
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the formation of cyclopentadiene in the C3H5˙ + C2H2 reaction†
Jordy
Bouwman
*a,
Andras
Bodi
b,
Jos
Oomens
ac and
Patrick
Hemberger
b
aRadboud University, Institute for Molecules and Materials, FELIX Laboratory, Toernooiveld 7c, NL-6525 ED Nijmegen, The Netherlands. E-mail: j.bouwman@science.ru.nl
bMolecular Dynamics Group, Paul Scherrer Institut, 5232 Villigen, Switzerland
cvan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, NL-1098 XH Amsterdam, The Netherlands
First published on 12th June 2015
Abstract
The reaction between the allyl radical (C3H5˙) and acetylene (C2H2) in a heated microtubular reactor has been studied at the VUV beamline of the Swiss Light Source. The reaction products are sampled from the reactor and identified by their photoion mass-selected threshold photoelectron spectra (ms-TPES) by means of imaging photoelectron photoion coincidence spectroscopy. Cyclopentadiene is identified as the sole reaction product by comparison of the measured photoelectron spectrum with that of cyclopentadiene. With the help of quantum-chemical computations of the C5H7 potential energy surface, the C2H2 + C3H5˙ association reaction is confirmed to be the rate determining step, after which H-elimination to form C5H6 is prompt in the absence of re-thermalization at low pressures. The formation of cyclopentadiene as the sole product from the allyl + acetylene reaction offers a direct path to the formation of cyclic hydrocarbons under combustion relevant conditions. Subsequent reactions of cyclopentadiene may lead to the formation of the smallest polycyclic aromatic molecule, naphthalene.
1 Introduction
Combustion processes are a major source of polycyclic aromatic hydrocarbons (PAH) and soot that are known to cause adverse health effects. Numerous PAH species formed in combustion processes have mutagenic and carcinogenic properties.1 Fine particulate matter, such as soot particles, has been linked to a variety of heart and lung diseases and their presence in the environment is thought to be responsible for more than a million premature deaths annually.2–4 Furthermore, soot plays an important role in global warming, because it strongly absorbs sunlight and affects cloud formation.5,6 A thorough understanding of the formation paths of PAHs and soot in combustion environments is essential in order to reduce their emissions.Resonantly stabilized radicals (RSRs) are considered important precursors in the formation of PAHs and soot.7–9 RSRs have delocalized unpaired electrons, leading to multiple resonance structures for the same atomic configuration. This results in an increased thermodynamic stability as well as reduced reaction rates with oxygen compared with non-resonantly stabilized radicals. As a consequence, the concentration of RSRs in combustion environments is increased compared to that of non-stabilized radicals. It is now widely believed that the main reaction path leading to the first aromatic ring species proceeds via the condensation of two resonantly stabilized propargyl (C3H3˙) radicals.10,11 However, this reaction alone cannot explain the formation rate of aromatic molecules, and other reactions, such as propargyl with acetylene and reactions involving other RSRs such as allyl (C3H5˙), also have to be considered.12–16
After the first ring has formed, PAH growth can proceed via subsequent addition of C2H2 groups in the hydrogen abstraction carbon addition (HACA) mechanism, or via the addition of larger hydrocarbon species, such as phenyl radicals.17–19 The products formed from the self-reaction of resonantly stabilized phenylpropargyl radicals have been investigated in a double resonance study.20 Reactions of the resonantly stabilized cyclopentadienyl (c-C5H5˙, CPDyl) radical with a variety of unsaturated hydrocarbons have gained increased attention.21,22 Based on theory and experiments, it has been suggested that the reaction between two CPDyl units yields the smallest polycyclic aromatic species, naphthalene.23–26 One possible path to form cyclopentadienyl is the allyl + acetylene reaction, followed by hydrogen abstraction from the thus formed cyclopentadiene.27 Subsequent reactions of CPDyl may be crucial in the production of large aromatic molecules that lead to soot formation in combustion environments.28,29
Nohara and Sakai30 studied the allyl + acetylene reaction in a flow tube at atmospheric pressure and employed gas chromatography mass spectrometry, flame ionization detectors and thermal conductivity detectors to identify the products. They determined the rate of the reaction relative to the allyl + allyl reaction rate by measuring the rate of formation of cyclopentadiene, C5H6 at temperatures ranging from 430 to 510 °C.30 Products were detected off-line, rather than sampled directly from the combustion environment and the computed energetics data for the reaction path were limited at the time. They concluded that the rate-limiting step was either cyclization to C5H7 or hydrogen loss to form C5H6. Because short-lived intermediates may remain undetected,31 off-line detection may also hamper the identification of the cyclopentadiene formation mechanism. In a different study, Isemer and Luther32 investigated the allyl + acetylene reaction in a shock tube. They employed time-resolved UV absorption spectroscopy to determine the reaction rate at combustion relevant temperatures (1060–1320 K). From analysis of the congested UV spectra it was suggested that under these experimental conditions, 95% of the products is in the form of cyclopentadiene rather than the stabilized C5H7 intermediate.
