
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmylin–Aβ oligomers at atomic resolution using molecular dynamics simulations: a link between Type 2 diabetes and Alzheimer's disease†
Michal
Baram
ab,
Yoav
Atsmon-Raz
ab,
Buyong
Ma
c,
Ruth
Nussinov
*cd and
Yifat
Miller
*ab
aDepartment of Chemistry, Ben-Gurion University of the Negev, Beér-Sheva 84105, Israel
bIlse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Beér-Sheva 84105, Israel. E-mail: ymiller@bgu.ac.il; Tel: +972-86428705
cBasic Science Program, Leidos Biomedical Research, Inc., Cancer and Inflammation Program, National Cancer Institute, Frederick, MD 21702, USA. E-mail: NussinoR@helix.nih.gov; Tel: +1 301 846 5579
dSackler Inst. of Molecular Medicine, Department of Human Genetics and Molecular Medicine, Sackler School of Medicine, Tel Aviv University, Tel Aviv 69978, Israel
First published on 24th August 2015
Abstract
Clinical studies have identified Type 2 diabetes (T2D) as a risk factor of Alzheimer's disease (AD). One of the potential mechanisms that link T2D and AD is the loss of cells associated with degenerative changes. Amylin1–37 aggregates (the pathological species in T2D) were found to be co-localized with those of Aβ1–42 (the pathological species in AD) to form the Amylin1–37–Aβ1–42 plaques, promoting aggregation and thus contributing to the etiology of AD. However, the mechanisms by which Amylin1–37 co-aggregates with Aβ1–42 are still elusive. This work presents the interactions between Amylin1–37 oligomers and Aβ1–42 oligomers at atomic resolution applying extensive molecular dynamics simulations for relatively large ensemble of cross-seeding Amylin1–37–Aβ1–42 oligomers. The main conclusions of this study are first, Aβ1–42 oligomers prefer to interact with Amylin1–37 oligomers to form single layer conformations (in-register interactions) rather than double layer conformations; and second, in some double layer conformations of the cross-seeding Amylin1–37–Aβ1–42 oligomers, the Amylin1–37 oligomers destabilize the Aβ1–42 oligomers and thus inhibit Aβ1–42 aggregation, while in other double layer conformations, the Amylin1–37 oligomers stabilize Aβ1–42 oligomers and thus promote Aβ1–42 aggregation.
Introduction
Type 2 diabetes (T2D) is one of the most common metabolic disorders and its prevalence increases with age. Clinical and epidemiological studies identified T2D as a risk factor of Alzheimer's disease (AD).1–3 Several studies have shown that there are many similarities between T2D and AD, and that both conditions underlie common physiological processes.3 AD is characterized by intracellular neurofibrillary tangles (NFTs), containing an abnormally hyperphosphorylated form of tau protein, and extracellular senile plaques, mainly composed of Amyloid β (Aβ) aggregates. Both Tau and Aβ aggregates which are the pathological hallmarks of AD are found in T2D.4,5 One of the potential mechanisms that link T2D and AD is the loss of cells associated with degenerative changes.1,2,6 AD is a neurodegenerative disease with extensive neuronal loss resulting from Tau and Aβ aggregation. T2D is also a degenerative disease that results from selective destruction of pancreatic β-cells and associated neuropathies,7–9 which are caused by aggregation of the neuroendocrine hormone named “Amylin”.Recently, Jackson et al.10 identified Amylin deposits in the temporal lobe gray matter – a major component of the central nervous system – of diabetes patients. In addition to the Amylin deposition in the human brain, Amylin aggregates are co-localized with Aβ aggregates to form the Amylin–Aβ plaques, promoting aggregation and thus contributing to the etiology of AD. Recent in vivo studies investigated the cross-seeding between Aβ and Amylin aggregates.11–13 Yet, the mechanisms by which Amylin co-aggregates with Aβ are still elusive. Both Aβ and Amylin are misfolded peptides. The direct interaction of misfolded peptides, a topic which to date has been poorly explored, could play a major role in the genesis and progression of several pathological conditions. Although not extensively studied, in vitro reports show cross-seeding interactions among several amyloidogenic proteins.14–20 One of these studies19 showed that Aβ1–42 acts as a good seed for Amylin1–37 oligomerization; however, Amylin1–37 aggregates slightly affect soluble Aβ1–42 oligomerization. A recent study applied electrospray ionization-ion mobility spectroscopy-mass spectroscopy to characterize the dynamics and the kinetics of Amylin1–37 oligomerization, Aβ1–40 oligomerization and Amylin1–37–Aβ1–40 oligomerization.21 The interactions between Amylin1–37 aggregates and Aβ1–42 aggregates at atomic resolution are still elusive.
Several studies proposed that the sequences of Aβ1–42 and Amylin1–37 have 25% identity and 50% similarity and thus some domains in Aβ and some in Amylin participate in the co-assembly of Aβ–Amylin.22–26 Yet, these studies do not provide the atomic resolution of the molecular structures of Aβ1–42–Amylin1–37 aggregates. Recently, Berhanu et al.27 investigated the molecular structures of Aβ15–40–Amylin10–35 oligomers at atomic resolution. They explored an Aβ15–40 oligomer fragment of the ssNMR model of Aβ17–42,28 not considering the toxic full-length Aβ1–42 oligomer, arguing that residues 1–16 in the N-terminus of Aβ are in a disordered domain and thus unlikely to play a role in aggregation. However, previous studies have shown that residues 1–16 in the N-terminus of Aβ can play important roles in fibrilization and form a well-organized β-strand structure.29–33 It is known that several mutations in the N-terminus accelerate amyloid formation, such as the English (H6R) mutation;34 in addition, mutating Ala2 to Thr or Val modifies the Aβ aggregation landscape.35–38 Amylin10–35 oligomers of one of the two structures proposed by the Eisenberg group,39 which differ in the orientation of the residues along the U-turn region and thus can strongly affect the interaction between Aβ and Amylin,39 were explored,27,40 but not those proposed by the Tycko group.41 Recently, our group illustrated that Amylin1–37 oligomers have four variant models that differ in the orientation of the residues along the β-strands and the turn region of the β-arch.42 The interactions between Aβ and Amylin may differ due to these different orientations. Although residues 1–7 in the N-terminus of Amylin are not part of the β-sheet of the experimental models and the disulfide bridge between Cys2 and Cys7 does not contribute to aggregate assembly, it may have an effect on Aβ–Amylin aggregation when considering single and double layer conformations. Residues Ala8 and Thr9 in Amylin that were overlooked in earlier studies27 may also contribute to Aβ–Amylin aggregation. Finally, the N-terminus of Aβ, residues Asp1–Lys16, may interact with Amylin and thus affect cross-seeding of Aβ–Amylin aggregation. Here we study the interactions between Aβ1–42 oligomers with each of the four models of the full-length Amylin1–37 oligomers at atomic resolution. Our results led to two important conclusions. First, all four variant models of the full-length Amylin1–37 oligomers prefer to interact with Aβ1–42 oligomers to form single layer conformations. Second, interactions between the cross-seeded species in the single layer and the double layer conformations affect differently the flexibility (or rigidity) of the turn region of the self-assembled β-arch amyloids. Finally, residues in the N-termini of Aβ and Amylin contribute to the cross-seeding Aβ–Amylin aggregation; therefore it is important to consider the full-length Aβ and Amylin.
