
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exceptional adsorption-induced cluster and network deformation in the flexible metal–organic framework DUT-8(Ni) observed by in situ X-ray diffraction and EXAFS†
Volodymyr
Bon
a,
Nicole
Klein‡
a,
Irena
Senkovska
a,
Andreas
Heerwig§
a,
Jürgen
Getzschmann
a,
Dirk
Wallacher
b,
Ivo
Zizak
c,
Maria
Brzhezinskaya
c,
Uwe
Mueller
d and
Stefan
Kaskel
*a
aInorganic Chemistry I, Technische Universität Dresden, Bergstrasse 66, 01062 Dresden, Germany. E-mail: stefan.kaskel@chemie.tu-dresden.de; Fax: +49 (351) 463 37287
bDepartment Sample Environments, Helmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner Platz 1, Berlin, Germany
cInstitute of Nanometer Optik and Technology, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Str. 15, 12489 Berlin, Germany
dMacromolecular Crystallography Group, Institute Soft Matter and Functional Materials, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Str. 15, 12489 Berlin, Germany
First published on 9th June 2015
Abstract
The “gate opening” mechanism in the highly flexible MOF Ni2(2,6-ndc)2dabco (DUT-8(Ni), DUT = Dresden University of Technology) with unprecedented unit cell volume change was elucidated in detail using combined single crystal X-ray diffraction, in situ XRD and EXAFS techniques. The analysis of the crystal structures of closed pore (cp) and large pore (lp) phases reveals a drastic and unique unit cell volume expansion of up to 254%, caused by adsorption of gases, surpassing other gas-pressure switchable MOFs significantly. To a certain extent, the structural deformation is specific for the guest molecule triggering the transformation due to subtle differences in adsorption enthalpy, shape, and kinetic diameter of the guest. Combined adsorption and powder diffraction experiments using nitrogen (77 K), carbon dioxide (195 K), and n-butane (272.5 K) as a probe molecules reveal a one-step structural transformation from cp to lp. In contrast, adsorption of ethane (185 K) or ethylene (169 K) results in a two-step transformation with the formation of intermediate phases. In situ EXAFS during nitrogen adsorption was used for the first time to monitor the local coordination geometry of the metal atoms during the structural transformation in flexible MOFs revealing a unique local deformation of the nickel-based paddle-wheel node.
Introduction
Metal–organic frameworks represent nowadays the record materials in terms of specific surface area and pore volume among all known porous solids.1,2 The simplicity of the guest exchange within the framework suggests a wide range of application fields.3–6 A feature of MOFs that is unique as compared to other porous crystalline materials is reversible structural switch-ability between non porous (closed pore structure) and porous (open pore structure) polymorphs.7 This novel (also termed 3rd) generation of coordination polymers8 is capable to perform a step-wise structural transformation as a response to external stimuli, such as guest molecules, electromagnetic radiation, temperature or pressure. Although more than 20.000 MOFs are known today only less than 100 of such switchable 3rd generation MOFs are structurally characterized and the theoretical elucidation of flexibility phenomenon is still in its infancy. Nevertheless, such MOFs already find a wide interest as switchable or highly selective gas adsorbents,9–12 threshold sensors,13,14 stimuli responsive drug delivery agents,15 switchable catalysts16etc.In order to establish a rational design, a detailed understanding of linker-cluster hinge deformation energies and atom rearrangement in the cluster nodes responsible for a ferroelastic deformation of the framework during the switching process is essential, as it represents a prerequisite for the effective prediction and recognition of the potential application fields as well as for understanding of driving forces, may it be derived from internal (adsorption induced) or external pressure. For such an understanding it is essential to elucidate crystal structures forming during the transformation induced by gas adsorption. However, the monitoring of the structural evolution during the “gate opening” in situ is challenging. So far only few groups have realized the parallelized collection of diffraction and adsorption data.17–21 In general, single crystal data during adsorption are very rare and sophisticated and thus powder diffraction is used giving good insight into structural framework deformation.22–24 For example, breathable MIL-53 family materials were successfully studied by in situ adsorption.21,25–27 However, the visualization of small local changes from Rietveld refinement even in combination with simulation is sometimes ambiguous. In this context the parallelized use of local probes is essential to confirm important structural details.
In the following we will for the first time show how a combination of single crystal and powder XRD with EXAFS elucidates precisely the structural changes of a highly flexible MOF, DUT-8(Ni), showing unprecedented unit cell volume expansion of up to 254% upon “gate opening”. Not only the structures of the terminal states of the compounds (so called large pore (lp) phase and the closed pore (cp) phase) could by visualized on the atomistic level, but also the gas filled structural phases of the material were solved from in situ data collected during the adsorption of gases. We have focused especially on gases being important in the fields of energy storage, separation and environmental applications, namely carbon dioxide (195 K), ethane (185 K), ethylene (169 K), n-butane (273 K), and nitrogen (77 K). The measurements reveal essential mechanistic insights and may have a high impact for the development of 3rd generation MOFs for technical applications.
