolyte materials on glass and metallic surfaces has in fact been widely used by several groups to assemble thin polymer films with various properties, and the above data show that this approach can be easily extended to other nanoscale surfaces.191–195
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000) as a flexible platform to prepare amphiphilic polymers to encapsulate various inorganic nanocrystals, including luminescent quantum dots and iron oxide nanoparticles (Fig. 9A).132 The polymer was initially adsorbed onto the hydrophobic QDs in an organic medium (e.g., chloroform solution). Addition of bis(6-aminohexyl) amine initiated the cross-linking of polymer capsules around the nanocrystals. After solvent evaporation, water was used to promote hydrolysis of the unreacted anhydride groups in the polymer capsule, resulting in the dispersion of the QDs in aqueous media; affinity to water is driven by the newly available carboxylic acids along the polymer (and in the capsules). In a follow up study, they coupled ATTO dye molecules pre-modified with the amino group to the alkyl-modified polymer backbone, and used these hybrid complexes to probe the dependence of the energy transfer interactions between QDs and dyes on the environmental conditions.204 In subsequent studies a few other groups expanded the above idea and introduced polyethylene glycol segments into the amphiphilic polymer structure to improve the bio-compatibility of QDs and reduce non-specific interactions. In one of those studies, Colvin and co-workers grafted lateral PEG chains onto the polymer prior to encapsulation of the nanocrystals (Fig. 9B).130 They first formed the amphiphilic polymer by reacting poly(maleic anhydride-alt-1-octadecene) with amine-modified methoxy-terminated poly(ethylene glycol) (NH2–PEG–OCH3, with PEG MW = 6000–20000). The nanocrystals (QDs or iron oxide NPs) were mixed with the polymer in chloroform, and following solution homogenization the solvent was evaporated. Addition of buffer to the medium facilitated dispersion of the materials and provided hydrophilic QDs. In another study Mulvaney and co-workers used an amphiphilic polymer, poly(styrene-co-maleic anhydride), Mw = 1700, which was synthesized via maleic anhydride coupling to either ethanolamine or the amino-PEG derivative Jeffamine M-1000 polyetheramine. The resulting water soluble nanocrystals simultaneously presented PEG as a solubilizing moiety and COOH reactive groups.205 The authors also reported that the experimental procedure described in ref. 130 (i.e. relying on reacting the PEG with the polymer precursor prior to encapsulation) could not be reproduced, whereas the use of Jeffamine M-1000 polyetheramine allowed easier implementation of chemical coupling followed by encapsulation of the nanocrystals.205 The authors subsequently introduced azide groups into the polymer structure and tested their ability to conjugate the resulting azide-functionalized QDs to cyclooctyne-modified proteins (see below). These studies clearly show that the maleic anhydride motif provides a flexible platform to prepare several tailor-made block co-polymers by introducing hydrophobic and/or hydrophilic moieties (e.g., alkyls and PEG moieties) along with the desired functionalities.
000–50000) as a flexible platform to prepare amphiphilic polymers to encapsulate various inorganic nanocrystals, including luminescent quantum dots and iron oxide nanoparticles (Fig. 9A).132 The polymer was initially adsorbed onto the hydrophobic QDs in an organic medium (e.g., chloroform solution). Addition of bis(6-aminohexyl) amine initiated the cross-linking of polymer capsules around the nanocrystals. After solvent evaporation, water was used to promote hydrolysis of the unreacted anhydride groups in the polymer capsule, resulting in the dispersion of the QDs in aqueous media; affinity to water is driven by the newly available carboxylic acids along the polymer (and in the capsules). In a follow up study, they coupled ATTO dye molecules pre-modified with the amino group to the alkyl-modified polymer backbone, and used these hybrid complexes to probe the dependence of the energy transfer interactions between QDs and dyes on the environmental conditions.204 In subsequent studies a few other groups expanded the above idea and introduced polyethylene glycol segments into the amphiphilic polymer structure to improve the bio-compatibility of QDs and reduce non-specific interactions. In one of those studies, Colvin and co-workers grafted lateral PEG chains onto the polymer prior to encapsulation of the nanocrystals (Fig. 9B).130 They first formed the amphiphilic polymer by reacting poly(maleic anhydride-alt-1-octadecene) with amine-modified methoxy-terminated poly(ethylene glycol) (NH2–PEG–OCH3, with PEG MW = 6000–20000). The nanocrystals (QDs or iron oxide NPs) were mixed with the polymer in chloroform, and following solution homogenization the solvent was evaporated. Addition of buffer to the medium facilitated dispersion of the materials and provided hydrophilic QDs. In another study Mulvaney and co-workers used an amphiphilic polymer, poly(styrene-co-maleic anhydride), Mw = 1700, which was synthesized via maleic anhydride coupling to either ethanolamine or the amino-PEG derivative Jeffamine M-1000 polyetheramine. The resulting water soluble nanocrystals simultaneously presented PEG as a solubilizing moiety and COOH reactive groups.205 The authors also reported that the experimental procedure described in ref. 130 (i.e. relying on reacting the PEG with the polymer precursor prior to encapsulation) could not be reproduced, whereas the use of Jeffamine M-1000 polyetheramine allowed easier implementation of chemical coupling followed by encapsulation of the nanocrystals.205 The authors subsequently introduced azide groups into the polymer structure and tested their ability to conjugate the resulting azide-functionalized QDs to cyclooctyne-modified proteins (see below). These studies clearly show that the maleic anhydride motif provides a flexible platform to prepare several tailor-made block co-polymers by introducing hydrophobic and/or hydrophilic moieties (e.g., alkyls and PEG moieties) along with the desired functionalities.
 | ||
| Fig. 9 Representative examples illustrating the phase transfer via encapsulation within amphiphilic block-copolymer micelles applied to: (A and B) semiconductor QDs and magnetic nanoparticles using amine-reactive poly(maleic anhydride). (C) A block-copolymer (PI-b-PEO) prepared via radical polymerization of reactive isoprene groups in the presence of the nanoparticles; the latter approach was also applied to QDs and magnetic nanoparticles.130,132,209 (Figures are reproduced from the above references with permission from the American Chemical Society.) | ||
In related approaches, Winnik and co-workers used PEG grafted polyethylenimine (PEI-g-PEG) and diblock copolymer poly(ethylene glycol-b-2-N,N-dimethylaminoethyl methacrylate) (PEG-b-PDMA) to promote the transfer of QDs to buffer media.206,207 The authors have, nonetheless, attributed this surface functionalization strategy to the removal of native ligands by ionic anchors present on the polymer. A similar tri-block copolymer construct made of poly(poly(ethylene glycol) methyl ether methacrylate)-block-poly(2-dimethylaminoethyl methacrylate)-block-poly(2-dimethylaminoethyl methacrylate-co-octyl methacrylate), [HOOC-PEGMA-b-PDMA-b-(PDMA-co-POMA)], has also been recently used by Gao and co-workers to encapsulate hydrophobic QDs.208 Weller and co-workers have recently described a few interesting developments in amphiphilic polymer design based on block-copolymers and their use to encapsulate individual or combinations of inorganic nanocrystals within a single capsule (e.g., QDs and iron oxide nanoparticles, see Fig. 9C).209–212 They further coupled these capsules to target molecules and used the resulting complexes for cellular imaging. In one study they detailed the use of a chemically designed triblock-copolymer to cap CdSe–ZnS QDs via partial ligand exchange. The polymer consists of a polyethyleneimine binding block (to promote interaction with the inorganic surface via amine binding), a hydrophobic polycaprolactone, and a polyethylene glycol block to facilitate dispersion of the nanoparticles in aqueous media.210 The authors explored the effects of varying the size of the three blocks and showed that 1H NMR could be used to track the polymer binding onto the QDs combined with a progressive removal of the native TOP/TOPO cap. They also found that changing the polymer-to-nanoparticle molar ratio can allow one to vary/control the number of nanocrystals (QDs, Fe3O4 nanoparticles or combination of both) per capsule; capsules containing either one or a few nanocrystals have been made using this route. In a more recent study, they employed in situ seeded emulsion polymerization in the presence of the hydrophobic nanocrystals (with their native cap) to prepare nanocrystals encapsulated within an amphiphilic polyisoprene-block-poly(ethylene oxide) diblock (PI-b-PEO) copolymer that are also reactive.213 With this in situ strategy, combinations of the surfactants, functional monomers, linkers and the radical initiator are sequentially introduced along with the nanocrystals to promote the encapsulation of one type or a combination of nanocrystals within the same capsule.213 In a subsequent report they detailed the synthesis of an amphiphilic miktoarm star copolymer made of two PEO and one PI chain, (PI-b-(PEO)2 star), with control over the arm size via changes in the precursor molecular weights.211 One of the key features of this polymer is its ability to provide an effective hydrophobic shielding around the nanocrystal surface, which drastically reduces diffusion/permeability of copper and iron ions to the QD surface. In particular, they showed that QDs encapsulated with an azide-modified block copolymer can be used to implement copper-catalyzed click reactions with minimal loss in the PL emission, circumventing previous limitations to using such coupling strategy when smaller surface ligands are used.211,214 Cyclic molecules such as calix[n]arenes (with n = 4, 6, and 8) containing carboxylic acid groups were also used to encapsulate luminescent QDs.215
4. Use of inorganic nanocrystals in targeted biological applications
4.1. Bioconjugation to target molecules
Conjugation of biomolecules (e.g., proteins and peptides) to the nanoparticle surfaces is critically important for the successful integration of such platforms in various biological systems. A few chemical coupling methodologies have been applied to conjugate hydrophilic nanoparticles (QDs, AuNPs and magnetic NPs) to proteins, peptides and DNAs. They are (1) avidin–biotin bridging; here, proteins or peptides can be pre-modified with biotin groups to facilitate interactions with streptavidin-functionalized QDs or vice versa.127,148,166 (2) 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxy succinimide) (EDC/NHS), or sulfo-NHS coupling between carboxyl groups on the NPs and amines on the biomolecules, and vice versa.10,127,148 (3) Thiol (–SH) reactive maleimide coupling to cysteine or (sulfhydryl)-modified proteins and peptides, starting with the transformation of the surface reactive groups on the nanocrystals.216,217 (4) Metal-affinity driven self-assembly between polyhsitidine-appended biomolecules and metal-rich nanocrystals;161,162,218 this method relies on the affinity between polyhistidine tags and certain transition metal ions (e.g., Ni and Zn), and requires direct interactions between the imidazole groups (on the tag) and the metal-rich surface of nanoparticles. (5) Azide–alkyne Huisgen cycloaddition (or “Click” reaction), which requires access to biomolecules pre-modified with either alkynes or azides, together with azide- or alkyne-functionalized nanoparticles.145,219–221 The use of avidin binding to the biotin molecule, EDC condensation as well as thiol-to-maleimide coupling strategies were more common in several of the early demonstrations, and this was due to the fact that these protocols have been well established and ubiquitous in biology.222 Metal-histidine driven self-assembly of QD-bioconjugates is extremely attractive, because it is relatively easy to implement (mixing reagents) and can benefit from the ubiquitous use of polyhistidine expression on proteins. Its use as a conjugation strategy is still somewhat limited, because it requires direct access of the imidazole residues to the inorganic surface of the nanocrystals, although it has been gaining interest in the past few years.159,162,223,224 The original azide–alkyne Huisgen cycloaddition reaction requires a copper catalyst in particular when using alkyne-modified molecules.225,226 It has been applied to non-fluorescent NPs such as Fe3O4 nanocrystals.143 However, recent developments have shown that following the ideas originally developed by Bertozzi's group,227,228 copper-free strain-promoted azide–alkyne cycloaddition (SPAAC) coupling can be effective without requiring the need for a copper catalyst. This advance has made “Click” reaction better suited for coupling onto luminescent QDs, since Cu ions can severely quench the QD PL.219 A few demonstrations applying this Cu-free click reaction to conjugate proteins to QDs have been reported over the past few years. Texier and co-workers applied this coupling strategy for conjugating cyclooctyne-functionalized QDs to azide-modified biomolecules.221 They first attached commercially-available carboxy-modified cyclooctyne to the amine-functionalized polyethylene glycol-coated QDs via EDC conjugation, and then allowed the resulting QD–cyclooctyne complexes to react with the azide-modified biomolecules at room temperature. This scheme allowed efficient coupling between QDs and biomolecules while preserving the fluorescence properties of the QDs, namely, quantum yield and spectral integrity. Bawendi and coworkers used norbornene–tetrazine to implement cycloaddition reaction and conjugate tetrazine-biomolecules to norbornene-modified QDs (Fig. 10).219 They first attached commercially available carboxylic-modified norbornene onto a polymeric imidazole ligand (introduced above), via