
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correlation of electrochromic properties and oxidation states in nanocrystalline tungsten trioxide†
S.
Darmawi
a,
S.
Burkhardt
a,
T.
Leichtweiss
b,
D. A.
Weber
b,
S.
Wenzel
b,
J.
Janek
b,
M. T.
Elm
*ab and
P. J.
Klar
*a
aJustus-Liebig-Universität Gießen, I. Physikalisches Institut, Heinrich-Buff-Ring 16, 35392 Gießen, Germany. E-mail: Matthias.Elm@phys.Chemie.uni-giessen.de; Peter.J.Klar@exp1.physik.uni-giessen.de; Tel: +49 641-9934544 Tel: +49 641-9933190
bJustus-Liebig-Universität Gießen, Physikalisch-Chemisches Institut, Heinrich-Buff-Ring 58, 35392 Gießen, Germany
First published on 13th May 2015
Abstract
Although tungsten trioxide (WO3) has been extensively studied since its electrochromic properties were first discovered, the mechanism responsible for the coloration or bleaching effect is still disputed. New insights into the coloration mechanism of electrochromic, nanocrystalline WO3 are provided in this paper by studying thin WO3 films combining the electrochemical and spectroscopic techniques. By employing in situ UV-Vis transmission spectroscopy at a fixed spectral band pass during electrochemical experiments, such as cyclic voltammetry, a two-step insertion process for both protons and lithium ions is identified, of which one step exhibits a significantly higher coloration efficiency than the other. To obtain a better understanding of the insertion process AxWO3 (A = H, Li,…) thin films were studied at different stages of intercalation using UV-Vis and X-ray photoelectron spectroscopy. The results show that the first step of the intercalation process represents the reduction from initial W6+ to W5+ and the second step the reduction of W5+ to W4+. We found that the blue coloration of this nanocrystalline tungsten trioxide is mainly due to the presence of W4+ rather than that of W5+.
1 Introduction
Electrochromism – the property of a thin film to reversibly change its transmission properties by electrochemical reduction or oxidation1–4 – has been intensively investigated during the last few decades.2,4,5 Studies on electrochromism were first published by Deb6 in 1969 and reports on energy efficient fenestration for buildings7,8 followed in 1984. Tungsten trioxide in particular is studied in more detail than any other chromogenic material for several reasons: it exhibits thermochromic, photochromic, gasochromic as well as electrochromic characteristics and is thus the most relevant material for chromogenic devices such as smart windows or gas sensors, etc.9–11 As other chromogenic oxides, such as MoO3, are somewhat similar in their properties, tungsten trioxide represents a widely used model system2 for examining the coloration processes.The general expression for the electrochromic reaction in tungsten trioxide is given by
| xA+ + xe− + WO3 ↔ AxWO3, | (1) |
Today two main mechanisms are usually discussed for explaining the color change in AxWO3: for crystalline systems, the effects are often discussed in terms of optical transitions or collective excitation of quasi-free electrons in not fully occupied bands in a one-electron band structure (rigid-band model), whereas in the case of amorphous films the effects are often described in terms of intervalence charge transfer (IVCT). The charge transfer involves W atoms in different oxidation states either alone or in conjunction with additional defects such as oxygen vacancies.13–18 The coloration and bleaching processes are difficult to describe in a single-particle model, i.e. for an appropriate theoretical description coupling of the carriers to the lattice as well as electron–electron interactions need to be accounted for at least to some degree. Therefore, details of the mechanisms are still controversially discussed and a unified model for the description of crystalline as well as amorphous films is not established yet. In the following, we summarize some of the current views.
Schirmer et al. presented a model for small polaron transitions between sites A and B, both occupied by tungsten, but of different oxidation states, i.e.:
| hν + W5+(A) + W6+(B) → W6+(A) + W5+(B). | (2) |
In bandstructure theory of crystalline materials, polarons go beyond the one-electron picture. They are technically dealt with as conduction band electrons accompanied by a spontaneous lattice deformation. This may be crudely incorporated into the one-electron picture as levels splitting off the bottom of the single-particle conduction band as a result of considering electron–phonon coupling as a perturbation. Electrons injected from the cathode due to insertion of alkali or hydrogen fill these states in the conduction band. Optical absorption is thus generated by optical transitions between occupied and unoccupied states.10,17
However, the details of the mechanisms are strongly debated: Zhang et al. also reported on small polaron absorption for amorphous WO3, but in this case with transitions between W5+ and W4+ sites.15 Broclawik et al. introduced a WO6 octahedra cluster model that bridges the gap between models for crystalline and amorphous WO3.17 Deb et al. proposed a new model, based on the presence of oxygen vacancies (analogous to F-centers) with associated W4+ ions.16 In a recent study, Limaye et al. performed in situ XANES on crystalline WO3 and found that coloration/bleaching is due to filling/unfilling of the W 5d–O 2p hybridized conduction band states. They also object to the intervalence transition between different tungsten sites and the formation of polarons. Based on their results, they could not verify a corresponding change in the nearest-neighbor W–O bond distances.19 Zhong et al. generally object to utilizing the rigid band model for charge transfer of the inserted electrons, i.e. a quick change in the crystal phase upon ion insertion destroys the validity of the rigid band model.20
Thus, to date it is not clear whether all mechanisms leading to the coloration in the various WO3 phases are known and to which extent they differ for different phases. With our study we add some additional experimental evidence which suggests that the presence of W4+ is essential for the coloration of nanocrystalline WO3. We analyze the electrochromic properties of nanocrystalline tungsten trioxide thin films prepared via a dip coating process. Dip coating is a versatile and cost-effective fabrication technique which due to its scalability is also of interest for industrial applications where a film deposition on large areas is required. By employing in operando optical transmission spectroscopy during both cyclic voltammetric and galvanostatic experiments we were able to identify a two-step insertion process for both protons and lithium ions, of which only one step causes significant coloration. To obtain further insight into the nature of this two-step mechanism, the oxidation state of tungsten as well as the crystallinity of the WO3 thin films prepared was analyzed as a function of the amount of the inserted charge.
2 Experimental details
2.1 Sample preparation
The preparation process for nanocrystalline WO3 thin films was adopted from Brezesinski et al.21 A solution consisting of 1.19 g of WCl6 in 4.8 g of ethanol was stirred for approximately 12 h. Approximately 10 mg of a block-copolymer (Pluronic 127) was used for reduction of the surface tension. Thin films were prepared by dip coating on FTO substrates (sheet resistance of 7 Ω sq−1) in a controlled air atmosphere (relative humidity < 10%) at a constant withdrawal rate of 10 mm s−1. After deposition, the films were dried for 1 h at 100 °C and 12 h at 300 °C (ramp 2 °C min−1).Annealing the samples in air at 450 °C for 30 minutes with a ramp rate of 5 °C h−1 resulted in nanocrystalline WO3. The resulting layers have a thickness of about 100 nm.