Results and discussions
DUT-8(Ni) is unique and anomalous among the pillar-layered compounds because of the huge pore volume expansion unattained in other pillared layer systems.28 The layers constructed of Ni paddle-wheels interconnected by 2,6-ndc linkers are pillared by dabco molecules, yielding a 3D framework with the α-Po topology. The as-made compound crystallizes in P4/n space group with 2 formula units per unit cell and contains square channels with limiting pore diameter of 9.0 Å (Fig. 1c left) filled with DMF and MeOH molecules. The analysis of crystallographic porosity reveals that 66.6% of the unit cell volume is accessible for solvent. Porosity calculations using Poreblazer 3.0 program29 on the crystal structure without solvent molecules results in the geometrical surface area of 2646 m2 g−1 and pore volume of 1.00 cm3 g−1.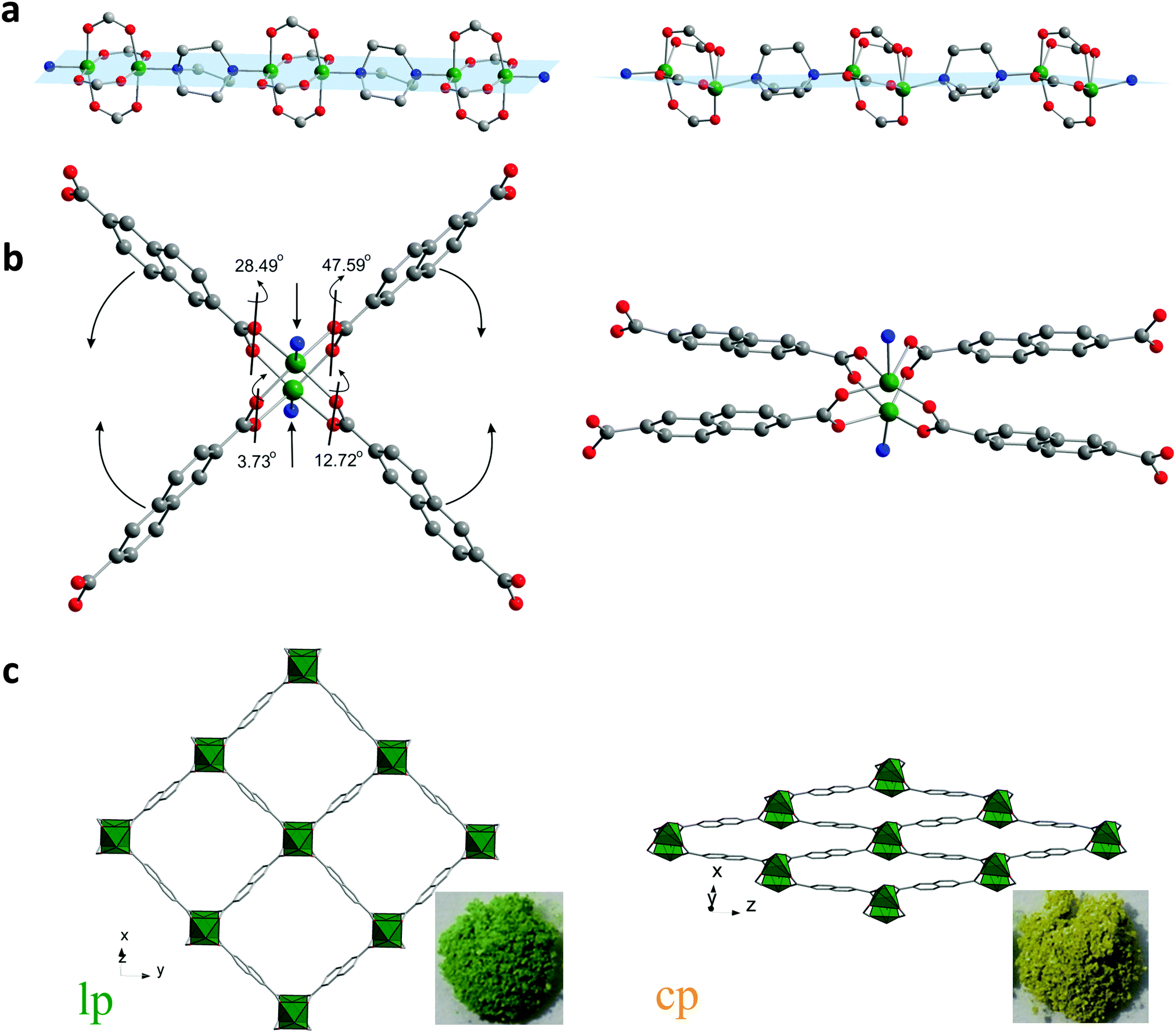 | ||
| Fig. 1 Structural transformation of DUT-8(Ni): the paddle-wheel chains pillared by dabco molecules (a); SBU environment (b); crystal structure along rectangular channels (c). | ||
During the desolvation, however, a transformation from the as made to the cp phase, takes place associated with a distinct color change from green to yellow, and the dense structure without nitrogen accessible porosity is formed (Fig. 1c right).
The crystal structure of the closed structure, DUT-8(Ni) cp, is described here for the first time, as it could only be solved combining single crystal XRD, powder X-ray diffraction and computational simulations. The poor diffraction data, collected on weakly scattering single crystals of DUT-8(Ni) cp phase revealed triclinic unit cell and the structure solution in the P1 space group provided the coordinates of the Ni atoms (see ESI,† Table S1). This initial model was used for the structure simulation, performed using the modelling and simulation software Material Studio 5.0.30 Subsequently, obtained structure was successfully refined against the powder X-ray diffraction data (ESI,† Fig. S1 and S2). The analysis of the crystal structure of cp DUT-8(Ni) shows a changing orientation of the pillaring ligand: dabco molecules, located along [001] direction in as-made phase, are located along [110] in the cp phase. The Ni2(2,6-ndc)2 layers parallel to the (110) plane in the as-made structure are parallel to (101) plane in the cp one.
Analysis of the atoms connectivity in both structures shows that no bond dissociation occurs during the phase transition but, unprecedented, within the paddle-wheel unit the coordination geometry of the Ni atom changes drastically during the evacuation: thus, in the as-made structure (space group P4/n), the N–Ni–Ni–N atoms of paddle-wheel and dabco molecules are positioned on a common axis forming a linear chain, with N–Ni–O angles of 95.43° (Fig. 1a). In the cp phase, however, the above mentioned angles are strongly distorted: the Ni–Ni–N angle is 154° causing the formation of Ni–Ni–N–N zigzag chains and the values of N–Ni–O angle vary significantly from 82.1° to 106.8° (Fig. 1a). The adjacent O–Ni–O angles deform even more dramatic from 89.5° in the solvent filled form to a wide region of 52.1–128.6° in the closed phase. However, the distance between two nickel atoms increases only slightly from 2.64 to 2.74 Å in the closed from.
Another soft feature of the structure is the interplanar angles between carboxylates and Ni paddle-wheel that vary from 3.73° to 47.59° (Fig. 1b). Thus, in contrast to MIL-53 (MIL – Matériaux de l'Institut Lavoisier), where breathing mechanism is associated purely with changes of the dihedral angle at the carboxylate hinge, structural transformation in DUT-8(Ni) is finally a synergetic effect of hinge deformation (knee-cap dihedral angle deformation) and a unique strong distortion of the square planar paddle-wheel node. Only the combination of cluster and hinge deformation can cause such an enormous change of the pore volume. More intriguing, such a combined mechanism of cluster and hinge deformation is challenging in terms of quantum chemical modelling due to the subtle interplay of van der Walls interactions, bond deformation, and changes of the electronic structure within the cluster. The contraction of the structure leads to drastic changes of the pore window: the channel aperture, measured as a diagonals between oppositely located Ni-paddle-wheels changes from 18.43 × 18.43 Å in the as-made phase to 23.66 × 6.95 Å in the cp phase. Such drastic changes of the pore system cause an extensive influence on the porosity. The PLATON provides no accessible void for the guest molecules that is confirmed by Poreblazer 3.0 showing zero surface area and pore volume for the cp structure.
Thus, the structural transformation from the as-made to cp phase results in a very strong contraction of the unit cell volume from 1595 Å3 to 647.7 Å3 (both recalculated with Z = 1). Consequently the crystallographic density of the framework increases from 0.686 g cm−3 to 1.687 g cm−3 (if both frameworks without guest molecules are considered). To our knowledge, this is the largest cell volume change induced by the adsorption of gas molecules in “gate pressure” MOFs ever observed.
