
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnharmonic simulations of the vibrational spectrum of sulfated compounds: application to the glycosaminoglycan fragment glucosamine 6-sulfate†
Loïc
Barnes
abc,
Baptiste
Schindler
abc,
Abdul-Rahman
Allouche
*abc,
Daniel
Simon
abc,
Stéphane
Chambert
abd,
Jos
Oomens
ef and
Isabelle
Compagnon
abcg
aUniversité de Lyon, F-69622, Lyon, France. E-mail: isabelle.compagnon@univ-lyon1.fr
bUniversité Lyon 1, Villeurbanne, France
cInstitut Lumière Matière, UMR5306 Université Lyon 1-CNRS, Université de Lyon 69622 Villeurbanne Cedex, France
dLaboratoire de Chimie Organique et Bioorganique, INSA Lyon, CNRS, UMR5246, ICBMS, Bât. J. Verne, 20 Avenue A. Einstein, 69621 Villeurbanne Cedex, France
eRadboud University, Institute for Molecules and Materials, FELIX Laboratory, Toernooiveld 7, 6525ED Nijmegen, The Netherlands
fVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098XH Amsterdam, The Netherlands
gInstitut Universitaire de France IUF, 103 Blvd St Michel, 75005 Paris, France
First published on 22nd June 2015
Abstract
Mid-infrared spectroscopy coupled with mass spectrometry is an appealing tool for the sequencing and structural elucidation of functional modifications in biopolymers, as it offers direct spectroscopic identification of the functionality where the traditional mass spectrometric approach is insufficient. Whereas the gas phase vibrational spectroscopy of peptides (and to a lesser extent saccharides) has been widely investigated, sulfation has attracted much less attention, despite its prevalence in natural polymers. The simulation of the vibrational spectra of such functionalized compounds is however notoriously challenging, which impairs the interpretation of spectroscopic data in terms of structure. Driven by a striking case of such a failure for a sulfated glycosaminoglycan fragment, we elaborate on an original hybrid GVPT2 anharmonic approach. This strategy offers a significantly improved accuracy in the description of the sulfate modes, without the recourse to empirical scaling factors, and with a greatly reduced computational cost which is otherwise prohibitive for molecules of this size. Alternatively, we propose a selection of reasonably accurate harmonic methods with adequate scaling factors optimized on a set of benchmark compounds.
Introduction
Sulfation and phosphorylation are among the most common post-translational modifications of biomolecules such as proteins and they control their biological function.1–4 Yet, their characterization remains a challenge in proteomics due to their lability, versatility and isobaricity. Indeed, fragile functional modifications tend to be lost upon traditional tandem mass spectrometry analysis, which is virtually blind to their structure. Functionalization is also a burning question in glycosciences, which have recently risen as a strategic priority.5 Elucidation of the structural features of saccharides is highly challenging due to their unique complexity among biomolecules, and the development of a suite of analytical tools analogous to these available for proteomics is timely. Namely, an accurate description of an oligosaccharide includes: the monosaccharide content, which is complicated by the frequent occurrence of isobaric monomers, the identification of the nature and position of functional modifications, and the branched structures. While mass spectrometry (MS) has proven its use as an accurate sequencing tool for other biopolymers (proteins, DNA), it is not straightforwardly transferred to the saccharides family due to their equivocal MS and MS/MS signatures.6–9 To date, the full development of glycomics is impaired by this ambiguity, and orthogonal structural tools are greatly sought after. Among them, the coupling of MS with ion mobility or laser spectroscopy appears to develop as very promising hyphenated methods, each offering direct structural information, with complementary resolutions. While the overall shape can be derived from ion mobility cross section measurements, laser spectroscopy combined with ab initio calculations can provide details of the local chemical arrangement. Recent examples of electronic spectroscopy,10 and vibrational spectroscopy11 coupled with mass spectrometry have demonstrated the relevance of gas phase spectroscopy for the structural characterization of isolated, mass selected carbohydrates ions.In this context, we are developing a suite of experimental and theoretical tools for the structural characterization of glycosaminoglycans (GAGs), a major class of sulfated carbohydrate polymers expressed in extracellular matrices and on cell surfaces. GAGs are involved in diverse processes essential to biomedical research (e.g. regulation of coagulation,12 development13 or cancer14). They are characterized by a large variety of structures originating from the different carbohydrate constituting monomers, the different possible linkages between them and also their modification patterns, including sulfation, which is essential to their biological activities. The understanding of those patterns is thus of considerable biological interest.15 We have recently used IR-MPD (InfraRed-Multiple Photon Dissociation) spectroscopy to distinguish between isobaric sulfated and phosphorylated monosaccharides isolated in an ion trap.16 In this previous work, we probed the OH and NH stretching vibrations accessible in the mid-IR region with a table-top tunable IR laser covering the frequency range between 2900 and 3700 cm−1. Despite their identical MS and MS/MS signatures we were able to unambiguously – although somewhat indirectly – identify the nature of the functional modification (sulfate vs. phosphate), and the conformation.
In order to obtain a direct signature of the sulfate pattern in the fingerprint region, and more generally to heavy atoms-containing functional modifications, it is desirable to extend the IR-MPD approach toward lower frequencies.17,18 From an experimental point of view, this is feasible using Free Electron Lasers. On the other hand, the interpretation of such experimental data raises acute theoretical questions. Firstly, the widely used harmonic approximation of ab initio electronic potentials typically yields overestimated simulated vibrational frequencies. While it is generally accepted to apply an empirical scaling factor to reduce the calculated frequencies and match experiments, the use of several empirical scaling factors to account for different families of vibrations presenting distinct anharmonic behavior is strongly debated. As we intend to investigate a wide IR spectroscopic range – including as various vibrational modes as stretchings, bendings and torsions involving light and heavy atoms – this issue cannot be ignored. Furthermore, we face a dramatic failure of traditional harmonic simulations in the case of our model glycosaminoglycan component 6-O-sulfated glucosamine19,20 in the fingerprint region, as described below.
