
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vibrational spectra and structures of SinC clusters (n = 3–8)†
Nguyen Xuan
Truong
a,
Marco
Savoca
a,
Dan J.
Harding
bc,
André
Fielicke
a and
Otto
Dopfer
*a
aInstitut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstraße 36, D-10623 Berlin, Germany. E-mail: dopfer@physik.tu-berlin.de
bInstitut für Physikalische Chemie, Georg-August-Universität Göttingen, Tammannstraße 6, D-37077 Göttingen, Germany
cDepartment of Dynamics at Surfaces, Max-Planck-Institut für Biophysikalische Chemie, Am Faßberg 11, D-37077 Göttingen, Germany
First published on 24th June 2015
Abstract
The effects of doping bare silicon clusters with carbon on their physical properties are of fundamental interest for the chemistry of the interstellar medium and the development of novel nanostructures in materials science. Carbon-doped silicon clusters (SinC, n = 3–8) are characterized in the gas phase with infrared-ultraviolet two-color ionization (IR-UV2CI) spectroscopy, mass spectrometry, and quantum chemical calculations. Structural identification is achieved by comparing the measured and calculated vibrational absorption spectra of the low-energy SinC isomers identified by global optimization algorithms. Except for planar Si3C, the most stable SinC clusters have three-dimensional configurations. While the Si3C and Si6C structures are uniquely assigned, several stable isomers of Si4C, Si5C, Si7C, and Si8C may co-exist under the present experimental conditions. Interestingly, some of the structures observed here are different from the ground state structures predicted previously. For the small neutral clusters (n ≤ 5), structures similar to those reported previously for the anions are observed. The highly stable Si3C unit with a nearly linear Si–C–Si motif is identified as characteristic building block in several of the most stable SinC structures. In all identified structures, a large negative charge of almost −2e is located on the C atom, indicating its role as electron donor in the Sin host moiety. The B3LYP/cc-pVTZ level proves reliable in finding the experimentally observed isomers.
I. Introduction
Silicon–carbon based nanotubes, nanowires, and clusters are of great interest as promising building blocks for the synthesis of novel materials at the nanoscale.1 In addition, small silicon–carbon clusters, e.g., SiCm with m = 1–4, were found in interstellar space.2–7 Hence understanding the nature of Si–C bonding might, on the one hand, open a way toward engineering of such nanostructures at the atomic level. On the other hand, it is crucial for several chemical processes and observations in the interstellar medium.8–11 Although located in the same main group of the periodic table, silicon and carbon exhibit very different physical and chemical properties. Silicon prefers multidirectional single bonds, while carbon can easily form single, double, and triple bonds. Generally, substituting a single Si atom with a C atom in a pure Sin cluster induces strong distortions, but the geometric topology maintains.12,13 When doped into silicon clusters, the C atom favors locations with more than threefold coordinated bonding in the ground state structures, making most SinCm clusters more stable than their pure silicon counterparts.13There have been numerous theoretical studies on silicon–carbon clusters.13–33 For small-sized clusters, for example, the ground state structures and vibrational spectra of Si3C and Si2C3 were determined using Hartree–Fock (HF) and second-order many body perturbation (MP2) theories.14,15 A clear trend of favorable geometries in the transition from pure carbon to pure silicon clusters was observed, i.e., the chain-like carbon-rich and three-dimensional silicon-rich clusters. This has also been observed for SinCm (n + m ≤ 8) cluster anions in both theory32 and experiments.12 Most close to our work, two lowest-energy Si4C structures were found at the CCSD(T) level, i.e., the minimum trigonal C3v pyramid and a distorted edge capped C2v pyramid (about 21 kJ mol−1 higher).33 At the HF level, however, the latter structure lies about 8 kJ mol−1 lower than the former. Additional calculations of infrared (IR) spectra, isotopic shifts, and dipole moments indeed hinted at their coexistence.33
In contrast to theory, spectroscopic characterization of silicon–carbon clusters is largely lacking. Most previous studies have concentrated on charged clusters isolated in the gas phase or neutrals in an inert matrix. For instance, photoelectron (PE) spectra of SinCm− (1 ≤ n ≤ 7, 1 ≤ m ≤ 5) cluster anions found that SinC− (3 ≤ n ≤ 7) and their Sin+1− counterparts have a similar topology.12 The PE spectrum of SiC3− shows electronic transitions, corresponding to both linear and cyclic forms.34 However, no conclusion about the true global minimum on the SiC3 potential energy surface could be drawn. With Fourier-transform infrared spectroscopy in Ar matrices combined with quantum chemical calculations, the molecular structures of small SinCm (n + m ≤ 5) clusters were determined, e.g., a linear centrosymmetric geometry for Si2C3 and a rhomboidal C2v structure for Si3C.35–38 Furthermore, Si3C2 is a pentagon, and Si2C4 has a linear symmetric geometry in the ground state.39 Similarly, C-rich clusters such as SiC4, SiC5, SiC7, and SiC9, were found to exhibit chain-like structures.38,40–43 For small SinCm clusters with n + m ≤ 4, the electronic structure has been studied by resonant photoionization.11,44,45 In general, little spectroscopic information is available for somewhat larger Si-rich SinCm clusters, which is the topic of the present work.
Through recent advances in the generation of intense IR laser radiation, novel spectroscopic methods for neutral clusters in the gas phase have been developed, such as IRMPD (infrared multiple photon dissociation),46,47 IR-REMPI (infrared resonance enhanced multiple photon ionization),48 and IR-UV2CI (infrared-ultraviolet two-color ionization).49 While IR-REMPI spectra are often broadened due to the absorption of several hundred IR photons, the IR-UV2CI spectra can be better resolved and more closely related to the linear IR absorption spectra. Employing the IR-UV2CI technique for neutral SinCm clusters, we recently observed the evolution from 3D to 1D chain-like configurations in SinC6−n clusters by sequentially substituting Si with C atoms in pure Si6.50 In addition, the influence of first-row dopant atoms on the properties of bare silicon clusters (Si6X with X = Be, B, C, N, O) has been determined.51 As expected, the geometric and electronic properties of silicon containing clusters can substantially be modified by changing the dopant atom.
Here we identify the geometric and electronic structures of SinC clusters with n = 3–8 via their IR-UV2CI spectra. Combining quantum chemical calculations with global optimization,52,53 the low-energy SinC isomers are determined. Structural identification is achieved through comparison of the measured IR-UV2CI spectra with the IR spectra predicted for the corresponding low-energy isomers. Results for Si5C and Si6C have been reported elsewhere,50,51 but are included here with additional insights for completeness. Electronic properties and charge distributions of selected low-energy isomers will also be discussed.
