
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-resolution X-ray absorption spectroscopy of iron carbonyl complexes†
Andrew J.
Atkins
a,
Matthias
Bauer
*b and
Christoph R.
Jacob
 *ac
*ac
aKarlsruhe Institute of Technology (KIT), Center for Functional Nanostructures and Institute of Physical Chemistry, Wolfgang-Gaede-Str. 1a, 76131 Karlsruhe, Germany
bUniversität Paderborn, Naturwissenschaftliche Fakultät, Department Chemie, Warburger Straße 100, 33098 Paderborn, Germany. E-mail: matthias.bauer@upb.de
cTU Braunschweig, Institute of Physical and Theoretical Chemistry, Hans-Sommer-Str. 10, 38106 Braunschweig, Germany. E-mail: c.jacob@tu-braunschweig.de
First published on 29th April 2015
Abstract
We apply high-energy-resolution fluorescence-detected (HERFD) X-ray absorption near-edge spectroscopy (XANES) to study iron carbonyl complexes. Mono-, bi-, and tri-nuclear carbonyl complexes and pure carbonyl complexes as well as carbonyl complexes containing hydrocarbon ligands are considered. The HERFD-XANES spectra reveal multiple pre-edge peaks with individual signatures for each complex, which could not be detected previously with conventional XANES spectroscopy. These peaks are assigned and analysed with the help of TD-DFT calculations. We demonstrate that the pre-edge peaks can be used to distinguish the different types of iron–iron interactions in carbonyl complexes. This opens up new possibilities for applying HERFD-XANES spectroscopy to probe the electronic structure of iron catalysts.
1 Introduction
Iron carbonyl complexes are the text-book example of coordination compounds following the 18 electron rule. The metal centre(s) coordinates to a set of ligands which will provide enough electrons to fill the valence shell.1–3 Here, the necessary 18 electrons can also be reached by forming a (formal) metal–metal bond in multinuclear carbonyls. Additionally, carbonyls are the prototypical example for synergetic effects by σ-donation and π-backdonation from the CO ligand to the metal centre(s) and vice versa. The ability to spectroscopically probe the changes in the electronic structure caused by the binding of CO ligands as well as by the formation of metal–metal bonds is key for understanding catalytic reactions featuring iron carbonyls or other types of iron clusters as active species.Examples of such reactions are the Fischer–Tropsch process4–10 or the CO oxidation reaction.11,12 Moreover, iron carbonyl complexes are of interest as model complexes to understand the working principles of catalysts.13–15 Finally, iron carbonyls have also been identified as potential substitutes to replace expensive catalysts based on rare elements such as ruthenium or platinum.16
The catalytic activity of iron carbonyls is usually attributed to two factors: First, the ease of the (photo-)dissociation of a carbonyl ligand in the iron carbonyl complexes,15,17,18 and second, the weak bonding character of the Fe–Fe contact that is easily broken.19 Both pathways lead to coordinatively unsaturated species which undergo fast interactions with reactants. However, it is often difficult—if not impossible—to study the bonding of CO and the iron–iron bond in the course of chemical reactions. Although several spectroscopic studies on iron carbonyls exist (see, e.g., ref. 18, 20 and 21), these spectroscopic methods can hardly be used under real reaction conditions (high pressure and temperatures) due to vacuum requirements for X-ray and ultraviolet photoelectron spectroscopy (XPS and UPS),22,23 due to low penetration depth or the interference of solvents for infrared (IR) spectroscopy, or simply the fact that window materials might not be transparent for the radiation used.
Hard X-ray absorption spectroscopy offers a unique opportunity to overcome these limitations by using high-energy radiation in an element specific manner.24 Previously, it was shown that valence-to-core X-ray emission spectroscopy (V2C-XES) can be used to identify details of the ligand coordination as well as the substitution of CO by hydrocarbons.25 However, since V2C-XES spectra are dominated by the ligand levels, details of the metal–metal interaction can barely be accessed.
X-ray absorption near edge structure (XANES) spectroscopy can provide information on the qualitative or comparative oxidation states in a fingerprinting way by comparing the absorption edge position of the sample with well-defined references. However, with respect to probing the electronic structure of iron carbonyls during catalytic reactions, much of the information on the geometric and electronic structure is contained in the pre-edge signal. In the case of iron K-edge spectra, pre-edge peaks are caused by 1s → 3d transitions,26–28 which thus probe the lowest unoccupied molecular orbitals (LUMOs) of metal catalysts. These unoccupied states reflect the geometric (coordination symmetry) and electronic (oxidation state) structure. However, extracting this information from conventional transition metal K-edge XANES spectra is complicated by the core-hole lifetime broadening of the 1s electron hole, which smears out features in the pre-edge region.
Eisenberger et al.29 showed that the lifetime broadening in the XANES region could be reduced by detecting the X-ray absorption spectrum using the intensity of the emitted X-ray fluorescence in a narrow energy bandwidth. This technique was called high-energy-resolution fluorescence detected XANES (HERFD-XANES).30–32 Previously we have shown that HERFD-XANES can reveal subtle changes in the electronic structure of iron complexes that are not detectable with V2C-XES spectroscopy.33,34 In this work, we want to establish the benefits of HERFD-XANES spectroscopy for the investigation of multinuclear metal complexes in general and iron carbonyls in particular.
To this end, we investigate the HERFD-XANES spectra of iron carbonyl with a special focus on the identification of signatures characteristic of CO-bonding and different types of Fe–Fe-bonds as well as their change due to ligand exchange. We use a test set of five iron carbonyl complexes consisting of (a) iron-pentacarbonyl Fe(CO)5, (b) di-iron-nonacarbonyl Fe2(CO)9, (c) tri-iron-dodecacarbonyl Fe3(CO)12, (d) iron-tricarbonyl-cyclooctadiene Fe(CO)3(C8H12), and (e) di-iron-tetracarbonyl-dicyclopentadiene Fe2(CO)4(Cp)2. The corresponding Lewis structures are shown in Fig. 1.
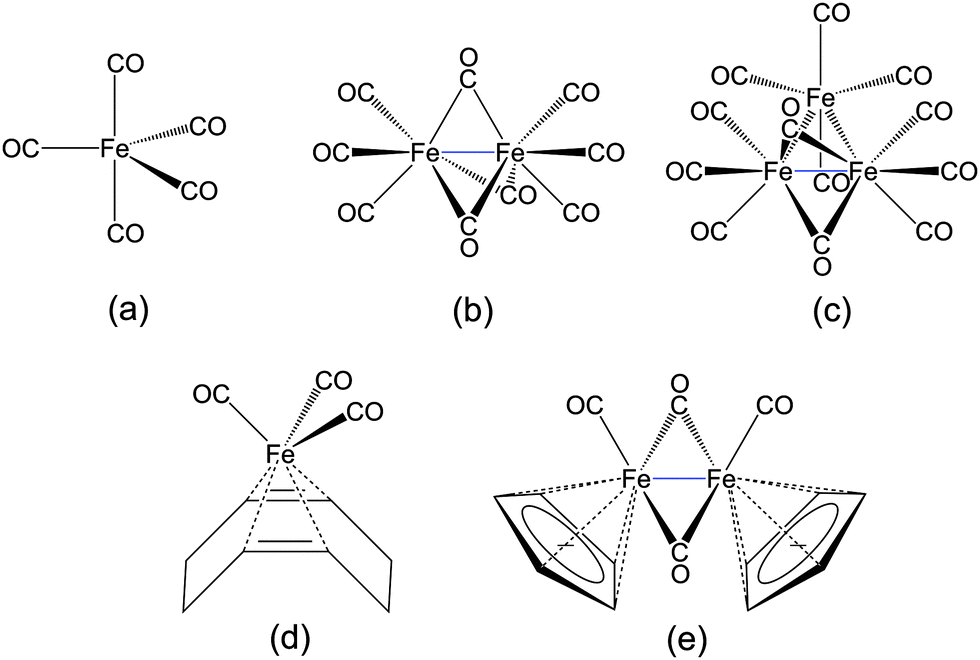 | ||
| Fig. 1 Lewis structures of the five iron carbonyl complexes considered in this work. (a) Fe(CO)5, (b) Fe2(CO)9, (c) Fe3(CO)12, (d) Fe(CO)3(C8H12), and (e) Fe2(CO)4(Cp)2. | ||
This work is organised as follows. In Section 2, we introduce the experimental and computational methodology used for recording and calculating the pre-edge region of the HERFD-XANES spectra. This is followed by a discussion of the electronic structure and spectra of Fe(CO)5 in Section 3. In Sections 4 and 5, the multinuclear iron carbonyls as well as substituted iron carbonyls are discussed, respectively. Finally, the results are summarised and conclusions are drawn in Section 6.