2.2 Electrochemical experiments
Proton insertion, using 0.1 M H2SO4 in H2O as an electrolyte, was carried out in a specially designed compact three-electrode cell (adapted from Wallys et al.22) consisting of a 3 M KCl Ag/AgCl reference electrode (E0 = 0.21 V vs. SHE, Driref-450 microreference, WPI) and a platinum counter electrode. The geometrical area of the WO3 working electrode was 0.07 mm2. Electrochemical tests were performed using a Jaissle IMP 83 PC potentiostat.Lithium insertion, using 0.2 M LiClO4 in propylene carbonate (PC) as an electrolyte, was carried out in a different, also specially designed compact three-electrode cell, consisting of a lithium reference electrode and a lithium counter electrode. The cell was assembled inside a glove box in an argon atmosphere with controlled H2O and O2 concentrations of less than 5 ppm and 0.1 ppm respectively. A Bio-logic sp300 potentiostat was used. The geometrical area of the WO3 working electrode was 0.07 mm2.
For transmission experiments, the films were illuminated by a halogen lamp. In the case of lithium insertion, the transmission was detected spectrally resolved using a spectrometer (LR1 spectrometer, ASEQ) whereas, in the case of hydrogen insertion, a subtractive double spectrometer (Spex 1680 b, Horiba) equipped with a CCD-Camera (pco1400) was used to detect light in a spectral window of 633 nm ± 15 nm.
Cyclic voltammetry experiments were carried out starting at the open circuit potential with a scan rate of 1 mV s−1 towards lower potentials. In the case of hydrogen insertion the investigated voltage range was between 1 V and −0.6 V vs. Ag/AgCl, while in the case of lithium insertion the voltage range was varied from 4 V to 1.7 V vs. Li/Li+.
2.3 Spectroscopic characterization
UV-Vis transmission spectroscopy was performed on a Perkin Elmer Lambda 900 spectrometer. The integration time was 0.16 s and the scan speed was 300 nm s−1 for the UV-Vis range and 375 nm s−1 for the NIR range with a spectral resolution of 5 nm.Samples for ex situ XPS and UV-Vis analyses were prepared as follows: the galvanostatic lithium insertion was carried out inside a glove box. A two-electrode setup inside a cuvette with lithium foil as a counter electrode was employed. Stainless steel screws were used as electric contacts and for fixing the counter electrode and the sample.
XPS measurements were performed using a PHI 5000 Versaprobe Scanning ESCA Microprobe (Physical Electronics, Inc.) equipped with a monochromatic aluminum X-ray source (beam area 200 μm × 1400 μm, X-ray power 100 W) at a chamber pressure of around 10−8 mbar. Pass energy for high resolution spectra was 23.5 eV with an energy step width of 0.1 eV. The spectra were analyzed using the CasaXPS software package (Version 2.3.17dev6.3y). The spectra were energy calibrated by shifting the binding energy of the adventitious carbon signal to a position of 284.8 eV.
2.4 Structural investigations
Structural investigations on pristine and lithium-inserted samples were carried out by X-ray diffraction (XRD), using a PANalytical Empyrean diffractometer with Cu Kα radiation (λ1 = 154.056 pm, λ2 = 154.539 pm, I(λ1/λ2 = 0.5) in Bragg–Brentano θ–θ geometry. Diffraction diagrams were recorded in a 2θ range from 10° to 90° with a step size of 0.013° and a counting time of 270 s per step. Additional powder samples (see Results section) were measured in a similar manner, but with a step size of 0.026°.3 Results
3.1 Cyclic voltammetry and transmission
For the discussion of the electrochromic properties of nanocrystalline WO3 films we use various representations of the spectro-electrochemical data, which are introduced in the following. In a typical cyclic voltammogram (CV) the amount of transferred charge per unit time (i.e. electric current) is analyzed as a function of the working electrode potential (WO3 thin film). It is worth mentioning that the cell current, due to the constant rate of change of potential E, i.e. a constant scan rate v = dE/dt [V s−1], is directly proportional to the differential capacity (dQ/dE) during a CV experiment:| I = dQ/dt = v·dQ/dE. | (3) |
| dT/dE = v−1·dT/dt. | (4) |
| η = d(OD)/dQ. | (5) |
As an example, Fig. 1(a) shows the first cycle of a cyclic voltammogram of a WO3 thin film in aqueous H2SO4. The cell current is plotted versus the applied working electrode potential E measured with respect to the Ag/AgCl reference electrode. Starting at the open circuit potential, around 0.29 V vs. Ag/AgCl, the potential is reduced at a rate of v = 1 mV s−1. The resulting negative current causes insertion of electrons and ions, in this case protons, into the tungsten trioxide film. This process results in a bluish coloration of WO3. At around −0.05 V vs. Ag/AgCl and −0.3 V vs. Ag/AgCl maxima of the negative current, “reduction peaks”, are observed. The strong current increase at −0.6 V vs. Ag/AgCl can be attributed to the unwanted side reactions at the substrate, such as hydrogen evolution or substrate decomposition. For comparison, a CV measurement which was performed for the substrate only is shown as a green curve. Fig. 1(b) shows the corresponding transmission characteristics of the same WO3 sample. The transmission decreases as the cathodic current increases to more negative values, meaning that with an increase in the amount of inserted hydrogen the coloration of the material increases. The coloration process is reversible, i.e. with decreasing cathodic current the transmission increases again. At −0.6 V vs. Ag/AgCl the scan direction is reversed and the potential is increased at a rate of 1 mV s−1. At approximately −0.4 V vs. Ag/AgCl, positive cell currents are observed that correspond to a net oxidation (hydrogen extraction) of the WO3 film.
