Methanol electro-oxidation on platinum modified tungsten carbides in direct methanol fuel cells: a DFT study
Received
9th April 2015
, Accepted 24th August 2015
First published on 25th August 2015
Abstract
In exploration of low-cost electrocatalysts for direct methanol fuel cells (DMFCs), Pt modified tungsten carbide (WC) materials are found to be great potential candidates for decreasing Pt usage whilst exhibiting satisfactory reactivity. In this work, the mechanisms, onset potentials and activity for electrooxidation of methanol were studied on a series of Pt-modified WC catalysts where the bare W-terminated WC(0001) substrate was employed. In the surface energy calculations of a series of Pt-modified WC models, we found that the feasible structures are mono- and bi-layer Pt-modified WCs. The tri-layer Pt-modified WC model is not thermodynamically stable where the top layer Pt atoms tend to accumulate and form particles or clusters rather than being dispersed as a layer. We further calculated the mechanisms of methanol oxidation on the feasible models via methanol dehydrogenation to CO involving C–H and O–H bonds dissociating subsequently, and further CO oxidation with the C–O bond association. The onset potentials for the oxidation reactions over the Pt-modified WC catalysts were determined thermodynamically by water dissociation to surface OH* species. The activities of these Pt-modified WC catalysts were estimated from the calculated kinetic data. It has been found that the bi-layer Pt-modified WC catalysts may provide a good reactivity and an onset oxidation potential comparable to pure Pt and serve as promising electrocatalysts for DMFCs with a significant decrease in Pt usage.
1. Introduction
Fuel cells via converting the chemical energy directly into electricity are a promising clean energy solution to replace traditional fossil fuel technologies.1–5 Liquid methanol as a fuel has some advantages over gaseous hydrogen in handling, transportation and storage. In direct methanol fuel cells (DMFCs), the ideal anodic reaction is the complete oxidation of methanol to CO2 with the release of 6 electrons per methanol molecule (CH3OH + H2O → CO2 + 6H+ + 6e−).1–11 Since the fuel cell devices have to operate in strongly basic or acidic electrolyte media, the corrosion of the electrode materials is problematic, resulting in the inevitable usage of novel metals such as Pt and Pd.12 However, the estimated global reserves for Pt-group metals are extremely low, and they are remarkably expensive for widespread application commercially.1–12 To overcome this challenge, the alternative low-cost catalytic materials are strongly desired to reduce the Pt loading in order to make fuel cells more commercially viable.
In the exploration of low-cost catalyst materials, transition metal carbides are found to have great potential for reducing Pt usage. Noticeably, tungsten carbide (WC) which has a similar electronic structure in the region of the Fermi level has attracted widespread attention.13 However, the surface states of tungsten carbide are considerably different from those of the Pt surface, and therefore the catalytic properties could not be compared directly with Pt.13 To date, surface modification of low-cost tungsten carbide by active metals has been extensively investigated.14–32 Many groups have prepared varieties of platinum modified tungsten carbide materials, which exhibit a higher reactivity than pure platinum, in hydrogen evolution reaction (HER), oxygen reduction reaction (ORR) and methanol oxidation reaction (MOR).14–32
To understand the mechanism of electrooxidation of methanol on platinum electrodes, a lot of effort has been made in experiments. Sun et al. and Herrero et al. studied methanol electrooxidation on a series of Pt(111), Pt(110) and Pt(100) facets in acidic electrolytes.33,34 These studies revealed the influence of the surface structures and the anions on the catalytic decomposition of methanol. Recently, density functional theory (DFT) calculations have been widely used to understand electrochemical catalytic reactions at the atomic level.35 Theoretical studies of the electrooxidation of methanol have been performed extensively.36–50 Zhang et al. showed that the decomposition of methanol could occur via both C–H bond and O–H bond dissociations on the closed-pack flat (111) surface.36 Greeley et al. carried out DFT calculations by investigating the reaction energy and activation barriers of the elementary steps for methanol decomposition to CO on Pt(111).38,39 Ferrin et al. showed the surface structure sensitivity of methanol electrooxidation on transition metals.40 Cao et al. studied the methanol decomposition on three well-defined low index platinum single crystal planes by combining the experimental and theoretical methods.41 The decomposition pathways in methanol oxidation were also calculated over some bimetallic surfaces such as PtAu, PtRu and PdIn.42–44 Kramer et al. presented a model of the surface kinetics of methanol dehydrogenation on transition metals.45 Stottlemyer et al. calculated the methanol activation on the Pt-modified WC(0001) surface via C–H and O–H bonds.48
In this work, in order to understand the catalytic performance of the Pt-modified WC catalysts in a DMFC, two key issues have been considered: (i) What are the reasonable effective theoretical models to describe the Pt-modified WC structures in reality? (ii) What are the possible pathways of methanol dehydrogenation and oxidation on the Pt-modified WC surfaces and what are the onset potentials and activities for the surface reactions? With the above questions in mind, we illustrated the surface energies of a series of Pt-modified WC models to determine the thermodynamic stability with an increase in the number of surface Pt atoms. The calculated results, including the mechanisms of methanol oxidation and C–H bond and O–H bond dissociations, are discussed in detail.
2 Theoretical methods
All the electronic structure calculations were performed using the Vienna Ab-initio Simulation Package (VASP) with the exchange–correlation functional of Perdew–Burke–Ernzerhof (PBE). The projector-augmented-wave (PAW) pseudopotentials were utilized to describe the core electron interaction. Geometry optimization was carried out using the BFGS algorithm.51–59 The cut-off energy was 400 eV and a 4 × 4 × 1 Monkhorst–Pack k-point sampling was used. The transition states were located using a constrained optimization approach with the force converge criterion below 0.05 eV Å−1 in modified VASP.60–62 Since the WC(0001) surface has been confirmed to be W-terminated which is more stable than C-terminated,48 three layers of W-terminated WC(0001) as the p(3 × 3) substrate including 27 W and 27 C atoms was used in the calculations with the bottom two layers being fixed while the top layer was allowed to relax during calculations to interact with Pt. The vacuum region was ∼12 Å to ensure that there is little interaction between slabs. For modeling the Pt-modified WC models, mono-, bi- and tri-layer Pt atoms were placed epitaxially on the W-terminated WC(0001) substrate with 9, 18 and 27 Pt atoms, respectively. All the Pt atoms were allowed to relax in the calculations. For each adsorbate, different binding sites (top, bridge, and hollow) were calculated to determine the most stable bonding configuration.
In this work, the adsorption energy was defined as
| | | Ead = Eadsorbate/slab − Eadsorbate − Eslab | (1) |
where
Eadsorbate/slab,
Eadsorbate, and
Eslab are the total energies of the adsorbate binding with the surface, the gaseous adsorbate and the clean surface, respectively.
For the calculation of the OH* formation potential,60–63 the reaction free energy change (ΔG) in the formation of OH* according to the water dissociation reaction of H2O → OH* + H+ + e− was calculated. The free energy was obtained using G = E + ZPE + TS, in which E is the total energy of species, S is the entropy and ZPE is the zero point energy at room temperature. Therefore, the free energy change of the reaction H2O → OH* + H+ + e− was derived as ΔG = G(OH*) + G(H+ + e−) − G(H2O). When the electrode potential is 0 V, pH = 0 ([H+] = 1 M), at 298 K, due to the equilibrium of H+ + e− → ½H2, we can use the free energy of gaseous ½H2 to replace that of H+ + e−. Two correction terms were introduced: the pH of the electrolyte (−pHkT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln10) and the electrode potential (eU) referring to the standard hydrogen electrode (SHE), resulting in G(H+ + e−) = G(½H2) − pHkTln10 + eU. When ΔG = 0, the reaction H2O → OH* + H+ + e− is in equilibrium and the OH* formation potential can be obtained.
ln10) and the electrode potential (eU) referring to the standard hydrogen electrode (SHE), resulting in G(H+ + e−) = G(½H2) − pHkTln10 + eU. When ΔG = 0, the reaction H2O → OH* + H+ + e− is in equilibrium and the OH* formation potential can be obtained.
For the calculation of the surface energy, the definition of eqn (2), proposed by Boettger et al.,64 was employed in this work:
| | | EPt,surf = 1/n(EPt/substrate − Esubstrate − nEPt,bulk) | (2) |
where
EPt/substrate is the total energy of the slab,
Esubstrate is the total energy of the substrate,
n is the number of Pt atoms, and
EPt,bulk is the bulk energy per Pt atom obtained from an independent bulk calculation. In the calculation of the surface energy, every possible position of Pt atoms was tested and the most stable structure was used.