Materials and methods
Molecular dynamics protocol
MD simulations of the solvated oligomers were performed in the NPT ensemble using NAMD43 with the CHARMM27 force field.44,45 The oligomers were energy minimized and explicitly solvated in a TIP3P water box46,47 with a minimum distance of 15 Å from each edge of the box. Each water molecule within 2.5 Å of the oligomers was removed. Counter ions were added at random locations to neutralize the charge of the oligomers. The Langevin piston method43,48,49 with a decay period of 100 fs and a damping time of 50 fs was used to maintain a constant pressure of 1 atm. The temperature (330 K) was controlled by a Langevin thermostat with a damping coefficient of 10 ps.43 The short-range van der Waals (VDW) interactions were calculated using the switching function, with a twin range cutoff of 10.0 and 12.0 Å. Long-range electrostatic interactions were calculated using the particle mesh Ewald method with a cutoff of 12.0 Å.50,51 The equations of motion were integrated using the leapfrog integrator with a step of 1 fs.The solvated systems were energy minimized for 2000 conjugated gradient steps, where the hydrogen bonding distance between the β-sheets in each oligomer is fixed in the range 2.2–2.5 Å. The counter ions and water molecules were allowed to move. Hydrogen atoms were constrained to the equilibrium bond using the SHAKE algorithm.52 The minimized solvated systems were energy minimized for 5000 additional conjugate gradient steps and 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 heating steps at 250 K, with all atoms allowed to move. Then, the systems were heated from 250 K to 300 K and then to 330 K for 300 ps and equilibrated at 330 K for 300 ps. Simulations ran for 30 ns for each variant model, with a total run of 960 ns for all models. The structures were saved every 10 ps for analysis. These conditions were applied to all models.
000 heating steps at 250 K, with all atoms allowed to move. Then, the systems were heated from 250 K to 300 K and then to 330 K for 300 ps and equilibrated at 330 K for 300 ps. Simulations ran for 30 ns for each variant model, with a total run of 960 ns for all models. The structures were saved every 10 ps for analysis. These conditions were applied to all models.
Generalized Born method with molecular volume (GBMV) and population analysis
To obtain the relative structural stability of the models, the trajectories of the last 5 ns were extracted from the explicit MD simulation excluding water molecules. The solvation energies of all systems were calculated using the generalized Born method with molecular volume (GBMV).53,54 In the GBMV calculations, the dielectric constant of water was set to 80.0. The hydrophobic solvent-accessible surface area (SASA) term factor was set to 0.00592 kcal mol−1.2 Each variant was minimized for 1000 cycles and the conformation energy was evaluated by grid-based GBMV. The minimization does not change the conformations of each variant, but only relaxed the local geometries due to thermal fluctuation which occurred during the MD simulations.A total of 16000 conformations (500 conformations for each of the 32 examined conformers) were used to construct the free energy landscape of the conformers and to evaluate the conformer probabilities by using Monte Carlo (MC) simulations. In the first step, one conformation of conformer i and one conformation of conformer j were randomly selected. Then, the Boltzmann factor was computed as e−(Ej−Ei)/kT, where Ei and Ej are the conformational energies evaluated using the GBMV calculations for conformations i and j, respectively, k is the Boltzmann constant and T is the absolute temperature (298 K used here). If the value of the Boltzmann factor was larger than the random number, then the move from conformation i to conformation j was allowed. After 1 million steps, the visited conformations for each conformer were counted. Finally, the relative probability of model n was evaluated as Pn = Nn/Ntotal, where Pn is the population of model n, Nn is the total number of conformations visited for model n, and Ntotal is the total steps. The advantages of using MC simulations to estimate conformer probability lie in their good numerical stability and the control that they allow for transition probabilities among several conformers.
Using all 32 models and 16000 conformations (500 for each model) generated from the MD simulations, we estimated the overall stability and populations for each conformer based on the MD simulations, with the energy landscape being computed with GBMV for these 32 models. The group that these 32 models are likely to represent may be only a very small percentage of the ensemble. Nevertheless, the carefully selected models cover the most likely structures. It should be noted here that the results obtained in this study depend on the initial structures and the initial conditions.