For example, the unit cell of MIL-53(Cr)31 changes from 1012.6 Å3 in the “narrow pore” phase to 1486.3 Å3 in the “large pore” one, corresponding to the 46.8%.31 The current record holder, MIL-88D32 shows a huge swelling effect during the liquid phase adsorption of solvent molecules, along with an increase of the unit cell volume in the “large pore” phase of 333% compared to the “closed pore” phase. However, MIL-88 materials exhibit this swelling only in liquid phase adsorption.
In order to explain the gating behavior during the adsorption of gases, parallelized adsorption and powder X-ray diffraction experiments were performed on DUT-8(Ni) at the Helmholtz Center Berlin.18 For this purpose, probe molecules with significant differences in polarity and kinetic diameter were chosen as adsorptives.
Adsorption of nitrogen at 77.4 K monitored in situ by XRD and EXAFS spectroscopy
Initially, in situ nitrogen physisorption at 77.4 K was studied, since the ex situ adsorption experiment shows a pronounced hysteresis in the adsorption isotherm (Fig. 2). The powder XRD of starting materials under vacuum shows predominately the presence of cp DUT-8 with a minor impurity of the lp phase (characteristic (110) reflection at 2θ = 6.8°). As described earlier, the activation procedure has a strong influence on the adsorption behavior below the gate opening pressure33 and on the phase purity of the compound. The cp![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :lp ratio determined from the powder XRD of activated DUT-8(Ni) is 98:2.
:lp ratio determined from the powder XRD of activated DUT-8(Ni) is 98:2.
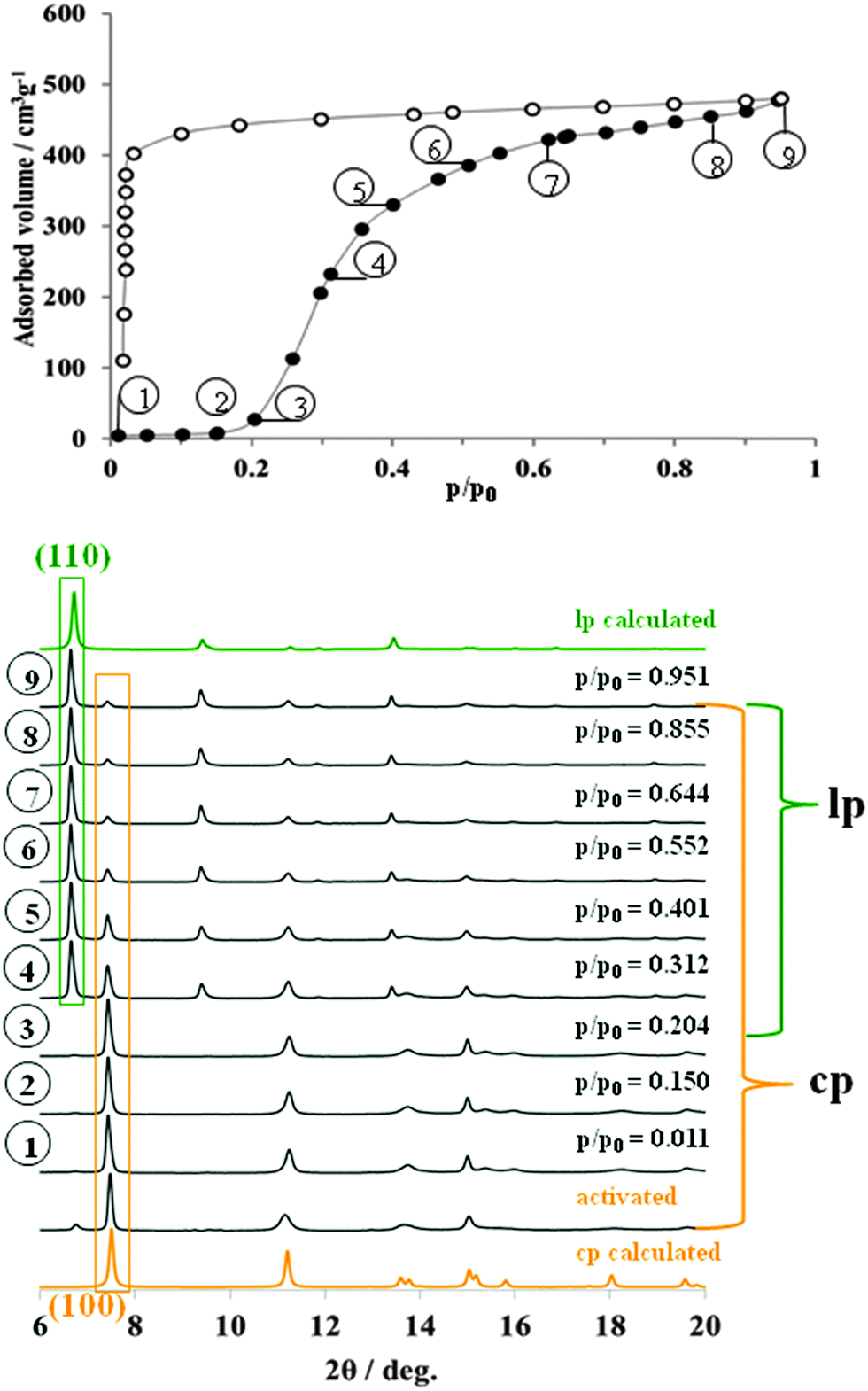 | ||
| Fig. 2 Adsorption N2 isotherm at 77 K (top) on DUT-8(Ni) monitored in situ by powder X-ray diffraction (bottom). | ||
Interestingly, during the adsorption of small quantities of nitrogen below the “gate pressure”, the intensity of lp diffraction peaks decreases, enhancing the ratio of cp:lp phase from 98:2 in the evacuated state to 100:0 at p/p0 = 0.15 (adsorption point 2, Fig. 2, see also ESI,† Table S4).
Such a phenomenon has been reported earlier and can be interpreted as a sort of re-equilibration in the presence of low gas concentrations indicating a dynamic equilibrium (for example for MIL-53(Cr) during the adsorption of water).31 At p/p0 = 0.2, the “gate pressure” is reached accompanied by strong adsorption of nitrogen. The most characteristic (110) reflection of N2@DUT-8(Ni) phase appears at 2θ = 6.8° in the diffraction patterns collected at relative pressures higher than 0.3. At the same time, the intensity of (100) reflection from the cp phase, continuously decreases with increasing pressure. The ratio of lp:cp phases derived from the PXRDs phase analysis at the highest measured relative pressure is 59:41 (ESI,† Table S4). The PXRD patterns of N2@DUT-8(Ni) collected at p/p0 = 0.95 does not exactly match the theoretical pattern of the tetragonal “as made” phase (see ESI,† Fig. S17). The indexing of the PXRD pattern results in a monoclinic cell (P21/m space group) with cell parameters quite similar to that of the as made phase, but with the monoclinic angle of 94.4° (Table 1, ESI,† Fig. S6). Interestingly, the unit cell volume of the N2@DUT-8(Ni) phase is slightly higher in comparison to the as-made tetragonal phase, making the unit cell expansion during the nitrogen adsorption even more drastic, reaching the level of 254%.
