
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmission spectroscopy of a ruthenium(II) polypyridyl complex adsorbed on calcium niobate lamellar solids and nanosheets†
Kazuhiko
Maeda
*,
Takayoshi
Oshima
and
Osamu
Ishitani
Department of Chemistry, Graduate School of Science and Engineering, Tokyo Institute of Technology, 2-12-1-NE-2 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: maedak@chem.titech.ac.jp
First published on 15th June 2015
Abstract
Ru(II) tris-diimine complexes are known to exhibit emission at around 630 nm as a result of 1MLCT photoexcitation. The emission is quenched in the presence of a suitable semiconductor solid due to electron injection from the excited state of a Ru(II) complex to the conduction band of the adjacent semiconductor. Here we investigated emission quenching behaviour of RuII{(4,4′-(CH3)2-bpy)2(4,4′-(CH2PO3H2)2-bpy)} (bpy = 2,2′-bipryridine) adsorbed on HCa2Nb3O10 solids having an ordered lamellar structure or a disordered nanostructure. Even though electron injection from the excited state of the Ru complex to the conduction band of nanostructured HCa2Nb3O10 is thermodynamically less favorable than that of layered HCa2Nb3O10, faster electron injection was observed using nanostructured HCa2Nb3O10. Experimental results highlighted that electron injection from the excited Ru complex takes place not only in the conduction band of HCa2Nb3O10 but also mid-gap states whose density is strongly dependent on both the morphological feature and the preparation method of HCa2Nb3O10.
Introduction
Electron injection from the excited state of dyes or metal complexes into a semiconductor metal oxide is fundamentally important as the primary process of dye-sensitized solar cells and photocatalytic hydrogen evolution.1–13 Electron injection efficiency is known to depend on the conduction band potential (ECB) of a metal oxide. For example, efficient electron injection from the excited state of N3 dye into TiO2 was observed, while that into ZrO2 having much more negative ECB was very slow.4 Maitani et al. recently observed different charge injection behaviours of the excited state of anthracene dyes on TiO2 nanocrystals, which originated from different facets of TiO2 with different energies.14We have studied metal oxide nanosheets as building blocks for dye-sensitized H2 evolution in combination with ruthenium(II) polypyridyl complexes as redox photosensitizers.10–13 Ruthenium(II) tris-diimine complexes exhibit emission at around 630 nm as a result of 1MLCT photoexcitation, and the efficiency of electron injection can be assessed by monitoring emission behaviour of the adsorbed Ru complex.11–13 According to our recent study on time-resolved emission spectroscopy, it was suggested that electron injection from the excited state of RuII{(4,4′-(CH3)2-bpy)2(4,4′-(CH2PO3H2)2-bpy)}, abbreviated as Ru for simplicity, occurs not only in the conduction band of HCa2Nb3O10 nanosheets but also mid-gap states.14 If this is the case, the charge injection process should depend on the structural feature of HCa2Nb3O10. It is fundamentally interesting to investigate emission behaviour of a ruthenium(II) polypyridyl complex on a semiconductor solid having different morphological features. While there have been several reports on emission spectroscopy of metal-complex/semiconductor hybrids focusing on different metal complexes and semiconductors,14–17 such structural effects of a metal oxide on the emission behaviour have not been investigated so far.
In this work, we investigated structural effects of HCa2Nb3O10 on the electron injection from the excited state of Ru by means of steady-state emission spectroscopy and time-resolved emission spectroscopy. Two types of HCa2Nb3O10 having distinct morphological features were synthesized: one is a submicron-order layered crystal and the other one is aggregated nanosheets with highly disordered structures, which was prepared by chemical exfoliation of the corresponding lamellar solid. In addition, we applied two different synthetic methods to prepare HCa2Nb3O10 having different physicochemical characteristics. The details of the materials preparation are included in the experimental section. Briefly, layered HCa2Nb3O10 was obtained by ion-exchange reaction of KCa2Nb3O10, which was prepared by a conventional solid-state reaction (SSR) or the polymerized complex (PC) method, with HNO3. Tetra(n-butyl)ammonium hydroxide (TBAOH), a bulky base molecule, was used to exfoliate layered HCa2Nb3O10 into unilamellar colloidal nanosheets. The TBA+-stabilized Ca2Nb3O10− nanosheets were flocculated by adding HCl, followed by washing with H2O and drying at 343 K overnight.
