
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriplet–triplet annihilation upconversion followed by FRET for the red light activation of a photodissociative ruthenium complex in liposomes†
Sven H. C.
Askes
a,
Miroslav
Kloz
b,
Gilles
Bruylants
c,
John T. M.
Kennis
b and
Sylvestre
Bonnet
 *a
*a
aLeiden Institute of Chemistry, Gorlaeus Laboratories, Leiden University, P.O. Box 9502, 2300 RA Leiden, The Netherlands. E-mail: bonnet@chem.leidenuniv.nl
bLaserlab Amsterdam, VU University Amsterdam, De Boelelaan 1081, 1081 HV Amsterdam, The Netherlands
cEngineering of Molecular NanoSystems, Université Libre de Bruxelles, 50 av. F.D. Roosevelt, 1050 Brussels, Belgium
First published on 22nd September 2015
Abstract
Upconversion is a promising way to trigger high-energy photochemistry with low-energy photons. However, combining upconversion schemes with non-radiative energy transfer is challenging because bringing several photochemically active components in close proximity results in complex multi-component systems where quenching processes may deactivate the whole assembly. In this work, PEGylated liposomes were prepared that contained three photoactive components: a porphyrin dye absorbing red light, a perylene moiety emitting in the blue, and a light-activatable ruthenium prodrug sensitive to blue light. Time-dependent spectroscopic studies demonstrate that singlet perylene excited states are non-radiatively transferred to the nearby ruthenium complex by Förster resonance energy transfer (FRET). Under red-light irradiation of the three-component membranes, triplet–triplet annihilation upconversion (TTA-UC) occurs followed by FRET, which results in a more efficient activation of the ruthenium prodrug compared to a physical mixture of two-component upconverting liposomes and liposomes containing only the ruthenium complex. This work represents a rare example where TTA-UC and Förster resonance energy transfer are combined to achieve prodrug activation in the phototherapeutic window.
Introduction
Light-sensitive ruthenium(II) polypyridyl compounds are classical tools in photochemistry that have been recently proposed as prodrugs in photoactivatable anticancer therapy (PACT).1–8 As shown in classical photodynamic therapy (PDT), the use of light to treat cancer allows for spatially and temporally controlling the release of a toxic species, which lowers side effects for cancer patients.9–12 Whereas PDT drugs rely on the photocatalytic generation of singlet oxygen to kill cancer cells, PACT exerts cytotoxic activity mainly via an oxygen-independent mechanism, which makes them suitable for hypoxic tumours.3,4,7,13 Meanwhile, loading anticancer drugs into drug carriers such as liposomes helps targeting the compounds to tumour tissues.9,14,15 Especially sterically hindered liposomes, i.e., those grafted with polyethylene glycol chains, have been recognized as versatile and biocompatible drug carriers for the treatment of various diseases because of their long lifetime in the blood circulation. With such PEGylated liposomes tumour uptake is increased because of the so-called enhanced permeability and retention (EPR) effect.16,17 In PACT, activation of, for example, ruthenium-functionalized liposomes could be realized using visible light.18 However, most ruthenium(II) polypyridyl compounds require activation with blue light (400–500 nm), which is outside the so-called “phototherapeutic window”, a range of wavelengths (600–1000 nm) that permeate mammalian tissues optimally. This drawback can be circumvented by using upconverting drug carriers: once in a tumour they locally convert red photons into blue photons that subsequently activate the phototherapeutic drug without having to travel over long distances in the tissue.Triplet–triplet annihilation upconversion (TTA-UC), a photophysical interplay of light and molecular dyes, is very promising for upconversion because it features strong red light absorption and high upconversion quantum yields at low irradiation power. In TTA-UC, low-energy photons are converted into higher-energy photons by means of a bimolecular mechanism involving a sensitizer and two annihilator molecules (Fig. 1).19–22 The sensitizer absorbs the low-energy light to generate a triplet state, which is transferred to an annihilator molecule by collisions. Further collision of two triplet annihilator molecules leads to triplet–triplet annihilation (TTA), whereby one annihilator molecule is promoted to the high energy-emitting singlet excited state, while the other falls back to the ground state. TTA-UC has been demonstrated in organic solvent,19,20,22 ionic liquid,23 polymer matrix,19,21,24–28 functionalized polymer,29,30 various water-soluble nanodevices,8,31–40 and in a solvent-free liquid.41 In all these systems further use of the upconverted light, for example to activate a prodrug,8 excite a quantum dot,38 control a soft actuator,42 or power photoelectrochemistry,43 relied exclusively on radiative energy transfer, because the molecule sensitive to high-energy light lies too far, at the nanoscale (>10 nm), from the annihilator. For example, Kwon et al. showed that TTA-UC in the core of oleic acid nanoparticles, encapsulated by a 12 nm thick silica layer, could trigger a photocatalytic reaction on CdS nanoparticles grafted on the silica shell, but that the shell was too thick to allow non-radiative energy transfer.38 Non-radiative energy transfer would be by far preferable as it is more efficient; however, it also requires a close contact, at the nanoscale, between the annihilator molecule and the functional molecule to be photoactivated.
 | ||
| Fig. 1 (a) Chemical structures of palladium tetraphenyltetrabenzoporphyrin (1), perylene (2), [Ru(tpy)(bpy)(SRR′)]2+ (32+), and [Ru(tpy)(bpy)(H2O)]2+ (42+), and the photochemical reaction from 32+ to 42+. (b) Cartoon showing the sequence of photochemical events demonstrated in this work: red light is absorbed by compound 1, after which triplet–triplet annihilation upconversion (TTA-UC) occurs, followed by non-radiative energy transfer from 2 to 32+, and finally the ruthenium prodrug photodissociates from the lipid bilayer. (c) Absorption spectrum of 32+ in chloroform (solid, left axis, εmax = 7700 at λmax = 456 nm) and area-normalized emission spectrum of compound 2 in PEGylated DMPC liposomes (dashed, right axis, λexc = 400 nm, 0.5 mol% of compound 2 with respect to the lipids). | ||
In this work PEGylated liposomes were used as a supramolecular scaffold to put palladium tetraphenyltetrabenzoporphyrin (1) and perylene (2) in close proximity to the cytotoxic ruthenium complex [Ru(tpy)(bpy)(SRR′)]2+ (32+, see Fig. 1a). When put together the red photosensitizer 1 and the blue emitter 2 are capable of red-to-blue TTA upconversion. On the other hand, 32+ dissociates, upon blue light irradiation, into the aqua species [Ru(tpy)(bpy)(H2O)]2+ (42+) and the free thioether ligand (Fig. 1a).8,18,44,45 The cytotoxicity of 32+ and its modification by blue light irradiation is a complex matter that will be reported in a separate paper.46 Here we realized that the metal-to-ligand charge transfer (MLCT) absorption band of 32+ ideally overlaps with the emission spectrum of 2 (Fig. 1c), which maximizes the distance at which non-radiative energy transfer from 2 to 32+ may take place. In liposomes containing all three components 1, 2, and 32+, the average distance between photochemically active components becomes shorter than 5–8 nm, for which non-radiative energy transfer may take place (see Fig. 1b).47 However, such proximity might also open unwanted quenching routes, such as energy back-transfer from the complex to either photosensitizer or annihilator, hetero triplet–triplet annihilation between any pair of triplets present in the membrane, or phase separation of one of the molecules. This article explores via steady-state and time-dependent spectroscopic studies how efficient non-radiative energy transfer from perylene to the ruthenium complex is, and whether the red-light triggered photosubstitution reaction in the three-component liposomes is more or less efficient than the (known) physical mixture of upconverting liposomes (containing only 1 and 2) and ruthenium-functionalized liposomes (containing only 32+).8
