
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of Pt NPs crystallinity and distribution on the photocatalytic activity of Pt–g-C3N4†
F.
Fina
a,
H.
Ménard
b and
J. T. S.
Irvine
*a
aSchool of Chemistry, University of St Andrews, St Andrews, KY16 9ST, Scotland, UK. E-mail: jtsi@st-andrews.ac.uk
bSasol Technology (UK) Ltd, St Andrews, KY16 9ST, Scotland, UK
First published on 30th April 2015
Abstract
Loading of a co-catalyst on the surface of a semiconductor photocatalyst is often carried out without considering the effect of the loading procedure on the final product. The present study looks in detail at the effect that the loading method has on the morphology and final composition of platinum-based nanoparticles by means of XPS and TEM analysis. Additionally, reduction pre-treatments are performed to investigate how the coverage, crystallinity and composition of the NPs affect the photocatalytic H2 evolution. The activity of Pt–g-C3N4 can significantly be enhanced by controlling the properties of the co-catalyst NPs.
Introduction
Solar energy is a green, abundant and renewable energy source and it can represent an alternative to the non-renewable fossil fuels used today. However efficient conversion of solar to chemical energy is still to be achieved.1 Natural photosynthesis inspires an artificial process where sunlight is exploited to produce green and renewable fuels without the emission of greenhouse gases. For instance hydrogen, together with oxygen, could be produced from pure water via overall water splitting and used as green fuel.Since Honda and Fujishima successfully produced hydrogen from water via photoelectrochemistry,2 much attention has been focused on the development of new semiconductor materials able to split water by using light. However, only few catalysts can produce both gases from water. Most of them are able to produce either H2 or O2. To complete the electron cycle an electron donor or an electron acceptor, respectively, are needed.3,4 In addition, most of the H2 producing catalysts require ultraviolet light which represents only 4% of the whole sunlight spectrum.3 Therefore, the development of visible light active catalysts is important.1,3,5–8 A suitable material must be able to absorb visible light by having a narrow Band Gap (<3.0 eV), but also wide enough (≥1.23 eV) to be able to drive the water splitting reaction.3
Graphitic carbon nitride (g-C3N4) has recently attracted significant attention because it fulfils all the above stated requirements.9–15 As reported by Wang et al. in 2009,9 g-C3N4 is a non-metallic semiconductor with a direct band gap of 2.7 eV allowing visible light absorption. Easily synthesised from cheap starting materials, g-C3N4 is a poly-conjugated polymer with a layered structure very similar to graphite.
Graphitic carbon nitride can produce 1 to 10 μmol h−1 g−1 of H2 under visible light irradiation;9 however, its performance can be significantly improved by the use of a co-catalyst. Noble metals such as Pt, Ru, Rh and transition metals such as Ni are often employed as co catalysts in energy storage processes.3,16–18 In the specific case of photocatalytic hydrogen evolution the co-catalyst is usually considered to act as an electron trap, preventing charge recombination from occurring and making electrons and holes more readily available to the photocatalytic process.3,9,10,13 In the case of g-C3N4, different transition metals have been investigated and the highest efficiencies were obtained with platinum;10,13 however, these can vary from 20 to 200 μmol h−1 g−1.9,10,13 There are several factors that may explain this difference in performances, the choice of the sacrificial agent used, the pH of the solution, the intensity and spectrum of the light source and the loading procedure of the co-catalyst. Here we focus on the behaviour of platinum loaded on g-C3N4; from the loading procedure to the interaction with light. In the literature, the standard loading procedure of platinum is either by thermal decomposition of the precursor after its impregnation on the surface of the support,8,10,19 or by photodeposition which involves adding the precursor under irradiation.10
In this study the two classic loading procedures are compared with a simple impregnation method, additionally the effect of reduction pre-treatments on the performance is also investigated. Since a systematic study of the effect that the loading procedure and light have on the co-catalyst has not been carried out, we approach the investigation of this active system in a detailed way. We believe that by fully understanding the process, it will not only be possible to further enhance the performance of platinum loaded g-C3N4, but also developing concepts that could be applied to enhance other photocatalytic systems.
