
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrong electron donation induced differential nonradiative decay pathways for para and meta GFP chromophore analogues†
Tanmay
Chatterjee
,
Mrinal
Mandal
,
Venkatesh
Gude
,
Partha Pratim
Bag
and
Prasun K.
Mandal
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER), Mohanpur, Kolkata, West-Bengal 741246, India. E-mail: prasunchem@iiserkol.ac.in
First published on 25th June 2015
Abstract
Z–E Isomerisation because of rotation around the exocyclic double bond (known as the τ-twist) and not any other internal conversion has been reported to be the major nonradiative decay channel for non-hydroxylic unconstrained para and meta GFP chromophore analogues. The equation Φf + 2ΦZE = 1 has been shown to hold well for both para and meta GFP chromophore analogues. If the above equation holds true, then upon reducing the extent of Z–E isomerisation (ΦZE), the fluorescence quantum yield (Φf) should increase. To probe the above proposition two sets of non-hydroxylic unconstrained para and meta GFP chromophore analogues were synthesized. Quite interestingly by introducing the strongly electron donating –NEt2 group to the benzenic moiety these para and meta GFP chromophore analogues were shown to exhibit differential optical behaviour w.r.t. the extent of the solvatochromic shift, Φf, ΦZE, and τf. For the first time it has been shown that the well accepted equation Φf + 2ΦZE = 1 does not hold at all for these non-hydroxylic unconstrained meta analogues. Although ΦZE has been shown to be <10%, Φf is much lower than the expected near unity value for these meta analogues. After detailed investigation into the nonradiative excited state decay channel, contrary to literature reports, energy gap law governed internal conversion and not Z–E isomerisation was shown to be the major nonradiative decay channel for these meta analogues. Two models are put forward to understand the differential optical behaviour of these para and meta GFP chromophore analogues. Support from X-ray crystal structures, NMR experiments, and computational calculations has also been provided.
Introduction
Z–E Isomerisation because of rotation around the exocyclic double bond (τ-twist, see Chart 1), and not any other internal conversion has been reported to be the major nonradiative decay channel, because of which the fluorescence quantum yield (Φf) of the GFP chromophore (p-HBDI, see Chart 1) in normal solvents is reduced.1–8 This proposition has been extended to other unconstrained GFP chromophore analogues.9–12 For example both p-HBDI and p-ABDI (Chart 1) have a Z–E isomerisation quantum yield (ΦZE) ∼ 0.5 and Φf ∼ 0.0001.9 However, meta analogues