2 Methodology
2.1 Experimental
The samples were purchased from Sigma Aldrich in high purity and used without further purification. The solid samples (b)–(e) were diluted with BN in a ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. For the liquid Fe(CO)5, BN was impregnated with a corresponding amount. All sample preparation was carried out under an inert atmosphere in a glove box.
:4. For the liquid Fe(CO)5, BN was impregnated with a corresponding amount. All sample preparation was carried out under an inert atmosphere in a glove box.
All spectra were recorded using a scanning Johann35,36 type spectrometer as available at the ESRF Grenoble, beamline ID26.37 Data acquisition was carried out in a horizontal plane, where the sample, crystal analysers and photon detector (avalanche photodiode) were arranged in a vertical Rowland geometry. An incident flux of ≈2 × 1013 photons per s is estimated at this experimental station. The incident energy was selected using the (311) reflection from a Si double crystal monochromator. To record HERFD-XANES spectra, the intensity of a single fluorescence channel, selected with the analyser crystals, is monitored with a resolution smaller than the lifetime broadening. Meanwhile, the incident energy of the double crystal monochromator is swept over the edge. Here, the emission energy of 7.056 keV was selected using the (620) reflection of five spherically bent Ge crystal analysers (with R = 1 m). The total fluorescence yield (TFY) was monitored by a photodiode. Measurements were carried out at 15 K using a He cryostat under vacuum conditions. To exclude radiation damage, fast measurements over the energy region 7110–7146 eV with a scan-time of four seconds were carried out before and after the HERFD-XANES acquisition. No radiation damage was detected in this procedure. Energy calibration of the monochromator was performed using Fe foil. The emission spectrometer was calibrated using the elastic scattering line. Selected RIXS planes were recorded to exclude off-diagonal signals that could lead to misinterpretations of the HERFD-XANES spectra.
Fig. 2 compares the conventional and HERFD-XANES spectra. The significant increase in resolution and the tremendously sharpened signals in the pre-edge region are obvious. Comparing the conventional spectra it is clear that here the pre-edge peaks look very similar. Only for the more ionic complex Fe2(CO)4(Cp)2 (e), with iron in the oxidation state +I, a more intense pre-edge peak is observed compared to all other complexes with a formal oxidation state of Fe(0). Nonetheless differences with respect to the electronic structure are hard to deduce from the experimental spectra alone, because, first, the pre-edge peak cannot be extracted in a reliable manner due to its strong overlap with the main edge, and, second, because the resolution of the experimental data is too low for a direct comparison with calculations.
 | ||
| Fig. 2 HERFD-XANES spectra (black solid line) of the compounds (a) Fe(CO)5, (b) Fe2(CO)9, (c) Fe3(CO)12, (d) Fe(CO)3(C8H12), and (e) Fe2(CO)4(Cp)2 in comparison to the according conventional XANES spectra (blue solid line). | ||
The differences in the pre-edge peaks of the carbonyl complexes (a)–(e) drastically increase in the HERFD-XANES data compared to the conventional XANES data. The spectra of all five compounds are now characterised by individual signatures that differ to a large extent. Beside the need for a quantitative understanding of the observed manifold of signals, they could also be used on a more simple basis in a fingerprint approach. However, in order to arrive at a clear assignment and to extract information on the unoccupied electronic states, quantum-chemical calculations need to be applied.
Deconvolution of the pre-edge region was carried out by a least square fit of the spectra using asymmetric Gaussian peak profiles in order to account for asymmetries in the RIXS plane from which the HERFD-XANES spectra are extracted. In the first iteration, an arctan step function representing the transition of the ejected photoelectron to the continuum was subtracted from the spectra. In the second step, the remaining pre-edge peaks as identified by inspection of the first two derivatives, were fitted. In Fig. 4, 7 and 9, the isolated and deconvoluted experimental pre-edge peaks are shown.
2.2 Computational
Geometry optimisations of the molecular complexes were performed with the ADF program package38,39 using the BP86 exchange–correlation functional40,41 in combination with the TZP Slater-type basis set from ADF's basis set library.42 For all considered compounds, the ground state is the closed-shell singlet spin state.The XANES spectra for the iron carbonyl complexes were then calculated using time-dependent density functional theory (TD-DFT). To selectively target core excitations, the TD-DFT calculations were restricted to excitations originating from the Fe 1s orbitals.43,44 Such restricted-channel TD-DFT calculations have been applied extensively for K-edge XANES spectra of transition metal complexes.45–50 The effect of the exchange–correlation functional was judged by performing the calculations with both the non-hybrid functional BP8640,41 and the hybrid functional B3LYP.40,51–53 The QZ4P Slater-type basis set42 was applied in all TD-DFT calculations. The intensities include second-order contributions due to the magnetic-dipole and electric-quadrupole transition moments, which are calculated using the origin-independent methodology described in ref. 54. Spin–orbit coupling, which could give rise to non-zero intensities of singlet–triplet transitions, has not been considered here.
The BP86 spectra were shifted by 180.62 eV and the B3LYP spectra by 150.08 eV for comparison to experiment. These shifts are chosen such that the energy of the first peak in the calculated spectrum of iron-pentacarbonyl agrees with the first peak in the experimental spectrum. While these shifts are rather large, they do not affect the relative position of the peaks. For Fe K-edge X-ray absorption spectroscopy, it has been demonstrated previously that despite the large absolute errors, the selective TD-DFT approach used here can provide relative excitation energies and intensities with an accuracy that allows for a direct comparison with experiment.55 The intensity of the calculated spectra of complexes containing more than one core excitation centre were divided by the number of iron atoms to normalise the spectra. For all the calculated spectra, the ionisation threshold is indicated by a blue vertical line in Fig. 4, 7 and 9. Excitations above this threshold, which is determined as the negative of the energy of the 1s core orbital, cannot be calculated reliably in conventional TDDFT calculations.56
It has previously been argued,45 that for molecules with equivalent core orbitals it is necessary to localise the core hole before performing the TD-DFT calculation. We note that in the present study, all calculations were run without considering the molecular symmetry, which results in localised core orbitals for the bi- and trimetallic complexes.
All orbital isosurface plots in Fig. 3, 5 and 8 were prepared with Molekel57 and use an isosurface value of ±0.025 e bohr−3. With the B3LYP hybrid functionals, some excitations are combinations of several orbital transitions. Fig. 5 and 8 shows the corresponding linear combination of unoccupied orbitals.33 This is possible because the excitations originate from the same localised Fe 1s core orbitals.