3. Results
3.1 Surface energies of Pt-modified WC(0001)
In this work, the models containing a three-layer W-terminated WC(0001) slab as the core and layers of Pt atoms as the shell were employed in the calculations. To quantify the thermodynamic stability of a series of Pt-modified WC(0001) surfaces, surface energies of a series of structures were calculated using eqn (2), with an increase of Pt loadings from mono-layer to tri-layers. Through comparing the calculated energy per Pt atom in the shell with an equivalent energy per Pt atom in the bulk as the reference, the stability of Pt atoms adsorbed on the substrate could be assessed effectively. Specifically, if the surface energy is lower than zero, the dispersed Pt atoms on WC(0001) would be favoured. On the other hand, if the surface energy is higher than 0, the dispersed Pt atoms would prefer to accumulate to form clusters. The assessments can be summarized as follows:
| EPt,surf < 0, Pt atoms → Pt layer |
| EPt,surf > 0, Pt atoms → Pt cluster |
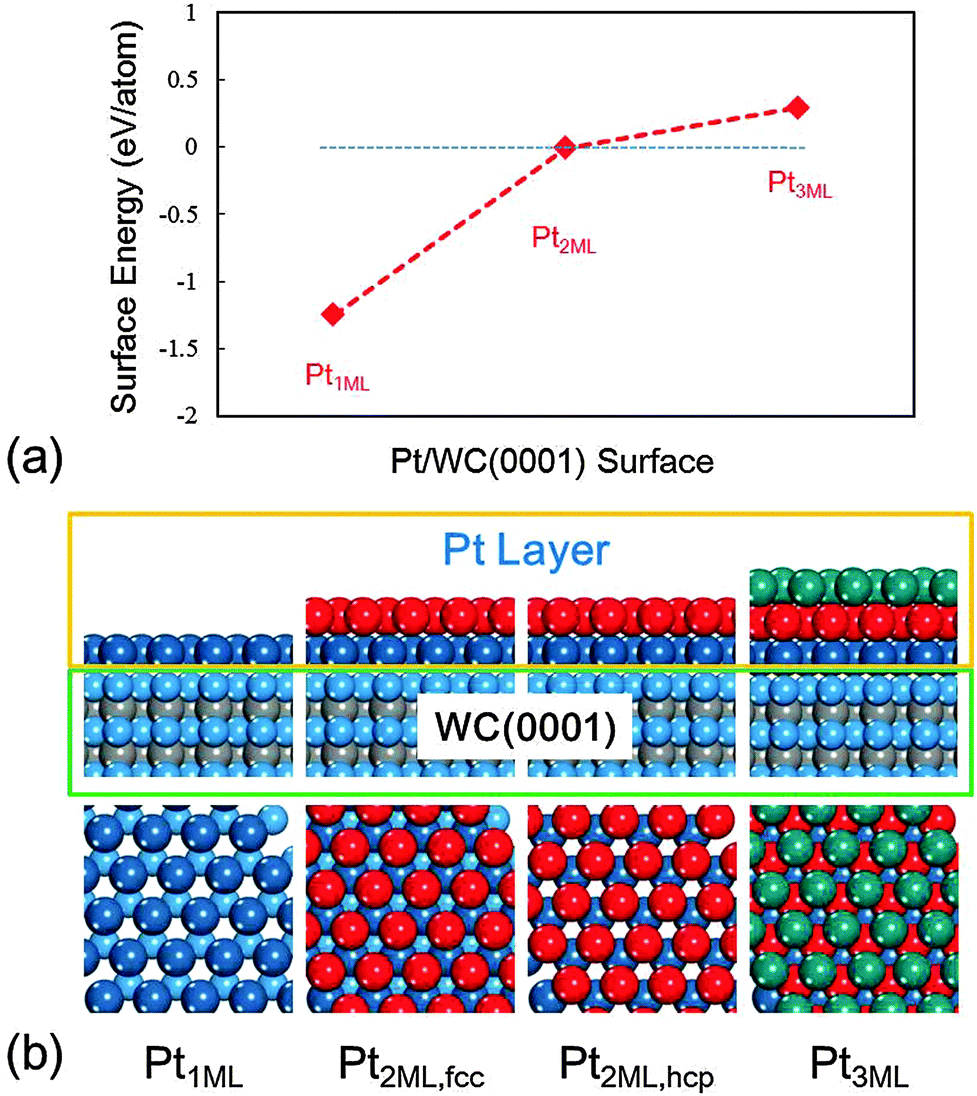
The calculated surface energies and the structures of each model with the top and side views at different Pt coverages are presented in Fig. 1. On mono-layer Pt-modified WC, the Pt1ML/WC(0001) surface, the Pt atoms favour to occupy the hcp sites rather than the fcc sites, and the calculated surface energy is −1.25 eV per atom, indicating that Pt atoms are favoured to be dispersed over W-terminated WC(0001) since the chemical bonds between the Pt atoms and W atoms are considerably strong. On the bi-layer Pt-modified WC surface, Pt2ML/WC(0001), Pt atoms could alternatively occupy the fcc or hcp sites on Pt1ML/WC(0001) with the same surface energy of −0.01 eV, which is close to zero, implying that both surface structures are likely to exist. With an increase of Pt atoms on tri-layers, the most stable structure would be such that the top layer Pt atoms occupy the fcc sites on Pt2ML,hcp/WC(0001). However, as shown in Fig. 1a, the calculated surface energy is noticeably higher than zero, indicating that the Pt atoms of the third layers cannot be effectively dispersed but tend to accumulate forming Pt clusters or particles on Pt2ML/WC(0001). According to these calculated results, we concluded that in reality mono- and bi-layer Pt-modified WC surfaces should commonly exist, depending on the amount of Pt loaded. Therefore, Pt1ML/WC(0001) and Pt2ML/WC(0001) as shown in Fig. 1b were used for the calculations in the study of the electrooxidation of methanol.
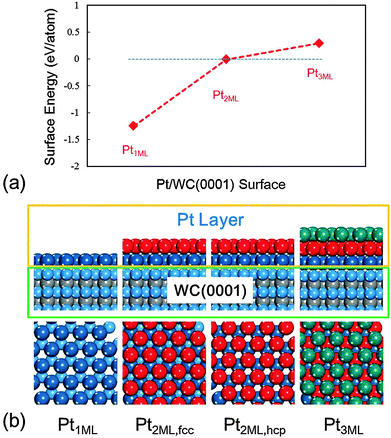 |
| | Fig. 1 (a) The calculated surface energies (in eV per atom) of a series of Pt-modified WC(0001) surfaces. (b) Top and side views of a series of Pt-modified WC(0001) surfaces with the increase of Pt atoms. Cyan: W; grey: C; blue: first layer Pt; red: second layer Pt; green: third layer Pt. (The same colors were used throughout this work.) | |
3.2 Methanol dehydrogenation
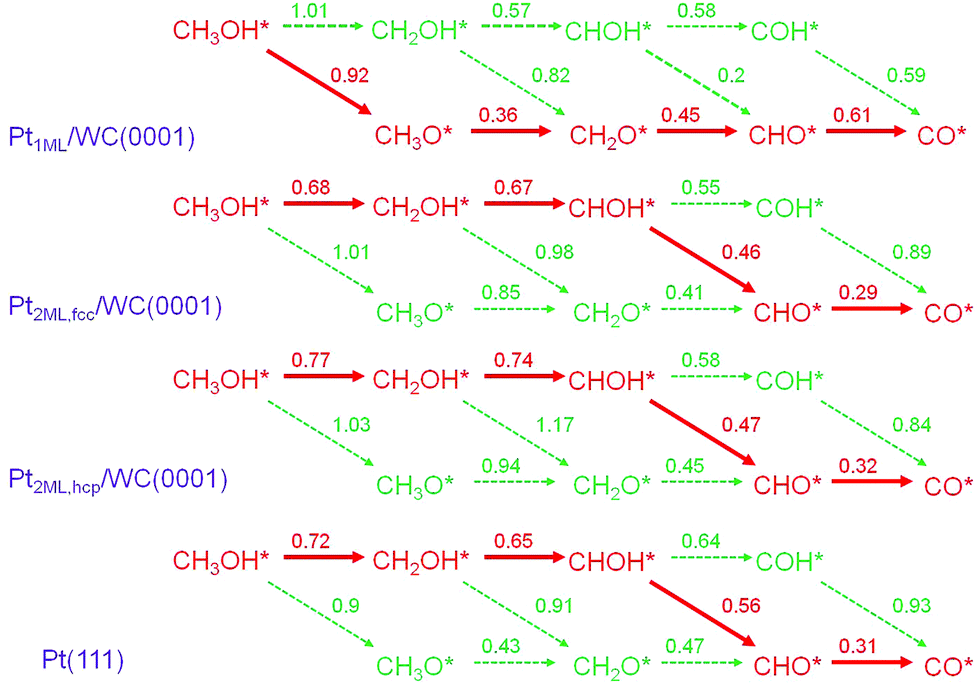
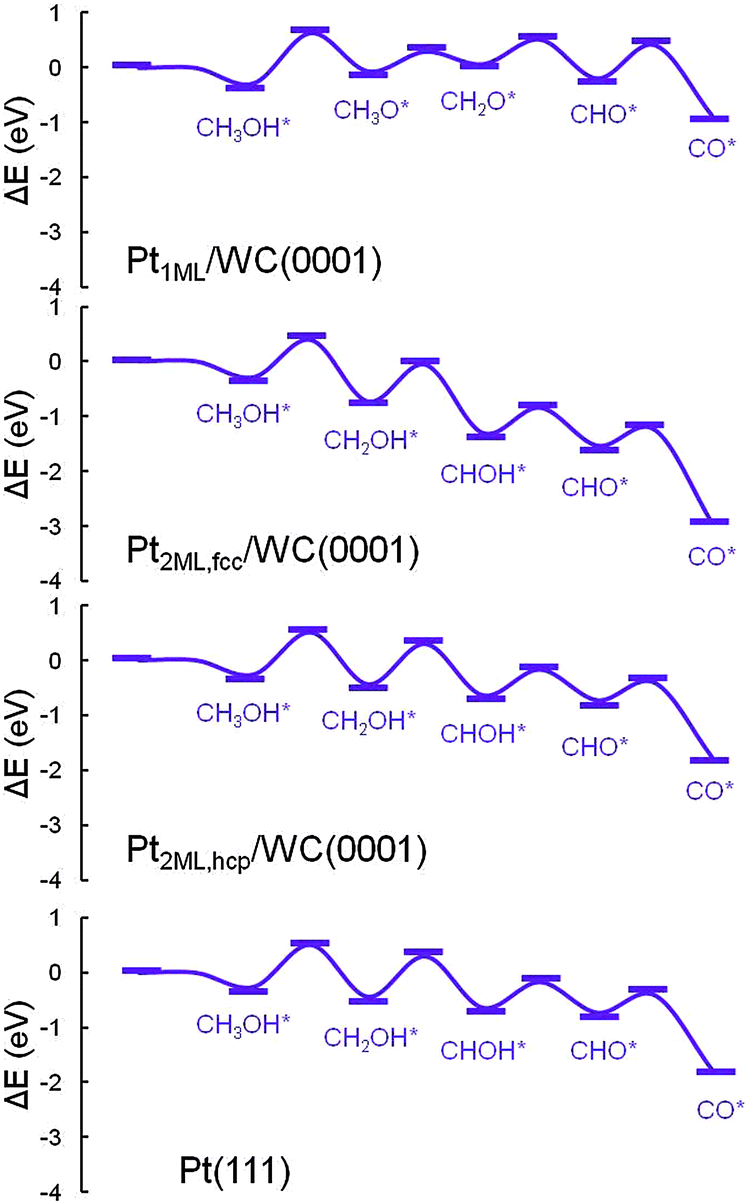
In the electrooxidation of methanol in DMFCs, the reactions could be divided into methanol dehydrogenation to surface CO adsorbates and the further CO oxidation in the presence of surface oxidants produced from water dissociation. Since in the dehydrogenation of CH3OH* to CO*, four C–H bonds and one O–H bond need to be cleaved subsequently, two main pathways are considered: C–H and O–H pathways. To highlight the dehydrogenation mechanisms, we mapped out an overall reaction network systematically as shown in Fig. 2, including all the possible paths in the methanol dehydrogenation involving 10 elementary reactions and 8 intermediates: methanol (CH3OH*), hydroxymethyl (CH2OH*), hydroxymethylene (CHOH*), hydroxymethylidyne (COH*), methoxy (CH3O*), formaldehyde (CH2O*), formyl (CHO*) and carbon monoxide (CO*). In this section, since there are two structures of bi-layer Pt-modified WC surfaces, the calculations in the methanol dehydrogenation were performed on both Pt2ML,fcc/WC(0001) and Pt2ML,hcp/WC(0001). In order to compare with the methanol dehydrogenation on pure Pt, Pt(111) was also employed. All the calculated data for the Pt1ML/WC(0001), Pt2ML,fcc/WC(0001), Pt2ML,hcp/WC(0001) and Pt(111), including kinetic barriers (Ea) and reaction energies (ΔE), are listed in Table 1. The most favoured reaction paths are highlighted in Fig. 2 with the kinetic data listed. The energy profiles for methanol dehydrogenation are presented in Fig. 3 whilst the optimized structures of intermediates and transition states involved in the reaction network are shown in Fig. 4.
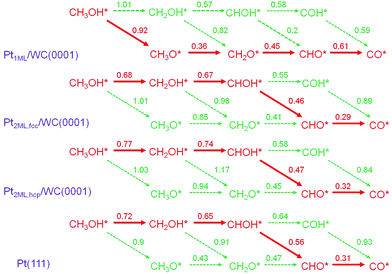 |
| | Fig. 2 Reaction networks and the calculated reaction barriers (in eV) for methanol dehydrogenation from CH3OH* to CO* + 4H* over Pt1ML/WC(0001), Pt2ML,fcc/WC(0001), Pt2ML,hcp/WC(0001) and Pt(111) surfaces. The paths in red color present the favored pathways in methanol dehydrogenation with the intermediates involved and the barriers shown. | |
Table 1 Calculated reaction barriers (Ea, in eV) and reaction energies (ΔE, in eV) of the elementary steps in methanol dehydrogenation to surface CO* and the further CO* oxidation over Pt1ML/WC(0001), Pt2ML,fcc/WC(0001), Pt2ML,hcp/WC(0001) and Pt(111) surfaces
| Surface reactions |
Pt1ML/WC |
Pt2ML,fcc/WC |
Pt2ML,hcp/WC |
Pt |
|
E
a
|
ΔE |
E
a
|
ΔE |
E
a
|
ΔE |
E
a
|
ΔE |
| CH3OH* → CH2OH* + H* |
1.01 |
0.29 |
0.68 |
−0.44 |
0.77 |
−0.18 |
0.72 |
−0.21 |
| CH3OH* → CH3O* + H* |
0.92 |
0.22 |
1.01 |
0.08 |
1.03 |
0.42 |
0.9 |
0.63 |
| CH2OH* → CHOH* + H* |
0.57 |
0.43 |
0.67 |
−0.57 |
0.74 |
−0.2 |
0.65 |
−0.17 |
| CH2OH* → CH2O* + H* |
0.82 |
0.07 |
0.98 |
0.29 |
1.17 |
0.53 |
0.91 |
0.51 |
| CH3O* → CH2O* + H* |
0.36 |
0.13 |
0.85 |
−0.23 |
0.94 |
−0.07 |
0.43 |
−0.33 |
| CHOH* → COH* + H* |
0.58 |
0.14 |
0.55 |
−0.95 |
0.58 |
−0.72 |
0.64 |
−0.51 |
| CHOH* → CHO* + H* |
0.2 |
−0.61 |
0.46 |
−0.23 |
0.47 |
−0.09 |
0.56 |
−0.11 |
| CH2O* → CHO* + H* |
0.45 |
−0.26 |
0.41 |
−1.1 |
0.45 |
−0.82 |
0.47 |
−0.79 |
| CHO* → CO* + H* |
0.61 |
−0.68 |
0.29 |
−1.4 |
0.32 |
−1.04 |
0.31 |
−0.73 |
| COH* → CO* + H* |
0.59 |
−1.43 |
0.89 |
−0.68 |
0.84 |
−0.42 |
0.93 |
−1.26 |
| CO* + OH* → COOH* |
0.49 |
0.04 |
0.58 |
−0.24 |
0.59 |
−0.24 |
0.43 |
−0.34 |
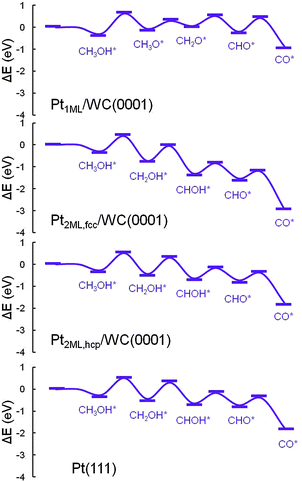 |
| | Fig. 3 Energy profiles for the dehydrogenation of methanol from CH3OH* to CO* + 4H* over Pt1ML/WC(0001), Pt2ML,fcc/WC(0001), Pt2ML,hcp/WC(0001) and Pt(111) surfaces. | |
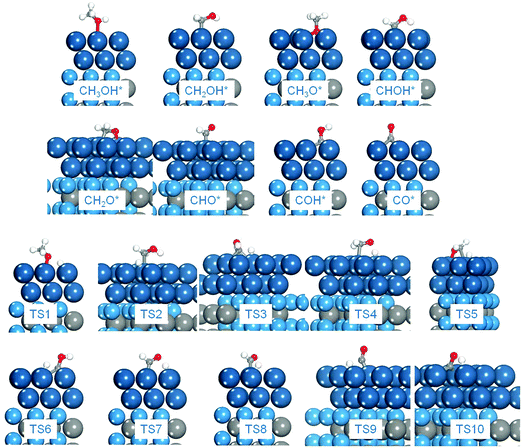 |
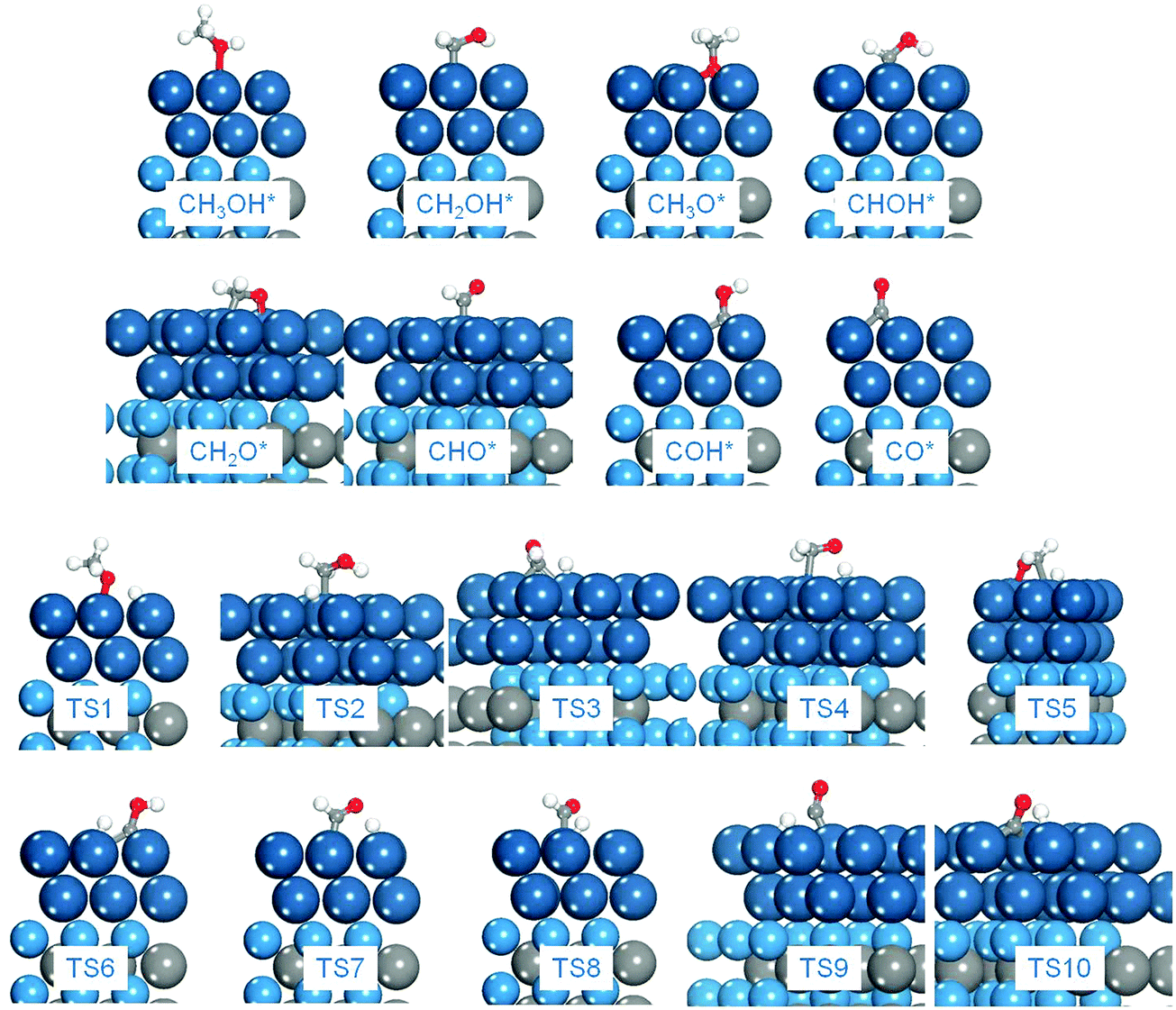
| | Fig. 4 Optimized structures of intermediates and transition states in methanol electro-oxidation on the Pt2ML,fcc/WC(0001) surface as an example. TS1: CH3OH* → CH3O* + H*; TS2: CH3OH* → CH2OH* + H*; TS3: CH2OH* → CHOH* + H*; TS4: CH2OH* → CH2O* + H*; TS5: CH3O* → CH2O* + H*; TS6: CHOH* → COH* + H*; TS7: CHOH* → CHO* + H*; TS8: CH2O* → CHO* + H*; TS9: CHO* → CO* + H*; TS10: COH* → CO* + H*, TS11: CO* + OH* → COOH*. Cyan: W; grey: C; blue: Pt; white: H; red: O. (The same colors were used throughout this work.) | |
