
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microhydrated dihydrogen phosphate clusters probed by gas phase vibrational spectroscopy and first principles calculations†
Shou-Tian
Sun
a,
Ling
Jiang
*bh,
J.W.
Liu
c,
Nadja
Heine
b,
Tara I.
Yacovitch‡
d,
Torsten
Wende§
b,
Knut R.
Asmis
*e,
Daniel M.
Neumark
*df and
Zhi-Feng
Liu
*ag
aDepartment of Chemistry and Centre for Scientific Modeling and Computation, Chinese University of Hong Kong, Shatin, Hong Kong, China
bFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4–6, D-14195 Berlin, Germany
cNational Supercomputing Center in Shenzhen, Shenzhen, China
dDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: dneumark@berkeley.edu
eWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, Linnéstrasse 2, D-04103 Leipzig, Germany. E-mail: knut.asmis@uni-leipzig.de
fChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
gShenzhen Research Institute, Chinese University of Hong Kong, No. 10, 2nd Yuexing Road, Shenzhen, China. E-mail: zfliu@cuhk.edu.hk
hState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: ljiang@dicp.a.cn
First published on 5th June 2015
Abstract
We report infrared multiple photon dissociation (IRMPD) spectra of cryogenically-cooled H2PO4−(H2O)n anions (n = 2–12) in the spectral range of the stretching and bending modes of the solute anion (600–1800 cm−1). The spectra cannot be fully understood using the standard technique of comparison to harmonic spectra of minimum-energy structures; a satisfactory assignment requires considering anharmonic effects as well as entropy-driven hydrogen bond network fluctuations. Aided by finite temperature ab initio molecular dynamics simulations, the observed changes in the position, width and intensity of the IRMPD bands with cluster size are related to the sequence of microsolvation. Due to stronger hydrogen bonding to the two terminal P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, these are hydrated before the two P–OH groups. By n = 6, all four end groups are involved in the hydrogen bond network and by n = 12, the cluster spectra show similarities to the condensed phase spectrum of H2PO4−(aq). Our results reveal some of the microscopic details concerning the formation of the aqueous solvation environment around H2PO4−, provide ample testing grounds for the design of model solvation potentials for this biologically relevant anion, and support a new paradigm for the interpretation of IRMPD spectra of microhydrated ions.
O groups, these are hydrated before the two P–OH groups. By n = 6, all four end groups are involved in the hydrogen bond network and by n = 12, the cluster spectra show similarities to the condensed phase spectrum of H2PO4−(aq). Our results reveal some of the microscopic details concerning the formation of the aqueous solvation environment around H2PO4−, provide ample testing grounds for the design of model solvation potentials for this biologically relevant anion, and support a new paradigm for the interpretation of IRMPD spectra of microhydrated ions.
Introduction
Dihydrogen phosphate, H2PO4−, is one of the anions in the dissociation equilibrium of phosphoric acid dissolved in water. It is the dominant anion under weakly acidic conditions. The vibrational spectra of hydrated phosphate ions are of interest owing to the presence of phosphate groups in key biological species including DNA and RNA, coenzymes, and ATP.3 Inorganic phosphates are critical in diverse cellular functions involving metabolism and energy consumption.4 The equilibrium between H2PO4− and HPO42− provides one of the two important biological buffer systems that stabilize the pH value in cell fluids. Hydrolysis of phosphates also produces phosphate ions as intermediate and biochemical signals,5 which can then be monitored using vibrational spectroscopy in reaction kinetics studies.6–10 Understanding the solvation dynamics of hydrated phosphate ions by computer simulations1,11–17 is of fundamental importance, as it provides the basis for the interpretation of spectroscopic data.18–24 These simulations require a detailed knowledge of the interaction between phosphate ions and water. Here, we gain such molecular level insights into the hydration behaviour of dihydrogen phosphate by studying the vibrational spectroscopy of size-selected H2PO4−(H2O)n clusters as a function of the number of water molecules.Gas phase mass spectrometry25,26 and cluster spectroscopy27–32 of microhydrated anions provide detailed energetic and structural information that is difficult to extract from measurement of bulk solutions. Measurements on isolated cluster ions are particularly attractive due to their size-selectivity and signal sensitivity. Even though the solvation environment in microhydrated cluster ions is not identical to that in a solution, e.g. there are typically no counter ions, the underlying solute–solvent interaction is the same and can be studied one water molecule at a time, providing ample testing grounds for theoretical modelling. Infrared (IR) photodissociation spectroscopy combined with cryogenic ion trap technology and the widely tuneable, intense radiation from an IR free electron laser represents one of the most powerful and generally applicable techniques currently available for studying the structure and stability of microhydrated anions.32–34
In previous studies on microhydrated dihydrogen phosphate ions, H2PO4−(H2O)n, hydration35–37 and electron-detachment energies38 were determined. Recently, we studied the infrared multiple photon dissociation (IRMPD) spectroscopy of the simplest member of this series, the monohydrated H2PO4−(H2O),2 from 500 to 4000 cm−1. The IRMPD spectrum shows considerable complexity as a result of large amplitude motion of the water molecule around the solute anion. In the O–H stretching region, strong hydrogen bonding between H2PO4− and H2O induces significant shifts and broadening in the spectral features, while in the fingerprint region the spectrum looks simpler than expected. These effects could not be fully rationalized using the standard harmonic analysis of stationary structures, but required performing ab initio molecular dynamics (AIMD) simulations. In general, it is becoming increasingly evident that consideration of anharmonic and dynamic effects in the vibrational spectra of microhydrated clusters, even when the clusters are thermalized to cryogenic temperatures, is critical for a satisfactory assignment of the IR vibrational spectra.2,39–41
Anion clusters comprising many water molecules can exhibit significant spectral congestion in the O–H stretching region. The fingerprint region (500–1800 cm−1) is typically more informative, as most fundamental vibrations of molecular solute ions fall into this region.32,33,42–48 These solute modes involve heavier atoms and are often better-resolved. Moreover, these features change significantly with the cluster size, yielding insights into the first steps of hydrogen bond (HB) network formation around a molecular anion and the underlying solvation interactions.
In this paper, we report the IRMPD spectra for the larger clusters H2PO4−(H2O)n, with n = 2–12, in the 600–1800 cm−1 region. Size-dependent trends observed in the experimental spectra are interpreted using AIMD simulations, overcoming the inadequacies of the harmonic analysis. Some spectral features are due to the contribution of multiple isomers that can interconvert into one another. In such cases, entropy becomes an important factor in determining their relative population. Shifting and broadening of spectral features are understood in the context of HB fluctuations. Based on the comparison between AIMD simulations and experimental results, a detailed picture for the evolution of the microsolvation dynamics as a function of cluster size n is obtained. In particular, we find that the terminal PO groups on the diphosphate anion are hydrated first, but by n = 6 all four end groups are involved in the HB network.
Methods
Experimental setup
IRMPD experiments are carried out using an ion trap/tandem mass spectrometer.44,45 Briefly, gas-phase ions are continuously produced in a Z-spray source from a 1 × 10−3 M aqueous solution of phosphoric acid in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/acetonitrile solvent. A beam of negative ions passes through a 4 mm diameter skimmer and is then collimated in a radio frequency (RF) decapole ion-guide. Parent ions are mass-selected in a quadrupole mass-filter, deflected by 90° in an electrostatic quadrupole deflector and focused into a gas-filled RF ring-electrode ion-trap. To allow for continuous ion loading and ion thermalization, the trap is continuously filled with He gas at an ion-trap temperature of 15 K. After filling the trap for 98 ms, all ions are extracted from the ion-trap and focused both temporally and spatially into the center of the extraction region of an orthogonally mounted linear time-of-flight (TOF) mass spectrometer. Here, the ion packet is irradiated with the IR laser pulse prior to the application of high-voltage pulses on the TOF electrodes and the subsequent measurement of the TOF mass spectrum.
:1 water/acetonitrile solvent. A beam of negative ions passes through a 4 mm diameter skimmer and is then collimated in a radio frequency (RF) decapole ion-guide. Parent ions are mass-selected in a quadrupole mass-filter, deflected by 90° in an electrostatic quadrupole deflector and focused into a gas-filled RF ring-electrode ion-trap. To allow for continuous ion loading and ion thermalization, the trap is continuously filled with He gas at an ion-trap temperature of 15 K. After filling the trap for 98 ms, all ions are extracted from the ion-trap and focused both temporally and spatially into the center of the extraction region of an orthogonally mounted linear time-of-flight (TOF) mass spectrometer. Here, the ion packet is irradiated with the IR laser pulse prior to the application of high-voltage pulses on the TOF electrodes and the subsequent measurement of the TOF mass spectrum.