Glucosamine 6-sulfate: a case of failure of harmonic simulations
It is noteworthy that the DFT B3LYP/6-311+G(d) harmonic frequencies, which previously supported an unambiguous structural assignment of glucosamine 6-sulfate in the high frequency range, utterly fail to account for its IR-MPD spectrum measured in the fingerprint region (Experimental detail in ESI†), as seen in Fig. 1.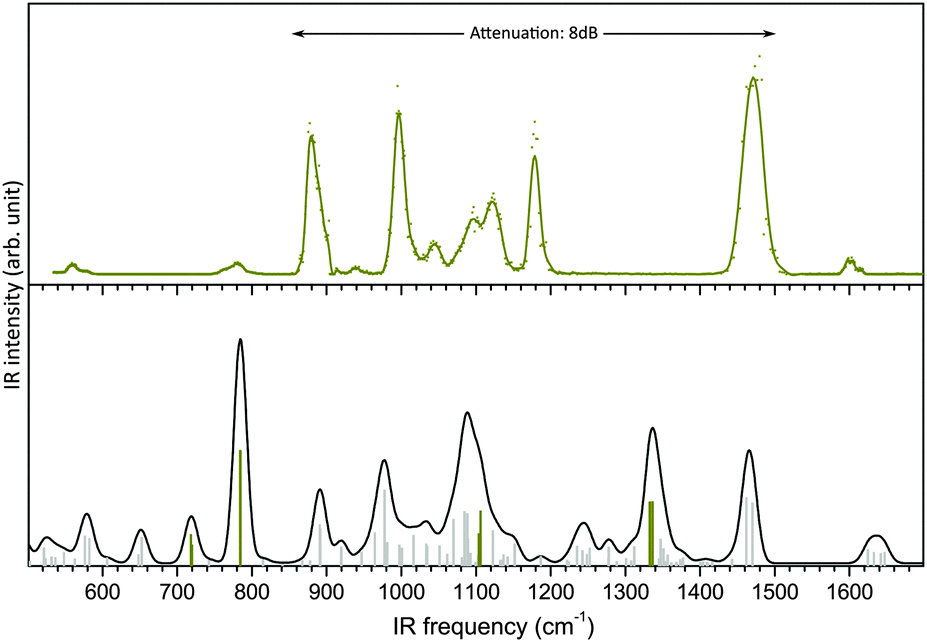 | ||
| Fig. 1 Mid-IR spectra of glucosamine 6-sulfate. Top panel: measured IR-MPD spectrum. Lower panel: B3LYP/6-311+G(d) harmonic frequencies scaled by 0.965 (sulfate modes are highlighted in dark yellow) and convoluted spectrum using a fwhm of 20 cm−1 (line). | ||
A most remarkable discrepancy is the absence of photofragmentation between 1200 and 1420 cm−1, while an intense SO2 asymmetric stretching mode is predicted at 1337 cm−1. Another inconsistency is the intense feature measured at 1180 cm−1, which is not accounted for in the simulations. Finally, the region below 850 cm−1, with only two weak bands measured at 560 cm−1 and 780 cm−1 is poorly reproduced.
This constitutes a striking case of impossible structural assignment, which is not due to an insufficient conformational exploration (the conformation was indeed established with confidence in ref. 16), but instead because of a thoroughly misleading simulated vibration pattern. Similar issues were encountered in the case of phosphorylated compounds, which remain to date a challenge for theoreticians.21 This suggests that the theoretical treatment of sulfation and phosphorylation is a general problem which critically impairs the structural characterization of a broad range of functionalized biomolecules. In this context, gas phase vibrational spectroscopy of peptides phosphorylation (and to a lesser extent saccharides phosphorylation) is widely investigated. On the other hand, harmonic and anharmonic simulations of the vibrational spectra of sulfate clusters related to aerosol seeding have attracted a great deal of attention.22,23 However, analogous studies addressing sulfated biomolecules are scarce despite their considerable biological relevance. Hence we believe that a strategy for the reliable simulation of the vibrational frequencies of sulfated biomolecules is of general interest.
Outline
In order to tackle the inconsistencies raised by sulfated vibrational frequencies, we report an extensive investigation of the accuracy of popular DFT functionals and basis sets for the prediction of the mid-IR harmonic frequencies of a series of benchmark species and we reckon suitable scaling factors. Then, the merits vs. computational costs of GVPT2 anharmonic corrections is assessed. The great advantage of the GVPT2 is that the anharmonic effect is taken into account without any empirical parameter. However, as the computational cost of the GVPT2 approach becomes prohibitive for larger species, we devised an original hybrid strategy, consisting of a large basis set for the calculation of the harmonic frequencies, combined with a smaller basis set for the calculations of the cubic and quartic terms of the potential. This method offers a significant reduction of the computational cost, bringing anharmonic correction to an accessible level for biomolecules without loss of precision and without empirical parameters. The performance of our hybrid method is emphasized in the first section for the case of glucosamine 6-sulfate, for which we show that the issue of the miscalculated vibrational pattern can be resolved. In the second section the elaboration of the hybrid method and a “how-to” are described in detail for facile implementation by others. Should one favor time-saving harmonic calculations, we also propose suitable combinations of functional and basis sets for sulfated compounds, together with appropriate scaling factors.Anharmonic simulations of glucosamine 6-sulfate
Influence of the functional and basis set on the harmonic frequencies
As shown in Fig. 1 and 2a, scaled harmonic B3LYP/6-311+G(d) simulations fail to account for the spectrum measured in the fingerprint region (see ESI† for Experimental details). This is particularly critical for the sulfate modes (dark yellow bars in Fig. 1) which do not match any experimental feature.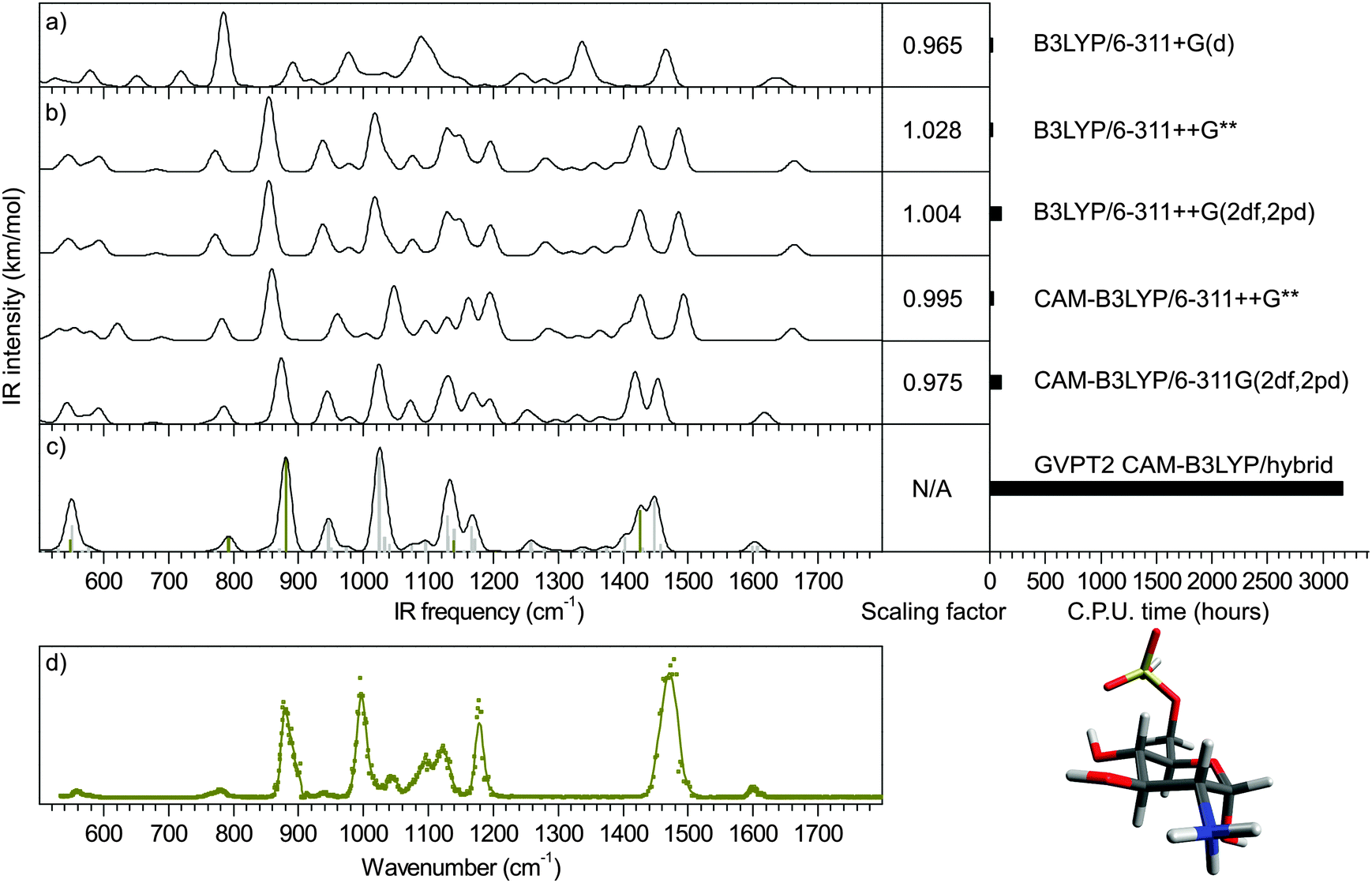 | ||
| Fig. 2 Mid-IR spectra of α-glucosamine 6-sulfate. (a) Convoluted harmonic spectrum calculated with B3LYP/6-311+G(d). Fwhm = 20 cm−1. Standard scaling factor: 0.965. (b) Convoluted harmonic spectra calculated with B3LYP/6-311++G**; B3LYP/6-311++G(2df,2pd); CAM-B3LYP/6-311++G** and CAM-B3LYP/6-311++G(2df,2pd). Fwhm = 20 cm−1. The computational times and our best scaling factors are given in the right panel. (c) GVPT2 CAM-B3LYP/hybrid anharmonic frequencies (sulfate modes in dark yellow) and convoluted spectrum with fwhm = 20 cm−1 (d) IR-MPD spectrum. | ||
First, in order to illustrate the influence of the choice of functional and basis set on the accuracy of the sulfate pattern, a series of harmonic calculations was performed for the conformer present in the experiment (a 4C1 chair, as previously established16), and scaled with our best scaling factors (reported in section Theoretical methods and How-to). The scaled harmonic spectra obtained with the widely used B3LYP functional and the most accurate CAM-B3LYP functional (see justification in section Theoretical methods and How-to); and a modest and a large basis sets (6-311++G** and 6-311++G(2df,2pd), respectively) are shown in Fig. 2b. Note that only the α-anomer is shown for clarity.‡