Experimental
Synthesis of the catalyst and co-catalyst loading
Graphitic carbon nitride was synthesised via thermal polycondensation in air of Melamine (Aldrich, 99% purity). Melamine was added into an alumina crucible covered with aluminium foil to create a closed system in order to restrict the gases evolved during the process to within the vessel. The organic precursor was heated up to 500 °C at a rate of 5 °C min−1. The temperature is then kept constant for 15 hours. After cooling down, the yellow product was ground to fine powder in an Agate mortar.Platinum was loaded using chloroplatinic acid hexahydrate (H2PtCl6·6H2O; Alfa Aesar, 99.9%) as platinum precursor. In all of the samples the platinum content was 1 wt% (1 mg of Pt for every 100 mg of g-C3N4). Three different loading procedures were followed: impregnation, impregnation followed by calcination and photodeposition. When the impregnation method was used, g-C3N4 was suspended in an aqueous solution of H2PtCl6·6H2O (1 g L−1). The suspension was then stirred overnight and subsequently heated at 90 °C under stirring to allow water evaporation. In a variation of this procedure thermal treatment of the impregnated sample was carried out. The powder was heated in a muffle furnace at 180 °C for 2 hours.8 The third loading procedure investigated was performed by in situ photodeposition: the solution of platinum precursor was directly added into the testing chamber and no pre-treatment of the catalyst was carried out.
Two different reduction procedures were applied. Reduction with 5% H2 was carried out in a tube furnace at 300 °C for 12, 24, 36 and 48 h.20 Sodium borohydride was used as reducing agent in the second procedure. Graphitic carbon nitride was suspended in an aqueous solution of H2PtCl6·6H2O (1 g L−1) and a fresh solution of NaBH4 (10 times excess)21 was added under argon atmosphere to the stirring solution. After 2 h the stirring was stopped and the powder was left depositing at the bottom of the flask. After removing the solution, fresh degassed water was added to dissolve residues of sodium borohydride. The sample was then dried at room temperature under flowing argon.
Characterisation
The microstructure was investigated by powder X-ray diffraction (XRD) using an Empyrean PANalytical series 2 diffractometer with a Cu Kα radiation source (λ = 1.5406 Å). Crystallite sise analysis is performed by applying the Scherrer and the Williamson–Hall equations.22,23To determine the absorbance and the Band Gap of the samples, ultraviolet-visible absorbance spectra were collected with a JASCO-V-650 spectrophotometer equipped with an integrated sphere coated with barium sulphate. The Band Gap energies were calculated via Kubelka–Munk transformation with a dedicated software (SpectraManager, JASCO).
High Resolution Transmission Electron Microscopy images (HRTEM) were obtained with a Jeol JEM-2011 operated at 200 KV using a Gatan 794 CCD camera to record digital images.
The chemical composition of the nanoparticles was investigated with X-ray photoelectron spectroscopy (XPS) performed on a Kratos AXIS Ultra DLD, with a monochromatic Al Kα (1486.6 eV) X-ray source and for peak calibration, adventitious C was used at 284.6 eV. The samples analysed after photocatalytic test were dried under slow argon flow to minimise exposition to air and possible changes in the oxidation state of the platinum species. They were, then, transferred in the XPS instrument under inert atmosphere using a glovebox.
Photocatalytic performance evaluation
Measurements of the photocatalytic hydrogen evolution were performed in a custom-made photocatalytic reactor with top irradiation through a quartz window. In a typical experiment 0.2 g of catalyst (1 g L−1) were suspended in 0.2 L of deionised water and oxalic acid (0.025 M) was used as sacrificial agent. During the investigation of the pre-reduction treatment, less catalyst was used (0.1 g in 0.1 ml of water) in order to prevent saturation of the GC with an excessive amount of hydrogen. The amount of oxalic acid was also doubled (0.05 M) to allow collection of more measurements during the steady state of the most active materials. The inner atmosphere was purged with Argon gas to remove any trace of air. Top irradiation was carried out with a 250 W iron-doped metal halide ultraviolet-visible lamp (≥290 nm; UV Light Technology Limited) with a cut-off filter (≥420 nm; Borosilicate Coated Glass, UQG Optics Ltd) to block UV-irradiations. Hydrogen evolution was monitored for an experimental duration of 20 hours using a gas chromatograph (Agilent 3000 Micro Gas Chromatograph).Results and discussion
The X-ray diffraction pattern of graphitic carbon nitride synthesised by thermal polycondensation of melamine at 500 C is illustrated in Fig. 1a. The characteristic peaks of graphitic-carbon nitride can be identified. The strong peak at 2θ = 27.5° corresponds to an inter-layer d-spacing of 0.324 nm, the (002) plane typical of graphitic materials. A smaller peak at 2θ = 13.0° is usually associated to an intra-layer distance of 0.680 nm, (100) plane.24 The ultraviolet-visible spectra of g-C3N4 (Fig. 1b) shows an absorption in the visible range and a band gap of 2.76 eV is calculated via Kubelka–Munk transformation of the absorbance curve (Fig. 1b, inset). | ||