IR spectra are measured using the radiation from the Free Electron Laser for Infrared eXperiments (FELIX)49 at the FOM Institute Rijnhuizen (The Netherlands). FELIX is operated at 10 Hz with a bandwidth of ∼0.2% RMS of the central wavelength and typical pulse energies of up to 30 mJ. The IRMPD cross section σIRMPD is obtained by normalising the frequency-dependent relative abundances of parent IP(ν) and fragment ions IF(ν) to the frequency-dependent laser pulse energy P(ν) (assuming a constant interaction area throughout the range of scanned wavelengths) using σIRMPD = −ln[IF/(IF + IP)]/P(ν).34
Computational details
Energy optimization for 0 K structures and the calculation of harmonic frequencies are performed with the Gaussian 03 package.50 Electronic structure is treated by the MP2 method, with a large basis set of aug-cc-pVDZ to account for the polarizability of hydrated anions. A scaling factor of 1.0418 is used in the region between 600–1800 cm−1.51 Calculated harmonic frequencies and intensities are convoluted using a Gaussian line shape function with a half-width of 4 cm−1.In AIMD simulations, the electronic energy and atomic forces are obtained within the framework of density functional theory, while the atomic motion is treated within Newtonian mechanics, as implemented in the CP2K package.52 The wave functions are expanded in a double zeta Gaussian basis set, while the electron density is expanded in Gaussians and auxiliary plane waves (GPW)53 with an energy cut-off at 280 Rydberg for the electron density. The atomic cores are modeled by the Goedecker–Teter–Hutter (GTH) type pseudopotentials.54–56 The exchange and correlation energies are calculated by the BLYP functional,57,58 with additional Grimme's dispersion corrections at the D3 level.59 A cluster ion is put at the center of a periodic cubic box, and the effects of the periodic charge density images are corrected by the decoupling technique developed by Martyna and Tuckerman.60 The box length is 16 Å for n = 2 and 20 Å for n = 4 and 6. The convergence criterion for the SCF electronic procedure is set to be 10−7 a.u. at each time step. For molecular dynamics, the temperature is controlled by a Nose–Hoover thermostat,61 with a time step of 0.5 fs. An equilibration period of up to 10 ps is performed first, with the temperature scaled to an interval of 20 K around the intended value. A data collection run is then followed in the microcanonical ensemble. At 20 K, when the structure is basically frozen around the equilibrium geometry, a short run of 10 ps is performed. At 140 K and 180 K, the duration of a trajectory is more than 100 ps. Each long trajectory is then cut into 10 ps interval for Fourier transformation and then added up to produce the dipole time-correlation function (DTCF) spectrum for a specific temperature.
Hydrated clusters are bound by HBs, which are relatively weak and therefore fairly flexible at finite temperature. Dynamic simulations are essential for sampling the solvation structures and for examining the thermal stability of a particular structure. More importantly, the fluctuations of HBs have strong effects on the vibrations that can be captured by AIMD simulations. A vibrational spectrum can be directly simulated by the Fourier transform of the DTCF,62
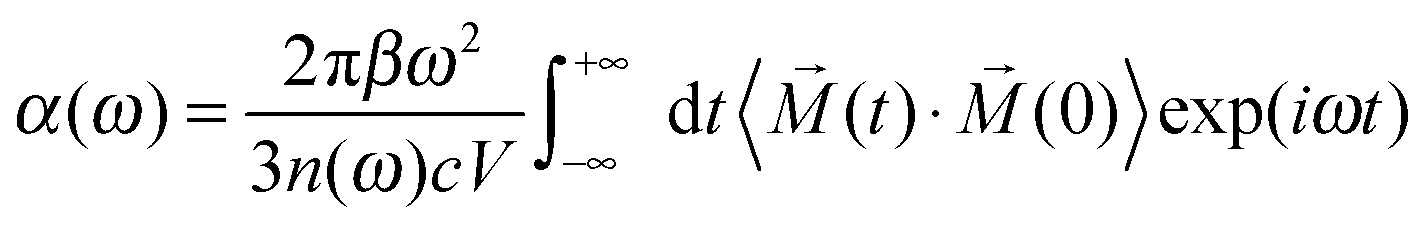
![[M with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_004d_20d1.gif) is the total dipole moment of the system, calculated by the polarization including both ionic and electronic contributions.
is the total dipole moment of the system, calculated by the polarization including both ionic and electronic contributions.
Results and discussion
Experimental IRMPD spectra
The IRMPD spectra of H2PO4−(H2O)n for n = 0–12 are shown in Fig. 1 in the spectral region between 520 and 1800 cm−1. Band positions and assignments are listed in Table 1. For convenience, we adopt the same labelling, i.e. capital letters (G–M), as in our previous study on the n = 0 and n = 1 species.2 Overall, at least eight unique absorption features are identified, of which six (G–L) have been previously observed in the n = 1 spectrum (see Fig. 1) and assigned to modes of the solute ion (H–L) and the solvent molecules (G). The intensity, position and width of these bands depend on the cluster size.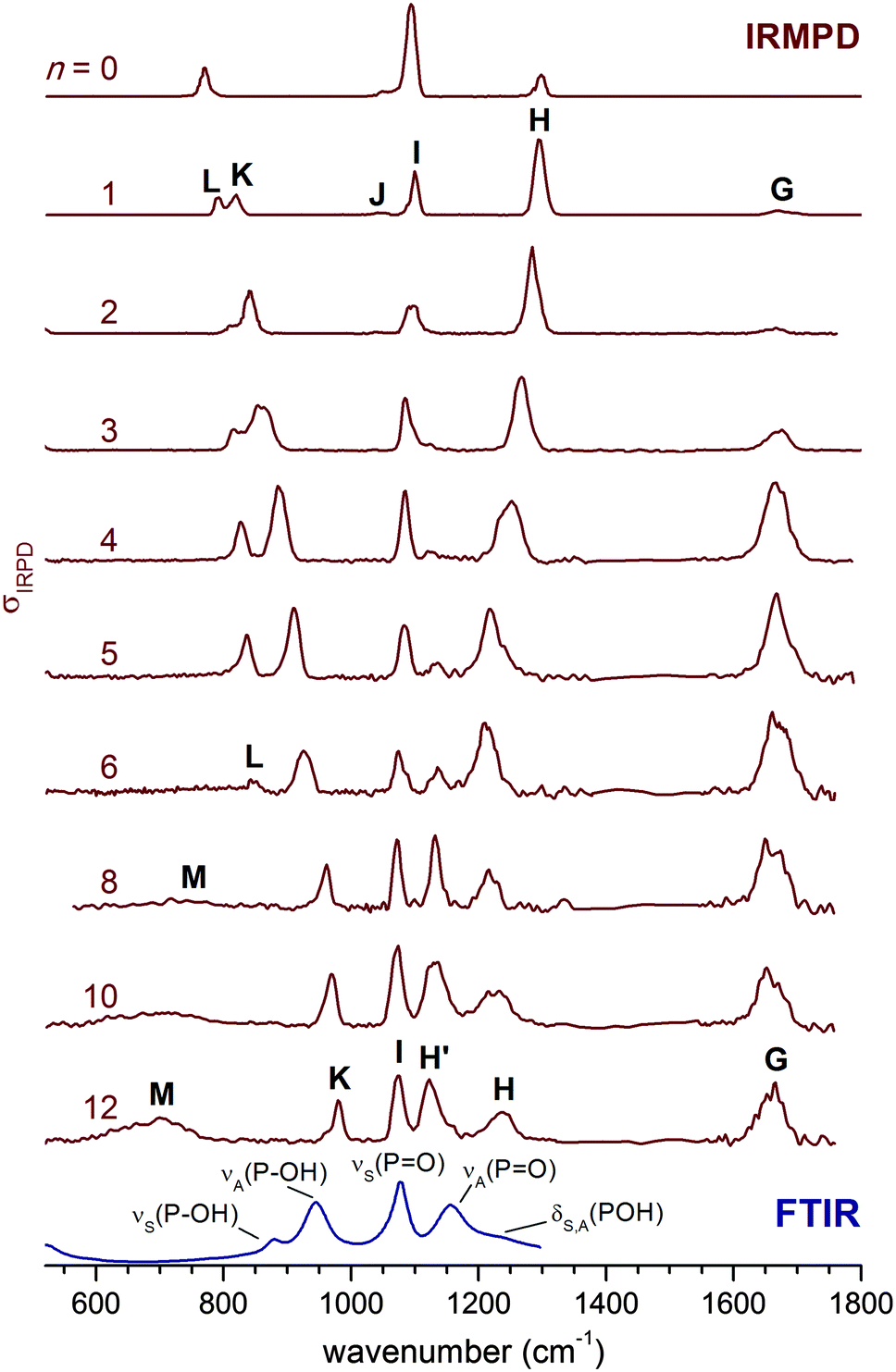 | ||
| Fig. 1 IRMPD spectra for H2PO4−(H2O)n from n = 0 to n = 12 in the 520–1800 cm−1 spectral region. The 300 K FTIR spectrum of a 0.1 M aqueous KH2PO4 solution1 is shown at the bottom. The n = 0 and n = 1 spectra are from ref. 2. | ||
| Band | n = 0a | n = 1a | n = 2 | n = 4 | n = 6 | n = 12 | n = ∞b | Symbol | Mode |
|---|---|---|---|---|---|---|---|---|---|
| a From ref. 2. b From ref. 1. | |||||||||
| G | 1671 | 1666 | 1664 | 1661 | 1663 | δ(H2O) | Water bend | ||
| H/H′ | 1299 | 1294 | 1284 | 1249 | 1207, 1138 | 1123 | 1156 |
ν
A(PO) |
Antisym. OPO stretch |
| H | 1234 | 1213 | δ(POH) | Solvated POH bending | |||||
| I | 1094 | 1099 | 1089 | 1086 | 1077 | 1073 | 1077 |
ν
S(PO) |
Sym. OPO stretch |
| J | 1049 | 1045 | δ(POH) | Unsolvated POH bending | |||||
| K | 770 | 820 | 837 | 885 | 924 | 979 | 944 | ν A(P–OH) | Antisym. P–OH stretch |
| L | 793 | 812 | 827 | 842 | 879 | ν S(P–OH) | Sym. P–OH stretch | ||
Band G, centered between 1660 and 1680 cm−1, corresponds to the water bending modes δ(H2O).2 For the smaller clusters (n ≤ 6), its relative intensity increases with n, as expected, while its position remains nearly unchanged. Its width, over 50 cm−1, is large even at n = 1, and remains similar up to n = 12. As in other studies on microhydrated ions,32δ(H2O) is thus rather insensitive to the details of the cluster structure, i.e., the particular binding site of the water molecules.