Experimental section
General
Palladium tetraphenyltetrabenzoporphyrin (1) was purchased from Frontier Scientific, Inc. (Logan, Utah, USA). Perylene (2) was purchased from Sigma-Aldrich Chemie BV (Zwijndrecht, The Netherlands). Sodium N-(carbonyl-methoxypolyethylene glycol-2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-MPEG-2000), and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) were purchased from Lipoid GmbH (Ludwigshafen, Germany) and stored at −18 °C. Dulbecco's phosphate buffered saline (DPBS) was purchased from Sigma Aldrich and had a formulation of 8 g L−1 NaCl, 0.2 g L−1 KCl, 0.2 g L−1 KH2PO4, and 1.15 g L−1 K2HPO4 with a pH of 7.1–7.5. All chemicals were used as received. The synthesis of 3[PF6]2 is described elsewhere.8 The concentration of 32+ in the liposome samples was measured with inductively coupled plasma optical emission spectroscopy (ICP-OES) after lysis of 100–500 μL of liposome suspension in 4 mL 65% nitric acid for 24 hours at 90 °C, and dilution to 10.0 mL. Regular UV-Vis absorption and emission spectra were recorded on a Varian Cary 50 UV-Vis spectrometer and a Shimadzu RF-5301PC spectrofluorimeter, respectively.Liposome assembly
All liposome formulations were prepared by the classical hydration–extrusion method. As an example, the preparation of L123 is described here. Aliquots of chloroform stock solutions containing the liposome constituents were added together in a flask to obtain a solution with 5.0 μmol DMPC, 0.20 μmol DSPE-MPEG-2000, 2.5 nmol 1, 25 nmol 2, and 0.20 μmol 3[PF6]2. The organic solvent was removed by rotary evaporation and subsequently under high vacuum for at least 30 minutes to create a lipid film. 1.0 mL DPBS buffer was added and the lipid film was hydrated by 4 cycles of freezing the flask in liquid nitrogen and thawing in warm water (50 °C). The resulting dispersion was extruded through a Whatman Nuclepore 0.2 μm polycarbonate filter at 40–50 °C at least 11 times using a mini-extruder from Avanti Polar Lipids, Inc. (Alabaster, Alabama, USA). The number of extrusions was always odd to prevent any unextruded material ending up in the final liposome sample. The extrusion filter remained practically colourless after extrusion, suggesting near-complete inclusion of the chromophoric compounds in the lipid bilayer. Liposomes were stored in the dark at 4 °C and used within 7 days. The average liposome size and polydispersity index (PDI) were measured with a Malvern Instruments Zetasizer Nano-S machine, operating with a wavelength of 632 nm. The size and PDI were typically 130–170 nm and 0.05–0.20, respectively.Time-correlated single photon counting (TCSPC)
For TCSPC experiments, a FluoTime 200 system from PicoQuant (Berlin, Germany) was used, operating with a PicoHarp 300 photon counting module, a PDL 800-B picosecond pulsed diode laser driver set at 10 MHz repetition rate, and a 440 nm LDH-P-C-440 laser diode. Samples were excited at 440 nm (6 μW laser power, 0.6 pJ per pulse) and detection was recorded at 474 nm. A 111-QS cuvette from Hellma GmbH & Co. KG (Müllheim, Germany) was used and was thermostated in the holder at either 293 K or 310 K with a TC 125 temperature controller from Quantum Northwest (Seattle, WA, USA). Samples were always allowed a minimum of 7 minutes of thermal equilibration before measurement. The samples were greatly diluted so that in all cases, [2] = 1 μM and the absorption at excitation and emission wavelengths was below 0.1 (for 10 mm path length). The data was fitted with Origin Pro 8.5 software.Transient absorption (TA) spectroscopy
Transient absorption spectroscopy was performed on a femtosecond laser setup described in detail elsewhere.48,49 A combined libra and legend laser system from coherent produced pulses at 40 fs duration and 1 kHz repetition rate. Long delays were achieved by using two individual femtosecond amplifiers for pump and probe. Due to the shared source of seed pulses among the two lasers (the legend was seeded from the libra seed), femtosecond time resolution and arbitrary long pump probe delays were achieved. The pump laser was fed into an automated optical parametric amplifier that allowed conversion of the input pulse into any wavelength in the region 480–1600 nm while keeping the pulse duration. In this case, the samples were excited with 400 nm light (20–60 μW, 20–60 nJ per pulse). The laser driving the probe beam was focused on a calcium fluoride plate to generate a super continuum spanning from 360–1200, which enabled spectrally resolved probing without the need for scanning. After passing through the sample, the probe beam was dispersed on a 256 element diode array, calculating pump-on pump-off difference signal on a shot to shot basis. The diode array was calibrated so that the spectrum was recorded from 430 to 730 nm. High concentration samples were used with [2] = 0.1 mM in a 110-QS cuvette from Hellma with a 1 mm optical path length so that A400 ≤ 0.6 (including sample scatter). The sample volume was 200 μL and the cuvette was mounted on a shaker to ensure mixing throughout the measurement. The acquisition time per sample was approximately 2 hours. It was confirmed with UV-Vis absorption spectroscopy before and after TA spectroscopy that the blue excitation did not cause any photochemical degradation of the samples within the measurement time. In TA experiments involving blue-light irradiated samples, 500 μl sample was irradiated with 10 mW 420 nm light (M420F2 fibre-coupled high-power LED from Thorlabs) for a minimum of 2 hours. The reaction was monitored with UV-Vis spectroscopy to control the reaction time until all 32+ had converted to 42+, at which point the spectrum no longer evolved.Photodissociation experiments with red light
Photodissociation experiments were conducted in a custom-built setup (Fig. S8, ESI†). All optical parts were connected with FC-UVxxx-2 (xxx = 200, 400, 600) optical fibres from Avantes (Apeldoorn, The Netherlands), with a diameter of 200–600 μm, respectively, and that were suitable for the UV-Vis range (200–800 nm). 1.5 mL of the diluted liposome sample was deoxygenated by bubbling argon through the sample with a rate of ∼2 bubbles per second for at least 30 minutes in an external ice-cooled pear-shaped flask. After this period, bubbling was stopped while maintaining the argon flow, and the sample was warmed in a water bath of approximately 40 °C for 10 minutes. Then, the sample was transferred by means of cannulation with argon pressure to a 111-OS macro fluorescence cuvette from Hellma in a CUV-UV/VIS-TC temperature-controlled cuvette holder from Avantes. The sample was allowed to equilibrate at 310 K for an additional 10 minutes while stirring. The sample was held under argon atmosphere at a constant temperature of 310 K and irradiated for 4 hours from the side with a 630 nm laser light beam from a clinical grade Diomed 630 nm PDT laser, set at a power of 30, 60, or 120 mW. The 630 nm light was filtered through a FB630-10, 630 nm band pass filter (Thorlabs, Dachau/Munich, Germany) put between the laser and the sample. The excitation power was controlled using a NDL-25C-4 variable neutral density filter (Thorlabs), and measured using a S310C thermal sensor connected to a PM100USB power meter (Thorlabs). The laser was collimated to a beam of 4 mm diameter to reach an intensity of 0.24, 0.48, or 0.95 W cm−2; in such conditions, a cylinder of approximately 0.13 cm3 was simultaneously excited by the laser (8% of the total sample volume). UV-Vis absorption spectra were measured using an Avalight-DHc halogen–deuterium lamp (Avantes) as light source and a 2048L StarLine spectrometer (Avantes) as detector, both connected to the cuvette holder at a 180° angle and both at a 90° angle with respect to the red laser irradiation direction. The filter holder between cuvette holder