| Fig. 1 (a) XRD pattern, (b) UV-Vis absorbance spectrum (inset): Kubelka–Munk, (c) XPS N1s and (d) C1s spectra of g-C3N4. | ||
The N 1s and C 1s XPS spectra of g-C3N4 are shown in Fig. 1c and d respectively. In the N 1s region the component at 398.7 eV identifies sp2 nitrogen with two neighbouring atoms within the triazine ring (C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C).24–28 This is further supported by the presence of a π-excitation signal at 404.5 eV.29 The components at 400.0 eV and 401.2 eV correspond to the bridging nitrogen between the aromatic rings and the amine groups respectively.25–28 In the carbon region, four components are identified. The main peak at 288.3 eV corresponds to sp2 carbon in the triazine ring of g-C3N4 (C–NC).26,29–32 This is confirmed by the presence in the C 1s spectrum of a component at 293.7 eV which is associated with the π-excitations characteristic of double bonds.29 The small peaks at 284.7 eV and 289.8 eV are assigned to C–C and C–O bonds respectively.33 These data confirm the successful synthesis of g-C3N4.24
C).24–28 This is further supported by the presence of a π-excitation signal at 404.5 eV.29 The components at 400.0 eV and 401.2 eV correspond to the bridging nitrogen between the aromatic rings and the amine groups respectively.25–28 In the carbon region, four components are identified. The main peak at 288.3 eV corresponds to sp2 carbon in the triazine ring of g-C3N4 (C–NC).26,29–32 This is confirmed by the presence in the C 1s spectrum of a component at 293.7 eV which is associated with the π-excitations characteristic of double bonds.29 The small peaks at 284.7 eV and 289.8 eV are assigned to C–C and C–O bonds respectively.33 These data confirm the successful synthesis of g-C3N4.24
Platinum is loaded onto the surface of the catalyst from H2PtCl6·6H2O by following three different procedures: simple impregnation, impregnation followed by calcination at 180 °C and photodeposition. The materials are tested for hydrogen evolution under visible light and their performances are compared to that of bare g-C3N4 in Table 1. Graphitic carbon nitride without the aid of the co-catalyst shows a hydrogen production of 2 μmol h−1 g−1. The loading of 1 wt% platinum brings a significant increase in the performance (Table 1). This increase is different depending on the loading procedure. After impregnation followed by calcination the hydrogen evolution rate is found to be ∼1100 μmol h−1 g−1. When no thermal treatment is carried out after impregnation of the precursor the hydrogen evolution rate is ∼1280 μmol h−1 g−1, showing an increase of 16%, well above the estimated experimental error of 5%. The third loading procedure investigated, photodeposition, sees the platinum precursor added directly into the testing chamber9,10 without any pre-treatment of the catalyst. After performance evaluation the hydrogen evolution rate is found being ∼732 μmol h−1 g−1, 43% lower than when simple impregnation is used (Table 1).
| g-C3N4 | Impregnation | Impregnation and calcination | Photodeposition | |||
|---|---|---|---|---|---|---|
| 1280 | 1100 | 732 | ||||
| H2 evolution rate (μmol h−1 g−1) | 2 | b.pc. | a.pc. | b.pc. | a.pc. | a.pc. |
| PtClx% | — | 51.6 | — | 54.8 | — | — |
| PtO% | — | 48.4 | 42.6 | 45.2 | 46.9 | 25.2 |
| Pt0% | — | — | 57.4 | — | 53.1 | 74.8 |
Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N (±0.010) :N (±0.010) |
— | 0.040 | 0.040 | 0.037 | 0.039 | 0.080 |
The oxidation state of platinum is determined via X-ray photoelectron spectroscopy (Table 1). The impregnated sample (Fig. 2a) is initially compared with the calcined sample (Fig. 2b) before photocatalysis. Fig. 2a shows the XP Pt 4f spectrum of as-made impregnated Pt–g-C3N4. The component at 72.8 eV is assigned to PtIIO,20,34,35 while the one at 75.0 eV corresponds to platinum chlorides, PtClx. The presence of chlorides is confirmed by the presence of a peak at 198.1 eV in the Cl 2p region (Fig. S1, ESI†). The relative amount of each platinum species is represented as a peak area ratio between the platinum species of interest and the total platinum. Therefore, 51.6% of platinum is present in the form of PtClx, while 48.4% in the form of PtO. A representation of the coverage of platinum species on the surface of graphitic carbon nitride is given by the Pt:N peak area ratio. After impregnation this ratio is found to be 0.040 ± 0.01. On the surface of the sample calcined at 180 °C, PtClx and PtO are found with relative amounts of 54.8% and 45.2% respectively (Fig. 1b and Fig. S1b, ESI†), similarly to the simply impregnated sample. The PtO:PtClx ratio for the two samples are 0.9 and 0.8 respectively. Additionally, the Pt:N ratio for the calcined sample is found to be 0.037, the same within error as the impregnated sample. These findings indicate that the additional calcination step in the loading procedure does not bring to major changes in the chemical composition of the co-catalyst.