The allyl + acetylene reaction has also been studied computationally. First, Dean33 reported a potential energy surface (PES) for the reaction and concluded that the reaction is an important source of cyclopentadiene. Later, Moskaleva and Lin34 studied the kinetics and PES of the C5H6 + H reaction. It could also be inferred from their C5H7 PES that cyclopentadiene can be formed from the allyl + C2H2 reaction.
Here we aim to identify the allyl + acetylene reaction mechanism and products unambiguously at a combustion relevant temperature. The reaction mixture is sampled directly from a pyrolysis microtubular reactor, and the constituents are identified isomer-selectively by threshold photoelectron spectroscopy in coincidence with photoion mass spectrometry using synchrotron radiation. Furthermore, the reaction is explored by computational chemistry and RRKM theory is applied to investigate whether thermalization can contribute to the stabilization of intermediates.
2 Experimental and computational methods
The reaction of the allyl radical with acetylene was studied in a pyrolysis tube reactor coupled to the iPEPICO instrument at the vacuum ultraviolet (VUV) beamline of the Swiss Light Source at Paul Scherrer Institute (PSI) in Villigen, Switzerland.35–37 Bending magnet synchrotron radiation was collimated, dispersed by a 150 grooves per mm grating in grazing incidence, and focused at the exit slit, which resulted in an energy resolution of about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1500. For the experiments reported here, the photon energy was tuned from 8.3 to 10.2 eV in step sizes of 20 or 25 meV. Higher order radiation was suppressed by a MgF2 window in this energy range.
:1500. For the experiments reported here, the photon energy was tuned from 8.3 to 10.2 eV in step sizes of 20 or 25 meV. Higher order radiation was suppressed by a MgF2 window in this energy range.
Allyl iodide was used as a radical precursor, as it was found to be a clean and efficient source of allyl radicals.38 A glass bubbler containing allyl iodide (98%, Sigma-Aldrich) was suspended in ice water to stabilize its vapor pressure at ∼9.5 mbar. Acetylene (>99.5% stabilized with acetone, Carbagas) or argon (99.999%, Messer) was bubbled through allyl iodide at a pressure of 2.5 bar to pick up the vapor. The resulting gas mixture of 0.5% allyl iodide in C2H2 (or Ar) was expanded through a 100 μm pinhole into the pyrolysis tube reactor.
The microtubular reactor consists of a 3 cm long SiC tube that is resistively heated by a DC power supply. The pressure before the gas flow reducing 100 μm pinhole is 0.3 bar and the pressure after the pinhole can be estimated to be several tens of mbars. The pressure at the outlet of the reactor tube is 5 × 10−4 mbar. The temperature of the reactor is monitored by a “type C” thermocouple and is varied from room temperature to 1073 K. A ±100 K uncertainty is assumed in the temperature reading due to the imperfect contact of the reactor to the thermocouple. Note that this temperature is measured on the outside of the tube reactor, and the absolute temperature inside is different. This temperature can be estimated from data reported in a recent modeling study by Guan et al.39 and is found to be around 700 K when the reactor surface is at 850 K. The residence time can also be estimated from the data in this study and is on the order of 10–100 μs.
The products exit the pyrolysis reactor and are subsequently skimmed by a 1 mm skimmer, through which the molecular beam enters the interaction region of the iPEPICO instrument. Typical pressures in the iPEPICO detection chamber are on the order of 10−6 mbar. Here, the molecular beam containing the reaction products is intersected by the ionizing VUV radiation from the beamline. Electrons are velocity map imaged on a RoentDek delay line detector and provide the start signal for the cation time-of-flight measurement (TOF). The ions are extracted in the same, constant 120 V cm−1 field and space focused at a Jordan-TOF MCP detector at the end of a linear time-of-flight (TOF) mass spectrometer. At an extraction field of 120 V cm−1, electrons with a maximum energy of 1 eV are imaged onto the detector.