With B3LYP functional, increasing the size of the basis set results in a blueshift of the sulfate modes (SO(C) stretch, SO(H) stretch and SO2 asymmetric stretch) from 760, 840 and 1420 cm−1 to 770, 860 and 1430 cm−1, respectively. Using the smaller basis set 6-311+G(d) and a standard scaling factor of 0.965, these modes were previously predicted at 720, 785 and 1337 cm−1, respectively and were thus beyond recognition (Fig. 2a). With the blueshift trend, it becomes plausible to associate the two red sulfate modes with the experimental features at 780 and 880 cm−1 (Fig. 2d). An other recognizable region is the NH3 scissor doublet at 1600 cm−1 in the IR-MPD spectrum, which is poorly reproduced. Overall, it is striking that B3LYP systematically underestimates the sulfate frequencies. This anomalous behavior is somewhat attenuated by a counter-intuitive scaling factor greater than 1.0, which in turn deteriorates other ordinary overestimated frequencies (such as the NH3 pattern in this example). This oddity is not present in CAM-B3LYP simulations, which are consequently scaled with more traditional scaling factors smaller than 1.0. The blueshift of sulfate modes is consistently reinforced. With the larger basis set SO(C) and SO(H) stretches reach near the experimental positions. The SO2 asymmetric stretch and the NH3 umbrella mode almost merge at 1420 and 1450 cm−1. Between 900 and 1030 cm−1, the two dominant carbohydrate modes predicted at 940 and 1020 cm−1 reasonably match the experimental pattern with features at 940 and 1000 cm−1 (note that the band measured at 940 cm−1 is very weak. This is due to the attenuation applied to record the most intense bands of the spectrum without saturation). In contrast, the 1030–1200 cm−1 range, which consists of partly unresolved bands, poorly accounts for the experimental pattern. The low frequency range (<700 cm−1) is equally unconvincing. The two NH scissor modes are predicted around 1610 cm−1, in good agreement with the experimental value. Finally, it appears that both the change of functional and the use of increased basis set improve the simulation of the sulfate pattern. Not unexpectedly, the improvement of the carbohydrate pattern is more debatable: as a matter of fact, B3LYP is very popular for its excellent performances for organic compounds. In fine, the CAM-B3LYP/6-311++G(2df,2pd) method provides the best simulation of the sulfate and NH scissor modes. The rest of the spectrum is equally satisfying between 900 and 1030 cm−1, and equally disappointing below 700 cm−1 and in the 1030–1200 cm−1 range regardless of the method.
Anharmonic corrections
Hybrid GVPT2 anharmonic simulation was performed in a second stage (Fig. 2c). It consists in the correction of the CAM-B3LYP/6-311++G(2df,2pd) harmonic spectrum at the GVPT2 CAM-B3LYP/6-311++G** level. With excellent prediction of the SO(C) and SO(H) stretching frequencies and significant alterations of the overall CAM-B3LYP/6-311++G(2df,2pd) harmonic pattern, the hybrid spectrum provides the most refined representation of the experimental data. Firstly, the series of weak bands predicted between 500 and 700 cm−1 narrows down to a single feature centered around 550 cm−1, which perfectly matches the single feature measured in this region. As already mentioned, the SO(C) and SO(H) stretching frequencies, predicted at 790 and 880 cm−1, are in excellent agreement with the corresponding experimental bands. The next bands observed at 940 and 1000 cm−1, which do not involve sulfate, are also well accounted for (942 and 1022 cm−1), although not better than with the harmonic approximation. The 1030–1200 cm−1 pattern is improved with the detachment of a band at 1170 cm−1 from the rest of the unresolved pattern, in good agreement with the experimental feature at 1180 cm−1. The rest of this range is dominated by a feature at 1130 cm−1 in both experimental and hybrid spectra but the shapes of the left part of this band mismatch. The hybrid spectrum shows a weak activity between 1200 and 1400 cm−1, which is not observed in the experimental spectrum. As for the low intensity of the band at 940 cm−1, this is reasonably justified by the experimental conditions. The NH3 umbrella mode and the SO2 asymmetric stretch have now fully merged into a broad band (fwhm = 40 cm−1) centered around 1435 cm−1, which is associated to the intense band measured at 1470 cm−1 with a fwhm of 45 cm−1 The shape of this broad feature is only correctly reproduced by the hybrid method, although its position is underestimated by 35 cm−1 which falls slightly off the experimental resolution. Finally, the doublet of NH3 scissor mode is perfectly reproduced.To summarize, the best harmonic approximation was obtained with the CAM-B3LYP/6-311++G(2df,2pd) method and the optimized scaling factor 0.975. Using the hybrid anharmonic approach, the IR spectrum is further refined without the recourse to any empirical scaling factor and reaches a close-to-perfect agreement with the experimental data, thus resolving the case of glucosamine 6-sulfate. The computational cost of the hybrid anharmonic corrections was 35 times greater than the harmonic frequencies: 3160 hCPU vs. 89 hCPU. In contrast the full anharmonic simulation at the CAM-B3LYP/6-311++G(2df,2pd) level was impractically long. We could however estimate its computational cost to 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 024 hCPU (extrapolated from the calculation of a reduced number of frequencies). This represent a gain of six, which is not only a quantitative gain, but brings otherwise prohibitive anharmonic corrections to an accessible level.
