
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual-fluorescence pH probe for bio-labelling†
C.
Richter
a,
C.
Schneider
b,
M. T.
Quick
a,
P.
Volz
b,
R.
Mahrwald
a,
J.
Hughes
c,
B.
Dick
d,
U.
Alexiev
*b and
N. P.
Ernsting
*a
aDepartment of Chemistry, Humboldt-Universität zu Berlin, 12489 Berlin, Germany. E-mail: nernst@chemie.hu-berlin.de
bDepartment of Physics, Freie Universität Berlin, 14195 Berlin, Germany. E-mail: ulrike.alexiev@fu-berlin.de
cInstitute for Plantphysiology, Justus-Liebig Universität, 35390 Giessen, Germany
dInstitute for Physical and Theoretical Chemistry, Universität Regensburg, 93053 Regensburg, Germany
First published on 19th October 2015
Abstract
Although seminaphtorhodafluor (SNARF) dyes are already widely used to measure pH in cells and at biofilms, their synthesis has low yield and results in an unspecific position of a carboxy-group. The separation of 5′- and 6′-carboxy-SNARF reveals a pKa difference of 0.15, calling into question pH measurements with the (commercially available) mixture. Here we replace the bulky external dicarboxyphenyl ring with a propionate group and evaluate the spectral properties of the new derivative. Proceeding to the ethyl-iodoacetamide, covalent linkage to cysteine protein sites is achieved efficiently as shown with a cyanobacterial phytochrome, extending the scarce application of SNARF in bio-labelling in the current literature. Application in fluorescence lifetime imaging is demonstrated both with the lifetime-based and ratiometric-yield method.
1. Introduction
Xanthene-based ratiometric pH-probes have been used extensively in the investigation of pH in cells and at biofilms.1 Fluorescein homologues are most commonly used, for example BCECF (Scheme 1) which was optimized for solubility.2–4 However, only their emission intensity and not the color is affected by protonation, so that ps-time-resolved measurements of the fluorescence decay are often needed in quantitative pH experiments. For imaging one would prefer a dual emission band which reflects the protonated and deprotonated populations more directly. This distinction can be achieved with “seminaphtorhodafluor” homologues like carboxy-SNARF.5–14 Advantages of these dyes include a wide excitation range, fluorescence from both protonated and deprotonated forms, and a pKa between them which is in the physiological range. | ||
| Scheme 1 Chemical structures of xanthene-based pH-probes. | ||
Several drawbacks currently prevent more widespread use of the SNARF probes mentioned above, however. (i) Their synthesis has low yield and results in an unspecific position (5′ or 6′) of a second carboxy-group.5–7 Note that the isomeric mixture is commonly used in the literature.13–15 (ii) Available spectral and photophysical data are less extensive than for fluorescein. For example, in the equivalent case of 5′- and 6′-carboxyfluorescein the differences have been described;16,17 such a distinction is still missing for rhodafluors. (iii) At higher pH the dicarboxyphenyl ring, being external to the electronic chromophore, causes a lactonic form and therefore renders a fraction of the dissolved dye useless.18–20
Here we report the new derivatives PAc- and EA–SNARF (Scheme 2) where the problematic dicarboxyphenyl ring has been replaced by a propionate or ethylamino group, respectively. For investigations of protonation dynamics near the entrance or exit site of proton channels in proteins, for example, covalent linkage of the dye to a target site is needed. A widely-used linker for cysteines is maleimide. The connection of maleimide to the SNARF-system was already described by I. Himachi and coworkers,7 but their chain is quite long and, correspondingly, the footprint of the tethered dye is too large for spatial resolution of the proton wire topology at the protein surface. To overcome this limitation we developed IA–SNARF (see also Scheme 2), described here, where the fluorescent core bears an ethyl chain with an iodacetamido group. This allows us to label a cyanobacterial phytochrome covalently at a free cysteine. Spectral and radiative properties of the label are described. This novel SNARF with dual pH-dependent fluorescence bands and well-separated fluorescence lifetimes is better suited for imaging than fluorescein derivatives.7,21–23
 | ||
| Scheme 2 New derivatives with propionate (PAc), ethyl-amino (EA) and iodoacet-amide (IA) functionality. Colors highlight the spectral difference. | ||
2. Experimental section
Syntheses are described in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dmso 20:1 (vol:vol before mixing) unless stated otherwise. For stationary measurements, standard buffer solutions from Merck were diluted with water 1:1. Note that the dmso cosolvent shifts the pH up by 0.1 on average. Buffers consisted of a disodium-hydrogenphosphate/potassiumdihydrogenphosphate mixture or of a citric acid/sodium hydroxyl/hydrochloric acid mixture (traceable to SRM from NIST and PTB.) The sodium salt of the carboxy-derivatives PAc- and 5′ (or 6′) C-SNARF dissolves excellently in water, but after acidification the absorption spectra at pH ∼ 5 showed amplitude variations if the peak absorbance of the protonated form exceeded 0.2 over 1 cm. The variations are possibly caused by very slow aggregation or other adsorption processes which we cannot account for. Thus it is challenging to obtain optical spectra with a well-defined isosbestic point. For time-resolved fluorescence measurements, the sodium salt of PAc–SNARF was first dissolved in Tris-citrate buffer (Sigma) at pH 10; subsequent titration down to pH 5 resulted in absorbance spectra with clear isosbestic points.