The photoelectron photoion coincidence mass spectra of the species exiting the microtubular reactor can be plotted using all electrons detected as coincidence start signal, irrespective of their energy. From here on, these are referred to as all-electron mass spectra. On the other hand, photoion mass selected threshold photoelectron spectra (ms-TPES) of the precursors, reactants and reactions products are obtained by taking into account only coincidences between electrons with less than 10 meV kinetic energy and cations. This is achieved by considering only the threshold electrons, which are imaged onto a small area in the center of the position-sensitive detector. However, kinetic energy electrons may also strike the center of the detector if their momentum is parallel with the extraction field, which results in “hot electron” contamination of the center signal. The hot electron contamination can be well approximated based on a ring around the center spot, and is subtracted from the center signal to obtain the TPES.40,41 The isomer specificity of the ms-TPES is generally much better than that of photoionization efficiency curves, in which the total ion yield in a specific mass channel is recorded as a function of photon energy.42,43
Quantum chemical computations have been performed using the Gaussian09 program44 to obtain insights into the C5H7 potential energy surface. Structures of intermediates, transition states, and products are located by identifying the internal coordinate that corresponds to the reaction coordinate in Question – a bond length most of the time – and scanning it while optimizing the rest of the coordinates at the B3LYP/6-311++G(d,p) level of theory. The minimum energy and transition state structures are then subjected to a CBS-QB3 calculation, in which their geometry is re-optimized. Accurate single point energies of the located products, transition states, and intermediates are thus obtained at the CBS-QB3 level of theory.45,46
3 Results and discussion
Mass spectra of the species sampled from the pyrolysis reactor have been recorded on the iPEPICO machine. Thereafter, photoion mass selected threshold photoelectron spectra (ms-TPES) have been recorded to identify the reaction products, and quantum chemical computations are also presented that give insights into the potential energy surface.3.1 Time-of-flight mass spectra
Mass spectra have been recorded at a set of photon energies and temperatures to study the product formation in the microtubular pyrolysis reactor. Products formed from the pyrolysis of each of the starting materials (C3H5I and C2H2) alone are identified in control experiments, which are presented in the ESI† along with a discussion reviewing several relevant references.31,41,47–56 Experiments on the pyrolysis of C3H5I in acetylene leading to the reaction products under investigation are discussed here.The time-of-flight mass spectra in Fig. 1 show the species resulting from the pyrolysis of a mixture of 0.5% allyl iodide in C2H2 containing traces of acetone at temperatures ranging from 582 to 1068 K and recorded at a photon energy of 9.5 eV. Most reaction products are also found in the control pyrolysis experiments involving only one of the reactants C3H5I or C2H2 (see ESI†). As the peak area ratios are unchanged with respect to the reference pyrolysis measurements, we assume that these species can be ruled out as significant reaction products of the allyl + acetylene reaction. Mass spectra recorded at 9.5 eV for pyrolysis of C3H5I in Ar (823 K), for C2H2 with traces of acetone (873 K) and for C2H2 with traces of acetone in the presence of allyl iodide (873 K) are shown in Fig. 2 for comparison.
 | ||
| Fig. 1 All-electron mass spectra of the pyrolysis of 0.5% allyl iodide in acetylene at temperatures of 582, 683, 777, 873, 981, and 1068 K, recorded at a photon energy of 9.5 eV. | ||