of these two molecules i.e. m-HBDI and m-ABDI possess a Φf of 0.0023 and 0.16 respectively.9–11 Their excited state fluorescence lifetimes (τf) are also at least one order of magnitude higher than that of the para analogues.9–11 Thus, enhancement of Φf and τf could be achieved by structural modification. It has been shown that the extent of charge transfer is much higher for the meta analogues in comparison to the para analogues.9,11 Thus, differential optical behaviour (w.r.t. Φf and τf values) has been shown for meta and para analogues.9–12 This difference has been explained on the basis of the “meta effect” in analogy with the behaviour observed for stilbene derivatives.10–18 Both for para and meta analogues in aprotic solvents it has been shown that9,11,12| Φf + 2ΦZE = 1. | (1) |
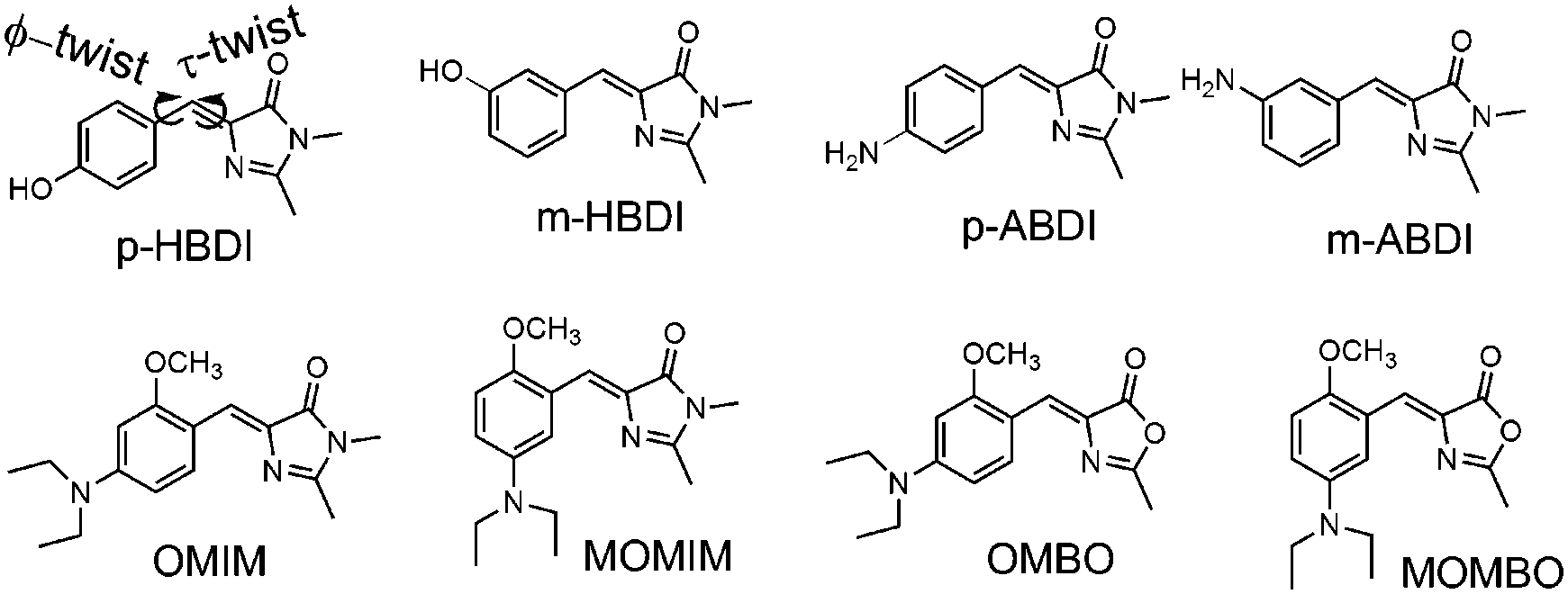 | ||
| Chart 1 Chemical structures of the different GFP chromophore analogues. | ||
Results and discussion
The extent of charge transfer reflected in the magnitude of solvatochromic shift has been observed to be much higher in meta analogues (141 nm for MOMIM and 176 nm for MOMBO from hexane to ACN) (see Fig. 1 for MOMIM and ESI† for MOMBO) in comparison to their para analogues (OMIM (∼30 nm) and OMBO (∼30 nm)). Similar observations for other meta analogues have been reported in literature.9,11 Also, Φf of MOMIM is much higher than OMIM. Moreover the fluorescence lifetime (τf) of the meta analogue MOMIM is ∼1000 times higher (2.78 ns in ACN) than the para analogue OMIM (1.9 ps in ACN) (see Fig. 1). Similar observations have been observed for MOMBO and OMBO (see ESI†). Thus, it is confirmed that the enhanced extent of charge transfer is reflected in high values of both Φf and τf in meta analogues in comparison to para analogues.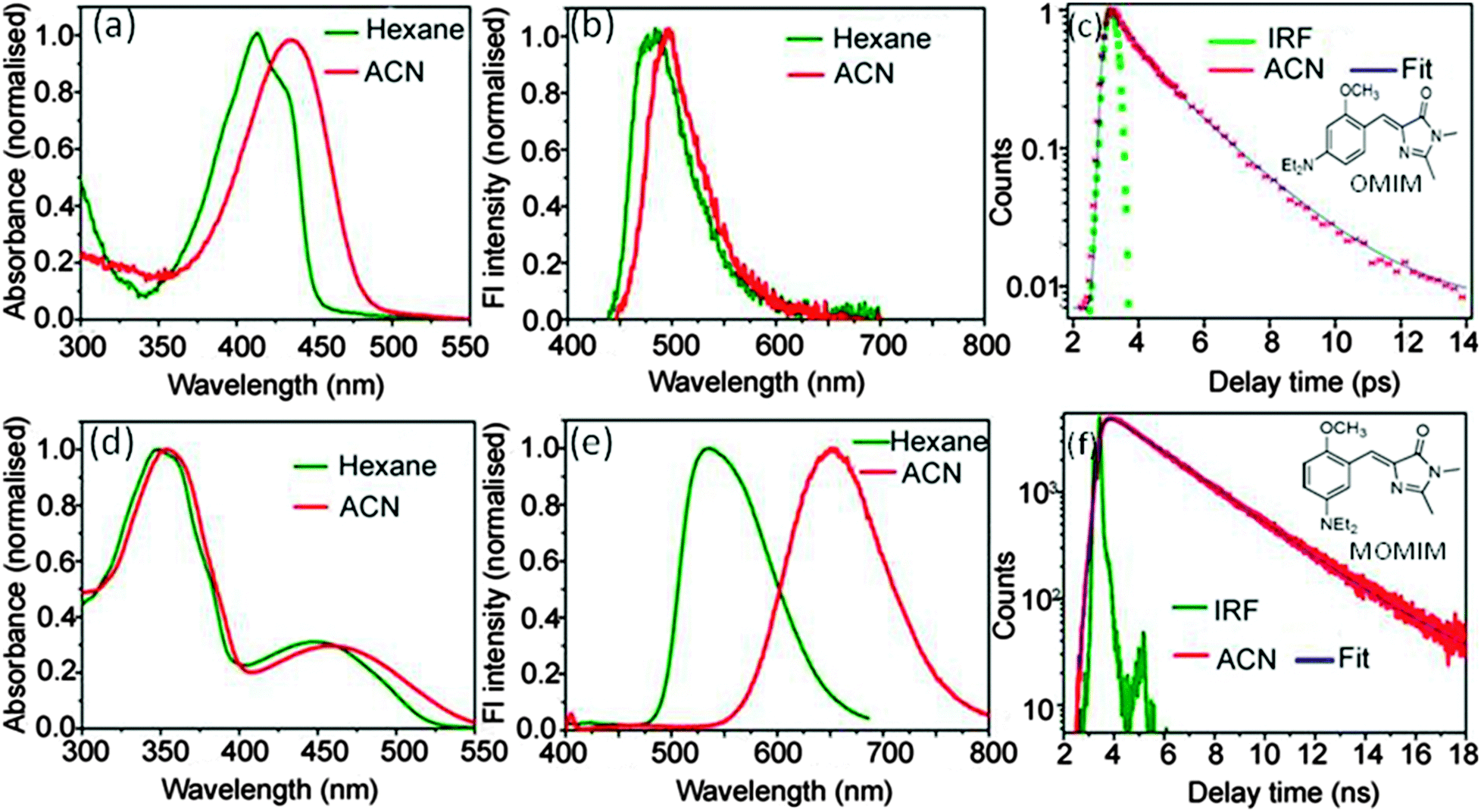 | ||
| Fig. 1 Differential steady state (absorption (left), emission (middle) and time resolved (right)) fluorescence behaviour of OMIM (para analogue, above) and MOMIM (meta analogue, below). | ||
As a next step it was necessary to calculate the ΦZE values of all four analogues in different solvents. Interestingly, unlike literature reports,9,11 the para and meta analogues exhibit quite different ΦZE values (see Fig. 2, Table 1 and ESI†). The ΦZE values for the para analogues (OMIM, OMBO) are ∼50%, however, the meta analogues (MOMIM and MOMBO) exhibit ΦZE values of ∼10% or less in all solvents. No significant solvent dependence (from CDCl3 to CD3CN, to DMSO-d6) was seen for either para or meta analogues. Thus, we can conclude that differential optical behaviour was observed between para and meta analogues (MOMIM and MOMBO) w.r.t. Φf, τf as well as ΦZE values.