024 hCPU (extrapolated from the calculation of a reduced number of frequencies). This represent a gain of six, which is not only a quantitative gain, but brings otherwise prohibitive anharmonic corrections to an accessible level.
Theoretical methods and how-to
The hybrid anharmonic approach was elaborated and validated on a set of benchmark systems of modest size (Chart 1). First, we assessed the accuracy of a series of functionals and basis sets for the prediction of sulfate vibrational frequencies. In order to improve the performances of the harmonic approximation, we reckoned pairs of scaling factor minimising the error on the ensemble of mid-IR modes. A cutoff was applied at 2800 cm−1 to best account for the high frequency modes (CH, NH and OH stretches) and the fingerprint modes, including the sulfate vibrations. Then, we assessed the precision of the functionals and basis sets for the GVPT2 corrections. Finally we devised a time-saving hybrid GVPT2 approach, thus making anharmonic corrections accessible for larger molecules. The method is fully illustrated on one of the benchmark species (i.e. dimethyl sulfate (4), the results obtained for the other benchmark species are shown in ESI†) for facile implementation by the interested reader. In order to verify the versatility of the hybrid method, it was also tested against phosphate frequencies – which are notoriously difficult to simulate – in the case of glucosamine 6-phosphate.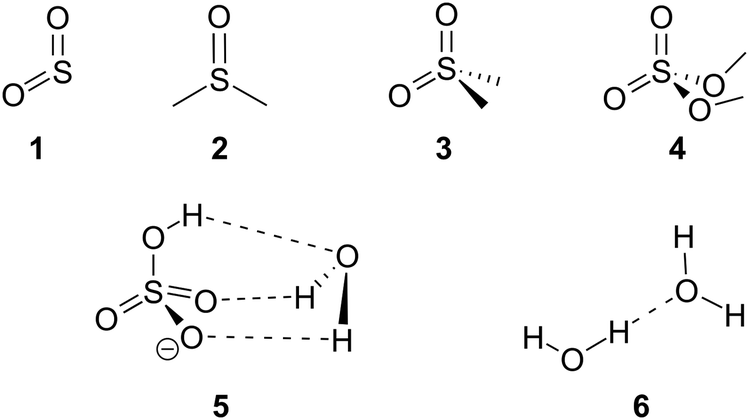 | ||
| Chart 1 Benchmark species: sulfur dioxide (1) dimethyl sulfoxide (2) dimethyl sulfone (3) dimethyl sulfate (4) hydrated bisulfate anion (5) and water dimer (6). | ||
Benchmark systems
The set of reference systems was selected to feature a sulfur atom in a variety of environments relevant to the context of our work on sulfated biomolecules, i.e. free and H-bonded S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, SOC and SOH groups. It includes sulfur dioxide (1), dimethyl sulfoxide (2), dimethyl sulfone (3) and dimethyl sulfate (4), as well as the hydrated bisulfate anion (5). The water dimer (6) was also included to examine the anharmonicity of H-bonded OH groups. The reference experimental IR spectra and geometries (shown in ESI†) were obtained from literature.24–27
O groups, SOC and SOH groups. It includes sulfur dioxide (1), dimethyl sulfoxide (2), dimethyl sulfone (3) and dimethyl sulfate (4), as well as the hydrated bisulfate anion (5). The water dimer (6) was also included to examine the anharmonicity of H-bonded OH groups. The reference experimental IR spectra and geometries (shown in ESI†) were obtained from literature.24–27
Harmonic simulations
The geometries were optimized with Gaussian0928 using seven functionals (BLYP,29,30 B3LYP,30–32 CAM-B3LYP,33 M06-2X,34,35 ωB97X-D,36 PBE0,37 LC-PBE38,39 with another range separation parameter, ω = 0.25 instead of 0.47, as per a previous study,40 referred to as LC-PBE), one post-Hartree Fock method (MP241) combined with eleven basis sets, including five triple-zeta Pople's basis (6-311+G(d), 6-311++G**, 6-311++G(df,pd), 6-311++G(2df,2pd) and 6-311++G(3df,3pd));42,43 four Dunning's basis sets (cc-pVDZ, cc-pVTZ, cc-pVQZ, aug-cc-pVTZ);44–46 and SNSD.47–49 Then, the harmonic frequencies were calculated at the same levels. The analysis of normal modes was carried out with Gabedit.50 To assess the reliability of the computed SO, SOC and SOH frequencies against the set of benchmark spectra, the signed errors (freqDFT–freqEXP) summed over all the concerned modes are reported in Fig. 3 (the list of experimental and computed frequencies is given in ESI†). It is expected that an accurate representation of the electronic potential yields harmonic frequencies greater than the experimental values. Thus, the negative errors obtained with BLYP, B3LYP with all basis sets and LC-PBE with most basis sets are anomalous and suggest a deficient representation of the electronic potential of sulfated compounds. More precisely, as the optimized geometries are not inaccurate, this indicates that the minimum of the electronic potential is realistic, while its curvature is underestimated. In contrast, ωB97X-D and M06-2X yield positive, physically acceptable errors for all basis sets with exception of the smallest one (SNSD). The behavior of CAM-B3LYP, PBE0 and MP2 is more basis set dependent: the inclusion of d and f orbitals is essential for Pople's basis sets to yield acceptable results and similarly, only the largest Dunning's basis sets are acceptable. Note that in this picture, an error close to zero might not be an indicator of quality. Instead, the methods yielding the best representations of the potential are likely to show errors “somewhere” above zero. Without a more quantitative criterion, we do not attempt to compare the accuracy of the methods in the positive domain at this stage.