3 Revisiting the electronic structure of Fe(CO)5
First, we consider the simplest iron carbonyl complex Fe(CO)5, which has a trigonal bipyramidal structure with D3h symmetry. As a starting point for the discussion of Fe(CO)5, we will consider a schematic representation of its molecular orbital (MO) diagram, which is shown in Fig. 3. For earlier computational investigations of the electronic structure of Fe(CO)5, see, e.g., ref. 58–61. In the D3h point group the d-orbitals of the iron atom belong to three irreducible representations (irreps). The iron dz2-orbital belongs to the totally symmetric A1′ irrep, whereas the dxy and dx2–y2 as well as dxz and dyz pairs belong to the two degenerate irreps E′ and E′′, respectively. These combine with the carbonyl ligand orbitals of the same symmetry. The carbonyl σ-orbitals form two combinations that belong to the irrep A1′, one combination that belongs to A2′′, and two combinations belonging to E′′. These carbonyl σ-orbitals are composed of the p-orbitals pointing along the carbonyl bond axes, and thus form the iron–carbonyl σ-bonds. The carbonyl π*-orbitals form two combinations that belong to the irreps A2′ and A2′′, respectively, four combinations belonging to the E′ irrep, and four combinations belonging to the E′′ irrep. These carbonyl π*-orbitals are responsible for the iron–CO π-backbonding.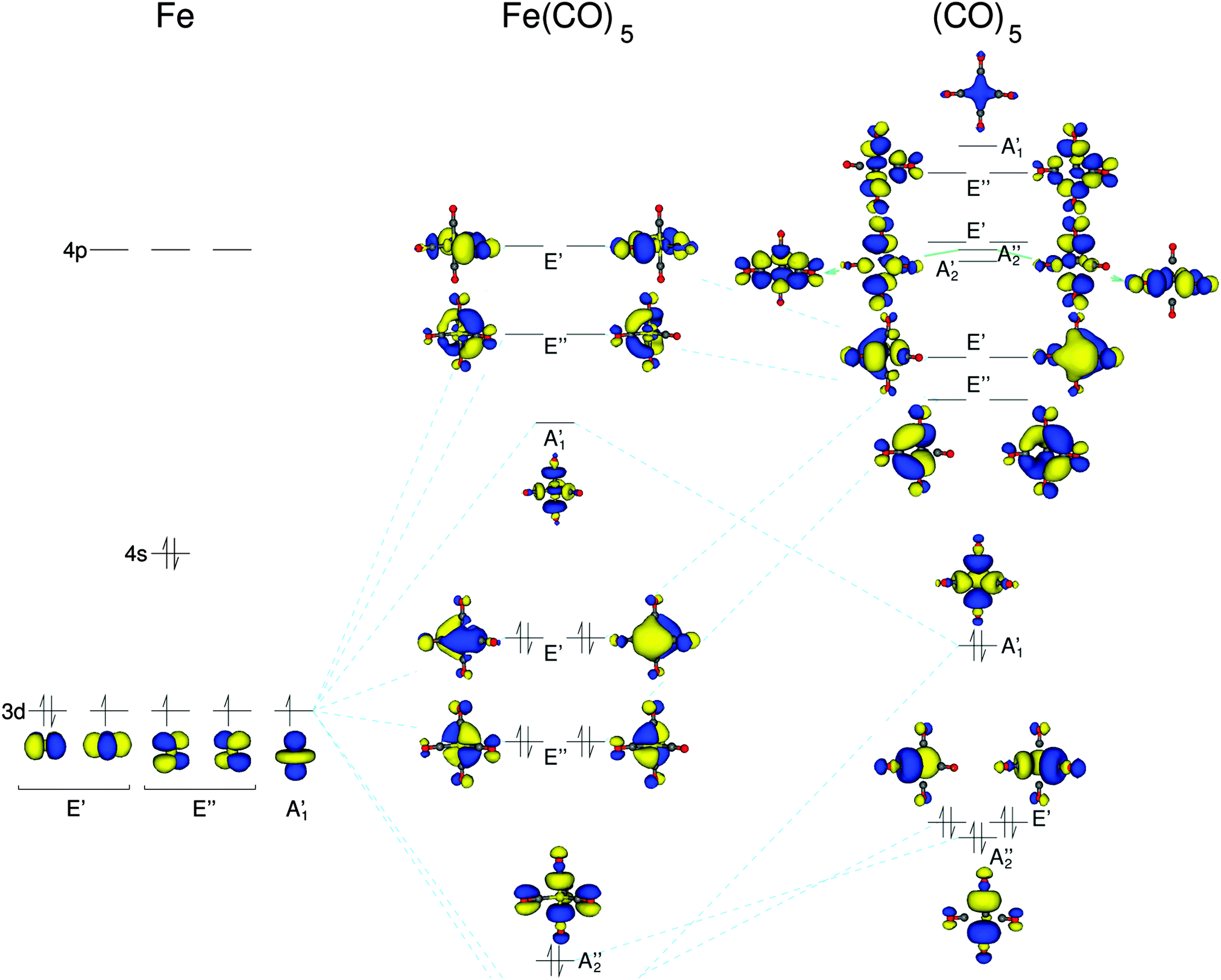 | ||
| Fig. 3 A schematic molecular orbital diagram of Fe(CO)5 constructed from density-functional theory calculations. The isosurface plots of the occupied and unoccupied orbitals have been obtained with BP86. The orbitals are arranged in the energetical order in which they appear within the B3LYP calculations. | ||
The five iron d-orbitals combine with the carbonyl σ- and π*-orbital combinations with the matching symmetry. The five highest occupied and the five lowest unoccupied orbitals each contain the five iron d-orbitals. These molecular orbitals are shown in the MO diagram of Fig. 3. The unoccupied orbitals are those that give rise to the pre-edge peaks in the HERFD-XANES spectra. The iron dz2-orbital interacts with the A1′ σ-carbonyl orbitals to form an anti-bonding combination with A1′ symmetry. The dxz- and dyz-orbitals interact only with the E′′ π-orbitals of the carbonyls, whereas the dxy- and dx2–y2-orbitals can interact with both σ and π carbonyl orbitals. This can be seen in the unoccupied orbitals of the E′ symmetry in Fig. 3 where there is some minor inclusion of a σ carbonyl orbital on the carbonyl ligand with no π contributions, whereas all other carbonyl ligand orbitals are pure π-orbitals.
Using the character table of point group D3h we can determine whether an X-ray transition from the iron 1s to these unoccupied orbitals will have dipole or quadrupole intensity. Transitions to the A1′ and E′′ MOs are only quadrupole-allowed, whereas those to E′ MOs are both quadrupole- and dipole-allowed.