139 THGC and EO65706 580 nm 1-9-13), or else with a long-pass filter (LP) ≥545 nm (HQ545, Chroma) or LP 715 nm (RG715, Schott). Fluorescence decay traces were collected in 1024 time channels with a channel width of 19.5 ps. The instrumental response function (IRF) was determined at the corresponding wavelengths with a colloidal silica solution as scattering material (LUDOX, Grace). The IRF of the system had 60–70 ps full width at half maximum. After deconvolution with the IRF the traces were fitted by a sum of exponentials.26,28
:dmso 20:1 (vol:vol before mixing) unless stated otherwise. For stationary measurements, standard buffer solutions from Merck were diluted with water 1:1. Note that the dmso cosolvent shifts the pH up by 0.1 on average. Buffers consisted of a disodium-hydrogenphosphate/potassiumdihydrogenphosphate mixture or of a citric acid/sodium hydroxyl/hydrochloric acid mixture (traceable to SRM from NIST and PTB.) The sodium salt of the carboxy-derivatives PAc- and 5′ (or 6′) C-SNARF dissolves excellently in water, but after acidification the absorption spectra at pH ∼ 5 showed amplitude variations if the peak absorbance of the protonated form exceeded 0.2 over 1 cm. The variations are possibly caused by very slow aggregation or other adsorption processes which we cannot account for. Thus it is challenging to obtain optical spectra with a well-defined isosbestic point. For time-resolved fluorescence measurements, the sodium salt of PAc–SNARF was first dissolved in Tris-citrate buffer (Sigma) at pH 10; subsequent titration down to pH 5 resulted in absorbance spectra with clear isosbestic points.
139 THGC and EO65706 580 nm 1-9-13), or else with a long-pass filter (LP) ≥545 nm (HQ545, Chroma) or LP 715 nm (RG715, Schott). Fluorescence decay traces were collected in 1024 time channels with a channel width of 19.5 ps. The instrumental response function (IRF) was determined at the corresponding wavelengths with a colloidal silica solution as scattering material (LUDOX, Grace). The IRF of the system had 60–70 ps full width at half maximum. After deconvolution with the IRF the traces were fitted by a sum of exponentials.26,28
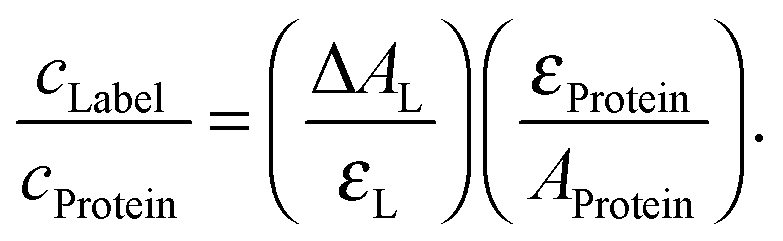 | (1) |
3. Results and discussion
3.1 Properties of PAc- and other SNARF derivatives
 | ||
| Fig. 1 Optical absorption (a) and fluorescence (b) spectra of PAc–SNARF in water/dmso (20:1 by volume, for buffer see text) as function of pH at 22 °C. Red and blue curves represent the species-associated spectra (SAS) of the pure deprotonated and protonated species, respectively, from global analysis. In stationary measurements, excitation wavelength λexc = 488 nm was chosen in order to enhance the fluorescence contribution by the protonated species. | ||
For analysis we may assume that the protonated and deprotonated forms are in equilibrium in the electronic ground state. The absorption spectra in Fig. 1a can therefore be decomposed into contributions from the two ground-state forms to yield the species-associated spectra (SAS). These are shown as red and blue lines, respectively, in panel (a). Their relative weight, corresponding to the mole fractions x of the two forms, is described by
| xred = 10−pKa/(10−pKa + 10−pH) and xblue = 1 − xred. | (2) |
The maximum extinction coefficient of the deprotonated form εred,max = 47100 ± 500 l mol−1 cm−1 was determined at pH 9.31 From Fig. 1a we find that upon protonation the maximum extinction coefficient drops to 58%, hence εblue,max = 27320 l mol−1 cm−1.