amide coupling, and used the resulting norbornene-modified polymer ligand to cap QDs. They then tested the ability of the resulting norbornene-modified QDs to react (via cycloaddition reaction) with the tetrazine derivative using one (in vitro) solution phase reaction along with another one involving cell membrane (in vivo) labeling. In the first example, they reacted the norbornene-modified QDs with a dye modified with a tetrazine derivative [3-(4-benzylamino)-1,2,4,5-tetrazine (BAT)]. They tested the effectiveness of the conjugation using a combination of optical absorption and energy transfer quenching, and indeed, they found that high levels of QD–dye coupling could be achieved with no drastic losses in QD PL since no copper ions were needed; rather large excess of BAT-dye with respect to QDs was required for the coupling, nonetheless. For cellular labeling they explored two configurations. In the first one, they modified the EGF (epidermal growth factor) with BAT, followed by reaction with norbornene-modified QDs to obtain EGF-coated QDs; the cycloaddition reaction was carried out at 37 °C. Then the resulting conjugates were incubated with A431 human carcinoma cells overexpressing EGF receptors (EGFRs) on their membranes. In the second setting, the BAT-modified EGF was first incubated with the A431 cells to provide BAT-presenting cells. These cells were then incubated with the norbornene-functionalized QDs. They found that the two approaches provided high levels of fluorescence labeling of the cells, compared with minimal QD fluorescence for control preparations. Liu and co-workers used a polyhistidine- and azide-modified block copolymer, starting from polymaleic anhydride, to cap luminescent QDs.145 They showed that following ligand exchange the azide groups were accessible for further conjugation to the Baculovirus pre-modified with cyclooctyne through metal-free “Click” reaction. These conjugates were tested in the intracellular uptake by the A549 cell line. The use of click coupling was also implemented by Mulvaney and co-workers.220 They first introduced azide groups into amphiphilic polymer capsules, then tested their ability to conjugate the azide-functionalized QDs to cyclooctyne-modified transferrin or Alexa Fluor 594, using strain-promoted azide–alkyne cycloaddition (SPAAC) (see Fig. 10).229 They further demonstrated the biological activities of SPACC-promoted QD-transferrin conjugates by monitoring their uptake in cells expressing a high level of transferrin-receptors on their membranes.220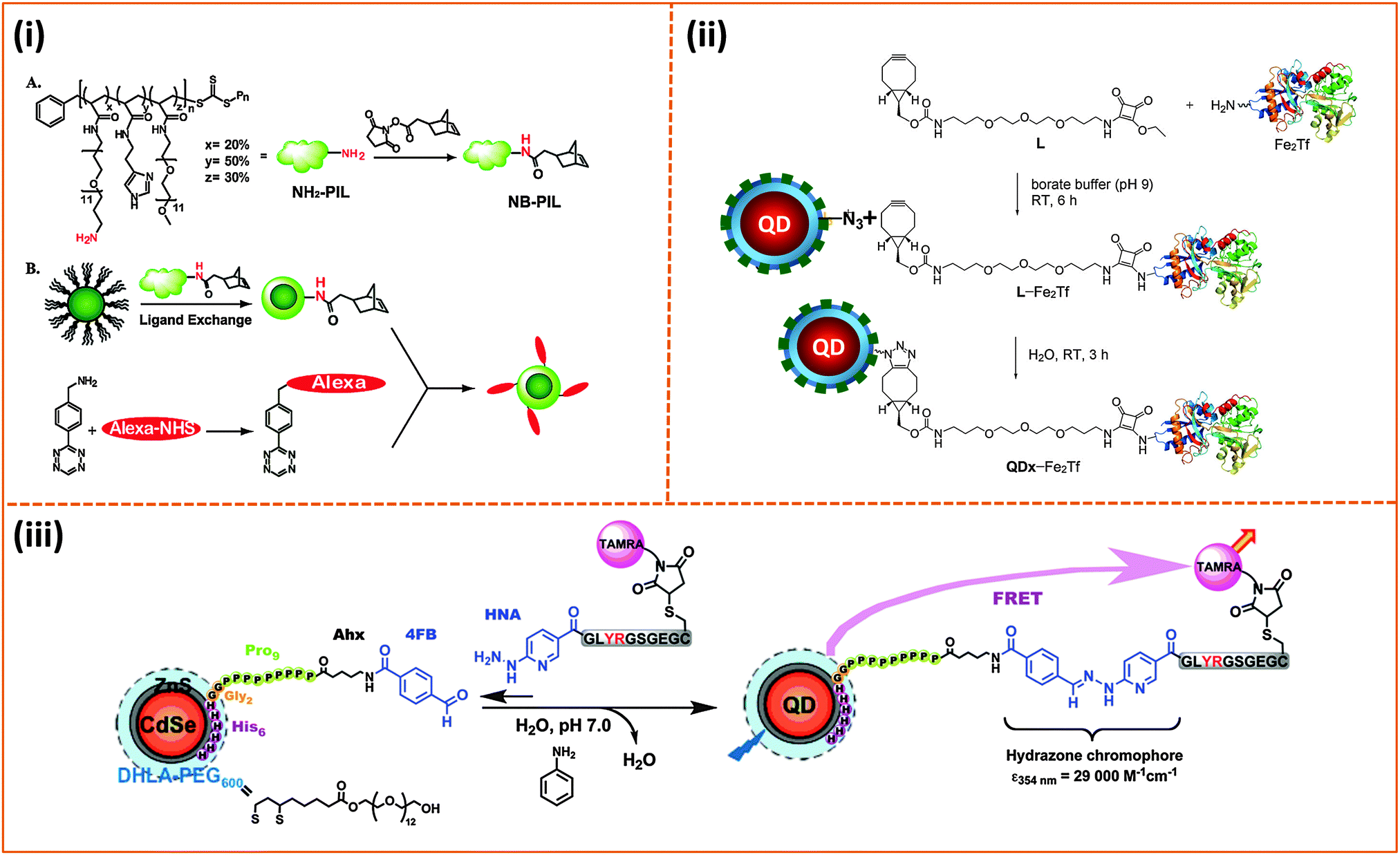 | ||
| Fig. 10 Three representative bio-conjugation strategies: (i) Coupling of norbornene to the NH2-functionalized imidazole polymer (A). Diels–Alder reaction between Alexa 594 pre-modified with 3-(4-benzylamino)-1,2,4,5-tetrazine (BAT) and norbornene-modified QDs (B). (ii) Strain-promoted copper-free azide–alkyne cycloaddition between azide-modified QDs and L-Fe2Tf (L-Fe2Tf denotes the product resulting from coupling of primary-amine Transferrin with ethyl squaramyl to provide a cyclooctyne conjugate used for the Click reaction). (iii) Hydrazone ligation of aldehyde-functionalized QDs with a peptide pre-modified with a HYNIC residue.219,220,230 (Figures are adapted from the above references with permission from the American Chemical Society and from Wiley.) | ||
Applying “Click” coupling to QDs and other NPs constitutes a major advance in promoting better integration of these nanoscale platforms into biological systems. One limitation of this approach, however, stems from the fact that an excess amount of target molecules is still needed to achieve saturation in the coupling efficiency. Aniline-catalyzed hydrazone ligation provides an alternative strategy. We have applied the scheme to couple aldehyde-functionalized QDs to a peptide modified with a 2-hydrazinonicotinoyl group (HYNIC); no polymer functionalized nanocrystals were used though.230 Starting with DHLA–PEG–QDs, the nanocrystals were subsequently self-assembled with a polyhistidine-terminated and aldehyde-modified peptide. The resulting conjugates were reacted with a second HYNIC-modified peptide, and the kinetics of the reaction were monitored optically by tracking the formation of the hydrazone chromophore at 354 nm with time (Fig. 10).230
4.2. Use of nanoparticles in biological imaging and sensing
The use of inorganic nanostructures in biology has focused on taking advantage of their unique physical and optical/spectroscopic properties either to improve the performance of more traditional materials or to develop new ideas that exploit their unique photophysical characteristics. These nanocrystals have a large surface area compared to molecular scale probes. Thus, a single nanoparticle can be easily coupled to several biomolecules with potential control over the orientation and spatial arrangements of the biomolecules in the resulting conjugates. This feature substantially enhances the affinity and biological activity of the resulting conjugates due to, for example, avidity effects. Applications of nanomaterials in biology have increased over the past decade and include use as fluorescent labels for live cells and tissue imaging, drug and gene delivery vehicles, detection of pathogens and soluble heavy metals, sensing of protein–protein, protein–DNA interactions as well as DNA hybridization. In this section, we will focus on a few representative examples where polymer-coated nanocrystals (QDs, AuNPs and magnetic nanoparticles) have been used as platforms for imaging and/or sensing.AuNPs and AuNRs are very effective fluorescence quenchers of dye and QD emission, with quenching efficiencies exceeding those predicted by the Förster dipole–dipole interaction formalism.234–236 However, use of polymer coated-AuNPs and AuNRs to develop bio-motivated sensors based on energy transfer has not been actively explored, presumably due to the fact that this surface-functionalization route can increase the separation distance and reduce the quenching efficiencies. Nonetheless, there have been a few reports on sensor design using the direct coordination of thiol-modified dye-labeled DNA and peptides.233,237,238 More recently the use of metal-histidine coordination to self-assemble fluorescent proteins on AuNPs with very high quenching efficiencies has been explored by a few groups.162,239 The combination of dyes, fluorescent proteins and QDs with AuNP- or AuNR-quenchers has been utilized by a few groups to develop sensing platforms for targeting protein–protein interactions and competitive binding assays.231,240,241
In an early demonstration, Kim and co-workers developed an inhibition assay to detect protein glycosylation based on changes in the PL quenching of QDs when assemblies of carbohydrate-conjugated QDs and lectin-conjugated AuNPs are formed; lectin is known to exhibit high-affinity to manno- and gluco-oligosaccharides.242 They first conjugated AuNPs to concanavalin A (conA). Then, amine-terminated co-polymer encapsulated QDs (provided from Invitrogen) were conjugated to dextran (polymerized glucose). Sensing of the saccharides was carried out using a competition assay format where the target molecules competed with the QD–dextran conjugate for interactions with conA on the AuNPs. The value of apparent binding constant (Ka) extracted from their titration procedure was similar to the previously reported value ≅ 1.0 × 107 M−1. This construct was then applied to probe differences between avidin and its non-glycosylated derivative, neutravidin, and between bovine serum albumin (BSA) and its chemically-modified neoglycosylated form, 22-MB (BSA–α-D-mannopyranosylphenyl isothiocyanate with 22 mannose units per BSA). In both cases, the inhibition of the QD-dextran binding to AuNP–ConA conjugates manifested in a substantial reduction in the QD PL quenching. Furthermore, a correlation between reduction in the PL quenching and the target concentration was observed only for the glycosylated proteins (avidin and 22-MB), which clearly proved that the specificity of the ConA protein on the AuNPs was maintained. They expanded the utility of this sensing scheme and tested its ability to differentiate between glycoproteins having different glycan density profiles per molecule. For this, they used recombinant glucose oxidases expressing different lengths of mannose glycans and found that changes in the PL emission signature closely traced the number of glycan groups present in the target protein.242 Rotello and co-workers explored the use of AuNPs capped with labile cationic ligands to assemble new platforms that can serve both as delivery vehicles and intercellular probes.243 These nanoparticles were partially functionalized with a small fraction of thiol-modified fluorescein (FITC-SH). They found that upon assembly on the NPs the fluorescence of the FITC was completely quenched, due to nonradiative energy transfer between FITC and the AuNPs. Addition of glutathione (GSH) to these assemblies promoted the release of the FITC dye from the AuNPs, resulting in the recovery of dye emission. This provided them with an analytical tool, based on changes in the fluorescence emission, to quantify the rate of ligand release from the NP surface. The GSH-induced and concentration-dependent release of the FITC from the AuNP surfaces combined with changes in the fluorescence signal provides a promising drug delivery and sensing platform with potential use in live cells using NPs decorated with a drug or a mixture of drug and fluorophores.243 In a follow up study, they assembled a new fluorescent protein (GFP)–AuNP conjugate exploiting electrostatic interactions of GFP with the cationic ligands on the gold NPs. This provided a flexible platform to detect specific soluble proteins in buffer media and in human serum, via competitive interactions with the surface bound GFP; this format exploits the weak electrostatic interactions driving the NP–GFP conjugate assembly. They applied this platform to sense five serum proteins (human serum albumin, immunoglobulin G, transferrin, fibrinogen and α-antitrypsin), both in buffer and when spiked into human serum. Combining this fluorescence assay with a linear discriminant analysis they were able to identify those soluble proteins with an identification accuracy of 100% in buffer and 97% in human serum.244
The large and tunable plasmonic absorption cross section of gold nanostructures (including spherical nanoparticles, nanorods, nanoshells and nanocages) from the visible to the NIR promotes controlled local heating driven by remote laser irradiation in the visible and/or NIR. This process has been exploited by a few groups to develop photothermal platforms, which can ablate cancerous tissues and malignous cells.245 We will describe three representative examples where this idea was tested in vitro and in vivo.
In one early study, El-Sayed and co-workers exploited the pronounced SPR peak of gold nanorods in the NIR to develop AuNR-based therapy platforms that can promote photothermally-induced death of cancer cells. Starting with poly(styrene sulfonate)-coated AuNRs (introduced above) they assembled a few copies of the anti-EGFR (epidermal growth factor receptor) antibody onto the NRs via electrostatic physisorption on the negatively charged nanorods. When the NR dispersions were incubated with two malignant epithelial cell lines (HOC 313 clone 8 and HSC 3), only anti-EGFR–AuNR conjugates bound to the membrane of malignant cells expressing EGFR. Exposure to irradiation at 800 nm provided by a CW laser induced a pronounced level of cell death (Fig. 11).190 They also found that to induce the same level of cell death, malignant cells required about one half of the laser power to induce a similar rate of cell death when the NRs were incubated with a culture of nonmalignant (HaCat) cells. This difference is attributed to the lack of specific interactions of the antibody-AuNRs with the control culture of non-malignant cells, thus producing much smaller concentration of AuNRs at their membrane and less thermal heating. In the second study, Murphy and co-workers combined layer-by-layer polyelectrolyte adsorption and CTAB-stabilized NRs to assemble a platform that allows remotely-controlled release of molecules embedded within the surface coating layer.186 To prove this concept, they integrated Rhodamine 6G within a polyelectrolyte coating multilayer using layer-by-layer self-assembly of poly(acrylic acid, sodium salt) (PAA) and poly(allylamine hydrochloride) (PAH) on CTAB–AuNRs. They then showed that upon laser irradiation with a NIR signal, a sizable and power-dependent dye release from the AuNR surfaces takes place. In addition, they found that the rate of dye release was correlated with the number of bilayers adsorbed onto the NR and the time of laser irradiation used (Fig. 12).186 This design can potentially be applied to promote the drug delivery in cell cultures and tissues. In the third example, Xia and co-workers used gold nanocages as a remote-controlled drug delivery system. They started with poly(vinylpyrrolidone) (PVP)-coated Au-nanocages, grown via a galvanic replacement reaction between truncated Ag nanocubes and chloroauric acid (HAuCl4).246,247 The PVP coating was exchanged with a poly(N-isopropylacrylamide) (pNIPAAm) polymer. Because the pNIPAAm copolymer exhibits thermo-sensitive properties (change in conformation with small temperature variations), the PINAM-encapsulated nanocages offer an efficient carrier for the delivery of cargo molecules (drugs and else) to target tissue, followed by remote release via laser irradiation. In one example, the authors loaded alizarin–PEG (Dye–PEG), or doxorubicin (Dox, a drug) inside the cage by mixing at 42 °C with continuous shaking. They found that NIR photo-irradiation (using a laser power of ∼10 mW cm−2) produced enough thermal energy to change the polymer conformation around the cages, releasing the embedded dye or drug. Such release was permitted by the thermally-induced change in the pNIPAAm conformation, opening pores along the cage walls and allowing diffusion of the drug into the surrounding medium (Fig. 13). Here too they measured laser power-dependent release of the drug in the medium.