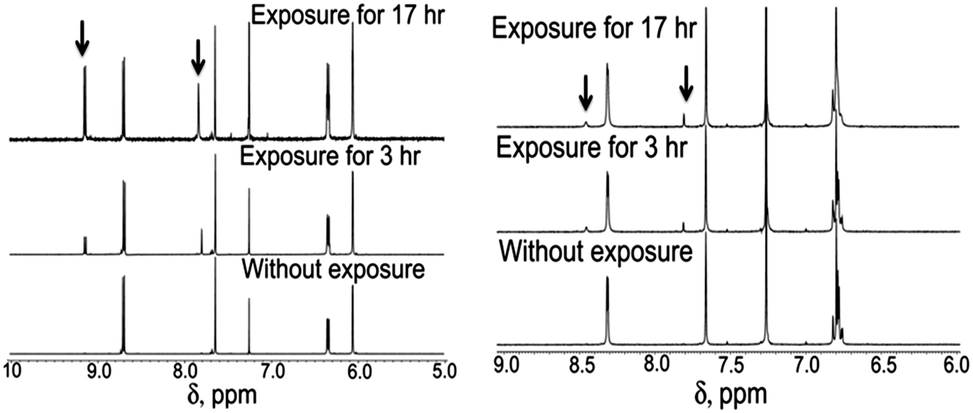 | ||
| Fig. 2 Differential Z–E isomerisation for OMIM (para analogue, left) and MOMIM (meta analogue, right). | ||
| Compound | Solvent | % of Z-isomera | % of E-isomerb | Φ f | Φ ZE | Φ f + 2ΦZE |
|---|---|---|---|---|---|---|
| a At the photo-stationary state (PSS). b NR: PSS could not be reached even after 17 h indicating significantly slower kinetics for meta analogues in comparison to para analogues. | ||||||
| OMIM | CDCl3 | 50 | 50 | <10−3 | 0.48 | 0.96 |
| CD3CN | 46 | 54 | <10−3 | 0.50 | 1.00 | |
| DMSO-d6 | 52 | 48 | <10−3 | 0.52 | 1.04 | |
| OMBO | CDCl3 | 38 | 62 | <10−3 | 0.60 | 1.20 |
| CD3CN | 48 | 52 | <10−3 | 0.50 | 1.00 | |
| DMSO-d6 | 45 | 55 | <10−3 | 0.55 | 1.10 | |
| MOMIM | CDCl3 | NR | NR | 0.04 | 0.08 | 0.20 |
| CD3CN | NR | NR | 0.006 | 0.05 | 0.11 | |
| DMSO-d6 | NR | NR | 0.006 | 0.04 | 0.09 | |
| MOMBO | CDCl3 | NR | NR | 0.02 | 0.13 | 0.28 |
| CD3CN | NR | NR | 0.002 | 0.04 | 0.09 | |
| DMSO-d6 | NR | NR | 0.002 | 0.06 | 0.13 | |
Moreover, as can be seen from Table 1, eqn (1) holds good for para analogues but the equation fails completely for the studied meta GFP chromophore analogues. Thus, an apparent correlation between reduced ΦZE and enhanced Φf could be drawn for meta analogues. However, had Z–E isomerisation been the major nonradiative decay channel for meta GFP chromophore analogues, its very small value (<10%) would have increased the Φf value to be close to unity. But this was not the case as Φf for MOMIM was only 12% in hexane and 0.6% in ACN. Thus, we can conclude that contrary to literature reports Z–E isomerisation is not the major nonradiative decay channel for these meta analogues. Thus it is necessary to know what kind of non-radiative decay is involved in the photophysics of these meta analogues.
In order to understand why eqn (1) fails completely for these meta GFP chromophore analogues and also to know what kind of major nonradiative decay could be taking place, we carried out structural analysis starting from ground state to excited state. We obtained single crystal X-ray structures of all four compounds. These crystal structures are depicted in Fig. 3. As can be seen from Fig. 3 all four compounds remain in a planar Z form with the –OMe group in the opposite direction to that of the imidazolidinone ring in the solid phase. As a next step, in order to know whether the same form persists in the solution phase, we carried out NMR measurements of all four compounds in different solvents. As can be seen from the NMR results (ESI†) even in the solution phase the ground state remains in the Z form. Thus, from single crystal X-ray and from NMR experiments we can conclude that the ground state structural form remains the same both in the solid and the solution phase for the meta analogues (and also for the para analogues). This means both meta and para analogues remain in same ground state structural form. Thus, we can make two important inferences: (i) that ground state structural heterogeneity does not exist; and (ii) that the differential optical behaviour is not due to ground state structural differences.