Analysis details
We examined the structural stability of the models by following the changes in the number of the hydrogen bonds between β-strands, with the hydrogen bond cut-off set to 2.5 Å. In addition we followed the root-mean square deviations (RMSDs) and root-mean square fluctuations (RMSFs) of all structures. The ψ and φ angles of each residue in the Amylin models were computed for the last 5 ns to estimate the secondary structure of the self-assembled models.Reaction coordinates for the formation of Amylin–Aβ oligomer structures
To investigate the stability of each soluble Amylin–Aβ oligomer structure, the conformational energies were computed for all Amylin oligomers and for the Aβ oligomer (Table S1, ESI†). The conformational energies for each model are based on the energy computed with the GBMV method. For each model, a total of 500 conformations from the last 5 ns of the simulations were used to evaluate the conformational energy.We estimated the relative stability of each Amylin–Aβ model by comparing its energy with the energies of its two types of components, the Amylin model and the Aβ oligomer, as illustrated by the following chemical “reaction”:
| (Amylin)n + (Aβ)n ⇔ (Amylin)n·(Aβ)n | (1) |
Obviously, single and double layer models may have different interaction types. Yet, as we previously demonstrated31–33,40,42,55,56 one can compare the relative conformational energies between the single and the double layer models to provide insight into these different interactions. The differences in the interaction types may explain the differences in the relative energies.
Results and discussion
Experiment-based Amylin1–37 oligomer model, Aβ1–42 oligomer model and Aβ1–42–Amylin1–37 oligomer model construction
Previously, we illustrated four models of Amylin1–37 that differ in the orientation of the residues along the backbone of the β-strands and along the turn domain of the β-arch structure.42 These four models (M1–M4, Fig. S1, ESI†) were based on the experimental structures of Tycko41 and Eisenberg.39 Herein, on the basis of Tycko's two-fold Aβ1–40 model,57 we extended the C-terminus by two residues to form Aβ1–42, which is more toxic than Aβ1–40. For each of the four Amylin1–37 oligomers we constructed eight models of Aβ1–42–Amylin1–37 oligomers, where each model consists of six Aβ1–42 monomers and six Amylin1–37 monomers. Four were single layer conformations and four double layer conformations. Therefore, we constructed a total of 32 Aβ1–42–Amylin1–37 models. Table 1 details all 32 Aβ1–42–Amylin1–37 models: B1–B8, C1–C8 and D1–D8. Fig. 1 illustrates the eight initial models of Aβ1–42–Amylin1–37 oligomers of one of the four Amylin1–37 models (model M1): B1–B8. Similarly we constructed the arrangements between Aβ1–42 oligomers and Amylin1–37 oligomers (models M2, M3 and M4). Fig. S2–S4 (ESI†) demonstrate the other 24 initial Aβ1–42–Amylin1–37 models and Fig. S5–S8 show the 32 simulated Aβ1–42–Amylin1–37 models. Finally, we simulated the Aβ1–42 oligomer model which is based on Tycko's two-fold Aβ1–40 model (Fig. S9a, ESI†).57 Interestingly, the simulated Aβ1–42 oligomer model illustrated a third β-stand at the C-termini of the monomers (Fig. S9b, ESI†) leading to a new structural model of the Aβ1–42 oligomer. Recently, a novel structural model of the Aβ11–42 oligomer provided further evidence for the highly polymorphic nature of the Aβ peptide fibril.58 Polymorphism was also obtained in some of the simulated Aβ1–42–Amylin1–37 models. Structural comparison suggests that the structural similarity between the Aβ11–42 oligomer and amylin oligomers is lower than with the model of the Aβ oligomer in the current study. It will be interesting to examine the cross-seeding Aβ1–42–Amylin1–37 oligomers using this new Aβ structure59 as well as with additional polymorphic states such as the triangular structure.60 Previous ssNMR studies have shown α-helical structures of Amylin and not cross-β structures.61,62 Recent ssNMR studies presented unstructured Aβ oligomers63 and Amylin oligomers that form large micelles,64 which may be a general phenomenon for natively unstructured Amylin. We did not apply these α-helical and the unstructured amylin, or the unstructured Aβ oligomers in the current study, because of the lack of the PDB coordinates. Future work would need to solve these oligomer structures in order to study the cross-seeding of Aβ1–42–Amylin1–37 oligomers. Finally, the RMSDs and the hydrogen bond analysis illustrate that the simulated Aβ1–42–Amylin1–37 models are structurally stable (Fig. S10–S15, ESI†).| Model | Amylin type | Orientations between Aβ and Amylin |
|---|---|---|
| B1 | M1 | Parallel |
| B2 | M1 | Antiparallel |
| B3 | M1 | Parallel |
| B4 | M1 | Antiparallel |
| B5 | M1 | N(Aβ)–N(Amylin) |
| B6 | M1 | N(Aβ)–C(Amylin) |
| B7 | M1 | C(Aβ)–N(Amylin) |
| B8 | M1 | C(Amylin)–C(Aβ) |
| C1 | M2 | Parallel |