 | ||
| Fig. 11 (A) Light scattering images of cells incubated for 30 min with: anti-EGFR–AuNPs (top), and anti-EGFR–AuNRs (bottom). (B) Typical examples of photothermal therapy of cancer cells (HSC and HOC malignant cells) incubated with anti-EGFR–AuNRs. The circles designate the area exposed to laser irradiation. At a laser power of 80 mW (10 W cm−2), the malignant cells are damaged while the HaCat normal cells are not affected. Higher powers (120 mW and 160 mW) are required to damage HaCat (normal) cells.190 (Figures are adapted from the above reference with permission from the American Chemical Society.) | ||
 | ||
| Fig. 12 (A) Schematic of the surface-coating and dye loading onto AuNRs using adsorption of a few polyelectrolyte layers. (B) Percentage of dye release vs. laser irradiation time for different samples: (a) AuNR + PAA + R6G + PAA (black); AuNR + PAA + R6G + PAA + PAH (red); AuNR + PAA + R6G + PAH (green); and AuNP + PAA + R6G + PAA (blue); (b) percentage of dye release vs. time for AuNR + PAA + R6G + PAA at a given temperature in a water bath. The black curve in the right panel is reproduced from the left panel and refers to the drug release under laser irradiation.186 (Figures are adapted from the above references with permission from the American Chemical Society.) | ||
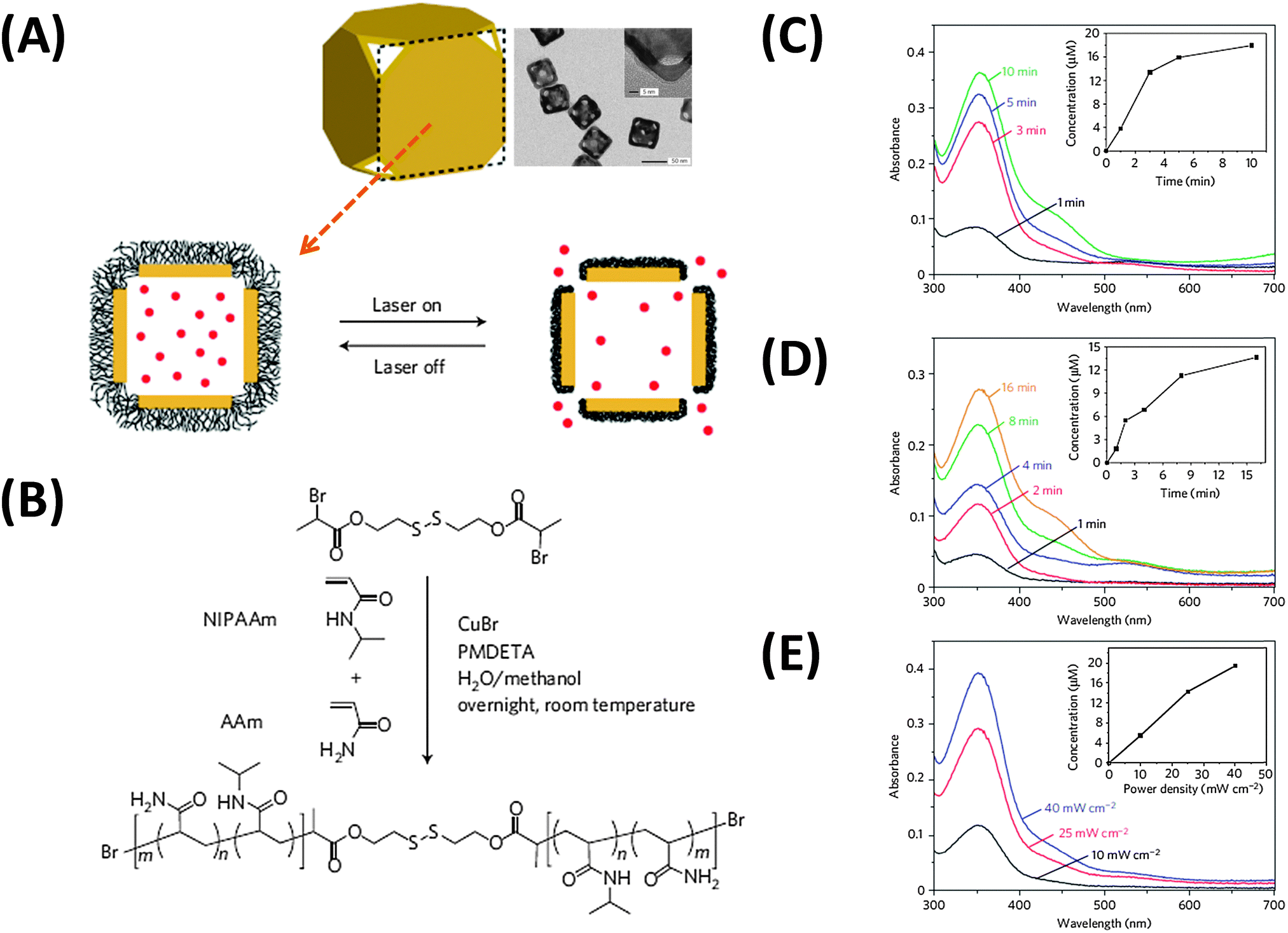 | ||
| Fig. 13 (A) Schematic illustration of the drug release from Au nanocages upon irradiation with a near-infrared laser source. The absorbed NIR photons are converted into heat, triggering the swelling of the amphiphilic polymer and release the drug molecules. When the laser is turned off, the polymer coating relaxes back to its initial conformation, preventing any further release of the drug. (B) Synthetic scheme of the amphiphilic polymer using RAFT. (C–E) represent the drug release rate under different conditions, namely, upon heating at 42 °C at different times (C), for a given laser power but different irradiation time (D), and for a given irradiation time but different laser power (E).247 (Figures are adapted from the above reference with permission from Nature Publishing Group.) | ||
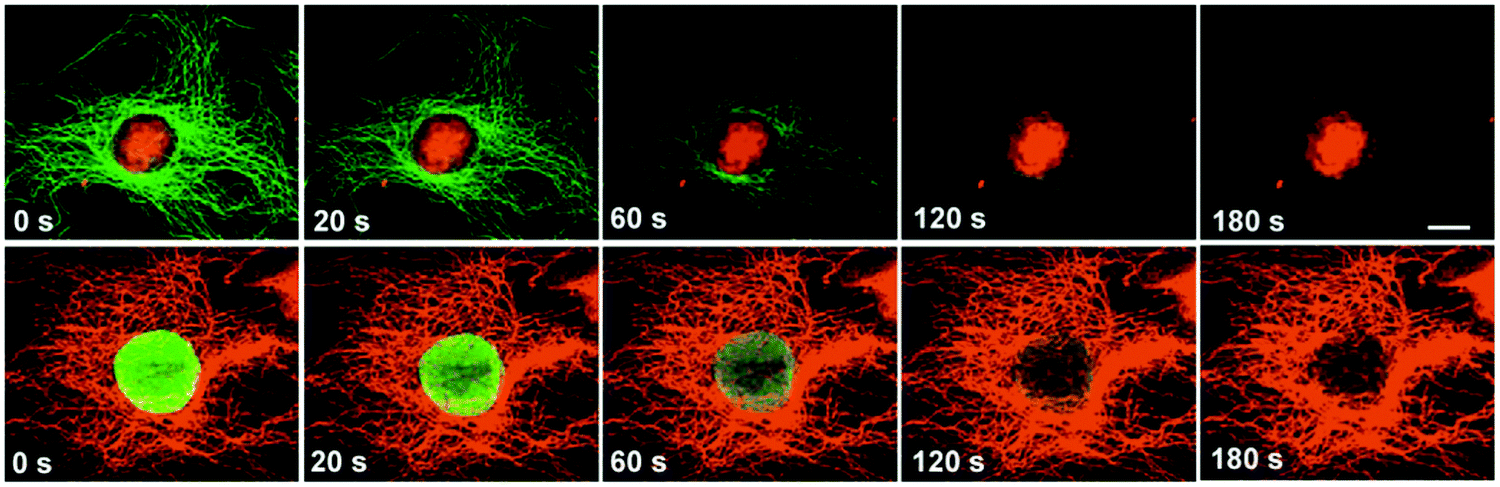 | ||
| Fig. 14 Side-by-side comparison of the photo-stability of QDs and Alexa 488. Top row: the Nuclei and microtubules of 3T3 cells were respectively labeled with QD 630–streptavidin (red) and Alexa 488 conjugated to anti-mouse IgG (green). Bottom row: microtubules were labeled with QD 630–streptavidin (red) and nuclear antigens were stained green with Alexa 488 conjugated to anti-human IgG.10 Scale bar, 10 μM. (Figures are adapted from the above reference with permission from the Nature Publishing Group.) | ||
However, the difficulties associated with the controlled delivery of QDs into the cytosols of live cells have limited the effective use of these materials to label and track protein receptors inside the cytosol. Much better success has been achieved in applications focusing on biological processes that occur at the outer surface of the cell (namely, membrane specific phenomena).200,248,249 Several attempts to achieve cytosolic delivery of QD-conjugates have been reported by various groups, albeit with little to modest success. In one example, Helms and co-workers relied on the ability of pH-dependent conformational change of a polymer coating on the QDs to initiate endosomal disruption and release of the nanocrystals in the cytosol. They first synthesized a cationic core–shell polymer colloid, where the core is made of a pH-buffering proton sponge using poly(ethylene glycol) dimethacrylate (PEGDMA, MW ∼ 330 Da) cross-linked poly(2-(diethylamino)ethyl methacrylate), PDEAEMA, and the shell is made of poly(2-aminoethyl methacrylate), PAEMA. Then streptavidin-coated QDs (605-SAQDs) were immobilized inside the polymeric colloids via multivalent electrostatic interactions between the ammonium ions on the polymer colloid shell and carboxylate functions of the streptavidin side chains. They showed that the QDs encapsulated within this polymer could be delivered into live cells after extended incubation (2–4 hours) via endocytosis.250 Once in the endosome the polymer–QD assemblies experience a chemical shock due to a pH change in acidic conditions, which substantially increases the polymer colloid dimensions, triggering the disruption of the endosomal compartments and measurable release of QDs into the cytosol. Recently, Bawendi and coworkers tried to address the problems associated with the inability to achieve direct delivery of QDs and QD-conjugates into the cytosol of live cells. They designed a microfluidic device that forces live cells to rapidly pass through a constriction in the microfluidic channel.251 This constriction transiently disrupts the membrane of the cell by opening a few pores and allowing exchange of materials with the extracellular matrix (Fig. 15A). Using this device combined with fluorescence microcopy they were able to track the slow diffusion of fluorescent materials across the cytoplasm (Fig. 15B and C).