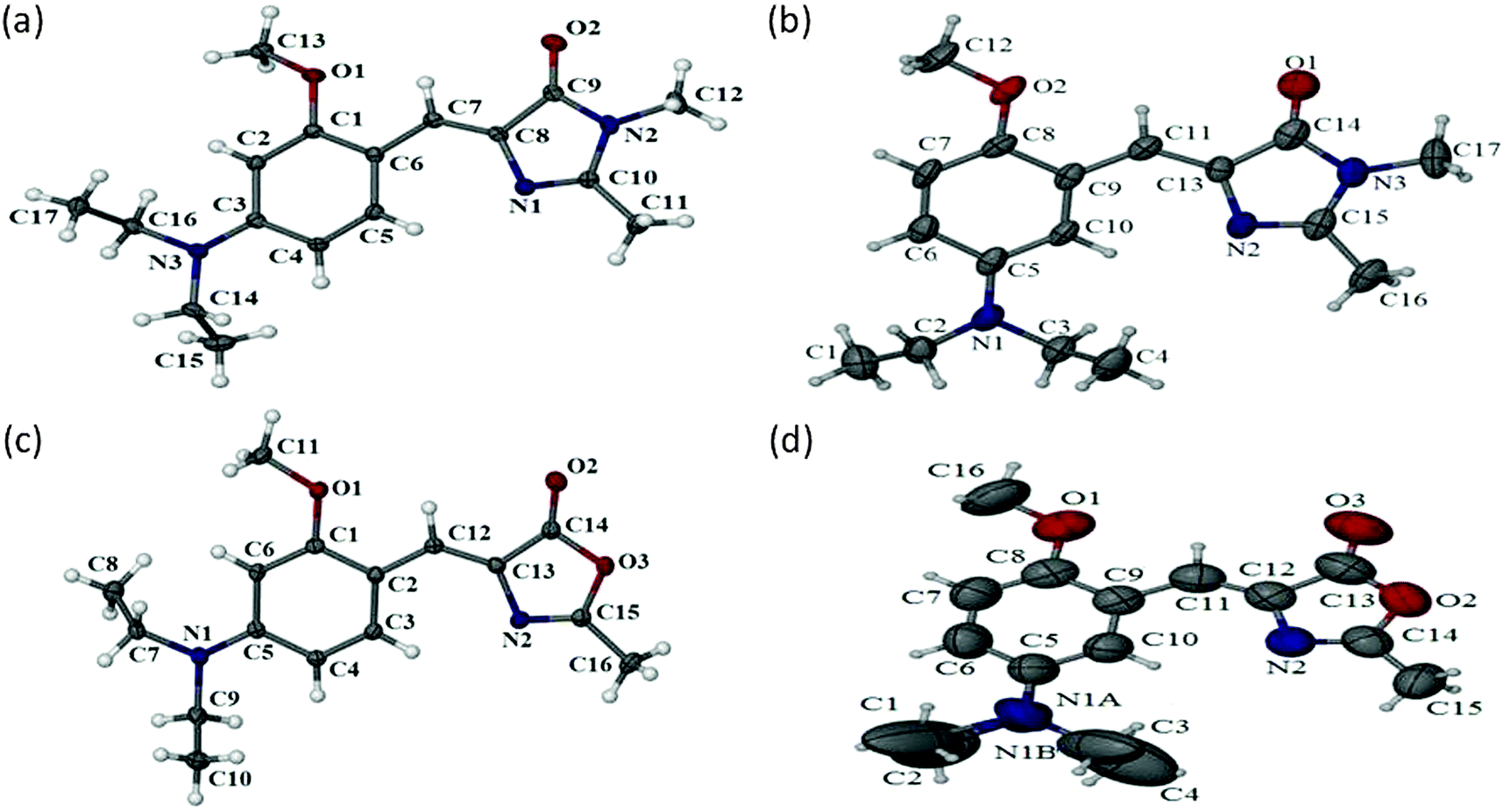 | ||
| Fig. 3 Single crystal X-ray structure of OMIM (a), MOMIM (b), OMBO (c), MOMBO (d). | ||
Ground state structural analysis could not provide significant clue regarding differential ΦZE values for para (∼50%) and meta analogues (<10%). Moreover, it is also not understood, why instead of having very low ΦZE values, the meta analogues have much lower (than unity) Φf. First, we tried to understand the reason behind the differential magnitude of ΦZE values for para (50%) and meta (<10%) analogues. Our experimental observation was that the extent of charge transfer (in the planar configuration) in the meta analogues is much higher than that of the para analogues. Computational calculation results (see Fig. 4) show that on going from the HOMO to the LUMO the extent of charge separation is much higher for meta analogues than para analogues. Hence, the emission spectra of the meta analogues should show bigger solvatochromic shifts than those of the para analogues. This is exactly what we obtained experimentally. Thus, the computational calculations support the experimental results.