| C2 | M2 | Antiparallel |
| C3 | M2 | Parallel |
| C4 | M2 | Antiparallel |
| C5 | M2 | N(Aβ)–N(Amylin) |
| C6 | M2 | N(Aβ)–C(Amylin) |
| C7 | M2 | C(Aβ)–N(Amylin) |
| C8 | M2 | C(Amylin)–C(Aβ) |
| D1 | M3 | Parallel |
| D2 | M3 | Antiparallel |
| D3 | M3 | Parallel |
| D4 | M3 | Antiparallel |
| D5 | M3 | N(Aβ)–N(Amylin) |
| D6 | M3 | N(Aβ)–C(Amylin) |
| D7 | M3 | C(Aβ)–N(Amylin) |
| D8 | M3 | C(Amylin)–C(Aβ) |
| E1 | M4 | Parallel |
| E2 | M4 | Antiparallel |
| E3 | M4 | Parallel |
| E4 | M4 | Antiparallel |
| E5 | M4 | N(Aβ)–N(Amylin) |
| E6 | M4 | N(Aβ)–C(Amylin) |
| E7 | M4 | C(Aβ)–N(Amylin) |
| E8 | M4 | C(Amylin)–C(Aβ) |
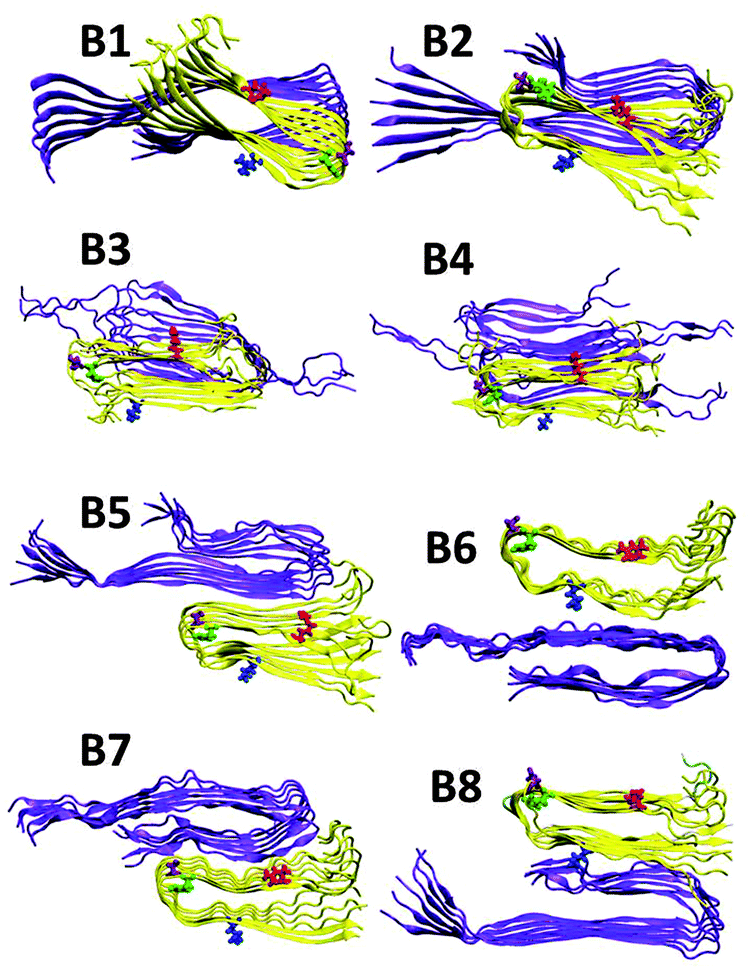 | ||
| Fig. 1 Constructed initial Aβ1–42–Amylin1–37 dodecamers. Aβ hexamer is based on Tycko's structure (ref. 51) and the Amylin hexamer M1 is obtained from Miller's structures (ref. 36) [based on Tycko's ssNMR (ref. 35) and Eisenberg's crystal structures (ref. 34)]. Models B1–B4 are single layer conformations and B5–B8 are double layer conformations. | ||
Single layer conformational arrangements of Aβ1–42–Amylin1–37 oligomer models are preferred over double layer conformational arrangements
In order to compare the 32 Aβ1–42–Amylin1–37 oligomers we generated 500 conformations for each arrangement by MD simulations and estimated the conformational energies and the populations (Table S1, ESI†). Fig. 2 demonstrates the populations of all 32 Aβ1–42–Amylin1–37 oligomers. One can see that single layer conformations of Aβ1–42–Amylin1–37 oligomers with parallel and antiparallel arrangements (B1, B2, C1, C2, D1, D2, E1 and E2) show the highest populations and thus are preferred over the two single layer conformations and all the double layer conformations. We previously showed that in the cross-seeded Aβ17–42-mutated Tau R2 oligomers the double layer conformations are preferred over the single layer conformations.56 The preference of the cross-seeding of some conformations over others may be due to the interactions between residues along the sequences of the various types of amyloids. The interactions that stabilize structurally and energetically the cross-seeding amyloid oligomers will yield preferred organizations.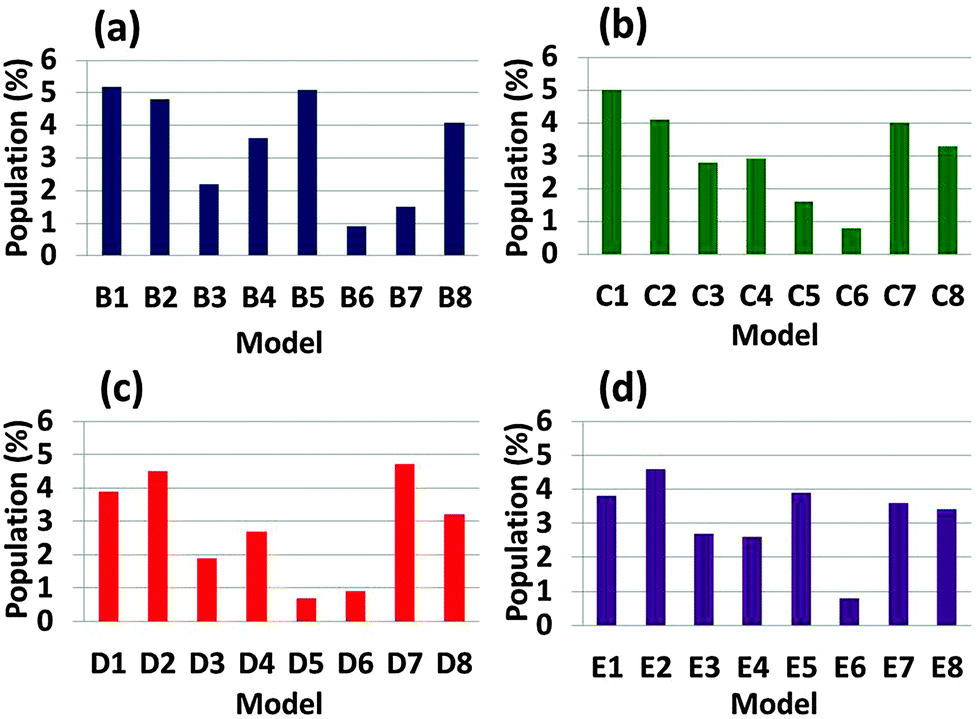 | ||
| Fig. 2 Populations of the simulated Aβ1–42–Amylin1–37 dodecamers, which are estimated by Monte-Carlo simulations. | ||
We computed the secondary structures of Aβ1–42 oligomers in all 32 cross-seeding Amylin1–37–Aβ1–42 oligomers (Fig. S16–S23, ESI†). Interestingly, one can see from Fig. S16–S21 (ESI†) that in the single layer simulated Aβ1–42–Amylin1–37 oligomer models and in the double layer simulated Aβ1–42–Amylin1–37 oligomer models (in which the C-termini of Aβ1–42 monomers do not interact with Amylin) residues Val39–Ala42 in the C-termini of Aβ1–42 showed formation of a third β-strand and a second turn region (residues Gly37–Gly38). The original experiment-based Aβ1–42 oligomer57 has two β-strands connected by a U-turn; however, herein our simulations demonstrate that the Aβ1–42 oligomer has three β-strands connected by two turns both when it does not interact with Amylin1–37 oligomers and when it does. A previous study has shown that isoforms of Tau repeats form triple-stranded and two-turn structures.65
Common mechanisms between various types of Amylin1–37 oligomers and Aβ1–42 oligomers
To investigate the mechanisms by which Amylin1–37 oligomers interact with Aβ1–42 oligomers to form the cross-seeded Amylin1–37–Aβ1–42 oligomers, we estimated the “reaction coordinate” in which Amylin1–37 oligomers interact with Aβ1–42 oligomers. To this aim, we computed the relative conformational energies of the separated oligomers and the cross-seeding Amylin1–37–Aβ1–42 oligomers using the GBMV method53,54 for each of the four Amylin1–37 oligomers (Fig. 3).42 We previously studied similarly the cross-seeding interactions between two types of amyloids55,56 and two types of Amylin1–37.42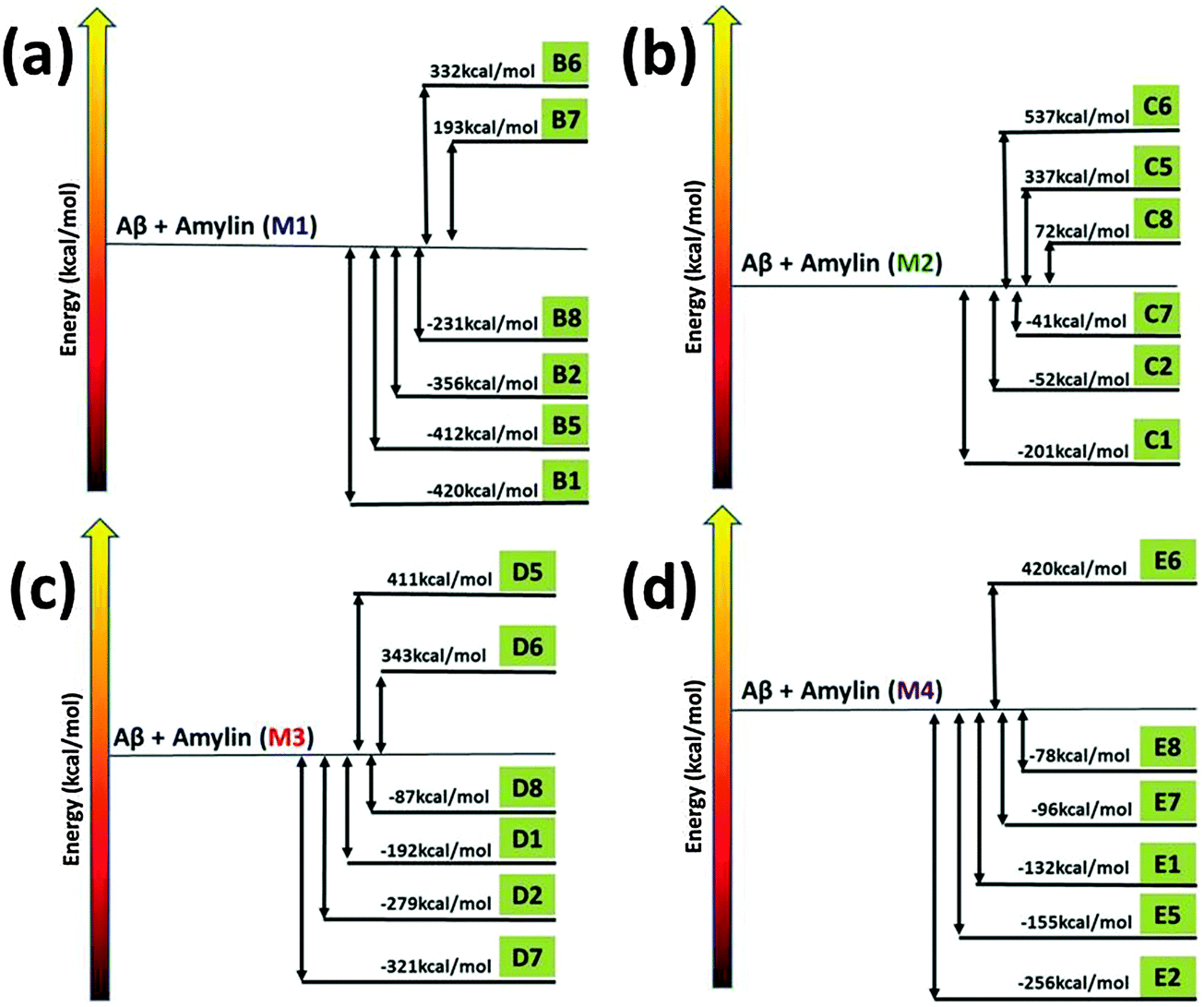 | ||
| Fig. 3 The relative conformational energies of separated Aβ1–42 hexamers and Amylin1–37 hexamers (M1–M4) and Aβ1–42–Amylin1–37 dodecamers. | ||
Interestingly, the common mechanisms by which Amylin1–37 oligomers interact with Aβ1–42 oligomers to form the cross-seeded Amylin1–37–Aβ1–42 oligomers illustrate that each of the four Amylin1–37 oligomers prefers to interact with an Aβ1–42 oligomer to form a single layer conformation in parallel and in antiparallel orientation, yielding an ‘exothermic reaction’. In some of the four Amylin1–37 oligomers there are other mechanisms that illustrate the formation of double layer conformations, but in all four Amylin1–37 oligomers the common mechanisms show the formation of the single layer conformation. Therefore, the preference of the single layer conformation indicates a strong cross-seeding tendency between Aβ1–42 and Amylin1–37 peptides.