 | ||
| Fig. 2 Comparison of mass spectra recorded for the pyrolysis of C3H5I in Ar at 823 K (bottom), pyrolysis of C2H2 at 873 K (middle) and pyrolysis of C2H2 in presence of C3H5I at 873 K (top). | ||
From Fig. 2 it is apparent that a strong signal arises at m/z = 66 when C2H2 and C3H5I are co-expanded into the heated reactor. This product corresponds to C5H6, which is formed via an addition-elimination reaction:
| C3H5˙ + C2H2 → C5H7˙‡ → C5H6 + H˙ |





3.2 Mass selected threshold photoelectron spectrum
A photoion mass selected threshold photoelectron spectrum of the C5H6 channel has been recorded at a temperature of ∼800 K in order to shed light on the identity of the product. The resulting ms-TPES is displayed in Fig. 3 together with the photoelectron spectrum of cyclopentadiene recorded in a separate unpublished set of experiments60 recorded on the low pressure flat flame burner apparatus coupled to the iPEPICO machine at the SLS.61 The strong transition observed at 8.57 eV matches the cyclopentadiene ionization energy of 8.57 eV reported in the literature very well.58 Hot bands of cyclopentadiene that are populated thermally in the pyrolysis reactor and by excess energy from the reaction cause the signal level to be non-zero before the sharp ionization onset.62 Cylopentadiene is by far the most stable reaction product, but the geometry relaxation upon ionization is relatively small in the other isomers as well (Table 1). Therefore, the origin transition is expected to have large intensity for all C5H6 isomers. It is conceivable that the origin transition of 1,2,4-pentatriene is hidden in the vibrational progression of the ground state band of cyclopentadiene at 8.9 eV. However, as will be shown later, the transition state to 1,2,4-pentatriene is actually the most energetic, and it is unlikely that it is produced in a measurable quantity and the other isomers are not. Therefore, it can be concluded that – within the experimental uncertainty – cyclopentadiene is the sole isomer formed in the reaction. It is also interesting to note that a signal grows in at m/z = 65 when the temperature is raised beyond 873 K (see Fig. 1). The intensity of this peak is larger than the background in the absence of the allyl precursor (see ESI†) and is thus likely formed via allyl chemistry. This product can be the resonantly stabilized cyclopentadienyl (C5H5˙) radical formed by thermal decomposition of cyclopentadiene,26 or by hydrogen abstraction from cyclopentadiene. As reported in the literature, both theoretical and experimental evidence suggests that cyclopentadienyl (CPDyl) radicals condense to form naphthalene (m/z = 128).23,25,26 At 9.5 eV, i.e. below the ionization energy of HI, we did not detect a peak at m/z = 128, which could be due to unfavorable temperatures or a low number density of cyclopentadienyl radicals in the reactor.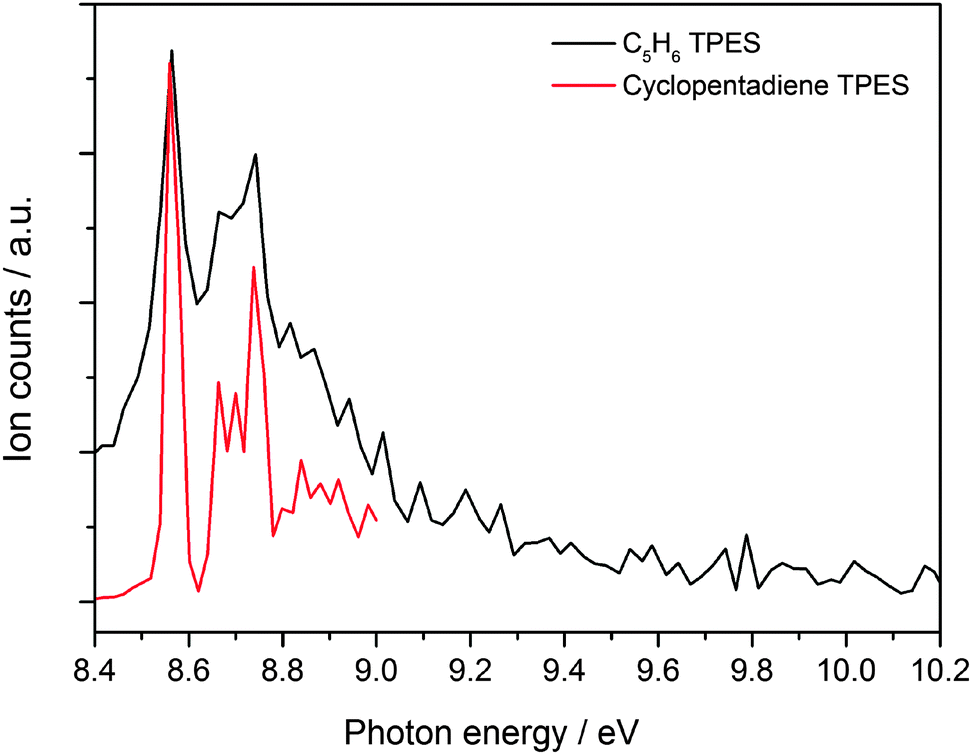 | ||
| Fig. 3 Mass selected threshold photoelectron spectrum of C5H6 formed in the reaction between C3H5˙ and C2H2 (in black) plotted together with a measured spectrum of cyclopentadiene (in red). | ||
3.3 C5H7 potential energy surface
Quantum chemical computations have been performed at the CBS-QB3 level of theory to rationalize experimental observations. The C5H7 potential energy surface has been mapped by constrained optimizations to locate intermediates (INT) and transition states (TS), which were subjected to CBS-QB3 calculations and the resulting PES is shown in Fig. 4. | ||
| Fig. 4 C5H7 potential energy surface obtained at the CBS-QB3 level of theory. All energies are shown in kJ mol−1 relative to the energy of C3H5 + C2H2. Products (red), intermediate states (black) and transition states (blue) are color coded. | ||