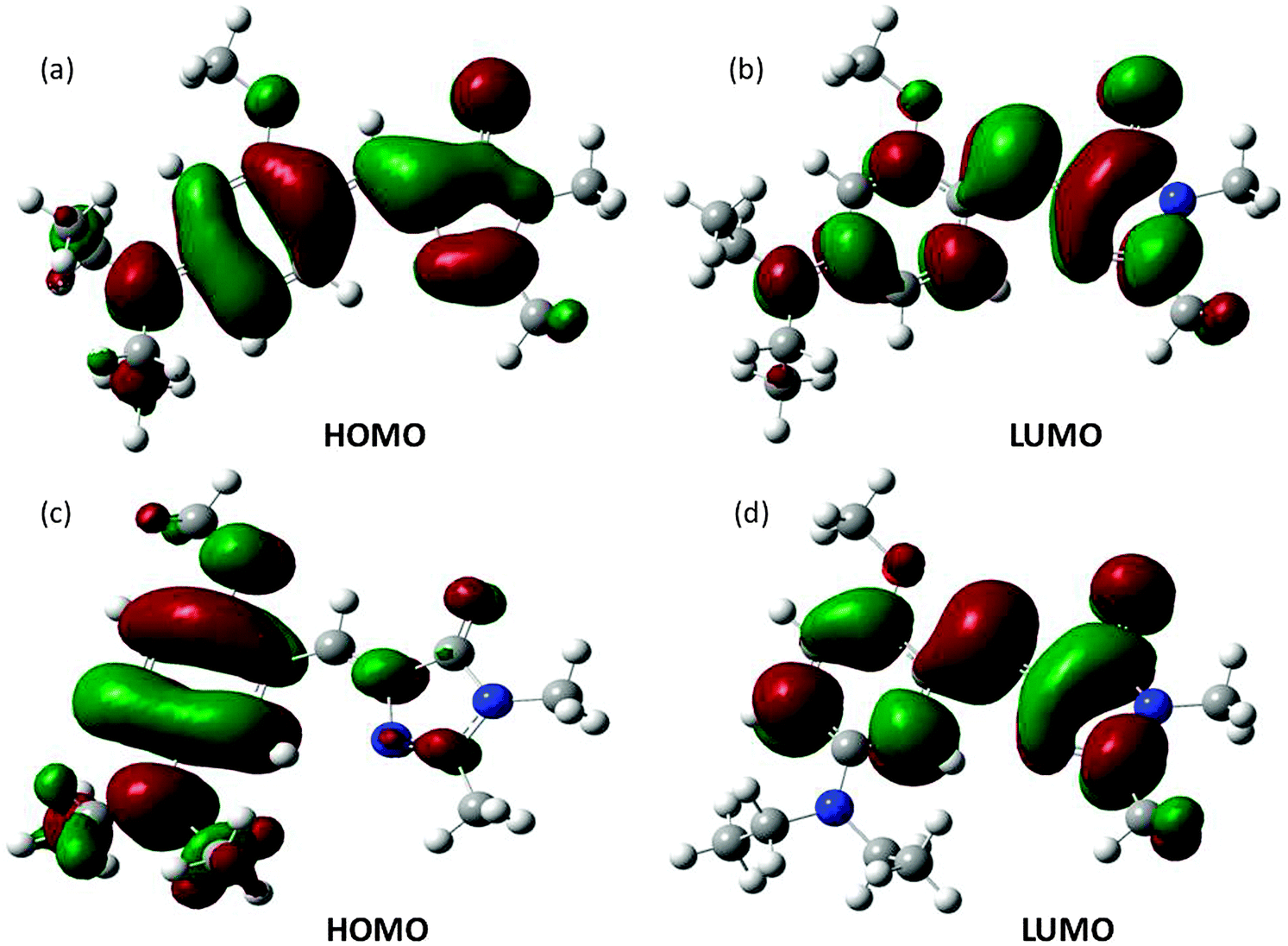 | ||
| Fig. 4 Frontier molecular orbitals of OMIM (a and b) and MOMIM (c and d) calculated at the B3LYP/6-31G++ level. | ||
After initial photo-excitation from the ground state Z form to the FC excited state, the excited state species could either go to the planar 1S* state or it could go to the τ-twisted 1P* state. Which process will take place depends on the height of the (excited state) barrier between the planar and twisted state. Lowering the energy of the 1P* twisted state relative to the planar 1S* state decreases the barrier height and vice versa. Applying a similar analogy to that proposed by Michl and Bonačić-Koutecký for simple alkenes, the 1P* state can be described as a combination of the biradical and charge transfer configuration.14,28,29
| ψ1P* = c1ψbiradical(A˙B˙) + c2ψCT(A+B−) + c3ψCT(A−B+) |
The stabilization of the 1S* state is much higher for meta analogues (as the polarity of the solvent increases), whereas according to the Michl and Bonačić-Koutecky model,28,29 stabilisation of the 1P* state is much higher for para analogues. Hence, for meta analogues the barrier height (from 1S* to 1P*) is much higher in comparison to para analogues (Scheme 1). Thus, meta analogues favour the planar rather than the twisted geometry in the excited state. This is why (solvatochromic) stabilisation of the planar 1S* state is much higher for meta analogues. Moreover, because of the strong electron donating –NEt2 group (in comparison to the –NH2 group) the solvatochromic shift (from hexane to ACN) of MOMIM is much larger (142 nm) in comparison to m-ABDI (83 nm) (see ESI†).
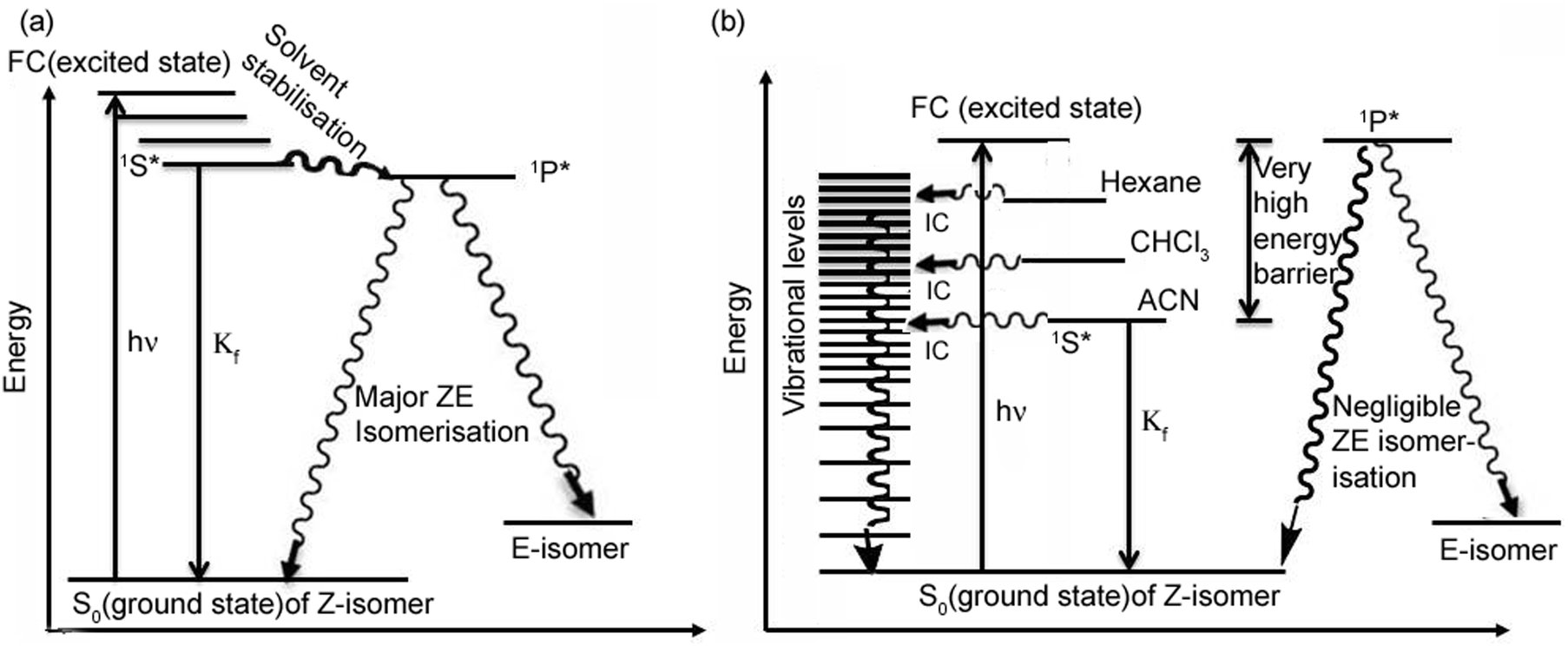 | ||
| Scheme 1 Photophysical processes of (a) para analogues (OMIM and OMBO) and (b) meta analogues (MOMIM and MOMBO). (Drawn in accordance with the Perrin–Jablonski diagram shown in ref. 30.) | ||
We have estimated the magnitude of stabilization of the charge transferred planar singlet state (1S*) of the meta analogues MOMIM and MOMBO to be 11.9 kcal mol−1 and 14.45 kcal mol−1 in comparison to that of para analogues (OMIM and OMBO respectively) from steady state emission in CHCl3. The same for 1P* state was not possible to calculate due to the non-fluorescent character of the state.
However, unlike meta analogues, for para analogues (Scheme 1a) the 1P* state is energetically lower than the FC or 1S* state. Thus, after initial excitation the molecule goes from the FC or 1S* state to the 1P* state for para analogues. Thus, the ΦZE value is much higher for para analogues (OMIM and OMBO) (ΦZE ∼ 50%). From the 1P* state the molecule relaxes to the ground electronic state in an ultrafast nonradiative fashion resulting in a much smaller Φf value (∼0.0001) as well as τf value (femto–picosecond) for para analogues. Whereas for meta analogues, since the energy of the 1P* state is comparable to the FC state or (much) higher than the 1S* state, there is less probability that the meta analogues go from the 1S* state to the 1P* state. Hence, the ΦZE value is much reduced (ΦZE < 10%) for meta analogues (MOMIM and MOMBO). Thus, for meta analogues, molecule decays from 1S* state, significantly radiatively resulting in much higher Φf value (∼0.12 in hexane) as well as high τf value (5.22 ns in hexane).
Schemes somewhat similar to Scheme 1 have been reported previously.9,11 However, that model can't explain the experimental results we obtained. Moreover, though the twisted 1P* state was conjectured to be much higher in energy than that of the planar 1S* state in m-ABDI, still much higher Z–E isomerisation (ΦZE = 0.45) in aprotic solvents has been reported.11 As Φf + 2ΦZE = 1 holds for both m-ABDI and p-ABDI in aprotic solvents, it was concluded that Z–E isomerisation is the only nonradiative decay channel available to both meta and para compounds in spite of the slow isomerisation kinetics of the former.9,11,12 Similar result was noted for m-HBDI by Tolbert et al. in nonaqueous solvents.10