The features labelled H and I are found between 1000 and 1400 cm−1 and are attributed to the antisymmetric and symmetric PO stretching modes, νA(PO) and νS(PO), respectively.2 They can couple markedly to the POH bending modes, δA(POH) and δS(POH), complicating a straightforward assignment. While the position of band I does not change much with n, band H is successively red-shifted from 1299 cm−1 (n = 0) to 1207 cm−1 (n = 6), after which it loses relative intensity and does not red-shift any further. Weak signals on both sides of band H are observed starting at n = 4. By n = 5, a new band H′ (at 1138 cm−1) emerges between I and H, and its intensity grows for n > 6. Bands K and L, observed at 820 and 793 cm−1, respectively, in the n = 1 spectrum, are assigned to P–OH stretching modes, νA(P–OH) and νS(P–OH). Their position is shifted to higher wavenumbers with increasing n. While band K remains prominent throughout all spectra shown in Fig. 1, band L broadens and drops in intensity such that it is difficult to identify for n > 6. Finally, a second new feature (M) is observed at smaller wavenumbers (<800 cm−1) for n > 6. It is very broad, and in analogy to the spectra of related ions,33,43,47,48 can be attributed to water librational modes. Hence, we observe five characteristic trends in the size-dependent IRMPD spectra shown in Fig. 1: (a) a continuous red-shift of band H up to n = 6, (b) the appearance and increase in cross section of band H′ for n ≥ 4, (c) a marked insensitivity of band I to the degree of microsolvation, (d) a monotonic blue-shift of band K up to n = 12, and (e) the appearance and increase in cross section of band M for n > 6.
The observed trends indicate significant variations in the solvation shell around H2PO4− as the cluster size increases. Up to n = 6, the solute bands mainly shift in position, while for the n > 6 spectra the relative intensities change significantly, such that these bands ultimately approach those observed in the condensed phase spectrum for n = 12. Indeed, comparison of the n = 12 spectrum to that of a 0.1 M aqueous KH2PO4 solution (see Fig. 1) supports our original assignment for bands I, K and L to νS(PO), νA(P–OH) and νS(P–OH). Band H′ then corresponds to excitation of the νA(PO) mode1 and band H to the bending modes δA(POH) and δS(POH), observed at 1156 cm−1 and 1213 cm−1 (see Table 1), respectively, in the condensed phase spectrum. However, in our previous study for n = 0 and n = 1 we assigned band H to νA(PO) and found evidence for δA(POH) and δS(POH) at lower energies around band I.2 Hence, upon hydration δ(POH) is blue-shifted while νA(PO) is red-shifted to such an extent that their energetic ordering is reversed. Note, the change in the relative intensities of bands H′ and H suggest that this reversal occurs between n = 6 and n = 8.
Simulated spectra for n = 2
We start our analysis with the n = 2 cluster which, as the smallest member of the current series, illustrates important factors that cannot be adequately reproduced by harmonic frequency calculations based on stationary 0 K structures. Four isomers labelled 2–1 to 2–4 are identified by structural optimization within 4 kJ mol−1. Their 0 K structures together with simulated harmonic IR spectra are shown in Fig. 2. Comparison to the experimental IRMPD spectrum indicates that the simulated spectra of 2–1 to 2–3 are in poor agreement with experiment. Only the spectrum of the fourth isomer 2–4, calculated to lie 3.6 kJ mol−1 higher in energy than 2–1, matches the experimental observation of just two bands (H and I) satisfactorily. For 2–4, band H and I mainly correspond to the antisymmetric and symmetric combinations of the terminal PO stretches νA(PO) and νS(PO), respectively, with the two POH bending modes, δA(POH) and δS(POH), also contributing to band I. Summarizing, the comparison to the predicted harmonic spectra indicates that 2–4 represents the dominant isomer, even though it is predicted to be highest in energy among the four minimum energy structures depicted in Fig. 2.
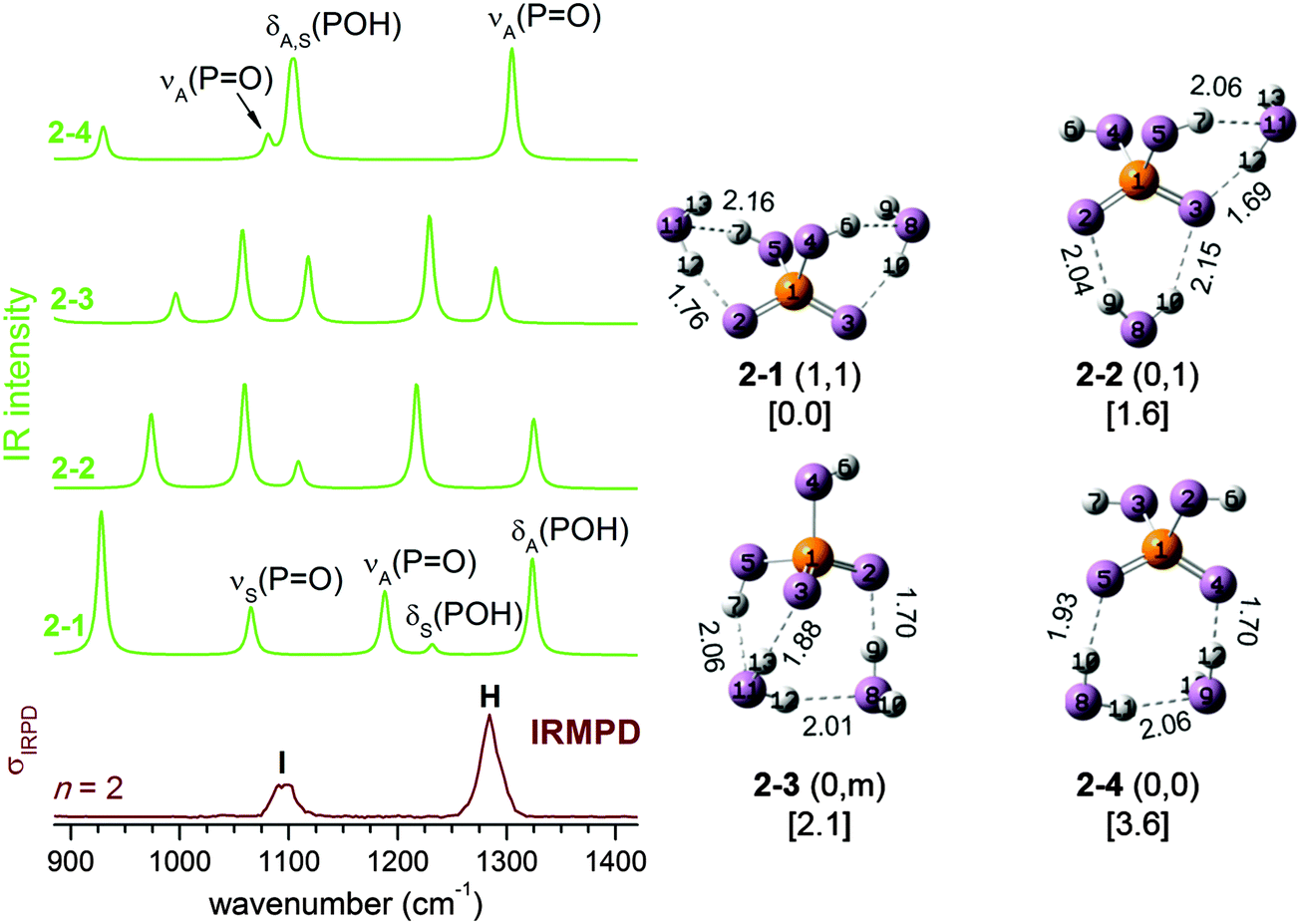 | ||
| Fig. 2 The harmonic IR spectra (left), as well as MP2/aug-cc-pVDZ minimum-energy structures (right), zero-point corrected relative energies (in kJ mol−1) and HB distances (in Å) for n = 2. The experimental IRMPD spectrum (bottom trace) is also shown. | ||
However, a good estimate of the experimental isomer population for hydrogen-bonded systems needs to include entropy, even at low temperatures. As we demonstrated in our previous study on the dihydrogen phosphate–water complex,2 the H2PO4−(H2O) potential energy surface (PES) is rather flat, allowing transformations between different conformers even at low temperatures. The entropy of non-rigid structures, as in the present case, cannot be reliably estimated from harmonic vibrational frequencies (e.g. harmonic analysis does not entropically favour 2–4) but needs to be determined by running AIMD simulations to sample the phase space directly, in which the cluster can undergo structural transformation continuously. At each time step along a trajectory, the presence of a HB is identified using a threshold distance value of 2.50 Å so that the configuration at this step is categorized as belonging to one of the four isomers. In this context, it will prove helpful to introduce an alternative classification scheme. In the following, each structure is labelled with (s,s), where the value of s indicates the extent of solvation of each POH group. Values of 0, 1, and m refer to a POH group which is either not solvated, solvated by a single water molecule, or by a water molecule connected to other water molecules, respectively. Within this scheme, isomer 2–1 in Fig. 2 is labelled as (1,1), 2–2 as (0,1), 2–3 as (0,m) and 2–4 as (0,0).