Fig. 4a contains both the calculated and experimental spectra of Fe(CO)5. Therein, the experimental spectrum has three distinct peaks, labeled A, B, and C. Previous experimental X-ray absorption spectra of Fe(CO)5 at the K-edge of iron show only one peak in the pre-edge spectra.62,63 This clearly demonstrates the power of HERFD-XANES to reveal the detailed structure of the pre-edge region for transition metal complexes. In the experimental spectrum, peaks A and B clearly belong to the pre-edge. However, peak C, with a significant increase in intensity compared to peaks A and B, is most likely due to dipole-allowed transitions to (Rydberg-like) states close to the absorption edge, which cannot be described with conventional exchange–correlation functionals.64,65 Moreover, the region close to and above the ionization threshold will be obscured by quasi-continuum states that cannot be described with finite atom-centered basis sets.56 Therefore, we will focus our discussion on peaks A and B in the following.
 | ||
| Fig. 4 X-ray absorption spectra of Fe(CO)5, Fe2(CO)9 and Fe3(CO)12 calculated with the BP86 and B3LYP exchange–correlation functionals and the experimental results. | ||
In the calculated spectra, BP86 shows one intense peak, whereas B3LYP provides a spectrum with three distinct features with lower intensities. Therefore, (qualitatively) we can conclude that the B3LYP exchange–correlation functional provides results that are in better agreement with the experimental spectrum. To further understand the differences between BP86 and B3LYP we must consider the transitions that make up the spectra. The single peak in the BP86 spectrum contains twelve contributing transitions lying very close together. Here, the lowest unoccupied orbital is the E′′, but the energy difference to the unoccupied A1′ orbital is only 0.1 eV. All twelve unoccupied orbitals contributing to the pre-edge transitions lie within 2.3 eV. Therefore, once the broadening of the transitions is taken into account, only one pre-edge peak remains. Clearly, such a picture of the electronic structure is contradicted by the experimental spectrum.
On the other hand, in the B3LYP spectrum two well-separated peaks appear. The lowest energy transition is to the A1′ orbitals (which can be assigned to peak A in the experimental spectrum), followed by transitions to the two degenerate representations E′′ and E′ (which can be assigned to peak B in the experimental spectrum). Here, the energy separation between A1′ and E′′ is 1.7 eV, and the difference between the first and last transition in the pre-edge is 5.4 eV. The isosurface plots of the (combinations of) unoccupied orbitals observed in these peaks are shown in Fig. 5a. These agree with the unoccupied orbitals obtained with BP86 (shown in the MO diagram in Fig. 3). However, as the spectrum obtained with B3LYP points to an electronic structure that is in better agreement with experiment, the orbital ordering extracted from the B3LYP spectrum has been used in the MO diagram of Fig. 3.
 | ||
| Fig. 5 Isosurface plots of the unoccupied orbitals probed by the pre-edge transitions as obtained from TD-DFT calculations with B3LYP/QZ4P, for the major contributions to the pre-edge peaks of Fe(CO)5 and Fe2(CO)9. Shown below the unoccupied orbitals are the isolated iron orbital contributions to them. The coloured dots indicate the iron atom (1s orbital) from which the transition originates. | ||
Table 1 shows the dipole and quadrupole contributions to the intensities of the transitions, separated into the transitions that contribute to each of the peaks. We can see that the contributions for Fe(CO)5 follow what is expected from symmetry considerations: Peak A (A1′) is composed entirely of quadrupole intensity and peak B (E′′ and E′) has a small dipole contribution due to the transition into the E′ orbitals. We note that earlier calculations66 of the Fe K-edge XAS spectra of Fe(CO)5 did not include quadrupole contributions to the intensities, and thus only showed a single pre-edge peak due to transitions to the E′ orbitals.
| Complex | Peak | Dipole (%) | Quadrupole (%) |
|---|---|---|---|
| (a) Fe(CO)5 | A | 0.0 | 100.0 |
| B-1 | 0.0 | 100.0 | |
| B-2 | 25.2 | 74.8 | |
| (b) Fe2(CO)9 | A | 96.3 | 3.7 |
| B | 100.0 | 0.0 | |
| (c) Fe3(CO)12 | A-1 | 92.7 | 7.3 |
| A-2 | 78.0 | 22.0 | |
| B-1 | 95.4 | 4.6 | |
| B-2 | 88.8 | 11.2 | |
| (d) Fe(CO)3(C8H12) | A | 87.1 | 12.9 |
| (e) Fe2(CO)4(Cp)2 | A-1 | 82.6 | 17.4 |
| A-2 | 96.0 | 4.0 | |
| B-1 | 97.6 | 2.4 | |
| B-2 | 91.5 | 8.5 | |
4 Bi- and trinuclear iron carbonyls
We now turn to the bi- and trinuclear iron carbonyl complexes Fe2(CO)9 and Fe3(CO)12, which have a molecular structures with D3h and C2v symmetry, respectively.19,67,68 In Fe2(CO)9, both iron centres have a pseudo-octahedral environment (consisting of five CO ligands and the other iron atom), and three bridging CO ligands connect the two iron centres, while each iron centre features three terminal CO ligands. The molecular structure of Fe3(CO)12 can be derived from Fe2(CO)9 by replacing one bridging CO ligand by an Fe(CO)4 moiety. Thus, all three iron centres are still in a pseudo-octahedral environment. However, the bonds between the iron atom of the Fe(CO)4 moiety and those of the Fe2(CO)8 backbone are not bridged by CO ligands anymore.Numerous computational studies of both the geometrical and the electronic structures of these bi- and trinuclear carbonyls have been performed,69–75 and the presence of direct metal–metal bonds has been discussed extensively. While applying the 18-electron rules clearly points towards Fe–Fe bonds, their presence is questioned by most theoretical studies for Fe–Fe bonds bridged by carbonyl ligands. For Fe2(CO)9, most computational studies rule out a direct Fe–Fe bond and attribute the stabilising interactions to the bridging carbonyl ligands69,70,73 or only point to a very weak direct metal–metal interaction.74 On the other hand, two of the three Fe–Fe bonds in Fe3(CO)12 are not bridged by carbonyl ligands and must thus be due to a direct metal–metal bonding interaction.71,72 Below, we will discuss to what extent our HERFD-XANES spectra can provide indications for these different kinds of metal–metal interactions.
For Fe2(CO)9, the experimental and calculated spectra are shown in Fig. 4b. There are three observable features in the experimental spectrum labelled A, B, and C. The conventional K-edge spectrum of Fe2(CO)9 shows only one distinct peak, again demonstrating the capability of HERFD-XANES. Within the experimental spectra we can assign both peaks A and B to the pre-edge whereas peak C is once again most likely due to transitions to Rydberg states or other states close to the absorption edge. Therefore, we will focus on peaks A and B in the following. If we compare the spectrum to the one of Fe(CO)5, we immediately notice that the pre-edge features have a much higher intensity and that all three peaks are shifted to higher energies.
First, we compare the spectra calculated with both the BP86 and B3LYP exchange–correlation functionals. BP86 calculates a spectrum with only two distinct features below the ionisation threshold, whereas B3LYP shows three distinct features. The latter appears to agree better with the experimental observations. Therefore, we will only discuss the B3LYP results in the following. The isosurface plots of the (combination of) unoccupied orbitals that are probed with a significant intensity contribution are plotted in the upper part of Fig. 5b. To highlight the contributions of the iron orbitals, the lower part of Fig. 5b shows isosurface plots that only cover a radius of 1 Å around the iron atoms.
These orbital plots show that peak A is comprised of transitions to an anti-bonding combination of the iron dz2-orbitals, which further combines with carbonyl π*-orbitals. The two contributing excitations originate from each of the two core orbitals to a combination of unoccupied orbitals. Both Fe(CO)5 and Fe2(CO)9 have the same point group and the major transitions in peak A are to orbitals containing the iron dz2-orbitals. The main reason for the large increase of peak A in intensity is that even though the molecular point group is the same, the symmetry of the iron centres has been reduced. In Fe2(CO)9 neither lie in the horizontal mirror plane and the lowest energy dz2-combination now belongs to the A2′′ irrep, whereas it belongs to the A1′ irrep in Fe(CO)5. Therefore, the corresponding transition becomes dipole-allowed, which can be seen in Table 1. The shift of peak A to higher energy can be rationalised by a combination of three factors: In Fe2(CO)9, the dz2-orbitals now interact with the π* ligand orbitals, which pushes it to higher energies. At the same time, the change in the ligand field of the iron centre from a pentagonal bipyramidal to a pseudo-octahedral ligand field (where the dz2-orbital is not involved in the bonding of the ligands) lowers the energy. Finally, the interaction of the dz2-orbitals of the two iron centres to form an anti-bonding combination increased the energy of the transition. The latter effect can be seen as an indication of direct metal–metal bonding, but is difficult to isolate from the other two (indirect) contributions.