Since the two sets of para and meta analogues exhibit widely different ΦZE values, quite contrary to previously reported results, Z–E isomerisation is not the major nonradiative decay channel for meta analogues in aprotic solvents and thus the reported models could not explain our experimental results. Thus, two different models need to be provided to understand the differential optical behaviour for para and meta analogues. These differential observations have been outlined in Scheme 1. From the scheme we could understand the electronic reason for such a reduced ΦZE value for meta analogues in comparison to the high ΦZE values for para analogues.
However, from Φf + 2ΦZE = 1, as the ΦZE value is <10%, the Φf value should have been close to 80% for the meta analogues. But the experimental observation says that the Φf value is at best 12% (in hexane). This means there exists another non-radiative decay channel for meta analogues. Thus, it is necessary to know what kind of nonradiative decay channels could possibly be the reason for deviation of Φf from its near unity value for the meta analogues. Because of enhanced charge transfer, the emission maximum of MOMIM is considerably red shifted (547 nm in hexane and 688 nm in ACN) in comparison to the other meta analogues (e.g. m-ABDI has an emission maximum of 495 nm in hexane and 578 nm in ACN) reported in the literature.9,11 The emission maxima of MOMIM and MOMBO are in the red whereas those for OMIM or OMBO as well as other reported meta analogues (m-HBDI and m-ABDI) are in the green region. Thus, the excited states of MOMIM and MOMBO are much more energetically stabilized in comparison to OMIM, OMBO and m-HBDI and m-ABDI. Thus, the energy difference between the stabilized excited state and the FC ground state is much smaller in case of MOMIM and MOMBO. Thus, following the energy gap law, nonradiative emission i.e. internal conversion from the stabilized excited state to the FC ground state becomes highly significant in the cases of MOMIM and MOMBO.31–35
However, the same is not so significant for para analogues or other meta analogues like m-ABDI.9,11,12 Thus, because of the facile energy gap law governed internal conversion, which is a nonradiative decay process, the Φf value deviates greatly from the expected 80% value. As the polarity of the solvent increases the energy gap between the stabilized/solvated excited state and ground state decreases and hence the nonradiative internal conversion becomes more facile. This also explains why Φf decreases with increasing polarity in the case of MOMIM (0.12 in hexane to 0.006 in ACN) and MOMBO (0.10 in hexane to 0.002 in ACN). On going from hexane to acetonitrile, the radiative decay rate constant (Kf = Φf/τf) decreases from 0.21 × 108 s−1 to 0.021 × 108 s−1 and the nonradiative decay rate constant (Knr = (1 − Φf)/τf) increases from 1.74 × 108 s−1 to 3.57 × 108 s−1, respectively, for MOMIM. Thus, the large extent of the charge transfer (strong ‘meta effect’) induced barrier in twisting makes Z–E isomerisation a minor nonradiative decay pathway for MOMIM and MOMBO. Rather, it is the energy gap law governed facile internal conversion which becomes the major nonradiative decay pathway for MOMIM and MOMBO. This observation is in complete contrast to that of the para analogues and the reported meta analogues (m-ABDI and m-HBDI) for which Z–E isomerisation is the only major nonradiative decay channel and for which any other internal conversion has been reported to be not at all important in aprotic solvents.