Results and discussion
Fig. 1A shows typical SEM images of layered HCa2Nb3O10 and the aggregated nanosheets prepared by the PC method. After exfoliation of lamellar HCa2Nb3O10 and subsequent restacking by HCl, the original plate-like layered structure was completely destroyed, giving aggregated solids with a disordered structure. This result is also supported by XRD (Fig. 1B), which indicated that long-range ordering in the stacking direction (reflections from (00n)) of the original layered material disappeared after the exfoliation-restacking process. Accompanied by this structural change, the specific surface area determined by nitrogen adsorption at 77 K was increased from 2.4 to 49 m2 g−1.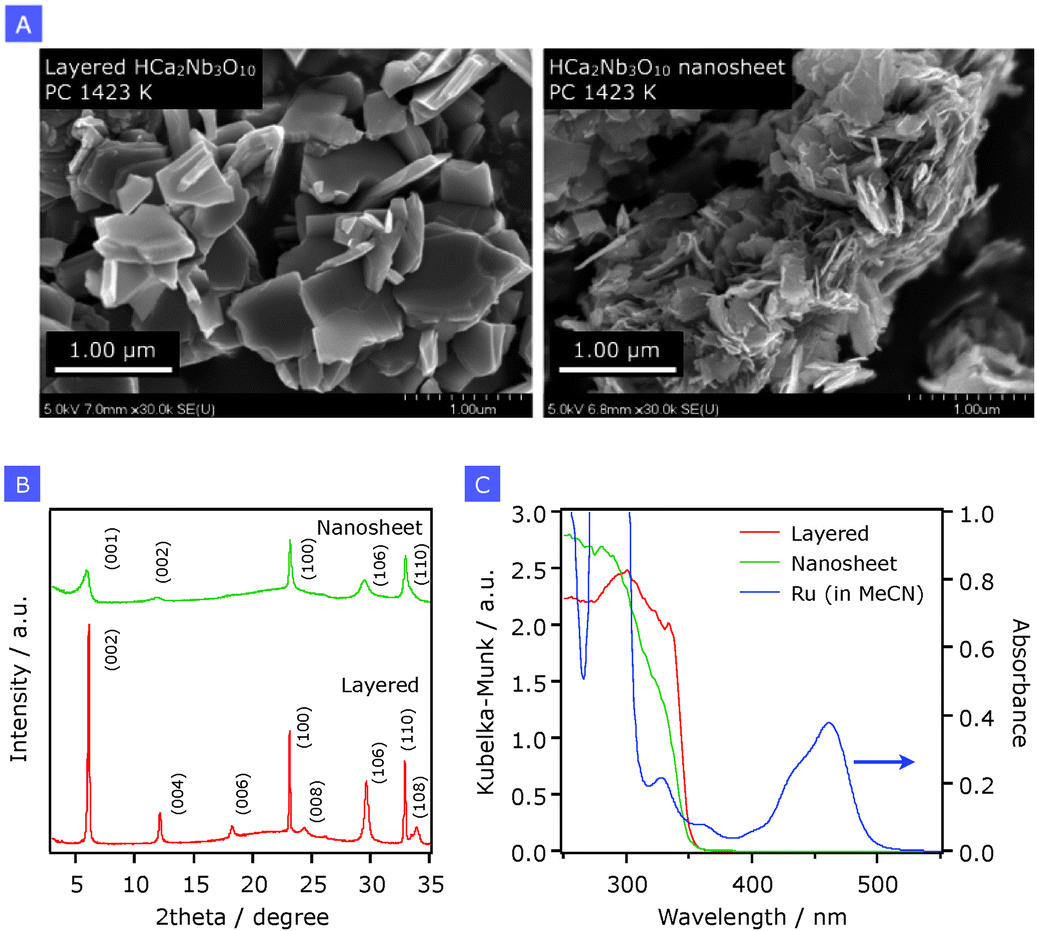 | ||
| Fig. 1 (A) SEM images, (B) X-ray diffraction patterns, and (C) UV-visible diffuse reflectance spectra of layered HCa2Nb3O10 and HCa2Nb3O10 nanosheets prepared by the PC method calcined at 1423 K. In panel (C), an absorption spectrum of Ru (24 μM) in acetonitrile (MeCN) is shown for comparison. | ||
Fig. 1C shows UV-visible diffuse reflectance spectra of the same samples, along with an absorption spectrum of a Ru solution. Layered HCa2Nb3O10 shows a steep absorption edge at around 350 nm, which is due to electron transition from the valence band formed by oxygen 2p orbitals to the conduction band that consists of empty orbitals of niobium 4d. On the other hand, there are at least two absorption edges in the aggregated HCa2Nb3O10 nanosheets, both of which are blue-shifted compared to that in the parent layered solid. This is most likely due to a quantum-confinement effect.18 As discussed in our previous paper, the generation of two absorption edges after the exfoliation–restacking process would result from an increase in the distortion of the two-dimensional nanosheet structure that consists of triple perovskite slabs having a nanosized thickness. This more pronounced distortion might alter the local structure of NbO6 octahedra in the perovskite block, although the long-range ordering in the perovskite block (shown in XRD) appears to be maintained. Although the precise determination of the band-edge positions of the aggregated HCa2Nb3O10 nanosheets appears to be difficult, one may think that the valence band maximum and the conduction band minimum of HCa2Nb3O10 are shifted downward and upward, respectively, thereby leading to band-gap widening, as the result of nanostructuring.19
Using the two structurally different materials but with almost the same composition, the adsorption of Ru was performed by dispersing the solid materials in an aqueous solution containing Ru (2.0 μmol g−1). UV-visible spectroscopy showed that the dissolved Ru complex was quantitatively adsorbed on the surface of both materials. Note that, however, intercalation of Ru into the gallery space of these solids does not occur, as revealed by our previous work.11,13 As shown in Fig. S1 (ESI†), modification of HCa2Nb3O10 material with Ru resulted in the generation of a new absorption band centred at around 460 nm, which is due to MLCT transition of Ru, identical to Ru in MeCN solution (Fig. 1C).