The accumulated statistics based on AIMD trajectories are listed in Table 2, which provides an estimate of the population for each isomer. The result is temperature dependent. At 140 K, the dominant isomer is 2–2 (0,1), while the population of 2–4 (0,0) is the second highest. When the temperature is raised to 180 K, 2–4 (0,0) becomes the dominant isomer (51%), while 2–2 (0,1) at 40% still makes a significant contribution. 2–1 (1,1), the most stable isomer, has a population of only 13% at 140 K, which reduces to 5% at 180 K. Hence, even though 2–4 (0,0) is the least stable in terms of energy, its presence is favoured by entropy and thus its population increases as the temperature is raised.
| n | 2 | 4 | 6 | |||
|---|---|---|---|---|---|---|
| T/K | 140 | 180 | 140 | 180 | 140 | 180 |
| (0,0) | 18 | 51 | 4 | 4 | 28 | 1 |
| (0,1) | 55 | 40 | 7 | 19 | 0 | 3 |
| (0,m) | 14 | 4 | 89 | 73 | 9 | 48 |
| (1,1) | 13 | 5 | 0 | 1 | 0 | 1 |
| (1,m) | 0 | 3 | 41 | 17 | ||
| (m,m) | 0 | 0 | 22 | 30 | ||
Based on the populations listed in Table 2 a mixture of two isomers, 2–2 (0,1) and 2–4 (0,0), mainly contributes to the n = 2 IRMPD spectrum. At 140 K, 2–2 (0,1) is the dominant isomer. But as the temperature is raised to 180 K, the contribution of 2–4 (0,0) increases. Note, the simulation temperatures cannot be directly compared to the experimental temperature of the ions. On one hand, the atomic motion in the AIMD simulations is treated by Newtonian mechanics and the temperature is calculated from the average kinetic energy, without considering zero point vibrational energy (ZPE). On the other hand, the measured ion trap temperature (15 K) only represents a lower limit for the temperature of the ions in the presence of RF heating as well as other collisional heating mechanisms. Thus, the average internal temperature of the ions is somewhat higher than the ion trap temperature, but considerably lower than the simulation temperature, since the latter needs to be increased in order to compensate for the lack of ZPE. The latter effect is quite substantial if one considers that the ZPE for the OH stretching modes are significantly larger (∼20 kJ mol−1) than the lowest isomerization barriers (4 kJ mol−1) for n = 2.
If the IRMPD spectrum is largely due to 2–2 (0,1) and 2–4 (0,0), there remains one problem: while the harmonic spectrum for 2–4 (0,0) is similar to the IRMPD result, the spectrum for 2–2 (0,1) is not. The difference is that in 2–4 (0,0), the POH groups are not solvated, and its IR spectrum (see Fig. 2) is thus quite similar to that of bare H2PO4−, with νS(PO), δS(POH) and δA(POH) close in energy and well separated from νA(PO). When there is a water molecule bound between a POH and a PO group, as in 2–2 (0,1) (and also in 2–1 (1,1) and 2–3 (0,m)), the coupling between the modes is enhanced, leading to four well separated bands (see Fig. 2).
At finite temperature, the lengths of the weaker HBs in H2PO4−(H2O)2 undergo much larger changes than the stronger covalent bonds. Such dynamic fluctuations influence the charge distribution and thus indirectly induce shifts in the vibrational frequencies of modes involving covalent bonds, which ultimately leads to a smearing out of the corresponding IR bands. In contrast to molecular dynamics simulations, harmonic analysis based on stationary structures cannot reproduce such effects. Dynamic effects can, in principle, be observed for any mode, but are typically most pronounced for moieties directly involved in hydrogen bonded networks, like the H2O librational modes in larger microhydrated clusters (band M in Fig. 1).
The mode related to the microhydrated POH group in 2–2 (0,1) provides a good example for such dynamic effects. As shown in Fig. 3, considerable fluctuations (2.0–5.5 Å) in the HPOH⋯Ow distance are predicted during AIMD runs at 140 K and 180 K. Dynamic fluctuations along this coordinate induce significant shifts in the harmonic frequency of the POH bending mode, which then leads to the observed broadening/smearing out of the IR band. Since the fluctuation of the HB length occurs on a much longer time scale than the PO–H stretching and POH bending vibrations, we can consider the cluster as vibrating under the perturbation of this HB. Approximately, its effect on the harmonic IR spectrum can be estimated by systematically varying the H7⋯O11 distance from 2.10 to 2.70 Å. A structure optimization is performed with H7⋯O11 constrained to a specific value, followed by a harmonic analysis shown in the right panel of Fig. 3. δA(POH) shifts appreciably to the red as the HB is lengthened, because the POH bending motion is not hindered any longer after the HB is broken. The νA(PO) band also shifts to the red, although not to the same extent.
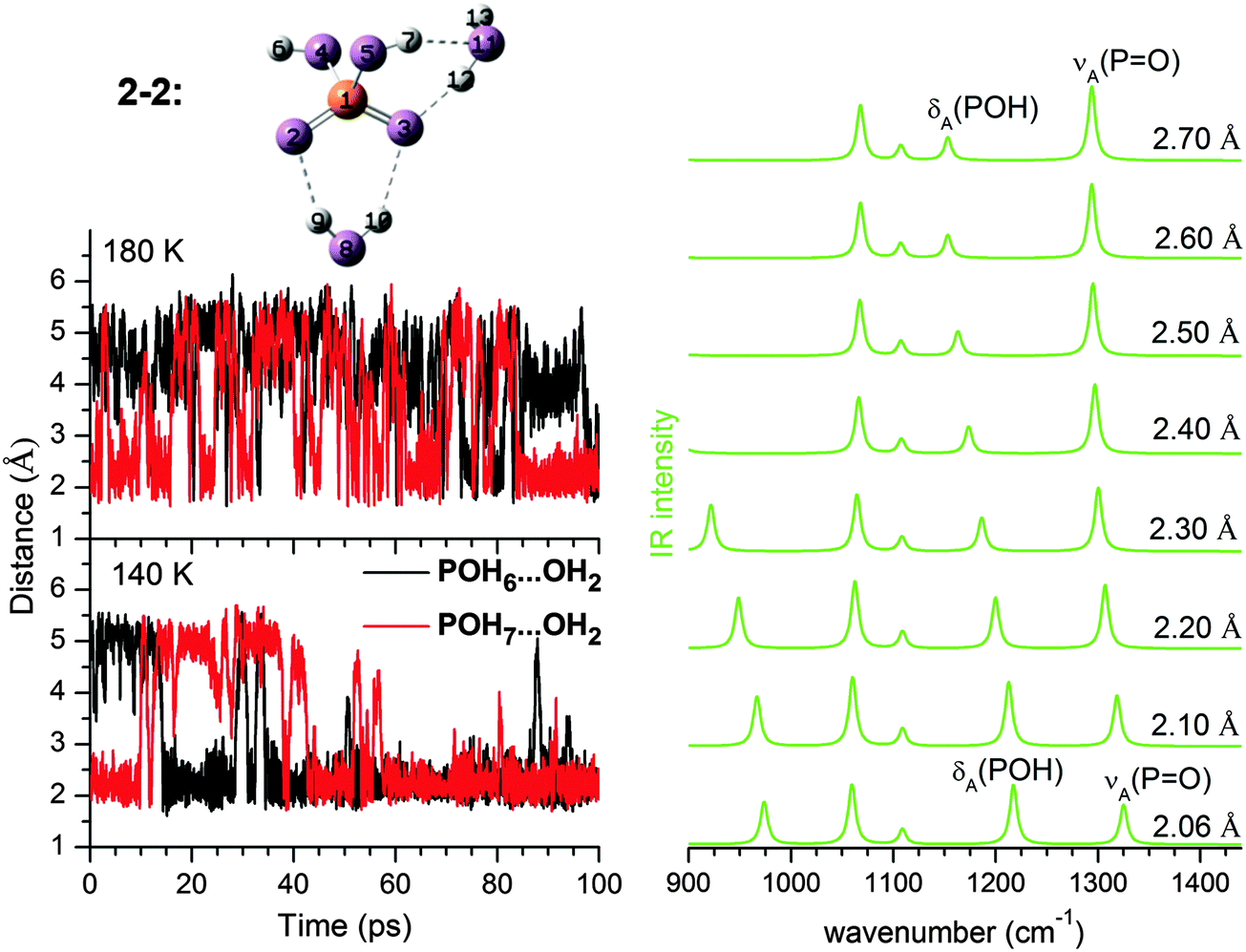 | ||
| Fig. 3 Left, fluctuation in the HPOH⋯Ow HB distance during AIMD simulation at 140 K and 180 K in 2–2 (0,1). Right, harmonic IR spectra derived from MP2/aug-cc-pVDZ calculations for 2–2 (0,1) and constraining the HPOH⋯Ow distance (H7⋯O11) to a value from 2.06 Å to 2.70 Å. The calculated energy difference between 2.06 Å to 2.70 Å is 2.13 kJ mol−1. | ||
DTCF spectra for n = 2 obtained from AIMD runs at three different temperatures are compared to the experimental IRMPD spectrum in Fig. 4. At 20 K, the initial structure, either 2–2 or 2–4, is maintained throughout the simulation (10 ps duration) and the vibrational motion is close to harmonic. At higher temperatures of 140 K and 180 K, the AIMD runs (300 ps duration) reveal substantial broadening of all IR-active features due to anharmonicities and interconversion between 2–2 and 2–4. At 180 K the combined population of 2–4 and 2–2 (see Table 2) amounts to more than 90% and the DTCF spectrum is quite similar to the experimental one. At 180 K, δS(POH) and νS(PO) merge into a single feature, corresponding to band I in the experimental spectrum. νA(PO) is associated with band H, while δA(POH) is smeared out due to the dynamic effects discussed above.