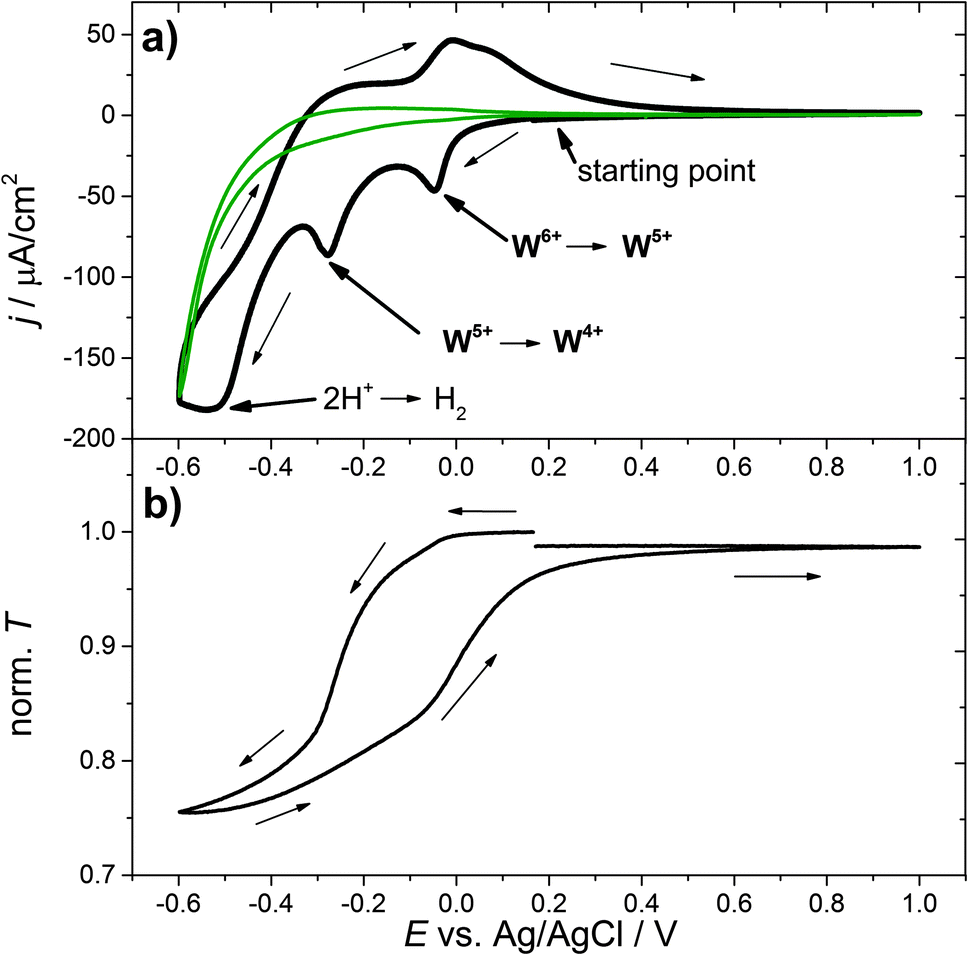 | ||
| Fig. 1 (a) Cyclic voltammogram of a WO3 film in a 0.1 M H2SO4 in H2O electrolyte with a scan rate of 1 mV s−1. The green curve shows the same measurement for the substrate alone. (b) Corresponding transmission characteristics during hydrogen insertion recorded for a spectral window of 633 nm ± 15 nm. Norm. T denotes the transmission T at the center wavelength 633 nm normalized to the corresponding transmission T0 at the beginning of the experiment. | ||
To further examine the electrochemical coloration process, the change in transmission with time (dT/dt) is analyzed. As a constant scan rate (v = 1 mV s−1) was used, this is equivalent to the change in transmission with potential (dT/dE). Fig. 2(a) shows the transmission change dT/dt vs. E and the corresponding cyclic voltammogram. It can be seen that the change in transmission, dT/dt, peaks at the same potential values as the current density j. The first current peak at E = −0.05 V is significantly smaller than the second one at E = −0.3 V (assuming a peak baseline of zero current). The corresponding transmission peaks show a similar behavior. The second current peak is more distinct and dT/dt is also significantly larger. At a first glance, a high cell current results in a high transmission change in the WO3 layer. To obtain more quantitative information we analyzed the differential transmission with respect to the coloration efficiency as a function of the electrode potential. The change in optical density with respect to the amount of inserted protons, i.e. electric charge, d(OD)/dQ vs. E, for the first coloration half-cycle is plotted in Fig. 2(b). It can be seen that for the first current peak at −0.05 V vs. Ag/AgCl the coloration efficiency d(OD)/dQ is smaller than for the current peak at −0.3 V, which is more distinct and has a value of about 0.15 cm2 C−1. The mechanism of hydrogen insertion and WO3 reduction seems to be a two-step process that takes place over a range of electrode potentials.
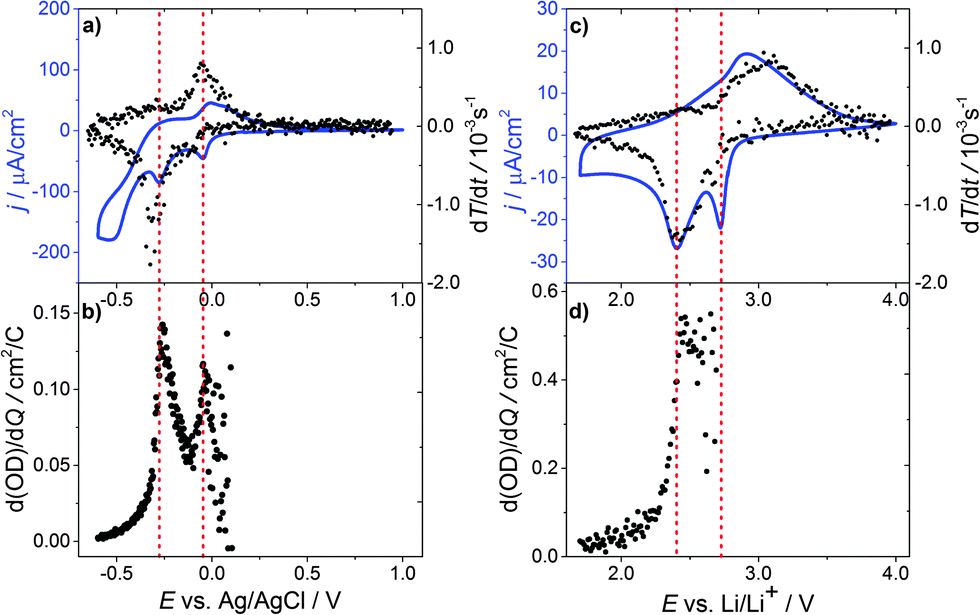 | ||
| Fig. 2 Transmission change dT/dt vs. E and the cyclic voltammogram of a WO3 film in: (a) 0.1 M H2SO4 in H2O and (c) 0.2 M LiClO4 in propylene carbonate. Change in the optical density with respect of the amount of (b) inserted protons/electric charge d(OD)/dQ vs. E and (d) inserted lithium ions/electric charge d(OD)/dQ vs. E. | ||
Fig. 3(a) displays the differential change in the optical density d(OD)/dE and dQ/dE vs. E, which in this case basically represents the cell current. Merely a shoulder in d(OD)/dE is visible at the first current peak (E = −0.05 V vs. Ag/AgCl), while at the second current peak (E = −0.3 V vs. Ag/AgCl) the main transmission change is observed.