As reported previously,13Ru on Al2O3 gives an emission peak at around 630 nm as a result of 1MLCT photoexcitation of Ru at 444 nm, with a quantum yield of ca. 5.6% (Fig. 2). However, the emission was almost completely quenched when Ru was loaded on HCa2Nb3O10 regardless of the structural feature. This indicates the occurrence of electron injection from the excited state of Ru into HCa2Nb3O10. Note that these experiments assessed the electron injection process not only from the 3MLCT excited-state but also from 1MLCT, which is known to be an ultrafast process that occurs within a timescale of several hundreds of fs.2,3
 | ||
| Fig. 2 Steady-state emission spectra of Ru on layered HCa2Nb3O10 and HCa2Nb3O10 nanosheets prepared by the PC method calcined at 1423 K, along with that of Ru/Al2O3 for reference. The spectra were obtained by 444 nm photoexcitation. Note that reductive quenching of the 3MLCT excited state of Ru by EDTA does not occur. | ||
The single-photon counting method was employed for measuring the decay of emission which was monitored at 630 nm after selective excitation of Ru at 444 nm. Fig. 3 shows decay curves of emission from Ru adsorbed on layered HCa2Nb3O10 and the nanosheets prepared by the PC method. As a reference, data on Ru/Al2O3 are also shown. Note that due to the time resolution of our apparatus (>200 ps), the observed emission decay is attributed to emission from the lowest 3MLCT excited state.20 Electron injection from the excited state of Ru into Al2O3 does not proceed because of the insulating nature of Al2O3. It is clear that the emission decay of Ru was more pronounced on the two HCa2Nb3O10 materials than on Al2O3, indicating the occurrence of electron injection from the excited state of Ru into the HCa2Nb3O10 materials, consistent with the result of steady-state emission spectroscopy (Fig. 2). However, the emission decay of the layered material was relatively slow, whereas the nanosheet material showed a faster decay profile. It should be stressed that this tendency was independent of the preparation condition and method of HCa2Nb3O10; that is, a faster decay profile was observed in the nanosheet system (Fig. S2, ESI†). The faster emission decay of the nanosheet material indicates the occurrence of faster electron injection. The faster electron injection of the nanosheet system compared to the layered one may seem unreasonable because one can expect an lowered driving force for electron injection due to the enlarged difference between the oxidation potential of the excited state (Eox*) and ECB upon exfoliation-restacking. Therefore, there is another pathway of electron injection from the excited state of Ru into HCa2Nb3O10. One plausible explanation is that part of electrons from the excited-state of Ru are injected not only into the conduction band of the aggregated HCa2Nb3O10 nanosheets but also into mid-gap states in the material.2,3 It is known that in an n-type semiconductor including the present niobates, there are mid-gap states located below the conduction band.3 Here the highly disordered morphological feature of the aggregated HCa2Nb3O10 nanosheets could contain more defects that give localized states working as electron-accepting levels, compared to the ordered lamellar structured one. This situation would contribute to more efficient electron injection from the excited-state of Ru into the aggregated HCa2Nb3O10 nanosheets.