On Pt1ML/WC(0001), methanol initially adsorbs on the surface via O–Pt bonding at the top site with an adsorption energy of −0.28 eV. Then, the adsorbed methanol CH3OH* prefers to break the O–H bond forming CH3O* with an activation energy of 0.92 eV; the O–H bond length is 1.50 Å at the transition state. On the other hand, C–H bond of CH3OH* is harder to be activated to form CH2OH* as a higher barrier of 1.01 eV is required with the C–H bond length of 1.64 Å at the transition state. The formation of both CH3O* and CH2OH* are endothermic by 0.22 eV and 0.29 eV, respectively. All in all, the initial dehydrogenation of CH3OH* via the O–H bond is favourable than via the C–H bond, both kinetically and thermodynamically. Therefore, the CH3O* adsorbed at the top site was identified to be a reactive intermediate for producing CH2O* with a low C–H bond dissociation barrier of 0.36 eV; the C–H bond length is 1.86 Å at the transition state. Once CH2O* is formed, it readily overcomes the 0.45 eV barrier to further dehydrogenate to CHO* with the C–H bond length being 1.53 Å at the transition state. The final dehydrogenation step of CHO* to CO* takes place with a barrier of 0.61 eV and the distance of the C–H bond being 1.41 Å at the transition state. Overall, the pathway for methanol dehydrogenation on Pt1ML/WC(0001) is as follows:
| CH3OH* → CH3O* → CH2O* → CHO* → CO*. |
For the dehydrogenation of methanol on Pt
2ML,fcc/WC(0001) and Pt
2ML,hcp/WC(0001), the reaction mechanisms are generally the same over the two surfaces, but are different from those over Pt
1ML/WC(0001). Herein, we take methanol dehydrogenation on Pt
2ML,fcc/WC(0001) as an example. In contrast to the initial activation of methanol
via the O–H bond on Pt
1ML/WC(0001), CH
3OH* is more easily dehydrogenated to CH
2OH*
via the C–H bond here with a reaction barrier of 0.68 eV instead of
via the O–H bond to CH
3O* for which the barrier would be much higher as 1.01 eV. At the transition state, the C–H bond length is 1.48 Å. The formation of CH
2OH* is exothermic by −0.44 eV, whilst the formation of CH
3O* is slightly endothermic by 0.08 eV. For the further dehydrogenation of CH
2OH* adsorbed at the top site, the C–H bond pathway was identified to be favoured with a reaction barrier of 0.67 eV and the C–H bond length is 1.43 Å at the transition state. The barrier is much lower than that of 0.98 eV
via the O–H bond pathway, where the length of the O–H bond is 1.90 Å at the transition state. The so formed CHOH* adsorbs at the bridge site and undergoes further dehydrogenation
via its O–H bond dissociation to yield CHO* with a lower barrier of 0.46 eV. If the C–H bond dissociation occurs to yield COH*, a higher barrier of 0.55 eV must be overcome; the formation of COH* is exothermic by −0.95 eV, whilst the formation of CHO* by −0.23 eV. The produced CHO* favours the top site and readily decomposes to CO* overcoming a small barrier of 0.29 eV with the C–H bond length of 1.39 Å at the transition state. On the other hand, if COH* is formed, it is considerably stable at the fcc site resulting in the difficulty in activating its O–H bond, which needs to overcome a barrier of 0.89 eV in forming CO* with the O–H bond length of 1.25 Å at the transition state. Therefore, the overall methanol dehydrogenation pathway on Pt
2ML,fcc/WC(0001) follows these steps:
| CH3OH* → CH2OH* → CHOH* → CHO* → CO*. |
Comparing the calculated data for methanol dehydrogenation over the Pt
2ML,hcp/WC(0001) and Pt
2ML,fcc/WC(0001) surfaces, the dehydrogenation barriers calculated on the hcp surface are only slightly higher than those on the fcc surface in the range 0.01–0.09 eV. For instance, the initial C–H bond dissociation barrier in CH
3OH* is 0.77 eV on Pt
2ML,hcp/WC(0001) and 0.68 eV on Pt
2ML,fcc/WC(0001); the O–H bond breaking barrier in CHOH* is 0.47 eV on Pt
2ML,hcp/WC(0001) and 0.46 eV on Pt
2ML,fcc/WC(0001). Therefore, the activity of methanol dehydrogenation on Pt
2ML,fcc/WC(0001) is only slightly higher than that on Pt
2ML,hcp/WC(0001).