Peak B has a more complicated assignment. From the isosurface plots in Fig. 5 we can see that one of the iron atom orbitals is dz2-like, while the other one belongs to the degenerate pair of iron dxz- and dyz-orbitals. The second transition is a mirror image of the first one, with the phase of the dz2-like orbitals reversed. The two transitions each originate from the iron atom upon which the dxz/yz-like orbital resides. Therefore, this can be considered a linear combination of transitions which occur from both the A and B peaks of Fe(CO)5.
Finally, we consider Fe3(CO)12 where the experimental and calculated spectra are shown in Fig. 4c. The experimental spectrum once more has three observable features, which we have labelled A, B, and C, and we focus on the pre-edge peaks A and B. If we compare the spectrum to the one of Fe2(CO)9, we can see that the features shift to lower energy and that the intensity of peak A is reduced, while peak B has a larger intensity. For the calculated spectra we will only discuss the B3LYP spectrum, which shows the better agreement with experiment.
The isosurface plots of the (combination of) unoccupied orbitals that are probed with a significant intensity contribution are plotted in Fig. 6, along with the corresponding iron orbital contribution. Peak A has four main contributing transitions. The first two transitions are from the two Fe2(CO)8 core orbitals to metal–metal anti-bonding σ*-orbitals between the Fe2(CO)8 backbone and the Fe(CO)4 moiety. The next transition originates from the 1s orbital of the Fe(CO)4 group to a completely anti-bonding orbital, in which all three possible metal–metal interactions are out of phase. These three transitions (labelled “1” in Fig. 6) all have a similar character, but originate from the three different iron core orbitals. The final transition (labelled “2”) also originates from the 1s of the Fe(CO)4 group, but goes to an orbital which resembles an antibonding combination of the iron d-orbital of the Fe(CO)4 group and of those of the Fe2(CO)8 backbone, while the d-orbitals of the iron centres of the Fe2(CO)8 backbone combine in a bonding fashion.
 | ||
| Fig. 6 Isosurface plots of the unoccupied orbitals probed by the pre-edge transitions as obtained from TD-DFT calculations with B3LYP/QZ4P, for the major contributions to the pre-edge peaks of Fe3(CO)12. Shown below the unoccupied orbitals are the isolated iron orbital contributions to them. The coloured dots indicate the iron atom (1s orbital) from which the transition originates. | ||
Peak B contains three main transitions, in which orbitals from the Fe2(CO)8 backbone are the main contributor. The first two transitions (labelled “1”) are to σ* anti-bonding metal–metal bonding orbitals of the backbone, with only a small orbital inclusion from the third iron centre. The third and final major contributing transition (labelled “2”) is to an orbital with a possible bonding interaction between the iron centres of the Fe2(CO)8 backbone.
To understand the changes in spectral intensity compared to Fe(CO)5 and Fe2(CO)9, we must note that symmetry of the molecule is reduced from D3h to C2v. This reduction in symmetry provides more possibilities for dipole-allowed transitions. Hence, the increase in intensity of peak B can be expected. However, the slight decrease in intensity of peak A compared to peak A of Fe2(CO)9, is not so easy to rationalise. If we look at the contributions of the transition intensities to the peak shown in Table 1 we can see that the main intensity contribution for all transitions is dipole. On the other hand, the amount of quadrupole intensity contribution to the peak has also increased relative to Fe2(CO)9.
The peaks also shift to lower energies than in Fe2(CO)9 and the reason is clear when we consider the molecular orbitals. On the introduction of the Fe(CO)4 group to the Fe2(CO)8 backbone, a third set of d-orbitals are introduced, which can interact with the σ/σ*-orbitals of the Fe2(CO)8 backbone. When these d-orbitals interact, they split into three orbitals, shifting the first available unoccupied orbitals to lower energies. This shift to lower energies is then reflected in the pre-edge peaks.
From all the observations discussed it becomes clear how the introduction of a second iron centre affects the electronic structure. A large part of the differences can be explained through a reduction of the symmetry of the individual iron centres and hence, their individual electronic structures. Moreover, we can see a correspondence between peak A of Fe(CO)5 and Fe2(CO)9 in that they both consist of iron dz2-orbitals. A subsequent correspondence can be made to peak A of Fe3(CO)12 in that this peak consists only of σ*-orbitals, the same as for peak A of Fe2(CO)9. All these features can be related to the unoccupied, anti-bonding (and partly bonding) combinations of the iron d-orbitals, even though it is difficult to isolate the contributions of a direct metal–metal interaction from indirect ones.
5 Substituted iron carbonyl complexes
In addition to the pure iron carbonyl complexes, we also consider the effect of substitution of one or more carbonyl groups on the pre-edge X-ray absorption spectra and thus, also the electronic structure. It was previously shown that the substitution of carbonyl ligands by hydrocarbons leads to clear changes in the V2C-XES spectra. Here, we want to investigate whether HERFD-XANES can provide complementary information.First, Fe(CO)3(C8H12) is a mononuclear iron complex, in which two of the CO groups in Fe(CO)5 have been replaced by an η4-coordinated cyclooctadiene ligand. In Fig. 7 we show the experimental and B3LYP-calculated spectra of Fe(CO)3(C8H12) in comparison to Fe(CO)5. In the experimental spectrum of Fe(CO)3(C8H12), we observe one pre-edge feature which has a large intensity increase as well as a shift to higher energies compared to Fe(CO)5. The calculated BP86 spectrum (see ESI†) shows one peak with a large intensity, while B3LYP leads to two distinct features. In comparison to the experiment, both provide the correct picture with one high-intensity peak. We note that neither exchange–correlation functional reproduces the shift to higher pre-edge peak energies.