 | ||
| Fig. 3 Emission decay curves of Ru on layered HCa2Nb3O10 and HCa2Nb3O10 nanosheets prepared by the PC method calcined at 1423 K. Excited at 444 nm and monitored at 630 nm, along with that of Ru/Al2O3 for reference. For each sample, the measurement was repeated until the signal count just after photoexcitation (at t = 0) reached 104. | ||
We also tried to resolve the decay profiles of the same samples. However, it was very difficult especially for the nanosheet material even using four exponential functions, suggesting that Ru on the nanosheet material had different adsorption forms such as protonation/deprotonation of the phosphonate groups in the ligand.
If the idea (electron injection into mid-gap states) is correct, HCa2Nb3O10 having more structural imperfections will accelerate electron injection from the excited state of Ru more efficiently, resulting in a more pronounced emission decay profile. In order to investigate this, we prepared lamellar HCa2Nb3O10 and the aggregated nanosheets at lower calcination temperature (1023 K). By lowering calcination temperature in the final step of the PC method, one can prepare less-crystallized lamellar HCa2Nb3O10 and the nanosheet with smaller lateral dimensions, as revealed by our previous work.12 Here XRD patterns and SEM images of the as-prepared lamellar HCa2Nb3O10 solids are given in the ESI† (Fig. S3 and S4).
Fig. 4 compares the emission decay profiles of Ru on two different HCa2Nb3O10 nanosheets prepared at 1023 and 1423 K. As expected, lowering the calcination temperature of the PC method from 1423 to 1023 K accelerated the emission decay. This result further supports our claim that Ru in the excited state injects an electron not only into the conduction band of HCa2Nb3O10 but also into mid-gap states.
 | ||
| Fig. 4 Emission decay curves of Ru on HCa2Nb3O10 nanosheets prepared by different methods. Excited at 444 nm and monitored at 630 nm, along with that of Ru/Al2O3 for reference. Each measurement was conducted by irradiating the sample cell with a fixed number of photons. | ||
The behaviour of emission decay of Ru on the aggregated HCa2Nb3O10 nanosheets was further investigated with respect to the preparation method of the nanosheet.21Fig. 4 also compares emission decays of Ru adsorbed on two different HCa2Nb3O10 nanosheets, which were prepared by SSR and PC methods at the same calcination temperature (1423 K). Interestingly, a faster decay curve was observed in the sample prepared by the SSR method. This strongly suggests that the SSR-derived material contains more mid-gap states than the PC material. However, this may seem to contradict the results of structural characterization; i.e., the intensity of diffraction peaks of the SSR sample is much stronger than that of the PC sample, indicating that more pronounced crystallization occurred in the former (Fig. S3 and S4, ESI†). Nevertheless, it is known that the PC method allows one to obtain metal oxides with lower density of defects and/or vacancies even at lower temperatures, compared to a conventional SSR method.22,23 Accordingly, we believe that a slower emission decay profile recorded in the PC sample is reasonable.