and detector was in a position without a filter (Fig. S8, ESI,† item 8). Luminescence emission spectra were measured using the same detector but with the UV-Vis light source switched off. To visualize the spectrum from 550 nm to 900 nm, while blocking the red excitation light, a Thorlabs NF-633 notch filter was used in the variable filter holder. To visualize the spectrum from 400 nm to 550 nm, an OD4 575 nm short pass filter (Edmund Optics, York, United Kingdom, part no. 84-709) was used. A UV-Vis absorption and two emission spectra (one for each filter) were measured every 15 min; each time the emission spectra were measured first by switching the filter holder to the appropriate position, then the laser was switched off, the halogen–deuterium lamp was turned on, the filter holder was switched to an open position, a UV-Vis absorption spectrum was recorded, the halogen–deuterium lamp was switched off, and the laser was switched on again. Each UV-Vis measurement took approximately 15 seconds in total. All spectra were recorded with Avasoft software from Avantes and further processed with Microsoft Office Excel 2010 and Origin Pro software. For each UV-Vis absorption spectrum, a baseline subtraction was performed followed by an evaporation correction to account for a slight loss of solvent as a result of the constant argon flow in the cuvette during the experiment. The emission spectra obtained from the two filters were stitched together at 550 nm to obtain a continuous spectrum from 400 to 900 nm. No correction was needed to seamlessly connect the spectra.Differential scanning calorimetry
Differential scanning calorimetry (DSC) was performed on a TA Instruments (DE, USA) nano-DSC III instrument in the range of 278 to 333 K with a scanning rate of 1 K min−1 at 3 atm. The capillary cell (V = 300 μL) was filled with the liposome solution (lipid bulk concentration of 5 mM) containing different concentrations of 1, 2 and 32+. The reference cell was filled with the corresponding liposome-free buffer solution. A blank measurement was performed with PBS buffer. The liposome dispersions were degassed for 10–15 minutes prior to measurement on a Nalgene degassing station. For each sample, at least two cycles of heating and cooling were performed with 10 minutes of thermal equilibration between the ramps. The machine was cleaned beforehand with 50% formic acid and rinsed thoroughly with Milli-Q water. The thermograms were processed and analysed using NanoAnalyze software from TA Instruments.Results and discussion
Liposome preparation and characterization
PEGylated liposomes were prepared as shown in Table 1 from a mixture of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 4 mol% sodium N-(carbonyl-methoxypolyethylene glycol-2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-MPEG-2000), and compound 1, 2, and/or 3(PF6)2, using a standard hydration–extrusion protocol in Dulbecco's phosphate buffered saline (DPBS). PEGylation prevented liposomes fusion and/or aggregation that would interfere with the experiments. The true concentration of 32+ in all liposome samples was determined experimentally by inductively coupled plasma optical emission spectroscopy (ICP-OES) to be on average 89% of the expected value (Fig. S1, ESI†). The average vesicle diameter and polydispersity index were determined by dynamic light scattering (DLS); typical values were 130–170 nm and 0.05–0.20, respectively.| Sample | [DMPC] (mM) | [DSPE-PEG-2000] (μM) | [1] (μM) | [2] (μM) | [32+] (μM) |
|---|---|---|---|---|---|
| L123 | 5.0 | 200 | 2.5 | 25 | 50–220 |
| L12 | 5.0 | 200 | 2.5 | 25 | — |
| L23 | 5.0 | 200 | — | 25 | 10–290 |
| L2 | 5.0 | 200 | — | 25 | — |
| L3 | 5.0 | 200 | — | — | 200 |
| L0 | 5.0 | 200 | — | — | — |
Because on the one hand, molecular diffusion plays an important role in TTA-UC, and on the other hand, phase separation of one of the dyes could impair the efficiency of the system, the gel-to-liquid phase transition temperature of a series of PEGylated DMPC liposomes was determined by differential scanning calorimetry (Fig. S2, ESI†). The transition temperature for PEGylated DMPC liposomes without any chromophores (L0) is 25.2 °C, with a pretransition peak at 14 °C. Functionalizing these liposomes with 0.05 mol% 1 and 0.5 mol% 2 (L12) caused a small decrease in the main transition peak height, but the main features of the thermogram remained, which indicates that the membrane was only slightly perturbed by the presence of the TTA-UC dyes and that the dyes are buried into the lipid bilayer. Next, a series of liposomes L123 was measured, which in addition to 0.05 mol% 1 and 0.5 mol% 2 also contained 1 to 4 mol% of the ruthenium complex. A progressive yet small decrease in the main transition temperature was observed compared to L12: from 25.1 °C for 0 mol% to 23.2 °C for 4 mol% 32+. Additionally, the main transition peak broadened and its intensity decreased upon increasing Ru concentration, while the pretransition peak disappeared already after inclusion of 1 mol% of the 32+ complex. These observations are compatible with the expected interaction of complex 32+ with the zwitterionic polar heads of the lipids. For this range of ruthenium concentrations however, no evidence was found that suggested phase separation of either 1, 2, or 32+. Overall, these DSC results show that at 20 °C and at 37 °C, all liposome formulations are in the gel phase and liquid crystalline phase, respectively.
Photodissociation experiments using red light
Red-light irradiation experiments were first conducted to evaluate whether L123 liposomes, which contained both dyes for TTA-UC plus the ruthenium complex 32+, would achieve higher photodissociation rates than a mixture of L12 and L3, in which the upconversion and the photosubstitution on the ruthenium complex are physically separated on two different liposomes.8 For all red light irradiation experiments the liposome samples were diluted with isotonic buffer so that the optical density due to the MLCT band of the Ru complex stayed low (A450–500 ≤ 0.25 with a 10 mm path length). Under such conditions, radiative energy transfer between the blue emitting perylene and the blue absorbing ruthenium complex is minimized, while the solution absorbance remains high enough for monitoring the experiments using UV-Vis absorption spectroscopy. The bulk concentrations of 1, of 2, and/or 32+ were kept equal in all experiments. Red light irradiation was realized for three hours at physiological temperature (310 K) and under anoxic conditions using a 630 nm clinical grade PDT laser set at low power (30, 60, or 120 mW, for an intensity of 0.24, 0.48, or 0.95 W cm−2). UV-Vis absorption and luminescence emission spectra were measured during irradiation every 15 minutes.The UV-Vis absorption spectrum of a L123 sample containing 3.5 mol% of 32+ evolved during red light irradiation at 120 mW power as shown in Fig. 2a. A band between 450 and 600 nm, typical of the aqua complex 42+, rises, while two isosbestic points are observed at 370 and 456 nm. The only photochemical reaction occurring in such conditions is thus the hydrolysis reaction of 32+ to 42+. The evolution of the absorbance at 490 nm (Fig. 2c) clearly showed that the reaction was finished after two hours of irradiation. For comparison, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume mixture of L12 and L3 was irradiated under the same conditions, where only radiative energy transfer may occur. Slower photodissociation kinetics was observed (Fig. 2c). Apparently, the optical density of the sample was high enough so that 32+ in L3 could reabsorb a significant amount of the blue photons upconverted by L12. When liposomes L23 were irradiated that contained only 2 and 32+, no TTA-UC could occur and very slow photodissociation was observed due to the low but non-zero molar absorption coefficient of 32+ at 630 nm (ε630 < 100 M−1 cm−1). Overall, our results clearly show that L123 achieved a higher photodissociation rate than a mere physical mixture of L12 and L3, and of course of L23.