It is worth pointing out that we have also calculated the reaction energies and barriers for the dehydrogenation of methanol on Pt(111) as a reference to compare with the calculated data on Pt-modified WC. On Pt(111), the reaction pathways are the same to those on bi-layer Pt-modified WC(0001) surfaces as CH3OH* → CH2OH* → CHOH* → CHO* → CO*. The initial activation of CH3OH* to form CH2OH* is endothermic by −0.21 eV with a reaction barrier of 0.72 eV. In the subsequent dehydrogenation processes, CH2OH* overcomes a barrier of 0.65 eV to produce CHOH* at the bridge site and then the so produced CHOH* continues to break the O–H bond with a barrier of 0.56 eV to yield CHO* rather than to break C–H bond with a barrier of 0.64 eV to yield COH*. The final dehydrogenation step of CHO* to CO* requires to overcome a small barrier of 0.31 eV to break the C–H bond of CHO* and it is exothermic by −0.73 eV.
3.3 Onset potential for surface oxidant formation and CO oxidation
It is well known that electrooxidation of surface CO* at high potentials requires the dissociation of water to form the active surface oxidant OH* to turn over CO* to CO2. Water dissociation involves the separation of a proton and an electron (H2O → OH* + H+ + e−). Therefore, the onset potential of OH* formation usually coincides with the onset potential of surface oxidation reaction.44,62 Herein, in order to estimate the onset potentials for the CO electrooxidation on pure Pt(111) and Pt-modified WC surfaces, we have calculated the OH* formation potentials on these surfaces. On Pt(111), the calculated OH* formation potential is 0.64 V (vs. SHE) at pH = 0. On Pt1ML/WC(0001), OH* forms at a very low onset potential of −0.26 V (vs. SHE), which implies that this surface is easily covered by OH*. On Pt2ML,fcc/WC(0001) and Pt2ML,hcp/WC(0001) surfaces, the calculated onset potentials for OH* formation are 0.67 V (vs. SHE) and 0.66 V (vs. SHE), respectively, which are very close to the potential (0.64 V) obtained on Pt(111), indicating that bi-layer Pt-modified WC surfaces have a similar ability to facilitate water dissociation as that of pure Pt(111). Table 2 lists the calculated onset potentials for OH* formation (UOH*, in V vs. SHE) on Pt1ML/WC(0001), Pt2ML,fcc/WC(0001), Pt2ML,hcp/WC(0001) and Pt(111) surfaces for comparison.
Table 2 The calculated one-set potentials for OH* formation (UOH*, in V vs. SHE), the barriers (Ea,1, in eV) of the rate-determining step in methanol dehydrogenation and the barriers (Ea,2, in eV) in CO oxidation on Pt1ML/WC(0001), Pt2ML,fcc/WC(0001), Pt2ML,hcp/WC(0001) and Pt(111) surfaces, respectively
| |
Pt1ML/WC |
Pt2ML,fcc/WC |
Pt2ML,hcp/WC |
Pt |
|
U
OH*
|
−0.26 |
0.67 |
0.66 |
0.64 |
|
E
a,1
|
0.92 |
0.68 |
0.77 |
0.72 |
|
E
a,2
|
0.49 |
0.58 |
0.59 |
0.4 |
For the surface oxidative reaction processes of CO* + OH*, CO* adsorbs at the fcc hollow site and OH* stays at the top site initially. At the transition state, CO* moves towards the top site to couple with OH* for producing COOH*, which is readily dehydrogenated to CO2 in the presence of OH*. The overall reaction barrier on Pt(111) is 0.43 eV, which is relatively low for the reaction to occur at room temperature. For mono- and bi-(fcc or hcp) layer Pt-modified WC surfaces, the calculated barriers are 0.49 eV on Pt1ML/WC(0001), 0.58 eV on Pt2ML,fcc/WC(0001) and 0.59 eV on Pt2ML,hcp/WC(0001); all of these are slightly higher than those on Pt(111) (0.40 eV) implying that CO* oxidation to CO2 still readily occurs in the presence of OH*.
We found that the surface CO* adsorption energy is −1.96 eV at the fcc site on Pt2ML,fcc/WC(0001) which is stronger than −1.76 eV obtained on Pt2ML,hcp/WC(0001), but a similar barrier is obtained on the fcc and hcp sites, i.e., 0.58 eV and 0.59 eV, respectively. According to a previous work, the geometry effect plays an important role in the total barriers.59 From the changes in the structures between the initial and transition states, it was evidenced that CO* diffused from the fcc to top sites for coupling but OH* still remained at the top sites. Therefore, we examined the CO* adsorption energy at the top sites and found that the adsorption energy is −1.61 eV on Pt2ML,fcc/WC(0001) and −1.43 eV on Pt2ML,hcp/WC(0001). The corresponding energy barriers for CO* diffusion over these two surfaces are 0.35 eV and 0.33 eV, respectively, which indicates that there is a similar CO* geometry effect on the two surfaces, the latter in turn results in similar CO* + OH* barriers on both surfaces. In addition, the reaction energy for the formation of COOH* on both surfaces is the same, −0.24 eV. According to the BEP relationship for bridging the kinetics and thermodynamics,58 two reactions with the same reaction energy usually encounter a similar kinetic barrier. Once COOH* forms, it can be readily transferred to CO2 in the presence of OH* via the proton transfer.67 Therefore, the CO adsorption energy hardly affects the CO oxidation rate on the Pt2ML/WC(0001) surface at higher potentials where the DMFC operates.
As listed in Table 2, compared with the barriers of 0.68 eV–0.92 eV in methanol dehydrogenation to produce CO*, the oxidative removal of CO* with the lower barriers of 0.4–0.59 eV should be much faster than the methanol dehydrogenation; therefore, in the overall methanol electrooxidation reaction, the rate-determining step should be the C–H bond activation of methanol but not CO oxidation. The calculated onset potentials (UOH*), reaction barriers (Ea,1) of the rate-determining step in methanol dehydrogenation and the barriers (Ea,2) in CO oxidation on Pt1ML/WC(0001), Pt2ML,fcc/WC(0001), Pt2ML,hcp/WC(0001) and Pt(111) surfaces are listed together in Table 2 for a clear comparison among the four surfaces.