II. Experimental and computational methods
The IR-UV2CI setup has been described elsewhere.47,49,50 Briefly, carbon-doped silicon clusters are produced by laser ablation of a pure silicon rod within a pulsed flow of He gas containing 1% CH4 and thermalized to about 100 K in a liquid-nitrogen cooled expansion channel. After passing through a skimmer, the neutral SinCm clusters are overlapped with counter-propagating IR radiation from the ‘Free Electron Laser for Infrared eXperiments’ (FELIX)54 and then post-ionized by an unfocused F2 laser (EF2 = 7.87 eV) in the extraction zone of a reflectron time-of-flight mass spectrometer. For clusters of specific sizes, with an ionization energy (IE) close to EF2, prior resonant excitation with IR photons from a FELIX pulse raises the internal energy and thereby enhances the efficiency for ionization by the UV laser pulse that is about 30 μs delayed. Resonant excitation of a vibrational mode is followed by rapid internal vibrational energy redistribution, leading to complete thermalization on this experimental timescale. The ionization efficiency usually follows a S-curve behavior as a function of excitation energy, with a slope depending on the Franck–Condon factor for ionization. An increase of the internal energy of the cluster upon IR absorption therefore results in an enhancement of the ionization yield and its IR wavelength dependence closely reflects the linear vibrational absorption spectrum of the neutral cluster. The IR-UV2CI spectra in Fig. 1 are obtained from the relative ionization enhancement normalized by the IR photon flux.55 The observed widths of the bands of 15–45 cm−1 arise from a combination of unresolved rotational structure, sequence hot band transitions involving low-frequency modes, the FELIX bandwidth (ca. 0.5–1% full width at half maximum (FWHM) of the central wavelength), and possibly the multiple photon absorption process.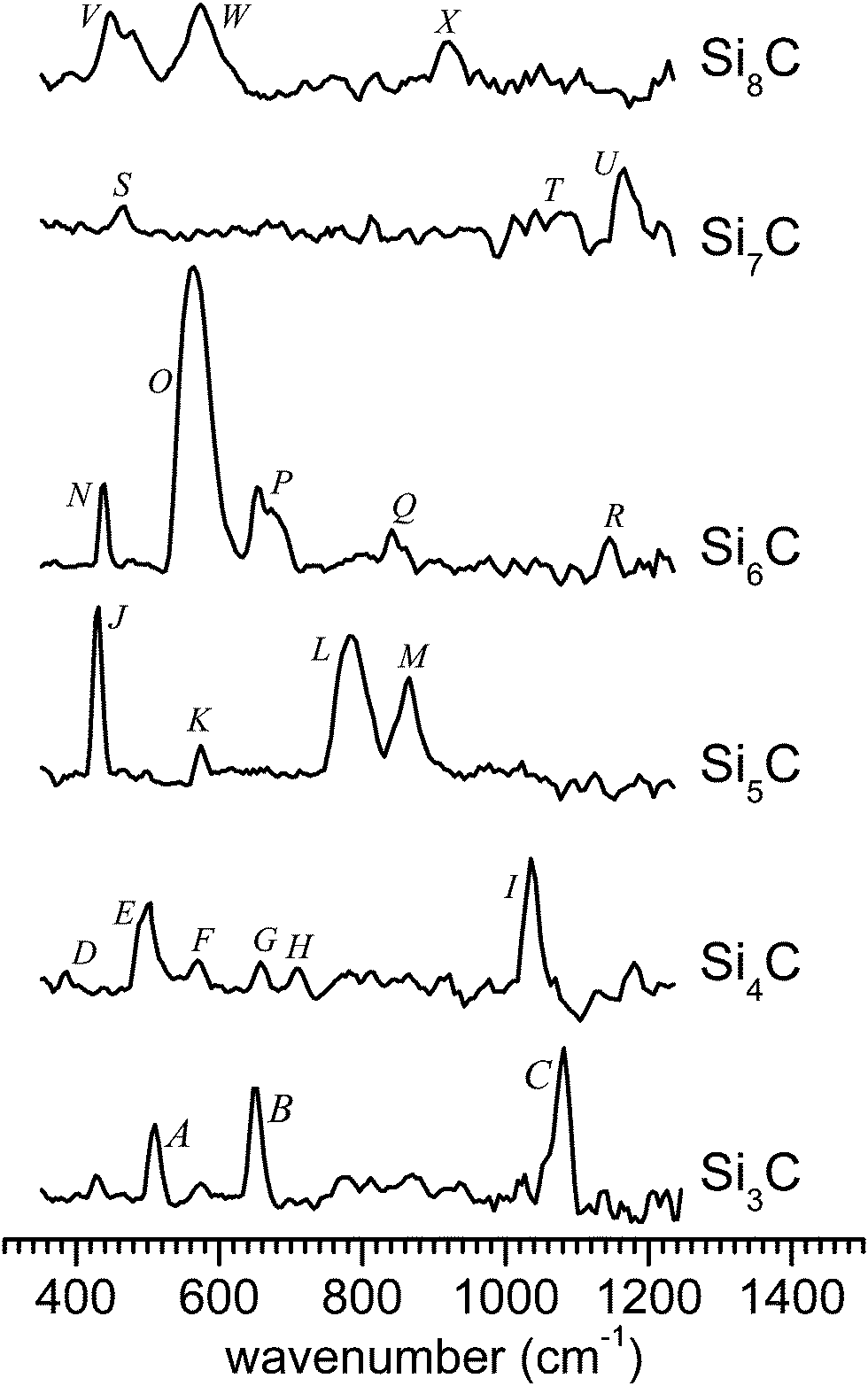 | ||
| Fig. 1 Measured IR-UV2CI spectra of SinC (with n = 3–8). All spectra are drawn to the same scale, according to the relative IR enhancement normalized by laser fluence. | ||
Quantum chemical calculations are performed for both singlet and triplet states of each cluster. In an effort to thoroughly explore the complex potential energy surface of the cluster, we employ global optimization (GO) techniques such as the genetic (GA) and basin hopping (BH) algorithms.56,57 Details of our BH implementation have been given elsewhere.51,52 Basically, there are two major steps. First, up to five thousand candidate structures are evaluated in terms of the total energy by the GO coupled with DFT calculations at the RI-BP86/def-SVP level as implemented in TURBOMOLE V6.3.1.58 Second, the first twenty nonequivalent lowest-energy isomers are tightly re-optimized at the B3LYP/cc-pVTZ level including the Grimme dispersion correction (D3)59 with the resolution-of-the-identity approximation as implemented in GAUSSIAN09.60 At this level, the linear IR absorption spectra, ionization and binding energies, and natural bond orbital (NBO) populations are obtained. To facilitate convenient comparison with the experimental spectra, the theoretical IR absorption stick spectra are convoluted with a 20 cm−1 FWHM Gaussian profile. The reported vibrational frequencies are unscaled. Similar calculations for selected clusters have been performed at the TPSS/cc-pVTZ and MP2/cc-pVTZ levels, which have also proven reliable for doped Si clusters.47,49,50 The vertical electronic transitions of selected SinC clusters are calculated at the second-order approximate coupled cluster RI-CC2/def2-TZVP and TD-DFT/cc-pVTZ levels using TURBOMOLE V6.3.1.58 As the excited-state calculation for high-symmetry structures (e.g., C3v, C4v, and C5v) is not supported at the CC2 level, the C1 point group is used for these clusters.
III. Results and discussion
The IR-UV2CI spectra measured for SinC (n = 3–8), the linear IR absorption spectra and the geometries of the respective lowest-energy isomers calculated at the B3LYP/cc-pTVZ level are shown in Fig. 1–7. The computational spectra are all drawn to the same linear intensity scale for direct comparison. The relative energies, ionization and binding energies are listed in Tables 1 and 2, respectively. Vertical excitations (Table 3) of selected SinC isomers are also provided. Natural bond orbital charges, vibrational and geometric data of all structures considered are available in Tables 1–3 in the ESI.† In the following discussion, the optimized structures obtained at the B3LYP level are used if not stated otherwise.| Cluster | Isomer | Symmetry (State) | Relative energy | Cluster | Isomer | Symmetry (State) | Relative energy | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| B3LYP | TPSS | MP2 | B3LYP | TPSS | MP2 | ||||||
| a Relaxing to a (C3v, 3A1) structure. b C 2v (1A1). | |||||||||||
| Si3C | 3a | C 2v (1A1) | 0.0 | 0.0 | 0.0 | Si6C | 6a | C 5v (1A1) | 0.0 | 0.0 | 0.0 |
| 3b | C 2v (3B1) | 116.6 | 117.1 | 187.0 | 6b | C s (1A′) | 12.2 | 36.8 | 86.5 | ||
| 3c | C s (3A′′) | 183.1 | 157.8a | 199.0 | 6c | C 1 (1A) | 29.3 | 56.1 | 105.5 | ||
| 6d | C s (1A′) | 47.2 | 59.9 | 86.5 | |||||||
| Si4C | 4a | C 2 (1A) | 0.0 | 19.9 | 24.2 | Si7C | 7a | C 1 (1A) | 0.0 | 7.2 | 21.0 |
| 4b | C 3v (1A1) | 1.1 | 0.0 | 0.0 | 7b | C 1 (1A) | 0.2 | 6.0 | 29.2 | ||
| 4c | C s (1A′) | 18.7 | 38.3 | 28.9b | 7c | C 3v (1A1) | 9.9 | 8.8 | 31.2 | ||
| 4d | C s (1A′) | 27.9 | 52.9 | 55.0 | 7d | C s (1A′) | 14.4 | 0.0 | 0.0 | ||
| Si8C | 8a | C s (1A′) | 0.0 | 0.0 | 64.6 | ||||||
| Si5C | 5a | C 2v (1A1) | 0.0 | →5b | →5b | 8b | C 1 (1A) | 3.1 | 7.1 | 0.0 | |