The effect of cross-seeding on the structural features of Amylin1–37 oligomers and Aβ1–42 oligomers
One of the interesting topics in studying the cross-seeding between amyloids is to investigate the effect of cross-seeding on the structural features of the amyloids. The cross-seeding between Amylin1–37 oligomers and Aβ1–42 oligomers is of particular interest, because there are four variant models of Amylin1–37 oligomers that differ in the orientation of the residues along the β-arch structures and thus we expect that the effect on the structural features of the various cross-seeding Amylin1–37–Aβ1–42 oligomers may be different.We first examined the effect on the β-strand of the β-arch structures of Aβ1–42 oligomers. The secondary structures of these oligomers in all 32 cross-seeding Amylin1–37–Aβ1–42 oligomers were computed (Fig. S16–S23, ESI†). In Aβ1–42 oligomers residues D1–F20 and residues A30–A42 show the properties of β-strand, but in the cases (models B6, C6, D6 and E6) where the N-termini of Aβ1–42 oligomers interact with the C-termini of Amylin1–37 oligomers to form double layer conformations these residues do not demonstrate β-strand properties. Also, in some cases (models C5 and D5) where the N-termini of Aβ1–42 oligomers interact with the N-termini of Amylin1–37 oligomers or in one case (model B7) where the C-termini of Aβ1–42 oligomers interact with the N-termini of Amylin1–37 oligomers these residues do not show β-strand properties. In such cases the cross-β structures that characterize the fibrillation of amyloids yield structurally less stable cross-seeding.
We then examined the fluctuation of the backbone of Aβ1–42 oligomers and Amylin1–37 oligomers for each of the 32 cross-seeding Amylin1–37–Aβ1–42 oligomers using RMSF calculations (Fig. 4 and 5). Interestingly, in cases (models B6, C6, D6 and E6) where the N-termini of Aβ1–42 oligomers interact with the C-termini of Amylin1–37 oligomers to form double layer conformations, the turn regions of Aβ1–42 oligomers fluctuate relatively more than in other cross-seeding models. These models demonstrated no β-strand properties, because the interactions in the double layer conformations do not allow structurally stable structures. One can see from the simulated models (Fig. S5–S8, ESI†) that Aβ1–42 oligomers do not show cross-β structures, and are structurally unstable oligomers. Finally, those simulated models that do not exhibit cross-β structures (models C5, D5 and B7) also fluctuate in the turn region of the Aβ1–42 oligomers.
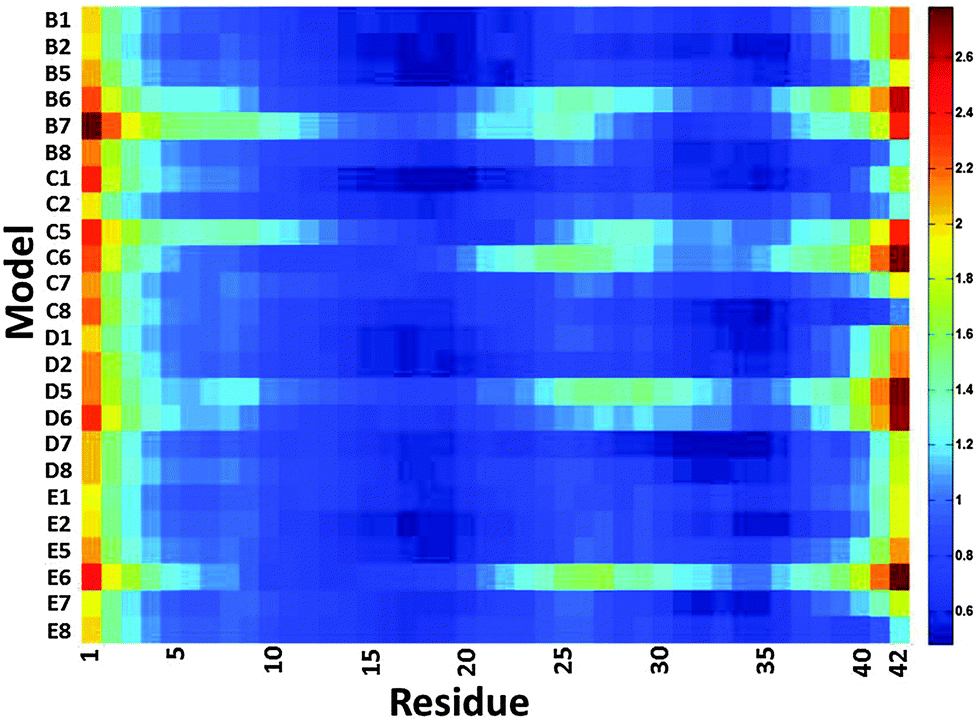 | ||
| Fig. 4 The averaged RMSF of residues of Aβ1–42 in the 32 models of Aβ1–42–Amylin1–37 dodecamers. | ||
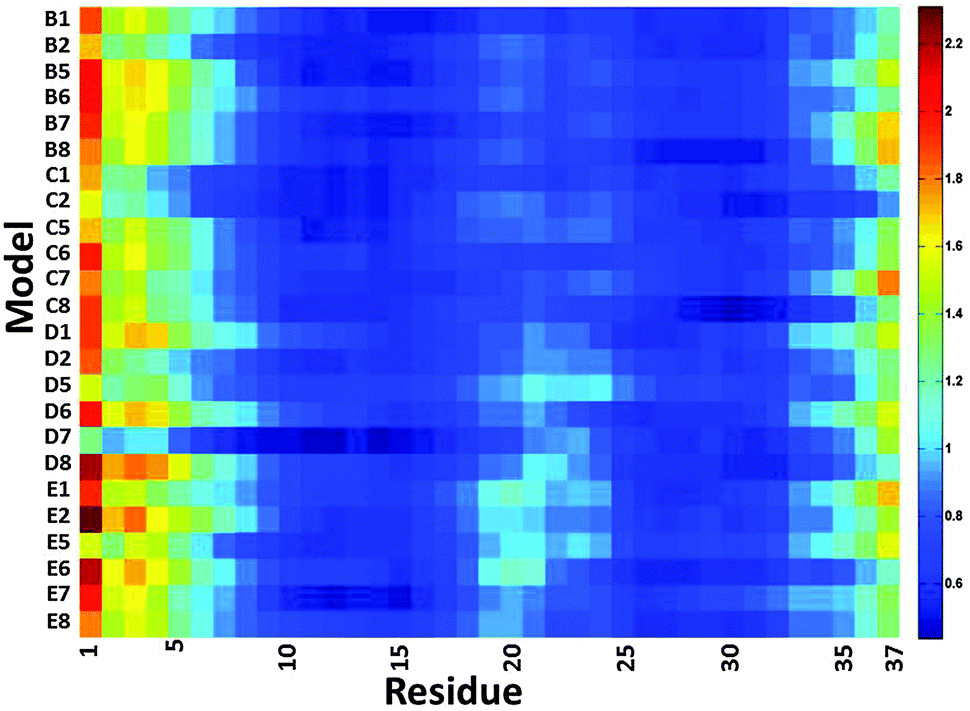 | ||
| Fig. 5 The averaged RMSF of residues of Amylin1–37 in the 32 models of Aβ1–42–Amylin1–37 dodecamers. | ||
Our results suggest that among the double layer conformations of the cross-seeding oligomers, the stability of the turn region domain in the Aβ1–42 oligomers may be affected by the interactions with all four Amylin1–37 oligomer variants suggesting that the turn region may affect the fibrillation (or the self-assembly process) of Aβ1–42 oligomers. In cases where the turn regions in Aβ1–42 oligomers are destabilized due to the interactions with Amylin1–37 oligomers, these interactions may inhibit aggregation of the Aβ1–42 oligomers. Zanni's group proposed an aggregation pathway for amylin in which the turn regions of amylin play a role as initial seeding for aggregation.66 We thus suggest that in some cases destabilization of the turn regions of Aβ1–42 oligomers may inhibit Aβ1–42 aggregation. However, in some cases the turn regions in Aβ1–42 oligomers are stabilized by these interactions and therefore we expect that these interactions will induce aggregation of Aβ1–42 oligomers.