3.4 CO oxidation at the interface
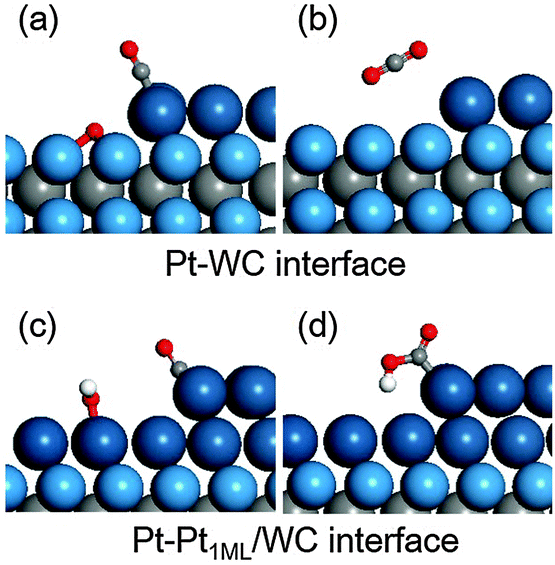
It is worth mentioning that, for understanding and screening improved Pt-based catalysts for methanol electrooxidation, the bi-functional mechanism is widely considered in a binary catalyst in which the surface Pt site catalyses the methanol dehydrogenation and the second element activates water to provide oxidants for surface CO* oxidation.62,65,66 Both W-terminated WC(0001) and Pt1ML/WC(0001) surfaces are easily oxidized and we therefore examined another possibility for the CO* oxidation at the interface between Pt and WC, in which WC(0001) or Pt1ML/WC(0001) provides surface oxidants (O*/OH* formed from water oxidation) and the edge of the Pt island adsorbs the CO* formed from methanol dehydrogenation. In this case, CO* oxidation occurs at the interface instead of the flat surface. For modelling the Pt–WC(0001) interface comprising the WC(0001) and Pt island, we expanded the Pt1ML/WC(0001) unit cell from p(3 × 3) to p(3 × 6) and removed half of the surface Pt atoms to create the interface. The same approach was used for building the Pt–Pt1ML/WC(0001) interface, and we found that the second Pt atom prefers to occupy the fcc sites at the interface. The two interface structures are shown in Fig. 5. At the Pt–WC(0001) interface, the bare W-terminated WC(0001) surface was identified to be occupied extremely easily by O* and the O binding energy is around −4.0 eV larger than that on pure Pt(111), indicating that once the clean WC(0001) is exposed to water or air, it would be oxidized immediately. The formation of CO2 from the CO*/Pt-edge and O*/WC(0001) is endothermic by 2.55 eV, mostly due to the overly strong adsorption of O*, which indicates that the adsorbed O* on WC(0001) is not reactive towards CO* oxidation. Therefore, this path is not energetically favourable. On the other hand, at the Pt–Pt1ML/WC(0001) interface, OH* is formed at a potential above −0.26 V (vs. SHE), also implying that the OH* binding energy is much stronger in comparison with that on Pt2ML/WC(0001) (where the onset potential for OH* formation is 0.66–0.67 V). Furthermore, CO* adsorption at the edge site is even stronger with an adsorption energy of −2.22 eV in comparison with −1.96 eV at the flat Pt2ML,fcc/WC(0001). Herein, CO* oxidation via the coupling of CO* and OH* is endothermic by 0.69 eV, indicating that CO* oxidation can hardly occur at this interface. The structures of the initial and final states at the two interfaces are shown in Fig. 5. Therefore, on the Pt modified WC catalyst, the bi-functional aspect for methanol electrooxidation is not feasible. We would suggest that the further modification on the Pt2ML/WC(0001) surface with an additional element like Ru or Sn may enable the bi-functional mechanism and further enhance the reactivity.
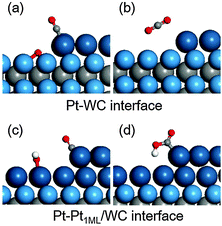 |
| | Fig. 5 Optimized structures for surface CO* oxidation at the Pt–WC(0001) and Pt–Pt1ML/WC(0001) interfaces: (a) CO*/Pt and O*/WC; (b) CO2 product; (c) CO*/Pt-edge and OH*/Pt1ML/WC; (d) COOH*/Pt-edge. | |
4. Discussion
4.1 Activity of the Pt-modified WC surfaces towards methanol oxidation
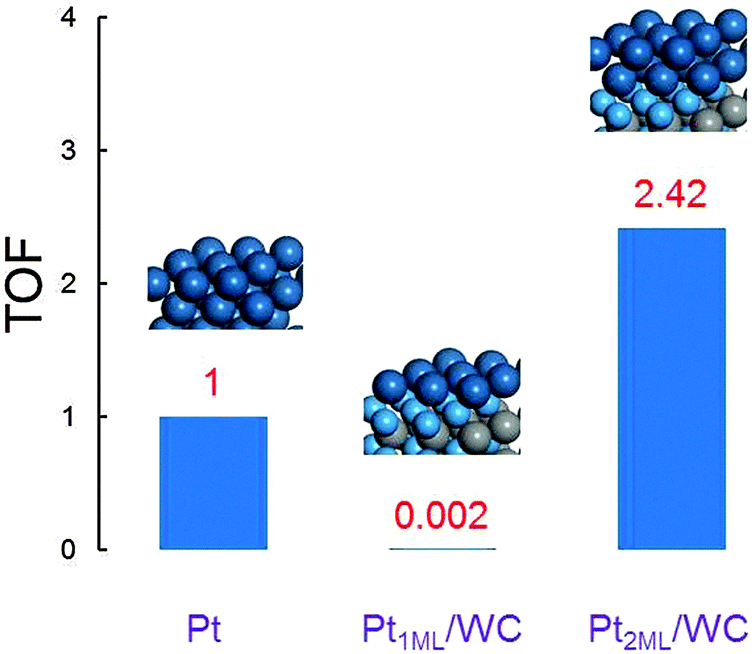
To understand the theoretical activity of these Pt-modified WC surfaces towards methanol oxidation under realistic conditions, we calculated the turnover frequency (TOF, in s−1) using the Arrhenius equation, TOF = kBT/hexp(−Ea/RT), to roughly estimate the catalyst/surface performance in the DMFC, where Ea is the activation energy for methanol oxidation to CO2, in which the initial activation barrier of methanol dehydrogenation is the rate determining step as illustrated above in the reaction pathways. Since the surface energies of Pt2ML,fcc/WC(0001) and Pt2ML,hcp/WC(0001) are the same, the positions of the second layer Pt atoms have the same possibility to occupy the fcc and hcp sites on Pt1ML/WC(0001). The TOF of methanol oxidation on bi- (fcc or hcp) layer Pt-modified WC(0001) is therefore calculated using the equation TOF2ML = ½TOF2ML,fcc + ½TOF2ML,hcp, considering the same contributions from the fcc and hcp surfaces to the total activity. The theoretical activity on mono- and bi-layer Pt-modified WC(0001) surfaces and Pt(111) are presented in Fig. 6. It can be seen from the figure that the activity on Pt1ML/WC(0001) is extremely low, having only 0.2% of the Pt(111) activity. It could be understood based on the fact that, due to the significantly strong chemical interactions between the Pt atoms and WC(0001) on Pt1ML/WC(0001), inferred from the lowest surface energy, the Pt atoms here are too stable to be active for methanol dehydrogenation. However, on bi- (fcc or hcp) layer Pt-modified WC(0001) surfaces, the activity is increased significantly to about 2.42 times that of Pt(111). The latter data suggest that the bi-layer Pt-modified WC materials could provide a better performance in the electrooxidation of methanol than pure Pt. These results are in fact supported by the recent experimental data which show that the onset potential is hardly changed, but the activity is increased significantly for the Pt/WC catalysts compared to that of pure Pt catalysts.26,32
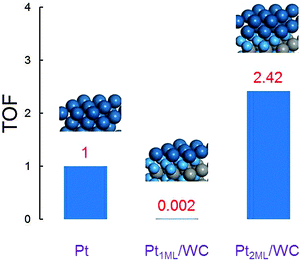 |
| | Fig. 6 The calculated normalized turnover frequency TOF in the electrooxidation of methanol on Pt(111), Pt1ML/WC(0001) and Pt2ML/WC(0001) surfaces, respectively. | |
4.2 The effect of potential
In methanol electrocatalysis, there are two elementary reactions involved in the first dehydrogenation process: (i) CH3OH* → CH2OH* + H* and (ii) H* → H+ + e−.68–70 It can be seen that the surface H* electrooxidative stripping in (ii) enables the removal of H* to refresh the surface with the current created. Liu's group has reported that the first reaction (i) is hardly affected by the potential, but the second step (ii) with the proton and electron transfer is indeed sensitive to the potential.68–70 The electrooxidative removal of H* in the electrolyte at potentials where the DMFC operates (e.g. above 0.20 V vs. SHE) is relatively quicker and the rate-determining step is (i), supported by the evidence observed in the isotope experiments.71 Therefore, we only compare the dehydrogenation barriers in (i) between the Pt modified WC and pure Pt for the methanol dehydrogenation.