Species-associated spectra were fitted by one or several functions
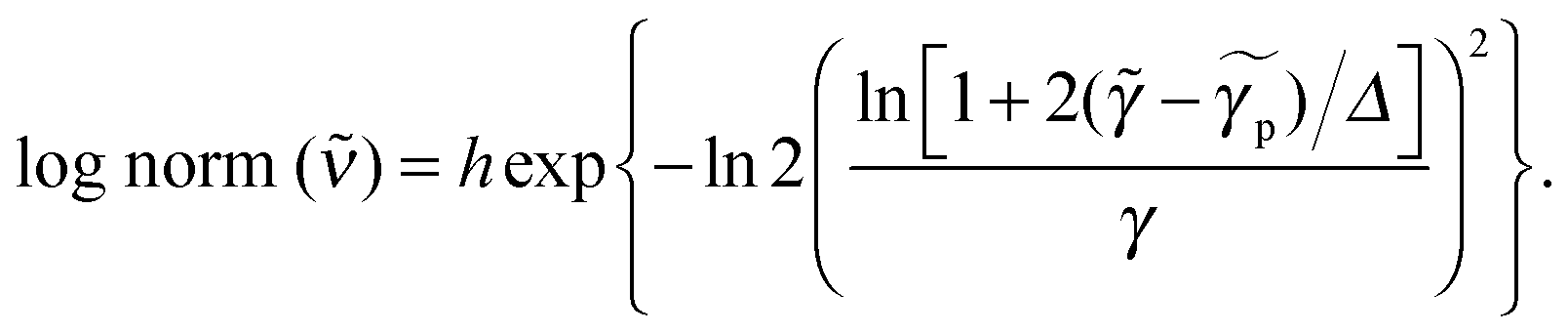 | (3) |
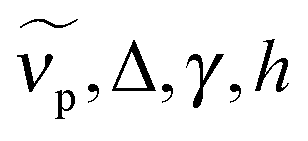 for peak wavenumber [cm−1], width [cm−1], asymmetry [−], and amplitude [−]. Note that in case of absorption, h refers to extinction coefficients (in l mol−1 cm−1). In case of fluorescence, quantum distributions over wavelengths were converted into distributions over wavenumbers, F(
for peak wavenumber [cm−1], width [cm−1], asymmetry [−], and amplitude [−]. Note that in case of absorption, h refers to extinction coefficients (in l mol−1 cm−1). In case of fluorescence, quantum distributions over wavelengths were converted into distributions over wavenumbers, F(![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ) = (∂Φ/(Φ)) ∝ (∂Φ/∂λ)λ2 and then set to 1 at the peak of the spectrum of the deprotonated form (Φ is the total number of photons counted). Lognorm parameters are collected in Table 1 and Tables S1–S5 (ESI†).
:1 vol:vol)
) = (∂Φ/(Φ)) ∝ (∂Φ/∂λ)λ2 and then set to 1 at the peak of the spectrum of the deprotonated form (Φ is the total number of photons counted). Lognorm parameters are collected in Table 1 and Tables S1–S5 (ESI†).
:1 vol:vol)
|
|
Δ/cm−1 | γ | h | |
|---|---|---|---|---|
|
a For 14300–20160 cm−1.
b 14300–22220 cm−1.
c dΦ/d.