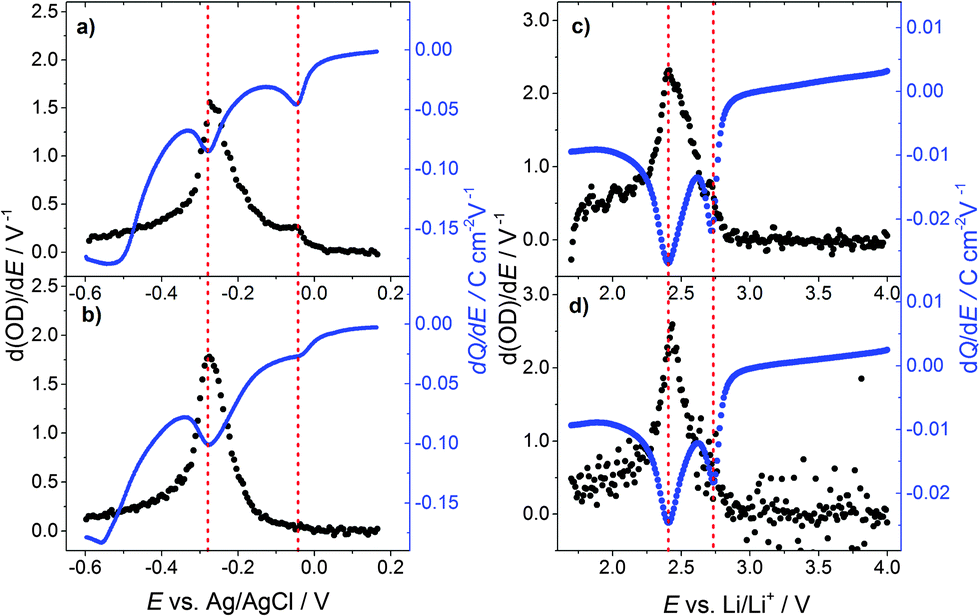 | ||
| Fig. 3 Differential change of the optical density d(OD)/dE vs. E and dQ/dE (cell current) vs. E of a WO3 film for the potential decreasing part of a cyclic voltammogram with a scan rate of 1 mV s−1 (a) in 0.1 M H2SO4 in H2O for the first cycle and (b) the 10th cycle; (c) in 0.2 M LiClO4 in PC for the first cycle and (d) the 10th cycle. | ||
Fig. 3(b) shows d(OD)/dE and dQ/dE vs. E after the 10th CV cycle of the sample. The first current peak is reduced significantly and the d(OD)/dE feature of this potential vanishes. The d(OD)/dE peak at −0.3 V vs. Ag/AgCl becomes narrower and higher. Analyzing the electrochromic properties of WO3 films using aqueous acid based electrolytes, however, is problematic for the following reasons. As can be seen from Fig. 1(a) substrate reduction and/or hydrogen evolution cause a high parasitic current and eventually lead to ageing of the films. Moreover, other side reactions, such as self-bleaching,23,24 lead to current-free discoloration of the films.
Thus, in order to overcome these problems, a similar series of measurements was carried out using lithium ions in a water and oxygen free environment instead of hydrogen. Fig. 2(c) shows the corresponding cyclic voltammogram and dT/dt vs. E. In the CV curve, two reduction peaks at 2.73 V vs. Li/Li+ and 2.4 V vs. Li/Li+ are observed. As for proton insertion, the peaks are about 0.3 V apart. Peak currents, however, are a factor of 10 smaller compared to the measurements in aqueous solutions, while the transmission change is about the same, showing that WO3 coloration via Li insertion is more effective. In addition, the reduction peaks for lithium insertion are narrower and more pronounced than the corresponding peaks for hydrogen, which can be interpreted in terms of a more well-defined phase transition for Li ions than for protons. After the major transmission change at 2.4 V vs. Li/Li+ the current almost relaxes towards zero.
The coloration efficiency d(OD)/dQ (Fig. 2(d)) for the first peak at 2.73 V vs. Li/Li+ is very small and hardly discernable as differentiating with respect to Q leads to strong scattering of the values for Q close to zero. A distinct peak occurs for the second current peak at 2.4 V vs. Li/Li+ with a value of about 0.6 cm2 C−1, which is a factor of 4 higher than for hydrogen insertion.
Fig. 3(c) shows d(OD)/dE vs. E and dQ/dE vs. E for the first cycle of lithium injection. As to the case of proton insertion, the main transmission change occurs in the vicinity of the second current peak. Again, the first current peak results in a comparably small differential transmission change only. After the tenth cycle (Fig. 3(d)), peak currents are reduced by approximately 20% for the first peak and 10% for the second one. The first peak in transmission change is also reduced but still clearly visible.
The results in Fig. 1–3 clearly indicate a two-step insertion process for both hydrogen and lithium insertion into nanocrystalline WO3 thin films, of which the first reduction step at a higher electrode potential is characterized by both a lower amount of the inserted electric charge and, more importantly, by a significantly lower coloration efficiency compared to the second step.
Cycling the films several times leads to an extinction of the bump in differential transmission change due to the first reduction peak in the case of hydrogen insertion, and to a noticeable decrease in the case of lithium insertion.
3.2 UV-Vis transmission spectroscopy
To obtain a better understanding of the insertion process, we analyzed the inserted WO3 thin films at different stages of lithiation, using UV-Vis, X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD) analysis. As already mentioned above, hydrogen insertion into WO3 is accompanied by self-bleaching and parasitic electrochemical processes of the FTO substrate and we therefore focus on Li inserted WO3 (LixWO3) samples in the following. Furthermore, instead of CV experiments, Li insertion was carried out by galvanostatic discharge. In galvanostatic experiments, ion insertion into the WO3 thin film is not as dynamic as in CV experiments with a preset rate. The potential saturates when a certain state is attained and during that time more charge, i.e. more ions are inserted at the same potential value.To make sure that the first insertion step is not accompanied by a significant transmission change in a different spectral window than the 633 nm monitored previously, UV-Vis transmission spectroscopy was employed in the entire range from 300 nm to 1300 nm. For this measurement the films were discharged by a constant current of −300 nA. Fig. 4(a) shows the curve for the galvanostatic discharge of WO3. Starting at the open circuit potential, at around E = 2.9 V, within the range of Q = 0 mC cm−2 to 2 mC cm−2 the curve displays an s-shaped form with a rapid potential drop. The cell potential of E = 2.76 V at around 1 mC cm−2 is described by a plateau-like potential curve. Then another s-shaped form of the potential develops – the potential decreases over a wider range with a more shallow slope until a second s-shaped form appears at Q = 2.5 mC cm−2 with a value of E = 2.55 V. The second plateau-like step spreads within a wider range than the first, i.e. from about 3 mC cm−2 to 7 mC cm−2. At 7 mC cm−2 the potential decreases again but with an even smaller slope. These two plateau-like potential curves correspond to the current peaks of the cyclic voltammograms.