Our study illustrates for the first time the importance of investigating the cross-seeding between full-length Aβ and full-length Amylin and that the N-termini play a role in some cases in the stabilization of the cross-seeding of Aβ–Amylin oligomers. One can see that interactions between residues in the N-termini of Aβ and Amylin (those that had not been considered earlier27) stabilize both single and double layer conformations (Fig. S24–S26, ESI†). On the other hand, interestingly, in some cases the interactions between the Val12 of Aβ (which is located in the N-terminus) and the C-terminus of Amylin (residues Ile26 and Leu27) destabilize the Aβ oligomers but do not affect the stabilization of Amylin (Fig. S27, ESI†). Therefore, this is the first study that illustrates the role of the interactions of the N-termini in cross-seeding Aβ–Amylin aggregation at atomic resolution. In some cases, the N-termini are favorable for cross-seeding and in some other cases the N-termini are unfavorable for cross-seeding.
Finally, we examined the effect of the interactions of Aβ1–42 oligomers on the fluctuation of the turn regions of Amylin1–37 oligomers. Interestingly, one can see from Fig. 5 that the interactions of Aβ1–42 oligomers with the variant models of Amylin1–37 oligomers (M3 and M4) result in fluctuations of the turn regions of Amylin1–37 oligomers, i.e. flexibility of the turn regions, which is in contrast with the interactions of Aβ1–42 oligomers with the variant models of Amylin1–37 oligomers (M1 and M2) which result in more rigid turn regions. Previously we showed that the variant models M1 and M2 of Amylin1–37 oligomers illustrated rigid turn regions, while with the variant models of Amylin1–37 oligomers M3 and M4 showed flexible turn regions.42 We thus suggest that the interactions of Aβ1–42 oligomers do not affect the structural features of Amylin1–37 oligomers; rather, in some cases Amylin1–37 oligomers affect the structural features and the stability of Aβ1–42 oligomers.
Conclusions
Many, though not all, clinical studies indicate that individuals with T2D are at higher risk of eventually developing AD or other dementia,1–3 but the connection between these two diseases is not understood. Recently, Amylin deposits were found in the temporal lobe gray matter – a major component of the central nervous system – of diabetes patients.10 In addition to the Amylin deposition in the human brain, Amylin aggregates were found to co-localize with Aβ aggregates to form Amylin–Aβ plaques, promoting aggregation and thus contributing to the etiology of AD. The mechanisms by which Amylin co-aggregates with Aβ are still elusive. Herein, we present for the first time the co-aggregation between the full-length Aβ1–42 oligomer and each of the four variants of the full-length Amylin1–37 oligomers that we have recently established.42 Here, we focus on the parallel β-sheet structure of the Aβ1–42 oligomer, although the antiparallel β-sheet structure of Aβ1–40 oligomers has also been considered as a toxic species.67 Yet, it is known that Aβ1–42 is more toxic than Aβ1–40. The toxicity of Aβ oligomers to neuronal cells has been demonstrated to occur via a two-step mechanism of membrane disruption.68Two important observations emerge from this study. First, all four variant models of the full-length Amylin1–37 oligomers preferred to interact with Aβ1–42 oligomers to form polymorphic single layer conformations. Second, the interactions between the cross-seeding Amylin1–37–Aβ1–42 oligomers both in single and in double layer conformations affect the structural features differently. In particular, the differences center on the flexibility/rigidity of the turn region and the order/disorder of the β-strands in the self-assembled β-arch amyloids. Finally, while studying cross-seeding Amylin1–37–Aβ1–42 oligomers, it is important to investigate the full-length of these amyloids because of the role that the terminal residues may play in the stabilization of the hetero-oligomers. Understanding the mechanisms and the range of structural features of the co-aggregates of Amylin1–37–Aβ1–42 oligomers is of crucial importance for effective drug design to reduce co-aggregate formation and maybe to prevent patients with T2D from developing AD in later life. A recent study demonstrated the cross-seeding effects of bacterial curli on semen enhancer of viral infection (SEVI), Aβ and Amylin.69 This experimental study reports important implications and it would be useful to further strengthen this study using MD simulations, as reported in the present study.
Acknowledgements
This project was supported by the FP7-PEOPLE-2011-CIG, research grant no. 303741, and in whole or in part by Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.All simulations were performed using the high-performance computational facilities of the Miller Lab in the BGU HPC computational center and the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). The support of the BGU HPC computational center staff is greatly appreciated.
References
- J. Gotz, L. M. Ittner and Y. A. Lim, Common features between diabetes mellitus and Alzheimer's disease, Cell. Mol. Life Sci., 2009, 66, 1321–1325 CrossRef CAS PubMed.
- Y. Yang and W. Song, Molecular links between Alzheimer's disease and diabetes mellitus, Neuroscience, 2013, 250, 140–150 CrossRef CAS PubMed.
- L. Li and C. Holscher, Common pathological processes in Alzheimer disease and type 2 diabetes: a review, Brain Res. Rev., 2007, 56, 384–402 CrossRef CAS PubMed.
- I. Churcher, Tau therapeutic strategies for the treatment of Alzheimer's disease, Curr. Top. Med. Chem., 2006, 6, 579–595 CrossRef CAS PubMed.
- C. G. Glabe, Common mechanisms of amyloid oligomer pathogenesis in degenerative disease, Neurobiol. Aging, 2006, 27, 570–575 CrossRef CAS PubMed.
- J. Gotz, Y. A. Lim and A. Eckert, Lessons from two prevalent amyloidoses − what amylin and Abeta have in common, Front. Aging Neurosci., 2013, 5, 38 Search PubMed.
- A. M. Brands, R. P. Kessels, E. H. de Haan, L. J. Kappelle and G. J. Biessels, Cerebral dysfunction in type 1 diabetes: effects of insulin, vascular risk factors and blood-glucose levels, Eur. J. Pharmacol., 2004, 490, 159–168 CrossRef CAS PubMed.
- M. Ristow, Neurodegenerative disorders associated with diabetes mellitus, J. Mol. Med., 2004, 82, 510–529 CrossRef PubMed.
- E. Roche, J. A. Reig, A. Campos, B. Paredes, J. R. Isaac, S. Lim, R. Y. Calne and B. Soria, Insulin-secreting cells derived from stem cells: clinical perspectives, hypes and hopes, Transplant Immunol., 2005, 15, 113–129 CrossRef CAS PubMed.
- K. Jackson, G. A. Barisone, E. Diaz, L. W. Jin, C. DeCarli and F. Despa, Amylin deposition in the brain: a second amyloid in Alzheimer disease?, Ann. Neurol., 2013, 74, 517–526 CrossRef CAS PubMed.
- M. E. Oskarsson, J. F. Paulsson, S. W. Schultz, M. Ingelsson, P. Westermark and G. T. Westermark, In vivo seeding and cross-seeding of localized amyloidosis: a molecular link between type 2 diabetes and Alzheimer disease, Am. J. Pathol., 2015, 185, 834–846 CrossRef CAS PubMed.