Crossing of the entrance barrier (TS1) leads to the adduct species INT1. A number of conformers exist for this adduct that are linked through internal rotations with associated barriers ranging from 8–12 kJ mol−1 and only the lowest energy structure for INT1 is shown in Fig. 4.34 From the initial adduct, crossing over TS2 leads to the formation of the very stable cyclic C5H7 radical (c-C5H7˙) species. The alternative products, 4-penten-1-yne (P2) and 1,2,4-pentatriene (P3) can only form from INT1 over quite energetic transition states TS4 and TS5, which explains why they are not detected in our experiments. The rate limiting step in the formation of cyclopentadiene (P1) from allyl + C2H2 is the formation of the initial acyclic C5H7 adduct (INT1), which involves a barrier of 58.8 kJ mol−1 and results in a low reaction rate at low temperatures. The c-C5H7 radical is formed from the adduct by crossing a barrier located at 6.3 kJ mol−1 with respect to the reactants. Once formed, the c-C5H7˙ can be stabilized by collisional de-excitation at elevated pressure, or it undergoes H-atom loss to form cyclopentadiene. H-atom loss from the non-cyclized intermediate INT1 leading to acyclic products (P2 and P3) requires at least 153.3 kJ mol−1 with respect to the reactants and is unlikely to compete with cyclization over the low-energy transition state TS2.
Collisional stabilization of c-C5H7˙ would manifest itself in the experimental mass spectra as a product at m/z = 67. While this peak is observed in the T = 981 and 1068 K spectra in Fig. 1, its intensity ratio to the m/z = 54 peak suggests that its sole source is the allyl iodide pyrolysis (see ESI†). The reason as to why the c-C5H7˙ intermediate in the allyl + acetylene reaction is not observed likely relates to the C5H7 species not being thermalized in the low-pressure environment of the microtubular reactor. In the absence of sufficient thermalization, it contains enough internal energy to proceed to the final products cyclopentadiene + H˙.
Based on the computed harmonic vibrational frequencies, we employed RRKM theory to further elucidate the possible stabilization of c-C5H7.63 The rate constant for the formation of INT2 from INT1 is found to be 3 × 109 s−1 at an energy of 58.8 kJ mol−1 relative to the reactants. Even the rate constant for the H-loss step, from INT2 to P1, is found to be 3 × 108 s−1 at this energy, despite the deep potential well and the resulting high density of states of the reactant. A typical collisional frequency under combustion relevant conditions (1000 K and 5 bar) is 1011 s−1, which allows for 100–1000 collisions at most, and suggest that INT1 and INT2 could only be thermalized and INT2 stabilized in high-pressure environments.
4 Conclusions
The products of the reaction between the resonantly stabilized allyl radical and acetylene have been identified using a pyrolysis reactor coupled to the iPEPICO system at the vacuum ultraviolet beamline of the Swiss Light Source. Control experiments have been performed to identify side reactions along with allyl + acetylene chemistry. Cyclopentadiene is unambiguously identified as the sole product of the reaction by recording the mass selected threshold photoionization spectrum of the C5H6 product and comparing it with a cyclopentadiene photoelectron spectrum.Quantum chemical computations of the reaction path rationalize the detection of a single product from the reaction. From the PES it is found that the formation of cyclopentadiene + H is lowest in energy and the barrier associated with the initial addition of allyl to C2H2 (58.8 kJ mol−1) is the rate-limiting step. The C5H7 adduct formed after cyclization of the initially formed acyclic adduct is more stable than cyclopentadiene + H by 125 kJ mol−1 and would require significant thermalization to be detected. The rate limiting barriers for the formation of 4-penten-1-yne + H and 1,2,4-pentatriene + H are 113.9 and 153.3 kJ mol−1 with respect to the reactants, respectively.