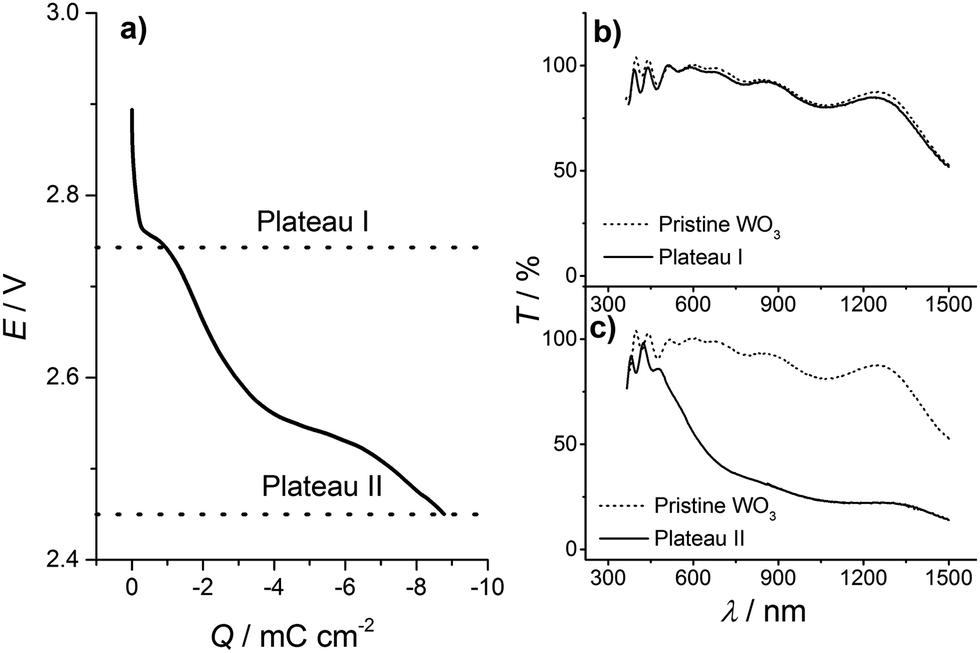 | ||
| Fig. 4 (a) Correlation of charge and potential for a galvanostatic insertion process of lithium ions into a WO3 thin film in 0.2 M LiClO4 in PC for a constant current of −300 nA. Ex situ UV-Vis spectra of WO3 films inserted up to (b) plateau I at 2.73 V and (c) plateau II at 2.45 V. | ||
For the ex situ UV-Vis transmission experiment, the discharge was stopped at 2.73 V (and held at that potential for 30 min) and 2.45 V, which are the ends of each insertion step indicated by the plateau-like potential curves at 2.76 V and 2.55 V, respectively, as displayed in Fig. 4(a). In the following, the potential values E = 2.73 V and 2.45 V, where the discharge was stopped, will be denoted plateau I and plateau II, respectively. After the insertion step, the samples were washed in dimethyl carbonate and dried in the argon atmosphere of the glove box. As a reference for the UV-Vis transmission spectroscopy, a FTO substrate was used. Transmission data for the states of the WO3 film corresponding to plateaus I (at 2.73 V) and II (at 2.45 V) are shown in Fig. 4(b) and (c) in comparison with the corresponding data for the pristine film. The transmission curves of the pristine film and the film in a state corresponding to plateau I look almost identical. A large transmission change appears for a film in a state corresponding to plateau II, where its transmission dramatically drops for wavelengths beyond 500 nm. The data support our findings that the first insertion step shows almost no electrochromic effect.
3.3 XPS analysis
Obviously a significant fraction of inserted lithium does not contribute to the coloration of the WO3 thin films. As ions are inserted together with electrons the insertion is essentially equivalent to the chemical reduction of the material and one may ask whether there is also a two-step process for the reduction of WO3. In order to answer this question with ex situ XPS measurements, films were prepared by galvanostatic insertion with a constant current of −300 nA. After stopping the insertion at 2.73 V (plateau I) and 2.45 V (plateau II), respectively, the LixWO3 samples and the pristine WO3 film were washed in dimethyl carbonate and dried in the argon atmosphere of the glove box. Afterwards, the films were transferred into the X-ray photoelectron spectrometer avoiding contact with the atmosphere. Besides a small carbon contamination from the sample preparation and/or electrochemical intercalation only tungsten and oxygen species were detected.Fig. 5 shows the W 4f core level spectra of the films corresponding to three different charge states. As expected, uncolored pristine WO3 exhibits a single W 4f doublet with peak positions of W 4f7/2 at 35.4 eV and W 4f5/2 at 37.6 eV which can be clearly attributed to the W6+ oxidation state. A weak emission originating from the W 5p3/2 level can be found at 41.2 eV.
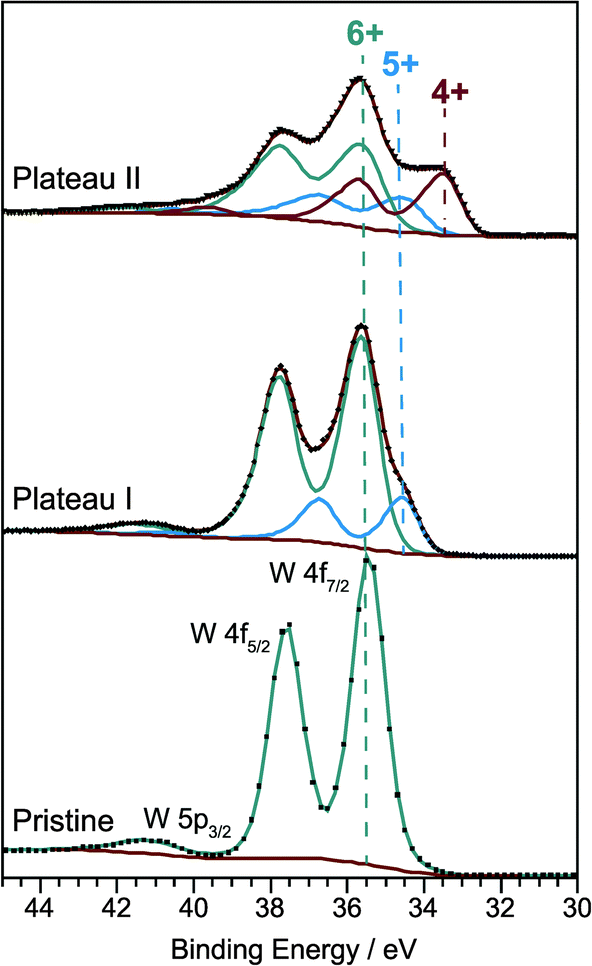 | ||
| Fig. 5 W 4f core level XP spectra of the pristine WO3 film. Lithium inserted WO3 films formed by galvanostatic discharge in 0.2 M LiClO4 in PC with a constant current of −300 nA down to the first plateau at 2.73 V and the second plateau at 2.45 V. | ||
Developing a consistent peak model for the W 4f core levels of tungsten trioxide with varying degrees of reduction is a major challenge mainly due to the overlap of the W 5p3/2 signal with the W 4f lines, but also due to the unknown degree of asymmetry caused by the interaction of core holes with conduction electrons and due to the unknown shape of the inelastic background. Moreover, electrochemical insertion of lithium could lead to a non-homogeneous depth distribution of oxidation states. Nevertheless, by applying constraints on peak area ratios (W 4f7/2 to W 4f5/2 = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, W 5p3/2 to W 4f7/2 ≈ 10:1 as determined for the pristine film), peak positions (ΔBE(W 4f5/2–W 4f7/2) = 2.1–2.2 eV) and line widths (FWHM(W 4f5/2) = FWHM(W 4f7/2) ± 0.05 eV) for each set of components, we were able to roughly estimate the relative amounts of different oxidation states. To account for the metallic character of the ion-intercalated samples the spectra were fitted with asymmetric line shapes with additional tail damping (LF function) as implemented in the CasaXPS software.25 The parameters obtained from the fitting procedure are listed in Table 1.