| 5b | C 4v (1A1) | 0.1 | 0.0 | 0.0 | 8c | C 1 (1A) | 17.6 | 13.8 | 54.4 | ||
| 5c | C s (1A′) | 33.2 | 44.1 | 76.5 | 8d | C s (1A′) | 22.1 | 10.6 | 0.0 | ||
| 5d | C s (3A′′) | 123.4 | 133.9 | 197.2 | 8e | C s (1A′) | 39.8 | 18.0 | 49.7 | ||
| Cluster | Isomer | VIE [eV] | AIE [eV] | BE [eV] |
|---|---|---|---|---|
| # calculations failed to converge. | ||||
| Si3C | 3a | 8.13; 8.21; 8.43 | 7.80; 8.00; 8.12 | 4.90; 5.14; 5.25 |
| 3b | 6.71; 6.89; 6.42 | 6.64; 6.83; 6.18 | 4.60; 4.83; 4.76 | |
| 3c | 7.16; 7.48; 7.10 | 5.91; 6.43; 6.06 | 4.43; 4.73; 4.73 | |
| Si4C | 4a | 7.90; 8.02; 8.54 | 7.30; 7.46; 7.44 | 4.75; 5.06; 5.15 |
| 4b | 8.15; 8.25; 8.72 | 7.94; 8.11; 8.37 | 4.75; 5.10; 5.20 | |
| 4c | 7.46; 7.60; 7.77 | 7.10; 7.27; 7.39 | 4.71; 5.02; 5.14 | |
| 4d | 7.32; 7.45; 7.64 | 7.17; 7.31; 7.50 | 4.69; 4.99; 5.08 | |
| Si5C | 5a | 8.19; 8.27; 8.72 | 7.83; 7.93; 8.61 | 4.86; 5.26; 5.48 |
| 5b | 8.19; 8.21; 8.72 | 7.95; 7.98; 8.61 | 4.86; 5.26; 5.48 | |
| 5c | 8.00; 8.03; 8.74 | 7.62; 7.67; # | 4.81; 5.19; 5.35 | |
| 5d | 7.51; 7.48; 8.69 | 6.62; 6.54; # | 4.65; 5.03; 5.14 | |
| Si6C | 6a | 7.85; 7.85; 8.16 | 7.56; 7.69; 8.16 | 4.73; 5.17; 5.40 |
| 6b | 8.00; 7.78; 8.00 | 7.70; 7.33; 7.36 | 4.72; 5.12; 5.28 | |
| 6c | 7.18; 7.58; 7.80 | 7.04; 7.13; 7.17 | 4.69; 5.09; 5.25 | |
| 6d | 7.42; 7.44; 7.68 | 7.05; 7.10; 7.41 | 4.66; 5.08; 5.28 | |
| Si7C | 7a | 7.59; 7.66; 8.18 | 7.32; 7.38; # | 4.69; 5.09; 5.27 |
| 7b | 7.60; 7.65; 8.17 | 7.31; 7.39; 7.91 | 4.69; 5.09; 5.26 | |
| 7c | 7.81; 7.84; 8.40 | 7.41; 7.43; 7.76 | 4.68; 5.09; 5.25 | |
| 7d | 7.52; 7.53; 8.06 | 7.07; 7.13; 7.45 | 4.67; 5.10; 5.30 | |
| Si8C | 8a | 7.52; 7.63; 8.24 | 7.12; 7.33; 7.73 | 4.69; 5.12; 5.29 |
| 8b | 7.75; 7.73; 8.23 | 7.48; 7.49; # | 4.69; 5.11; 5.37 | |
| 8c | 7.47; 7.44; 8.49 | 7.24; 7.25; # | 4.67; 5.10; 5.30 | |
| 8d | 7.47; 7.48; 8.23 | 7.24; 7.28; 8.08 | 4.66; 5.11; 5.37 | |
| 8e | 7.41; 7.42; 7.67 | 7.18; 7.24; 7.37 | 4.64; 5.10; 5.31 | |
| Cluster | Isomer (point group, state) | Transition energy [eV] | |
|---|---|---|---|
| RI-CC2/def2-TZVP | B3LYP/cc-pVTZ | ||
| a For structures 4b (C3v), 5b (C4v), and 6a (C5v), calculations with C1 symmetry are performed at the RI-CC2 level. Numbers in parentheses are the oscillator strengths in 10−3. | |||
| Si3C | 3a (C2v, 1A1) | 2.08 (1B2, 3.21) | 1.87 (1B2, 1.54) |
| 2.83 (1A2, 0.00) | 3.08 (1B2, 27.82) | ||
| 3.09 (1A2, 0.00) | 3.77 (1B2, 17.18) | ||
| Si4C | 4a (C2, 1A) | 2.25 (1B, 17.7) | 2.20 (1B, 10.39) |
| 2.49 (1A, 8.65) | 2.39 (1B, 8.03) | ||
| 2.63 (1B, 4.19) | 2.40 (1A, 5.54) | ||
| 4b (C3v, 1A1) | 2.77 (1A, 10.6) | 2.57 (1A1, 2.62) | |
| 2.79 (1A, 0.00) | 2.63 (1E, 38.28) | ||
| 2.83 (1A, 1.56) | 3.19 (1E, 12.43) | ||
| Si5C | 5a (C2v, 1A1) | 3.40 (1A2, 0.00) | 3.17 (1A1, 0.15) |
| 3.51 (1A2, 0.00) | 3.28 (1A1, 0.18) | ||
| 3.57 (1A1, 0.00) | 3.42 (1B2, 0.03) | ||
| 5b (C4v, 1A1) | 3.38 (1A, 0.00) | 3.18 (1A1, 0.24) | |
| 3.49 (1A, 0.00) | 3.49 (1E, 2.65) | ||
| 3.55 (1A, 0.00) | 3.77 (1E, 26.42) | ||
| Si6C | 6a (C5v, 1A1) | 2.59 (1A, 0.00) | 3.26 (1E1, 23.87) |
| 3.47 (1A, 0.00) | 3.44 (1A1, 12.62) | ||
| 3.51 (1A, 10.17) | 3.56 (1E1, 2.80) | ||
| Si7C | 7a (C1, 1A) | 2.03 (1A, 2.01) | 2.22 (1A, 0.77) |
| 2.24 (1A, 0.34) | 2.39 (1A, 0.67) | ||
| 2.43 (1A, 0.39) | 2.49 (1A, 1.36) | ||
| 7b (C1, 1A) | 2.38 (1A, 0.81) | 2.42 (1A, 0.66) | |
| 2.52 (1A, 0.35) | 2.59 (1A, 0.41) | ||
| 2.68 (1A, 1.91) | 2.68 (1A, 0.71) | ||
| 7c (C3v, 1A1) | 2.09 (1A, 0.36) | 1.94 (1E, 0.05) | |
| 2.48 (1A, 0.00) | 2.43 (1E, 0.56) | ||
| 2.50 (1A, 0.07) | 2.78 (1E, 1.08) | ||
| Si8C | 8a (Cs, 1A′) | 2.19 (1A′, 1.38) | 1.99 (1A′, 0.37) |
| 2.28 (1A′′, 14.69) | 2.17 (1A′′, 0.10) | ||
| 2.55 (1A′, 11.90) | 2.34 (1A′′, 2.40) | ||
| 8b (C1, 1A) | 2.15 (1A, 0.00) | 2.12 (1A, 0.53) | |
| 2.32 (1A, 2.29) | 2.24 (1A, 2.86) | ||
| 2.51 (1A, 8.64) | 2.47 (1A, 1.71) | ||
| 8c (C1, 1A) | 1.65 (1A, 0.54) | 2.07 (1A, 2.34) | |
| 2.08 (1A, 2.91) | 2.29 (1A, 4.29) | ||
| 2.14 (1A, 2.83) | 2.42 (1A, 1.22) | ||
A. Vibrational spectra and geometries
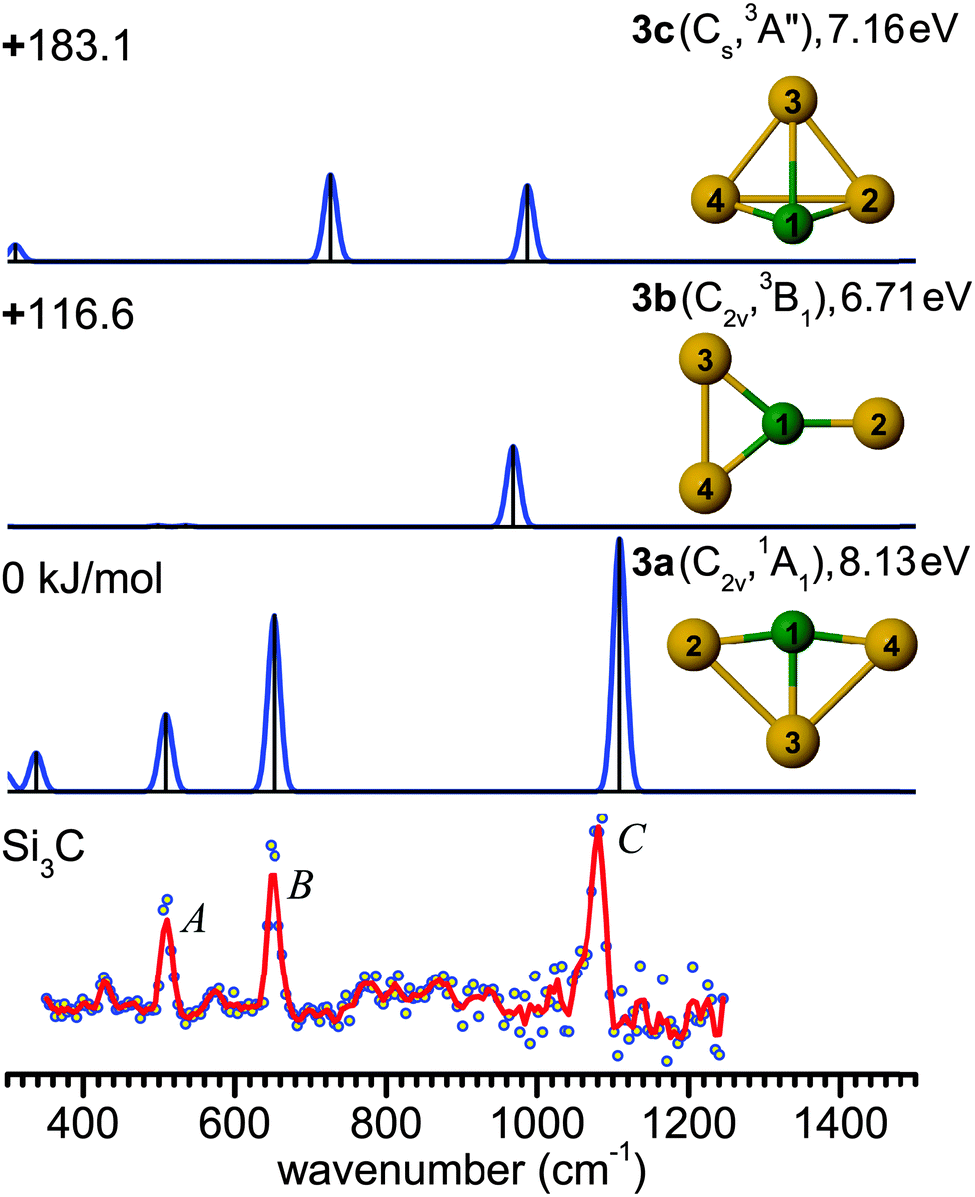 | ||
| Fig. 2 Comparison of the experimental IR-UV2CI spectrum of Si3C (bottom) with linear IR spectra of the three lowest-energy isomers (3a–3c) calculated at the B3LYP/cc-pVTZ level. Molecular symmetries and electronic states are given in parentheses, while vertical ionization energies are given in eV. Relative energies are given in kJ mol−1. The red line is the three-point adjacent average of the original data (circles). The experimental peak positions (in cm−1) are 509 (A), 651 (B) and 1080 (C). | ||
Structure 3a has been predicted as the ground state of Si3C.13,14,22,61,62 While the ground state of neutral Si4 has a D2h (1Ag) rhomboidal geometry,63–653a is a distorted rhombus (C2v, 1A1) with shorter and stronger C–Si bonds and a nearly linear Si–C–Si bridge (∠4Si–1C–2Si = 164.9°). The calculated C–Si bond lengths of 1.764 and 1.944 Å are close to the previous results.44 The anion counterpart Si3C− has been concluded to be a distorted planar rhombus (C2v, 2A2), due to the similarity between the PE spectra of Si3C− and Si4−.11,12,61 The good agreement observed between the measured and predicted frequencies of Si3C in Fig. 2 supports the use of the B3LYP/cc-pVTZ level for the structural and vibrational assignment of the larger carbon-doped silicon clusters described below. The next two stable isomers 3b (C2v, 3B1) and 3c (Cs, 3A′′) are triplet states with relative energies well above 100 kJ mol−1 (Fig. 2). Isomer 3b exhibits a planar Y-shaped geometry, while 3c is a distorted triangular pyramid. At the MP2 level, isomer 3c relaxes to a C3v (3A1) distorted tetrahedron.