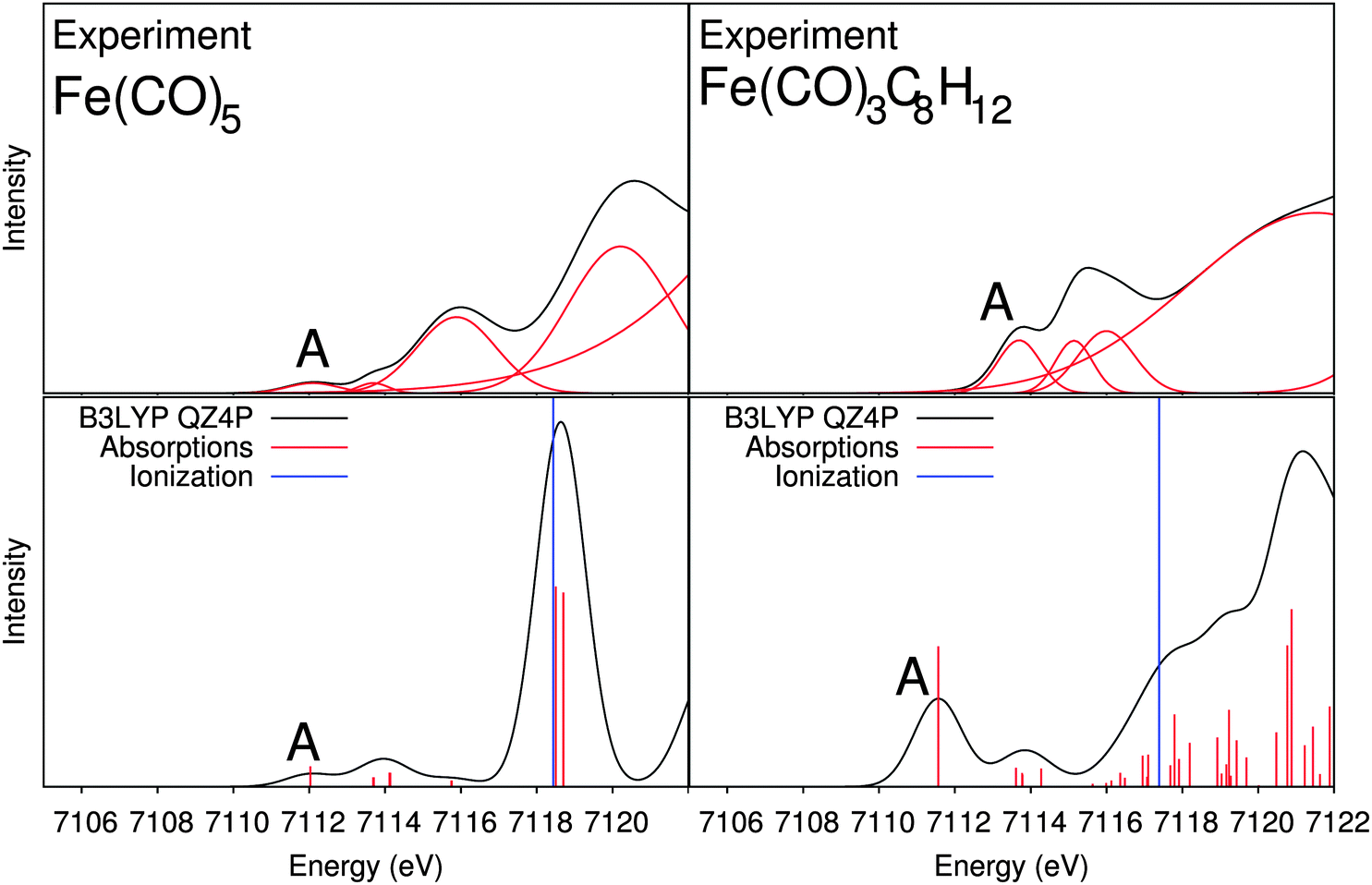 | ||
| Fig. 7 X-ray absorption spectra for Fe(CO)5 and Fe(CO)3(C8H12). Top: experimental spectra; bottom: B3LYP calculated spectra. | ||
To understand what changes occur on substitution of the two carbonyl groups for the C8H12, we consider the isosurface plots of the unoccupied orbitals that are probed with a significant intensity contribution. These are plotted in Fig. 8 along with the corresponding isolated iron contribution. Peak A of Fe(CO)3(C8H12) contains a dxz/yz-like orbital, which is an anti-bonding combination of this iron orbital and π*-orbitals of the C8H12 ligand. For Fe(CO)5, the transition to the corresponding dxz/yz-orbitals were contained in the second pre-edge peak (peak B). This can be used to rationalise the shift to a higher energy, even though this shift is not reproduced correctly by the calculations. The loss of the degeneracy of the dxz/yz-orbitals can be explained through its interaction with the C8H12 ligand, because only one of the two dxz/yz-orbitals will be in the correct orientation to interact with the π*-orbitals of the C8H12 ligand. The large increase in intensity can be attributed to the reduction of the symmetry at the iron centre. We can see that in Table 1 that the primary intensity contribution is now due to electric-dipole contributions.
 | ||
| Fig. 8 Isosurface plots of the unoccupied orbitals probed by the pre-edge transitions as obtained from TD-DFT calculations with B3LYP/QZ4P, for the major contributions to the pre-edge peaks of Fe(CO)3(C8H12) and Fe2(CO)4(Cp)2. Shown below the unoccupied orbitals are the isolated iron orbital contributions to them. The coloured dots indicate the iron atom (1s orbital) from which the transition originates. | ||
We also considered a bimetallic substituted iron carbonyl system, Fe2(CO)4(Cp)2, where five of the CO groups in Fe2(CO)9 have been replaced by two cyclopentadiene groups (one on each iron centre). Fig. 9 shows the experimental and B3LYP-calculated spectra of Fe2(CO)4(Cp)2 and of Fe2(CO)9 for comparison. In the experimental spectrum, we observe two distinguishable pre-edge features, labelled A and B. Compared to the spectrum of Fe2(CO)9 no significant shift in the peak positions is observed, but there is a significant increase in the intensity of both features. Again, the spectra calculated with B3LYP are more accurate compared to the experimental spectrum than the ones obtained with BP86 (see ESI†).
 | ||
| Fig. 9 X-ray absorption spectra for Fe2(CO)9 and Fe2(CO)4(Cp)2. Top: experimental spectra; bottom: B3LYP calculated spectra. | ||
The isosurface plots of the unoccupied orbitals that are probed with a significant intensity contribution are plotted in Fig. 8 along with the corresponding isolated iron orbital contribution. Peak A is comprised of four transitions, two from each core orbital. The first two transitions (labelled “1”) are to orbitals which are antibonding combinations of dxz/yz-like orbitals on the two iron centres interacting in an anti-bonding fashion with π*-orbitals of the Cp-ligand at one of the iron atoms. The other two transitions (labelled “2”) are once again to dxz/yz-like iron orbitals interacting in an anti-bonding fashion with other Cp π*-orbitals, which are rotated by approximately 90 degrees around the Cp ring. Peak B of Fe2(CO)4Cp2 also contains four main transitions, two from each core orbital. The first two transitions (labelled “1”) are to orbitals with anti-bonding characteristics between a dxz/yz-like orbital and the Cp π*-orbitals on one of the irons. However, unlike in peak A, the other iron atom now has a dz2-like appearance. The final two transitions (labelled “2”) are dx2−y2/xy-like for the iron orbitals and from Fig. 8 appear to be anti-bonding orbitals for the metal–CO bonds.
The large increase in the intensity of both pre-edge peaks in relation to the pre-edge peaks of Fe2(CO)9 can be explained through the decrease in the symmetry of the complex. The same as was observed for Fe(CO)3(C8H12). We can see in Table 1 that the intensity is mainly due to dipole contributions. However, as the Fe2(CO)9 peaks were also primarily composed of dipole transitions, only the loss of symmetry and thus, relaxation of the selection rules for the dipole mechanism can account for the increase in the intensity.
In conclusion, we can see that substitution of carbonyl groups for a group containing π-orbitals reduces the symmetry and thus, the intensity of the spectra increases. In addition, the inclusion of π-orbital containing substituents increases the intensity of transitions to the dxz/yz-like iron orbitals, which can combine with the ligand π*-orbitals, compared to the dz2-like iron orbital(s) that dominated for the pure iron carbonyl complex spectra.