| SG | a (Å) | b (Å) | c (Å) | α (deg) | β (deg) | γ (deg) | V (Å3) | Z | Void (Å3) (% of as made lp phase) | |
|---|---|---|---|---|---|---|---|---|---|---|
| DUT-8(Ni) cp | P1 | 6.947(1) | 8.181(1) | 12.172(1) | 91.14(1) | 103.87(1) | 104.55(1) | 647.7(1) | 1 | 0 (0) |
| C2H6@DUT-8(Ni) IP1 184.6 K | P1 | 9.478(1) | 11.066(1) | 12.694(1) | 101.54(1) | 92.06(1) | 100.59(1) | 1278.5(1) | 1 | 729.2 (57.0) |
| C2H4@DUT-8(Ni) IP2 169.4 K | P1 | 9.611(1) | 11.254(1) | 12.556(1) | 103.50 (1) | 94.53(1) | 98.32(1) | 1296.6(1) | 1 | 736.2 (56.8) |
| DUT-8(Ni) as made 298 K | P4/n | 18.431(1) | 18.431(1) | 9.391(1) | 90 | 90 | 90 | 3190.0(1) | 2 | 2124.6 (66.6) |
| N2@DUT-8(Ni) lp 77 K | P21/m | 18.657(1) | 18.736(1) | 9.433(1) | 90 | 94.42(1) | 90 | 3287.5(1) | 2 | 2196.0 (66.8) |
| CO2@DUT-8(Ni) lp 195 K | P21/m | 18.475(1) | 18.604(1) | 9.431(1) | 90 | 95.45(1) | 90 | 3226.9(1) | 2 | 2156.4 (66.8) |
| C4H10@DUT-8(Ni) lp 273 K | P21/m | 18.514(1) | 18.179(1) | 9.409(1) | 90 | 95.62(1) | 90 | 3151.0(8) | 2 | 2080.3 (66.0) |
| C2H4@DUT-8(Ni) lp 169.4 K | P21/m | 20.405(1) | 16.497(1) | 9.347(1) | 90 | 94.12(1) | 90 | 3138.2(1) | 2 | 2070.8 (66.0) |
The crystallographic data collected in whole range of relative pressures above the “gate pressure”, indicate a step-like transition from cp phase to the N2@DUT-8(Ni) phase. Peaks belonging to an intermediate (partially opened) structure are not detected. It is remarkable that the nitrogen uptake of 480 cm3 g−1, obtained at p/p0 = 0.95 during the in situ experiment (and consistently the corresponding pore volume of 0.743 cm3 g−1) amount to 71.4% of the theoretical values calculated for “empty” as-made structure (pore volume 1.040 cm3 g−1). In this case, the nitrogen adsorption uptake can to some extent be used as an indicator for the degree of crystal structure transformation.
The changes in the local Ni coordination geometry during the adsorption of nitrogen at 77 K, especially bond lengths in the first coordination shell, were monitored by in situ EXAFS measurements. In the Fig. 3, the fits of the first coordination shell of the Ni scatterers for cp and lp phases are shown. For the cp phase the fit shows a good correlation for the Ni–Ni single scattering (SS) path. Thus, according to the crystallographic data the Ni–Ni distance in the cp phase is 2.736 Å, whereas EXAFS fit suggests 2.759 Å. Because of the low symmetry of the cp structure, as well as a very small spread in Ni–O and Ni–N distances, it becomes complicated from the physical point of view to fit each SS path separately because of drastically increasing number of parameters. Therefore three Ni–O SS paths with the similar lengths were fitted as one with degeneracy parameter of 3. The fourth Ni–O SS path with smallest distance as well as Ni–N SS path were fitted separately. Even in this case, the Debye–Waller factors for these paths refine to the negative values indicating the low reliability of the data. The EXAFS spectrum of the nitrogen filled sample was fitted to the corresponding structural model. Due to the higher symmetry of the Ni-paddle-wheel, only one Ni–O path was used with the degeneracy parameter of 4. Both Ni–N and Ni–Ni single scattering paths were fitted in the same manner. In addition, the high symmetry of the SBU as well as high quality of the spectrum allows to involve Ni–Ccarboxylate path in the fit. As a result, the Ni–O SS path was fitted with the distance of 2.017 Å that is 0.158 Å higher in comparison with crystallographic data, reported for as made phase. Opposite, the Ni–Ni, Ni–N and Ni–C SS paths are fitted with the values very close to the crystallographic ones (ESI,† Table S2).