 | ||
| Fig. 2 XP spectra of the Pt 4f region for Pt–g-C3N4 (a) impregnated and (b) calcined before photocatalysis; (c) impregnated and (d) photodeposited after photocatalysis. | ||
After photocatalysis the simply impregnated Pt–g-C3N4 is compared with the photodeposited sample. The XP spectra of the Pt 4f region of the two samples are illustrated in Fig. 2c and d respectively. After 20 h of visible light irradiation the peak at 75.0 eV disappears from the impregnated sample indicating the absence of chlorides; this is accompanied by the formation of Pt0 characterised by a binding energy of 70.8 eV with a relative amount of 57.4%.19,20 The PtO:Pt0 in this case is 0.8 (1:1.3) and the Pt:N is 0.040, not affected by exposure to light. (For comparison, the values obtained for the calcined after photocatalysis are listed in Table 1 and shown in Fig. S2, ESI.†)
The same species are present on the surface of the photodeposited sample (Fig. 2d). The relative amount of PtO is 25.2% and that of Pt0 is 74.8%, with a PtO:Pt0 ratio of 0.3 (1:3). An increase in the Pt:N ratio can also be observed, this is found having a value of 0.080, indicating a higher coverage of the catalyst surface by the platinum. These preliminary results show that the photoreduction of the co-catalyst occurs in all cases and that the loading procedure can affect the oxidation state of the co-catalyst together with the coverage of the catalyst's surface.
The morphology of the platinum co-catalyst is investigated via Transmission Electron Microscopy (TEM and HRTEM, Fig. 3). The impregnated sample is first compared to the calcined before photocatalysis (Fig. 3a–d). When the platinum precursor is simply impregnated the size of nanoparticles varies between 3 and 10 nm (Fig. 3a). The high resolution TEM images (Fig. 3b) reveal lattice fringes of value 0.27 nm which can be attributed to the (002) face of PtO.36 This is of further support to the XPS results which identified PtO as one of the main platinum species after impregnation. PtClx can be expected to be present as a monolayer and therefore it would not appear in the form of nanoparticles. After calcination at 180 °C, lattice fringes characteristic of PtO (0.27 nm) can still be observed, however the particle size of the nanoparticles increases and agglomerates from 20 to 40 nm size are present (Fig. 3c). The difference in PtO particle size provides an explanation for the lower activity of the calcined Pt–g-C3N4 compared to the simply impregnated one. Big particle size reduce the number of available active sites on the platinum. The TEM and HRTEM images of the impregnated sample after photocatalysis are compared with those of the photodeposited in Fig. 3e–h. Due to the higher number of visible nanoparticles compared to the samples before test, it is possible, in this case, to determine the statistical particle size distribution for the samples (Fig. 3e and g insets). When photodeposition is used as the loading procedure, nanoparticles with an average diameter of 1.3 ± 0.6 nm are obtained. However, when the precursor is only impregnated the average diameter increases to 2.3 ± 1.2 nm with a wider distribution and sporadic clusters formation. These value are coherent with the results obtained from the XPS analysis which showed a higher Pt:N ratio for the photodeposited sample compared to the impregnated one. This can now be explained by the smaller size of the particles. High resolution TEM images (Fig. 3i and h) reveal that after photocatalytic experiments the lattice fringes for both samples are 0.23 nm which is characteristic of Pt0.37 This confirms the formation of platinum metal on the surface of graphitic carbon nitride after photocatalytic test. It is also important to report that for the samples before photocatalytic test, TEM specimen preparation can easily cause the removal of the nanoparticles from the catalyst surface, indicating a weak interaction between the nanoparticles and graphitic carbon nitride. However, when the same preparation method is used for the samples after the photocatalytic process, the removal of the nanoparticles does not occur. The reason for this anchoring of the nanoparticles seems to be the stronger interaction created between the catalyst (in particular the nitrogen atoms) and the platinum metal during the photoreduction of the co-catalyst precursor.28,35
 | ||
| Fig. 3 TEM (a, c, e, g) and HRTEM (b, d, f, h) images of different Pt–g-C3N4 samples before (a–d) and after (e–h) photocatalytic process. Images of platinum a, (b) impregnated on g-C3N4 and (c, d) after calcination treatment. Images of (e, f) impregnated and (g, h) photodeposited Pt–g-C3N4 after 20 hours of visible irradiation. Insets e and g: statistical nanoparticle size distribution. | ||
From these results it is possible to state that the different loading methods affect not only the oxidation state and relative amount of the platinum species, but also the morphology. The differences between the samples result in differences in activity for hydrogen evolution. At this point two factors can be considered having an effect on the activity: the coverage of the co-catalyst on the catalyst and the relative amounts of metal and metal oxide.