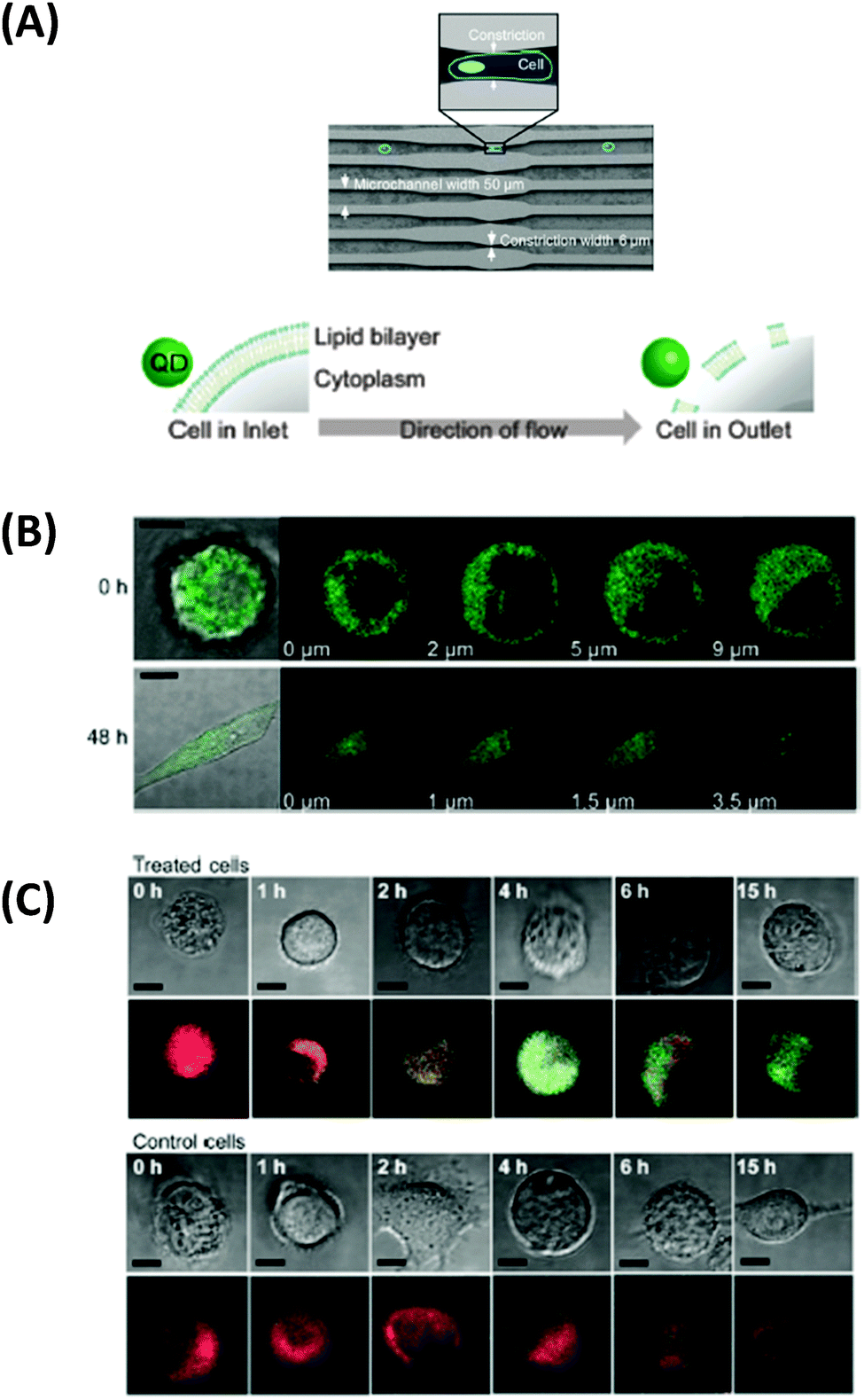 | ||
| Fig. 15 (A) Schematic diagram of the microfluidic device used for the cell uptake (top), along with the hypothesized method of intracellular entry of QDs (bottom). (B) Overlay of DIC and confocal fluorescence image of representative cells, along with a series of z-section fluorescence images of cells with delivered QDs; shown images are immediately after treatment and after 48 h. (C) Confocal microscopy images of live treated and control cells. The observed diffuse staining is limited to the cytoplasm, with no QD fluorescence emanating from the nucleus (dark region within the cell).251 Scale bar is 10 μm. (Figures are adapted from the above reference with permission from the American Chemical Society.) | ||
Raymo and co-workers explored the ideas of achieving intracellular photo-cleavage of surface bound groups. They started with an amphiphilic polymer ligand presenting multiple copies of DHLA anchoring groups and reactive PEG chains, described in an earlier report,150 to assemble QD platforms decorated with photo-cleavable 2-nitrobenzyl groups.252 The nitrobenzyl groups were attached onto the polymer coating using covalent carbodiimide chemistry. They first tested these assemblies in solution, and found that following QD–nitrobenzyl formation the nanocrystal fluorescence signal was drastically quenched, due to electron transfer interactions between the QD and proximal nitrobenzyl groups. Following photolysis of the 2-nitrobenzyl appendages, they measured a large recovery of the QD emission (by more than 60%), a process attributed to a reduction in the rate of electron transfer between nanocrystal and nitrobenzyl groups promoted by the photoinduced release of the organic chromophores from the QDs. These assemblies were then tested in live cells, where they showed that following cellular uptake (via endocytosis) and photo-irradiation of the culture, the intracellular fluorescence signal emanating from the QDs increases by ∼80% after photo-cleavage of the 2-nitrobenzyl quenchers.252
The influence of the QD surface charge distribution on the in vivo binding and transport of these materials was recently investigated by Bawendi and coworkers.163 For this, they compared the interactions of QDs surface ligated with two sets of zwitterion polymers that share the same imidazole anchoring groups: (1) sulfobetaine-functionalized poly(imidazole) ligand, and (2) carboxybetaine-functionalized poly(imidazole) ligand. Here they were able to control the fraction and nature of exposed charges on the QD surfaces. They evaluated the influence of the spatial charge configuration presented on the QDs coated with these two sets of polymer ligands on the QD interactions with cultured HeLa cells. They found that even for nanocrystals that are neutral or slightly charged, the number of spatially exposed amines (essentially positively charged surface groups) play an important role in determining the level of uncontrolled nonspecific binding to the cells. For instance, they measured significantly higher degrees of nonspecific binding to cells for QDs with coating that possess unconverted tertiary amines. In comparison, polymer coatings that endow QDs with non-exposed amines exhibit little to no binding to cells; these QDs still present a small negative charge on their surfaces. These findings highlight the importance of charge distribution and access of those charges to receptors on cell surfaces. They further complemented their findings by carrying out tumor transport measurements, and showed that non-ionic QDs extravasate from vessels into tumor tissues much faster (an order of magnitude faster) than QDs displaying zwitterionic coating with a net negative surface charge distribution. Overall, this study suggests that effective spatial screening of positively charged groups on the nanoparticles is the best route to minimize nonspecific interactions of such materials with biological media.163
The use of QDs as platforms (mostly as exciton donor) to design sensing assemblies based on energy transfer has been explored by several groups over the past decade. Several groups explored the design of sensors to detect enzymatic activity, protein–protein binding, small molecule detection, DNA hybridization and telemorization, using primarily QDs capped with small molecules (compact), as the FRET (fluorescence resonance energy transfer) process requires close proximity between dots and dyes in order to be effective.160,253,254 However, the use of polymer capped or encapsulated QDs for sensing based on energy transfer interaction has been less explored, since polymer capping has often produced large hydrophilic QDs.29,255 Nonetheless, a few groups have managed to prepare compact hydrophilic QDs, mainly relying on ligand exchange using multi-coordinating polymers.7,152,255 For example, Nocera, Bawendi and co-workers have explored two pH sensor designs based on FRET and QD donors. In one example, they conjugated QDs encapsulated within a modified amphiphilic poly(acrylic acid) polymer to a squaraine dye known to exhibit a pH-dependent absorption profile, via EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) condensation. This promoted proximal FRET interactions with high efficiency that vary as a function of the environmental pH. In particular, the authors showed that modulation of the FRET efficiency by varying the solution pH values below and above the pKa of the dye (∼8.5) produced net ratiometric dependence between the QD and dye emissions. This also provided a simple means to measure the solution pH, by analyzing the ratio of the QD and dye peak intensities, or comparing them to the value at the isosbestic point. Because the ratiometric measurements are potentially not sensitive to fluctuations in the overall collected signals, such an approach can be more accurate and more reliable than ‘conventional’ chemo- or bio-sensors that utilize one signal response (i.e., either brightening or darkening). This design was further expanded by Snee and co-workers who used a blue emitting CdS–ZnS QD paired with a fluorescein dye to create a ratiometric pH sensor, similar to the one described above. They used a surface-functionalization scheme based on encapsulating the nanoparticles within a thiolated amphiphilic polyacrylic-octylamine ‘raft’ block-copolymer to promote water transfer of the QDs. Because the thiol on the polymer did not interact directly with the nanoparticle surface, it offered a free target site for attaching a maleimide-activated dye (such as fluorescein or BODIPY), which then provided an acceptor with pH-dependent absorption properties. The resulting QD–polymer–dye construct exhibited a pH-dependent rate of energy transfer between QD and dye. Subsequently Bawendi and co-workers showed that QDs capped with poly-PEG-imidazole (PEGPIL) exhibit compact lateral extension and the resulting fluorescent platforms were suitable for implementing FRET interactions with proximal dyes. In a recent study, they used PEGPIL–QDs to assemble a FRET-based pH sensing platform.255 To build the NC-based pH sensor, they coupled a carboxy-X-rhodamine (Rox) to the 3′ overhanging end of a cytosine-rich 35-mer oligonucleotides sequence. This sequence was hybridized with a 16-mer oligonucleotide appended at the 3′-terminus with a thiol group, which could act as a point of attachment for polymer-coated NCs. The use of cytosine-rich oligonucleotide sequences allowed them to exploit a known property of these systems to undergo folding or unfolding in response to changes in pH, depending on the protonation state of the cytosine imino group. They verified that successful conjugation of the oligonucleotide on the QDs is accompanied by changes in the optical absorption and in particular the fluorescence signature of the conjugates compared to QDs and dye alone. When the pH of the buffer solution was varied from 8 to 6, they measured a pronounced change in the relative intensities of the QD and Rox. In particular, they found that at high pH values, low FRET interactions produce a strong emission from the QD combined with a weak contribution from the dye, which is consistent with reduced FRET efficiency and with the expected unfolded configuration of the oligonucleotide hybrid, keeping the donor and acceptor far apart. Conversely, when the buffer pH value is lowered, folding of the oligonucleotide hybrid brings the QD in close proximity with the Rox and enhances the energy transfer efficiency. This produces a composite fluorescence spectrum that shows an increasingly quenched QD signal concomitant with enhancement in the Rox contribution, with a pH-dependent change in the QD and dye contributions to the measured fluorescence. They further applied this construct to probe intracellular pH changes.
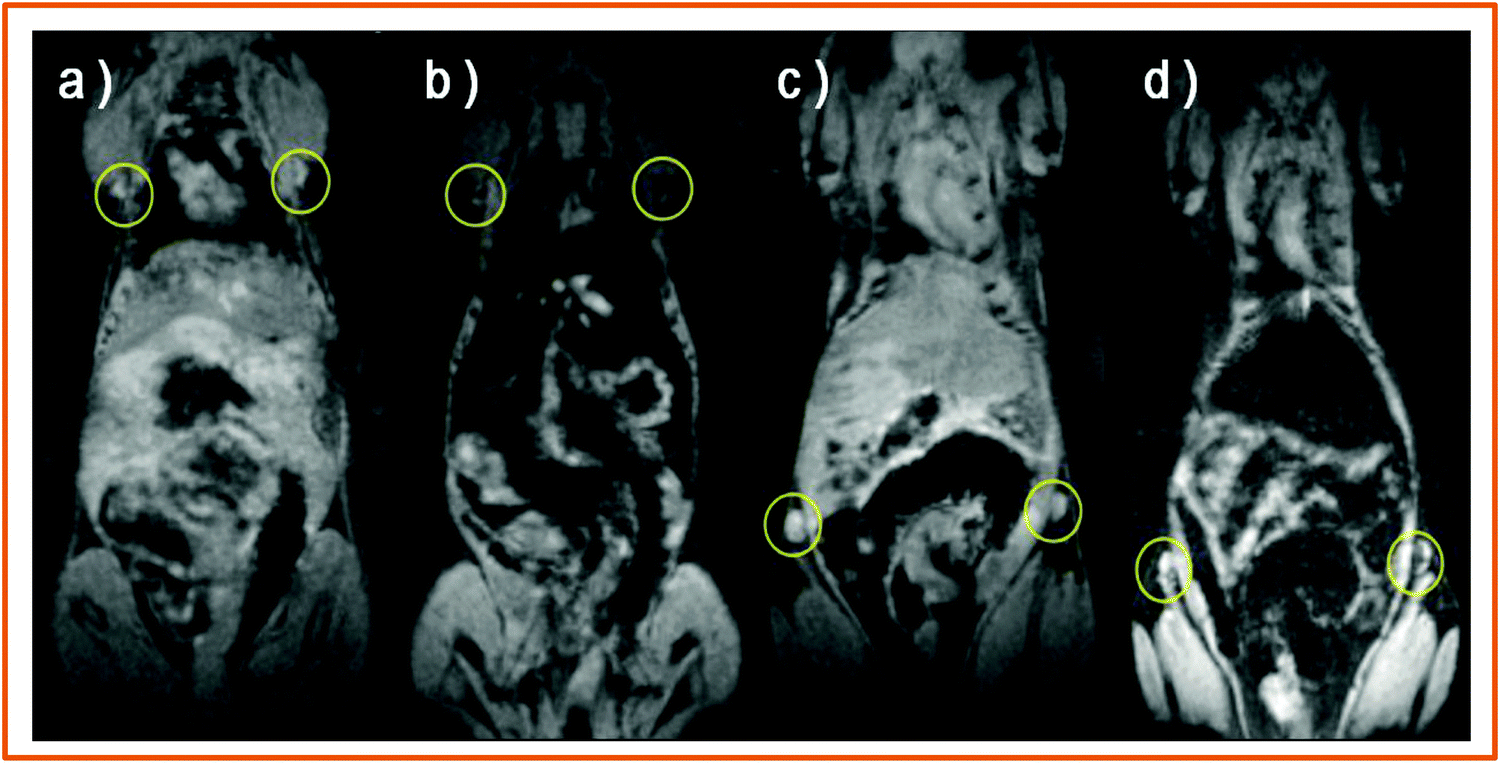 | ||
| Fig. 16 Comparison of T2-weighted MR images of brachial lymph node (a, b) and inguinal lymph node (c, d) in nude mouse. Shown images are collected before intravenous injection of MIL2-functionalized Fe3O4 nanoparticles (a, c) and after 24 h after injection (b, d).173 (Figures are adapted from the above reference with permission from Wiley.) | ||
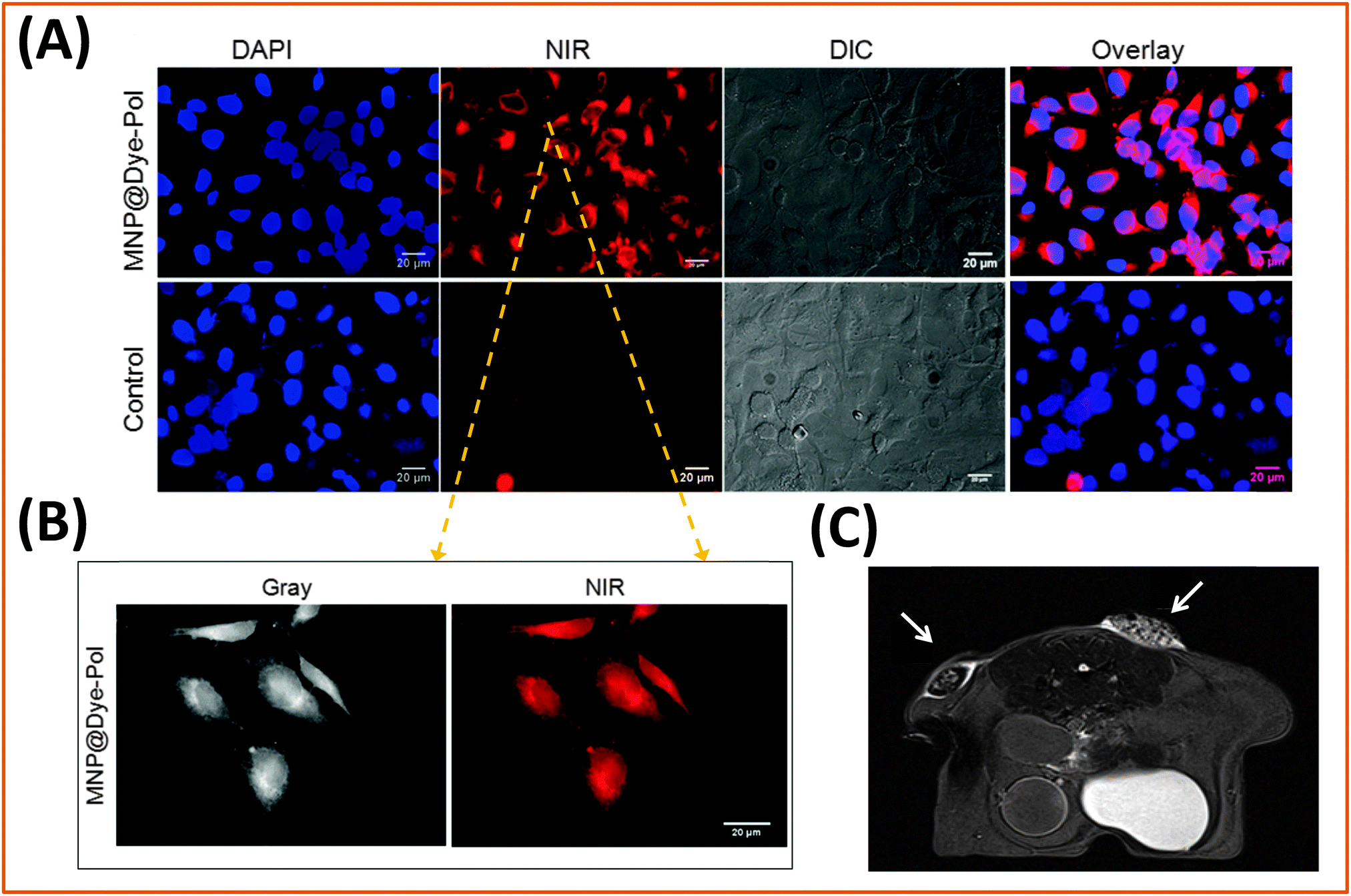 | ||
| Fig. 17 (A) Labeling of HeLa cells incubated for 2 h with 50 μL of magnetic nanoparticles encapsulated with a dye-conjugated-polymer, MNP@Dye-Pol, (1 mg mL−1). Lower panel shows images of untreated cells (control); (B) higher magnification images. Scale bar: 20 μm; (C) T2-weighted MR imaging of a murine model after subcutaneous injection of 0.7 mM MNP@Dye-Pol fixed in 0.8% agarose. The two arrows indicate distinct sites with a sizable contrast difference; this was attributed to the difference in the molecular weight of the encapsulating polymer used.257 (Figures are adapted from the above reference with permission from the American Chemical Society.) | ||