6 Conclusions
The HERFD-XANES spectra of iron carbonyl model complexes reported here demonstrate the unique sensitivity of the experimental method to changes both in the geometric and in the electronic structure at the iron centre(s). By comparison with quantum-chemical calculations, the pre-edge region of the spectra can be analysed in detail, which provides a clear assignment of the observed spectral features and allows us to relate changes in the pre-edge peaks to changes in the electronic structure of the complexes.We note here that the two exchange–correlation functionals considered, BP86 and B3LYP, lead to XANES spectra that differ significantly. Similar observations have been made before by us and others.33,49,76 For the example at hand, we found that B3LYP was sufficiently accurate to allow for a qualitative assignment and analysis of the experimental spectra. However, significant deviations between the spectra calculated with B3LYP and the experimental spectra remain, both for relative energies and relative intensities when comparing different complexes. Therefore, we have not performed a quantitative comparison of the experimental and calculated spectra. This situation is clearly not satisfactory, and calls for a more detailed assessment of various (TD-)DFT approaches and of different exchange–correlation functionals for the calculation of X-ray absorption spectra. Moreover, the development of accurate wave-function based methods for X-ray spectroscopy that are still applicable to multi-nuclear transition metal complexes appears promising.77,78
Nevertheless, the combination of the experimental HERFD-XANES spectra and our TD-DFT calculations using the B3LYP exchange–correlation functional demonstrates the advantages of HERFD-XANES for spectroscopic studies on multinuclear iron complexes. First, it is possible to distinguish the different types of iron–iron bonding interactions. For the pure iron carbonyl complexes, we have shown that the pre-edge region shows different signatures for the mono-, bi-, and trimetallic iron carbonyls. Interestingly, the V2C-XES spectra of these compounds show only subtle differences.25 Our assignment shows that the first pre-edge peak A is in all cases due to dz2-like orbitals, while the second peak B is due to dxy/yz-like orbitals. For the bi- and trinuclear complexes, the d-orbitals of the different iron centres form bonding and anti-bonding combinations (which are also mediated by the interaction with the ligand π*-orbitals), which modify their orbital energies and the corresponding intensities. Therefore, these peaks both reflect the change in the coordination environment of the iron centres and the change in the iron–iron interactions. An isolated iron centre in Fe(CO)5, a CO-bridged iron–iron bond in Fe2(CO)9 and the direct iron–iron bonds in Fe3(CO)12 can be clearly distinguished, even though the iron has the same oxidation state in all three complexes.
Such a distinction remains possible if some of the carbonyl ligands are replaced by hydrocarbons (cyclopentadienyl and cyclooctadienyl). In this case, the intensities of the pre-edge peaks increase, but they are largely unmodified with respect to the underlying pure carbonyl complexes. In contrast, in the V2C-XES spectra25 such an exchange of carbonyl ligands leads to the appearance of new peaks, i.e., it can clearly detect the identity of the ligands. Thus, HERFD-XANES and V2C-XES provide complementary information that, in combination, will make it possible to detect the coordination environment, different types of iron–iron interactions, and the identity of the ligands. Therefore, these methods offer a unique potential for in situ studies following both the ligand environment and the electronic structure of iron carbonyls during catalytic processes.
Acknowledgements
We thank the ESRF and staff of beamline ID26 for provision and support during the measurements. The Deutsche Forschungsgemeinschaft is acknowledged for funding through FOR 1405 (MB) and the DFG-Center for Functional Nanostructures at KIT (AJA and CRJ).References
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley-Interscience, New York, 6th edn, 1999 Search PubMed.
- N. Wiberg, E. Wiberg and A. F. Holleman, Lehrbuch der Anorganischen Chemie, Gruyter, Berlin, New York, Auflage: 102., stark umgearb. u. verb. edn, 2007 Search PubMed.
- C. Elschenbroich, Organometallics, Wiley-VCH, Weinheim, 3rd edn, 2006 Search PubMed.
- A. Chen, M. Kaminsky, G. L. Geoffroy and M. A. Vannice, J. Phys. Chem., 1986, 90, 4810–4819 CrossRef CAS.
- M. Kollár, A. De Stefanis, H. E. Solt, M. R. Mihályi, J. Valyon and A. A. G. Tomlinson, J. Mol. Catal. A: Chem., 2010, 333, 37–45 CrossRef PubMed.
- C. Li, P. Wang, W. Ying and D. Fang, Pet. Sci. Technol., 2011, 29, 1546–1554 CrossRef CAS PubMed.
- H. P. Withers, K. F. Eliezer and J. W. Mitchell, Ind. Eng. Chem. Res., 1990, 29, 1807–1814 CrossRef CAS.
- S. Lögdberg, M. Lualdi, S. Järs, J. C. Walmsley, E. A. Blekkan, E. Rytter and A. Holmen, J. Catal., 2010, 274, 84–98 CrossRef PubMed.
- C. Zhang, G. Zhao, K. Liu, Y. Yang, H. Xiang and Y. Li, J. Mol. Catal. A: Chem., 2010, 328, 35–43 CrossRef CAS PubMed.
- B. H. Davis, Catal. Today, 2009, 141, 25–33 CrossRef CAS PubMed.
- R. Schoch, H. Huang, V. Schünemann and M. Bauer, ChemPhysChem, 2014, 15, 3768–3775 CrossRef CAS PubMed.
- M. Tepluchin, M. Casapu, A. Boubnov, H. Lichtenberg, D. Wang, S. Kureti and J.-D. Grunwaldt, ChemCatChem, 2014, 6, 1763–1773 CrossRef CAS PubMed.
- H. Kuroda, Pure Appl. Chem., 1992, 64, 14491460 CrossRef.
- P. Johnston, G. J. Hutchings, N. J. Coville, K. P. Finch and J. R. Moss, Appl. Catal., A, 1999, 186, 245–253 CrossRef CAS.
- F. Gärtner, B. Sundararaju, A.-E. Surkus, A. Boddien, B. Loges, H. Junge, P. H. Dixneuf and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 9962–9965 CrossRef PubMed.
- R. Jennerjahn, R. Jackstell, I. Piras, R. Franke, H. Jiao, M. Bauer and M. Beller, ChemSusChem, 2012, 5, 734–739 CrossRef CAS PubMed.
- M. Wrighton, Chem. Rev., 1974, 74, 401–430 CrossRef CAS.
- P. T. Snee, C. K. Payne, S. D. Mebane, K. T. Kotz and C. B. Harris, J. Am. Chem. Soc., 2001, 123, 6909–6915 CrossRef CAS.
- E. Hunstock, C. Mealli, M. J. Calhorda and J. Reinhold, Inorg. Chem., 1999, 38, 5053–5060 CrossRef CAS PubMed.
- W. F. Edgell, W. E. Wilson and R. Summitt, Spectrochim. Acta, 1963, 19, 863–872 CrossRef CAS.
- L. Bañares, T. Baumert, M. Bergt, B. Kiefer and G. Gerber, Chem. Phys. Lett., 1997, 267, 141–148 CrossRef.
- C. Crotti, E. Farnetti, T. Celestino and S. Fontana, J. Electron Spectrosc. Relat. Phenom., 2003, 128, 141–157 CrossRef CAS.
- C. Crotti, E. Farnetti, T. Celestino, M. Stener and S. Fontana, Organometallics, 2004, 23, 5219–5225 CrossRef CAS.
- M. Bauer and H. Bertagnolli, in Methods in Physical Chemistry, ed. R. Schäfer and P. C. Schmidt, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 231–269 Search PubMed.
- M. U. Delgado-Jaime, S. DeBeer and M. Bauer, Chem. – Eur. J., 2013, 19, 15888–15897 CrossRef CAS PubMed.
- M. F. Ruiz-Lopez, M. Loos, J. Goulon, M. Benfatto and C. R. Natoli, Chem. Phys., 1988, 121, 419–437 CrossRef CAS.
- M. Bauer, T. Kauf, J. Christoffers and H. Bertagnolli, Phys. Chem. Chem. Phys., 2005, 7, 2664–2670 RSC.