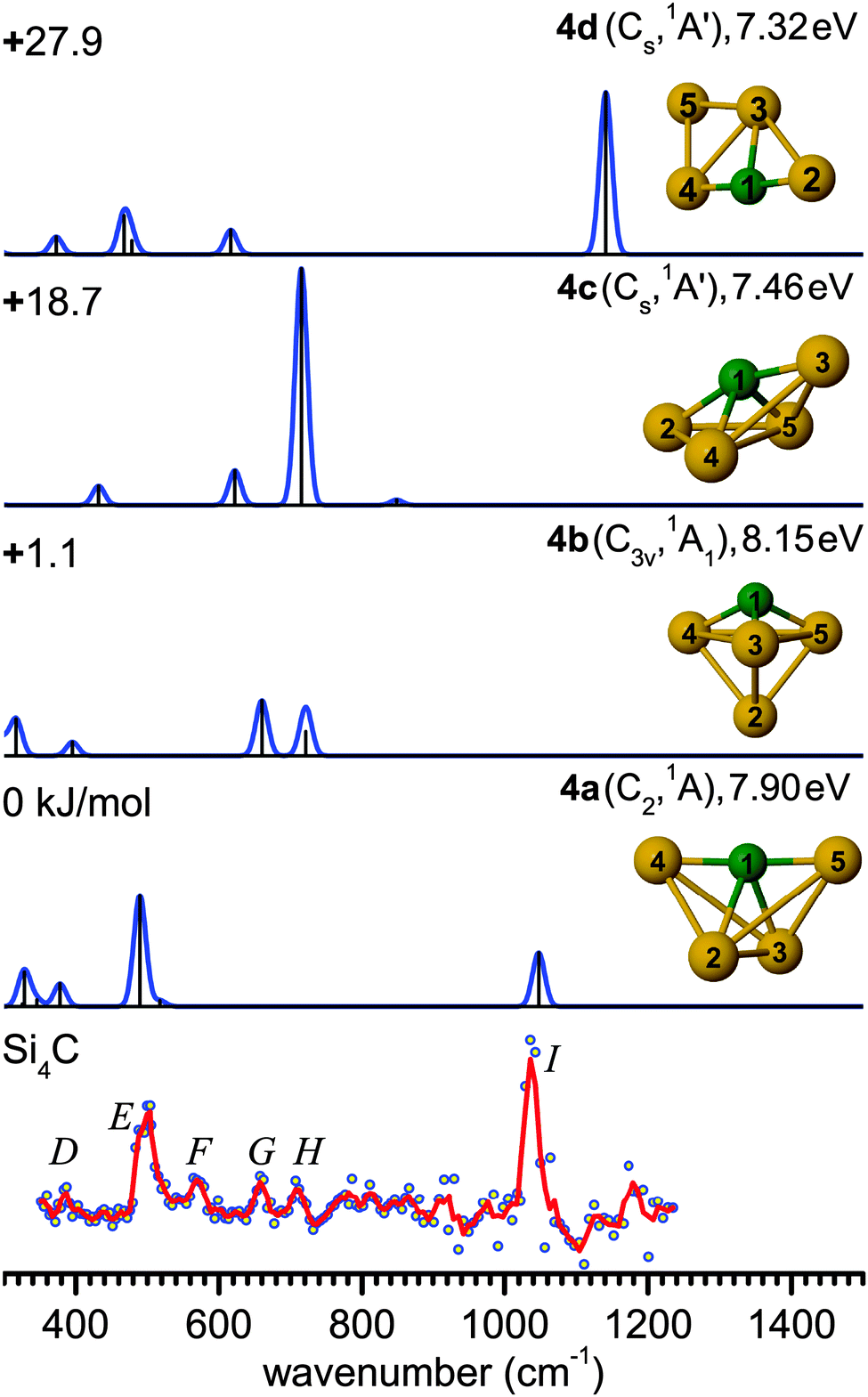 | ||
| Fig. 3 Comparison of the experimental IR-UV2CI spectrum of Si4C (bottom) with linear vibrational spectra of the four lowest-energy isomers (4a–4d) calculated at the B3LYP/cc-pVTZ level. Molecular symmetries and electronic states are given in parentheses. Relative energies are given in kJ mol−1, while vertical ionization energies are given in eV. The red line is the three-point adjacent average of the original data (circles). The experimental peak positions (in cm−1) are 388 (D), 499 (E), 571 (F), 659 (G), 710 (H), and 1037 (I). | ||
Interestingly, isomer 4b (C3v, 1A1) is often predicted as the ground state structure of Si4C,12,13,61,64 as also obtained here with the TPSS and MP2 methods (Table 1). This structure is formed by replacing the Si atom at one of the apexes of the trigonal bipyramidal Si5 (D3h, 1A1′) cluster.66 However, the energy difference between 4a and 4b is known to be strongly dependent on the theoretical level.33 For example, the present B3LYP calculations predict a lower-symmetry ground state structure 4a (C2, 1A), and the next stable structures are 1.1 (4b), 18.7 (4c, Cs, 1A′), and 27.9 kJ mol−1 (4d, Cs, 1A′) higher in energy. Structure 4a with an estimated IE of about 7.9 eV (Table 2) is expected to yield a somewhat different IR-UVC2I enhancement than 4b with a slightly higher IE (8.15 eV). Nonetheless, the observed relative band intensities in Fig. 3 suggest that the population of both isomers are of similar order of magnitude. The ground state structure of Si4C (4a) is similar to the most stable structure of its anion counterpart Si4C− (C2v, 2A1) predicted at the MP2/6-31G* level.12,61 A significant contribution of 4c and 4d to the IR-UV2CI spectrum is ruled out, because of their substantially higher energies and the mismatch of their predicted IR spectrum with the measured one. Finally, we note that the Si4C structures assigned from the IR-UV2CI spectrum are different from those postulated from the analysis of the photodetachment experiments of the corresponding anions.61 This example illustrates the importance of the direct spectroscopic study of the neutral clusters for the reliable experimental determination of the true ground state.