For the most active material (simply impregnated) the oxidation state of platinum during the photocatalytic test was monitored via XPS. Fig. 4a shows the profile of the hydrogen evolution rate against experimental time. The rate increases until reaching a steady state (330 μmol h−1) after ∼6 h. It is important to specify that five experiments were carried out covering different experimental lengths to allow characterisation by XPS, the results of which are shown in Fig. 4b. The metal content increases in the first few hours and reaches a plateau with a ∼1:1 PtII to Pt0 ratio. This profile correlates strongly with the trend observed for the H2 evolution rate (Fig. 4a) suggesting that during the first few hours of the photocatalytic process, part of the excited electrons are employed for the photoreduction of platinum to achieve a constant and optimum PtII/Pt0 ratio of 1:1. From the results obtained so far it seems that a high proportion of Pt0 could relate to the lower activity of photodeposited Pt–g-C3N4 (1:3 PtII/Pt0 ratio) which produces 43% less hydrogen than when impregnation is used as the loading procedure. In order to confirm this hypothesis, reduction pre-treatments were carried out on the simply impregnated Pt–g-C3N4.
 | ||
Fig. 4 (a) H2 evolution rate (μmol h−1) of five different experiments: ( ) 3 hours, ( ) 3 hours, ( ) 5 hours, ( ) 5 hours, ( ) 10 hours, (◇) 12 hours, (○) final 10 hours. Test conditions: 0.2 g of catalyst in 0.2 L of 0.025 M aqueous solution of oxalic acid, visible light (λ ≥ 420 nm). (b) Pt0% of total platinum content on Pt–g-C3N4 as obtained from XPS analysis (for XP spectra see Fig. S3, ESI†). ) 10 hours, (◇) 12 hours, (○) final 10 hours. Test conditions: 0.2 g of catalyst in 0.2 L of 0.025 M aqueous solution of oxalic acid, visible light (λ ≥ 420 nm). (b) Pt0% of total platinum content on Pt–g-C3N4 as obtained from XPS analysis (for XP spectra see Fig. S3, ESI†). | ||
Two different reduction procedures are compared: reduction with 5% H2/Ar20 and chemical reduction with NaBH4.21 For the reduction under 5% H2/Ar the temperature is set at 300 °C. After reducing the sample for various lengths of time, testing for hydrogen evolution (Fig. S4, ESI†) and analysing with XPS (Table S1, ESI†) the products, the optimum reduction duration is found to be 24 h. After this length of time no significant difference can be observed in the hydrogen evolution rate (Fig. S3, ESI†) or the platinum metal content (Table 1). For the chemical reduction, NaBH4 is used as the reducing agent in a molar excess of 10.21 The H2 evolution rates at the steady state for the reduced samples are compared in Table 2.
| 5% H2/Ar | NaBH4 | NaBH4 + 5% H2/Ar | |
|---|---|---|---|
| H2 evolution rate (μmol h−1 g−1) | 3900 | 515 | 1721 |
| PtClx% | — | 3.9 | — |
| PtO% | 44.2 | 20.2 | 16.6 |
| Pt0% | 55.8 | 75.9 | 83.4 |
| Pt:N (±0.010) |
0.051 | 0.282 | 0.102 |
| TEM particle size (nm) | 4.9 ± 4.7 | 3.7 ± 2.9 | 10 ± 9.6 |
| S crystallite size (nm) | 16 | 8 | 20 |
| W–H crystallite size (nm) | 15 | 5 | 19 |
| W–H strain | −2.25 × 10−4 | −4.98 × 10−3 | −2.67 × 10−4 |
Reduction under hydrogen gas brings to a significant increase in performance with an H2 evolution rate of 3900 μmol h−1 g−1, corresponding to an increase of a factor 3 compared to the not reduced sample. XPS analysis reveals a platinum metal content of 55.8% (Table 2), while the remaining 44.2% of platinum is identified as PtO, resulting in a 1:1.3 PtO:Pt0 ratio. The Pt:N ratio after reduction shows no variation compared to the impregnated sample with a value of 0.051. These values are comparable with those of the impregnated sample after photocatalysis, therefore the higher activity cannot be explained by the composition of the platinum. The XRD pattern in Fig. 5a-I shows sharp peaks at 2θ = 39.8°, 46.3°, 67.6° and 81.4°, characteristic of the (111), (200), (220) and (311) planes of Pt0 (PDF #04-0802).34,35,38 By applying the Scherrer equation22 and the Williamson–Hall plot23 the crystallite size and the level of crystallinity of the platinum nanoparticles are estimated (Table 2 and Fig. 5b). The two equations give very similar values: 16 nm and 15 nm respectively, suggesting a high level of crystallinity, which is confirmed by the very small slope in the Williamson–Hall plot (Table 2 and Fig. 5b). The average particle size obtained from TEM images (Fig. S5, ESI†) results being 4.9 ± 4.7 nm, significantly smaller than the values obtained from the XRD pattern. This is probably due to the size dependence of the XRD technique, more sensitive to bigger and more crystallised particles. Nonetheless, the crystallite/particle size is bigger than the not reduced sample indicating that the calcination at 300 °C brings to a coalescence of the particles, while the unaffected Pt:N ratio suggests that the particles do not grow in depth.