:1 volume mixture of L12 and L3 was irradiated under the same conditions, where only radiative energy transfer may occur. Slower photodissociation kinetics was observed (Fig. 2c). Apparently, the optical density of the sample was high enough so that 32+ in L3 could reabsorb a significant amount of the blue photons upconverted by L12. When liposomes L23 were irradiated that contained only 2 and 32+, no TTA-UC could occur and very slow photodissociation was observed due to the low but non-zero molar absorption coefficient of 32+ at 630 nm (ε630 < 100 M−1 cm−1). Overall, our results clearly show that L123 achieved a higher photodissociation rate than a mere physical mixture of L12 and L3, and of course of L23.
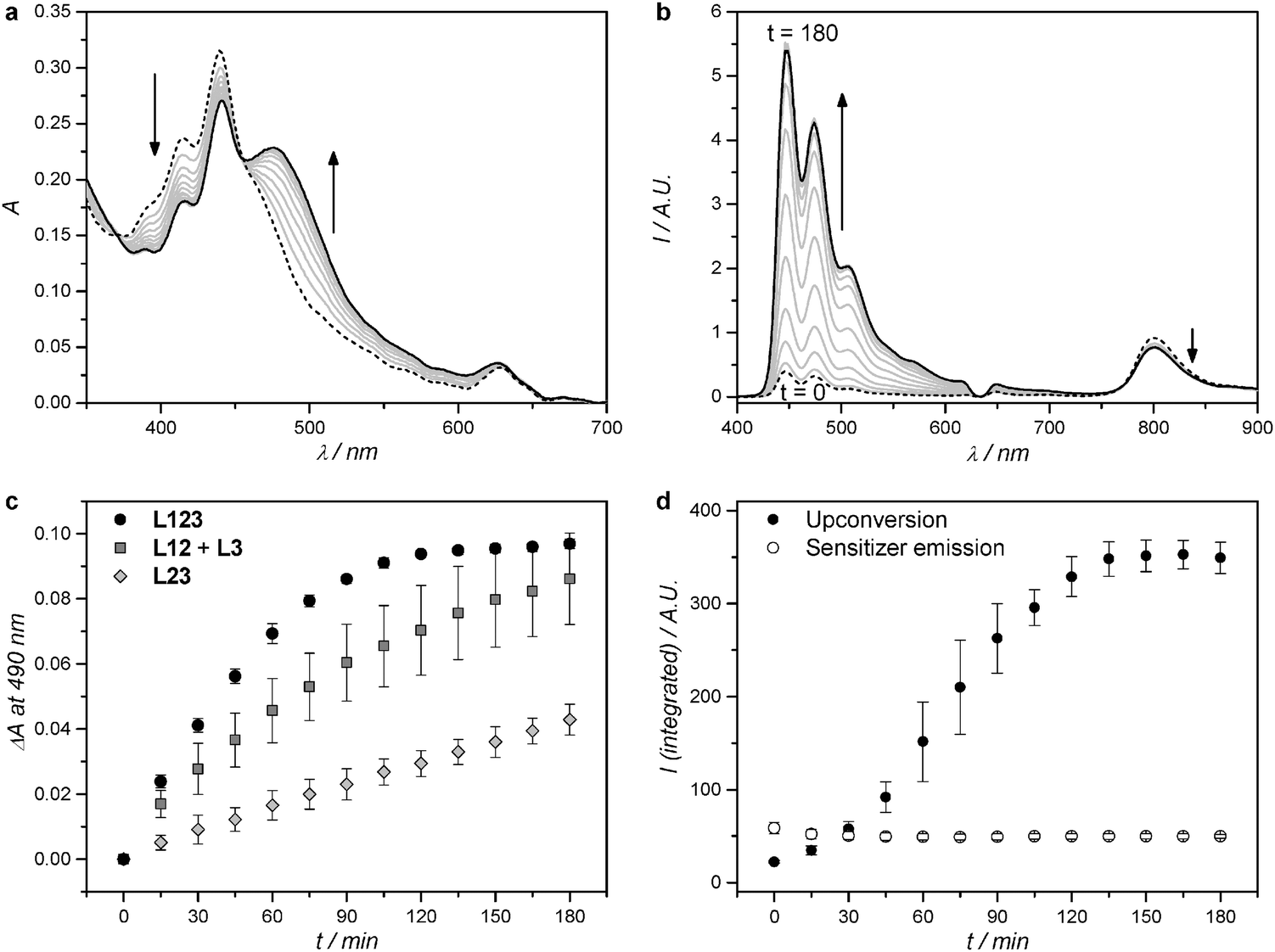 | ||
| Fig. 2 Absorption (a) and luminescence emission (b) spectra of L123 during red-light irradiation (630 nm) of liposomes functionalized with 32+. Dashed line: spectrum at t = 0; black solid line: spectrum at t = 180 minutes; grey lines: spectra measured every 15 minutes. (c) Difference in absorbance at 490 nm, after baseline correction, during red-light irradiation (630 nm) of L123 (black filled circles), a 1:1 volume mixture of L12 and L3 (dark-grey filled squares), and L23 (light-grey filled diamonds). Error bars represent standard deviation of three independent experiments. (d) Integrated upconversion emission (400–600 nm, black filled circles) and integrated sensitizer emission (750–900 nm, empty circles) during red-light irradiation of L123. Error bars represent standard deviation of three independent experiments. Irradiation conditions: power 120 mW, beam diameter 4 mm, intensity 0.95 W cm−2, T = 310 K, sample volume 1.5 ml, 8% of sample volume simultaneously irradiated. Liposome dispersions used in these experiments were prepared as in Table 1, and then diluted with PBS buffer prior to measurement so that every time [1] = 0.25 μM, and [2] = 2.5 μM. The bulk concentration of 32+ was experimentally determined with ICP-OES and was 19 ± 1 μM, 21 ± 3 μM, and 20 ± 2 μM for L123, L23, and L3, respectively. | ||
When the red light-induced photodissociation reaction of L123 was followed by luminescence emission spectroscopy (Fig. 2b), a peak at 800 nm, corresponding to the phosphorescence of 1, and a broad structured emission band ranging from 400 to 600 nm, corresponding to the upconverted emission of 2, could clearly be identified.8 Interestingly, the upconverted emission intensity evolved a lot during irradiation. It was initially very weak compared to the upconversion emission of L12 alone (Fig. S3, ESI†), but increased roughly 15-fold during the course of the reaction. No further evolution was observed after two hours of irradiation, which closely matches the reaction time observed by UV-Vis absorption spectroscopy. The low initial upconversion emission intensity, combined with the observation by UV-Vis spectroscopy that the photosubstitution clearly occurred, suggested that the upconverted energy may be transferred non-radiatively from 2 to 32+. As the reaction proceeded, and 32+ was progressively liberated from the membrane to diffuse in solution, non-radiative energy transfer from perylene to the ruthenium complex becomes less efficient, thus explaining the recovery of the blue emission when all ruthenium complex were detached from the membrane. To prove this hypothesis, we studied the non-radiative energy transfer between 2 and 32+ by direct excitation of perylene with violet or blue light, and studied the evolution of the system with time-dependent absorption and emission spectroscopy.