- L. M. Yan, A. Velkova and A. Kapurniotu, Molecular characterization of the hetero-assembly of beta-amyloid peptide with islet amyloid polypeptide, Curr. Pharm. Des., 2014, 20, 1182–1191 CrossRef CAS PubMed.
- W. Q. Qiu, M. Wallack, M. Dean, E. Liebson, M. Mwamburi and H. Zhu, Association between amylin and amyloid-beta peptides in plasma in the context of apolipoprotein E4 allele, PLoS One, 2014, 9, e88063 Search PubMed.
- L. K. Clinton, M. Blurton-Jones, K. Myczek, J. Q. Trojanowski and F. M. LaFerla, Synergistic interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline, J. Neurosci., 2010, 30, 7281–7289 CrossRef CAS PubMed.
- J. P. Guo, T. Arai, J. Miklossy and P. L. McGeer, Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer's disease, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1953–1958 CrossRef CAS PubMed.
- M. R. Krebs, L. A. Morozova-Roche, K. Daniel, C. V. Robinson and C. M. Dobson, Observation of sequence specificity in the seeding of protein amyloid fibrils, Protein Sci., 2004, 13, 1933–1938 CrossRef CAS PubMed.
- P. K. Mandal, J. W. Pettegrew, E. Masliah, R. L. Hamilton and R. Mandal, Interaction between Abeta peptide and alpha synuclein: molecular mechanisms in overlapping pathology of Alzheimer's and Parkinson's in dementia with Lewy body disease, Neurochem. Res., 2006, 31, 1153–1162 CrossRef CAS PubMed.
- R. Morales, L. D. Estrada, R. Diaz-Espinoza, D. Morales-Scheihing, M. C. Jara, J. Castilla and C. Soto, Molecular cross talk between misfolded proteins in animal models of Alzheimer's and prion diseases, J. Neurosci., 2010, 30, 4528–4535 CrossRef CAS PubMed.
- B. O'Nuallain, A. D. Williams, P. Westermark and R. Wetzel, Seeding specificity in amyloid growth induced by heterologous fibrils, J. Biol. Chem., 2004, 279, 17490–17499 CrossRef PubMed.
- I. F. Tsigelny, L. Crews, P. Desplats, G. M. Shaked, Y. Sharikov, H. Mizuno, B. Spencer, E. Rockenstein, M. Trejo, O. Platoshyn, J. X. Yuan and E. Masliah, Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer's and Parkinson's diseases, PLoS One, 2008, 3, e3135 Search PubMed.
- L. M. Young, R. A. Mahood, J. C. Saunders, L. H. Tu, D. P. Raleigh, S. E. Radford and A. E. Ashcroft, Insights into the consequences of co-polymerisation in the early stages of IAPP and Abeta peptide assembly from mass spectrometry, Analyst, 2015 10.1039/C5AN00865D.
- M. A. Schroer, Y. Zhai, D. C. Wieland, C. J. Sahle, J. Nase, M. Paulus, M. Tolan and R. Winter, Exploring the piezophilic behavior of natural cosolvent mixtures, Angew. Chem., 2011, 50, 11413–11416 CrossRef CAS PubMed.
- W. A. Banks, A. J. Kastin, L. M. Maness, W. Huang and J. B. Jaspan, Permeability of the blood–brain barrier to amylin, Life Sci., 1995, 57, 1993–2001 CrossRef CAS PubMed.
- N. Ida, T. Hartmann, J. Pantel, J. Schroder, R. Zerfass, H. Forstl, R. Sandbrink, C. L. Masters and K. Beyreuther, Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay, J. Biol. Chem., 1996, 271, 22908–22914 CrossRef CAS PubMed.
- T. Sanke, T. Hanabusa, Y. Nakano, C. Oki, K. Okai, S. Nishimura, M. Kondo and K. Nanjo, Plasma islet amyloid polypeptide (Amylin) levels and their responses to oral glucose in type 2 (non-insulin-dependent) diabetic patients, Diabetologia, 1991, 34, 129–132 CrossRef CAS PubMed.
- L. M. Yan, M. Tatarek-Nossol, A. Velkova, A. Kazantzis and A. Kapurniotu, Design of a mimic of nonamyloidogenic and bioactive human islet amyloid polypeptide (IAPP) as nanomolar affinity inhibitor of IAPP cytotoxic fibrillogenesis, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2046–2051 CrossRef CAS PubMed.
- W. M. Berhanu, F. Yasar and U. H. Hansmann, In silico cross seeding of Abeta and amylin fibril-like oligomers, ACS Chem. Neurosci., 2013, 4, 1488–1500 CrossRef CAS PubMed.
- T. Luhrs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert and R. Riek, 3D structure of Alzheimer's amyloid-beta(1-42) fibrils, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef CAS PubMed.
- Y. Miller, B. Ma, C. J. Tsai and R. Nussinov, Hollow core of Alzheimer's Abeta42 amyloid observed by cryoEM is relevant at physiological pH, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14128–14133 CrossRef CAS PubMed.
- Y. Miller, B. Ma and R. Nussinov, Polymorphism in Alzheimer Abeta amyloid organization reflects conformational selection in a rugged energy landscape, Chem. Rev., 2010, 110, 4820–4838 CrossRef CAS PubMed.
- B. Ma and R. Nussinov, Polymorphic triple beta-sheet structures contribute to amide hydrogen/deuterium (H/D) exchange protection in the Alzheimer amyloid beta42 peptide, J. Biol. Chem., 2011, 286, 34244–34253 CrossRef CAS PubMed.
- B. Ma and R. Nussinov, Polymorphic C-terminal beta-sheet interactions determine the formation of fibril or amyloid beta-derived diffusible ligand-like globulomer for the Alzheimer Abeta42 dodecamer, J. Biol. Chem., 2010, 285, 37102–37110 CrossRef CAS PubMed.
- L. Xu, S. Shan, Y. Chen, X. Wang, R. Nussinov and B. Ma, Coupling of Zinc-Binding and Secondary Structure in Nonfibrillar Abeta40 Peptide Oligomerization, J. Chem. Inf. Model., 2015, 55, 1218–1230 CrossRef CAS PubMed.
- Y. Hori, T. Hashimoto, Y. Wakutani, K. Urakami, K. Nakashima, M. M. Condron, S. Tsubuki, T. C. Saido, D. B. Teplow and T. Iwatsubo, The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Abeta fibril formation without increasing protofibril formation, J. Biol. Chem., 2007, 282, 4916–4923 CrossRef CAS PubMed.
- M. Messa, L. Colombo, E. del Favero, L. Cantu, T. Stoilova, A. Cagnotto, A. Rossi, M. Morbin, G. Di Fede, F. Tagliavini and M. Salmona, The peculiar role of the A2V mutation in amyloid-beta (Abeta) 1-42 molecular assembly, J. Biol. Chem., 2014, 289, 24143–24152 CrossRef CAS PubMed.
- I. Benilova, R. Gallardo, A. A. Ungureanu, V. Castillo Cano, A. Snellinx, M. Ramakers, C. Bartic, F. Rousseau, J. Schymkowitz and B. De Strooper, The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-beta (Abeta) aggregation, J. Biol. Chem., 2014, 289, 30977–30989 CrossRef CAS PubMed.
- P. Das, B. Murray and G. Belfort, Alzheimer's protective A2T mutation changes the conformational landscape of the Abeta(1)(-)(4)(2) monomer differently than does the A2V mutation, Biophys. J., 2015, 108, 738–747 CrossRef CAS PubMed.
- P. H. Nguyen, B. Tarus and P. Derreumaux, Familial Alzheimer A2 V mutation reduces the intrinsic disorder and completely changes the free energy landscape of the Abeta1–28 monomer, J. Phys. Chem. B, 2014, 118, 501–510 CrossRef CAS PubMed.