Cyclopentadiene is observed as the sole product formed in the pyrolysis tube under our experimental conditions and no evidence is found for stabilization of the C5H7 adduct. The detection of cyclopentadiene as the only product of the reaction is in agreement with the study by Nohara and Sakai.30 The non-detection of the C5H7 adduct is in accordance with the study by Isemer and Luther32 and points to a prompt ring closure in the energized adduct followed by a fast subsequent unimolecular dissociation forming C5H6 + H. Hence, the rate limiting step in the formation of cyclopentadiene from the C3H5 + C2H2 reaction is indeed, as predicted based on computations, the formation of the initial adduct. This is in contrast with findings by Nohara and Sakai, who argued that not the initial addition, but either ring closure or the loss of an H-atom would be the rate limiting step.30
At elevated temperatures in combustion environments, the allyl radical can undergo thermal decomposition to allene + H, or isomerization to the vinyl radical.64,65 At temperatures and acetylene densities for which the allyl + acetylene reaction competes with dissociation, the reaction of the allyl radical with acetylene yields solely cyclopentadiene. Resonance stabilized CPDyl radicals can subsequently form by hydrogen abstraction from cyclopentadiene; a reaction that is also observed in our experiments, albeit at elevated reactor temperatures. Both theory and experiments suggest that the reaction of two cyclopentadienyl radicals (CPDyl) yields naphthalene.23–26 This emphasizes the importance of the C3H5 + C2H2 reaction in the formation of aromatic molecules in combustion environments at temperatures where bimolecular reactions of allyl with acetylene outcompete unimolecular dissociation of allyl.
Acknowledgements
JB acknowledges the Netherlands Organisation for Scientific Research for a VENI grant (Grant number 722.013.014). JO acknowledges the Netherlands Organisation for Scientific Research for a VICI grant (Grant number 724.011.002). This work was further supported by NWO Exacte Wetenschappen (Physical Sciences) for the use of the supercomputer facilities at SurfSara. The iPEPICO experiments were performed at the VUV beamline of the Swiss Light Source (Paul Scherrer Institut, Villigen, Switzerland). AB and PH acknowledge funding by the Swiss Federal Office for Energy (BFE Contract number 101969/152433).References
- J. L. Durant, W. F. Busby, A. L. Lafleur, B. W. Penman and C. L. Crespi, Mutat. Res., Genet. Toxicol., 1996, 371, 123–157 CrossRef CAS.
- K. Katsouyanni, G. Touloumi, E. Samoli, A. Gryparis, A. Le Tertre, Y. Monopolis, G. Rossi, D. Zmirou, F. Ballester and A. Boumghar, et al. , Epidemiology, 2001, 12, 521–531 CrossRef CAS PubMed.
- C. A. Pope, R. T. Burnett, M. J. Thun, E. E. Calle, D. Krewski, K. Ito and G. D. Thurston, JAMA, J. Am. Med. Assoc., 2002, 287, 1132–1141 CrossRef CAS.
- J. Londahl, E. Swietlicki, E. Lindgren and S. Loft, Atmos. Chem. Phys., 2010, 10, 9441–9449 CrossRef.
- U. Poschl, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef PubMed.
- O. L. Hadley and T. W. Kirchstetter, Nat. Clim. Change, 2012, 2, 437–440 CrossRef CAS PubMed.
- J. A. Miller, M. J. Pilling and E. Troe, Proc. Combust. Inst., 2005, 30, 43–88 CrossRef PubMed.
- C. S. McEnally, L. D. Pfefferle, B. Atakan and K. Kohse-Hoinghaus, Prog. Energy Combust. Sci., 2006, 32, 247–294 CrossRef CAS PubMed.
- N. Hansen, J. A. Miller, S. J. Klippenstein, P. R. Westmoreland and K. Kohse-Hoinghaus, Combust., Explos. Shock Waves, 2012, 48, 508–515 CrossRef.
- R. D. Kern and K. Xie, Prog. Energy Combust. Sci., 1991, 17, 191–210 CrossRef CAS.
- J. A. Miller and C. F. Melius, Combust. Flame, 1992, 91, 21–39 CrossRef CAS.
- C. J. Pope and J. A. Miller, Proc. Combust. Inst., 2000, 28, 1519–1527 CrossRef CAS.
- H. Richter and J. B. Howard, Prog. Energy Combust. Sci., 2000, 26, 565–608 CrossRef CAS.
- M. Frenklach, Phys. Chem. Chem. Phys., 2002, 4, 2028–2037 RSC.
- T. M. Selby, G. Meloni, F. Goulay, S. R. Leone, A. Fahr, C. A. Taatjes and D. L. Osborn, J. Phys. Chem. A, 2008, 112, 9366–9373 CrossRef CAS PubMed.