|
||||
| Absorption (Fig. 1a) | ||||
| Deprotonateda | 16309 |
971 | −0.42 | 3425 |
| 17602 |
1576 | 0.19 | 47100 |
|
| 19033 |
1675 | 0.49 | 15926 |
|
| Protonatedb | 16934 |
1450 | −0.58 | 563 |
| 18206 |
974 | −0.07 | 13403 |
|
| 19130 |
1696 | −0.25 | 23321 |
|
| 20385 |
2012 | 0.53 | 13808 |
|
| Emissionc from 488 nm excitation (Fig. 1b) | ||||
| Deprotonated | 15844 |
1605 | −0.12 | 1.000 |
| 14174 |
1434 | −0.25 | 0.193 | |
| Protonated | 17283 |
1219 | 0.03 | 0.316 |
| 15953 |
1604 | −0.26 | 0.247 | |
| 14242 |
1562 | −0.42 | 0.044 | |

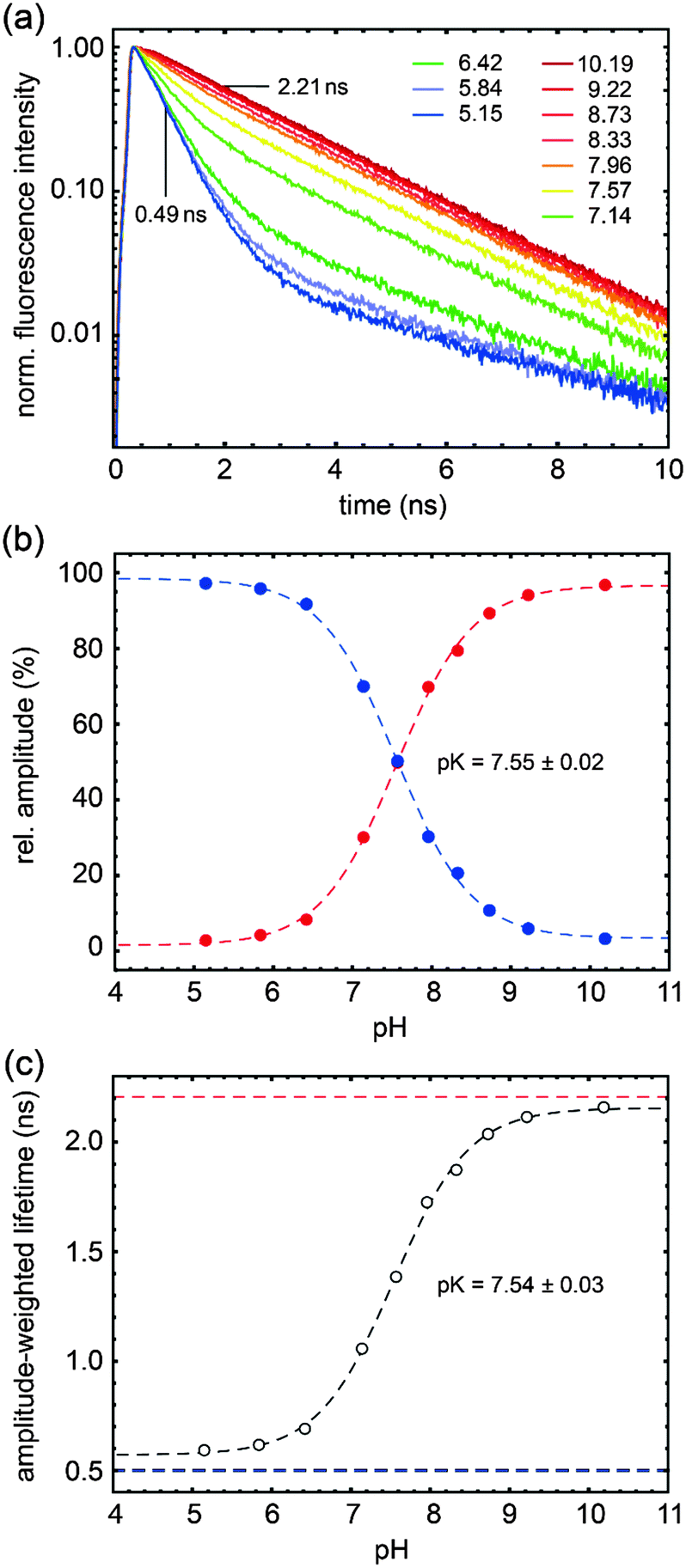 | ||
| Fig. 2 Time-resolved fluorescence of PAc–SNARF as a function of pH. (a) Decay traces at various pH values as indicated by the different colors. Red and blue curves refer to the pure deprotonated and protonated species; the two lifetimes from global analysis are indicated. (b) Relative amplitudes of the decay components due to the protonated (blue) and deprotonated form (red curve). (c) Amplitude-weighted lifetimes. Fit lines and pKa values are also given. Conditions: 10 mM Tris Citrate, 190 mM NaCl, 20:1 water:dmso volume fraction, 20 °C. λexc = 526 nm, and emission was detected with long pass filter HQ 545. | ||
The dependence of fluorescence lifetimes on excitation and emission wavelengths is shown in Fig. 3. At pH 10 the deprotonated form is detected easily as a pure species with 2.21 ns lifetime (λex = 526 nm, λem = 545 nm long pass). The same observation is made even upon excitation at the “blue” side of the absorption spectrum (λex = 488 nm), by choosing an appropriate emission filter (e.g. longpass 715 nm or bandpass 630 nm). At pH 5 the protonated form, when excited at the isosbestic point or at 488 nm, gives fluorescence decay traces with a main decay component of 0.49 ns (96–97%). The remaining percentages can be attributed to the residual amount of the deprotonated form, in particular when detecting in the “red” emission band (λex = 488 nm, λem = 630 nm band pass). The existence of a minor additional protonated species with a lifetime of ∼4.0 ns (λex = 526 nm, λem = 545 nm long pass) is also conceivable.