4.3 Water effect
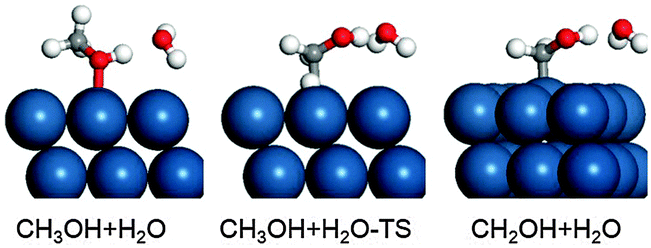
Since the methanol surface reactions occur in aqueous solution, we further investigated the effect of water on the initial dehydrogenation of methanol, which is the rate-determining step. The simulation of water structures is still a huge challenge in electrocatalysis, the main reason being that the water structure is always fluctuating at the dynamics scale. Usually, the choice of a statistic water structure is a good approximation for generally understanding the role of water.60,72–74 Here we compare the adsorption energy and dehydrogenation barrier through the [CH3OH⋯H2O] complex structure. All the calculated data, including the adsorption energy, the bond distance and the dehydrogenation barrier, are listed in Table 3. The complex is trapped on the Pt2ML,fcc/WC(0001) surface at the top site with an adsorption energy of −0.82 eV, and the optimized structure with a H-down configuration of H2O is shown in Fig. 7. It has been found that the O–Pt bond is shortened, from 2.46 Å without H2O to 2.29 Å due to the water effect, indicating that water increases the interaction between methanol and the surface Pt site. On Pt(111), this phenomenon is also observed where the O–Pt bond, which is originally 2.38 Å in the absence of water, decreases to 2.28 Å in the presence of H2O, yielding a significant adsorption energy of −0.75 eV. Looking at all the three surfaces studied, the adsorption energy of the [CH3OH⋯H2O] complex has increased by 0.49–0.52 eV in comparison with CH3OH adsorption in the absence of water, indicating that the water effect on the adsorption of methanol on the three surfaces studied is very similar.
Table 3 The calculated adsorption energy (Eads, in eV), the distance of the O–Pt bond (in Å), the reaction barrier (Ea, in eV) and the distance of the C–H bond at the transition state of the [CH3OH⋯H2O] complex on Pt2ML,fcc/WC(0001), Pt2ML,hcp/WC(0001) and Pt(111) surfaces, respectively. The values in parentheses are the data obtained without the H2O effect
| |
Pt2ML,fcc/WC |
Pt2ML,hcp/WC |
Pt |
|
E
ads
|
−0.82 (−0.3) |
−0.76 (−0.27) |
−0.75 (−0.24) |
|
d(O–Pt) |
2.29 (2.46) |
2.30 (2.48) |
2.28 (2.38) |
|
E
a
|
0.72 (0.68) |
0.79 (0.77) |
0.75 (0.72) |
|
d(C–H) |
1.44 (1.47) |
1.46 (1.49) |
1.45 (1.47) |
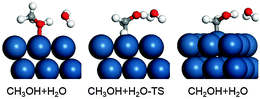 |
| | Fig. 7 Optimized structures in the dehydrogenation of the [CH3OH⋯H2O] complex. | |
In terms of the dehydrogenation barrier of [CH3OH⋯H2O]* to [CH2OH⋯H2O]*, on Pt2ML,fcc/WC(0001), the barrier increased from 0.68 eV to 0.72 eV and the C–H bond distance at the transition state elongated from 1.44 Å to 1.47 Å due to the presence of water; on Pt2ML,hcp/WC(0001), the barrier also increased slightly from 0.77 eV to 0.79 eV; on Pt(111), the barrier increased from 0.72 eV to 0.75 eV. From the data listed in Table 3, it can be seen clearly that the barrier only increased by around 0.02–0.04 eV, showing that water only slightly slows down the C–H bond dissociation rate. Because the C–H bond is relatively hydrophobic and the water which forms hydrogen bonds with the OH group hardly affects the C–H bond dissociation barriers, the slight influence of water is thought not to be unreasonable. More importantly, it provides evidence that water could equally affect the adsorption and kinetic barrier for the methanol electrooxidation on Pt(111) and Pt/WC(0001) surfaces. All in all, our data suggest that the role of water is indeed significant in determining the absolute adsorption and kinetic values in electrocatalysis, but it does not significantly affect the assessment of the general trend in the inherent activity of catalysts.
5. Conclusions
In this work, the electrooxidation of methanol in a DMFC has been investigated on a series of Pt-modified WC(0001) model surfaces. Firstly, through the calculation of surface energies, we found that the most stable Pt coverage is the mono- or bi- (fcc or hcp) layer but not the tri-layer Pt-modified WC(0001) which is considerably unstable thermodynamically. Then, on the three possible surfaces of Pt1ML/WC(0001), Pt2ML,fcc/WC(0001) and Pt2ML,hcp/WC(0001), we calculated the reaction network in the methanol dehydrogenation involving the intermediates CH3OH*, CH2OH*, CHOH*, COH*, CH3O*, CH2O*; CHO* and CO* via the four C–H and one O–H bond dissociations subsequently. It has been found that the favourable methanol dehydrogenation paths on Pt1ML/WC(0001) are CH3OH* → CH3O* → CH2O* → CHO* → CO* but those on Pt2ML/WC(0001) and Pt(111) are CH3OH* → CH2OH* → CHOH* → CHO* → CO*. The initial activation of methanol was found to be the rate-determining step. Subsequently CO* can be readily oxidized to CO2 in the presence of surface OH* at high potentials where the DMFC operates. The on-set potentials for the surface OH* formation through water dissociation were calculated for the Pt-modified WC surfaces and it has been found that bi- (fcc or hcp) layer Pt-modified WC catalysts have similar onset potentials compared with that on pure Pt(111), but exhibit up to 2.4 times higher reactivity compared to that of pure Pt. Our theoretical data were supported by recent experimental results and suggests that bi-layer Pt-modified WC catalysts could be a promising low cost high performance electrocatalysts for DMFC application with the benefit of reducing Pt usage.
Acknowledgements
Financial support from the EPSRC (EP/I013229/1), as part of the RCUK Energy Programme, the NSFC (21361140374 and 21321062) and International Science & Technology Cooperation Programme of China (2010DFB63680) is acknowledged. X.L. and C.M. thank QUB for the awards of the titles of Visiting Research Associate and Visiting Professor, respectively.
References
- M. Baldauf and W. Preidel, J. Power Sources, 1999, 84, 161 CrossRef CAS.
- C. Lamy, A. Lima, V. LeRhun, F. Delime, C. Coutanceau and J. M. Leger, J. Power Sources, 2002, 105, 283 CrossRef CAS.
- Z. G. Shao, W. F. Lin, P. A. Christensen and H. M. Zhang, Int. J. Hydrogen Energy, 2006, 31, 1914 CrossRef CAS PubMed.
- Z. G. Shao, F. Y. Zhu, W. F. Lin, P. A. Christensen and H. M. Zhang, J. Power Sources, 2006, 161, 813 CrossRef CAS PubMed.
- U. B. Demirci, J. Power Sources, 2007, 169, 239 CrossRef CAS PubMed.
- Z. G. Shao, F. Y. Zhu, W. F. Lin, P. A. Christensen, H. M. Zhang and B. L. Yi, J. Electrochem. Soc., 2006, 153, A1575 CrossRef CAS PubMed.
- Z. G. Shao, W. F. Lin, F. Y. Zhu, P. A. Christensen, H. M. Zhang and B. L. Yi, J. Power Sources, 2006, 160, 1003 CrossRef CAS PubMed.
- Z. G. Shao, W. F. Lin, F. Y. Zhu, P. A. Christensen and H. M. Zhang, Phys. Chem. Chem. Phys., 2006, 8, 2720 RSC.
- H. X. Liu, N. Tian, M. P. Brandon, J. Pei, Z. Huangfu, C. Zhan, Z. Y. Zhou, C. Hardacre, W. F. Lin and S. G. Sun, Phys. Chem. Chem. Phys., 2012, 14, 16415 RSC.
- W. F. Lin, P. A. Christensen and A. Hamnett, Phys. Chem. Chem. Phys., 2001, 3, 3312 RSC.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS.
- S. A. Grigoriev, P. Millet and V. N. Fateev, J. Power Sources, 2008, 177, 281 CrossRef CAS PubMed.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547 CAS.
- D. V. Esposito and J. G. Chen, Energy Environ. Sci., 2011, 4, 3900 CAS.
- H. H. Hwu and J. G. Chen, Chem. Rev., 2005, 105, 185 CrossRef CAS PubMed.
- M. B. Zellner and J. G. Chen, Surf. Sci., 2004, 569, 89 CrossRef CAS PubMed.
- D. V. Esposito, S. T. Hunt, A. L. Stottlemyer, K. D. Dobson, B. E. McCandless, R. W. Birkmire and J. G. Chen, Angew. Chem., Int. Ed., 2010, 49, 9859 CrossRef CAS PubMed.
- D. V. Esposito, S. T. Hunt, Y. C. Kimmel and J. G. Chen, J. Am. Chem. Soc., 2012, 134, 3025 CrossRef CAS PubMed.