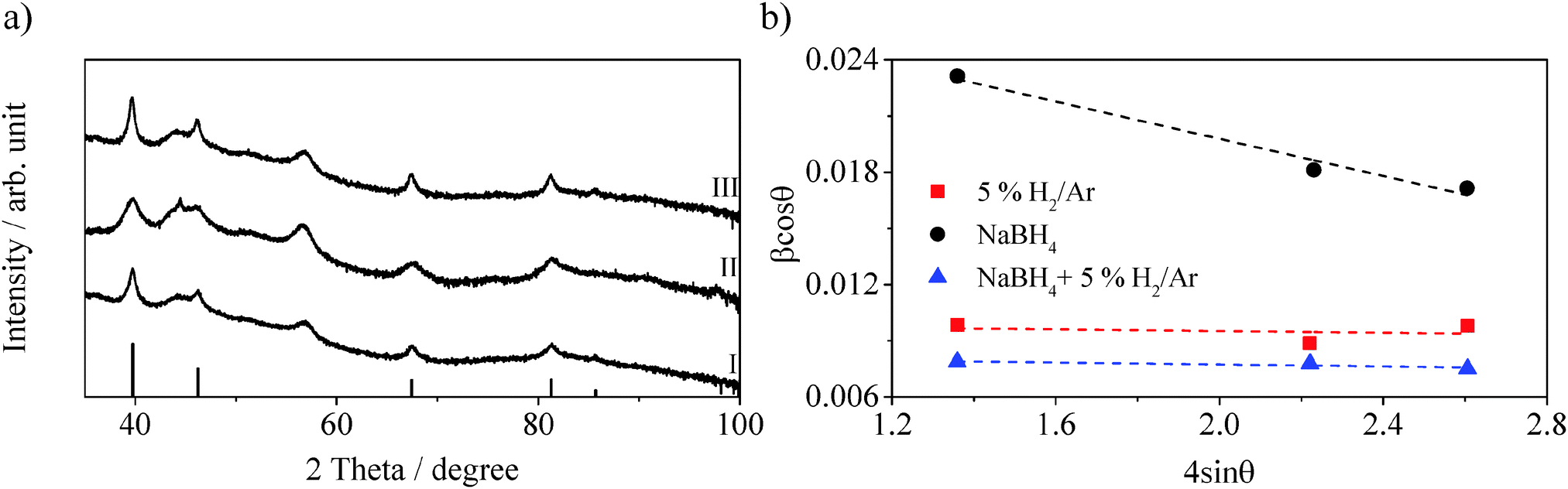 | ||
| Fig. 5 (a) XRD patterns of Pt–g-C3N4 reduced under 5% H2/Ar (I), NaBH4 (II) and double reduced (III), bars Pt XRD pattern as from PDF card #04-0802; (b) Williamson–Hall plots of the three reduced samples. | ||
Chemical reduction via NaBH4 causes a decrease of a factor 2.5 in the H2 evolution rate, with a value of 515 μmol h−1 g−1. XPS analysis shows that after chemical reduction the relative amount of platinum metal is 75.9%. The sample is now characterised by a PtO:Pt0 ratio of 1:3.8. The coverage of the nanoparticles over g-C3N4 (Pt:N) is also found increased to a value of 0.282, suggesting that smaller or thinner nanoparticles are formed. Fig. 5a-II shows the XRD pattern for the chemically reduced Pt–g-C3N4. In this case the reflections characteristic of fcc Pt0 appear broad. The crystallite size analysis via Scherrer equation reveals a value of 8 nm while the Williamson–Hall equation reveals a value of 5 nm. The difference between these values is explained by the low crystallinity of the metal nanoparticles indicated by the negative slope of the Williamson–hall plot (Fig. 5b and Table 2), suggesting a higher level of strain than the previous sample. Nonetheless, the low crystallite size confirms the high Pt:N ratio obtained from XPS analysis (Table 2). From TEM images (Fig. S6, ESI†), the average particle size is found to be 3.7 ± 2.9 nm, in line with the results from the XRD analysis.