 | ||
| Fig. 3 Fluorescence decay analysis of protonated and deprotonated forms of PAc–SNARF for different excitation and emission wavelengths, as indicated. (a) Fluorescence decay traces and their multi-exponential fits. (b) Fit residuals. (BP-band pass; LP-long pass filter). | ||
000–25000 cm−1). Thus we find oscillator strengths fosc,blue = 0.65 and fosc,red = 0.83. Radiative lifetimes are calculated with the Strickler–Berg formula, resulting in τrad,blue = 6.03 ns and τrad,red = 5.08 ns. Compare this with the measured fluorescence lifetimes of τblue = 0.49 ns and τred = 2.21 ns (Fig. 3). Looking only at the red form at pH > 9, we predict a fluorescence quantum yield QYred = 44%. Instead our direct measurement (with rhodamine 101/methanol as reference) gave a value of 35%. The experimental value being lower is consistent with the idea that a higher electronically excited state contributes to the first absorption band of the red form.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H, see Scheme 2) in water are presented in Table 2. The lowest-energy transition of the protonated form is calculated at ΔE = 21290 cm−1 with oscillator strength f = 0.49. It is accompanied by a moderate change in dipole moment, Δμ = 4.3 D (=Debye). For the deprotonated form the lowest transition at 20080 cm−1 is predicted to be strong (f = 0.75). The energy separation of the lowest excited electronic states and the relative strengths of the corresponding transitions are in good agreement with observations. Note that the TDDFT method also finds a charge transfer transition (Δμ = 25.1 Debye) with zero oscillator strength. This is due to a well-known artifact of DFT methods which overestimate the stability of charge-transfer states. Molecular orbitals are shown in the ESI.†
H) in water, calculated with TD-DFT/B3LYP/cc-pVDZ for the first three electronic transitions
H, see Scheme 2) in water are presented in Table 2. The lowest-energy transition of the protonated form is calculated at ΔE = 21290 cm−1 with oscillator strength f = 0.49. It is accompanied by a moderate change in dipole moment, Δμ = 4.3 D (=Debye). For the deprotonated form the lowest transition at 20080 cm−1 is predicted to be strong (f = 0.75). The energy separation of the lowest excited electronic states and the relative strengths of the corresponding transitions are in good agreement with observations. Note that the TDDFT method also finds a charge transfer transition (Δμ = 25.1 Debye) with zero oscillator strength. This is due to a well-known artifact of DFT methods which overestimate the stability of charge-transfer states. Molecular orbitals are shown in the ESI.†
H) in water, calculated with TD-DFT/B3LYP/cc-pVDZ for the first three electronic transitions
| ΔE/cm−1 | f | Δμ/D | |
|---|---|---|---|
| Deprotonated | 20080 |
0.752 | 7.43 |
| 22460 |
0.000 | 25.12 | |
| 23910 |
0.008 | 6.96 | |
| Protonated | 21290 |
0.491 | 4.27 |
| 25140 |
0.158 | 10.99 | |
| 27130 |
0.027 | 8.01 | |
:dmso 5:1 in this case, in order to avoid solubility problems at pH ∼5. Species-associated spectra of absorption and fluorescence are almost identical to those of PAc–SNARF at the same cosolvent concentration; however the pKa of 5′C- is larger by 0.15 compared to 6′C- (see ESI†). In precision measurements of pH it is important to use only one of the isomers, because if a mixture is used, protonated and deprotonated forms coexist in a broader range and resolution is lost correspondingly.
3.2 Labelling of cyanobacterial phytochrome Cph1Δ2
The function of light-activated proteins such as bacteriorhodopsin, visual rhodopsin, channelrhodopsin or phytochrome is associated with protonation changes in the protein and subsequent proton release/uptake events.32 A pH-dependent dye for covalent linkage to the protein surface often used in the investigation of these proteins is iodoacetamido-fluorescein, IA-F.33 Instead we test here the covalent binding of IA–SNARF to a protein surface, using the cyanobacterial phytochrome Cph1Δ230 as example.Under the conditions described in the Experimental Section, about 0.8 mol of IA–SNARF was bound per mol Cph1Δ2. The 2VEA crystal structure29 implies that only C371 in the sensor module is exposed and thus is the most likely target for the label. Fig. 4 shows the absorption spectra of Cph1Δ2 in the Pr form of phytochrome. For pH 6 and pH 8.8, spectra before and after labelling are compared (dashed and solid line, respectively). When SNARF is bound to the protein, the absorption band has its maximum at λmax = 529 ± 1 nm in the protonated form and at λmax = 565 ± 1 nm in the deprotonated form. The protonated form is slightly red-shifted compared to that of PAc–SNARF (cf.Fig. 1a). The same holds true for the isosbestic point, which is now observed at 541 nm. The fluorescence lifetimes of IA–SNARF are τblue = 0.49 ns and τred = 2.10 ns, very similar to PAc–SNARF. As the peak absorbance of phytochrome in the Pr form is located at 658 nm, an excitation of the SNARF dye results in minimal phytochrome activation. This is one prerequisite for using the SNARF dye with photoreceptor proteins.