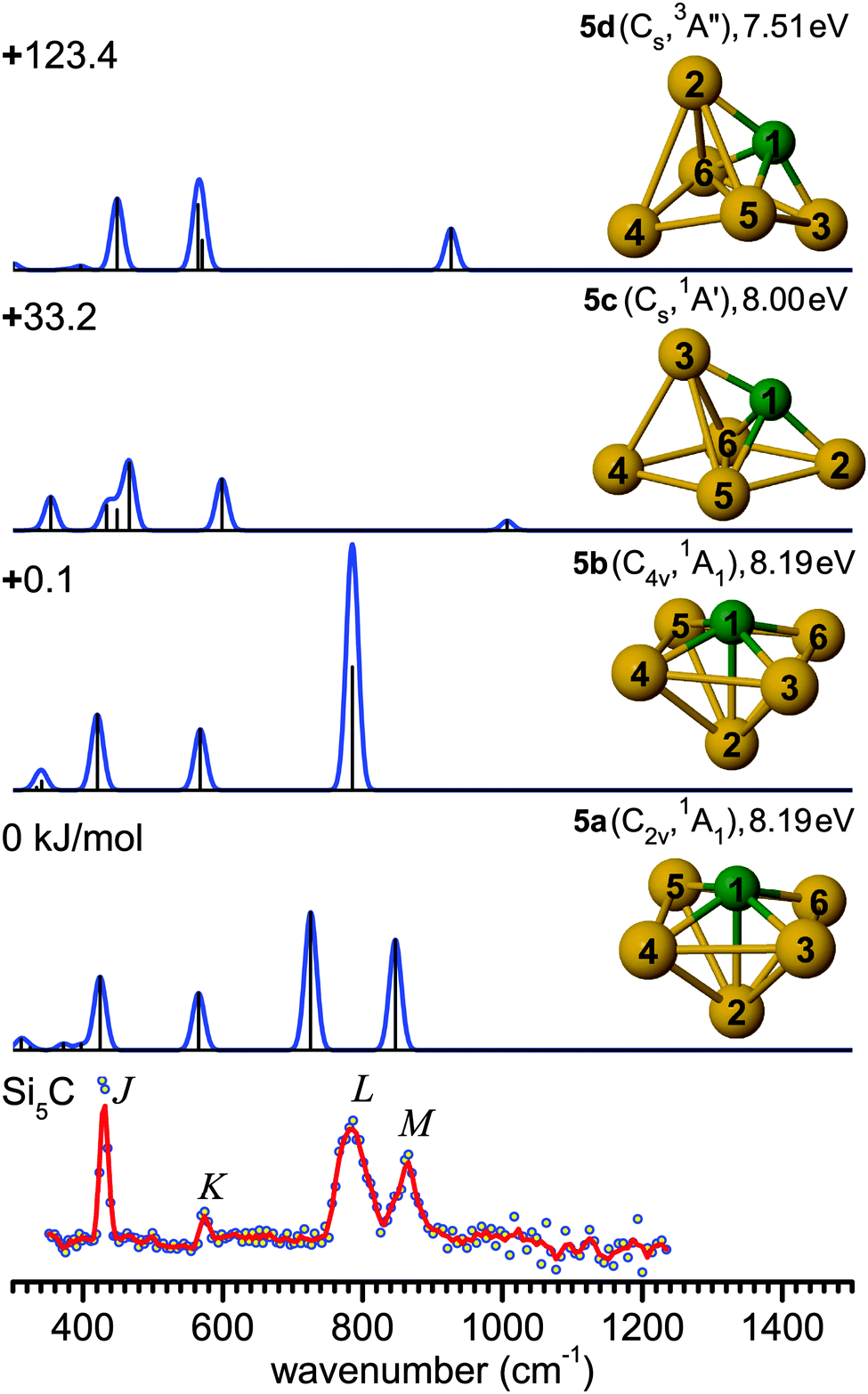 | ||
| Fig. 4 Comparison of the experimental IR-UV2CI spectrum of Si5C (bottom)50 with linear vibrational spectra of the four lowest-energy isomers (5a–5d) calculated at the B3LYP/cc-pVTZ level. Molecular symmetries and electronic states are given in parentheses, while vertical ionization energies are given in eV. Relative energies are given in kJ mol−1. The red line is the three-point adjacent average of the original data (circles). The experimental peak positions (in cm−1) are 430 (J), 574 (K), 786 (L), and 863 (M). | ||
The B3LYP/cc-pVTZ level suggests an alternative scenario. In contrast to the MP2 and TPSS levels, B3LYP yields a ground state structure 5a (C2v, 1A1) resulting from a distortion of the C4v cage 5b, along with an energy lowering of only 0.1 kJ mol−1, while 5b is a low-lying transition state, leading to a fluxional structure. Similar to 5a, the low frequency bands J and K can again be assigned to deformation and breathing modes at 424 (a1) and 565 cm−1 (a1). While in the higher frequency range, the 5b spectrum shows a single intense degenerate peak at 785 cm−1 (e) in excellent agreement with band L, 5a exhibits two split vibrational modes at 726 cm−1 (b1, asymmetric Si–C stretching of 4Si–1C–6Si) and 847 cm−1 (b2, asymmetric Si–C stretching of 3Si–1C–5Si). These may be assigned to bands L and M, however with unusually large redshifts of up to 50 cm−1 from experiment, which possibly arise from unusual anharmonicity along the distortion coordinate.
Similar findings were noted for isovalent Si6.47,49 Two possible ground state structures have also been proposed for Si6 showing the best agreement with the measured IR-UV2CI spectra, i.e., a C2v (1A1) structure found by DFT and a D4h (1A1g) structure predicted with MP2. This difference has been reasoned as distortions in Si6 induced by a pseudo Jahn–Teller effect.67,68 It was impossible to draw a firm conclusion on the true ground state structure of Si6, as a potential band splitting for the C2v structure was unresolved experimentally. For Si5C, the splitting is larger (∼120 cm−1) and hence observable. More theoretical efforts need to be devoted to the pseudo Jahn–Teller effects in Si6 and Si5C to fully understand the measured IR spectra. Such efforts are, however, beyond the scope of the present work.
For comparison, quantum chemical calculations for Si5C− have indicated a C2v (2A1) ground state for the anion, but the PE spectrum has been interpreted to mainly originate from a Cs (2A′) anion into a Cs (1A′) neutral state (corresponding to 5c).12,61 Similar to Si4C, the structures of the neutral Si5C clusters derived from the IR and PE spectra are different, due to the vertical transitions favored by the Franck–Condon principle.