Conclusions
In summary, even though the conduction band potential of aggregated HCa2Nb3O10 nanosheets is slightly more negative than that of lamellar HCa2Nb3O10, electron injection from the excited state of Ru is faster in the former than in the latter. Mid-gap states in the HCa2Nb3O10 nanosheets are suggested to accept electrons from the excited state of Ru, thereby facilitating the emission decay of Ru. Although it was believed that the ECB of a semiconductor primarily determines the driving force for electron injection from the excited state of a redox photosensitizer, this is the first experimental result that strongly suggests significant contribution of mid-gap states existing in a semiconductor to the charge injection process. Mid-gap states that originate from defects and/or vacancies in a semiconductor solid are known to have a strong impact on photocatalytic activity of the semiconductor.24,25 Therefore, the present result may be useful as a probe to assess the density of defects and/or vacancies. Our research is now under way along this line.Experimental section
Preparation of layered HCa2Nb3O10
First, KCa2Nb3O10 was synthesized by the polymerized complex method according to our previous papers.12,13 The final calcination temperature of the PC method varied from 1023 to 1423 K in order to control the size of lateral dimensions of 2D sheets. The detailed characterization data can be found in our previous paper. Then, the as-prepared KCa2Nb3O10 was subject to proton exchange with 1 M HNO3 (100 mL) for 6–7 days, followed by centrifugation to separate the resulting solid, which was washed with pure water until the pH of the supernatant became neutral.KCa2Nb3O10 was also prepared by a solid-state reaction method according to a previous report.18 K2CO3 (≥99.5%, Kanto Chemical Co.), CaCO3 (≥99.5%, Kanto Chemical Co.), and Nb2O5 (≥99.95%, Kanto Chemical Co.) were mixed with an agate mortar and pestle at a molar ratio of K/Ca/Nb = 1.1/2/3, and the mixture was calcined in air at 1123 K for 1 h. An excess of K2CO3 (10 mol% excess K) was added to compensate for loss due to volatilization. After cooling to room temperature, the sample was mixed again, and was calcined in air at 1423 K for 10 h. The as-prepared material was subject to proton-exchange in the same manner.
Elemental analysis by means of energy-dispersive X-ray spectroscopy (EDS) showed that approximately 95% of K+ ions in the interlayer space were exchanged with protons. For simplicity, the obtained products will be referred to as HCa2Nb3O10.