 | ||
| Fig. 4 Covalent binding of IA–SNARF to cyanobacterial phytochrome Cph1Δ2. Absorption spectra are shown before (–––) and after (–) labelling at pH 8.8 (a) and pH 6.0 (b). Arrows indicate the absorption maximum of the deprotonated (565 nm at pH 8.8) and the protonated (529.5 nm at pH 6) form of bound SNARF. Inset in (a): left – photographs of the fractions after gel filtration containing SNARF (red fluorescence in fraction F2) and phytochrome (blue absorption in fraction F2); right – SDS PAGE of IA–SNARF in the fluorescence image (lane 1: molecular weight marker, lane 2: Cph1Δ2-A-SNARF). Inset in (b): absorption spectrum of IA–SNARF at pH 6.0. | ||
4. Outlook
Besides investigations on the molecular level, monitoring the pH inside living cells is important since intracellular pH changes are critical for cell and tissue activities, including phagocytosis, endocytosis, ion transport, signaling cascades, and synaptic transmission. Both PAc–SNARF and IA–SNARF are able to enter the cell freely.To validate the applicability of our SNARF derivatives as dual fluorescence probe for quantitative pH measurements in imaging experiments, we conducted two proof-of-principle experiments using a confocal raster scanning fluorescence microscope equipped with TCSPC detection for fluorescence lifetime experiments.29
The first experiment (see Fig. 5) demonstrates the amplitude-weighted-lifetime method for determining pH. Two drops of PAc–SNARF solution with different pH values (6.5 and 7.8) are placed on a cover slip. Panel (a) shows the false-coded FLIM image according to the different fluorescence lifetime characteristics of the image, clearly indicating the two different drops in the image. The corresponding fluorescence decay curves and lifetimes are shown in panel (b). The calibration curve from the TCSPC measurements (Fig. 2c) was used to obtain the pH values as shown in Fig. 5c, which match the target pH values. Since the lifetime is used in the pH calibration curve, it does not matter whether the calibration was obtained in a spectroscopic or microscopic setup as long as the excitation and emission parameter are identical.
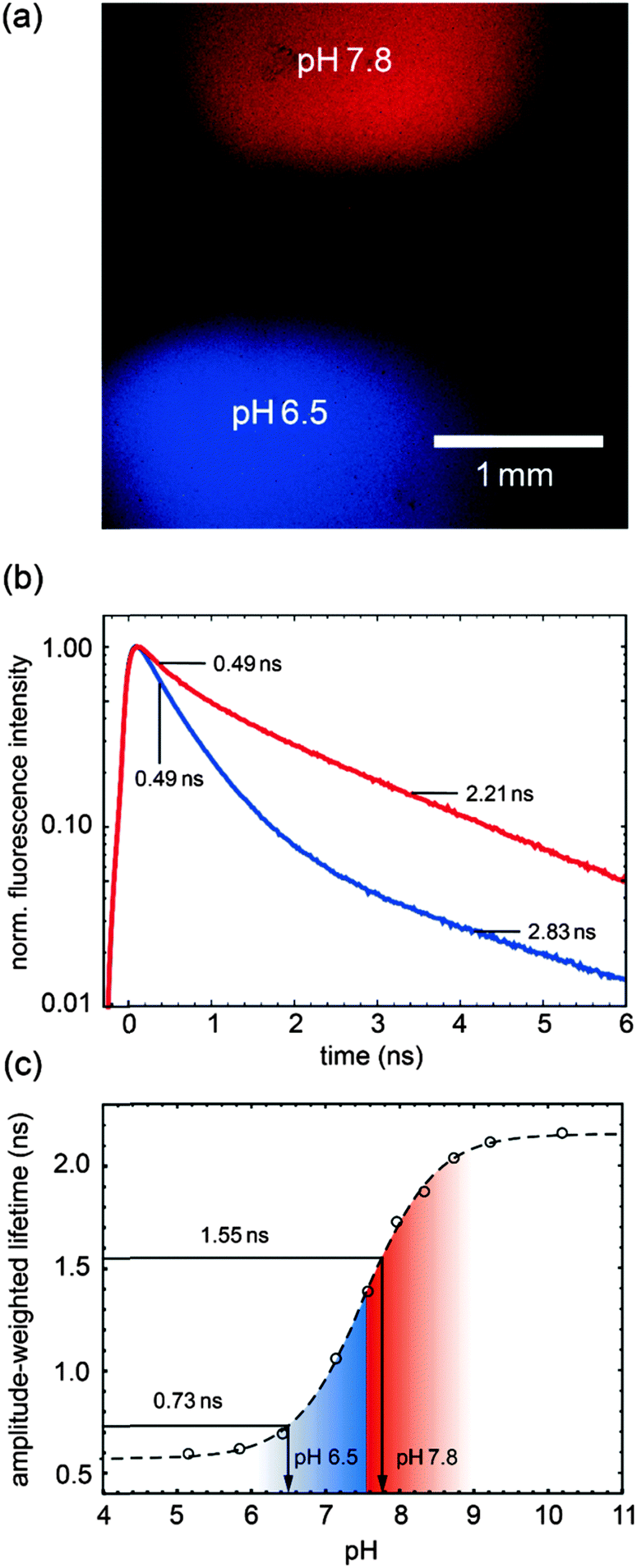 | ||
| Fig. 5 FLIM based determination of pH values. (a) FLIM image of PAc–SNARF in acidic (blue) and basic (red) environment. Conditions: 4× objective, excitation at 526 nm and emission filter HQ545LP. (b) Fluorescence decay traces of protonated (blue) and deprotonated (red) PAc–SNARF of (a). (c) pH calibration curve obtained by TCSPC cuvette measurements using the same excitation wavelength and emission filter. The determined amplitude-weighted lifetimes (0.73 & 1.55 ns) of the traces in (b) and respective determined pH values (6.5 & 7.8) are shown. | ||