:3, W 5p3/2 to W 4f7/2 ≈ 10:1 as determined for the pristine film), peak positions (ΔBE(W 4f5/2–W 4f7/2) = 2.1–2.2 eV) and line widths (FWHM(W 4f5/2) = FWHM(W 4f7/2) ± 0.05 eV) for each set of components, we were able to roughly estimate the relative amounts of different oxidation states. To account for the metallic character of the ion-intercalated samples the spectra were fitted with asymmetric line shapes with additional tail damping (LF function) as implemented in the CasaXPS software.25 The parameters obtained from the fitting procedure are listed in Table 1.
| Sample | Formal W oxidation state | Binding energy W 4f7/2 (eV) | FWHM W 4f7/2 (eV) | Asymmetry index | Relative amount (%) |
|---|---|---|---|---|---|
| Pristine | 6+ | 35.4 | 1.0 | 0 | 100 |
| Plateau I | 6+ | 35.5 | 1.1 | 0.13 | 80 |
| 5+ | 34.5 | 1.0 | 0.13 | 20 | |
| Plateau II | 6+ | 35.4 | 1.3 | 0.3 | 50 |
| 5+ | 34.3 | 1.3 | 0.3 | 20.4 | |
| 4+ | 33.3 | 1.1 | 0.3 | 29.6 |
The degree of asymmetry is given by the asymmetry index which is defined as 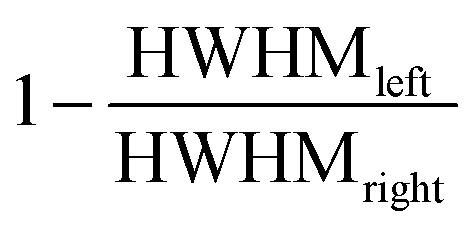 where HWHMleft and HWHMright denote the peak width at half-maximum for the respective side. It should be noted that in order to fulfill the area constraints on the high binding energy (BE) side of the W 4f spectra the Shirley type background for the pristine and the first inserted sample corresponding to plateau I was blended with a spline function.
where HWHMleft and HWHMright denote the peak width at half-maximum for the respective side. It should be noted that in order to fulfill the area constraints on the high binding energy (BE) side of the W 4f spectra the Shirley type background for the pristine and the first inserted sample corresponding to plateau I was blended with a spline function.
After the first insertion step the W 4f core level spectrum of the sample corresponding to plateau I displays a second doublet peak with a maximum position at 34.5 eV for the W 4f7/2 line and a fractional area of 20% of the total W 4f signal. This doublet can be assigned to W5+.26–30 Further ion insertion, up to the second potential plateau, then leads to an even more pronounced shoulder on the low binding energy side of the W 4f signal. For fitting the resulting spectrum a third doublet line at 33.3 eV has to be added to the model. This component can be attributed to W4+ species and has a relative peak area of 29.6% (W6+: 50%, W5+: 20.4%).26–28 Thus the XPS results prove that the lithium insertion is accompanied by a two-step reduction of tungsten. In the first step W6+ is reduced to W5+, and in the second step W4+ is formed in addition.
From our findings, we conclude that the formation of W4+ is responsible for the electrochromic behavior of nanocrystalline tungsten trioxide. Samples containing W5+ exhibit only a very small degree of coloration.
It should be remembered that XPS is a surface sensitive analysis and has an information depth for W 4f electrons in WO3 of about 7 nm. However, our sol–gel based films are porous exhibiting crystallites with sizes of 20 nm and without a preferred orientation. Thus our XPS analysis to a large degree also gives information about the bulk of the crystallites.
The coloration in WO3 associated with the existence of W4+ has been proposed in the literature, however, there are significant differences to our findings.
In the work of Deb et al.16 the presence of W4+ was correlated to the presence of an oxygen vacancy meaning that equal concentrations of oxygen vacancies and W4+ are present. For our findings, i.e. 30% W4+ after intercalation, this would require 10% of oxygen sites being vacant (WO2.7). Such a large amount of oxygen vacancies in crystalline WO3 is very unlikely. In addition, our XPS analysis clearly shows that the pristine films are almost stoichiometric, as solely W6+ is found. From this we can conclude that the formation of W4+ does not require oxygen vacancies in crystalline WO3, but results from electrochemical reduction via lithium insertion (formation of lithium interstitials, Lii):
| WO3 + 2Li → WW′′ + 2Lii + 3OxO. | (6) |
| hν + W4+(A) + W5+(B) → W5+(A) + W4+(B) | (7) |
In contrast to the findings of Lee et al., our results clearly show that our pristine nanocrystalline WO3 is almost stoichiometric as only W6+ is found, and a notable coloration is only possible due to the existence of W4+, most likely performing the following bipolaronic transition
| hν + W4+(A) + W6+(B) → W6+(A) + W4+(B), | (8) |
In contrast to this interpretation, Zhong et al. concluded from their XRD experiments that the potential plateaus in galvanostatic insertion experiments correspond to phase transitions of the crystal structure from monoclinic to tetragonal and from tetragonal to cubic.20 As shown in the next section, our ex situ XRD analyses indeed show increasing symmetry of the films as lithium is inserted, however, a distinct phase transition at the galvanostatic potential plateaus/CV peaks is only found for the second plateau at 2.45 V.
3.4 X-ray diffraction
In order to classify the quality of our synthesis procedure, preliminary XRD measurements on two powder samples were carried out. These powders were prepared by heating the sol–gel precursor to 300 °C and 450 °C for 8 h. Interestingly, unlike other groups,21 we already found a nanocrystalline phase with an average crystallite size of 7 nm in the sample heated at 300 °C, which is an unusually low temperature for the preparation of crystalline samples from sol–gel precursors. The observed broad reflections could be attributed to the orthorhombic high-temperature phase of WO3 (usual range: 467 °C to 680 °C, space group Pnma),34 and Rietveld refinement (see ESI,† S1) shows excellent agreement of the measured data with the theoretical structural model. Hence, the chimie douce approach does indeed promote formation of the kinetically stable instead of the thermodynamically stable product, which is a well-described phenomenon.35 The sample heated at 450 °C revealed typical reflections of monoclinic WO3 (space group P21/n) with an average crystallite size of 23 nm. The results of the Rietveld refinement can be found in the ESI,† S2. These results clearly document the identity of our WO3 samples.Structural changes and the evolution of crystallinity in the films during insertion of lithium will be discussed in the following. Fig. 6 shows the diffraction diagrams of the three thin film samples on FTO-coated SiO2, corresponding to the charge states of the pristine WO3 film and intercalated films up to plateau I and II, respectively. Reflections that can be attributed to the FTO substrate are marked by vertical gray dashed lines. Considering the shape and size of the strongest reflections at 2θ ≈ 24°, a salient effect of the preferential orientation in the samples can be determined. Away from these reflections, broad and minor features are found, revealing the nanocrystalline nature of the samples. In contrast to the common literature, reporting similar methods for the preparation of WO3 thin films,19,21 our pristine sample does not reveal the expected monoclinic structure. Instead, indexation of the few mostly very poor reflections in the diffraction diagram points to tetragonal symmetry.