Apart from imaging, iron oxide nanoparticles (in particular, those rendered hydrophilic and functional using a polymer coating) have also been used to develop magnetic sensors based on changes in the T2-relaxivity; they were further applied to detect full size biomolecules (such as antibody and proteins), as well as small target molecules in solution. For example, Weissleder and co-workers used commercially-available, aminated cross-linked iron oxide nanoparticles (CLIO-NH2) and conjugated them to either a synthetic oligonucleotide complementary to TTAGGG telomeric repeats, or to a polyclonal anti-hTERT antibody, to provide sensing platforms capable of simultaneously detecting protein levels and enzymatic activities in solution.259,260 They showed that mixing the two magnetic platforms in the same solution can allow the detection of different amounts of telomerase protein and measure telomerase activity in various cancer and normal cell lines, and furthermore assess the contribution of phosphorylation to the telomerase activity. The NP–oligonucleotide conjugates were prepared by attaching the complementary sequence 5′-CCC-TAA-CCC-TAA-CCC-TAA-3′ directly to the amine groups on the polymer coating of the NPs. Conversely, the NP–anti-hTERT antibody conjugates were formed by coupling protein G (bridging protein) onto the amine groups on the NPs via EDC coupling, followed by incubation with a required amount of anti-hTERT. For both constructs interactions of the magnetic platforms with the target molecules (antigen or telomerase) resulted in a sizable decrease in the T2 relaxation of water molecules. In particular, they applied these conjugates to various cell lysates (including breast cancer, lymphoma, HCC, liver metastasis, prostate, insulinoma, melanoma, and melanocytes) obtained from tumor and normal cell lines. They were able to estimate the protein concentrations from changes in T2 values for each set of lysates (Fig. 18). Combining these two sets of relaxation data collected using both magnetic platforms, they found that the HCC cell line had the highest telomerase activity, while the melanoma cell line had the highest amount of telomerase protein. More recently Perez and co-workers extended this sensing idea to calculate the dissociation constant (KD) for a broad range of protein–ligand (e.g., small drug molecule) interactions in solution. They first showed that binding of a protein to a ligand immobilized on a Fe3O4 NP resulted in an increase in the T2 relaxation times of water protons in solution, indicating that magnetic relaxation nanosensors, capable of sensing the presence of target proteins (from increases in the water T2 values upon binding to the NPs), can be easily assembled. Furthermore, this transduction mechanism is fully suitable for competition assay format in solution, where increase in the concentration of competing ligand progressively disrupts the binding of specific target protein to the magnetic conjugates; this results in a smaller increase in the T2 MR signal. Thus, mixing increasing concentration of competing ligand with the nanosensor assembly containing the target molecule ligand, a quantitative titration curve can be built to provide an accurate estimate for the value of KD. They used this sensing scheme to measure the dissociation constant for several protein-to-ligand pairs, including avidin–biotin, protein G–IgG, dextran–concavalin A, and the folic acid–folate receptor. In particular, they showed that for certain target ligands sub-femtomolar concentrations can be measured.261
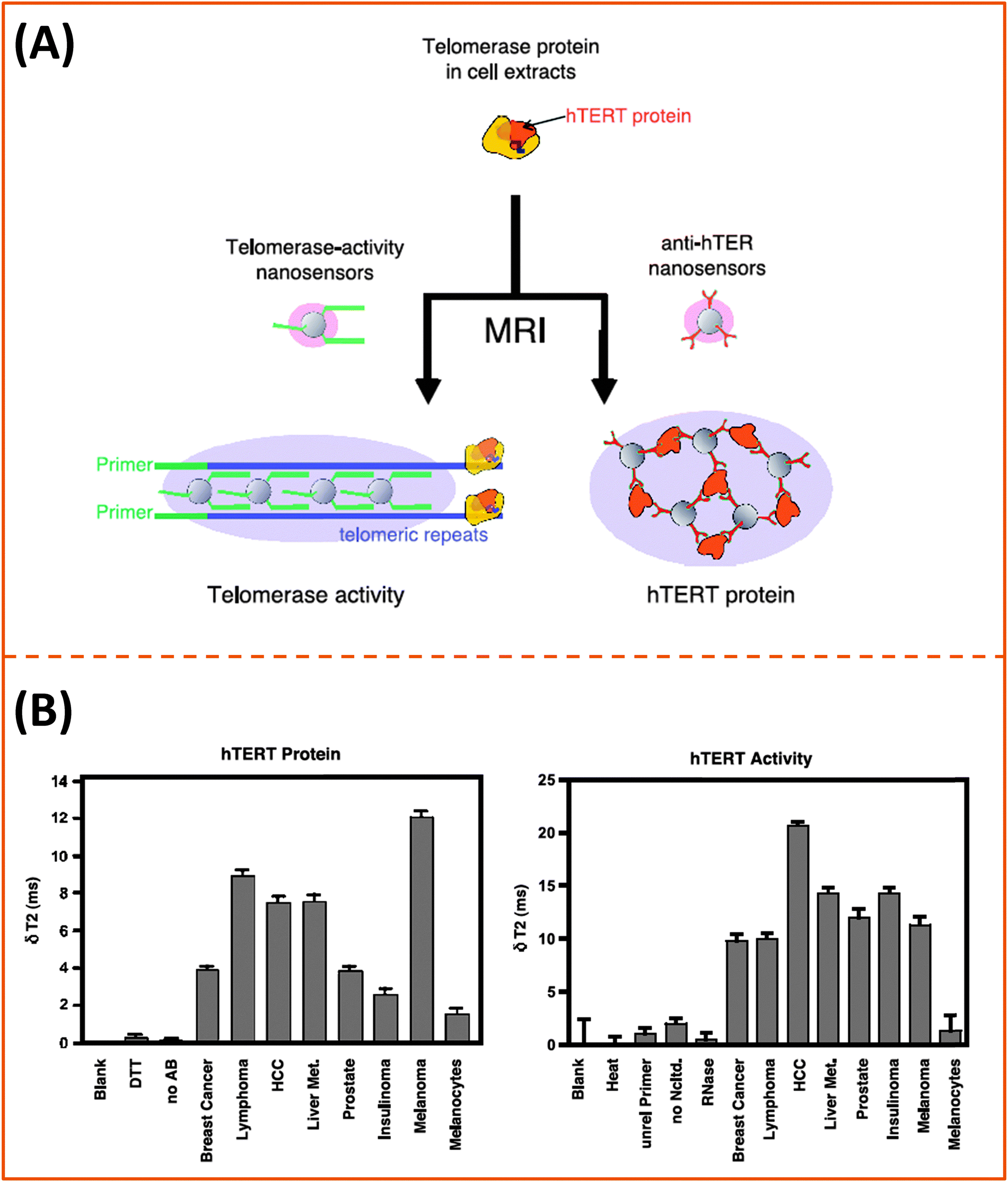 | ||
| Fig. 18 (A) Schematic diagram of the magnetic nanosensor. (B) Detection of the amounts of telomerase protein (left), and telomerase activity (right) in various cell lysates using the telomerase magnetic nanosensors. Data were collected using a bench top relaxometer.259 (Figures are adapted from the above reference with permission from Neoplasia Press.) | ||
5. Conclusion
We have provided an overview of the strategies developed over the past decade for the surface-functionalization of metal, metal oxide and semiconductor nanostructures with amphiphilic polymers, via either encapsulation or cap exchange strategies. We discussed the methods developed to functionalize several types of inorganic nanoparticles with polymeric materials, along with the most commonly used conjugation strategies. We then provided a few representative examples where such polymer-coated inorganic platforms have been used to develop new sensors and for the imaging of cells and/or tissue based on fluorescence or MR contrast. Examples introducing the use of gold nanoparticles and nanorods as laser activated platforms to treat cancerous cells via photo-thermal therapy have also been provided.The remarkable flexibility offered by block-copolymers in terms of size, stereochemistry and conformational control can easily allow their use to surface functionalize inorganic nanocrystals, via either of the above strategies. For instance, amphiphilic block copolymers as encapsulating platforms provide the ability to control the micelle size, with extremely low critical micelle concentration easily reachable. Amphiphilic block copolymers also allow one to optimize the hydrophobic block, maximizing the entropy-driven interdigitation of the polymer with the native hydrophobic ligands on the nanoparticles, as well the nature and size of the hydrophilic block for enhanced water affinity. Conversely, the ability to chemically insert large but controllable numbers of metal-coordinating groups with high affinity to a specific metal, metal oxide or metal chalcogenide of choice, can easily provide multidentate ligands with more stable ligand-nanoparticle constructs. Here, the metal-coordinating groups can competitively displace the native ligands on the nanocrystals, while the hydrophilic block promotes water compatibility and reactivity. Thus, as a strategy polymer-coating of nanoparticles can provide dispersions of nanoparticles with great colloidal stability and easy conjugation.
We would also like to provide a critical comparison of the two polymer coating routes. Encapsulation of the inorganic nanocrystals within amphiphilic block copolymers tends to better preserve the photo-physical characteristics (e.g., PL quantum yield for QDs), because it preserves the native ligands. This is beneficial for use in fluorescence imaging, for example. However, preserving such hydrophobic cap can potentially have undesirable effects such as inducing toxicity to biological systems. Furthermore, encapsulation substantially increases the hydrodynamic size of the final hydrophilic nanocrystals. In comparison, ligand exchange provides more compact hydrophilic nanocrystals, as multi-coordination imposes a more extended thinner polymer coating of the nanocrystal surfaces. This is proven by the ability to implement FRET sensing using polymer-capped QDs prepared via ligand exchange. However, ligand exchange often results in a slight alteration of the physical properties of the native materials (e.g., lower PL quantum yield of QDs). Also, the chemical design of the polymer ligands can be tedious for some precursors.
Given the wealth of possibilities offered by polymeric materials, we expect that polymer-coating as a strategy to develop fluorescent, magnetic and plasmonic platforms will substantially grow in the future. Groups will continue to explore the use of various amphiphilic polymers to eventually reduce the lateral size of the hydrating layer on the nanoparticles, and introduce multiple orthogonally reactive functions, which should permit easier interfacing with biological systems, such as cells and tissues. One promising design involves the use of zwitterion moieties as the hydrophilic moieties in the amphiphilic polymers. These moieties are much smaller than PEG chains, and tend to exhibit better compatibility with various biological media and reduced nonspecific interactions with serum proteins.
References
- J. Park, K. J. An, Y. S. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- P. Alivisatos, Nat. Biotechnol., 2004, 22, 47–52 CrossRef CAS PubMed.
- X. Michalet, F. Pinaud, L. Bentolila, J. Tsay, S. Doose, J. Li, G. Sundaresan, A. Wu, S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- P. Reiss, M. Protiere and L. Li, Small, 2009, 5, 154–168 CrossRef CAS PubMed.
- D. V. Talapin, J. S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- M. H. M. Dias and P. C. Lauterbur, Magn. Reson. Med., 1986, 3, 328–330 CrossRef CAS.
- P. T. Snee, R. C. Somers, G. Nair, J. P. Zimmer, M. G. Bawendi and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 13320–13321 CrossRef CAS PubMed.
- H. Mattoussi, G. Palui and H. B. Na, Adv. Drug Delivery Rev., 2012, 64, 138–166 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. N. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- X. Y. Wu, H. J. Liu, J. Q. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. F. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41–46 CrossRef CAS PubMed.
- A. M. Alkilany, S. E. Lohse and C. J. Murphy, Acc. Chem. Res., 2013, 46, 650–661 CrossRef CAS PubMed.
- F. Aldeek, M. A. H. Muhammed, G. Palui, N. Q. Zhan and H. Mattoussi, ACS Nano, 2013, 7, 2509–2521 CrossRef CAS PubMed.
- Y.-w. Jun, Y.-M. Huh, J.-s. Choi, J.-H. Lee, H.-T. Song, S. Kim, S. Kim, S. Yoon, K.-S. Kim, J.-S. Shin, J.-S. Suh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 5732–5733 CrossRef CAS PubMed.
- J. H. Lee, Y. M. Huh, Y. Jun, J. Seo, J. Jang, H. T. Song, S. Kim, E. J. Cho, H. G. Yoon, J. S. Suh and J. Cheon, Nat. Med., 2007, 13, 95–99 CrossRef CAS PubMed.