Is there non-radiative energy transfer from 2 to 32+?
Assuming that the same singlet excited state of 2 is reached by blue light excitation of 2 in absence of 1 and by red-light excitation of 1 in presence of 2, a series of L23 liposomes was prepared with a fixed amount of 2 (0.5 mol%), a varying amount (0.4–3.5 mol%) of 32+, but no red photosensitizer (1). The aim of this study was to determine with time-dependent spectroscopy whether the singlet excited state of 2 could transfer its energy non-radiatively to the ruthenium complex. Control liposomes were also prepared that contained only perylene (L2) or only 3.3 mol% of 32+ (L3). All liposomes were first studied by ultrafast transient absorption (TA) spectroscopy with 400 nm excitation (40 fs pulse duration, 20–60 nJ per pulse) while recording the transient absorption spectrum from 430 to 730 nm in the ps to ns range. The TA data were globally fitted using the software package Glotaran.50 Full data set and analysis is detailed in Fig. 3, Table 2, and in the ESI.† In short, the TA spectrum of L2 1.0 ns after a 400 nm excitation pulse (Fig. S4, ESI†) closely matched literature reports for perylene in cyclohexane. It features negative signals from 430 to 550 nm due to ground state bleach and stimulated emission, and a strong positive band centred at 700 nm due to excited state absorption.51 Global analysis using Glotaran gave an excited state lifetime of 6.00 ± 0.10 ns for 2 in L2, which also corresponds to literature values.51,52 For L3 the transient absorption spectrum 1.0 ps after the excitation pulse at 400 nm showed a negative band ranging from 400 to 500 nm and a weaker featureless positive band from 500 nm to 800 nm. The former coincides with the MLCT absorption band in the steady state absorption spectrum (Fig. 1c) and can be attributed to ground state bleaching of the ruthenium complex. The latter is attributed to excited state absorption. The time evolution of the transient spectrum was best fitted with Glotaran to give an excited state lifetime of 0.52 ns for 32+. UV-Vis spectroscopy before and after the TA experiment showed that in such conditions negligible photodissociation occurred (see ESI†).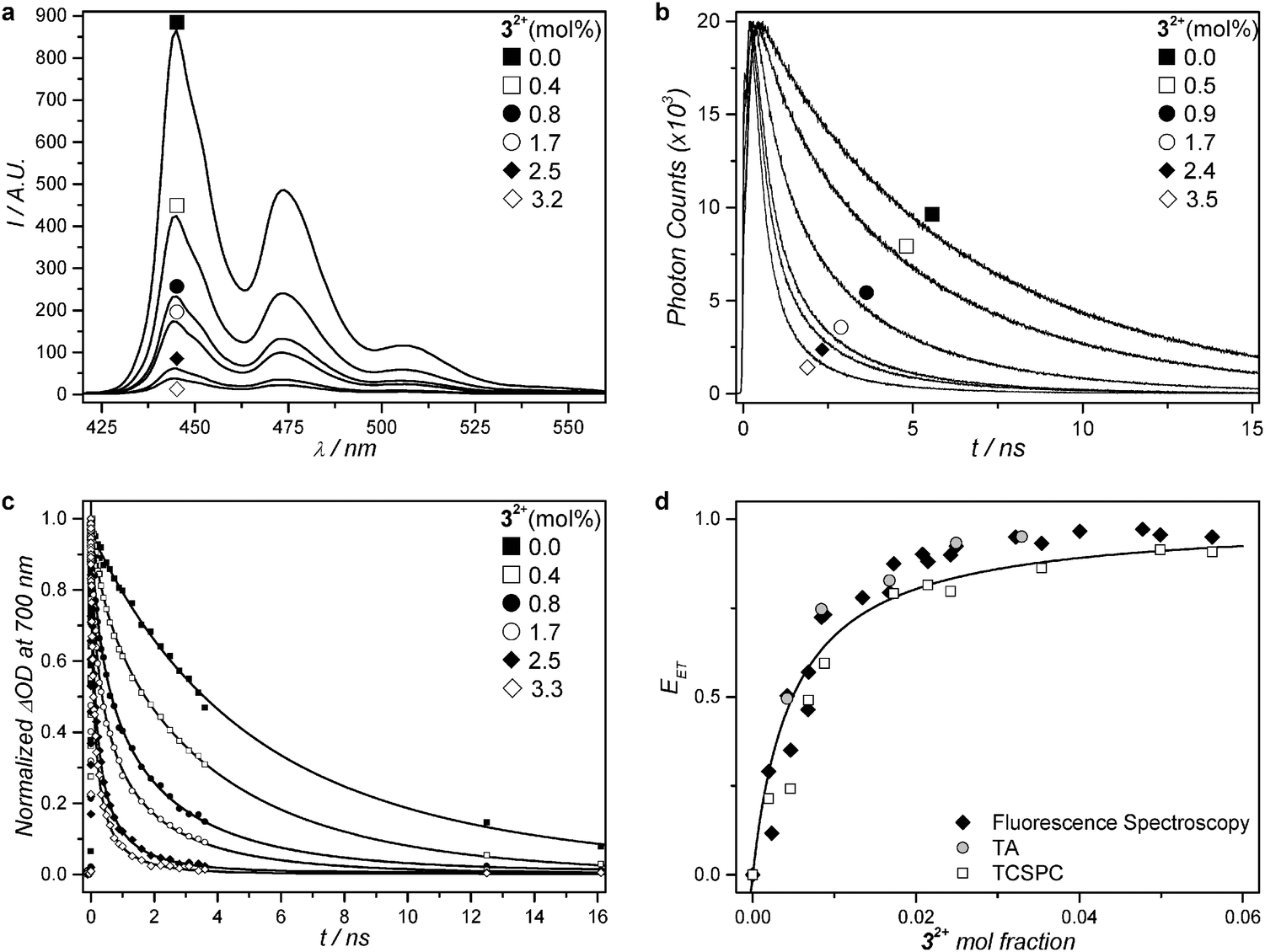 | ||
| Fig. 3 Steady-state fluorescence spectra (a) and excited state time decays of 2 (b and c) for a series of PEGylated (4 mol%) DMPC liposomes containing a fixed amount of compound 2 (0.5 mol%) and 0 to 4 mol% of compound 32+ at 293 K. (a) Steady-state fluorescence spectra (λexc = 400 nm, [2] = 1 μM). (b) Fluorescence decay curves from time-correlated single photon counting (λexc = 440 nm, λemi = 474 nm, [2] = 1 μM). (c) Normalized transient absorption kinetic traces at 700 nm. Best-fit curves drawn as solid lines for samples with addition of 32+ according to a three-dimensional Förster decay model (see ESI†). For the sample without addition of 32+, the fit curve represents a bi-exponential decay model. (d) Energy transfer efficiency (EET) as a function of the mole fraction of 32+ at 293 K, calculated from time-correlated single photon counting data (empty squares), transient absorption data (grey filled circles), and steady state fluorescence spectroscopy (black filled diamonds). Best-fit curve according to a Stern–Volmer model (see ESI†). | ||
| Molar percentage of 32+ in PEGylated DMPC liposomes | ||||||
|---|---|---|---|---|---|---|
| 0.0 | 0.4 | 0.8 | 1.7 | 2.5 | 3.3 | |
| TA spectroscopy at 293 K | ||||||
| τ | 6.00 ± 0.10 | 3.03 ± 0.02 | 1.52 ± 0.01 | 1.04 ± 0.01 | 0.40 ± 0.00 | 0.29 ± 0.00 |
| E ET (%) | 0 | 50 | 75 | 83 | 93 | 95 |
| 32+ molar percentage in PEGylated DMPC liposomes | ||||||
|---|---|---|---|---|---|---|
| 0.0 | 0.5 | 0.9 | 1.7 | 2.4 | 3.5 | |
| TCSPC at 293 K | ||||||
| τ | 6.17 ± 0.01 | 4.67 ± 0.01 | 2.50 ± 0.05 | 1.29 ± 0.02 | 1.25 ± 0.05 | 0.85 ± 0.06 |
| E ET (%) | 0 | 24 | 60 | 79 | 80 | 86 |
| TCSPC at 310 K | ||||||
| τ | 5.69 ± 0.01 | 4.00 ± 0.01 | 2.37 ± 0.07 | 1.14 ± 0.00 | 0.84 ± 0.00 | 0.45 ± 0.00 |
| E ET (%) | 0 | 30 | 58 | 80 | 85 | 92 |