- J. A. Miller, S. J. Klippenstein, Y. Georgievskii, L. B. Harding, W. D. Allen and A. C. Simmonett, J. Phys. Chem. A, 2010, 114, 4881–4890 CrossRef CAS PubMed.
- M. Frenklach, D. W. Clary, W. C. Gardiner Jr. and S. E. Stein, Proc. Combust. Inst., 1985, 20, 887–901 CrossRef.
- B. Shukla and M. Koshi, Phys. Chem. Chem. Phys., 2010, 12, 2427–2437 RSC.
- D. S. N. Parker, R. I. Kaiser, T. P. Troy and M. Ahmed, Angew. Chem., Int. Ed., 2014, 53, 7740–7744 CrossRef CAS PubMed.
- K. H. Fischer, J. Herterich, I. Fischer, S. Jaeqx and A. M. Rijs, J. Phys. Chem. A, 2012, 116, 8515–8522 CrossRef CAS PubMed.
- G. da Silva, J. A. Cole and J. W. Bozzelli, J. Phys. Chem. A, 2010, 114, 2275–2283 CrossRef CAS PubMed.
- C. Cavallotti, D. Polino, A. Frassoldati and E. Ranzi, J. Phys. Chem. A, 2012, 116, 3313–3324 CrossRef CAS PubMed.
- C. F. Melius, M. E. Colvin, N. M. Marinov, W. J. Pit and S. M. Senkan, Proc. Combust. Inst., 1996, 26, 685–692 CrossRef.
- E. Ikeda, R. S. Tranter, J. H. Kiefer, R. D. Kern, H. J. Singh and Q. Zhang, Proc. Combust. Inst., 2000, 28, 1725–1732 CrossRef CAS.
- A. M. Mebel and V. V. Kislov, J. Phys. Chem. A, 2009, 113, 9825–9833 CrossRef CAS PubMed.
- A. M. Scheer, C. Mukarakate, D. J. Robichaud, G. B. Ellison and M. R. Nimlos, J. Phys. Chem. A, 2010, 114, 9043–9056 CrossRef CAS PubMed.
- A. M. Dean, J. Phys. Chem., 1985, 89, 4600–4608 CrossRef CAS.
- M. M. Lu and J. A. Mulholland, Chemosphere, 2004, 55, 605–610 CrossRef CAS PubMed.
- M. R. Djokic, K. M. Van Geem, C. Cavallotti, A. Frassoldati, E. Ranzi and G. B. Marin, Combust. Flame, 2014, 161, 2739–2751 CrossRef CAS PubMed.
- D. Nohara and T. Sakai, Ind. Eng. Chem. Fundam., 1980, 19, 340–344 CAS.
- S. Liang, P. Hemberger, N. M. Neisius, A. Bodi, H. Grützmacher, J. Levalois-Grützmacher and S. Gaan, Chem. – Eur. J., 2015, 21, 1073–1080 CrossRef CAS PubMed.
- S. J. Isemer and K. Luther, in Shock Waves, ed. Z. Jiang, Springer, Berlin, Heidelberg, 2005, ch. 91, pp. 609–614 Search PubMed.
- A. M. Dean, J. Phys. Chem., 1990, 94, 1432–1439 CrossRef CAS.
- L. V. Moskaleva and M. C. Lin, Proc. Combust. Inst., 2002, 29, 1319–1327 CrossRef CAS.
- A. Bodi, M. Johnson, T. Gerber, Z. Gengeliczki, B. Sztaray and T. Baer, Rev. Sci. Instrum., 2009, 80, 034101 CrossRef PubMed.
- M. Johnson, A. Bodi, L. Schulz and T. Gerber, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 610, 597–603 CrossRef CAS PubMed.
- D. W. Kohn, H. Clauberg and P. Chen, Rev. Sci. Instrum., 1992, 63, 4003–4005 CrossRef CAS PubMed.
- H. J. Deyerl, I. Fischer and P. Chen, J. Chem. Phys., 1999, 111, 3441–3448 CrossRef CAS PubMed.
- Q. Guan, K. N. Urness, T. K. Ormond, D. E. David, G. B. Ellison and J. W. Daily, Int. Rev. Phys. Chem., 2014, 33, 447–487 CrossRef CAS PubMed.
- B. Sztaray and T. Baer, Rev. Sci. Instrum., 2003, 74, 3763–3768 CrossRef CAS PubMed.