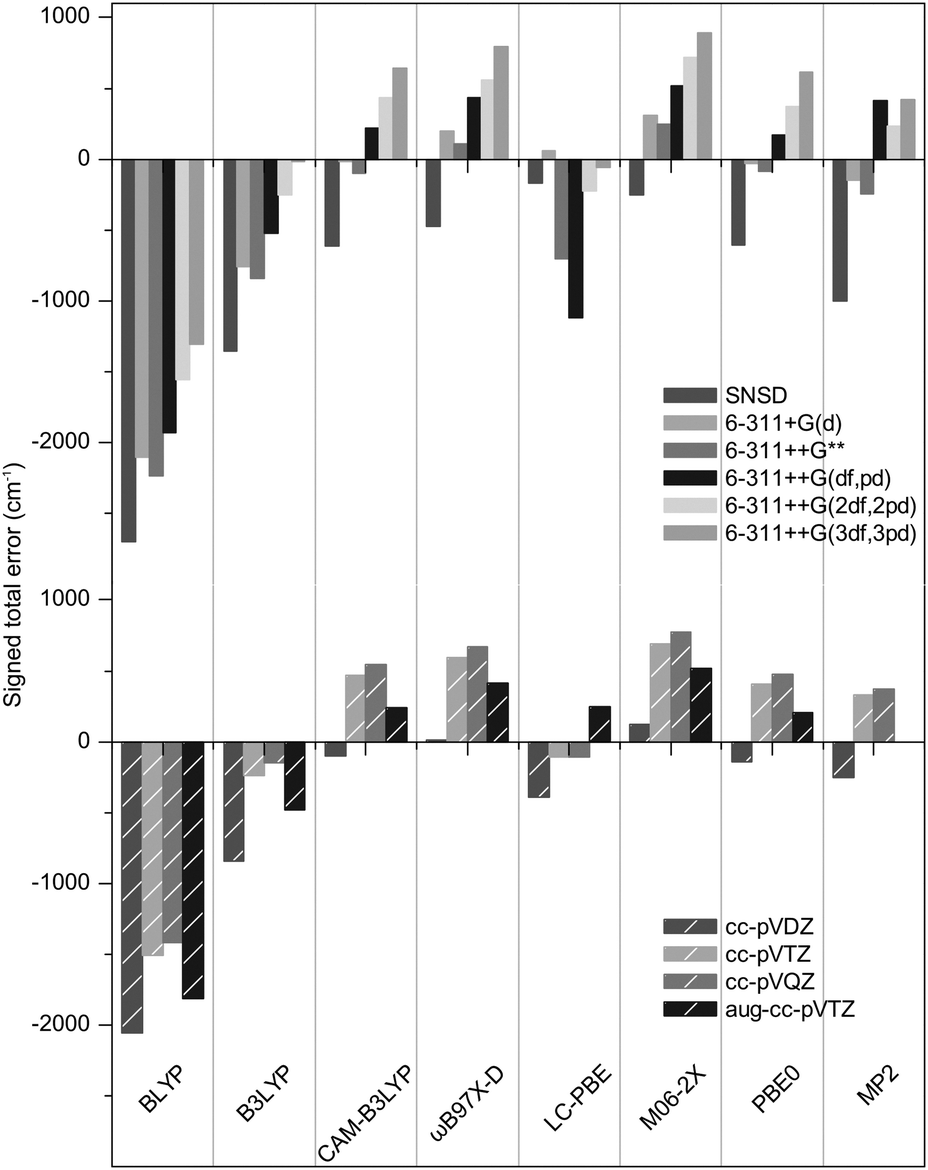 | ||
| Fig. 3 Signed total error on the SO, SOC and SOH frequencies of the benchmark species. | ||
Optimization of the scaling factors
The explicit simulation of the anharmonic terms of the electronic potential is not a widely used procedure to date. Instead, it is generally accepted to use an empirical correcting scaling factor which reduces the values of the harmonic frequencies. To further quantify the performances of the selected functionals and basis sets for sulfated compounds, we have reckoned pairs of scaling factors minimizing the RMS (root-mean squared) deviation over all frequencies of the benchmark set (the list of experimental and computed frequencies is given in ESI†) in the high frequency and low frequency ranges. The cutoff was applied at 2800 cm−1 to best account for the CH, NH and OH stretches region and the fingerprint region. The minimal RMS deviations obtained with our optimized scaling factors are shown in Fig. 4. and the scaling factors are given in Table 1.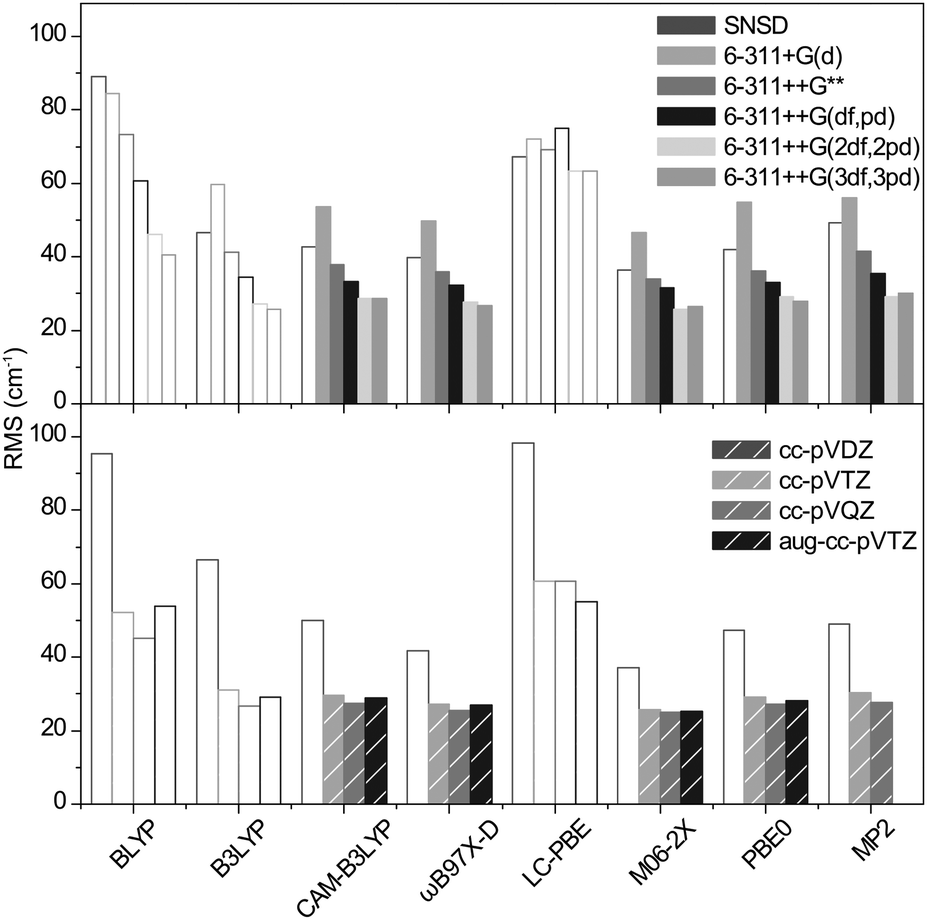 | ||
| Fig. 4 RMS deviations of the harmonic frequencies of the benchmark set scaled with our optimized scaling factors. Empty bars are used to fade out the methods consistently underestimating the sulfate frequencies. | ||
| BLYP | B3LYP | CAM-B3LYP | ωB97X-D | LC-PBE | M06-2X | PBE0 | MP2 | |
|---|---|---|---|---|---|---|---|---|
| SNSD | 1.049/0.996 | 1.049/0.962 | 1.018/0.953 | 1.011/0.945 | 0.997/0.978 | 1.003/0.945 | 1.018/0.95 | 1.036/0.95 |
| 6-311+G(d) | 1.049/1.004 | 1.015/0.968 | 0.984/0.958 | 0.974/0.951 | 0.981/0.984 | 0.97/0.949 | 0.983/0.956 | 0.988/0.949 |
| 6-311++G** | 1.049/0.994 | 1.028/0.961 | 0.995/0.952 | 0.986/0.946 | 1.029/0.976 | 0.982/0.944 | 0.994/0.949 | 1/0.942 |
| 6-311++G(df,pd) | 1.049/0.994 | 1.014/0.962 | 0.982/0.953 | 0.973/0.946 | 1.049/0.993 | 0.971/0.943 | 0.984/0.95 | 0.971/0.94 |
| 6-311++G(2df,2pd) | 1.049/0.994 | 1.004/0.962 | 0.975/0.953 | 0.969/0.946 | 1.009/0.978 | 0.963/0.946 | 0.977/0.95 | 0.983/0.945 |
| 6-311++G(3df,3pd) | 1.049/0.996 | 0.994/0.963 | 0.966/0.955 | 0.96/0.947 | 1.002/0.979 | 0.957/0.946 | 0.967/0.952 | 0.976/0.947 |
| cc-pVDZ | 1.049/1.011 | 1.021/0.972 | 0.992/0.962 | 0.987/0.952 | 1.002/0.986 | 0.984/0.953 | 0.994/0.958 | 0.997/0.947 |
| cc-pVTZ | 1.049/0.999 | 1.002/0.965 | 0.972/0.956 | 0.966/0.948 | 1.002/0.98 | 0.964/0.948 | 0.975/0.953 | 0.978/0.948 |
| cc-pVQZ | 1.049/0.998 | 0.999/0.965 | 0.97/0.956 | 0.964/0.947 | 1.004/0981 | 0.962/0.948 | 0.973/0.953 | 0.978/0.949 |
| aug-cc-pVTZ | 1.049/0.999 | 1.015/0.966 | 0.983/0.957 | 0.975/0.949 | 0.984/0.982 | 0.973/0.949 | 0.984/0.955 |
Conforming to the previous section, the methods consistently underestimating the SO, SOC and SOH frequencies require scaling factors greater than 1.0. Not only is this an arbitrary way to correct for the incorrect representation of the sulfate modes, it also leads to a poorer match for the other modes (as illustrated in Fig. 2b for NH scissor modes). The RMS's of these methods are not further discussed and are plotted with empty bars in Fig. 4 for clarity.