- S. H. Choi, H. Bin Na, Y. I. Park, K. An, S. G. Kwon, Y. Jang, M. Park, J. Moon, J. S. Son, I. C. Song, W. K. Moon and T. Hyeon, J. Am. Chem. Soc., 2008, 130, 15573–15580 CrossRef CAS PubMed.
- T. Hyeon, S. S. Lee, J. Park, Y. Chung and H. B. Na, J. Am. Chem. Soc., 2001, 123, 12798–12801 CrossRef CAS PubMed.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- H. Mattoussi and J. Cheon, Inorganic nanoprobes for biological sensing and imaging, Artech House, Boston, 2009 Search PubMed.
- P. Zrazhevskiy, M. Sena and X. H. Gao, Chem. Soc. Rev., 2010, 39, 4326–4354 RSC.
- Y. C. Yeh, B. Creran and V. M. Rotello, Nanoscale, 2012, 4, 1871–1880 RSC.
- A. L. Rogach, L. Katsikas, A. Kornowski, D. S. Su, A. Eychmuller and H. Weller, Ber. Bunsen-Ges., 1996, 100, 1772–1778 CrossRef CAS.
- N. Gaponik, D. V. Talapin, A. L. Rogach, K. Hoppe, E. V. Shevchenko, A. Kornowski, A. Eychmüller and H. Weller, J. Phys. Chem. B, 2002, 106, 7177–7185 CrossRef CAS.
- A. L. Rogach, T. Franzl, T. A. Klar, J. Feldmann, N. Gaponik, V. Lesnyak, A. Shavel, A. Eychmüller, Y. P. Rakovich and J. F. Donegan, J. Phys. Chem. C, 2007, 111, 14628–14637 CAS.
- F. Aldeek, L. Balan, J. Lambert and R. Schneider, Nanotechnology, 2008, 19, 475401 CrossRef PubMed.
- D. K. Kim, Y. Zhang, W. Voit, K. V. Rao and M. Muhammed, J. Magn. Magn. Mater., 2001, 225, 30–36 CrossRef CAS.
- D. Maity and D. C. Agrawal, J. Magn. Magn. Mater., 2007, 308, 46–55 CrossRef CAS PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- F. Aldeek, C. Mustin, L. Balan, T. Roques-Carmes, M. P. Fontaine-Aupart and R. Schneider, Biomaterials, 2011, 32, 5459–5470 CrossRef CAS PubMed.
- I. Medintz, H. Uyeda, E. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- C. M. Tyrakowski and P. T. Snee, Phys. Chem. Chem. Phys., 2014, 16, 837–855 RSC.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS PubMed.
- N. Hadjichristidis, S. Pispas and G. Floudas, Block Copolymers: Synthetic Strategies, Physical Properties, and Applications, Wiley, 2003 Search PubMed.
- H. Weller, U. Koch, M. Gutierrez and A. Henglein, Ber. Bunsen-Ges., 1984, 88, 649–656 CrossRef CAS.
- L. Brus, J. Phys. Chem., 1986, 90, 2555–2560 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- A. L. Efros, M. Rosen, M. Kuno, M. Nirmal, D. J. Norris and M. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 4843–4856 CrossRef CAS.
- D. J. Norris and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 16338–16346 CrossRef CAS.
- D. J. Norris, A. L. Efros, M. Rosen and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 16347–16354 CrossRef CAS.
- V. I. Klimov, Nanocrystal quantum dots, CRC Press, Boca Raton, 2nd edn, 2010 Search PubMed.
- A. L. Washington, M. E. Foley, S. Cheong, L. Quffa, C. J. Breshike, J. Watt, R. D. Tilley and G. F. Strouse, J. Am. Chem. Soc., 2012, 134, 17046–17052 CrossRef CAS PubMed.
- J. H. Yu, S. H. Kwon, Z. Petrasek, O. K. Park, S. W. Jun, K. Shin, M. Choi, Y. Il Park, K. Park, H. B. Bin Na, N. Lee, D. W. Lee, J. H. Kim, P. Schwille and T. Hyeon, Nat. Mater., 2013, 12, 359–366 CrossRef CAS PubMed.
- J. H. Yu, X. Y. Liu, K. E. Kweon, J. Joo, J. Park, K. T. Ko, D. Lee, S. P. Shen, K. Tivakornsasithorn, J. S. Son, J. H. Park, Y. W. Kim, G. S. Hwang, M. Dobrowolska, J. K. Furdyna and T. Hyeon, Nat. Mater., 2010, 9, 47–53 CrossRef CAS PubMed.
- V. I. Klimov, A. A. Mikhailovsky, S. Xu, A. Malko, J. A. Hollingsworth, C. A. Leatherdale, H. J. Eisler and M. G. Bawendi, Science, 2000, 290, 314–317 CrossRef CAS.
- M. Kuno, D. P. Fromm, H. F. Hamann, A. Gallagher and D. J. Nesbitt, J. Chem. Phys., 2001, 115, 1028–1040 CrossRef CAS PubMed.
- K. T. Shimizu, R. G. Neuhauser, C. A. Leatherdale, S. A. Empedocles, W. K. Woo and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 205316 CrossRef.
- L. S. Li, J. T. Hu, W. D. Yang and A. P. Alivisatos, Nano Lett., 2001, 1, 349–351 CrossRef CAS.
- L. Manna, D. J. Milliron, A. Meisel, E. C. Scher and A. P. Alivisatos, Nat. Mater., 2003, 2, 382–385 CrossRef CAS PubMed.
- N. R. Jana and X. G. Peng, J. Am. Chem. Soc., 2003, 125, 14280–14281 CrossRef CAS PubMed.
- C. Bouet, B. Mahler, B. Nadal, B. Abecassis, M. D. Tessier, S. Ithurria, X. Xu and B. Dubertret, Chem. Mater., 2013, 25, 639–645 CrossRef CAS.
- S. Ithurria, M. D. Tessier, B. Mahler, R. P. S. M. Lobo, B. Dubertret and A. L. Efros, Nat. Mater., 2011, 10, 936–941 CrossRef CAS PubMed.
- A. I. Ekimov and A. A. Onushchenko, Sov. Phys. Semicond.-USSR, 1982, 16, 775–778 Search PubMed.
- A. I. Ekimov and A. A. Onushchenko, JETP Lett., 1981, 34, 345–349 Search PubMed.
- A. L. Efros and A. L. Efros, Sov. Phys. Semicond.-USSR, 1982, 16, 772–775 Search PubMed.
- N. F. Borrelli, D. W. Hall, H. J. Holland and D. W. Smith, J. Appl. Phys., 1987, 61, 5399–5409 CrossRef CAS PubMed.
- D. W. Hall and N. F. Borrelli, J. Opt. Soc. Am. B, 1988, 5, 1650–1654 CrossRef CAS.
- A. Fojtik, H. Weller, U. Koch and A. Henglein, Ber. Bunsen-Ges., 1984, 88, 969–977 CrossRef CAS.
- L. Brus, J. Phys. Chem., 1986, 90, 2555–2560 CrossRef CAS.
- Z. A. Peng and X. G. Peng, J. Am. Chem. Soc., 2001, 123, 183–184 CrossRef CAS.
- L. H. Qu, Z. A. Peng and X. G. Peng, Nano Lett., 2001, 1, 333–337 CrossRef CAS.
- M. V. Kovalenko, M. I. Bodnarchuk, J. Zaumseil, J. S. Lee and D. V. Talapin, J. Am. Chem. Soc., 2010, 132, 10085–10092 CrossRef CAS PubMed.
- D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, Nano Lett., 2001, 1, 207–211 CrossRef CAS.
- T. Pons, E. Pic, N. Lequeux, E. Cassette, L. Bezdetnaya, F. Guillemin, F. Marchal and B. Dubertret, ACS Nano, 2010, 4, 2531–2538 CrossRef CAS PubMed.
- C. R. Bullen and P. Mulvaney, Nano Lett., 2004, 4, 2303–2307 CrossRef CAS.
- S. Sapra, A. L. Rogach and J. Feldmann, J. Mater. Chem., 2006, 16, 3391–3395 RSC.
- T. Pons, N. Lequeux, B. Mahler, S. Sasnouski, A. Fragola and B. Dubertret, Chem. Mater., 2009, 21, 1418–1424 CrossRef CAS.
- M. D. Regulacio and M. Y. Han, Acc. Chem. Res., 2010, 43, 621–630 CrossRef CAS PubMed.
- W. J. Cai, L. M. Jiang, D. M. Yi, H. Z. Sun, H. T. Wei, H. Zhang, H. C. Sun and B. Yang, Langmuir, 2013, 29, 4119–4127 CrossRef CAS PubMed.
- E. Cassette, T. Pons, C. Bouet, M. Helle, L. Bezdetnaya, F. Marchal and B. Dubertret, Chem. Mater., 2010, 22, 6117–6124 CrossRef CAS.
- I. S. Liu, H.-H. Lo, C.-T. Chien, Y.-Y. Lin, C.-W. Chen, Y.-F. Chen, W.-F. Su and S.-C. Liou, J. Mater. Chem., 2008, 18, 675–682 RSC.
- C. Bullen and P. Mulvaney, Langmuir, 2006, 22, 3007–3013 CrossRef CAS PubMed.
- B. O. Dabbousi, J. RodriguezViejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS.
- M. A. Hines and P. Guyot-Sionnest, J. Phys. Chem., 1996, 100, 468–471 CrossRef CAS.
- P. Reiss, J. Bleuse and A. Pron, Nano Lett., 2002, 2, 781–784 CrossRef CAS.
- X. G. Peng, M. C. Schlamp, A. V. Kadavanich and A. P. Alivisatos, J. Am. Chem. Soc., 1997, 119, 7019–7029 CrossRef CAS.
- B. Mahler, P. Spinicelli, S. Buil, X. Quelin, J. P. Hermier and B. Dubertret, Nat. Mater., 2008, 7, 659–664 CrossRef CAS PubMed.
- X. Y. Wang, X. F. Ren, K. Kahen, M. A. Hahn, M. Rajeswaran, S. Maccagnano-Zacher, J. Silcox, G. E. Cragg, A. L. Efros and T. D. Krauss, Nature, 2009, 459, 686–689 CrossRef CAS PubMed.
- H. B. Shen, H. Z. Wang, X. M. Li, J. Z. Niu, H. Wang, X. Chen and L. S. Li, Dalton Trans., 2009, 10534–10540 RSC.
- J. R. Dethlefsen and A. Dossing, Nano Lett., 2011, 11, 1964–1969 CrossRef CAS PubMed.
- H. B. Shen, H. Z. Wang, Z. J. Tang, J. Z. Niu, S. Y. Lou, Z. L. Du and L. S. Li, CrystEngComm, 2009, 11, 1733–1738 RSC.
- Y. Zhang, Q. Q. Dai, X. B. Li, Q. Z. Cui, Z. Y. Gu, B. Zou, Y. D. Wang and W. W. Yu, Nanoscale Res. Lett., 2010, 5, 1279–1283 CrossRef CAS PubMed.
- J. Park and S. W. Kim, J. Mater. Chem., 2011, 21, 3745–3750 RSC.
- X. S. Tang, K. A. Yu, Q. H. Xu, E. S. G. Choo, G. K. L. Goh and J. M. Xue, J. Mater. Chem., 2011, 21, 11239–11243 RSC.
- A. J. Morris-Cohen, M. D. Donakowski, K. E. Knowles and E. A. Weiss, J. Phys. Chem. C, 2009, 114, 897–906 Search PubMed.
- G. Ennas, A. Musinu, G. Piccaluga, D. Zedda, D. Gatteschi, C. Sangregorio, J. L. Stanger, G. Concas and G. Spano, Chem. Mater., 1998, 10, 495–502 CrossRef CAS.
- D. Thapa, V. R. Palkar, M. B. Kurup and S. K. Malik, Mater. Lett., 2004, 58, 2692–2694 CrossRef CAS PubMed.
- A. N. Christensen, T. R. Jensen, C. R. H. Bahl and E. DiMasi, J. Solid State Chem., 2007, 180, 1431–1435 CrossRef CAS PubMed.
- S. H. Tolbert, P. Sieger, G. D. Stucky, S. M. J. Aubin, C. C. Wu and D. N. Hendrickson, J. Am. Chem. Soc., 1997, 119, 8652–8661 CrossRef CAS.
- F. X. Geng, Z. G. Zhao, H. T. Cong, J. X. Geng and H. M. Cheng, Mater. Res. Bull., 2006, 41, 2238–2243 CrossRef CAS PubMed.
- T. Douglas and M. Young, Nature, 1998, 393, 152–155 CrossRef CAS PubMed.
- G. Salazar-Alvarez, M. Muhammed and A. A. Zagorodni, Chem. Eng. Sci., 2006, 61, 4625–4633 CrossRef CAS PubMed.
- C. Pascal, J. L. Pascal, F. Favier, M. L. E. Moubtassim and C. Payen, Chem. Mater., 1999, 11, 141–147 CrossRef CAS.
- J. H. Bang and K. S. Suslick, J. Am. Chem. Soc., 2007, 129, 2242–2243 CrossRef CAS PubMed.
- S. H. Sun and C. B. Murray, J. Appl. Phys., 1999, 85, 4325–4330 CrossRef CAS PubMed.
- C. R. K. C. B. Murray and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef CAS.
- F. X. Redl, C. T. Black, G. C. Papaefthymiou, R. L. Sandstrom, M. Yin, H. Zeng, C. B. Murray and S. P. O'Brien, J. Am. Chem. Soc., 2004, 126, 14583–14599 CrossRef CAS PubMed.
- Y. W. Jun, Y. Y. Jung and J. Cheon, J. Am. Chem. Soc., 2002, 124, 615–619 CrossRef CAS PubMed.
- Y.-w. Jun, J.-H. Lee and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 5122–5135 CrossRef CAS PubMed.
- V. F. Puntes, K. M. Krishnan and P. Alivisatos, Appl. Phys. Lett., 2001, 78, 2187–2189 CrossRef CAS PubMed.
- V. F. Puntes, P. Gorostiza, D. M. Aruguete, N. G. Bastus and A. P. Alivisatos, Nat. Mater., 2004, 3, 263–268 CrossRef CAS PubMed.
- N. R. Jana, Y. Chen and X. Peng, Chem. Mater., 2004, 16, 3931–3935 CrossRef CAS.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2003, 126, 273–279 CrossRef PubMed.