- M. Bauer and C. Gastl, Phys. Chem. Chem. Phys., 2010, 12, 5575–5584 RSC.
- P. Eisenberger, P. M. Platzman and H. Winick, Phys. Rev. Lett., 1976, 36, 623–626 CrossRef CAS.
- W. M. Heijboer, P. Glatzel, K. R. Sawant, R. F. Lobo, U. Bergmann, R. A. Barrea, D. C. Koningsberger, B. M. Weckhuysen and F. M. F. de Groot, J. Phys. Chem. B, 2004, 108, 10002–10011 CrossRef CAS.
- O. V. Safonova, M. Tromp, J. A. van Bokhoven, F. M. F. de Groot, J. Evans and P. Glatzel, J. Phys. Chem. B, 2006, 110, 16162–16164 CrossRef CAS PubMed.
- P. Glatzel, M. Sikora, G. Smolentsev and M. Fernández-García, Catal. Today, 2009, 145, 294–299 CrossRef CAS PubMed.
- A. J. Atkins, Ch. R. Jacob and M. Bauer, Chem. – Eur. J., 2012, 18, 7021–7025 CrossRef CAS PubMed.
- A. J. Atkins, M. Bauer and Ch. R. Jacob, Phys. Chem. Chem. Phys., 2013, 15, 8095–8105 RSC.
- H. H. Johann, Z. Phys., 1931, 69, 185–206 CrossRef CAS.
- T. Johansson, Z. Phys., 1933, 82, 507–528 CrossRef CAS.
- C. Gauthier, V. A. Solé, R. Signorato, J. Goulon and E. Moguiline, J. Synchrotron Radiat., 1999, 6, 164–166 CrossRef CAS PubMed.
- Theoretical Chemistry, Vrije Universiteit Amsterdam, ADF, Amsterdam density functional program, http://www.scm.com.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- M. Stener, G. Fronzoni and M. de Simone, Chem. Phys. Lett., 2003, 373, 115–123 CrossRef CAS.
- K. Ray, S. DeBeer George, E. I. Solomon, K. Wieghardt and F. Neese, Chem. – Eur. J., 2007, 13, 2783–2797 CrossRef CAS PubMed.
- S. DeBeer George, T. Petrenko and F. Neese, Inorg. Chim. Acta, 2008, 361, 965–972 CrossRef CAS PubMed.
- P. Banerjee, S. Sproules, T. Weyhermüller, S. DeBeer George and K. Wieghardt, Inorg. Chem., 2009, 48, 5829–5847 CrossRef CAS PubMed.
- P. Chandrasekaran, S. C. E. Stieber, T. J. Collins, J. Lawrence Que, F. Neese and S. DeBeer, Dalton Trans., 2011, 40, 11070–11079 RSC.
- C. C. Scarborough, S. Sproules, T. Weyhermüller, S. DeBeer and K. Wieghardt, Inorg. Chem., 2011, 50, 12446–12462 CrossRef CAS PubMed.
- M. Roemelt, M. A. Beckwith, C. Duboc, M.-N. Collomb, F. Neese and S. DeBeer, Inorg. Chem., 2012, 51, 680–687 CrossRef CAS PubMed.
- C. C. Scarborough, S. Sproules, C. J. Doonan, K. S. Hagen, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2012, 51, 6969–6982 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- S. Bernadotte, A. J. Atkins and Ch. R. Jacob, J. Chem. Phys., 2012, 137, 204106 CrossRef PubMed.
- S. DeBeer George, T. Petrenko and F. Neese, J. Phys. Chem. A, 2008, 112, 12936–12943 CrossRef CAS PubMed.
- E. Epifanovsky, I. Polyakov, B. Grigorenko, A. Nemukhin and A. I. Krylov, J. Chem. Theory Comput., 2009, 5, 1895–1906 CrossRef CAS.
- U. Varetto, Molekel, Swiss National Supercomputing Centre, Manno (Switzerland), 2009 Search PubMed.
- E. J. Baerends, C. Oudshoorn and A. Oskam, J. Electron Spectrosc. Relat. Phenom., 1975, 6, 259–267 CrossRef CAS.
- M. Kotzian, N. Roesch, H. Schroeder and M. C. Zerner, J. Am. Chem. Soc., 1989, 111, 7687–7696 CrossRef CAS.
- D. Guenzburger, E. Baggio-Saitovitch, M. A. D. Paoli and H. Manela, J. Chem. Phys., 1984, 80, 735–744 CrossRef CAS PubMed.
- R. Fukuda, M. Ehara, H. Nakatsuji, N. Kishimoto and K. Ohno, J. Chem. Phys., 2010, 132, 084302 CrossRef PubMed.
- B. Ahr, M. Chollet, B. Adams, E. M. Lunny, C. M. Laperle and C. Rose-Petruck, Phys. Chem. Chem. Phys., 2011, 13, 5590–5599 RSC.
- M. Bauer, T. Stalinski and E. F. Aziz, ChemPhysChem, 2011, 12, 2088–2091 CrossRef CAS PubMed.
- R. van Leeuwen and E. J. Baerends, Phys. Rev. A: At., Mol., Opt. Phys., 1994, 49, 2421–2431 CrossRef CAS.
- P. R. T. Schipper, O. V. Gritsenko, S. J. A. van Gisbergen and E. J. Baerends, J. Chem. Phys., 2000, 112, 1344–1352 CrossRef CAS PubMed.
- G. Fronzoni, P. Decleva and A. Lisini, Chem. Phys., 1993, 174, 57–70 CrossRef CAS.
- F. A. Cotton and J. M. Troup, J. Am. Chem. Soc., 1974, 96, 4155–4159 CrossRef CAS.
- J. H. Jang, J. G. Lee, H. Lee, Y. Xie and H. F. Schaefer, J. Phys. Chem. A, 1998, 102, 5298–5304 CrossRef CAS.
- W. Heijser, E. J. Baerends and P. Ros, Faraday Symp. Chem. Soc., 1980, 14, 211–234 RSC.
- C. W. Bauschlicher, Jr., J. Chem. Phys., 1986, 84, 872–875 CrossRef PubMed.
- J. Li and K. Jug, Inorg. Chim. Acta, 1992, 196, 89–95 CrossRef CAS.
- D. Braga, F. Grepioni, E. Tedesco, M. J. Calhorda and P. E. M. Lopes, J. Chem. Soc., Dalton Trans., 1995, 3297–3306 RSC.
- J. Reinhold, A. Barthel and C. Mealli, Coord. Chem. Rev., 2003, 238–239, 333–346 CrossRef CAS.
- J. Reinhold, O. Kluge and C. Mealli, Inorg. Chem., 2007, 46, 7142–7147 CrossRef CAS PubMed.
- P. Hirva, M. Haukka, M. Jakonen and M. A. Moreno, J. Mol. Model., 2008, 14, 171–181 CrossRef CAS PubMed.
- F. A. Lima, R. Bjornsson, T. Weyhermüller, P. Chandrasekaran, P. Glatzel, F. Neese and S. DeBeer, Phys. Chem. Chem. Phys., 2013, 15, 20911–20920 RSC.
- J. Wenzel, M. Wormit and A. Dreuw, J. Comput. Chem., 2014, 35, 1900–1915 CrossRef CAS PubMed.
- J. Wenzel, M. Wormit and A. Dreuw, J. Chem. Theory Comput., 2014, 10, 4583–4598 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional calculated X-ray absorption spectra. See DOI: 10.1039/c5cp01045d |
| This journal is © the Owner Societies 2015 |