- A. Bodi and P. Hemberger, Phys. Chem. Chem. Phys., 2014, 16, 505–515 RSC.
- P. Hemberger, A. J. Trevitt, E. Ross and G. da Silva, J. Phys. Chem. Lett., 2013, 4, 2546–2550 CrossRef CAS.
- A. Bodi, P. Hemberger, D. L. Osborn and B. Sztaray, J. Phys. Chem. Lett., 2013, 4, 2948–2952 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian, Inc., 2009, Wallingford CT Search PubMed.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822–2827 CrossRef CAS PubMed.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 2000, 112, 6532–6542 CrossRef CAS PubMed.
- I. Fischer, T. Schuessler, H.-J. Deyerl, M. Elhanine and C. Alcaraz, Int. J. Mass Spectrom., 2007, 261, 227–233 CrossRef CAS PubMed.
- Nist Chemistry Webbook. NIST Standard Reference Database Number 69, National Institute of Standards and Technology, 2009.
- P. Wolkoff, J. L. Holmes and F. P. Lossing, Can. J. Chem., 1980, 58, 251–257 CrossRef CAS.
- E. E. Rennie, A.-M. Boulanger, P. M. Mayer, D. M. P. Holland, D. A. Shaw, L. Cooper and L. G. Shpinkova, J. Phys. Chem. A, 2006, 110, 8663–8675 CrossRef CAS PubMed.
- C. A. Taatjes, D. L. Osborn, T. M. Selby, G. Meloni, H. Fan and S. T. Pratt, J. Phys. Chem. A, 2008, 112, 9336–9343 CrossRef CAS PubMed.
- M. Bobeldijk, W. J. van der Zande and P. G. Kistemaker, Chem. Phys., 1994, 179, 125–130 CrossRef CAS.
- S. H. Mousavipour and P. D. Pacey, J. Phys. Chem., 1996, 100, 3573–3579 CrossRef CAS.
- J. P. Senosiain, S. J. Klippenstein and J. A. Miller, J. Phys. Chem. A, 2006, 110, 5772–5781 CrossRef CAS PubMed.
- J. H. Kiefer and W. A. Vondrasek, Int. J. Chem. Kinet., 1990, 22, 747–786 CrossRef CAS PubMed.
- C. Saggese, N. E. Sanchez, A. Frassoldati, A. Cuoci, T. Faravelli, M. U. Alzueta and E. Ranzi, Energy Fuels, 2014, 28, 1489–1501 CrossRef CAS.
- J. Bouwman, F. Goulay, S. R. Leone and K. R. Wilson, J. Phys. Chem. A, 2012, 116, 3907–3917 CrossRef CAS PubMed.
- G. Bieri, F. Burger, E. Heilbronner and J. P. Maier, Helv. Chim. Acta, 1977, 60, 2213–2233 CrossRef CAS PubMed.
- J. Bouwman, M. Fournier, I. R. Sims, S. R. Leone and K. R. Wilson, J. Phys. Chem. A, 2013, 117, 5093–5105 CrossRef CAS PubMed.
- T. Bierkandt, E. Alkyildiz, T. Kasper, P. Hemberger, P. Oßwald, M. Köhler, 2014, private communication.
- P. Oßwald, P. Hemberger, T. Bierkandt, E. Akyildiz, M. Köhler, A. Bodi, T. Gerber and T. Kasper, Rev. Sci. Instrum., 2014, 85, 025101 CrossRef PubMed.
- B. K. C. de Miranda, C. Alcaraz, M. Elhanine, B. Noller, P. Hemberger, I. Fischer, G. A. Garcia, H. Soldi-Lose, B. Gans and L. A. V. Mendes, et al. , J. Phys. Chem. A, 2010, 114, 4818–4830 CrossRef PubMed.
- B. Sztaray, A. Bodi and T. Baer, J. Mass Spectrom., 2010, 45, 1233–1245 CrossRef CAS PubMed.
- R. X. Fernandes, B. R. Giri, H. Hippler, C. Kachiani and F. Striebel, J. Phys. Chem. A, 2005, 109, 1063–1070 CrossRef CAS PubMed.
- B. S. Narendrapurapu, A. C. Simmonett, H. F. Schaefer, J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2011, 115, 14209–14214 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Results of control experiments to map the side reactions of acetylene and allyl iodide pyrolysis in order to identify the acetylene + allyl chemistry. See DOI: 10.1039/c5cp02243f |
| This journal is © the Owner Societies 2015 |