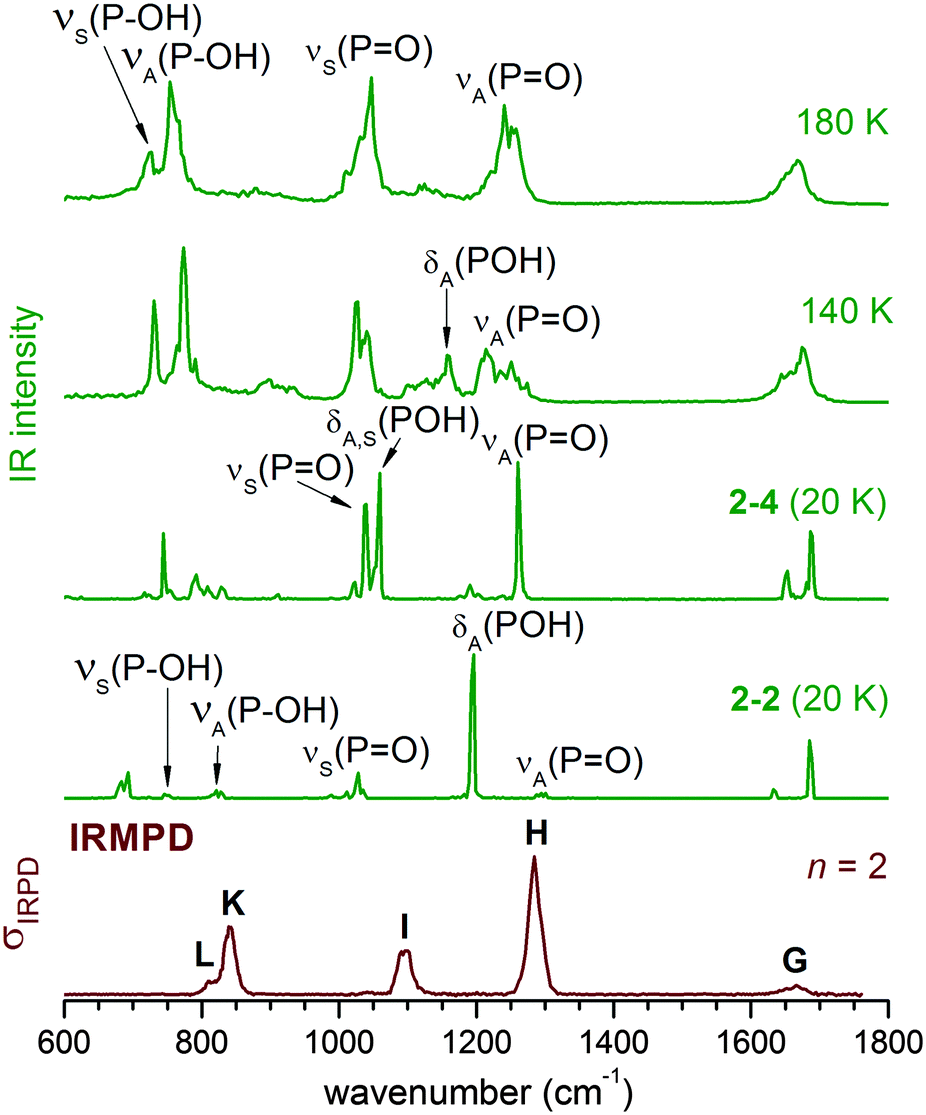 | ||
| Fig. 4 Comparison of DTCF spectra for n = 2 at 20 K, 140 K and 180 K, based on AIMD simulations, to the experimental IRMPD spectrum. | ||
The overall shape of the 180 K DTCF spectrum agrees quite well with the experimental IRMPD spectrum. Note, some discrepancies between experiment and simulation can also be expected due to the multiple photon nature of the photodissociation process, which is not explicitely considered in the AIMD simulations.34,47 However, these effects do not seem to be dominant here and the AIMD simulations do capture the underlying physics relevant for the important features observed in the IRMPD spectra.
Simulated spectra for n = 4 and n = 6
As n increases, the number of possible isomers (see ESI†) increases rapidly. As is typical for microhydrated clusters, there are many ways to arrange the HBs between solute and solvent molecules. Given the small energy differences among the isomers, it is not easy to identify the minimum energy structure with confidence, which in any case is not necessarily the most populated conformer at finite temperature as shown in Table 2. The key to understanding the IRMPD spectra is the extent of solvation around the two POH groups; this not only provides a way to classify these structures, but also turns out to be a good indication of its harmonic spectra. Our results show that the harmonic IR spectra for the same type of solvation motifs, such as (0,0), (0,1), or (1,1), but different n, are fairly similar (see ESI†). The changes observed in the IRMPD spectra as n increases are thus mainly related to how the POH groups are solvated.At n = 4, the 4–3 (0,m) structure (see Fig. 5) becomes dominant. In 4–3, one POH group is free, while the other one (PO5H7) is part of a HB network involving a four-membered water ring and the two PO groups. The population of 4–3 in the AIMD simulations is 89% at 140 K and decreases slightly to 73% at 180 K (see Table 2). A (0,m) structure is also identified for n = 2 as 2–3, although it is very unstable and easily interconverts to 2–2 during the AIMD runs. Hence, the free energy of the (0,m) structures becomes more favourable as more water molecules are added, accompanied by a strengthening of the HPOH⋯Ow HB (O9⋯H7 in Fig. 5). Its fluctuation around 2.00 Å during the 140 and 180 K AIMD runs is less pronounced than that observed for 2–2 (0,1), indicating a better dynamic stability.
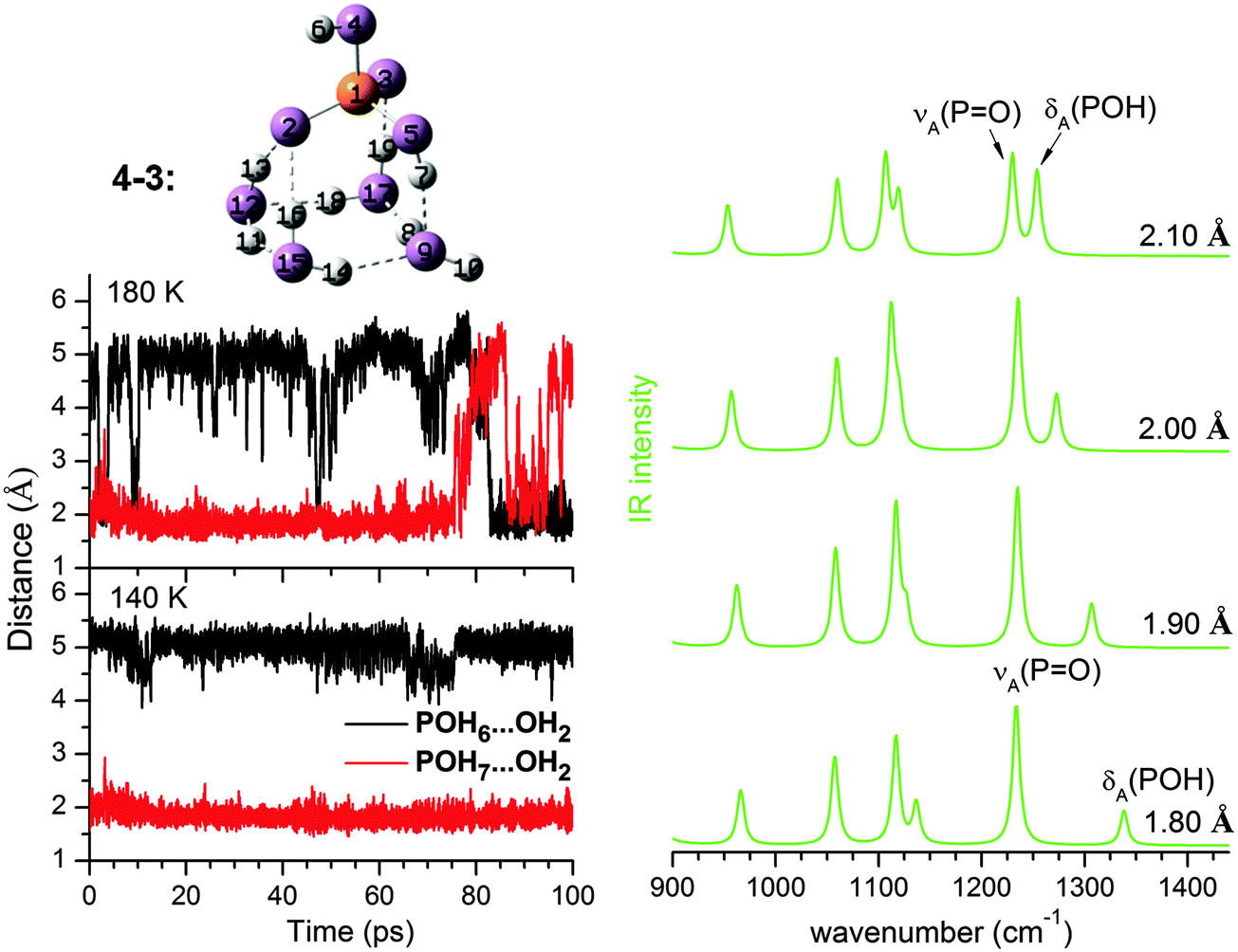 | ||
| Fig. 5 Left, fluctuation in the HPOH⋯Ow HB distance during AIMD simulations at 140 K and 180 K for the (0,m) structure at n = 4. Right, harmonic IR spectra derived from MP2/aug-cc-pVDZ calculations for 4–3 and constraining the HPOH⋯Ow distance to a value from 1.80 Å to 2.10 Å. The calculated energy difference between 2.10 Å and 1.90 Å is 1.55 kJ mol−1. | ||
The harmonic IR spectrum of the (0,m) structure (see Fig. 6) shows new features compared to the predicted spectra of the (0,1) and (0,0) structures. The microhydrated POH group (PO5H7 in Fig. 5) is now part of a HB network and separated from the next PO group by at least two water molecules. The dependence of the POH angle on the PO stretching mode is reduced. As a result, δ(POH) of the solvated POH group is decoupled from νA(PO) and becomes the highest energy feature in the spectrum, while νA(PO) is considerably red-shifted (∼100 cm−1) compared to νA(PO) of 2–2 (0,1) shown in Fig. 3.