Conclusions
In conclusion, we synthesized two sets of non-hydroxylic unconstrained para and meta GFP chromophore analogues which exhibited differential optical properties in aprotic solvents w.r.t. Φf, ΦZE, and τf values. For the meta analogues because of the strong “meta effect” the extent of charge transfer was much greater in comparison to the para analogues. Φf, and τf values of the meta analogues are ∼1000 times higher than those of the para analogues. ΦZE values for para analogues are ∼50% and for meta analogues are <10%. The well accepted equation Φf + 2ΦZE = 1 holds good for para analogues but fails completely for meta analogues. In contrast to existing literature reports, Z–E isomerization has been shown not to be the major nonradiative decay channel for meta analogues. Rather, because of enhanced charge transfer and hence a reduced energy gap between the stabilised excited state and ground state, the energy gap law governed facile internal conversion was shown to be the major nonradiative decay channel for meta analogues. These observations for meta analogues are in stark contrast to the existing literature of unconstrained non-hydroxylic GFP chromophore analogues. Two models have been provided which successfully explain the differential optical behaviour of para and meta GFP chromophore analogues. Thus, by structural variation the extent of charge transfer can be modulated and thus the nature of optical behaviour as well as the pathway of nonradiative excited state decay could be controlled.Experimental details
Calculation of Z–E isomerisation efficiency
To measure the Z–E isomerisation quantum yield (ΦZE), solutions of low (mM) concentration range were used and irradiated with 370 nm light (8 W mercury lamp) without applying any special filter. The irradiation was followed by 1H NMR at different time intervals. Since distinctive 1H NMR signals are obtained for Z and E isomers, integration of the NMR peak corresponding to each isomer allowed us to know the isomer ratio at a certain time of irradiation. p-HBDI served as the reference standard (ΦZE = 0.48 in ACN).9 The isomerisation quantum yield was calculated with the following equation:9,36where C is the concentration of the substrate, P is the amount (%) of the initial Z-isomer that undergoes conversion to the E-isomer after irradiation, V is the volume of the solution, t is the irradiation time, and the subscripts r and s stand for the reference standard (p-HBDI) and the substrate (OMIM, OMBO, MOMIM and MOMBO) respectively. P was determined from the integrated intensity of the 1H NMR peak for the E-isomer. The sample of ∼10−3 M concentration was prepared in an NMR tube (Sigma Aldrich) and kept in a UV chamber for irradiation with a 370 nm light source. The back isomerisation in the ground state was also monitored by NMR. It was observed that back isomerisation after exposure was possible in the presence of light for OMIM and OMBO but not for MOMIM or MOMBO. However, in the dark the ground state back isomerisation from the E-isomer to the initial Z-isomer did not happen over a duration of three days for either of the derivatives. So, to keep ground state back isomerisation out of the calculation of ΦZE, the NMR tube was wrapped with aluminium foil after removal from the irradiation chamber. For the ΦZE value calculation, the sample was irradiated for a short time so that the irradiated sample containing a mixture of E and Z isomers was far from reaching a photo-stationary state (PSS).
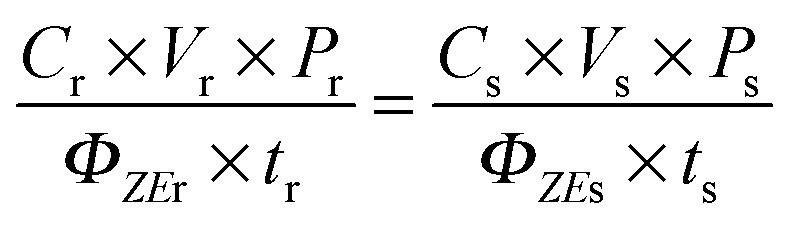
Abbreviations used
Acronyms like p-HBDI, m-HBDI, p-ABDI, m-ABDI have previously been used in the literature.9,10| GFP | green fluorescence protein |
| OMIM | o-methoxy imidazolidinone |
| OMBO | o-methoxybenzoxazolidinone |
| MOMIM | meta diethylamino o-methoxy imidazolidinone |
| MOMBO | meta diethylamino o-methoxy benzoxazolidinone |
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
PKM thanks IISER-Kolkata for financial help and instrumental facilities. Support from the Fast-Track Project (SR/FT/CS-52/2011) of DST-India is gratefully acknowledged. PKM acknowledges Prof. P. Ramamurthy (NCUFP) for his help in femtosecond decay measurements. TC, and PPB thank CSIR, MM and VG thank IISER-Kolkata for their respective Fellowship.References
- W. Weber, V. Helms, J. A. McCammon and P. W. Langhoff, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6177–6182 CrossRef CAS.