- J. J. Wiltzius, S. A. Sievers, M. R. Sawaya, D. Cascio, D. Popov, C. Riekel and D. Eisenberg, Atomic structure of the cross-beta spine of islet amyloid polypeptide (amylin), Protein Sci., 2008, 17, 1467–1474 CrossRef CAS PubMed.
- R. Qi, Y. Luo, B. Ma, R. Nussinov and G. Wei, Conformational distribution and alpha-helix to beta-sheet transition of human amylin fragment dimer, Biomacromolecules, 2014, 15, 122–131 CrossRef CAS PubMed.
- S. Luca, W. M. Yau, R. Leapman and R. Tycko, Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR, Biochemistry, 2007, 46, 13505–13522 CrossRef CAS PubMed.
- V. Wineman-Fisher, Y. Atsmon-Raz and Y. Miller, Orientations of residues along the beta-arch of self-assembled amylin fibril-like structures lead to polymorphism, Biomacromolecules, 2015, 16, 156–165 CrossRef CAS PubMed.
- L. Kale, R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan and K. Schulten, NAMD2: greater scalability for parallel molecular dynamics, J. Comput. Phys., 1999, 151, 283–312 CrossRef CAS.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, Charmm – a Program for Macromolecular Energy, Minimization, and Dynamics Calculations, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, All-atom empirical potential for molecular modeling and dynamics studies of proteins, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- M. W. Mahoney and W. L. Jorgensen, A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions, J. Chem. Phys., 2000, 112, 8910–8922 CrossRef CAS.
- S. E. Feller, Y. H. Zhang, R. W. Pastor and B. R. Brooks, Constant-Pressure Molecular-Dynamics Simulation – the Langevin Piston Method, J. Chem. Phys., 1995, 103, 4613–4621 CrossRef CAS.
- K. Tu, D. J. Tobias and M. L. Klein, Constant pressure and temperature molecular dynamics simulation of a fully hydrated liquid crystal phase dipalmitoylphosphatidylcholine bilayer, Biophys. J., 1995, 69, 2558–2562 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, Particle Mesh Ewald – an N·log(N) Method for Ewald Sums in Large Systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, A Smooth Particle Mesh Ewald Method, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical-Integration of Cartesian Equations of Motion of a System with Constraints – Molecular-Dynamics of N-Alkanes, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- M. S. Lee, M. Feig, F. R. Salsbury and C. L. Brooks, New analytic approximation to the standard molecular volume definition and its application to generalized born calculations, J. Comput. Chem., 2003, 24, 1348–1356 CrossRef CAS PubMed.
- M. S. Lee, F. R. Salsbury and C. L. Brooks, Novel generalized Born methods, J. Chem. Phys., 2002, 116, 10606–10614 CrossRef CAS.
- Y. Miller, B. Ma and R. Nussinov, Synergistic interactions between repeats in tau protein and Abeta amyloids may be responsible for accelerated aggregation via polymorphic states, Biochemistry, 2011, 50, 5172–5181 CrossRef CAS PubMed.
- Y. Raz and Y. Miller, Interactions between Abeta and mutated Tau lead to polymorphism and induce aggregation of Abeta-mutated tau oligomeric complexes, PLoS One, 2013, 8, e73303 CAS.
- A. T. Petkova, W. M. Yau and R. Tycko, Experimental constraints on quaternary structure in Alzheimer's beta-amyloid fibrils, Biochemistry, 2006, 45, 498–512 CrossRef CAS PubMed.
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Abeta(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease, Nat. Struct. Mol. Biol., 2015, 22, 499–505 CAS.
- B. Ma and R. Nussinov, Selective molecular recognition in amyloid growth and transmission and cross-species barriers, J. Mol. Biol., 2012, 421, 172–184 CrossRef CAS PubMed.
- J. X. Lu, W. Qiang, W. M. Yau, C. D. Schwieters, S. C. Meredith and R. Tycko, Molecular structure of beta-amyloid fibrils in Alzheimer's disease brain tissue, Cell, 2013, 154, 1257–1268 CrossRef CAS PubMed.
- R. P. Nanga, J. R. Brender, S. Vivekanandan and A. Ramamoorthy, Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment, Biochim. Biophys. Acta, 2011, 1808, 2337–2342 CrossRef CAS PubMed.
- J. R. Brender, K. Hartman, R. P. Nanga, N. Popovych, R. de la Salud Bea, S. Vivekanandan, E. N. Marsh and A. Ramamoorthy, Role of zinc in human islet amyloid polypeptide aggregation, J. Am. Chem. Soc., 2010, 132, 8973–8983 CrossRef CAS PubMed.
- S. A. Kotler, J. R. Brender, S. Vivekanandan, Y. Suzuki, K. Yamamoto, M. Monette, J. Krishnamoorthy, P. Walsh, M. Cauble, M. M. Holl, E. N. Marsh and A. Ramamoorthy, High-resolution NMR characterization of low abundance oligomers of amyloid-beta without purification, Sci. Rep., 2015, 5, 11811 CrossRef PubMed.
- J. R. Brender, J. Krishnamoorthy, M. F. Sciacca, S. Vivekanandan, L. D'Urso, J. Chen, C. La Rosa and A. Ramamoorthy, Probing the sources of the apparent irreproducibility of amyloid formation: drastic changes in kinetics and a switch in mechanism due to micellelike oligomer formation at critical concentrations of IAPP, J. Phys. Chem. B, 2015, 119, 2886–2896 CrossRef CAS PubMed.
- X. Yu, Y. Luo, P. Dinkel, J. Zheng, G. Wei, M. Margittai, R. Nussinov and B. Ma, Cross-seeding and conformational selection between three- and four-repeat human Tau proteins, J. Biol. Chem., 2012, 287, 14950–14959 CrossRef CAS PubMed.
- S. H. Shim, R. Gupta, Y. L. Ling, D. B. Strasfeld, D. P. Raleigh and M. T. Zanni, Two-dimensional IR spectroscopy and isotope labeling defines the pathway of amyloid formation with residue-specific resolution, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6614–6619 CrossRef CAS PubMed.
- J. Nasica-Labouze, P. H. Nguyen, F. Sterpone, O. Berthoumieu, N. V. Buchete, S. Cote, A. De Simone, A. J. Doig, P. Faller, A. Garcia, A. Laio, M. S. Li, S. Melchionna, N. Mousseau, Y. Mu, A. Paravastu, S. Pasquali, D. J. Rosenman, B. Strodel, B. Tarus, J. H. Viles, T. Zhang, C. Wang and P. Derreumaux, Amyloid beta Protein and Alzheimer's Disease: When Computer Simulations Complement Experimental Studies, Chem. Rev., 2015, 115, 3518–3563 CrossRef CAS PubMed.
- M. F. Sciacca, D. Milardi, G. M. Messina, G. Marletta, J. R. Brender, A. Ramamoorthy and C. La Rosa, Cations as switches of amyloid-mediated membrane disruption mechanisms: calcium and IAPP, Biophys. J., 2013, 104, 173–184 CrossRef CAS PubMed.
- K. Hartman, J. R. Brender, K. Monde, A. Ono, M. L. Evans, N. Popovych, M. R. Chapman and A. Ramamoorthy, Bacterial curli protein promotes the conversion of PAP248-286 into the amyloid SEVI: cross-seeding of dissimilar amyloid sequences, PeerJ, 2013, 1, e5 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp03338a |
| This journal is © the Owner Societies 2016 |