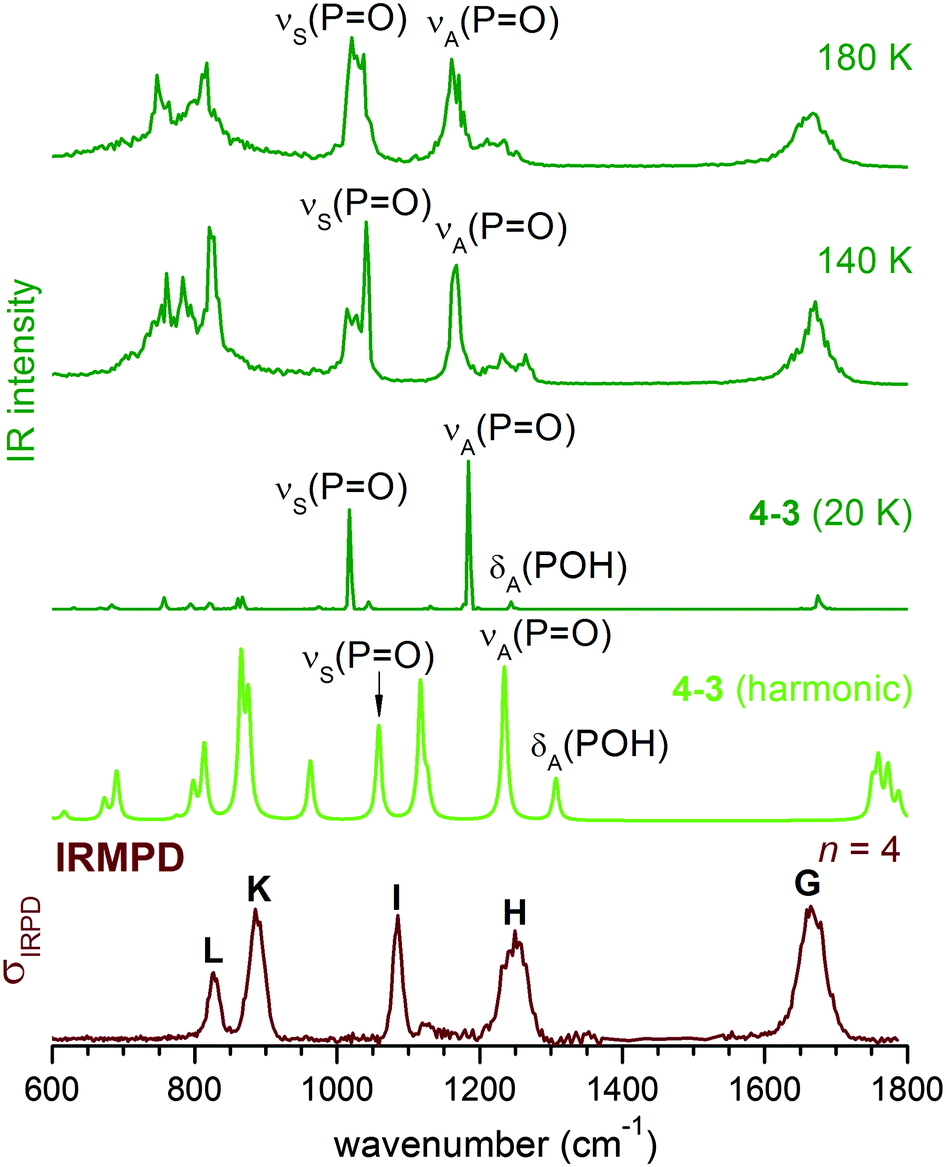 | ||
| Fig. 6 Comparison of the experimental IRMPD spectrum for n = 4 (bottom trace) to the MP2 harmonic IR spectrum for 4–3 (0,m), the dominant structure at n = 4, and the DTCF spectra at 20 K, 140 K and 180 K. The duration of simulation is 10 ps at 20 K, and 300 ps at 140 and 180 K. | ||
As the HPOH⋯Ow distance varies from 1.80 to 2.10 Å in 4–3 (0,m), it is again δA(POH) that changes significantly in position, while νA(PO) moves little (Fig. 5). Considering this dynamic effect, the IR spectrum should contain a νA(PO) band, red-shifted compared to the νA(PO) band in 2–2 (0,1), and a broader feature on its high energy side, which can be attributed to δA(POH) of the solvated POH group. This trend is indeed reproduced in the DTCF spectra at 140 and 180 K and is also observed in the experimental IRMPD spectrum (see Fig. 6). At 20 K, the minimum energy structure is maintained throughout the simulation and the DTCF spectrum basically resembles the harmonic spectrum, although the band positions are red-shifted relative to the MP2 and experimental results, due to the DFT method used in the AIMD simulations. The dynamic effect also broadens the features related to νS(PO) as well as δ(POH) of the free POH group, which both merge into a single band and correspond to band I in the experimental spectrum. The shift of band H to lower energies as n increases from two to four is attributed to the enhanced presence of (0,m) structures, i.e. the solvation of one of the two POH groups.
In contrast to the n = 4 cluster, for which mainly a single isomer, (0,m), is predicted, a mixture of isomers is found to be populated during AIMD simulations for n = 6 (see Table 2), including significant contributions from (0,m), (m,m) and (1,m) structures. The second POH group is thus solvated starting at n = 6 (see 6–1 and 6–3 in Fig. 7). This leads to further changes to the harmonic IR spectrum. Upon incorporation of both POH groups into the HB network, both POH bending modes are shifted to higher energies and are effectively decoupled from the PO stretching modes. The two bands at highest energy around 1300 cm−1 are now mainly from combinations of the two POH bending modes, while the PO stretching modes are predicted between 1050 and 1150 cm−1.
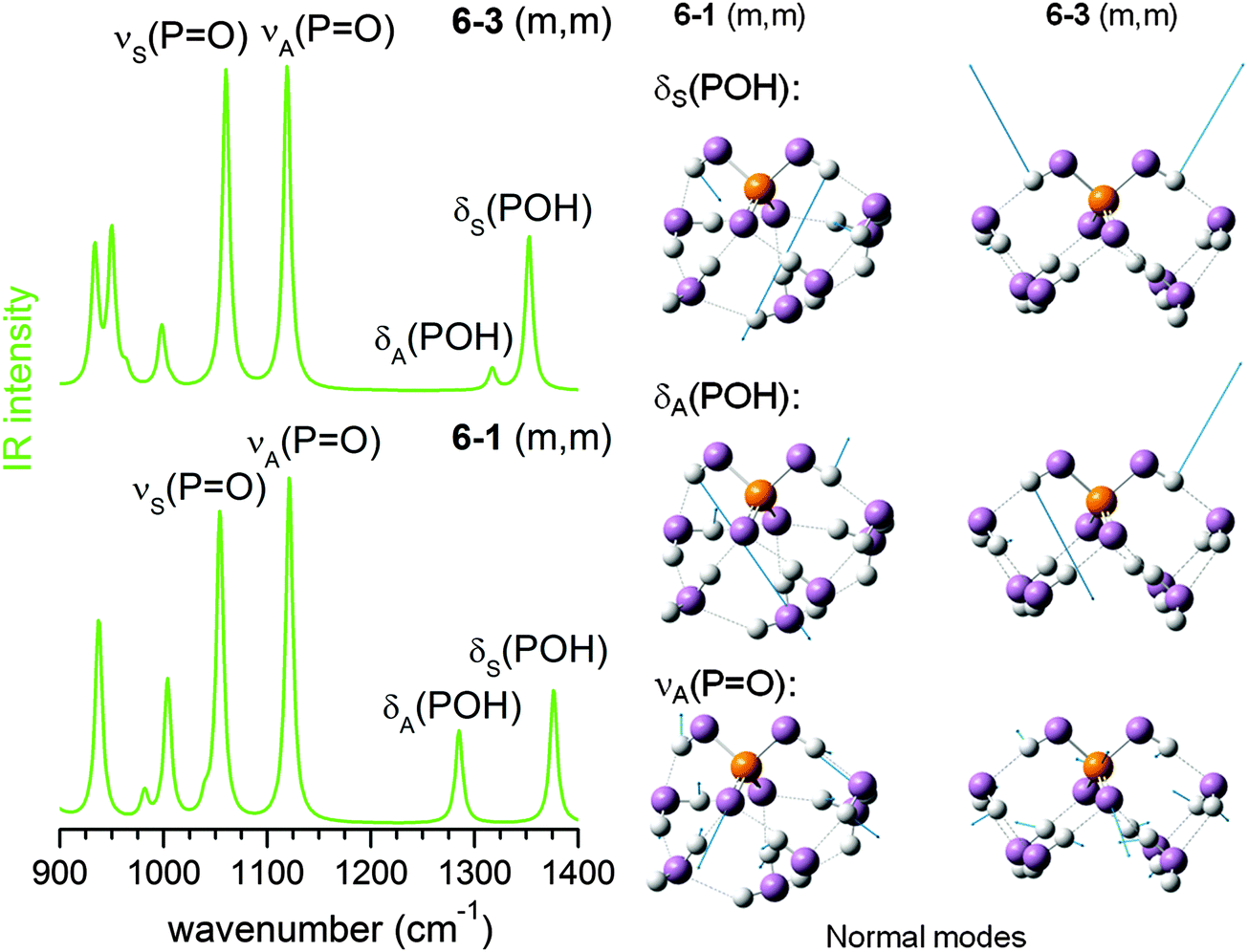 | ||
| Fig. 7 MP2/aug-cc-pVDZ harmonic IR spectra (left) and displacement vectors (blue arrows) of three harmonic vibrational modes (right) for 6–1 (m,m) and 6–3 (m,m). | ||
The experimental IRMPD spectrum can be assigned by considering such a mixture, as shown in Fig. 8. Band I is attributed to νS(PO), which is not particularly sensitive to structural changes. Band H is assigned to the νA(PO) modes of the (0,0), (0,m) and (1,m) structures. The first appearance of band H′ between I and H is then linked to the νA(PO) mode of the (m,m) structures, which were not significantly populated for n < 6, but are thermodynamically favoured for larger n.