The TA data of liposomes L23 were qualitatively very similar to that of L2, and global fitting did not allow detecting any feature reminiscent of 32+ as in L3. However, the average excited state lifetime (τ) of 2 decreased strongly as a function of the mole fraction of 32+ (Fig. 3c and Table 2): from τ = 6.0 ns without any ruthenium complex in the membrane (L2) addition of up to 3.3 mol% ruthenium complex (L23) lowered the lifetime of 2 down to 0.3 ns, which shows non-radiative quenching of the excited state of 2 by 32+. These results were confirmed by steady state fluorescence spectroscopy and time-correlated single photon counting (TCSPC). Because 32+ is not emissive at all, 2 is observed selectively using fluorescence techniques. The steady-state fluorescence spectra of the series of liposomes L23 clearly showed a decrease in fluorescence intensity of 2 with increasing concentration of 32+ in the membrane (Fig. 3a). TCSPC was performed with 440 nm excitation (0.6 pJ per pulse), while monitoring the emission at 474 nm on the nanosecond scale. The TCSPC results were very similar to TA data: the singlet excited state lifetime of 2 was found to be τ = 6.2 ns in absence of 32+, and decreased to τ = 0.9 ns in presence of 3.5 mol% of 32+ (see Fig. 3b and Table 2). Clearly, the singlet state of 2 is quenched by the nearby ruthenium complex. Although no rising of a Ru-based excited state could be detected by TA spectroscopy in L23, probably due to its low and rather broad features as observed in L3, the increased rate of the photosubstitution reaction in liposomes L123 irradiated with red light, the recovery of the upconverted emission when the ruthenium aqua complex leaves the membrane, and the quenching of 2 by 32+ observed in L23, conclude to non-radiative energy transfer occurring from 2 to 32+ both in L23 and in L123.
From TA, steady state fluorescence, and TCSPC data, the non-radiative energy transfer efficiency (EET) was calculated for each composition of the liposomes L23 following eqn (1) (see also Fig. 3d, Table 2, and ESI†):
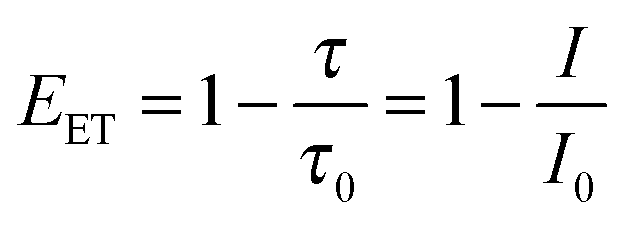 | (1) |
Does the aqua photoproduct quench the perylene excited state as well?
Theoretically, when the ruthenium complex hydrolyses to the aqua species it is no longer attached to the cholesterol anchor and as the membrane is made of neutral lipids 42+ will diffuse away from the membrane.18 After photodissociation 2 should thus regain its unquenched lifetime of ca. 6 ns. To probe this hypothesis, liposomes L23 with 4 mol% of 32+ (2.5 mM DMPC bulk concentration) were irradiated with blue light (420 nm) until full conversion of 32+ to 42+ was detected by UV-Vis spectroscopy. No change in size or polydispersity of the liposomes was observed as judged by dynamic light scattering, indicating that the liposomes were unaffected by the light treatment. The lifetime of 2 was then measured with TA spectroscopy again, and indeed increased from 0.3 ns prior to irradiation to 3.9 ns after irradiation, owing to the decreased concentration of the energy acceptor 32+ in the membrane. However, the lifetime did not rise to the unquenched value of 6.0–6.2 ns found in L2. To explain this observation, new L23 samples were prepared using the same relative composition of the membrane, but with an 8 times higher lipid bulk concentration. In such concentrated liposomes the lifetime after the photoreaction rose to only 1.7 ns, which clearly demonstrated that the excited state lifetime of compound 2 depends on the bulk concentration of the aqua complex 42+. Our interpretation of this observation is that at higher concentrations the chance of finding 42+ in proximity of the membrane, and thus of 2, increases and that non-radiative energy transfer can occur as well. Overall, the rise in the lifetime of 2 upon blue light irradiation of the L23 samples confirms the results from the red light photodissociation experiments on L123: when the energy acceptor leaves the membrane, 2 no longer performs as much non-radiative energy transfer as when the complex is bound to the membrane, but instead loses its energy radiatively, which explains the increased blue emission at the end of the photoreaction (Fig. 2b).What is the mechanism of energy transfer?