The second experiment (see Fig. 6) demonstrates the ratiometric-yield method with a SNARF-labelled protein. Here the two emission bands from only one excitation wavelength are exploited. Using the standard set of emission filters that exists in many confocal laser scanning microscopes, we measure the stationary fluorescence intensity of the SNARF-labelled phytochrome in the “red” and “green” emission channel (green: λem = 545–560 nm, red: λem > 590 nm). The corresponding images are shown in panel (a). The calibration curve (b) was obtained with the confocal microscope using the same settings as for the ratiometric pH determination of the pH at the protein surface of phytochrome. The resulting pH value is 7.7.
 | ||
| Fig. 6 Laser scanning image for ratiometric intensity based determination of pH values. (a) Image of SNARF-labelled phytochrome in the two detection channels. Conditions: 40× objective, excitation at 526 nm and dual detection mode using a dichroic filter 560 dclp. In the “green” and “red” channel an additional HQ545LP respectively OG590 longpass filter was used. (b) pH calibration curve obtained by the fluorescence intensity ratio r = Ired/Igreen of both channels measuring PAc–SNARF calibration samples at different pH values. | ||
The results of these proof-of-principle experiments show clearly that the new SNARF dye offers the opportunity for tracking spatio-temporal pH changes inside living cells, and to elucidate the function of proteins to which the SNARF dye is attached to.
Acknowledgements
This project was supported by the Deutsche Forschungsgemeinschaft through grants to UA and NPE (Collaborative Research Center 1078 “Protonation Dynamics in Protein Function”, projects A2 and A3) and to UA (Collaborative Research Center 1112 “Nanocarriers: Architecture, Transport, and Topical Application of Drugs for Therapeutic Use”, project B03). We would like to thank Dr Katharina Achazi for providing HeLa cells.Notes and references
- J. Han and K. Burgess, Chem. Rev., 2010, 110, 2709–2728 CrossRef CAS PubMed.
- A. M. Paradiso, R. Y. Tsien and T. E. Machen, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 7436–7440 CrossRef CAS.
- X. Wang, A. J. Levi and A. P. Halestrap, Am. J. Physiol.: Heart Circ. Physiol., 1994, 267, H1759–H1769 CAS.
- M. J. Fehr, M. A. McCloskey and J. W. Petrich, J. Am. Chem. Soc., 1995, 117, 1833–1836 CrossRef CAS.
- R. P. Haugland and J. Whitaker and U.S. Pat., 4945171, 1990 Search PubMed.
- J. Liu, Z. Diwu and W.-Y. Leung, Bioorg. Med. Chem. Lett., 2001, 11, 2903–2905 CrossRef CAS PubMed.
- E. Nakata, H. Wang and I. Hamachi, ChemBioChem, 2008, 9, 25–29 CrossRef CAS PubMed.
- A. Keppler, H. Pick, C. Arrivoli, H. Vogel and K. Johnsson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9955–9959 CrossRef CAS PubMed.
- E. Nakata, Y. Nazumi, Y. Yukimachi, Y. Uto, H. Maezawa, T. Hashimoto, Y. Okamoto and H. Hori, Bioorg. Med. Chem. Lett., 2011, 21, 1663–1666 CrossRef CAS PubMed.