- J. Cheon and J. H. Lee, Acc. Chem. Res., 2008, 41, 1630–1640 CrossRef CAS PubMed.
- J. R. Lai, K. V. P. M. Shafi, A. Ulman, K. Loos, N. L. Yang, M. H. Cui, T. Vogt, S. Estournes and D. C. Locke, J. Phys. Chem. B, 2004, 108, 14876–14883 CrossRef CAS.
- R. Qiao, C. Yang and M. Gao, J. Mater. Chem., 2009, 19, 6274–6293 RSC.
- S. Sun, Adv. Mater., 2006, 18, 393–403 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 55–75 RSC.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- X. P. Sun, S. J. Dong and E. K. Wang, Polymer, 2004, 45, 2181–2184 CrossRef CAS PubMed.
- A. Pucci, M. Bernabo, P. Elvati, L. I. Meza, F. Galembeck, C. A. D. Leite, N. Tirelli and G. Ruggeri, J. Mater. Chem., 2006, 16, 1058–1066 RSC.
- G. Maiorano, S. Sabella, B. Sorce, V. Brunetti, M. A. Malvindi, R. Cingolani and P. P. Pompa, ACS Nano, 2010, 4, 7481–7491 CrossRef CAS PubMed.
- N. Wangoo, C. R. Suri and G. Shekhawat, Appl. Phys. Lett., 2008, 92, 133104 CrossRef PubMed.
- D.-H. Tsai, F. W. DelRio, A. M. Keene, K. M. Tyner, R. I. MacCuspie, T. J. Cho, M. R. Zachariah and V. A. Hackley, Langmuir, 2011, 27, 2464–2477 CrossRef CAS PubMed.
- E. Oh, K. Susumu, R. Goswami and H. Mattoussi, Langmuir, 2010, 26, 7604–7613 CrossRef CAS PubMed.
- M. A. Muhammed, F. Aldeek, G. Palui, L. Trapiella-Alfonso and H. Mattoussi, ACS Nano, 2012, 6, 8950–8961 CrossRef CAS PubMed.
- S. E. Lohse, J. R. Eller, S. T. Sivapalan, M. R. Plews and C. J. Murphy, ACS Nano, 2013, 7, 4135–4150 CrossRef CAS PubMed.
- X. Ye, L. Jin, H. Caglayan, J. Chen, G. Xing, C. Zheng, V. Doan-Nguyen, Y. Kang, N. Engheta, C. R. Kagan and C. B. Murray, ACS Nano, 2012, 6, 2804–2817 CrossRef CAS PubMed.
- T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126, 8648–8649 CrossRef CAS PubMed.
- H. Hakkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- D. A. Fleming and M. E. Williams, Langmuir, 2004, 20, 3021–3023 CrossRef CAS.
- H. Hiramatsu and F. E. Osterloh, Chem. Mater., 2004, 16, 2509–2511 CrossRef CAS.
- O. C. Compton and F. E. Osterloh, J. Am. Chem. Soc., 2007, 129, 7793–7798 CrossRef CAS PubMed.
- S. Liu, G. Y. Chen, P. N. Prasad and M. T. Swihart, Chem. Mater., 2011, 23, 4098–4101 CrossRef CAS.
- W. H. Liu, A. B. Greytak, J. Lee, C. R. Wong, J. Park, L. F. Marshall, W. Jiang, P. N. Curtin, A. Y. Ting, D. G. Nocera, D. Fukumura, R. K. Jain and M. G. Bawendi, J. Am. Chem. Soc., 2010, 132, 472–483 CrossRef CAS PubMed.
- W. C. W. Chan and S. M. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS.
- H. T. Uyeda, I. L. Medintz, J. K. Jaiswal, S. M. Simon and H. Mattoussi, J. Am. Chem. Soc., 2005, 127, 3870–3878 CrossRef CAS PubMed.
- W. Liu, M. Howarth, A. B. Greytak, Y. Zheng, D. G. Nocera, A. Y. Ting and M. G. Bawendi, J. Am. Chem. Soc., 2008, 130, 1274–1284 CrossRef CAS PubMed.
- M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS.
- H. Mattoussi, J. M. Mauro, E. R. Goldman, G. P. Anderson, V. C. Sundar, F. V. Mikulec and M. G. Bawendi, J. Am. Chem. Soc., 2000, 122, 12142–12150 CrossRef CAS.
- W. W. Yu, E. Chang, J. C. Falkner, J. Y. Zhang, A. M. Al-Somali, C. M. Sayes, J. Johns, R. Drezek and V. L. Colvin, J. Am. Chem. Soc., 2007, 129, 2871–2879 CrossRef CAS PubMed.
- F. Zhang, E. Lees, F. Amin, P. R. Gil, F. Yang, P. Mulvaney and W. J. Parak, Small, 2011, 7, 3113–3127 CrossRef CAS PubMed.
- T. Pellegrino, L. Manna, S. Kudera, T. Liedl, D. Koktysh, A. L. Rogach, S. Keller, J. Radler, G. Natile and W. J. Parak, Nano Lett., 2004, 4, 703–707 CrossRef CAS.
- A. N. Lukyanov and V. P. Torchilin, Adv. Drug Delivery Rev., 2004, 56, 1273–1289 CrossRef CAS PubMed.
- R. Levy, N. T. K. Thanh, R. C. Doty, I. Hussain, R. J. Nichols, D. J. Schiffrin, M. Brust and D. G. Fernig, J. Am. Chem. Soc., 2004, 126, 10076–10084 CrossRef CAS PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- J. M. Kogot, H. J. England, G. F. Strouse and T. M. Logan, J. Am. Chem. Soc., 2008, 130, 16156–16157 CrossRef CAS PubMed.
- P. Dash and R. W. J. Scott, Chem. Commun., 2009, 812–814 RSC.
- X. Zhang, M. R. Servos and J. Liu, J. Am. Chem. Soc., 2012, 134, 7266–7269 CrossRef CAS PubMed.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- K. O. Aruda, M. Tagliazucchi, C. M. Sweeney, D. C. Hannah, G. C. Schatz and E. A. Weiss, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4212–4217 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- J. R. Reimers, Y. Wang, B. O. Cankurtaran and M. J. Ford, J. Am. Chem. Soc., 2010, 132, 8378–8384 CrossRef CAS PubMed.
- H. B. Na, G. Palui, J. T. Rosenberg, X. Ji, S. C. Grant and H. Mattoussi, ACS Nano, 2012, 6, 389–399 CrossRef CAS PubMed.
- N. Chan, M. Laprise-Pelletier, P. Chevallier, A. Bianchi, M.-A. Fortin and J. K. Oh, Biomacromolecules, 2014, 15, 2146–2156 CrossRef CAS PubMed.
- P. Zhang, S. Liu, D. Gao, D. Hu, P. Gong, Z. Sheng, J. Deng, Y. Ma and L. Cai, J. Am. Chem. Soc., 2012, 134, 8388–8391 CrossRef CAS PubMed.
- G. P. Mitchell, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1999, 121, 8122–8123 CrossRef CAS.
- Z.-J. Zhu, Y.-C. Yeh, R. Tang, B. Yan, J. Tamayo, R. W. Vachet and V. M. Rotello, Nat. Chem., 2011, 3, 963–968 CrossRef CAS PubMed.
- K. Susumu, H. T. Uyeda, I. L. Medintz, T. Pons, J. B. Delehanty and H. Mattoussi, J. Am. Chem. Soc., 2007, 129, 13987–13996 CrossRef CAS PubMed.
- J. S. Kang and T. A. Taton, Langmuir, 2012, 28, 16751–16760 CrossRef CAS PubMed.
- I. Yildiz, E. Deniz, B. McCaughan, S. F. Cruickshank, J. F. Callan and F. M. Raymo, Langmuir, 2010, 26, 11503–11511 CrossRef CAS PubMed.
- I. Yildiz, B. McCaughan, S. F. Cruickshank, J. F. Callan and F. M. Raymo, Langmuir, 2009, 25, 7090–7096 CrossRef CAS PubMed.
- G. Palui, H. B. Na and H. Mattoussi, Langmuir, 2012, 28, 2761–2772 CrossRef CAS PubMed.
- L. Liu, X. H. Guo, Y. Li and X. H. Zhong, Inorg. Chem., 2010, 49, 3768–3775 CrossRef CAS PubMed.
- E. Giovanelli, E. Muro, G. Sitbon, M. Hanafi, T. Pons, B. Dubertret and N. Lequeux, Langmuir, 2012, 28, 15177–15184 CrossRef CAS PubMed.
- M. Sun, L. Yang, P. Jose, L. Wang and J. Zweit, J. Mater. Chem. B, 2013, 1, 6137–6146 RSC.
- G. Palui, T. Avellini, N. Zhan, F. Pan, D. Gray, I. Alabugin and H. Mattoussi, J. Am. Chem. Soc., 2012, 134, 16370–16378 CrossRef CAS PubMed.
- G. Bucher, C. Y. Lu and W. Sander, ChemPhysChem, 2005, 6, 2607–2618 CrossRef CAS PubMed.
- X. J. Chen, J. Lawrence, S. Parelkar and T. Emrick, Macromolecules, 2013, 46, 119–127 CrossRef CAS.
- K. E. Sapsford, T. Pons, I. L. Medintz, S. Higashiya, F. M. Brunel, P. E. Dawson and H. Mattoussi, J. Phys. Chem. C, 2007, 111, 11528–11538 CAS.
- A. R. Clapp, I. L. Medintz, J. M. Mauro, B. R. Fisher, M. G. Bawendi and H. Mattoussi, J. Am. Chem. Soc., 2004, 126, 301–310 CrossRef CAS PubMed.
- K. E. Sapsford, T. Pons, I. L. Medintz, S. Higashiya, F. M. Brunel, P. E. Dawson and H. Mattoussi, J. Phys. Chem. C, 2007, 111, 11528–11538 CAS.
- F. Aldeek, M. Safi, N. Zhan, G. Palui and H. Mattoussi, ACS Nano, 2013, 7, 10197–10210 CrossRef CAS PubMed.
- H. S. Han, J. D. Martin, J. Lee, D. K. Harris, D. Fukumura, R. K. Jain and M. Bawendi, Angew. Chem., 2013, 52, 1414–1419 CrossRef CAS PubMed.
- Y. Sahoo, H. Pizem, T. Fried, D. Golodnitsky, L. Burstein, C. N. Sukenik and G. Markovich, Langmuir, 2001, 17, 7907–7911 CrossRef CAS.
- S.-W. Kim, S. Kim, J. B. Tracy, A. Jasanoff and M. G. Bawendi, J. Am. Chem. Soc., 2005, 127, 4556–4557 CrossRef CAS PubMed.
- H. Wei, N. Insin, J. Lee, H.-S. Han, J. M. Cordero, W. Liu and M. G. Bawendi, Nano Lett., 2011, 12, 22–25 CrossRef PubMed.
- L. Sandiford, A. Phinikaridou, A. Protti, L. K. Meszaros, X. Cui, Y. Yan, G. Frodsham, P. A. Williamson, N. Gaddum, R. M. Botnar, P. J. Blower, M. A. Green and R. T. M. de Rosales, ACS Nano, 2012, 7, 500–512 CrossRef PubMed.
- C. J. Xu, K. M. Xu, H. W. Gu, R. K. Zheng, H. Liu, X. X. Zhang, Z. H. Guo and B. Xu, J. Am. Chem. Soc., 2004, 126, 9938–9939 CrossRef CAS PubMed.
- E. Amstad, T. Gillich, I. Bilecka, M. Textor and E. Reimhult, Nano Lett., 2009, 9, 4042–4048 CrossRef CAS PubMed.
- J. Xie, K. Chen, H.-Y. Lee, C. Xu, A. R. Hsu, S. Peng, X. Chen and S. Sun, J. Am. Chem. Soc., 2008, 130, 7542–7543 CrossRef CAS PubMed.
- K. Cheng, S. Peng, C. Xu and S. Sun, J. Am. Chem. Soc., 2009, 131, 10637–10644 CrossRef CAS PubMed.
- W. Wang, X. Ji, H. B. Na, M. Safi, A. Smith, G. Palui, J. M. Perez and H. Mattoussi, Langmuir, 2014, 30, 6197–6208 CrossRef CAS PubMed.
- D. Ling, W. Park, Y. I. Park, N. Lee, F. Li, C. Song, S.-G. Yang, S. H. Choi, K. Na and T. Hyeon, Angew. Chem., Int. Ed., 2011, 50, 11360–11365 CrossRef CAS PubMed.
- C. C. Lu, L. R. Bhatt, H. Y. Jun, S. H. Park and K. Y. Chai, J. Mater. Chem., 2012, 22, 19806–19811 RSC.
- A. Graillot, D. Bouyer, S. Monge, J. J. Robin, P. Loison and C. Faur, J. Hazard. Mater., 2013, 260, 425–433 CrossRef CAS PubMed.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS PubMed.
- C. Tassa, S. Y. Shaw and R. Weissleder, Acc. Chem. Res., 2011, 44, 842–852 CrossRef CAS PubMed.
- Y. J. Kang and T. A. Taton, Angew. Chem., Int. Ed., 2005, 44, 409–412 CrossRef CAS PubMed.
- Y. Kang and T. A. Taton, Macromolecules, 2005, 38, 6115–6121 CrossRef CAS.
- Y. Chen, J. Cho, A. Young and T. A. Taton, Langmuir, 2007, 23, 7491–7497 CrossRef CAS PubMed.
- W. K. Li, S. Q. Liu, R. H. Deng, J. Y. Wang, Z. H. Nie and J. T. Zhu, Macromolecules, 2013, 46, 2282–2291 CrossRef CAS.
- B. A. Kairdolf and S. M. Nie, J. Am. Chem. Soc., 2011, 133, 7268–7271 CrossRef CAS PubMed.
- D. H. Kim, A. Wei and Y.-Y. Won, ACS Appl. Mater. Interfaces, 2012, 4, 1872–1877 CAS.
- A. Elbakry, A. Zaky, R. Liebkl, R. Rachel, A. Goepferich and M. Breunig, Nano Lett., 2009, 9, 2059–2064 CrossRef CAS PubMed.