- A. Follenius-Wund, M. Bourotte, M. Schmitt, F. Iyice, H. Lami, J. J. Bourguignon, J. Haiech and C. Pigault, Biophys. J., 2003, 85, 1839–1850 CrossRef CAS.
- M. E. Martin, F. Negri and M. Olivucci, J. Am. Chem. Soc., 2004, 126, 5452–5464 CrossRef CAS PubMed.
- P. Altoe, F. Bernardi, M. Garavelli, G. Orlandi and F. Negri, J. Am. Chem. Soc., 2005, 127, 3952–3963 CrossRef CAS PubMed.
- S. Olsen and S. C. Smith, J. Am. Chem. Soc., 2007, 129, 2054–2065 CrossRef CAS PubMed.
- S. A. Olsen, J. Chem. Theory Comput., 2010, 6, 1089–1103 CrossRef CAS.
- G. J. Huang and J. S. Yang, Chem. – Asian J., 2010, 5, 2075–2085 CrossRef CAS PubMed.
- A. Baldridge, S. R. Samanta, N. Jayaraj, V. Ramamurthy and L. M. Tolbert, J. Am. Chem. Soc., 2010, 132, 1498–1499 CrossRef CAS PubMed.
- J.-S. Yang, G.-J. Huang, Y.-H. Liu and S.-M. Peng, Chem. Commun., 2008, 1344–1346 RSC.
- J. Dong, K. M. Solntsev, O. Poizat and L. M. Tolbert, J. Am. Chem. Soc., 2007, 129, 10084–10085 CrossRef CAS PubMed.
- C. W. Cheng, G. J. Huang, H. Y. Hsu, C. Prabhakar, Y. P. Lee, E. W. G. Diau and J. S. Yang, J. Phys. Chem. B, 2013, 117, 2705–2716 CrossRef CAS PubMed.
- G. J. Huang, J. H. Ho, C. Prabhakar, Y. H. Liu, S. M. Peng and J. S. Yang, Org. Lett., 2012, 14, 5034–5037 CrossRef CAS PubMed.
- E. M. Crompton and F. D. Lewis, Photochem. Photobiol. Sci., 2004, 3, 660–668 CAS.
- F. D. Lewis, S. R. Kalgutkar and J. S. Yang, J. Am. Chem. Soc., 1999, 121, 12045–12053 CrossRef CAS.
- H. K. Sinha and K. Yates, J. Am. Chem. Soc., 1991, 113, 6062–6067 CrossRef CAS.
- H. E. Zimmerman, J. Am. Chem. Soc., 1995, 117, 8988–8991 CrossRef CAS.
- H. E. Zimmerman, J. Phys. Chem. A, 1998, 102, 5616–5621 CrossRef CAS.
- K. M. Solntsev, O. Poizat, J. Dong, J. Rehault, Y. Lou, C. Burda and L. M. Tolbert, J. Phys. Chem. B, 2008, 112, 2700–2711 CrossRef CAS PubMed.
- L. Wu and K. Burgess, J. Am. Chem. Soc., 2008, 130, 4089–4096 CrossRef CAS PubMed.
- A. Baldridge, K. M. Solntsev, C. Song, T. Tanioka, J. Kowalik, K. Hardcastle and L. M. Tolbert, Chem. Commun., 2010, 46, 5686–5688 RSC.
- M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Slipchenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, J. Am. Chem. Soc., 2012, 134, 6025–6032 CrossRef CAS PubMed.
- C. C. Hsieh, P. T. Chou, C. W. Shih, W. T. Chuang, M. W. Chung, J. Lee and T. Joo, J. Am. Chem. Soc., 2011, 133, 2932–2943 CrossRef CAS PubMed.
- K. P. Kent and S. G. Boxer, J. Am. Chem. Soc., 2011, 133, 4046–4052 CrossRef CAS PubMed.
- K. Do and S. G. Boxer, J. Am. Chem. Soc., 2011, 133, 18078–18081 CrossRef CAS PubMed.
- A. Baldridge, S. Feng, Y. T. Chang and L. M. Tolbert, ACS Comb. Sci., 2011, 13, 214–217 CrossRef CAS PubMed.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 CrossRef CAS PubMed.
- T. Chatterjee, D. Roy, A. Das, A. Ghosh, P. P. Bag and P. K. Mandal, RSC Adv., 2013, 3, 24021–24024 RSC.
- V. Bonačić-Koutecký, J. Köhler and J. Michl, Chem. Phys. Lett., 1984, 104, 440–443 CrossRef.
- V. Bonačić-Koutecký and J. Michl, J. Am. Chem. Soc., 1985, 107, 1765–1766 CrossRef.
- B. Valeur, Molecular Fluorescence – Principles and Applications, WILEY-VCH Verlag GmbH, Weinheim, 2002 Search PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern molecular photochemistry of organic molecules, University Science Books, Sausalito, CA, 2010 Search PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS PubMed.
- C. Liu, K. C. Tang, H. Zhang, H. A. Pan, J. Hua, B. Li and P. T. Chou, J. Phys. Chem. A, 2012, 116, 12339–16345 CrossRef CAS PubMed.
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
- M. Bixon and J. Jortner, J. Phys. Chem., 1994, 98, 1289–1294 CrossRef.
- G. J. Huang, C. W. Cheng, H. Y. Hsu, C. Prabhakar, Y. P. Lee, G. E. W. Diau and J. S. Yang, J. Phys. Chem. B, 2013, 117, 2695–2704 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis and characterization of GFP chromophore analogues, additional NMR data and stack plot, crystallographic table, additional steady state and time resolved spectroscopic data, etc. CCDC 1001355 and 1001357–1001359. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cp03086b |
| This journal is © the Owner Societies 2015 |