The comparison of the results obtained from the two reduced samples indicates that three factors could be responsible for the difference in H2 evolution rate:
• platinum species relative amount;
• platinum nanoparticles coverage (Pt:N);
• platinum nanoparticles crystallinity.
To further investigate the effects of the three factors, the sample is initially reduced with NaBH4 and subsequently exposed to reduction under 5% H2/Ar at 300 °C. The H2 evolution rate of the double reduced sample is now 1721 μmol h−1 g−1, three times higher than the chemically reduced only. XPS analysis of the sample shows that the double treatment produces a Pt0 content of 83.4% (Table 2) which is comparable to that of the chemically reduced sample. On the other hand, the thermal treatment brings a decrease of the Pt:N ratio, now only 0.102, about 3 times less that the chemically reduced sample (Table 2). This can be associated to a coalescence of the nanoparticles due to the calcination step. The XRD pattern (Fig. 5a-III) shows the peaks at 2θ = 39.8°, 46.3°, 67.6° and 81.4° characteristic of the cubic structure of platinum metal.34,35,38 The Scherrer and the Williamson–Hall analyses reveal crystallite sizes with values of 20 nm and 19 nm respectively for the double reduced sample. Additionally the slope of the Williamson–hall plot indicate a level of crystallinity comparable to the sample reduced with 5% H2/Ar. The average particle size obtained from TEM images (Fig. S7, ESI†) is 10 ± 9.6 nm, consistent with the results from the XRD analysis.
The results indicate that a high metal content is not associated to an enhancement in activity for hydrogen evolution. However, crystallinity and coverage are shown to have an effect on the hydrogen evolution rate. An increase in crystallinity is accompanied by an increase in activity. Well crystallised nanoparticles may have an improved interaction with the surface of the catalyst compared to less crystallised NPs. Additionally the thermal treatment may present different exposed facets. In the literature it has been reported that different crystal facets, (111), (100) etc., may have different catalytic activity.39,40 The coverage of the catalyst surface by the nanoparticles has the most important effect on the hydrogen evolution. A decrease of about a factor three in the coverage (Pt:N of the double reduced sample) results in an increase of a factor three in the performance (Table 2). The beneficial effect of a lower coverage (Pt:N) can be explained by a higher exposure of the catalyst surface to light absorption or more sites available for the oxidation of the sacrificial agent (oxalic acid). Additionally, a small Pt:N ratio indicates bigger particles and it has been reported that the activity of metal nanoparticles can increase with particle size.41,42 Crystallinity as well plays, even though in smaller measure, a role in enhancing the activity of Pt–g-C3N4.
Conclusions
The effect of the loading procedure and different reduction treatments on the activity of Pt–g-C3N4 for photocatalytic hydrogen evolution was investigated by mean of XPS, XRD and TEM. A simple impregnation method was found to be the most effective loading procedure between the ones that were investigated. The investigation of the platinum oxidation state during photocatalytic test revealed that the metal content increases with light exposure due to the photoreduction of the platinum precursor by the excited electrons.Reduction pre-treatments provided a better understanding of the effect on the performance of the relative amount of Pt0, the coverage of the nanoparticles with respect to g-C3N4 and the crystallinity of the Pt0 NPs. It was demonstrated that the oxygen content of the co-catalyst does not seem to affect the performances. From the study, it emerged that the main factor affecting the H2 evolution rate of the materials is the coverage, represented as the Pt:N ratio, with a lower coverage being more beneficial. Following the coverage, crystallinity of the metal nanoparticles also plays a role in the catalytic activity. Pt–g-C3N4 with particles characterised by a high crystallinity results in a high H2 evolution.
Acknowledgements
We thank EPSRC for support through the EPSRC/NSF chemistry programme and the Royal Society for a Wolfson Merit award.Notes and references
- K. Takanabe and K. Domen, Green, 2011, 1, 313–322 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625–627 CrossRef CAS PubMed.
- A. Kudo, Int. J. Hydrogen Energy, 2007, 32, 2673–2678 CrossRef CAS PubMed.
- D. Mitoraj and H. Kisch, Angew. Chem., Int. Ed., 2008, 47, 9975–9978 CrossRef CAS PubMed.