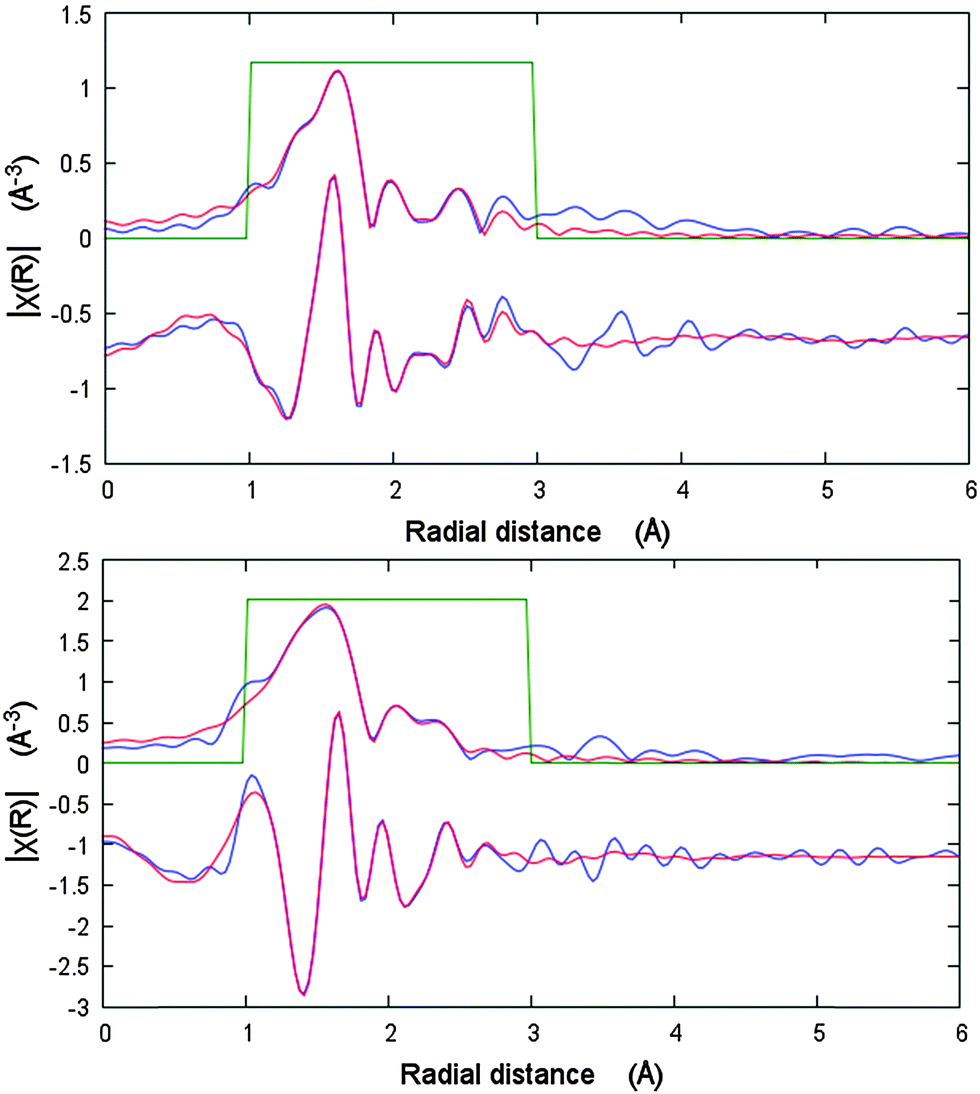 | ||
| Fig. 3 EXAFS data analysis of DUT-8(Ni) cp (bottom) and N2@DUT-8(Ni) lp (top) phases in R-space showing modulus and imaginary part. Blue lines – measured data, red line – fit. | ||
Adsorption of CO2 at 195 K monitored in situ adsorption by powder XRD
Compared to nitrogen, the kinetic diameter of carbon dioxide molecules is smaller and the quadrupole moment is higher. This usually results in stronger interaction with the host framework reflected in a higher adsorption enthalpy.Tuning the adsorption behavior of MOFs for specific gases such as CO2 is essential for the application in separation units. Switchable MOFs show typically guest specific gate opening pressures with significant differences in magnitude for CO2 and N2.34 Such selectivity is also characteristic for DUT-8(Ni). The relative gate opening pressure for CO2 is ca. 0.4 at 195 K and for N2ca. 0.2 (at 77 K). In the high pressure region (up to 50 bar at 298 K), only CO2 is able to induce the framework opening (gate opening pressure 30 bar).28 The adsorption of CO2 on DUT-8(Ni) at high pressure was investigated earlier by in situ-13C NMR spectroscopy.35 A certain degree of ordering was observed for all the CO2 molecules adsorbed inside DUT-8(Ni) and a tilt angle of 49° between the symmetry axis and the rotation axis of the molecules adsorbed in the flexible DUT-8(Ni) was postulated.
Surprisingly, from the structural point of view, the DUT-8(Ni) framework shows at 195 K carbon dioxide adsorption behavior very similar to that observed during the adsorption of nitrogen at 77 K (ESI,† Fig. S11). The evacuated sample contains predominantly cp DUT-8 (ESI,† Table S5). After adsorbing of small CO2 amount in the pre-gate region, the (110) peak belonging to lp phase at 2θ = 6.8° disappears indicating an increase of the cp phase amount in the bulk material. The (110) reflection of the CO2@DUT-8(Ni) phase appears at p/p0 = 0.61 and constantly grows in intensity with increasing CO2 pressure. At the same time the intensity of (100) peak of cp decreases steadily. According to the powder XRD pattern at p/p0 = 0.99, the majority of the sample exist in the large pore CO2@DUT-8(Ni) phase (ESI,† Table S5). However both, evaluation of the pore volume at p/p0 = 0.99 as well as quantitative phase analysis of PXRD, suggest the presence of nearly 25% of closed phase in the sample. The indexing of the CO2@DUT-8(Ni) PXRD pattern collected at p/p0 = 0.99 results in a monoclinic cell (space group P21/m) with monoclinic angle of 95.4°, close to that of N2@DUT-8(Ni) phase (Table 1, ESI,† Fig. S7). Thus, the structural changes during the adsorption of CO2 at 195 K occur in one step and in the similar way as compared to the adsorption of N2 at 77 K.
Adsorption of n-butane at 273 K monitored in situ by powder XRD
In order to prove the interaction of non-polar hydrophobic adsorptive with the DUT-8 framework, the pore opening process was studied during the adsorption of n-butane at 273 K. At low relative pressures no significant adsorption of n-butane was detected (Fig. 4).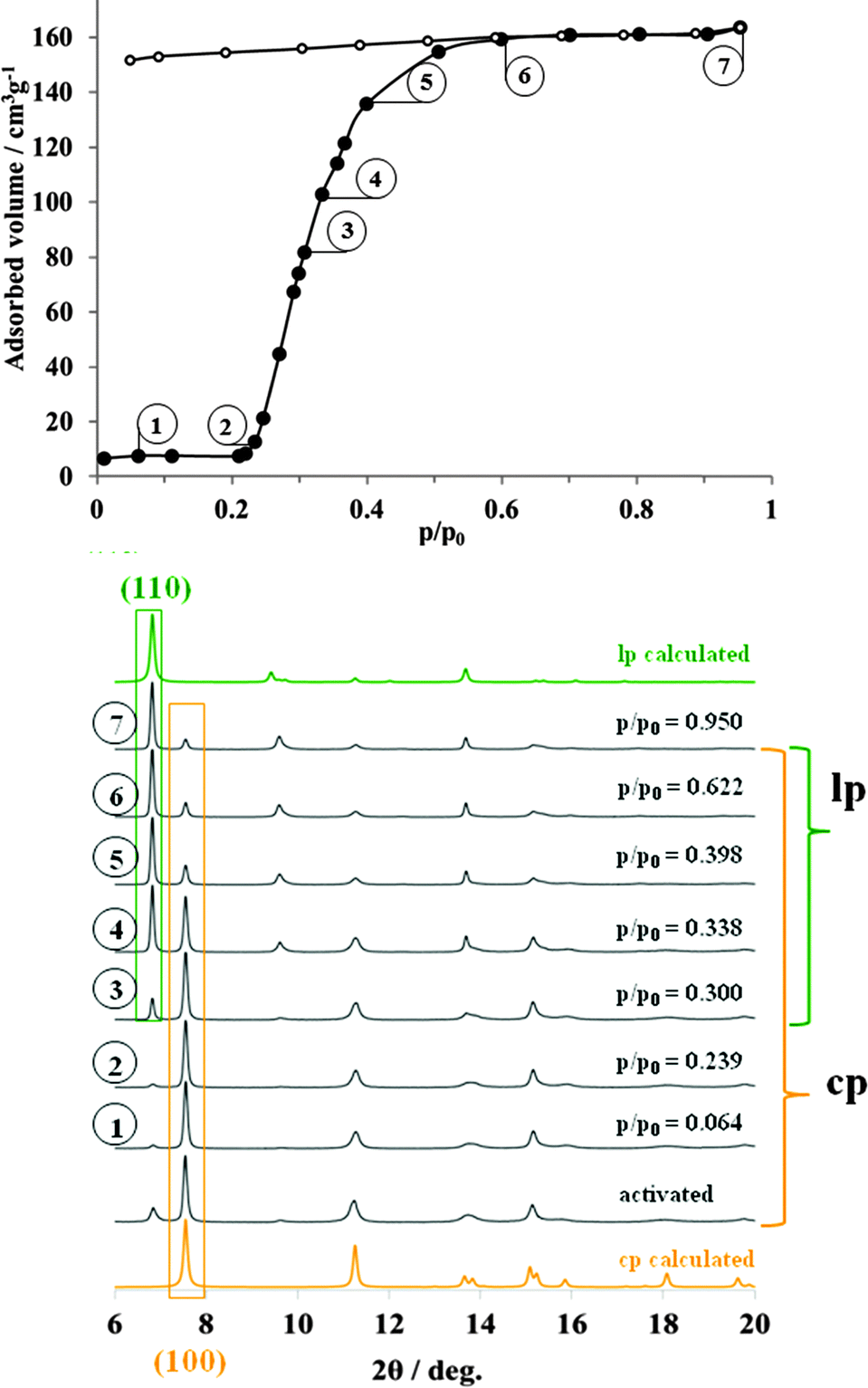 | ||
| Fig. 4 Adsorption of n-butane (272.5 K) on DUT-8(Ni) monitored in situ-XRD. | ||