For the methods consistently yielding errors in the positive domain, the minimized RMS deviations are hardly functional dependent. For Pople's basis sets, the precision steadily increases with the size of the basis set and the best results are obtained with the 6-311++G(2df,2pd) and the 6-311++G(3df,3pd) basis sets (RMS = 26 to 35 cm−1). The Dunning's basis sets show similar performances with RMS deviations ranging from 25 to 30 cm−1. In conclusion, the precision of the harmonic frequencies corrected with our optimized scaling factors ranges from 25 to 35 cm−1, which is slightly below the experimental resolution (typically 20 cm−1 in the fingerprint region and 10 cm−1 in the high frequency range).
Anharmonic corrections
The cubic and quartic terms of the potential were calculated with Gaussian09 using the GVTP2 method.51 The RMS deviations thus obtained are shown in Fig. 5. In contrast to scaled harmonic simulations, it is remarkable that the result is highly functional dependent. In particular, the precision obtained with ωB97X-D and M06-2X (RMS = 50 to 400 cm−1) deteriorates dramatically. This is also true, to a lesser extent for MP2. With CAM-B3LYP and PBE0, the trend is opposite: the RMS is reduced to 18 and 19 cm−1, respectively, with 6-311++G(2df,2pd). For Dunning's basis sets, it ranges from 22 to 17 cm−1 as the size of the basis set increases. With a systematic improvement of 10 cm−1, the precision now falls very near the experimental resolution.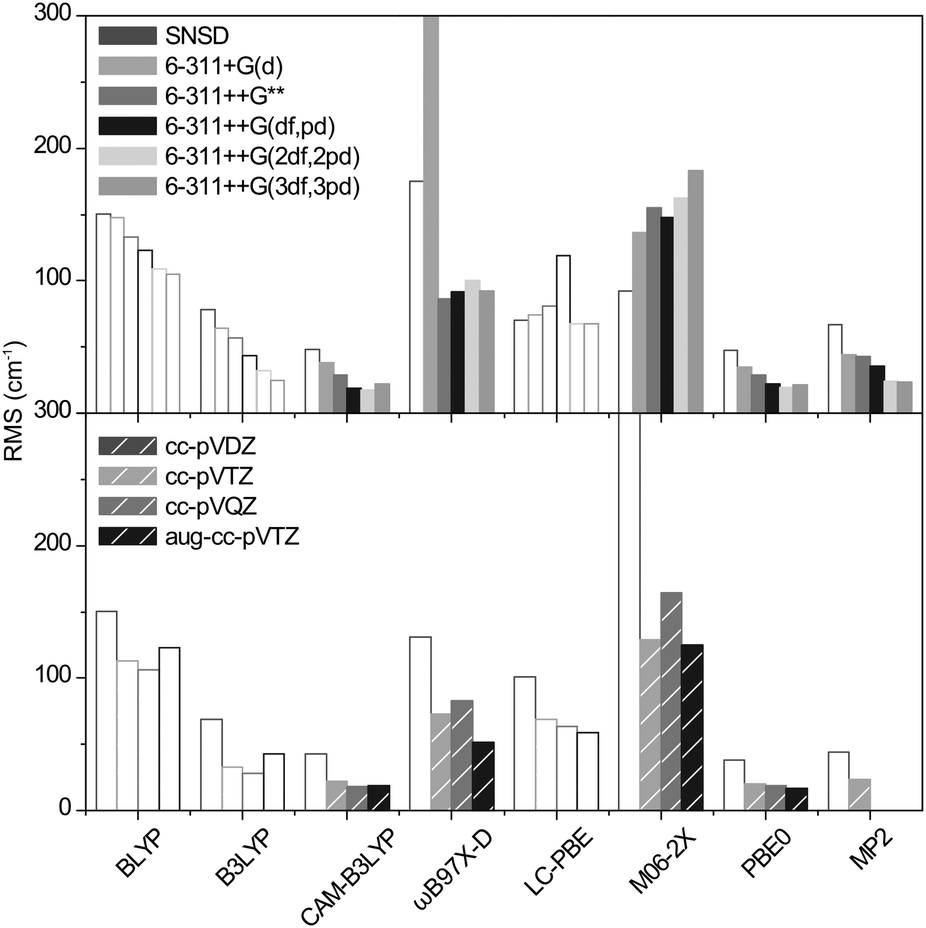 | ||
| Fig. 5 RMS deviations of the anharmonic frequencies of the benchmark set. The y scale is cropped at 300 cm−1 for clarity (for ωB97X-D/6-311+G(d) RMS = 400). Empty bars are used to fade out the methods consistently underestimating the sulfate frequencies. | ||
The computational cost of 6-311++G(2df,2pd) calculation is comparable for both CAM-B3LYP and PBE0 functionals (292 and 203 hCPU for dimethyl sulfate (4), for instance). For Dunning's basis sets, the cost is similar for cc-pVTZ but increases by a factor of 10 for cc-pVQZ and aug-cc-pVTZ.
Considering the gain in precision vs. the computational cost associated with the anharmonic corrections, we select a combination of 6-311++G(2df,2pd) with either CAM-B3LYP or PBE0 for the best representation of vibrational frequencies in sulfated compounds.
The hybrid method
Furthermore, in the prospect of extending the application of the GVPT2 approach to more complex molecules, we attempted to introduce a hybrid method, aiming at reducing the computational cost of the anharmonic corrections with minimal loss of precision.The hybrid method consists of computing the harmonic frequencies with the best combination of functional and basis set (i.e. CAM-B3LYP or PBE0 with 6-311++G(2df,2pd)), followed by calculation of the time consuming cubic and quartic terms of the potential with a smaller basis set. The procedure is explained in ESI.† In short, the normal modes obtained in the harmonic simulations at the 6-311++G(2df,2pd) level and at the lower level are matched. Then, hybrid anharmonic frequency is obtained by adding the low level anharmonic correction to the corresponding high level harmonic frequency.