Preparation of aggregated HCa2Nb3O10 nanosheets
Layered HCa2Nb3O10 samples were treated with an aqueous TBAOH solution to exfoliate the layered structure. The molar ratio of TBA+ cations to exchangeable cations was 1 by mole. After shaking the suspension for 6–7 days, the resulting colloidal solution was allowed to stand for 1 day to precipitate the unreacted portion. After removal of the unreacted solids, the resulting nanosheet suspension was reassembled by adding HCl, followed by washing with H2O in a similar manner.Adsorption of Ru complexes
Ru{(4,4′-(CH3)2-bpy)2(4,4′-(CH2PO3H2)2-bpy)}(PF6)2 (bpy = 2,2′-bipyridine), abbreviated as Ru in this work, was synthesized according to the previous literature with some modifications.26,27 It was confirmed by 1H-NMR spectroscopy, electrospray ionization-mass spectroscopy (ESI-MS) and elemental analysis that the complexes were successfully synthesized.The as-prepared HCa2Nb3O10 lamellar solid or aggregated nanosheet (20 mg) was suspended in acetonitrile containing Ru (total volume, 10 mL). After magnetically stirring overnight in the dark, the suspension was filtered, and the resulting pale-orange powder was collected. Finally, the collected powder was dried in an oven at 343 K overnight. The amount of adsorbed Ru sensitizers onto a given substrate was estimated by using the following equation:
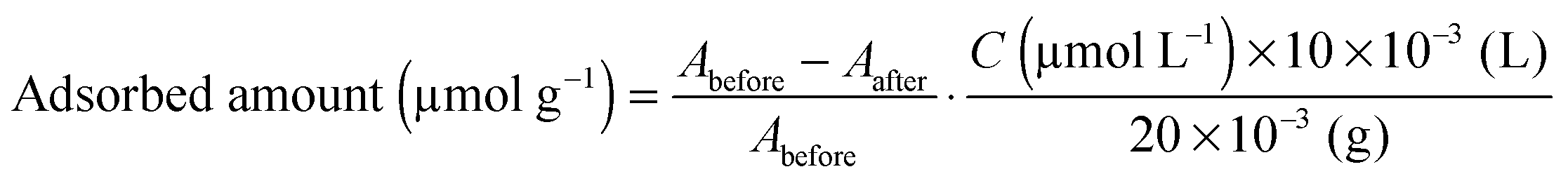
Steady-state emission spectroscopy
Steady-state emission spectra were acquired using a multichannel spectrometer attached to a calibrated integrating sphere (C9920-02G, Hamamatsu Photonics) under 444 nm excitation at room temperature. The measurements were conducted by dispersing a 5.0 mg of the powdered sample adsorbed with Ru (2.0 μmol g−1) in 3.0 mL of 10 mM EDTA aqueous solution under an Ar atmosphere.Time-resolved emission spectroscopy
Emission decay monitored at 630 nm, corresponding to the emission from Ru, was measured by the time-dependent single photon counting method using a FluoroCube 1000U-S spectrofluorometer under 444 nm photoexcitation (NanoLED-440L, HORIBA) with a TBX-04 detector at room temperature. A 5.0 mg of Ru/HCa2Nb3O10 powder was dispersed in 3.0 mL of 10 mM EDTA aqueous solution with continuous magnetic stirring under an Ar atmosphere.Acknowledgements
This work was supported by the ENEOS Hydrogen Trust Fund, Grant-in-Aid for Scientific Research on Innovative Areas (Project No. 25107512; AnApple) and the PRESTO/JST program “Chemical Conversion of Light Energy”. The authors would like to thank Prof. Takashi Hisatomi and Kazunari Domen (The University of Tokyo) for assistance in SEM observations and EDS measurements. K.M. acknowledges The Noguchi Institute for the financial support.Notes and references
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed
, and references therein.
- R. Huber, S. Sebastian Spörlein, J. E. Moser, M. Grätzel and J. Wachtveitl, J. Phys. Chem. B, 2000, 104, 8995 CrossRef CAS
- A. Furube, R. Katoh, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2003, 107, 4162 CrossRef CAS
- R. Katoh, A. Furube, T. Yoshihara, K. Hara, G. Fujihashi, S. Takano, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 4818 CrossRef CAS
- V. H. Houlding and M. Grätzel, J. Am. Chem. Soc., 1983, 105, 5695 CrossRef CAS
- Y.-I. Kim, S. Salim, M. J. Huq and T. E. Mallouk, J. Am. Chem. Soc., 1991, 113, 9561 CrossRef CAS
- E. Bae, W. Choi, J. Park, H. S. Shin, S. B. Kim and J. S. Lee, J. Phys. Chem. B, 2004, 108, 14093 CrossRef CAS
- Q. Li, Z. Jin, Z. Peng, Y. Li, S. Li and G. Lu, J. Phys. Chem. C, 2007, 111, 8237 CAS
- R. Abe, K. Shinmei, K. Hara and B. Ohtani, Chem. Commun., 2009, 3577 RSC
- K. Maeda, M. Eguchi, W. J. Youngblood and T. E. Mallouk, Chem. Mater., 2008, 20, 6770 CrossRef CAS