The predominating phase in activated sample, according to X-ray diffraction patterns, is the close pore phase. At p/p0 = 0.25, the “gate opening” pressure is reached and DUT-8(Ni) the adsorption starts, accompanied by the corresponding structural changes. Thus, powder XRD patterns, measured in p/p0 range from 0.3 to 0.4 indicate the presence of both phases (cp and butane@DUT-8(Ni)) with different ratios (ESI,† Table S3). Even at relative pressure, close to the condensation point, weak (100) reflection belonging to the cp phase is still distinguishable in the powder XRD pattern. As in both previous cases, indexing of the XRD pattern, measured from the n-butane filled material results in the monoclinic cell with similar unit cell constants (ESI,† Fig. S8). In order to evaluate the conversion of cp to lp, the pore volume in the point 7 of the isotherm (p/p0 = 0.95) was compared with the result of the phase analysis at the same point. The pore volume of 0.71 cm3 g−1 (67.9% of the theoretical value) indirectly indicates that not all crystallites undergo the phase transition to the lp phase. As in the case of nitrogen adsorption, the pore volume of the sample strongly correlates with the ratio of lp:cp, obtained from the quantitative phase analysis of the powder XRD pattern (lp:cp = 67:33 (ESI,† Table S3)).
Adsorption of C2H6 at 185 K and C2H4 at 169 K monitored in situ by powder XRD
Alkene/alkane separations are of high economic importance for chemical industry. In order to understand selectivity of DUT-8(Ni) for ethane vs. ethylene, adsorption studies were carried out. The in situ experiments were performed at the saturation temperature for each gas. The changes in the structure start for both gases at nearly the same relative pressure around p/p0 = 0.2. But the network transformation induced by ethane adsorption proceeds along a different structural trajectory in comparison with the probe molecules discussed above. Increasing the pressure, the peaks of the cp phase decrease in intensity and simultaneously a new reflection at 2θ = 7.1° characteristic for the open structure emerges instead of peaks at 2θ = 6.8°. Obviously, increasing kinetic diameter of the probe molecule leads to changes in the opening mechanism of DUT-8(Ni) and an intermediate phase C2H6-IP1 forms first.The formation of intermediate MOF phases and associated multistep adsorption was also recognized previously.36,37 Indexing of the powder XRD pattern measured at p/p0 = 0.61 (point 8 on the adsorption isotherm in Fig. 5) leads to a triclinic unit cell with commensurate DUT-8(Ni) cell axes and angles (Table 1, ESI,† Fig. S3). The lattice parameters were used for the geometric optimization of the cp structure using Material Studio 5.0. The resulting model, containing nine ethane molecules per unit cell, was subjected to Rietveld refinement (ESI,† Fig. S3). The number of ethane molecules was estimated from the adsorption isotherm.
 | ||
| Fig. 5 Adsorption of C2H6 (184.6 K) on DUT-8(Ni) monitored in situ by powder XRD. | ||
The refinement of the crystal structure revealed, that IP1 is based on a distorted paddle-wheel SBU (Fig. 6a). The O–Ni–O angles lie in the narrower range from 79.6(1)° to 98.8(1)° in comparison with cp of DUT-8. The Ni–Ni–N angle (characteristic for the linearity of paddle-wheel – dabco axis) evolves from 153.9(1)° (bent) in the cp to nearly linear (173.3(1)°) in IP1. It should be mentioned that hinges connected with the paddle-wheel unit play a key role in the pore opening mechanism. The dihedral angles between carboxylate groups and O–Ni–Ni–O fragments of the paddle-wheel cover a range from 121.1 to 152.9° (Fig. 6a).