With the highest gain in computational time and a preserved precision (RMS = 19 cm−1), the 6-311++G** basis set gave the best results.
Illustration: dimethyl sulfate
Using the CAM-B3LYP functional, the performance of the hybrid approach in terms of precision and speed is illustrated in Fig. 6 for dimethyl sulfate (4) (all other benchmark species are shown in ESI†). Both the position and relative intensities of the vibrational modes calculated with the CAM-B3LYP/6-311++G(2df,2pd) and CAM-B3LYP/hybrid (Fig. 6c and d) are in close agreement with the experimental spectrum (Fig. 6e), the latter offering a substantial gain of a factor of 11 in computational time without significant loss of precision. For comparison, two scaled harmonic spectra are presented: (i) a popular combination of B3LYP with an augmented Dunning's basis set17,27 with a standard scaling factor of 0.96824 which yields unacceptably underestimated sulfate frequencies (Fig. 6a), as previously discussed; and (ii) CAM-B3LYP/6-311++G(2df,2pd) (Fig. 6b) with our optimized scaling factor of 0.975, which offers the most accurate harmonic approximation of the experimental spectrum. The gain of the anharmonic calculations is twofold: an increase in precision on individual frequencies, and a consequential alteration of the harmonic pattern, in better agreement with the experimental spectrum. The results obtained with the PBE0 functional are shown in ESI.† Additionally, the versatility of the hybrid method is illustrated in ESI† in the case of glucosamine 6-phosphate.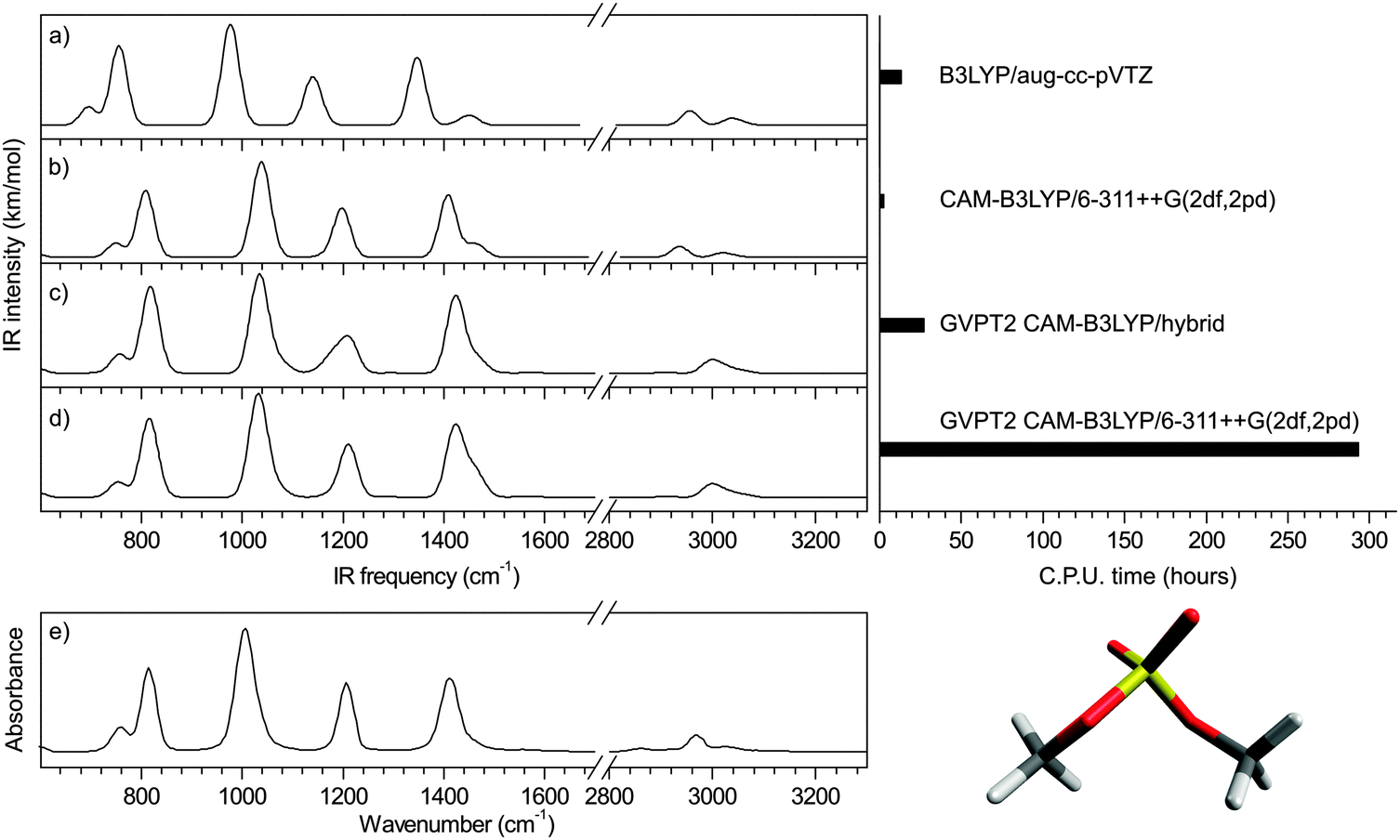 | ||
| Fig. 6 Illustration: dimethyl sulfate (4) (a) scaled B3LYP harmonic spectrum with NIST scaling factor, (b) scaled CAM-B3LYP harmonic spectrum with optimized scaling factor, (c) hybrid anharmonic spectrum, (d) anharmonic spectrum and (e) experimental spectrum from NIST. | ||
Conclusion
A striking case of failure of DFT harmonic simulation of sulfate vibrational frequencies was observed in the case of our model glycosaminoglycan fragment glucosamine 6-sulfate, critically impairing its structural characterization and somewhat echoing similar situations reported for phosphorylated compounds. Driven by this observation, we have explored the accuracy of 79 methods for predicting the sulfate frequencies of a set of benchmark species. We report the anomalous behavior of the BLYP, B3LYP and LC-PBE functionals, as well as of small basis sets, yielding atypical underestimated frequencies. With an adequate choice of method and scaling factor, we show that a precision of ca. 30 cm−1 can be obtained in the harmonic approximation, approaching the experimental resolution. In a second step, we explored the performance of GVPT2 anharmonic corrections. The best results were obtained with CAM-B3LYP/6-311++G(2df,2pd), offering a precision of 18 cm−1 within the spectroscopic resolution in the fingerprint region. Finally, as the computational cost of explicit anharmonic corrections becomes prohibitive for species of increasing size, we devised an alternative “hybrid” strategy which brings down the computational cost to an accessible level for functionalized biomolecules, without significant loss of precision. Using this original approach, we could solve the case of glucosamine 6-sulfate. The robustness of the hybrid method was further illustrated on a phosphorylated species. We expect that this novel theoretical tool will unlock the potential of mid-IR spectroscopy coupled to mass spectrometry for the sequencing and structural characterization of functionalized biopolymers.Acknowledgements
This work was supported by the LABEX iMUST (ANR-10-LABX-0064) of Université de Lyon, within the program “Investissements d'Avenir” (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR). Experiments in the mid-IR region were performed at the free electron laser facility FELIX in Netherlands with the skillful assistance of the FELIX staff, in particular Dr Britta Redlich and Lex van der Meer. Travel costs to the Netherlands were supported by the Dutch-French Van Gogh program. In this work, we were granted access to the HPC resources of the FLMSN, “Féderation Lyonnaise de Modélisation et Sciences Numeriques”, partner of EQUIPEX EQUIP@MESO and the HPC Resources from GENCI-CINES (Grant 2013–2014 [087025]).Notes and references
- A. Alonso, J. Sasin, N. Bottini, I. Friedberg, I. Friedberg, A. Osterman, A. Godzik, T. Hunter, J. Dixon and T. Mustelin, Cell, 2004, 117, 699–711 CrossRef CAS PubMed.