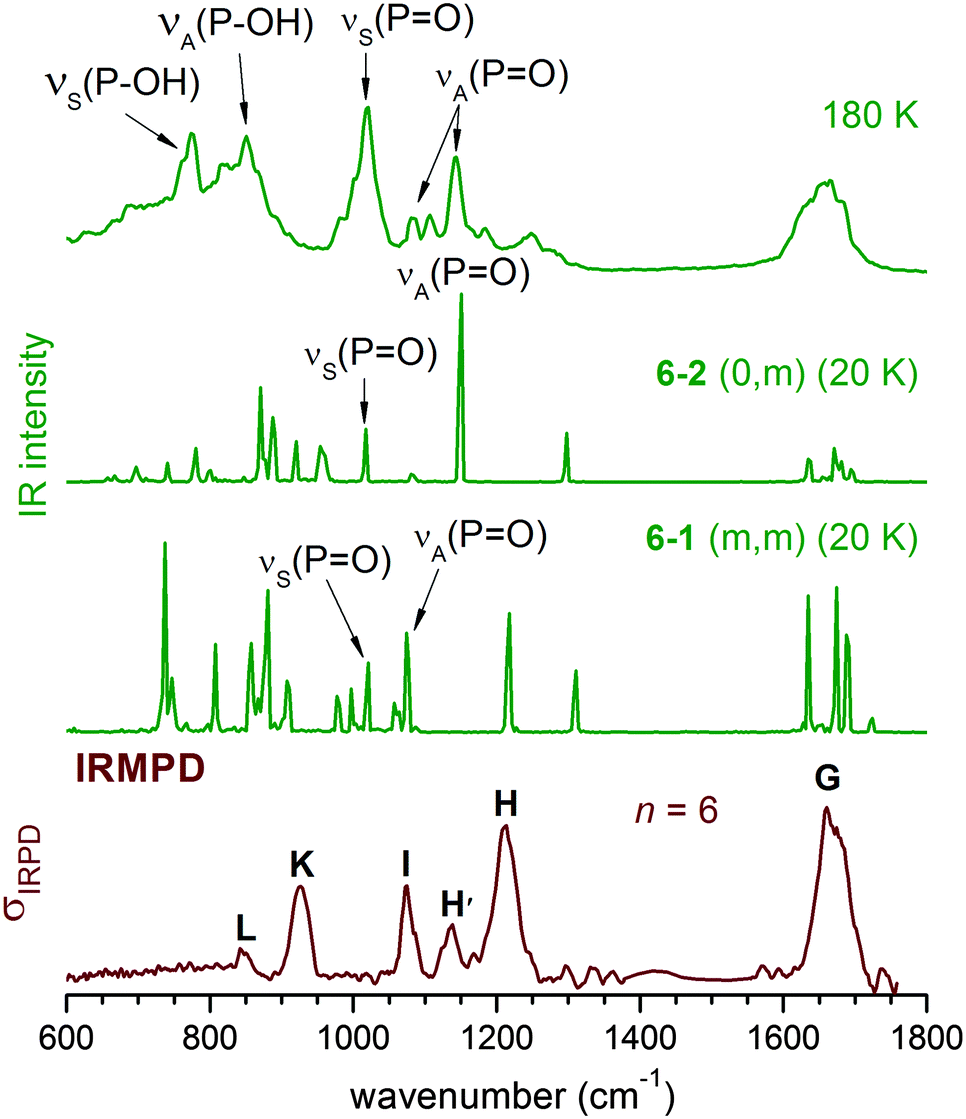 | ||
| Fig. 8 DTCF spectra for n = 6 at 20 K, and 180 K, based on AIMD simulation, in comparison with the experimental IRMPD spectrum. At 20 K, the structure is maintained throughout the 10 ps simulation, and the DTCF spectrum is basically the harmonic spectrum, although the peak positions are red-shifted relative to the MP2 and experimental results, due to the DFT method used in the AIMD simulations. At 180 K, the simulation duration is 300 ps. | ||
Overall Trends
Based on the analysis above, the trends observed in the experimental spectra shown in Fig. 1 are directly linked to site-specific microhydration of the solute H2PO4− anions. The first water molecules bind to the PO groups instead of the POH groups, because the terminal O-atoms are more negatively charged.1 Up to n = 2, the POH groups are free or involved in a single HB to a H2O molecule. This HB between a water molecule and a POH group is weaker than the corresponding HB involving a PO group, and at finite temperature the weaker HB with the POH group is more easily disrupted. Hence, the experimental IRMPD spectra in the PO stretching/POH bending region are dominated by the contribution from (0,0) structures.
By n = 4, there are enough water molecules so that one of the POH groups forms a HB to a unit of four water molecules solvating the two PO groups. The formation of such a (0,m) structure changes the vibrational signature. The bending mode of the solvated POH group is no longer coupled to the PO stretching modes, and its position shifts to higher energies. Furthermore, its intensity smears out over a broader energy range as a result of HB dynamics, leading to the weak signals on the high energy tail of band H. At the same time, νA(PO) shifts to lower energies, which is the main reason for the observed red-shift of band H with increasing n.
By n = 6, solvation of the second POH group is favoured and (m,m) structures significantly contribute to the IR spectrum. The energy of the νA(PO) mode for the (m,m) structures is lowered to 1100 cm−1, corresponding to band H′, while both POH bending bands are blue-shifted and their intensity is smeared out due to the dynamic effect. However, the mixture of structures at n = 6 is not yet dominated by (m,m) structures, and the νA(PO) vibration of other structures contributes to band H. As n increases, band H′ gains in intensity, because more water molecules are available and population of the (m,m) structure is favoured. In aqueous solution, band H′ becomes dominant, while the intensity of band H is smaller and contributes, together with the bending of solvated POH, to the observed shoulder above 1200 cm−1.16
Observations in the region below 1000 cm−1 provide further information. Bands L and K are assigned to the symmetric and antisymmetric P–OH stretching modes, respectively. The blue-shifts in their positions with increasing n are related to the extent of POH solvation. For example, among the (0,0) structures, the calculated νA(P–OH) harmonic frequencies shift from 833 cm−1 at n = 2 to 850 cm−1 at n = 4 and 867 cm−1 at n = 6, none of which is big enough to account for the more than 100 cm−1 blue-shift observed in the experiment. At n = 6, only (m,m) structures exhibit νA(P–OH) vibrational frequencies above 900 cm−1. The position of band K for n = 8, 10 and 12 again indicates the predominance of (m,m) structures, which also explains the almost fixed position of band K at these sizes. Dynamic effects are also important. Many water wagging modes fall into this region. Their vibrational frequencies are very sensitive to HB network fluctuations, resulting in the broad absorption feature in-between 600 cm−1 and 1000 cm−1. A full table of experimental band positions and assignments is provided in the ESI.†
Conclusions
H2PO4−(H2O)n clusters represent a useful model system for studying solute–solvent interactions, one water molecule at a time, by way of vibrational action spectroscopy combined with AIMD simulations. However, the IRMPD spectra of cold H2PO4−(H2O)n anions with n = 2–12 in the spectral range of the stretching and bending modes of the solute anion cannot be fully understood by comparison to harmonic spectra of minimum-energy structures, requiring consideration of anharmonic and dynamic effects. The abundance of a particular isomer is not determined simply by relative energy. Rather, entropy plays a significant role already at lower temperatures. Furthermore, the fluctuation of the HB network, in general, and individual HB bond distances, in particular, induces changes in the charge distribution, which result in vibrational frequency shifts and hence characteristic broadening of spectral features.Finite temperature AIMD simulations that directly sample the phase space qualitatively recover the observed changes in the position, width and intensity of the IRMPD bands with cluster size and allow relating them to the sequence of microhydration. The first water molecules bind to the two PO groups and only when these have been incorporated into a HB network are the two POH groups hydrated. Starting with n = 8, the gas phase cluster spectra begin to show similarities to the solution phase spectrum, indicating that the first hydration shell is nearing completion.
The present results show that anharmonic and dynamic effects are important in understanding these biologically relevant interactions. The consideration of such effects may also be important in the interpretation of the vibrational spectra of many other microhydrated ions, most of which have, up to date, been interpreted on the basis of a harmonic analysis of 0 K structures.
Acknowledgements
We would like to thank the Stichting voor Fundamenteel Onderzoek der Materie (FOM) for beam time at FELIX and the FELIX-staff for excellent support and assistance. L. Jiang thanks the Alexander von Humboldt Foundation for a postdoctoral scholarship. DMN and TY acknowledge support from the Air Force Office of Scientific Research under Grant No. FA9550-12-1-0160. AIMD simulations reported in this paper were performed at National Supercomputing Center in Shenzhen. ZFL acknowledges support from National Natural Science Foundation of China under Project No. 21473151.Notes and references
- M. Klahn, G. Mathias, C. Kotting, M. Nonella, J. Schlitter, K. Gerwert and P. Tavan, J. Phys. Chem. A, 2004, 108, 6186–6194 CrossRef.