In photochemistry, non-radiative energy transfer such as that between 2 and 32+ is either attributed to a Dexter or to a Förster mechanism. The Förster mechanism relies on long-range dipole–dipole interactions and can be efficient up to a distance of 10 nm for spectrally well-matching donor–acceptor pairs. In the Dexter mechanism, direct orbital overlap is required between donor and acceptor, and energy transfer efficiency decays quickly beyond distances of 1 nm between the donor and the acceptor. For Dexter type energy transfer, high mobility of both the donor and acceptor is imperative to realize energy transfer within the nanosecond timeframe in which 2 is in its singlet excited state. However, the DMPC bilayer at room temperature does not support such mobility: diffusion coefficients for fluorescent probes in DMPC membranes are typically 0.01 μm2 s−1 at 292 K,53 which leads to a negligible displacement of 5 × 10−3 Å2 in the 6 ns excited state lifetime of 2. In other words, in the timeframe in which 2 is in the singlet excited state the lipid bilayer is practically frozen. Also, below (293 K) and above (310 K) the phase transition temperature at which the lipid bilayer transforms from a gel-like to liquid crystalline structure, there is no significant difference in quenching efficiency (vide supra), which advocates for a diffusion-independent quenching mechanism. Finally, 2 is located in the hydrophobic interior of the lipid bilayer, while the positively charged centre of 32+, responsible for accepting the energy, is dangling at the bilayer–water interface.54 Therefore, it is very unlikely that 2 and 32+ can come in close contact to realize orbital overlap. For these reasons, we conclude that the most likely mechanism of energy transfer is Förster resonance energy transfer (FRET). Based on this assumption, and following the work of Holmes et al.,55 the decay curves from TA and TCSPC data were satisfactorily fitted with a three-dimensional Förster decay function (see ESI†). From the fitting parameters, the experimental Förster distance R0, for which FRET occurs with 50% efficiency, was calculated to be 29 Å. This value fits rather well the theoretical value (41 Å) that can be calculated form the spectra overlap shown in Fig. 1c (see ESI†). Both values also match the thickness of a DMPC lipid bilayer (36 Å),53 which explains why FRET between 2 and 32+ is so efficient in this system.Overall efficiency of the sequential combination of TTA-UC and FRET
Based on the evidence presented above, we propose that the enhanced photosubstitution rate when the three photoactive components 1, 2, and 32+ are present in the same membrane (L123) and irradiated with red light, results from the combination of TTA-UC between 1 and 2, and FRET from 2 to 32+. To the best of our knowledge, this is one of the rare system in which TTA-UC and FRET are combined to activate a photosensitive molecule.56Fig. 4 shows a qualitative Jablonski diagram of this system: instead of realizing upconverted blue emission, the energy that is stored in the singlet excited perylene molecule is primarily transferred to the ruthenium complex by FRET, to generate the singlet MLCT excited state. This excited state in turn leads to the selective substitution of the thioether ligand by a water molecule via the classical mechanism involving intersystem crossing to the 3MLCT state followed by thermal promotion of a triplet metal-centered state (3MC). In such a sequence, the photosubstitution quantum efficiency Etotal under red light irradiation, defined as the total number of photosubstitution reactions divided by the total number of red photons absorbed, is the product of the individual efficiencies, see eqn (2):| Etotal = ΦUCEETΦRu | (2) |
 | ||
| Fig. 4 Qualitative Jablonski diagram for a sequential combination of TTA-UC, FRET, and photosubstitution in liposomes L123 under red light irradiation. Abbreviations: intersystem crossing (ISC), triplet–triplet energy transfer (TTET), triplet–triplet annihilation (TTA), Förster resonance energy transfer (FRET), metal to ligand charge transfer (MLCT) state, metal-centred (MC) state. | ||
In principle, at such power densities the quantum efficiency of photodissociation reaction under red light irradiation should not depend on the photon flux. It was indeed shown that the TTA upconversion quantum yield does not depend on light intensity above a certain intensity threshold ITH.57 For the 2-component liposomes L12 we demonstrated recently58 that indeed the TTA-UC intensity linearly scales with irradiation intensity above ITH = 0.05 W cm−2, while a quadratic dependence of the upconversion intensity on irradiation power is observed at lower intensities, i.e., between 50 and 8 mW cm−2 (see also Fig. S11, ESI†). When 3-component liposomes L123 were irradiated with 30, 60, or 120 mW power, i.e., 0.24, 0.48, and 0.95 W cm−2, respectively, the decrease in photosubstitution reaction rate was found almost proportional to light intensity (Fig. S12, ESI†). In other words, at these intensities the overall efficiency of the photosubstitution reaction was indeed found, within experimental errors, independent on the red light intensity (Fig. S13, ESI†). Clinical PDT frequently reports light intensities of 1 W cm−2, and recent work even used intensities as high as 79 W cm−2,59 which underlines the biological applicability of TTA-UC in liposomes as a prodrug activation strategy.
Conclusion
This work is one of the first demonstrations that TTA-UC can be combined with FRET.56 By putting the three photochemically active components 1, 2, and 32+ inside a single lipid bilayer highly efficient non-radiative energy transfer between the upconverted perylene excitation and the ground state of complex 32+ occurs. Under red light excitation from a commercial PDT laser the photosubstitution reaction of 32+ to 42+ was much faster in the combined liposomes L123 than using radiative energy transfer, i.e., in a physical mixture of L12 and L3. In principle, this strategy can be generalized to any blue-light absorbing (pro)drug with good spectral overlap with the perylene emission. Light activation triggered by TTA-UC upconversion and FRET is especially attractive for the treatment of hypoxic tumours, in which classical PDT lose efficacy. For example, a system combining TTA-UC and PACT would function optimally. In the field of anticancer prodrug activation lanthanide-doped upconverting nanoparticles have shown promising results,13,46,60,61 but the upconversion is poorly efficient in water and it requires laser powers in the multi-Watt regime that can be detrimental to cell viability.62 Adding TTA-UC in the palette of the medicinal photochemistry offers a significant alternative to the upconverting strategy: more light is absorbed, higher upconversion efficiencies are achieved at intensities as low as 10 mW cm−2, and the molecular nature of the photoactive components allows for studying the mechanism of energy transfer in great detail. Recent work from our lab has shown that the problem of the sensitivity of TTA-upconversion schemes to dioxygen can be addressed using for example sodium sulfite.58 These approaches may be combined to achieve red-to-blue TTA-upconversion and prodrug activation in cellulo.Acknowledgements
Prof. Dr Lies Bouwman is kindly acknowledged for support and scientific discussion. NWO (The Netherlands Organization for Scientific Research) is acknowledged for a VENI grant to M.K., a VIDI grant to S.B., a VICI grant to J.T.M.K., and a Middelgroot investment grant to J.T.M.K. The European Research Council is acknowledged for an ERC starting grant to S.B., G. B. acknowledges the FNRS (FRFC 2010: 2.4592.10F) and the Van Buuren Foundation for the funding of the microcalorimetry equipment. COST action CM1005 (Supramolecular Chemistry in Water) is acknowledged for an STSM travel grant to S.H.C.A. COST action CM1105 (Functional metal complexes that bind to biomolecules) is acknowledged for stimulating scientific discussion.References
- L. Zayat, C. Calero, P. Alborés, L. Baraldo and R. Etchenique, J. Am. Chem. Soc., 2003, 125, 882–883 CrossRef CAS PubMed.
- P. J. Bednarski, F. S. Mackay and P. J. Sadler, Adv. Anticancer Agents Med. Chem., 2007, 7, 75–93 CrossRef CAS.
- N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 10690–10701 RSC.
- U. Schatzschneider, Eur. J. Inorg. Chem., 2010, 1451–1467 CrossRef CAS PubMed.
- S. L. H. Higgins and K. J. Brewer, Angew. Chem., Int. Ed., 2012, 51, 11420–11422 CrossRef CAS PubMed.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- M. A. Sgambellone, A. David, R. N. Garner, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2013, 135, 11274–11282 CrossRef CAS PubMed.
- S. H. C. Askes, A. Bahreman and S. Bonnet, Angew. Chem., Int. Ed., 2014, 53, 1029–1033 CrossRef CAS PubMed.
- A. Master, M. Livingston and A. Sen Gupta, J. Controlled Release, 2013, 168, 88–102 CrossRef CAS PubMed.
- S. G. Bown, J. Natl. Compr. Cancer Network, 2012, 10(suppl 2), S69–S74 Search PubMed.
- A. M. Bugaj, Photochem. Photobiol. Sci., 2011, 10, 1097 CAS.
- D. W. Felsher, Nat. Rev. Cancer, 2003, 3, 375–379 CrossRef CAS PubMed.
- E. Ruggiero, A. Habtemariam, L. Yate, J. Mareque Rivas and L. Salassa, Chem. Commun., 2014, 50, 1715–1718 RSC.
- R. R. Sawant and V. P. Torchilin, Soft Matter, 2010, 6, 4026–4044 RSC.
- R. Weijer, M. Broekgaarden, M. Kos, R. van Vught, E. A. J. Rauws, E. Breukink, T. M. van Gulik, G. Storm and M. Heger, J. Photochem. Photobiol., C, 2015, 23, 103–131 CrossRef CAS PubMed.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- S. Bonnet, B. Limburg, J. D. Meeldijk, R. J. M. K. Gebbink and J. A. Killian, J. Am. Chem. Soc., 2010, 133, 252–261 CrossRef PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS PubMed.