- C. Ma, J. Sheng, N. Brandon, C. Zhang and G. Li, Int. J. Hydrogen Energy, 2007, 32, 2824 CrossRef CAS PubMed.
- M. Wu, P. K. Shen, Z. D. Wei, S. Q. Song and M. Nie, J. Power Sources, 2007, 166, 310 CrossRef CAS PubMed.
- D. J. Ham, R. Ganesan and J. S. Lee, Int. J. Hydrogen Energy, 2008, 33, 6865 CrossRef CAS PubMed.
- Y. Hara, N. Minami, H. Matsumoto and H. Itagaki, Appl. Catal., A, 2007, 332, 289 CrossRef CAS PubMed.
- M. K. Jeon, K. R. Lee, W. S. Lee, H. Daimon, A. Nakahara and S. I. Woo, J. Power Sources, 2008, 185, 927 CrossRef CAS PubMed.
- D. J. Ham, Y. K. Kim, S. H. Han and J. S. Lee, Catal. Today, 2008, 132, 117 CrossRef CAS PubMed.
- M. Nie, P. K. Shen, M. Wu, Z. Wei and H. Meng, J. Power Sources, 2006, 162, 173 CrossRef CAS PubMed.
- R. Wang, Y. Xie, K. Shi, J. Wang, C. Tian, P. Shen and H. Fu, Chem. – Eur. J., 2012, 18, 7443 CrossRef CAS PubMed.
- M. D. Obradovic, B. M. Babic, V. R. Radmilovic, N. V. Krstajic and S. L. Gojkovic, Int. J. Hydrogen Energy, 2012, 37, 10671 CrossRef CAS PubMed.
- Z. J. Mellinger, T. G. Kelly and J. G. Chen, ACS Catal., 2012, 2, 751 CrossRef CAS.
- C. K. Poh, S. H. Lim, Z. Tian, L. Lai, Y. P. Feng, Z. Shen and J. Lin, Nano Energy, 2013, 2, 28 CrossRef CAS PubMed.
- H. Meng and P. K. Shen, Chem. Commun., 2005, 4408 RSC.
- C. Ma, W. Liu, M. Shi, X. Lang, Y. Chu, Z. Chen, D. Zhao, W. Lin and C. Hardacre, Electrochim. Acta, 2013, 114, 133 CrossRef CAS PubMed.
- Z. Y. Chen, C. A. Ma, Y. Q. Chu, J. M. Jin, X. Lin, C. Hardacre and W. F. Lin, Chem. Commun., 2013, 49, 11677 RSC.
- S. G. Sun and J. Clavilier, J. Electroanal. Chem., 1987, 236, 95 CrossRef CAS.
- E. Herrero, K. Franaszczuk and A. Wieckowski, J. Phys. Chem., 1994, 98, 5074 CrossRef CAS.
- B. Braunchweig, D. Hibbitts, M. Neurock and A. Wieckowski, Catal. Today, 2013, 202, 197 CrossRef CAS PubMed.
- C. J. Zhang and P. Hu, J. Chem. Phys., 2001, 115, 7182 CrossRef CAS PubMed.
- S. K. Desai, M. Neurock and K. Kourtakis, J. Phys. Chem. B, 2002, 106, 2559 CrossRef CAS.
- J. Greeley and M. Mavrikakis, J. Am. Chem. Soc., 2002, 124, 7193 CrossRef CAS PubMed.
- J. Greeley and M. Mavrikakis, J. Am. Chem. Soc., 2004, 126, 3910 CrossRef CAS PubMed.
- P. Ferrin and M. Mavrikakis, J. Am. Chem. Soc., 2009, 131, 14381 CrossRef CAS PubMed.
- D. Cao, G. Q. Lu, A. Wieckowski, S. A. Wasileski and M. Neurock, J. Phys. Chem. B, 2005, 109, 11622 CrossRef CAS PubMed.
- D. W. Yuan, X. G. Gong and R. Q. Wu, J. Chem. Phys., 2008, 128, 64706 CrossRef PubMed.
- J. Ye, C. Liu and Q. Ge, Phys. Chem. Chem. Phys., 2012, 14, 16660 RSC.
- B. Y. Liu, J. M. Jin, X. Lin, C. Hardacre, P. Hu, C. A. Ma and W. F. Lin, Catal. Today, 2015, 242, 230 CrossRef CAS PubMed.
- Z. C. Kramer, X. K. Gu, D. Y. Zhou, W. X. Li and R. T. Skodje, J. Phys. Chem. C, 2014, 118, 12364 CAS.
- R. Jiang, W. Guo, M. Li, D. Fu and H. Shan, J. Phys. Chem. C, 2009, 113, 4188 CAS.
- A. Morgan, R. Kavanagh, W. F. Lin, C. Hardacre and P. Hu, Phys. Chem. Chem. Phys., 2013, 15, 20170 RSC.
- A. L. Stottlemyer, P. Liu and J. G. Chen, J. Chem. Phys., 2010, 133, 104702 CrossRef PubMed.
- C. Ma, T. Liu and L. Chen, Appl. Surf. Sci., 2010, 256, 7400 CrossRef CAS PubMed.
- D. D. Vasic, I. A. Pasti and S. V. Mentus, Int. J. Hydrogen Energy, 2013, 38, 5009 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115 CrossRef CAS.
- G. Kresse and J. Furthmuler, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 CrossRef CAS.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
-
(a) G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS;
(b) A. Alavi, P. Hu, T. Deutsch, P. L. Silvestrelli and J. Hutter, Phys. Rev. Lett., 1998, 80, 3650 CrossRef CAS.
-
(a) J. P. Pedrew and K. Burke, Phys. Rev. Lett., 1996, 77, 3865 CrossRef;
(b) A. Michadelides, Z. P. Liu, C. J. Zhang, A. Alavi, D. A. King and P. Hu, J. Am. Chem. Soc., 2003, 125, 3704 CrossRef PubMed.
-
(a) J. P. Pedrew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 Search PubMed;
(b) Z. P. Liu and P. Hu, J. Am. Chem. Soc., 2003, 125, 1958 CrossRef CAS PubMed.
- J. Rossmeisl, J. K. Norskov, C. D. Taylor, M. J. Janik and M. Neurock, J. Phys. Chem. B, 2006, 110, 21833 CrossRef CAS PubMed.
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 1520 CrossRef.
- J. M. Jin, T. Sheng, X. Lin, R. Kavanagh, P. Hamer, P. Hu, C. Hardacre, A. Martinez-Bonastre, J. Sharman, D. Thompsett and W. F. Lin, Phys. Chem. Chem. Phys., 2014, 16, 9432 RSC.
- T. Sheng, W. F. Lin, C. Hardacre and P. Hu, Phys. Chem. Chem. Phys., 2014, 16, 13248 RSC.
- J. C. Boettger, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 16, 798 Search PubMed.
- P. Ferrin, A. U. Nilekar, J. Greeley, M. Mavrikakis and J. Rossimeisl, Surf. Sci., 2008, 602, 3424 CrossRef CAS PubMed.
- J. Rossimeisl, P. Ferrin, G. A. Tritsaris, A. U. Nilekar, S. Koh, S. E. Bae, S. R. Brankovic, P. Strasser and M. Mavrikakis, Energy Environ. Sci., 2012, 5, 8335 Search PubMed.
- X. Q. Gong, P. Hu and R. Raval, J. Chem. Phys., 2003, 119, 6324 CrossRef CAS PubMed.
- Y. H. Fang and Z. P. Liu, J. Am. Chem. Soc., 2010, 132, 18214 CrossRef CAS PubMed.
- Y. H. Fang and Z. P. Liu, Surf. Sci., 2015, 631, 42 CrossRef CAS PubMed.
- Y. H. Fang, G. F. Wei and Z. P. Liu, J. Phys. Chem. C, 2013, 117, 669 Search PubMed.
- J. Ren, Y. Y. Yang, B. W. Zhang, N. Tian, W. B. Cai, Z. Y. Zhou and S. G. Sun, Electrochem. Commun., 2013, 37, 49 CrossRef CAS PubMed.
- Y. H. Fang, G. F. Wei and Z. P. Liu, Catal. Today, 2013, 202, 98 CrossRef CAS PubMed.
- X. Sun, X. Cao and P. Hu, Sci. China: Chem., 2015, 58, 55 Search PubMed.
- T. Sheng, W. F. Lin, C. Hardacre and P. Hu, J. Phys. Chem. B, 2014, 118, 5762 CAS.
Footnote |
| † Current address: Peterhouse, Trumpington Street, University of Cambridge, Cambridge CB2 1RD, UK. |
|
| This journal is © the Owner Societies 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a