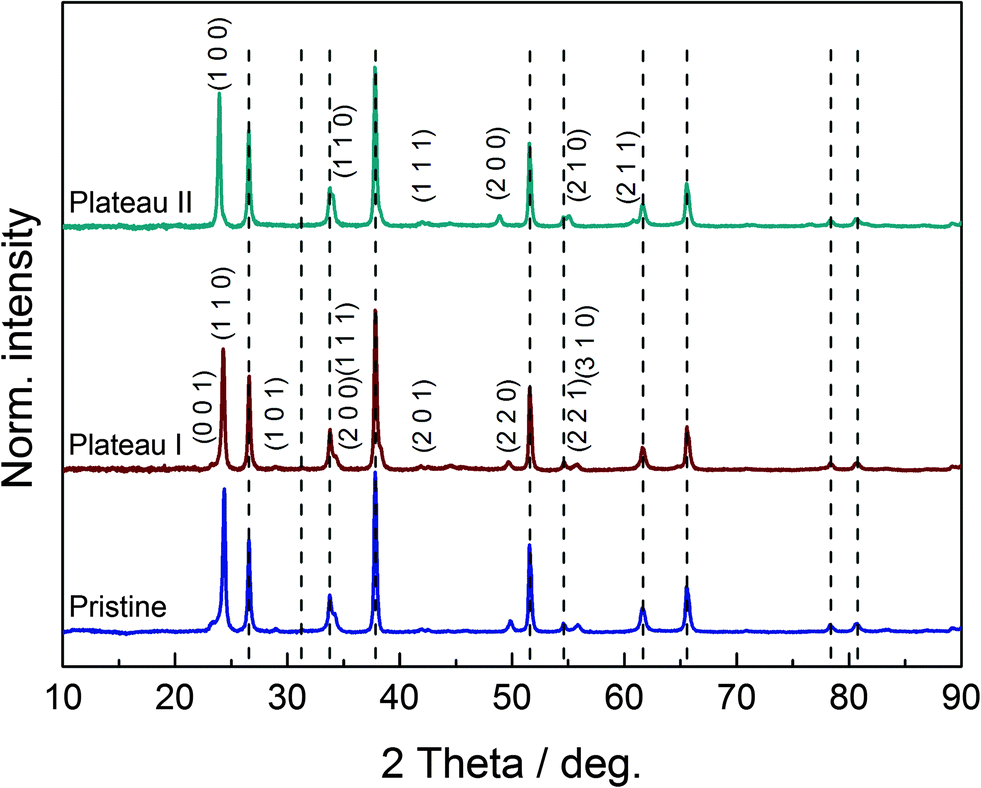 | ||
| Fig. 6 Diffraction diagrams of the pristine WO3 film (bottom trace) and the lithium-inserted films formed by galvanostatic discharge down to plateau I at 2.73 V (center trace) and plateau II at 2.45 V (top trace). FTO substrate reflections are marked by gray dashed lines. Indices for reflections attributable to the tetragonal (pristine, plateau I) and cubic phases (plateau II) are depicted. | ||
Furthermore, a direct comparison of the diffraction diagrams of the pristine sample and the sample at the first plateau at 2.73 V gives strong evidence for the similarity of the two corresponding crystal structures. According to the literature,20 the LixWO3 sample at the first plateau reveals a tetragonal crystal lattice, which matches our findings. Profile refinements for both samples, leaving out the reflections of the FTO substrate, led to decent results. Hence, both samples show tetragonal symmetry and the first insertion step obviously proceeds without a phase transformation. However, we cannot fully exclude that the first plateau-like potential curve is due to the coexistence of a tetragonal WO3 and a tetragonal LixWO3 phase. It may be concluded that this lack of structural alteration entails minor insertion of lithium ions/electric charge only, and therefore a poor transmission change results in the vicinity of the first peak. Looking at the diffraction diagram of the LixWO3 sample at the second plateau at 2.45 V, a structural change becomes visible. In agreement with the literature, a phase transformation to the cubic structure of LixWO3 is observed. Certain reflections of the tetragonal system disappear due to extinction rules, as can be seen from the indices of the tetragonal and cubic lattice in Fig. 6. In this particular case, one could deduce that the phase transformation promotes the insertion of charge carriers and leads to a higher state of coloration for the second peak. The unit cell volume of the samples increases with increasing lithium content/further discharge level, as expected. While the pristine film has a unit cell volume of 51.20(2) × 106 pm3 per formula unit, the unit cell volume of the sample at the first plateau is 52.65(3) × 106 pm3 per formula unit. For the sample at the second plateau, a unit cell volume of 52.94(1) × 106 pm3 is found.
The average crystallite size in the thin films was calculated via Williamson–Hall analysis, using LaB6 powder (NIST Standard Reference Material 660B) as an external standard for instrumental line broadening and omitting the FTO substrate reflections. Using this method, an increase in the average crystallite size from 11 nm for the pristine WO3 film to 20 nm for the sample at the first plateau and 22 nm for the sample at the second plateau is observed. Apparently, besides inducing structural changes, insertion of lithium leads to crystallite growth. In order to check on the reversibility of this behavior, a sample that was oxidized to the deintercalated (tetragonal) state at 4 V vs. Li/Li+ after 10 cycles was analyzed. For this sample, an average crystallite size of 26 nm is found, which is even bigger than the value identified for the cubic sample during the first cycle. Hence, the nanocrystalline samples do not fully recover and with growing crystallite size, extraction of Li becomes slow due to the reduced surface area. This observation is in line with effects described by Limaye et al.,19 who state that the number of ions trapped inside the chains between WO6 octahedra increases with the cycle number, resulting in structural disorder and degradation of the electrochromic properties of WO3. This interpretation could provide an explanation for the decrease in differential transmission change we observed for the first reduction peak during lithium insertion after several cycles.
However, one should keep in mind that the data presented here should be regarded with caution, as values for crystallinity and unit cell volume are, in general, difficult to obtain for thin film samples.