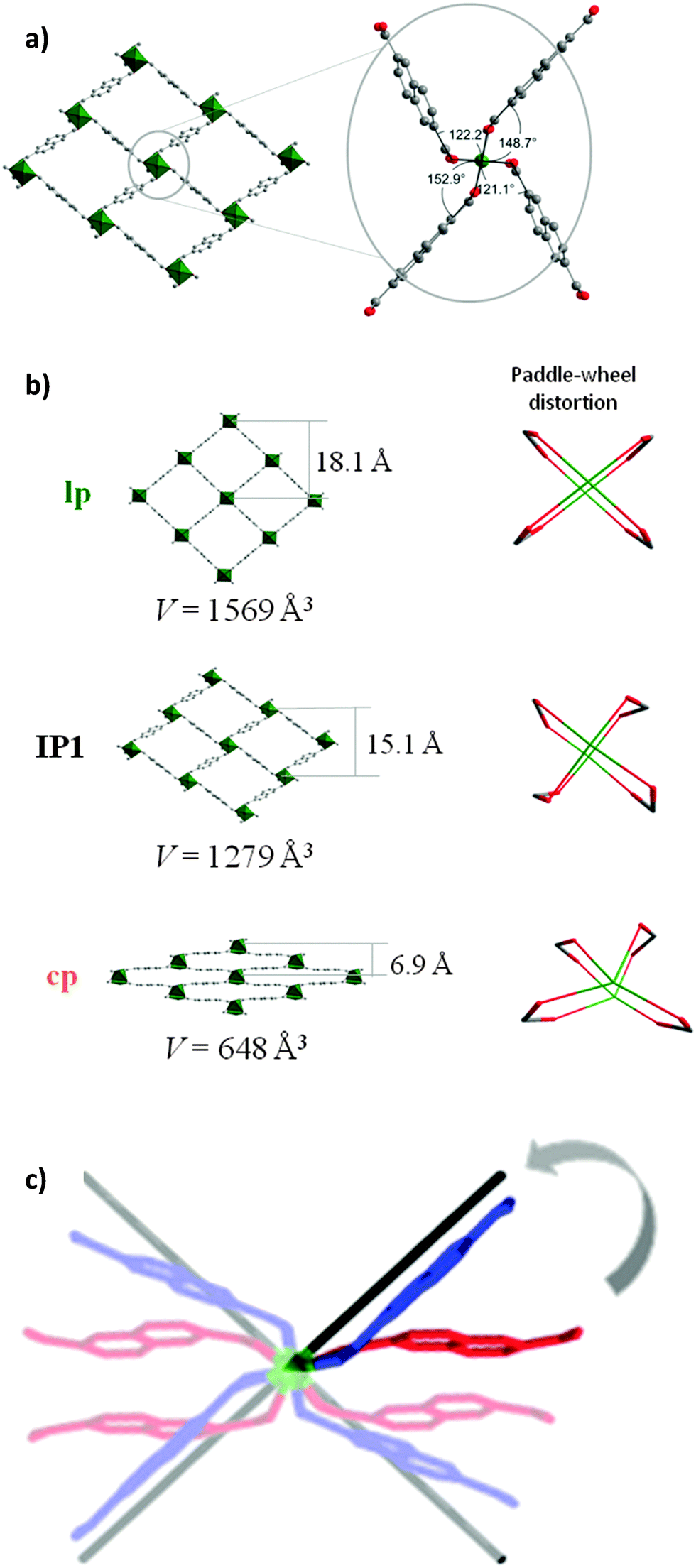 | ||
| Fig. 6 (a) Coordination geometry of the cluster in IP1 (top). (b) Evolution of the crystal structure and paddle-wheel distortion in DUT-8 cp, IP1 and lp; (c) Superposition of the paddle-wheels: cp (red), IP1 (blue) and lp (black). | ||
During the adsorption, the aperture of the rectangular channels changes from 6.9 × 23.7 Å in cp phase via 15.1 × 18.4 Å in the IP1 up to 18.1 × 18.6 Å in C2H6@DUT-8(Ni) structure (Fig. 6b and c). According to the powder XRD data, the intermediate phase IP1 is stable at ethane relative pressures from 0.31 to 0.99. Applying the Poreblazer 3.0 Software on IP1 a geometrical surface area of 1888 m2 g−1 is estimated, a pore volume of 0.524 g cm−3, and a pore limiting diameter of 4.91 Å. The characteristic peak of the lp C2H6@DUT-8(Ni) phase appears in the powder XRD at p/p0 = 0.61 increasing in intensity with increasing relative pressure. It should be mentioned that even at p/p0 = 0.99 the phase analysis of powder XRD patterns shows that only 36.1% of lp is detected in the sample (ESI,† Table S6), indicating the weak host–guest interactions in the C2H6@DUT-8(Ni) system. The direct comparison of PXRDs of cp, IP1 and lp is given in ESI,† Fig. S12.
Since the ethylene adsorption isotherm at 185 K is, at a first glance, very similar to the ethane adsorption at 169 K, we assumed the existence of the similar intermediate phase during the gate opening.
However, the analysis of the powder diffraction patterns measured during the adsorption indicates the appearance of a new peak starting at p/p0 = 0.202. At the same time, the positions of the new peaks do not match neither the position of the peaks of the lp phases nor the position of the peaks corresponding to the IP1 phase (ESI,† Fig. S13 and S16).
According to the ethylene adsorption isotherm, an unexpected, novel C2H4-IP2 (intermediate phase 2) is present in the mixture with cp at 0.20 ≤ p/p0 ≤ 0.25. In the range of 0.25 ≤ p/p0 ≤ 0.55, IP2 co-exists in the equilibrium with large C2H4@DUT-8(Ni) (ESI,† Fig. S16). Because of the strong overlap of the reflections from all three phases, powder XRDs measured in this range of pressure were not suitable for indexing and further structural analysis. In contrast to all other experiments, complete structural transition from cp to lp phase is achieved at high relative pressures that confirmed by absence of characteristic reflection of cp at 2θ = 7.6°. The C2H4@DUT-8(Ni) lp phase is solved in the P21/m space group and shows slightly different cell parameters but very similar to the other guest loaded phases crystal structure (see ESI,† Fig. S5). Interestingly, the reverse structural transition from lp to IP2 proceeds quantitatively and therefore, the powder XRDs, measured during the desorption in the range 0.006 ≤ p/p0 ≤ 0.095, contain mainly peaks from the IP2. The indexing of one powder XRD in this range resulted in a triclinic unit cell with the lattice parameters and cell volume, very close to that of C2H6-IP1 (Table 1, ESI,† Fig. S4 and S13). Further desorption of ethylene leads to the formation of cp phase (ESI,† Fig. S13). Thus, despite the similarities of ethane and ethene adsorption isotherms, subtle structural differences are detected regarding the intermediates formed indicating significant differences in the packing of the small molecules and pore–wall interactions.
Conclusions
Summarizing, we have shown the importance of combined EXAFS, single crystal and powder XRD measurements parallelized with adsorption methods to reveal the complex structural changes and dynamics of the switchable coordination polymer DUT-8(Ni) in the presence of probe molecules differing in kinetic diameter, polarity and adsorption enthalpy. Especially the strong cluster deformation during N2 and n-butane adsorption is unprecedented and has so far only been observed in one case.24 For nitrogen and carbon dioxide adsorption, DUT-8(Ni) shows comparable direct structural transformations from cp to lp phase with a huge expansion of the unit cell volume during the gas adsorption as high as 250%. In the case of n-butane, the complete structural transformation to the open phase is hindered. In contrast, ethane and ethene lead to a stepwise adsorption behavior, confirmed by the structural transformations, where intermediate phases could be clearly identified and solved from the powder XRDs, measured at well-defined pressure. Certainly this extent of complex switchability is unique for MOFs. The structural complexity described here for only one MOF (DUT-8(Ni)) demonstrates that through the eye of highly developed in situ-analytical methods there is much more structural chemistry and physics to discover in future. Recognizing soft porous solids as systems in which the structure of the solid cannot be considered without specifying the continuous surrounding phase opens a new way of interpreting structure property relationships.Acknowledgements
BMBF (German Federal Ministry of Education and Research) is acknowledged for the financial support (Projects 05K10OD3 and 05K13OD3). We acknowledge the Helmholtz-Zentrum Berlin for provision of travel grants and synchrotron radiation beamtime at beamlines MagS, KMC-2 and MX BL14.2 of BESSY II.References
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2011, 112, 782–835 CrossRef PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2011, 112, 869–932 CrossRef PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724–781 CrossRef PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2011, 112, 1105–1125 CrossRef PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6069 RSC.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- K. Nakagawa, D. Tanaka, S. Horike, S. Shimomura, M. Higuchi and S. Kitagawa, Chem. Commun., 2010, 46, 4258–4260 RSC.