- X. Xu, G. Liu, C. Randorn and J. T. S. Irvine, Int. J. Hydrogen Energy, 2011, 36, 13501–13507 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940–4947 CAS.
- L. Ge, Mater. Lett., 2011, 65, 2652–2654 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- S.-W. Cao, Y.-P. Yuan, J. Barber, S. C. J. Loo and C. Xue, Appl. Surf. Sci., 2014, 319, 344–349 CrossRef CAS PubMed.
- S. Cao and J. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef CAS.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- J. Yu, K. Wang, W. Xiao and B. Cheng, Phys. Chem. Chem. Phys., 2014, 16, 11492–11501 RSC.
- Y. Zhong, Z. Wang, J. Feng, S. Yan, H. Zhang, Z. Li and Z. Zou, Appl. Surf. Sci., 2014, 295, 253–259 CrossRef CAS PubMed.
- B. Ohtani, K. Iwai, S.-i. Nishimoto and S. Sato, J. Phys. Chem. B, 1997, 101, 3349–3359 CrossRef CAS.
- F. Zhang, J. Chen, X. Zhang, W. Gao, R. Jin, N. Guan and Y. Li, Langmuir, 2004, 20, 9329–9334 CrossRef CAS PubMed.
- Y. Mei, G. Sharma, Y. Lu, M. Ballauff, M. Drechsler, T. Irrgang and R. Kempe, Langmuir, 2005, 21, 12229–12234 CrossRef CAS PubMed.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS PubMed.
- V. D. Mote, Y. Purushotham and B. N. Dole, J. Theory Appl. Phys., 2012, 6, 1–8 CrossRef.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- Q. Guo, Y. Xie, X. Wang, S. Zhang, T. Hou and S. Lv, Chem. Commun., 2004, 26–27 RSC.
- M. Kim, S. Hwang and J.-S. Yu, J. Mater. Chem., 2007, 17, 1656–1659 RSC.
- J. T. Titantah and D. Lamoen, Diamond Relat. Mater., 2007, 16, 581–588 CrossRef CAS PubMed.
- A. Zamudio, A. L. Elías, J. A. Rodríguez-Manzo, F. López-Urías, G. Rodríguez-Gattorno, F. Lupo, M. Rühle, D. J. Smith, H. Terrones, D. Díaz and M. Terrones, Small, 2006, 2, 346–350 CrossRef CAS PubMed.
- A. P. Dementjev, A. de Graaf, M. C. M. van de Sanden, K. I. Maslakov, A. V. Naumkin and A. A. Serov, Diamond Relat. Mater., 2000, 9, 1904–1907 CrossRef CAS.
- B. Yue, Q. Li, H. Iwai, T. Kako and J. Ye, Sci. Technol. Adv. Mater., 2011, 12, 034401 CrossRef.
- Q. Guo, Q. Yang, L. Zhu, C. Yi, S. Zhang and Y. Xie, Solid State Commun., 2004, 132, 369–374 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS.
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- Z. Q. Tian, S. P. Jiang, Y. M. Liang and P. K. Shen, J. Phys. Chem. B, 2006, 110, 5343–5350 CrossRef CAS PubMed.
- B. Yue, Y. Ma, H. Tao, L. Yu, G. Jian, X. Wang, X. Wang, Y. Lu and Z. Hu, J. Mater. Chem., 2008, 18, 1747–1750 RSC.
- J. R. McBride, G. W. Graham, C. R. Peters and W. H. Weber, J. Appl. Phys., 1991, 69, 1596–1604 CrossRef CAS PubMed.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- L. J. Durndell, C. M. A. Parlett, N. S. Hondow, K. Wilson and A. F. Lee, Nanoscale, 2013, 5, 5412–5419 RSC.
- A.-X. Yin, X.-Q. Min, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2011, 133, 3816–3819 CrossRef CAS PubMed.
- N. Markovic, H. Gasteiger and P. N. Ross, J. Electrochem. Soc., 1997, 144, 1591–1597 CrossRef CAS PubMed.
- G. L. Bezemer, J. H. Bitter, H. P. C. E. Kuipers, H. Oosterbeek, J. E. Holewijn, X. Xu, F. Kapteijn, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2006, 128, 3956–3964 CrossRef CAS PubMed.
- O. M. Wilson, M. R. Knecht, J. C. Garcia-Martinez and R. M. Crooks, J. Am. Chem. Soc., 2006, 128, 4510–4511 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional XP spectra and TEM images can be found. See DOI: 10.1039/c5cp00560d |
| This journal is © the Owner Societies 2015 |