4 Conclusions
We successfully synthesized nanocrystalline tungsten trioxide thin films via a sol–gel based dip coating process. Electrochemical insertion of hydrogen or lithium results in a transmission change, which yields a blue color to the film. In CV experiments two distinct redox reaction peaks are observed, which correspond to a two-step reduction of the material. We observe this two-step insertion process for both hydrogen and lithium. By combining optical spectroscopy with electrochemical experiments in situ we were able to show that the first reduction step at high electrode potential is characterized by both a lower amount of the inserted electric charge and more importantly a much lower decrease in optical transmission compared to the second step. Hence coloration of nanocrystalline WO3 at low potential is more efficient than at higher potentials. Using XPS we showed that these two reduction steps denote the reduction from initial W6+ to W5+ at −0.05 V vs. Ag/AgCl and 2.73 V vs. Li/Li+ and from W5+ to W4+ at −0.3 V vs. Ag/AgCl and 2.45 V vs. Li/Li+. UV-Vis spectroscopy showed that there is no noticeable transmission change from W6+ to W5+, whereas the transmission decreases strongly for W4+. In essence, we conclude that electrochromism in nanocrystalline WO3 is mainly caused by the formation of W4+. That is, nanocrystalline WO3 shows only a significant transmission change when it is reduced to a W4+ state. A possible transition could be W4+ → W5+, but due to the large amount of W6+ (50%) a bipolaronic transition, W4+ → W6+, is more likely. XRD measurements showed that the electrochemical reduction is accompanied by structural alteration of the film towards higher symmetry. However, a two-step mechanism is not observed here. Only one structural transition, i.e. the transition from initial tetragonal to cubic WO3, is found when W4+ is formed. XRD measurements also prove that the nanocrystalline WO3 experiences grain growth by conventional Ostwald ripening.Acknowledgements
We thank B. M. Smarsly, J. Sann, D. Krüger and C. Schneider for experimental support and fruitful discussions. This work was funded by the Hessen State Ministry of Higher Education, Research and the Arts (LOEWE initiative, STORE-E).Notes and references
- G. Niklasson and C. Granqvist, J. Mater. Chem., 2007, 17, 127–156 RSC.
- K. Bange, Sol. Energy Mater. Sol. Cells, 1999, 58, 1–131 CrossRef CAS.
- C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2012, 99, 1–13 CrossRef CAS PubMed.
- P. M. S. Monk, Crit. Rev. Solid State Mater. Sci., 1999, 24, 193–226 CrossRef CAS PubMed.
- C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2000, 60, 201–262 CrossRef CAS.
- S. K. Deb, Appl. Opt., 1969, 3, 192 Search PubMed.
- C. M. Lampert, Sol. Energy Mater., 1984, 11, 1–27 CrossRef CAS.
- J. S. E. M Svensson and C. G. Granqvist, Sol. Energy Mater., 1984, 11, 29–34 CrossRef.
- T. Tesfamichael, M. Arita, T. Bostrom and J. Bell, Thin Solid Films, 2010, 518, 4791–4797 CrossRef CAS PubMed.
- D. T. Gillaspie, R. C. Tenent and A. C. Dillon, J. Mater. Chem., 2010, 20, 9585–9592 RSC.
- D. R. Rosseinsky and R. J. Mortimer, Adv. Mater., 2001, 13, 783–793 CrossRef CAS.
- B. W. Faughnan and R. S. Crandall, Top. Appl. Phys., 1980, 40, 181–211 CAS.
- S.-H. Lee, M. J. Seong, H. M. Cheong, E. Ozkan, E. C. Tracy and S. K. Deb, Solid State Ionics, 2003, 156, 447–452 CrossRef CAS.
- O. F. Schirmer and E. Salje, Solid State Commun., 1980, 33, 333–336 CrossRef CAS.
- J.-G. Zhang, D. K. Benson, C. E. Tracy, S. K. Deb and A. W. Czanderna, J. Electrochem. Soc., 1997, 144, 2022–2026 CrossRef CAS PubMed.
- S. K. Deb, Sol. Energy Mater. Sol. Cells, 2008, 92, 245–258 CrossRef CAS PubMed.
- E. Broclawik, A. Góra, P. Liguzinski, P. Petelenz and H. A. Witekb, J. Chem. Phys., 2006, 124, 054709 CrossRef CAS PubMed.
- B. W. Faughnan, R. S. Crandall and M. A. Lampert, Appl. Phys. Lett., 1975, 27, 275 CrossRef CAS PubMed.
- M. V. Limaye, J. S. Chen, S. B. Singh, Y. C. Shao, Y. F. Wang, C. W. Pao, H. M. Tsai, J. F. Lee, H. J. Lin, J. W. Chiou, M. C. Yang, W. T. Wu, J. S. Chen, J. J. Wu, M. H. Tsai and W. F. Pong, RSC Adv., 2014, 4, 5036–5045 RSC.
- Q. Zhong, J. R. Dahn and K. Colbow, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 4 Search PubMed.
- T. Brezesinski, D. Fattakhova Rohlfing, S. Sallard, M. Antonietti and B. A. Smarsly, Small, 2006, 2, 1203–1211 CrossRef CAS PubMed.
- J. Wallys, J. Teubert, F. Furtmayr, D. M. Hofmann and M. Eickhoff, Nano Lett., 2012, 12, 6180 CrossRef CAS PubMed.
- H. Kamal, A. A. Akl and K. Abdel-Hady, Physica B, 2004, 349, 192–205 CrossRef CAS PubMed.
- A. Georg, A. Georg, U. O. Krasovec and V. Wittwer, J. New Mater. Electrochem. Syst., 2005, 8, 317–325 Search PubMed.
- J. I. Jeong, J. H. Hong, J. H. Moon and J.-S. Kang, J. Appl. Phys., 1996, 79, 1996 CrossRef PubMed.
- M. Stolze, B. Camina, F. Galbertb, U. Reinholzc and L. K. Thomas, Thin Solid Films, 2002, 409, 254–264 CrossRef CAS.
- L. Su and Z. Lu, J. Phys. Chem. Solids, 1998, 59, 1175–1180 CrossRef CAS.
- A. Henningsson, A. Stashans, A. Sandell, H. Rensmo, S. Södergren, H. Lindström, L. Vayssieres, A. Hagfeldt, S. Lunell and H. Siegbahn, Adv. Quantum Chem., 2004, 47, 23–36 CAS.
- A. Siokou, S. Ntais, S. Papaefthimiou, G. Leftheriotis and P. Yianoulis, Surf. Sci., 2004, 566–568(part 2), 1168–1173 CrossRef CAS PubMed.
- A. Romanyuk and P. Oelhafen, Sol. Energy Mater. Sol. Cells, 2006, 90, 1945–1950 CrossRef CAS PubMed.
- S.-H. Lee, H. M. Cheong, J.-G. Zhang, C. E. Tracy, A. Mascarenhas, D. K. Benson and S. K. Deb, Electrochim. Acta, 1999, 44, 3111 CrossRef CAS.
- S.-H. Lee, H. M. Cheong, J.-G. Zhang, A. Mascarenhas, D. K. Benson and S. K. Deb, J. Appl. Phys., 1999, 74, 242 CAS.
- S.-H. Lee, H. M. Cheong, C. E. Tracy, A. Mascarenhas, A. W. Czanderna and S. K. Deb, J. Appl. Phys., 1999, 75, 1541 CAS.
- E. Salje, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 574–577 CrossRef.
- R. J. Cohen and G. B. Benedek, J. Phys. Chem., 1982, 86, 3696–3714 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp02482j |
| This journal is © the Owner Societies 2015 |