- R. El Osta, A. Carlin-Sinclair, N. Guillou, R. I. Walton, F. Vermoortele, M. Maes, D. de Vos and F. Millange, Chem. Mater., 2012, 24, 2781–2791 CrossRef CAS.
- J. Kim, W. Y. Kim and W.-S. Ahn, Fuel, 2012, 102, 574–579 CrossRef CAS PubMed.
- F. Zhang, X. Zou, X. Gao, S. Fan, F. Sun, H. Ren and G. Zhu, Adv. Funct. Mater., 2012, 22, 3583–3590 CrossRef CAS PubMed.
- Q. Chen, Z. Chang, W.-C. Song, H. Song, H.-B. Song, T.-L. Hu and X.-H. Bu, Angew. Chem., Int. Ed., 2013, 52, 11550–11553 CrossRef CAS PubMed.
- R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, P. C. J. Kepert and M. R. Hill, Angew. Chem., Int. Ed., 2013, 52, 3695–3698 CrossRef CAS PubMed.
- A. C. McKinlay, J. F. Eubank, S. Wuttke, B. Xiao, P. S. Wheatley, P. Bazin, J. C. Lavalley, M. Daturi, A. Vimont, G. De Weireld, P. Horcajada, C. Serre and R. E. Morris, Chem. Mater., 2013, 25, 1592–1599 CrossRef CAS.
- R. K. Das, A. Aijaz, M. K. Sharma, P. Lama and P. K. Bharadwaj, Chem. – Eur. J., 2012, 18, 6866–6872 CrossRef CAS PubMed.
- S. Bureekaew, H. Sato, R. Matsuda, Y. Kubota, R. Hirose, J. Kim, K. Kato, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2010, 49, 7660–7664 CrossRef CAS PubMed.
- V. Bon, I. Senkovska, D. Wallacher, A. Heerwig, N. Klein, I. Zizak, R. Feyerherm, E. Dudzik and S. Kaskel, Microporous Mesoporous Mater., 2014, 188, 190–195 CrossRef CAS PubMed.
- T. R. Jensen, T. K. Nielsen, Y. Filinchuk, J.-E. Jorgensen, Y. Cerenius, E. M. Gray and C. J. Webb, J. Appl. Crystallogr., 2010, 43, 1456–1463 CrossRef CAS PubMed.
- M. Lange, M. Kobalz, J. Bergmann, D. Lassig, J. Lincke, J. Mollmer, A. Moller, J. Hofmann, H. Krautscheid, R. Staudt and R. Glaser, J. Mater. Chem. A, 2014, 2, 8075–8085 CAS.
- P. L. Llewellyn, P. Horcajada, G. Maurin, T. Devic, N. Rosenbach, S. Bourrelly, C. Serre, D. Vincent, S. Loera-Serna, Y. Filinchuk and G. Férey, J. Am. Chem. Soc., 2009, 131, 13002–13008 CrossRef CAS PubMed.
- S. R. Miller, P. A. Wright, T. Devic, C. Serre, G. R. Férey, P. L. Llewellyn, R. Denoyel, L. Gaberova and Y. Filinchuk, Langmuir, 2009, 25, 3618–3626 CrossRef CAS PubMed.
- P. K. Allan, B. Xiao, S. J. Teat, J. W. Knight and R. E. Morris, J. Am. Chem. Soc., 2010, 132, 3605–3611 CrossRef CAS PubMed.
- J. Seo, C. Bonneau, R. Matsuda, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2011, 133, 9005–9013 CrossRef CAS PubMed.
- C. Serre, S. Bourrelly, A. Vimont, N. A. Ramsahye, G. Maurin, P. L. Llewellyn, M. Daturi, Y. Filinchuk, O. Leynaud, P. Barnes and G. Ferey, Adv. Mater., 2007, 19, 2246–2251 CrossRef CAS PubMed.
- P. L. Llewellyn, G. Maurin, T. Devic, S. Loera-Serna, N. Rosenbach, C. Serre, S. Bourrelly, P. Horcajada, Y. Filinchuk and G. Ferey, J. Am. Chem. Soc., 2008, 130, 12808–12814 CrossRef CAS PubMed.
- L. Hamon, P. L. Llewellyn, T. Devic, A. Ghoufi, G. Clet, V. Guillerm, G. D. Pirngruber, G. Maurin, C. Serre, G. Driver, W. V. Beek, E. Jolimaître, A. Vimont, M. Daturi and G. R. Férey, J. Am. Chem. Soc., 2009, 131, 17490–17499 CrossRef CAS PubMed.
- N. Klein, C. Herzog, M. Sabo, I. Senkovska, J. Getzschmann, S. Paasch, M. R. Lohe, E. Brunner and S. Kaskel, Phys. Chem. Chem. Phys., 2010, 12, 11778–11784 RSC.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 37, 1248–1257 CrossRef CAS PubMed.
- Material Studio, Accelrys Software Inc., San Diego, Release 5.0 edn, 2009 Search PubMed.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- F. Štěpánek, M. Kubíček, M. Marek, M. Šoóš, P. Rajniak and R. T. Yang, Chem. Eng. Sci., 2000, 55, 431–440 CrossRef.
- H. C. Hoffmann, B. Assfour, F. Epperlein, N. Klein, S. Paasch, I. Senkovska, S. Kaskel, G. Seifert and E. Brunner, J. Am. Chem. Soc., 2011, 133, 8681–8690 CrossRef CAS PubMed.
- J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS PubMed.
- H. Hoffmann, M. Debowski, P. Müller, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Materials, 2012, 5, 2537–2572 CrossRef CAS PubMed.
- F. Salles, G. Maurin, C. Serre, P. L. Llewellyn, C. Knofel, H. J. Choi, Y. Filinchuk, L. Oliviero, A. Vimont, J. R. Long and G. Ferey, J. Am. Chem. Soc., 2010, 132, 13782–13788 CrossRef CAS PubMed.
- V. Bon, I. Senkovska, D. Wallacher, D. M. Többens, I. Zizak, R. Feyerherm, U. Mueller and S. Kaskel, Inorg. Chem., 2014, 53, 1513–1520 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1034317, 1034322, 1056823 and 1034320. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cp02180d |
| ‡ Present address: Fraunhofer Institute for Material and Beam Technology, IWS, Winterbergstraße 28, 01277 Dresden, Germany. |
| § Present address: Physical Chemistry, Measurement and Sensor Technology, Technische Universität Dresden, Eisenstuckstr. 5, 01069 Dresden, Germany. |
| This journal is © the Owner Societies 2015 |