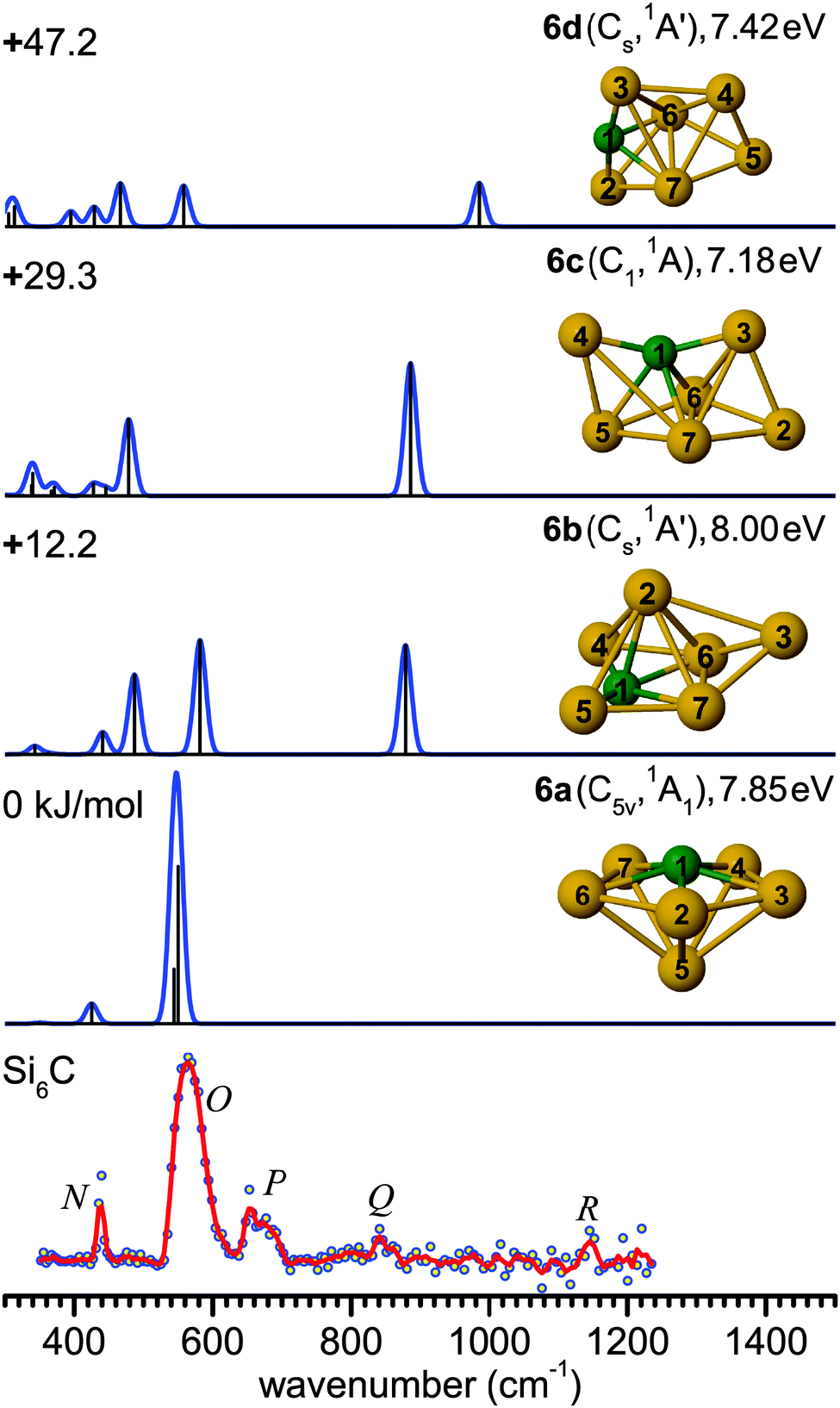 | ||
| Fig. 5 Comparison of the experimental IR-UV2CI spectrum of Si6C (bottom) with linear vibrational spectra of the four lowest-energy isomers (6a–6d) calculated at the B3LYP/cc-pVTZ level. Molecular symmetries and electronic states are given in parentheses. Relative energies are given in kJ mol−1, while vertical ionization energies are given in eV. The red line is the three-point adjacent average of the original data (circles). The experimental peak positions (in cm−1) are 438 (N), 566 (O), 664 (P), 842 (Q), and 1137 (R). | ||
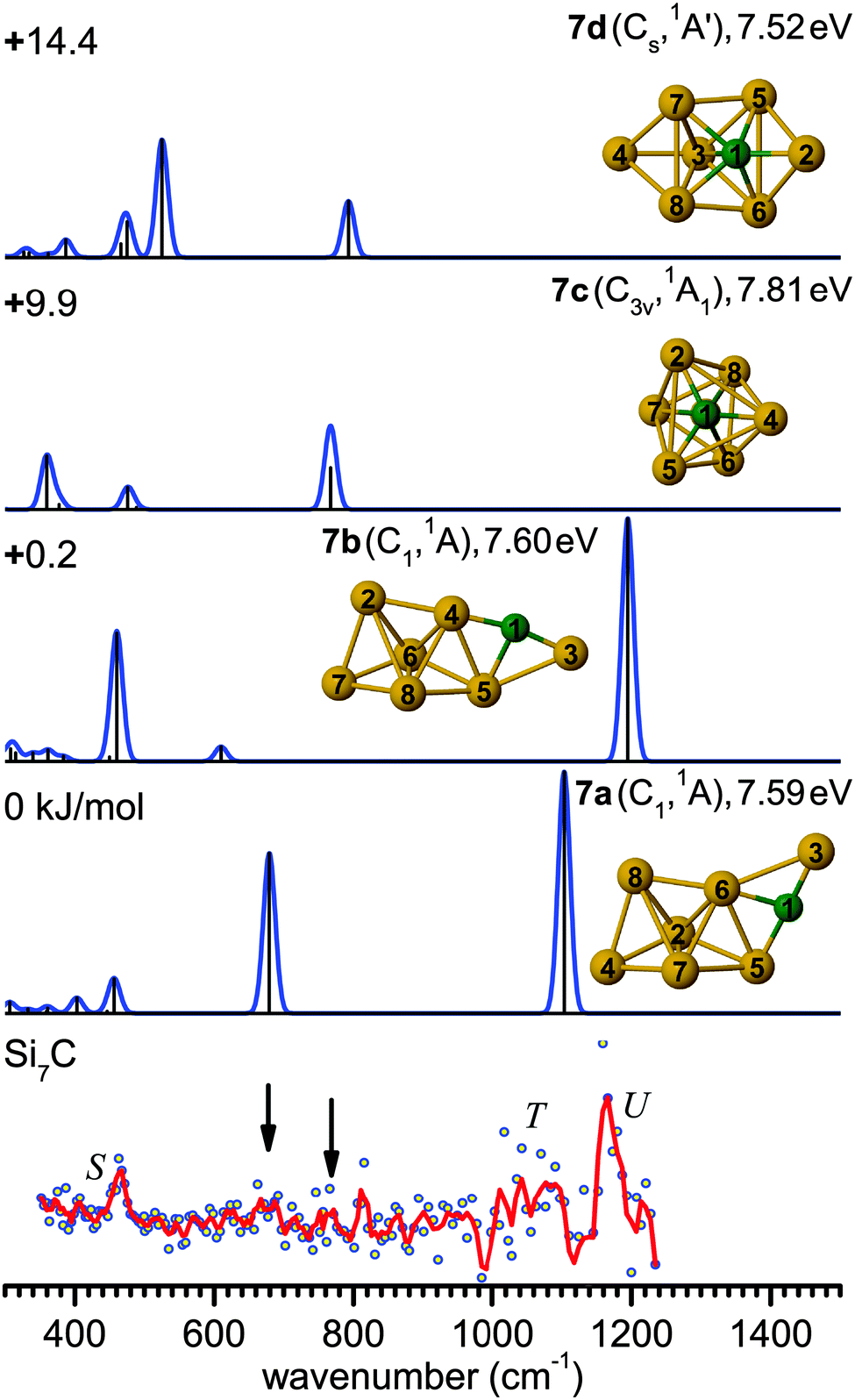 | ||
| Fig. 6 Comparison of the experimental IR-UV2CI spectrum of Si7C (bottom) with linear vibrational spectra of the four lowest-energy isomers (7a–7d) calculated at the B3LYP/cc-pVTZ level. Molecular symmetries and electronic states are given in parentheses. Relative energies are given in kJ mol−1, while vertical ionization energies are given in eV. The red line is the three-point adjacent average of the original data (circles). The experimental peak positions (in cm−1) are 464 (S), 1075 (T) and 1163 (U). Arrows indicate weak bands at 678 and 769 cm−1, which might be attributed to the modes at 679 (7a) and 768 cm−1 (7c), respectively. | ||
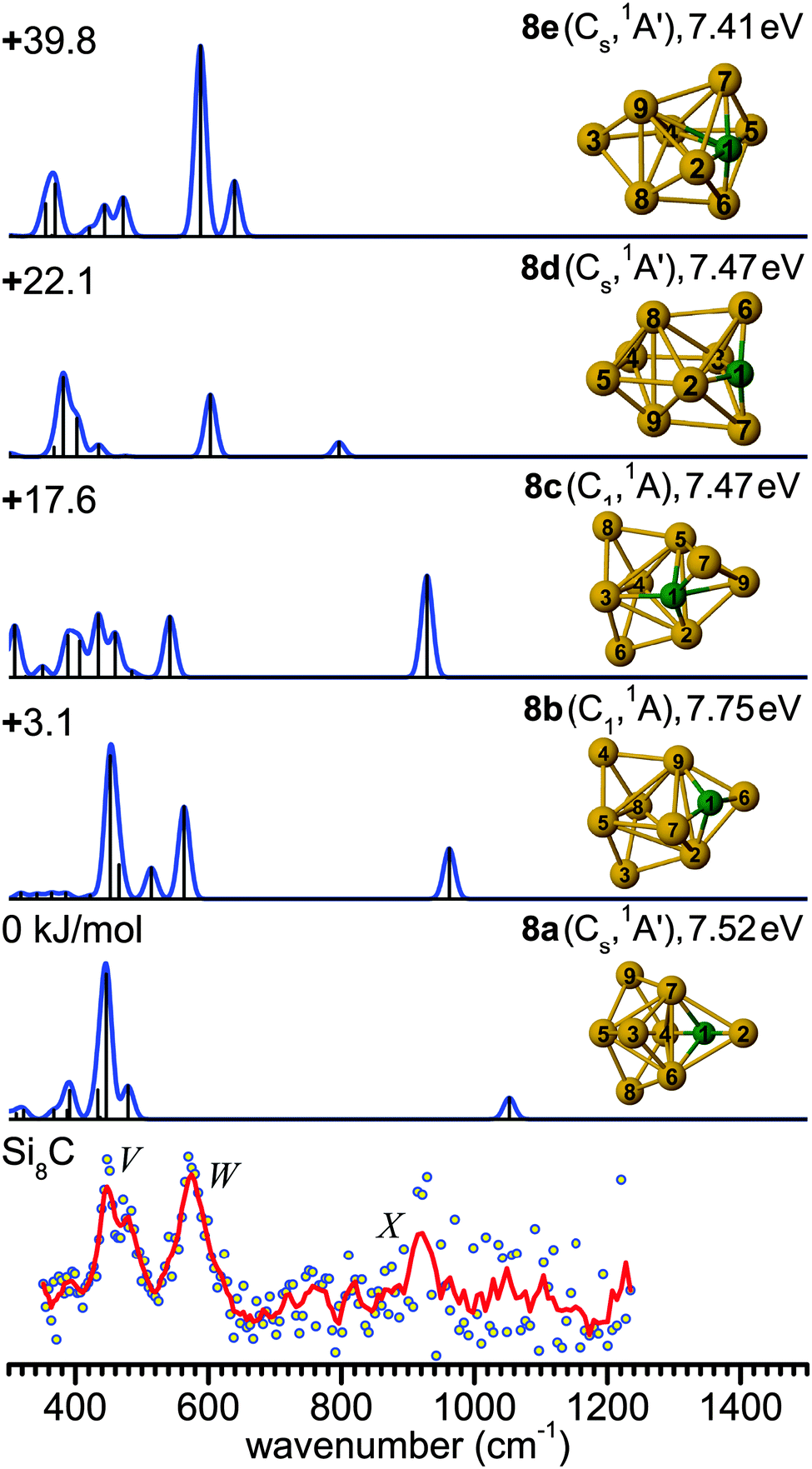 | ||
| Fig. 7 Comparison of the experimental IR-UV2CI spectrum of Si8C (bottom) with linear vibrational spectra of the five lowest-energy isomers (8a–8e) calculated at the B3LYP/cc-pVTZ level. Molecular symmetries and electronic states are given in parentheses. Relative energies are given in kJ mol−1, while vertical ionization energies are given in eV. The red line is the three-point adjacent average of the original data (circles). The experimental peak positions (in cm−1) are 449 (V), 574 (W), and 922 (X). | ||
The main feature in the IR spectrum of isomer 8a at 446 (a′) coincides with band V. Interestingly, structure 8a also contains the linear Si–C–Si block with ∠4Si–1C–2Si = 179.5°. The corresponding asymmetric Si–C–Si stretching mode at 1051.7 cm−1 (a′) is, however, weak. Therefore, a contribution of this isomer to the measured spectrum cannot be ruled out from the calculated IR spectrum. However, its calculated IE value is rather low (7.52 eV), so that the IR-UV2CI enhancement using 7.87 eV photons is probably weak. Similar conclusions apply to all higher energy isomers (7.41–7.47 eV for 8c–8e) with the notable exception of the identified isomer 8b (7.75 eV). Hence, our favored assignment of the measured spectrum is mainly to isomer 8b, while the presence of further isomers in the beam cannot be excluded. No other experimental information about Si8C is available for comparison.