- A. Elbakry, E. C. Wurster, A. Zaky, R. Liebl, E. Schindler, P. Bauer-Kreisel, T. Blunk, R. Rachel, A. Goepferich and M. Breunig, Small, 2012, 8, 3847–3856 CrossRef CAS PubMed.
- J. Huang, K. S. Jackson and C. J. Murphy, Nano Lett., 2012, 12, 2982–2987 CrossRef CAS PubMed.
- A. Gole and C. J. Murphy, Chem. Mater., 2005, 17, 1325–1330 CrossRef CAS.
- K. S. Mayya, B. Schoeler and F. Caruso, Adv. Funct. Mater., 2003, 13, 183–188 CrossRef CAS.
- H.-C. Huang, S. Barua, D. B. Kay and K. Rege, ACS Nano, 2009, 3, 2941–2952 CrossRef CAS PubMed.
- X. H. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- J. B. Schlenoff, H. Ly and M. Li, J. Am. Chem. Soc., 1998, 120, 7626–7634 CrossRef CAS.
- L. L. Rouhana, M. D. Moussallem and J. B. Schlenoff, J. Am. Chem. Soc., 2011, 133, 16080–16091 CrossRef CAS PubMed.
- W. B. Stockton and M. F. Rubner, Macromolecules, 1997, 30, 2717–2725 CrossRef CAS.
- C. Tedeschi, H. Möhwald and S. Kirstein, J. Am. Chem. Soc., 2001, 123, 954–960 CrossRef CAS PubMed.
- S. K. Chakraborty, J. A. J. Fitzpatrick, J. A. Phillippi, S. Andreko, A. S. Waggoner, M. P. Bruchez and B. Ballou, Nano Lett., 2007, 7, 2618–2626 CrossRef CAS PubMed.
- B. C. Lagerholm, M. M. Wang, L. A. Ernst, D. H. Ly, H. J. Liu, M. P. Bruchez and A. S. Waggoner, Nano Lett., 2004, 4, 2019–2022 CrossRef CAS.
- D. R. Larson, W. R. Zipfel, R. M. Williams, S. W. Clark, M. P. Bruchez, F. W. Wise and W. W. Webb, Science, 2003, 300, 1434–1436 CrossRef CAS PubMed.
- S. Courty, C. Luccardini, Y. Bellaiche, G. Cappello and M. Dahan, Nano Lett., 2006, 6, 1491–1495 CrossRef CAS PubMed.
- M. Dahan, S. Levi, C. Luccardini, P. Rostaing, B. Riveau and A. Triller, Science, 2003, 302, 442–445 CrossRef CAS PubMed.
- D. S. Lidke, P. Nagy, R. Heintzmann, D. J. Arndt-Jovin, J. N. Post, H. E. Grecco, E. A. Jares-Erijman and T. M. Jovin, Nat. Biotechnol., 2004, 22, 198–203 CrossRef CAS PubMed.
- M. J. Roberti, M. Morgan, G. Menendez, L. I. Pietrasanta, T. M. Jovin and E. A. Jares-Erijman, J. Am. Chem. Soc., 2009, 131, 8102–8107 CrossRef CAS PubMed.
- X. H. Gao, Y. Y. Cui, R. M. Levenson, L. W. K. Chung and S. M. Nie, Nat. Biotechnol., 2004, 22, 969–976 CrossRef CAS PubMed.
- M. T. Fernández-Argüelles, A. Yakovlev, R. A. Sperling, C. Luccardini, S. Gaillard, A. Sanz Medel, J.-M. Mallet, J.-C. Brochon, A. Feltz, M. Oheim and W. J. Parak, Nano Lett., 2007, 7, 2613–2617 CrossRef PubMed.
- E. E. Lees, T. L. Nguyen, A. H. A. Clayton, P. Mulvaney and B. W. Muir, ACS Nano, 2009, 3, 1121–1128 CrossRef CAS PubMed.
- M. F. Wang, M. Zhang, J. Li, S. Kumar, G. C. Walker, G. D. Scholes and M. A. Winnik, ACS Appl. Mater. Interfaces, 2010, 2, 3160–3169 CAS.
- M. F. Wang, N. Felorzabihi, G. Guerin, J. C. Haley, G. D. Scholes and M. A. Winnik, Macromolecules, 2007, 40, 6377–6384 CrossRef CAS.
- J. Qian and X. H. Gao, ACS Appl. Mater. Interfaces, 2013, 5, 2845–2852 CAS.
- E. Pöselt, C. Schmidtke, S. Fischer, K. Peldschus, J. Salamon, H. Kloust, H. Tran, A. Pietsch, M. Heine, G. Adam, U. Schumacher, C. Wagener, S. Förster and H. Weller, ACS Nano, 2012, 6, 3346–3355 CrossRef PubMed.
- E. Poselt, S. Fischer, S. Foerster and H. Weller, Langmuir, 2009, 25, 13906–13913 CrossRef CAS PubMed.
- J. Ostermann, J. P. Merkl, S. Flessau, C. Wolter, A. Kornowksi, C. Schmidtke, A. Pietsch, H. Kloust, A. Feld and H. Weller, ACS Nano, 2013, 7, 9156–9167 CrossRef CAS PubMed.
- H. Kloust, E. Poselt, S. Kappen, C. Schmidtke, A. Kornowski, W. Pauer, H. U. Moritz and H. Weller, Langmuir, 2012, 28, 7276–7281 CrossRef CAS PubMed.
- H. Kloust, C. Schmidtke, A. Feld, T. Schotten, R. Eggers, U. E. A. Fittschen, F. Schulz, E. Pöselt, J. Ostermann, N. G. Bastús and H. Weller, Langmuir, 2013, 29, 4915–4921 CrossRef CAS PubMed.
- H. Kloust, C. Schmidtke, J. P. Merkl, A. Feld, T. Schotten, U. E. A. Fittschen, M. Gehring, J. Ostermann, E. Poselt and H. Weller, J. Phys. Chem. C, 2013, 117, 23244–23250 CAS.
- T. Jin, F. Fujii, E. Yamada, Y. Nodasaka and M. Kinjo, J. Am. Chem. Soc., 2006, 128, 9288–9289 CrossRef CAS PubMed.
- E. Oh, K. Susumu, J. B. Blanco-Canosa, I. L. Medintz, P. E. Dawson and H. Mattoussi, Small, 2010, 6, 1273–1278 CrossRef CAS PubMed.
- M. Zhou, E. Nakatani, L. S. Gronenberg, T. Tokimoto, M. J. Wirth, V. J. Hruby, A. Roberts, R. M. Lynch and I. Ghosh, Bioconjugate Chem., 2007, 18, 323–332 CrossRef CAS PubMed.
- A. R. Clapp, E. R. Goldman and H. Mattoussi, Nat. Protoc., 2006, 1, 1258–1266 CrossRef CAS PubMed.
- H.-S. Han, N. K. Devaraj, J. Lee, S. A. Hilderbrand, R. Weissleder and M. G. Bawendi, J. Am. Chem. Soc., 2010, 132, 7838–7839 CrossRef CAS PubMed.
- C. Schieber, A. Bestetti, J. P. Lim, A. D. Ryan, T.-L. Nguyen, R. Eldridge, A. R. White, P. A. Gleeson, P. S. Donnelly, S. J. Williams and P. Mulvaney, Angew. Chem., Int. Ed., 2012, 51, 10523–10527 CrossRef CAS PubMed.
- A. Bernardin, A. Cazet, L. Guyon, P. Delannoy, F. Vinet, D. Bonnaffe and I. Texier, Bioconjugate Chem., 2010, 21, 583–588 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate techniques, Academic Press, San Diego, 1996 Search PubMed.
- I. L. Medintz, T. Pons, K. Susumu, K. Boeneman, A. M. Dennis, D. Farrell, J. R. Deschamps, J. S. Melinger, G. Bao and H. Mattoussi, J. Phys. Chem. C, 2009, 113, 18552–18561 CAS.
- J. M. Kogot, H. J. England, G. F. Strouse and T. M. Logan, J. Am. Chem. Soc., 2008, 130, 16156–16157 CrossRef CAS PubMed.
- R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS.
- F. Amblard, J. H. Cho and R. F. Schinazi, Chem. Rev., 2009, 109, 4207–4220 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed.
- P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821–1826 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- J. B. Blanco-Canosa, I. L. Medintz, D. Farrell, H. Mattoussi and P. E. Dawson, J. Am. Chem. Soc., 2010, 132, 10027–10033 CrossRef CAS PubMed.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- Z. Li, R. C. Jin, C. A. Mirkin and R. L. Letsinger, Nucleic Acids Res., 2002, 30, 1558–1562 CrossRef CAS PubMed.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078–1081 CrossRef CAS.
- C. J. Breshike, R. A. Riskowski and G. F. Strouse, J. Phys. Chem. C, 2013, 117, 23942–23949 CAS.
- F. Aldeek, X. Ji and H. Mattoussi, J. Phys. Chem. C, 2013, 117, 15429–15437 CAS.
- T. Pons, I. L. Medintz, K. E. Sapsford, S. Higashiya, A. F. Grimes, D. S. English and H. Mattoussi, Nano Lett., 2007, 7, 3157–3164 CrossRef CAS PubMed.
- E. Dulkeith, M. Ringler, T. A. Klar, J. Feldmann, A. M. Javier and W. J. Parak, Nano Lett., 2005, 5, 585–589 CrossRef CAS PubMed.
- Y. Liu, K. Yehl, Y. Narui and K. Salaita, J. Am. Chem. Soc., 2013, 135, 5320–5323 CrossRef CAS PubMed.
- E. Petryayeva and U. J. Krull, Langmuir, 2012, 28, 13943–13951 CrossRef CAS PubMed.
- J. Griffin, A. K. Singh, D. Senapati, P. Rhodes, K. Mitchell, B. Robinson, E. Yu and P. C. Ray, Chem. – Eur. J., 2009, 15, 342–351 CrossRef CAS PubMed.
- W. Q. Li, C. Y. Sun, F. Wang, Y. C. Wang, Y. W. Zhai, M. Liang, W. J. Liu, Z. M. Liu, J. Wang and F. Sun, Nano Lett., 2013, 13, 2477–2484 CrossRef CAS PubMed.
- E. Oh, D. Lee, Y. P. Kim, S. Y. Cha, D. B. Oh, H. A. Kang, J. Kim and H. S. Kim, Angew. Chem., Int. Ed., 2006, 45, 7959–7963 CrossRef CAS PubMed.
- A. Chompoosor, G. Han and V. M. Rotello, Bioconjugate Chem., 2008, 19, 1342–1345 CrossRef CAS PubMed.
- M. De, S. Rana, H. Akpinar, O. R. Miranda, R. R. Arvizo, U. H. F. Bunz and V. M. Rotello, Nat. Chem., 2009, 1, 461–465 CrossRef CAS PubMed.
- X. Q. An, F. Zhan and Y. Y. Zhu, Langmuir, 2013, 29, 1061–1068 CrossRef CAS PubMed.
- S. E. Skrabalak, L. Au, X. D. Li and Y. N. Xia, Nat. Protoc., 2007, 2, 2182–2190 CrossRef CAS PubMed.
- M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. Xie, C. Kim, K. H. Song, A. G. Schwartz, L. V. Wang and Y. Xia, Nat. Mater., 2009, 8, 935–939 CrossRef CAS PubMed.
- N. Anikeeva, T. Lebedeva, A. R. Clapp, E. R. Goldman, M. L. Dustin, H. Mattoussi and Y. Sykulev, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16846–16851 CrossRef CAS PubMed.
- F. Pinaud, X. Michalet, G. Iyer, E. Margeat, H. P. Moore and S. Weiss, Traffic, 2009, 10, 691–712 CrossRef CAS PubMed.
- A. R. Bayles, H. S. Chahal, D. S. Chahal, C. P. Goldbeck, B. E. Cohen and B. A. Helms, Nano Lett., 2010, 10, 4086–4092 CrossRef CAS PubMed.
- J. Lee, A. Sharei, W. Y. Sim, A. Adamo, R. Langer, K. F. Jensen and M. G. Bawendi, Nano Lett., 2012, 12, 6322–6327 CrossRef CAS PubMed.
- S. Impellizzeri, B. McCaughan, J. F. Callan and F. M. Raymo, J. Am. Chem. Soc., 2012, 134, 2276–2283 CrossRef CAS PubMed.
- I. L. Medintz, A. R. Clapp, H. Mattoussi, E. R. Goldman, B. Fisher and J. M. Mauro, Nat. Mater., 2003, 2, 630–638 CrossRef CAS PubMed.
- R. C. Somers, R. M. Lanning, P. T. Snee, A. B. Greytak, R. K. Jain, M. G. Bawendi and D. G. Nocera, Chem. Sci., 2012, 3, 2980–2985 RSC.
- E. R. Kay, J. Lee, D. G. Nocera and M. G. Bawendi, Angew. Chem., Int. Ed., 2013, 52, 1165–1169 CrossRef CAS PubMed.
- N. Lee and T. Hyeon, Chem. Soc. Rev., 2012, 41, 2575–2589 RSC.
- S. K. Yen, D. Jańczewski, J. L. Lakshmi, S. B. Dolmanan, S. Tripathy, V. H. B. Ho, V. Vijayaragavan, A. Hariharan, P. Padmanabhan, K. K. Bhakoo, T. Sudhaharan, S. Ahmed, Y. Zhang and S. Tamil Selvan, ACS Nano, 2013, 7, 6796–6805 CrossRef CAS PubMed.
- M. Satpathy, L. Wang, R. Zielinski, W. Qian, M. Lipowska, J. Capala, G. Y. Lee, H. Xu, Y. A. Wang, H. Mao and L. Yang, Small, 2014, 10, 544–555 CrossRef CAS PubMed.
- J. M. Perez, J. Grimm, L. Josephson and R. Weissleder, Neoplasia, 2008, 10, 1066–1072 CAS.
- J. Grimm, J. M. Perez, L. Josephson and R. Weissleder, Cancer Res., 2004, 64, 639–643 CrossRef CAS.
- O. J. Santiesteban, C. Kaittanis and J. M. Perez, Angew. Chem., Int. Ed., 2012, 51, 6728–6732 CrossRef CAS PubMed.
Footnote |
| † Contributed equally to the manuscript assembly. |
| This journal is © The Royal Society of Chemistry 2015 |