- K. Maeda, M. Eguchi, S.-H. A. Lee, W. J. Youngblood, H. Hata and T. E. Mallouk, J. Phys. Chem. C, 2009, 113, 7962 CAS
- K. Maeda, M. Eguchi, W. J. Youngblood and T. E. Mallouk, Chem. Mater., 2009, 21, 3611 CrossRef CAS
- K. Maeda, G. Sahara, M. Eguchi and O. Ishitani, ACS Catal., 2015, 5, 1700 CrossRef CAS
- M. M. Maitani, K. Tanaka, D. Mochizuki and Y. Wada, J. Phys. Chem. Lett., 2011, 2, 2655 CrossRef CAS
- K. Hashimoto, M. Hiramoto, T. Sakata, H. Muraki, H. Takemura and M. Fujihira, J. Phys. Chem., 1987, 91, 6198 CrossRef CAS
- W. E. Ford, J. M. Wessels and M. A. J. Rodgers, Langmuir, 1996, 12, 3449 CrossRef CAS
- C.-W. Chang, C. K. Chou, I. Chang, Y.-P. Lee and E. W. Diau, J. Phys. Chem. C, 2007, 111, 13288 CAS
- T. Oshima, O. Ishitani and K. Maeda, Adv. Mater. Interfaces, 2014, 1, 1400131 Search PubMed
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294 RSC
- T. Yui, H. Takeda, Y. Ueda, K. Sekizawa, K. Koike, S. Inagaki and O. Ishitani, ACS Appl. Mater. Interfaces, 2014, 6, 1992 CAS
- Here, the integration of the signal count profile from t = 0 (just after photoexcitation) to t = ∞ corresponds, in principle, to the emission quantum yield. Therefore, time-resolved emission measurement should be done in this way. Due to different morphological characteristics of different materials, however, the y-axis count depends on the material employed. To avoid this complication, we normalized data obtained using different materials by repeating the measurement until the signal count reached 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (Fig. 3). Nevertheless, we should be able to apply this way (irradiation with a fixed number of photons) when we compare materials with similar structural features. That is why we used different representations in the y axis shown in Fig. 4. Actually, the nanosheet materials shown in Fig. 4 have rough surface structures with almost the same surface area (approximately 50 m2 g−1). We wish to point out that the direct measurement of emission strength cannot be employed for determining the emission quantum yield because light scattering by the nanosheet should cause disunity of the number of photons absorbed by the Ru complex.
000 (Fig. 3). Nevertheless, we should be able to apply this way (irradiation with a fixed number of photons) when we compare materials with similar structural features. That is why we used different representations in the y axis shown in Fig. 4. Actually, the nanosheet materials shown in Fig. 4 have rough surface structures with almost the same surface area (approximately 50 m2 g−1). We wish to point out that the direct measurement of emission strength cannot be employed for determining the emission quantum yield because light scattering by the nanosheet should cause disunity of the number of photons absorbed by the Ru complex. - M. Kakihana, J. Sol-Gel Sci. Technol., 1996, 5, 7 CrossRef
- S. Ikeda, M. Hara, J. N. Kondo and K. Domen, Chem. Mater., 1998, 10, 72 CrossRef CAS
- S. Ikeda, N. Sugiyama, S.-y. Murakami, H. Kominami, Y. Kera, H. Noguchi, K. Uosaki, T. Torimoto and B. Ohtani, Phys. Chem. Chem. Phys., 2003, 5, 778 RSC
- K. Maeda, N. Murakami and T. Ohno, J. Phys. Chem. C, 2014, 118, 9093 CAS
- B. Gholamkhass, H. Mametsuka, K. Koike, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326 CrossRef CAS PubMed
- M. R. Norris, J. J. Concepcion, C. R. K. Glasson, Z. Fang, A. M. Lapides, D. L. Ashford, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2013, 52, 12492 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Additional spectroscopic and characterization data. See DOI: 10.1039/c5cp02050f |
| This journal is © the Owner Societies 2015 |