B. Structural and electronic properties
The evolution of structures with increasing size for SinC (n = 3–8) is summarized in the left part of Fig. 8 showing the isomers that explain the major features of the experimental spectra. Most of these structures contain a Si3C substructure with a (nearly) linear Si–C–Si unit, except Si5C and Si6C. Interestingly, the Si3C cluster (along with Si2C) has been identified as a particular stable size in fragmentation experiments.72,73 This stability, thermodynamically and as structural element, may be explained in terms of its valence structure as shown in Fig. 8 (right). Boys localization of the eight valence orbitals reveals the presence of three lone pairs, each at one of the Si atoms, two Si–C σ-bonds in the Si–C–Si unit, as well as three delocalized bonds, one of them being a π-bond delocalized over the entire cluster. The resulting formal bond orders range from 0.53 between neighboring Si atoms to 1.53 within the linear Si–C–Si unit. The high bond order in the Si–C–Si unit goes along with relatively short Si–C bond lengths of 176 pm as compared to the third Si–C bond, which is 194 pm long and has a formal bond order of only 0.87. The larger clusters also have similar bond lengths within the Si–C–Si bridge unit. The average of the corresponding Si–C distances in this unit is a measure of its strength and can be directly related with the frequency of the asymmetric Si–C–Si stretching mode observed experimentally. For the structures in which we identify this element (3a, 4a, 7b, 8b) this particular mode is found at 1080, 1037, 1163 and 922 cm−1, respectively, and the average Si–C distances are 176, 178, 174 and 183 pm, i.e. there is a clear anti-correlation between bond length and stretching frequency.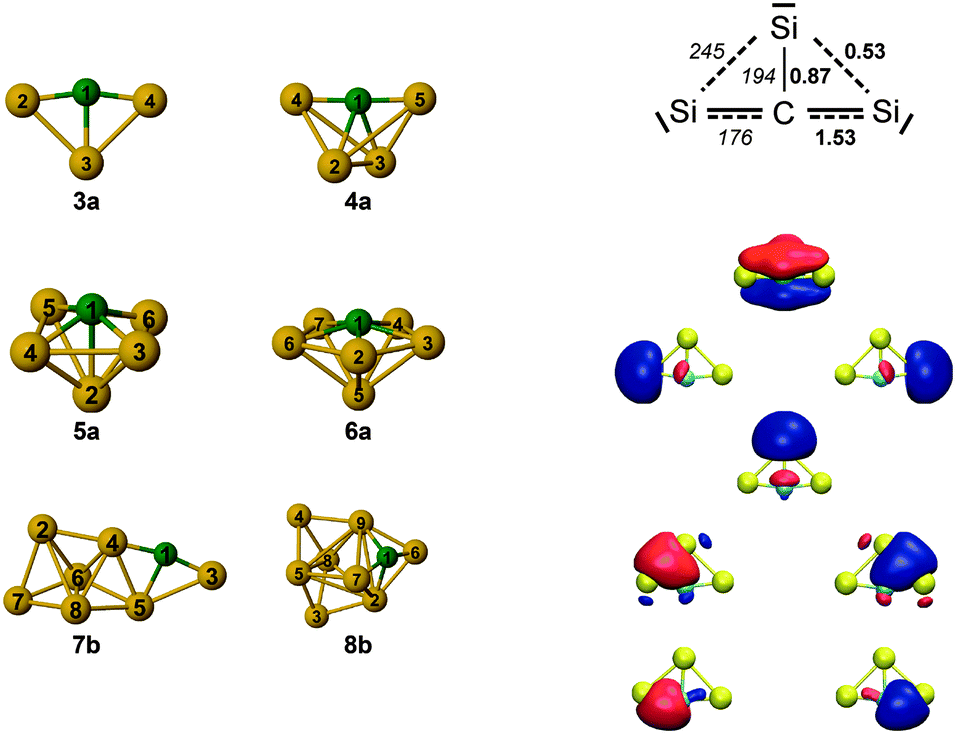 | ||
| Fig. 8 (left) Overview of the structures observed for SinC (n = 3–8). Most of these structures contain a Si3C substructure with a (nearly) linear Si–C–Si unit, except Si5C and Si6C. (right) Valence structure and localized molecular orbitals for Si3C as obtained from a Boys localization. Formal bond orders (bold) are compared to the bond lengths in pm (italics). | ||
The NBO charge analysis is increasingly used for a view of local charge instead of the unreliable Mulliken population analysis. As expected for SinC clusters, a large negative charge is located at the C atom (almost −2e, Table S1 in ESI†), which is more electronegative than Si. The positive part is mainly shared by the Si atoms directly bound to the C atom. This result suggests that these Si atoms are very good electron donors, as observed previously for the first-row element doped silicon clusters.51
Vertical excitations of selected stable clusters calculated with the second-order approximate coupled cluster and TD-DFT methods are listed in Table 3 for the first three transitions. The first electronic transition energies for the considered clusters are relatively high (>1.65 eV).
IV. Conclusions
Carbon-doped silicon clusters are characterized with IR-UV2CI spectroscopy combined with quantum chemical simulations. The structures of the SinC (n = 3–8) clusters are determined by comparison of the measured IR-UV2CI spectra with linear IR absorption spectra of the low-energy isomers calculated at the B3LYP/cc-pVTZ level. Optimized stable structures are found employing global optimization algorithms coupled with electronic structure (DFT) methods. This technique is robust and reliable in searching for the ground state structures of such type of clusters, in which electronic effects might play an important role. For Si3C and Si6C, only the lowest-energy isomers are present in the prepared cluster sample, while for Si4C, Si5C, Si7C, and Si8C multiple stable structures may co-exist. The nearly linear motif (Si–C–Si) is identified as characteristic building block in several of the most stable SinC structures (n = 3–5, 7, 8). The NBO analysis shows that most charge is located on the C atom and its nearest-neighbor Si atoms. Although the theoretical approaches used in this work agree well on predicting low-energy isomers, the B3LYP method proves to be the most reliable one in finding the ground state structures.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft within the research unit FOR 1282 (DO 729/5, FI 893/4). We gratefully acknowledge the support of the Stichting voor Fundamenteel Onderzoek der Materie (FOM) in providing beam time of FELIX and the FELIX staff for their skillful assistance, in particular, B. Redlich and A. F. G. van der Meer. The project has received funding from the European Community's Seventh Framework Programme (FP7/2007–2013) under Grant Agreement No. 226716. We thank J. Langer and A. Lagutschenkov for their initial contribution.References
- P. Melinon, B. Masenelli, F. Tournus and A. Perez, Nat. Mater., 2007, 6, 479–490 CrossRef CAS PubMed.
- I. Cherchneff, Y. H. Le Teuff, P. M. Williams and A. G. G. M. Tielens, Astron. Astrophys., 2000, 357, 572–580 CAS.
- K. P. Lim and F. W. Lampe, Int. J. Mass Spectrom., 1990, 101, 245–256 CrossRef CAS.
- S. K. Mandal and H. W. Roesky, Chem. Commun., 2010, 46, 6016–6041 RSC.
- D. S. N. Parker, A. V. Wilson, R. I. Kaiser, N. J. Mayhall, M. Head-Gordon and A. G. G. M. Tielens, Astrophys. J, 2013, 770, 33 CrossRef.
- P. Thaddeus, S. E. Cummins and R. A. Linke, Astrophys. J., 1984, 283, L45–L48 CrossRef CAS PubMed.
- M. Frenklach, C. S. Carmer and E. D. Feigelson, Nature, 1989, 339, 196–198 CrossRef CAS PubMed.
- F. Combes, M. Boquien, C. Kramer, E. M. Xilouris, F. Bertoldi, J. Braine, C. Buchbender, D. Calzetti, P. Gratier, F. Israel, B. Koribalski, S. Lord, G. Quintana-Lacaci, M. Relano, M. Rollig, G. Stacey, F. S. Tabatabaei, R. P. J. Tilanus, F. van der Tak, P. van der Werf and S. Verley, Astron. Astrophys., 2012, 539, A67 CrossRef PubMed.
- K. Furuya, Y. Aikawa, K. Tomida, T. Matsumoto, K. Saigo, K. Tomisaka, F. Hersant and V. Wakelam, Astrophys. J., 2012, 758, 86 CrossRef.
- M. Satta, T. Grassi and F. A. Gianturco, Mon. Not. R. Astron. Soc., 2013, 429, 269–274 CrossRef CAS PubMed.
- M. Steglich and J. P. Maier, Astrophys. J., 2015, 801, 119 CrossRef.
- A. Nakajima, T. Taguwa, K. Nakao, M. Gomei, R. Kishi, S. Iwata and K. Kaya, J. Chem. Phys., 1995, 103, 2050 CrossRef CAS PubMed.
- Q. Y. Chu, B. X. Li and J. Yu, J. Mol. Struct., 2007, 806, 67–76 CrossRef CAS PubMed.
- C. M. L. Rittby, J. Chem. Phys., 1992, 96, 6768–6772 CrossRef CAS PubMed.
- C. M. L. Rittby, J. Chem. Phys., 1994, 100, 175–180 CrossRef CAS PubMed.