- J. Zhao, S. Ji and H. Guo, RSC Adv., 2011, 1, 937–950 RSC.
- Y. C. Simon and C. Weder, J. Mater. Chem., 2012, 22, 20817–20830 RSC.
- Y. Y. Cheng, B. Fückel, T. Khoury, R. L. G. C. R. Clady, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, J. Phys. Chem. A, 2011, 115, 1047–1053 CrossRef CAS PubMed.
- Y. Murakami, T. Ito and A. Kawai, J. Phys. Chem. B, 2014, 118, 14442–14451 CrossRef CAS PubMed.
- C. Wohnhaas, K. Friedemann, D. Busko, K. Landfester, S. Baluschev, D. Crespy and A. Turshatov, ACS Macro Lett., 2013, 2, 446–450 CrossRef CAS.
- R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652–12653 CrossRef CAS PubMed.
- P. B. Merkel and J. P. Dinnocenzo, J. Lumin., 2009, 129, 303–306 CrossRef CAS PubMed.
- A. Monguzzi, R. Tubino and F. Meinardi, J. Phys. Chem. A, 2009, 113, 1171–1174 CrossRef CAS PubMed.
- T. N. Singh-Rachford, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2009, 131, 12007–12014 CrossRef CAS PubMed.
- P. C. Boutin, K. P. Ghiggino, T. L. Kelly and R. P. Steer, J. Phys. Chem. Lett., 2013, 4, 4113–4118 CrossRef CAS.
- S.-H. Lee, Á. Sonseca, R. Vadrucci, E. Giménez, E. J. Foster and Y. Simon, J. Inorg. Organomet. Polym. Mater., 2014, 24, 898–903 CrossRef CAS.
- A. Turshatov, D. Busko, S. Baluschev, T. Miteva and K. Landfester, New J. Phys., 2011, 13, 083035 CrossRef.
- J.-H. Kang and E. Reichmanis, Angew. Chem., Int. Ed., 2012, 51, 11841–11844 CrossRef CAS PubMed.
- J.-H. Kim and J.-H. Kim, J. Am. Chem. Soc., 2012, 134, 17478–17481 CrossRef CAS PubMed.
- Q. Liu, B. Yin, T. Yang, Y. Yang, Z. Shen, P. Yao and F. Li, J. Am. Chem. Soc., 2013, 135, 5029–5037 CrossRef CAS PubMed.
- K. Tanaka, H. Okada, W. Ohashi, J.-H. Jeon, K. Inafuku and Y. Chujo, Bioorg. Med. Chem., 2013, 21, 2678–2681 CrossRef CAS PubMed.
- C. Wohnhaas, V. Mailänder, M. Dröge, M. A. Filatov, D. Busko, Y. Avlasevich, S. Baluschev, T. Miteva, K. Landfester and A. Turshatov, Macromol. Biosci., 2013, 13, 1422–1430 CrossRef CAS PubMed.
- Y. C. Simon, S. Bai, M. K. Sing, H. Dietsch, M. Achermann and C. Weder, Macromol. Rapid Commun., 2012, 33, 498–502 CrossRef CAS PubMed.
- O. S. Kwon, J.-H. Kim, J. K. Cho and J.-H. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 318–325 CAS.
- M. Penconi, P. L. Gentili, G. Massaro, F. Elisei and F. Ortica, Photochem. Photobiol. Sci., 2014, 13, 48–61 CAS.
- J.-H. Kim and J.-H. Kim, ACS Photonics, 2015, 2, 633–638 CrossRef CAS.
- P. Duan, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2013, 135, 19056–19059 CrossRef CAS PubMed.
- Z. Jiang, M. Xu, F. Li and Y. Yu, J. Am. Chem. Soc., 2013, 135, 16446–16453 CrossRef CAS PubMed.
- R. S. Khnayzer, J. Blumhoff, J. A. Harrington, A. Haefele, F. Deng and F. N. Castellano, Chem. Commun., 2012, 48, 209–211 RSC.
- R. E. Goldbach, I. Rodriguez-Garcia, J. H. van Lenthe, M. A. Siegler and S. Bonnet, Chem. – Eur. J., 2011, 17, 9924–9929 CrossRef CAS PubMed.
- A. Bahreman, B. Limburg, M. A. Siegler, R. Koning, A. J. Koster and S. Bonnet, Chem. – Eur. J., 2012, 18, 10271–10280 CrossRef CAS PubMed.
- Y. Dai, H. Xiao, J. Liu, Q. Yuan, P. a. Ma, D. Yang, C. Li, Z. Cheng, Z. Hou, P. Yang and J. Lin, J. Am. Chem. Soc., 2013, 135, 18920–18929 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Science + Business Media, LLC, New York, NY, USA, 3rd edn, 2006 Search PubMed.
- R. Berera, R. Grondelle and J. M. Kennis, Photosynth. Res., 2009, 101, 105–118 CrossRef CAS PubMed.
- J. Ravensbergen, F. F. Abdi, J. H. van Santen, R. N. Frese, B. Dam, R. van de Krol and J. T. M. Kennis, J. Phys. Chem. C, 2014, 118, 27793–27800 CAS.
- S. L. Joris J. Snellenburg, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- Y. H. Meyer and P. Plaza, Chem. Phys., 1995, 200, 235–243 CrossRef CAS.
- A. S. Holmes, D. J. S. Birch and T. Salthammer, J. Fluoresc., 1993, 3, 77–84 CrossRef CAS PubMed.
- D. Marsh, Handbook of Lipid Bilayers, Taylor & Francis Group, LLC, Boca Raton, FL, USA, 2nd edn, 2013 Search PubMed.
- A. Bahreman, M. Rabe, A. Kros, G. Bruylants and S. Bonnet, Chem. – Eur. J., 2014, 20, 7429–7438 CrossRef CAS PubMed.
- A. S. Holmes, K. Suhling and D. J. S. Birch, Biophys. Chem., 1993, 48, 193–204 CrossRef CAS.
- W. Wang, Q. Liu, C. Zhan, A. Barhoumi, T. Yang, R. G. Wylie, P. A. Armstrong and D. S. Kohane, Nano Lett., 2015 DOI:10.1021/acs.nanolett.1025b01325.
- A. Haefele, J. Blumhoff, R. S. Khnayzer and F. N. Castellano, J. Phys. Chem. Lett., 2012, 3, 299–303 CrossRef CAS.
- S. H. C. Askes, N. L. Mora, R. Harkes, R. I. Koning, B. Koster, T. Schmidt, A. Kros and S. Bonnet, Chem. Commun., 2015, 51, 9137–9140 RSC.
- S. G. Bown, A. Z. Rogowska, D. E. Whitelaw, W. R. Lees, L. B. Lovat, P. Ripley, L. Jones, P. Wyld, A. Gillams and A. W. R. Hatfield, Gut, 2002, 50, 549–557 CrossRef CAS.
- Y. Min, J. Li, F. Liu, E. K. L. Yeow and B. Xing, Angew. Chem., Int. Ed., 2014, 53, 1012–1016 CrossRef CAS PubMed.
- E. Ruggiero, J. Hernández-Gil, J. C. Mareque-Rivas and L. Salassa, Chem. Commun., 2015, 51, 2091–2094 RSC.
- Y.-F. Wang, G.-Y. Liu, L.-d. Sun, J.-W. Xiao, J.-C. Zhou and C.-H. Yan, ACS Nano, 2013, 7, 7200–7206 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp04352b |
| This journal is © the Owner Societies 2015 |