- R. Martinez-Zaguilan, G. M. Martinez, F. Lattanzio and J. R. Gillies, Am. J. Phys., 1991, 260, 297–307 Search PubMed.
- M. Frasconi, R. Tel-Vered, J. Elbaz and I. Willner, J. Am. Chem. Soc., 2010, 132, 2029–2036 CrossRef CAS PubMed.
- M. A. Schroeder, M. A. Ali, A. Hulikovam, C. T. Supuran, K. Clarke, R. D. Vaughan-Jones, D. J. Tyler and P. Swietach, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E958–E967 CrossRef CAS PubMed.
- M. P. Weekes, S. Y. Tan, E. Poole, S. Talbot, R. Antrobus, D. L. Smith, C. Montag, S. P. Gygi, J. H. Sinclair and P. J. Lehner, Science, 2013, 340, 199–202 CrossRef CAS PubMed.
- S. Schlafer, J. E. Garcia, M. Greve, M. K. Raarup, B. Nyvad and I. Dige, Appl. Environ. Microbiol., 2015, 81, 1267–1273 CrossRef PubMed.
- K. J. Buckler and R. D. Vaughan-Jones, Pflugers Arch., 1990, 417, 234–239 CrossRef CAS PubMed.
- M. V. Kvach, D. A. Tsybulsky, A. V. Ustinov, I. A. Stepanova, S. L. Bondarev, S. V. Gontarev, V. A. Korshun and V. V. Shmanai, Bioconjugate Chem., 2007, 18, 1691–1696 CrossRef CAS PubMed.
- I. Johnson and M. T. Z. Spence, The Molecular Probes Handbook, 11th edn, 2010, ch 1.5, Fluorescein, Oregon Green and Rhodamine Green Dyes Search PubMed.
- Y. Kaneko and D. C. Neckers, J. Phys. Chem. A, 1998, 102, 5356–5363 CrossRef CAS.
- J. Karpiuk, J. Phys. Chem. A, 2004, 108, 11183–11195 CrossRef CAS.
- J. Karpiuk, Z. R. Grabowski and F. C. De Schryver, J. Phys. Chem., 1994, 98, 3247–3256 CrossRef CAS.
- A. Galant, R. P. Koester, E. A. Ainsworth, L. M. Hicks and J. M. Jez, New Phytol., 2012, 194, 220–229 CrossRef CAS PubMed.
- T. Kreisig, R. Hoffmann and T. Zuchner, Anal. Chem., 2011, 83, 4281–4287 CrossRef CAS PubMed.
- M. Misheva, G. Kaur, K. R. W. Ngoei, Y. Y. Yeap, I. H. W. Ng, K. M. Wagstaff, D. C. H. Ng, D. A. Jans and M. A. Bogoyevitch, Biochim. Biophys. Acta, 2014, 1843, 253–264 CrossRef CAS PubMed.
- A. A. Granovsky, GAMESS Firefly version 8, (http://classic.chem.msu.su/gran/firefly/index.html) which is partly based on, M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis, J. A. Montgomery, J. Comput. Chem., 14, 1993, 1347–1363.
- K. H. Drexhage, J. Res. NBS, Sect. A: Phys. Chem., 1976, 80, 421–428 Search PubMed.
- U. Alexiev, I. Rimke and T. Pöhlmann, J. Mol. Biol., 2003, 328, 705–719 CrossRef CAS PubMed.
- T.-Y. Kim, K. Winkler and U. Alexiev, Photochem. Photobiol., 2007, 83, 378–385 CrossRef CAS PubMed.
- U. Alexiev and D. L. Farrens, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 694–709 CrossRef CAS PubMed.
- A. Boreham, T.-Y. Kim, V. Spahn, C. Stein, L. Mundhenk, A. D. Gruber, R. Haag, P. Welker, K. Licha and U. Alexiev, ACS Med. Chem. Lett., 2011, 2, 724–728 CrossRef CAS PubMed.
- L. O. Essen, J. Mailliet and J. Hughes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14709–14714 CrossRef CAS PubMed.
- The storage form (see ESI†) was weighed in since it can be purified better. Reproducible results were obtained when milligram quantities were dissolved directly in the appropriate final liquid volume.
- J. J. van Thor, B. Borucki, W. Crielaard, H. Otto, T. Lamparter, J. Hughes, K. J. Hellingwerf and M. P. Heyn, Biochemistry, 2001, 40, 11460–11471 CrossRef CAS PubMed.
- U. Alexiev, P. Scherrer, T. Marti, H. G. Khorana and M. P. Heyn, FEBS Lett., 1995, 373, 81–84 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of all compounds can be found. See DOI: 10.1039/c5cp05454k |
| This journal is © the Owner Societies 2015 |