- R. S. Grev and H. F. Schaefer, J. Chem. Phys., 1984, 80, 3552–3555 CrossRef CAS PubMed.
- R. S. Grev and H. F. Schaefer, J. Chem. Phys., 1985, 82, 4126–4130 CrossRef CAS PubMed.
- R. S. Grev and H. F. Schaefer, Chem. Phys. Lett., 1985, 119, 111–118 CrossRef CAS.
- I. L. Alberts, R. S. Grev and H. F. Schaefer, J. Chem. Phys., 1990, 93, 5046–5052 CrossRef CAS PubMed.
- Z. Y. Jiang, X. H. Xu, H. S. Wu, F. Q. Zhang and Z. H. Jin, J. Mol. Struct., 2002, 589, 103–109 CrossRef.
- Z. Y. Jiang, X. H. Xu, H. S. Wu, F. Q. Zhang and Z. H. Jin, Chin. J. Struct. Chem., 2003, 22, 459–463 CAS.
- Z. Y. Jiang, X. H. Xu, H. S. Wu and Z. H. Jin, J. Phys. Chem. A, 2003, 107, 10126–10131 CrossRef CAS.
- Z. Y. Jiang, X. H. Xu, H. S. Wu, F. Q. Zhang and Z. H. Jin, Chem. Phys., 2003, 290, 223–231 CrossRef CAS.
- Z. Y. Jiang, X. H. Xu, H. S. Wu, F. Q. Zhang and Z. H. Jin, J. Mol. Struct., 2003, 624, 61–67 CrossRef CAS.
- Z. Y. Jiang, X. H. Xu, H. S. Wu, F. Q. Zhang and Z. H. Jin, J. Mol. Struct., 2003, 621, 279–284 CrossRef CAS.
- J. Hou and B. Song, J. Chem. Phys., 2008, 128, 154304 CrossRef PubMed.
- B. Song, Y. Yong, J. Hou and P. He, Eur. Phys. J. D, 2010, 59, 399–406 CrossRef CAS.
- J. Zhang, W. C. Lu, Q. J. Zang, L. Z. Zhao, C. Z. Wang and K. M. Ho, J. Phys.: Condens. Matter, 2011, 23, 205305 CrossRef PubMed.
- S. Erkoc and L. Turker, Physica E, 2000, 8, 50–56 CrossRef CAS.
- M. Cui, M. Ge, J. Feng, H. Zhang, W. Tian and C. Sun, J. Mol. Struct., 1999, 492, 241–256 CrossRef CAS.
- Q. X. Li, W. C. Lu, Q. J. Zang, L. Z. Zhao, C. Z. Wang and K. M. Ho, Comput. Theor. Chem., 2011, 963, 439–447 CrossRef CAS PubMed.
- S. Hunsicker and R. O. Jones, J. Chem. Phys., 1996, 105, 5048–5060 CrossRef CAS PubMed.
- A. D. Zdetsis, G. Froudakis, M. Muhlhauser and H. Thumnel, J. Chem. Phys., 1996, 104, 2566–2573 CrossRef CAS PubMed.
- G. E. Davico, R. L. Schwartz and W. C. Lineberger, J. Chem. Phys., 2001, 115, 1789–1794 CrossRef CAS PubMed.
- J. D. Presilla-Marquez, S. C. Gay, C. M. L. Rittby and W. R. M. Graham, J. Chem. Phys., 1995, 102, 6354–6361 CrossRef CAS PubMed.
- J. D. Presilla-Marquez and W. R. M. Graham, J. Chem. Phys., 1994, 100, 181–185 CrossRef CAS PubMed.
- J. D. Presilla-Marquez and W. R. M. Graham, J. Chem. Phys., 1992, 96, 6509–6514 CrossRef CAS PubMed.
- P. A. Withey and W. R. M. Graham, J. Chem. Phys., 1992, 96, 4068–4072 CrossRef CAS PubMed.
- J. D. Presilla-Marquez, C. M. L. Rittby and W. R. M. Graham, J. Chem. Phys., 1996, 104, 2818–2824 CrossRef CAS PubMed.
- T. H. Le, C. M. L. Rittby and W. R. M. Graham, J. Chem. Phys., 2014, 140, 064314 CrossRef CAS PubMed.
- X. D. Ding, S. L. Wang, C. M. L. Rittby and W. R. M. Graham, J. Phys. Chem. A, 2000, 104, 3712–3717 CrossRef CAS.
- X. D. Ding, S. L. Wang, C. M. L. Rittby and W. R. M. Graham, J. Chem. Phys., 1999, 110, 11214–11220 CrossRef CAS PubMed.
- J. D. Presilla-Marquez, C. M. L. Rittby and W. R. M. Graham, J. Chem. Phys., 1997, 106, 8367–8373 CrossRef CAS PubMed.
- N. J. Reilly, M. Steglich, D. L. Kokkin, J. P. Maier, J. F. Stanton and M. C. McCarthy, J. Mol. Spectrosc., 2015, 310, 135–140 CrossRef CAS PubMed.
- J. F. Stanton, J. Dudek, P. Theule, H. Gupta, M. C. McCarthy and P. Thaddeus, J. Chem. Phys., 2005, 122, 124314 CrossRef PubMed.
- P. Gruene, D. M. Rayner, B. Redlich, A. F. G. van der Meer, J. T. Lyon, G. Meijer and A. Fielicke, Science, 2008, 321, 674–676 CrossRef CAS PubMed.
- M. Haertelt, J. T. Lyon, P. Claes, J. de Haeck, P. Lievens and A. Fielicke, J. Chem. Phys., 2012, 136, 064301 CrossRef PubMed.
- G. von Helden, D. van Heijnsbergen and G. Meijer, J. Phys. Chem. A, 2003, 107, 1671–1688 CrossRef CAS.
- A. Fielicke, J. T. Lyon, M. Haertelt, G. Meijer, P. Claes, J. de Haeck and P. Lievens, J. Chem. Phys., 2009, 131, 171105 CrossRef PubMed.
- M. Savoca, A. Lagutschenkov, J. Langer, D. J. Harding, A. Fielicke and O. Dopfer, J. Phys. Chem. A, 2013, 117, 1158–1163 CrossRef CAS PubMed.
- N. X. Truong, M. Savoca, D. J. Harding, A. Fielicke and O. Dopfer, Phys. Chem. Chem. Phys., 2014, 16, 22364–22372 RSC.
- D. J. Harding, C. Kerpal, G. Meijer and A. Fielicke, J. Phys. Chem. Lett., 2013, 4, 892–896 CrossRef CAS.
- R. Gehrke and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 085412 CrossRef.
- D. Oepts, A. F. G. van der Meer and P. W. van Amersfoort, Infrared Phys. Technol., 1995, 36, 297–308 CAS.
- M. Haertelt, A. Fielicke, G. Meijer, K. Kwapien, M. Sierka and J. Sauer, Phys. Chem. Chem. Phys., 2012, 14, 2849–2856 RSC.
- S. Heiles and R. L. Johnston, Int. J. Quantum Chem., 2013, 113, 2091–2109 CrossRef CAS PubMed.
- D. J. Wales, Energy landscapes, Cambridge University Press, Cambridge, 2003 Search PubMed.
- E. M. Fernández, J. M. Soler, I. L. Garzón and L. C. Balbás, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 165403 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN09, Rev.D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- R. Kishi, M. Gomei, A. Nakajima, S. Iwata and K. Kaya, J. Chem. Phys., 1996, 104, 8593–8604 CrossRef CAS PubMed.
- P. S. Yadav, R. K. Yadav, S. Agrawal and B. K. Agrawal, Physica E, 2006, 33, 249–262 CrossRef CAS PubMed.
- K. Raghavachari and V. Logovinsky, Phys. Rev. Lett., 1985, 55, 2853–2856 CrossRef CAS.
- K. Raghavachari, Abstr. Pap. Am. Chem. Soc., 1986, 192, 96 Search PubMed.
- K. Raghavachari, J. Chem. Phys., 1986, 84, 5672–5686 CrossRef CAS PubMed.
- S. Li, R. J. Vanzee, W. Weltner and K. Raghavachari, Chem. Phys. Lett., 1995, 243, 275–280 CrossRef CAS.
- A. D. Zdetsis, J. Chem. Phys., 2007, 127, 014314 CrossRef PubMed.
- P. Karamanis, D. Zhang-Negrerie and C. Pouchan, Chem. Phys., 2007, 331, 417–426 CrossRef CAS PubMed.
- E. C. Honea, A. Ogura, C. A. Murray, K. Raghavachari, W. O. Sprenger, M. F. Jarrold and W. L. Brown, Nature, 1993, 366, 42–44 CrossRef CAS PubMed.
- K. Raghavachari and C. M. Rohlfing, J. Chem. Phys., 1988, 89, 2219–2234 CrossRef CAS PubMed.
- G. Meloni and K. A. Gingerich, J. Chem. Phys., 2001, 115, 5470 CrossRef CAS PubMed.
- B. W. Ticknor and M. A. Duncan, Chem. Phys. Lett., 2005, 405, 214–219 CrossRef CAS PubMed.
- M. Pellarin, C. Ray, P. Melinon, J. Lerme, J. L. Vialle, P. Keghelian, A. Perez and M. Broyer, Chem. Phys. Lett., 1997, 277, 96–104 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp02588e |
| This journal is © the Owner Societies 2015 |