- T. Pawson and J. D. Scott, Science, 1997, 278, 2075–2080 CrossRef CAS.
- W. B. Huttner, Nature, 1982, 299, 273–276 CrossRef CAS PubMed.
- A. Tempez, M. Ugarov, T. Egan, J. A. Schultz, A. Novikov, S. Della-Negra, Y. Lebeyec, M. Pautrat, M. Caroff, V. S. Smentkowski, H.-Y. J. Wang, S. N. Jackson and A. S. Woods, J. Proteome Res., 2005, 4, 540–545 CrossRef CAS PubMed.
- Transforming Glycoscience: A Roadmap for the Future, The National Academies Press, Washington, DC, 2012 Search PubMed.
- M. J. Kailemia, L. R. Ruhaak, C. B. Lebrilla and I. J. Amster, Anal. Chem., 2014, 86, 196–212 CrossRef CAS PubMed.
- D. J. Harvey, Mass Spectrom. Rev., 1999, 18, 349–450 CrossRef CAS.
- J. Zaia, OMICS: J. Integr. Biol., 2010, 14, 401–418 CrossRef CAS PubMed.
- J. Zaia, Mass Spectrom. Rev., 2004, 23, 161–227 CrossRef CAS PubMed.
- A. Racaud, R. Antoine, L. Joly, N. Mesplet, P. Dugourd and J. Lemoine, J. Am. Soc. Mass Spectrom., 2009, 20, 1645–1651 CrossRef CAS PubMed.
- E. B. Cagmat, J. Szczepanski, W. L. Pearson, D. H. Powell, J. R. Eyler and N. C. Polfer, Phys. Chem. Chem. Phys., 2010, 12, 3474–3479 RSC.
- B. Casu, M. Guerrini and G. Torri, Curr. Pharm. Des., 2004, 10, 939–950 CrossRef CAS.
- G. K. Dhoot, M. K. Gustafsson, X. Ai, W. Sun, D. M. Standiford and C. P. Emerson Jr, Science, 2001, 293, 1663–1666 CrossRef CAS PubMed.
- R. Sasisekharan, Z. Shriver, G. Venkataraman and U. Narayanasami, Nat. Rev. Cancer, 2002, 2, 521–528 CrossRef CAS PubMed.
- S. M. Muthana, C. T. Campbell and J. C. Gildersleeve, ACS Chem. Biol., 2012, 7, 31–43 CrossRef CAS PubMed.
- B. Schindler, J. Joshi, A.-R. Allouche, D. Simon, S. Chambert, V. Brites, M.-P. Gaigeot and I. Compagnon, Phys. Chem. Chem. Phys., 2014, 16, 22131–22138 RSC.
- A. L. Patrick, C. N. Stedwell, B. Schindler, I. Compagnon, G. Berden, J. Oomens and N. C. Polfer, Int. J. Mass Spectrom., 2015, 379, 26–32 CrossRef CAS PubMed.
- R. Paciotti, C. Coletti, N. Re, D. Scuderi, B. Chiavarino, S. Fornarini and M. E. Crestoni, Phys. Chem. Chem. Phys., 2015 10.1039/C5CP01409C.
- L. Wang, J. R. Brown, A. Varki and J. D. Esko, J. Clin. Invest., 2002, 110, 127–136 CrossRef CAS.
- X. Ai, J. Cell Biol., 2003, 162, 341–351 CrossRef CAS PubMed.
- A. Sharma, G. Ohanessian and C. Clavaguéra, J. Mol. Model., 2014, 20, 1–9 Search PubMed; F. Turecek, C. L. Moss, I. Pikalov, R. Pepin, K. Gulyuz, N. Polfer, M. F. Bush, J. Brown, J. Williams and K. Richardson, Int. J. Mass Spectrom., 2013, 354–355, 249–256 CrossRef CAS PubMed.
- C. J. Johnson and M. A. Johnson, J. Phys. Chem. A, 2013, 117, 13265–13274 CrossRef CAS PubMed.
- Y. Miller, G. M. Chaban, J. Zhou, K. R. Asmis, D. M. Neumark and R. Benny Gerber, J. Chem. Phys., 2007, 127, 094305 CrossRef PubMed.
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, National Institute of Standards and Technology, 2005.
- T. I. Yacovitch, T. Wende, L. Jiang, N. Heine, G. Meijer, D. M. Neumark and K. R. Asmis, J. Phys. Chem. Lett., 2011, 2, 2135–2140 CrossRef CAS.
- R. Kalescky, W. Zou, E. Kraka and D. Cremer, Chem. Phys. Lett., 2012, 554, 243–247 CrossRef CAS PubMed.
- A. Borba, A. Gómez-Zavaglia, P. N. N. L. Simões and R. Fausto, Spectrochim. Acta, Part A, 2005, 61, 1461–1470 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc, Wallingford, CT, USA, 2009 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540–3544 CrossRef CAS PubMed.
- L. Barnes, S. Abdul-Al and A. R. Allouche, J. Phys. Chem. A, 2014, 118, 11033–11046 CrossRef CAS PubMed.
- C. Moller and M. S. Plesset, Phys. Rev., 1934, 46, 0618–0622 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS PubMed.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1994, 100, 2975–2988 CrossRef CAS PubMed.
- V. Barone and P. Cimino, Chem. Phys. Lett., 2008, 454, 139–143 CrossRef CAS PubMed.
- V. Barone, P. Cimino and E. Stendardo, J. Chem. Theory Comput., 2008, 4, 751–764 CrossRef CAS.
- V. Barone and P. Cimino, J. Chem. Theory Comput., 2009, 5, 192–199 CrossRef CAS.
- A.-R. Allouche, J. Comput. Chem., 2011, 32, 174–182 CrossRef CAS PubMed.
- V. Barone, J. Chem. Phys., 2005, 122, 014108 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods; cpu time for dimethyl sulfate; experimental frequencies and theoretical variation; geometry of the benchmark species; vibrational spectra of the benchmark species; anharmonic PBE0 spectrum of dimethyl sulfate; application of the hybrid method to glucosamine 6-phosphate. See DOI: 10.1039/c5cp02079d |
| ‡ The two pyranoside anomers (α and β) co-exist in the measured sample. NMR experiments were performed in a deuterated water/methanol (1:1) mixture to measure the anomeric ratio (method and spectra in ESI†). The sugar was found predominantly in the alpha configuration immediately after the preparation of the solution sample, it reached an equilibrium after few hours with a ratio of 70% of the α-anomer for 30% of the β-anomer. We have verified that introduction of a fraction of β-anomer does not significantly affect the shape of the simulated spectra. |
| This journal is © the Owner Societies 2015 |