- L. Jiang, S. T. Sun, N. Heine, J. W. Liu, T. I. Yacovitch, T. Wende, Z. F. Liu, D. M. Neumark and K. R. Asmis, Phys. Chem. Chem. Phys., 2014, 16, 1314–1318 RSC.
- F. H. Westheimer, Science, 1987, 235, 1173–1178 CAS.
- E. Takeda, Y. Taketani, N. Sawada, T. Sato and H. Yamamoto, BioFactors, 2004, 21, 345–355 CrossRef CAS PubMed.
- I. R. Vetter and A. Wittinghofer, Science, 2001, 294, 1299–1304 CrossRef CAS PubMed.
- H. Takeuchi, H. Murata and I. Harada, J. Am. Chem. Soc., 1988, 110, 392–397 CrossRef CAS.
- C. Kotting, A. Kallenbach, Y. Suveyzclis, A. Wittinghofer and K. Gerwert, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6260–6265 CrossRef PubMed.
- C. Kotting, M. Blessenohl, Y. Suveyzdis, R. S. Goody, A. Wittinghofer and K. Gerwert, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13911–13916 CrossRef PubMed.
- E. L. Karjalainen, A. Hardell and A. Barth, Biophys. J., 2006, 91, 2282–2289 CrossRef CAS PubMed.
- A. Barth, J. Biol. Chem., 1999, 274, 22170–22175 CrossRef CAS PubMed.
- A. B. Pribil, T. S. Hofer, B. R. Randolf and B. M. Rode, J. Comput. Chem., 2008, 29, 2330–2334 CrossRef CAS PubMed.
- E. Tang, D. Di Tommaso and N. H. de Leeuw, J. Chem. Phys., 2009, 130, 234502 CrossRef PubMed.
- E. Tang, D. Di Tommaso and N. H. de Leeuw, Phys. Chem. Chem. Phys., 2010, 12, 13804–13815 RSC.
- T. B. Lee and M. L. McKee, Phys. Chem. Chem. Phys., 2011, 13, 10258–10269 RSC.
- E. Pluharova, M. Oncak, R. Seidel, C. Schroeder, W. Schroeder, B. Winter, S. E. Bradforth, P. Jungwirth and P. Slavicek, J. Phys. Chem. B, 2012, 116, 13254–13264 CrossRef CAS PubMed.
- J. VandeVondele, P. Troester, P. Tavan and G. Mathias, J. Phys. Chem. A, 2012, 116, 2466–2474 CrossRef CAS PubMed.
- C. C. Pye and M. R. Michels, Can. J. Anal. Sci. Spectrosc., 2005, 50, 70–86 CAS.
- K. A. Syed, S. F. Pang, Y. Zhang and Y. H. Zhang, J. Chem. Phys., 2013, 138, 024901 CrossRef PubMed.
- K. A. Syed, S. F. Pang, Y. Zhang, G. Zeng and Y. H. Zhang, J. Phys. Chem. A, 2012, 116, 1558–1564 CrossRef CAS PubMed.
- C. C. Pye and W. W. Rudolph, J. Phys. Chem. A, 2003, 107, 8746–8755 CrossRef CAS.
- P. E. Mason, J. M. Cruickshank, G. W. Neilson and P. Buchanan, Phys. Chem. Chem. Phys., 2003, 5, 4686–4690 RSC.
- Y. Kameda, K. Sugawara, T. Hosaka, T. Usuki and O. Uemura, Bull. Chem. Soc. Jpn., 2000, 73, 1105–1112 CrossRef CAS.
- M. Smiechowski, E. Gojlo and J. Stangret, J. Phys. Chem. B, 2009, 113, 7650–7661 CrossRef CAS PubMed.
- K. Vonburg and P. Delahay, Chem. Phys. Lett., 1981, 78, 287–290 CrossRef CAS.
- A. T. Blades, J. S. Klassen and P. Kebarle, J. Am. Chem. Soc., 1995, 117, 10563–10571 CrossRef CAS.
- P. Kebarle and U. H. Verkerk, Mass Spectrom. Rev., 2009, 28, 898–917 CrossRef CAS PubMed.
- G. Markovich, S. Pollack, R. Giniger and O. Cheshnovsky, J. Chem. Phys., 1994, 101, 9344–9353 CrossRef CAS PubMed.
- O. M. Cabarcos, C. J. Weinheimer, J. M. Lisy and S. S. Xantheas, J. Chem. Phys., 1999, 110, 5–8 CrossRef CAS PubMed.
- X. B. Wang, X. Yang and L. S. Wang, Int. Rev. Phys. Chem., 2002, 21, 473–498 CrossRef CAS PubMed.
- W. H. Robertson and M. A. Johnson, Annu. Rev. Phys. Chem., 2003, 54, 173–213 CrossRef CAS PubMed.
- D. M. Neumark, Mol. Phys., 2008, 106, 2183–2197 CrossRef CAS PubMed.
- K. R. Asmis and D. M. Neumark, Acc. Chem. Res., 2012, 45, 43–52 CrossRef CAS PubMed.
- J. Zhou, G. Santambrogio, M. Brummer, D. T. Moore, G. Meijer, D. M. Neumark and K. R. Asmis, J. Chem. Phys., 2006, 125, 111102 CrossRef PubMed.
- N. Heine and K. R. Asmis, Int. Rev. Phys. Chem., 2015, 34, 1–34 CrossRef CAS PubMed.
- R. G. Keesee and A. W. Castleman, J. Am. Chem. Soc., 1989, 111, 9015–9018 CrossRef CAS.
- A. T. Blades, Y. H. Ho and P. Kebarle, J. Am. Chem. Soc., 1996, 118, 196–201 CrossRef CAS.
- A. T. Blades, J. S. Klassen and P. Kebarle, J. Am. Chem. Soc., 1996, 118, 12437–12442 CrossRef CAS.
- X. B. Wang, E. R. Vorpagel, X. Yang and L. S. Wang, J. Phys. Chem. A, 2001, 105, 10468–10474 CrossRef CAS.
- N. Heine, M. R. Fagiani, M. Rossi, T. Wende, G. Berden, V. Blum and K. R. Asmis, J. Am. Chem. Soc., 2013, 135, 8266–8273 CrossRef CAS PubMed.
- S. Jaeqx, J. Oomens, A. Cimas, M. P. Gaigeot and A. M. Rijs, Angew. Chem., Int. Ed., 2014, 53, 3663–3666 CrossRef CAS PubMed.
- V. Brites, J. M. Lisy and M. P. Gaigeot, J. Phys. Chem. A, 2015, 119, 2468–2474 CrossRef CAS PubMed.
- E. Garand, T. Wende, D. J. Goebbert, R. Bergmann, G. Meijer, D. M. Neumark and K. R. Asmis, J. Am. Chem. Soc., 2010, 132, 849–856 CrossRef CAS PubMed.
- D. J. Goebbert, E. Garand, T. Wende, R. Bergmann, G. Meijer, K. R. Asmis and D. M. Neumark, J. Phys. Chem. A, 2009, 113, 7584–7592 CrossRef CAS PubMed.
- D. J. Goebbert, G. Meijer and K. R. Asmis, AIP Conf. Proc., 2009, 1104, 22–29 CrossRef CAS PubMed.
- D. J. Goebbert, T. Wende, R. Bergmann, G. Meijer and K. R. Asmis, J. Phys. Chem. A, 2009, 113, 5874–5880 CrossRef CAS PubMed.
- L. Jiang, T. Wende, R. Bergmann, G. Meijer and K. R. Asmis, J. Am. Chem. Soc., 2010, 132, 7398–7404 CrossRef CAS PubMed.
- Y. Miller, G. M. Chaban, J. Zhou, K. R. Asmis, D. M. Neumark and R. B. Gerber, J. Chem. Phys., 2007, 127, 094305 CrossRef PubMed.
- T. I. Yacovitch, T. Wende, L. Jiang, N. Heine, G. Meijer, D. M. Neumark and K. R. Asmis, J. Phys. Chem. Lett., 2011, 2, 2135–2140 CrossRef CAS.
- D. Oepts, A. F. G. Vandermeer and P. W. Vanamersfoort, Infrared Phys. Technol., 1995, 36, 297–308 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven Jr., K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, revision D.01, Gaussian, Inc., Pittsburgh, PA, 2005 Search PubMed.
- J. P. Merrick, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 11683–11700 CrossRef CAS PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS PubMed.
- G. Lippert, J. r. Hutter and M. Parrinello, Theor. Chem. Acc., 1999, 103, 124–140 CrossRef CAS.
- M. Krack, Theor. Chem. Acc., 2005, 114, 145–152 CrossRef CAS.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. J. Martyna and M. E. Tuckerman, J. Chem. Phys., 1999, 110, 2810–2821 CrossRef CAS PubMed.
- S. Nose, J. Chem. Phys., 1984, 81, 511–519 CrossRef CAS PubMed.
- D. A. McQuarrie, Statistic Mechanics, Harper-Collins Publishers, New York, 1976 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Assignments (Table S1), isomer populations (Table S2), n = 4 (Fig. S1) and n = 6 (Fig. S2) 0 K structures, harmonic spectra for n = 2, 4, 6 (Fig. S3 and S4). See DOI: 10.1039/c5cp02253c |
| ‡ Current address: Aerodyne Research, Inc., Billlerica, MA 01821 USA. |
| § Current address: Physical and Theoretical Chemistry Laboratory, South Parks Road, Oxford OX1 3QZ, UK. |
| This journal is © the Owner Societies 2015 |