
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced molecular dissociation and photoinduced recombination mediated by superfluid helium nanodroplets
Andreas
Kautsch
,
Markus
Koch
* and
Wolfgang E.
Ernst
*
Graz University of Technology, Institute of Experimental Physics, NAWI Graz, Petersgasse 16, A-8010 Graz, Austria. E-mail: markus.koch@tugraz.at; wolfgang.ernst@tugraz.at
First published on 14th April 2015
Abstract
We have investigated photoinduced chemical reaction dynamics of cold, isolated Cr2 molecules in helium nanodroplets (HeN), exploiting the quantum state specific spatial separation of solvated and surface locations on the droplet. The molecules are excited to achieve dissociation to a ground state (a7S3) and a metastable state (a5S2) atom. State specific spatial separation, in combination with efficient translational cooling to avoid ejection, causes the ground state atom to be solvated inside the droplet while the metastable atom migrates to the surface. A barrier between the two reactants formed by the HeN prevents recombination. We apply a resonance-enhanced multiphoton ionization scheme including the  transition of the surface atom as well as a two-laser scheme including the
transition of the surface atom as well as a two-laser scheme including the  transition of the solvated atom in order to verify the locations and separation of the dissociation products. Furthermore, ionization of the a5S2 surface atom triggers solvation followed by geminate recombination with the a7S3 atom, which is verified by the detection of Cr2+ molecular ions. For small Cr clusters, our results indicate that they may be composed of chromium dimers that exhibit the same dissociation behavior.
transition of the solvated atom in order to verify the locations and separation of the dissociation products. Furthermore, ionization of the a5S2 surface atom triggers solvation followed by geminate recombination with the a7S3 atom, which is verified by the detection of Cr2+ molecular ions. For small Cr clusters, our results indicate that they may be composed of chromium dimers that exhibit the same dissociation behavior.
1 Introduction
Helium nanodroplets (HeN) represent a well-established matrix for the spectroscopic investigation of isolated, cold (0.4 K) atoms, molecules, and clusters1 and offer a unique method for cold chemistry on a single molecule level. The spatial separation of surface located and solvated dopants in HeN provides a promising approach to control and monitor chemical reactions, especially if the location is state specific and can thus be changed by photoexcitation. In this article we demonstrate a complete cycle of photoinduced molecular dissociation, spatial separation of the fragments to a fully solvated and a stable, surface bound state and finally, photoinduced geminate recombination of the fragments.Increasing interest in exploring chemical reactions in HeN originates from the ability to stabilize and investigate radical reaction intermediates2–7 or confine dissociation fragments to the volume of the droplet.8,9 High cooling rates permit efficient relaxation of excited molecules,1,8 the formation and stabilization of metastable clusters,10,11 or aggregation of weak van der Waals bound complexes (see, e.g., ref. 12). Shallow energy barriers along reaction pathways can lead to trapping in local reactive potential-energy surface minima4 which can be overcome by photo-activation to trigger chemical reactions.13,14 The location of a dopant is dictated by the interaction with helium (the pair potential) and can be estimated by the dimensionless Ancilotto parameter.15 While the majority of atoms and molecules in their ground state are located inside the droplet, an electronic transition from the ground state to an excited state can initiate migration from inside to the droplet surface. This has been observed, for example, for Ag,16–18 Cu,19,20 Cr,21–23 CF3,24,25 and NO.26 Usually, surface migration is followed by desorption from the droplet, only for NO* there was indication that the excited molecule remains in a surface-bound state.26 For droplets doped with both surface located and solvated species the helium matrix can form a barrier that prevents molecule formation or clustering, even in the presence of long range van der Waals interaction.27 On the other side, van der Waals forces between solvated noble gas atoms and surface-located alkaline earth metal atoms can be exploited to overcome the separating character of HeN.28,29 Transition from a surface location to solvation was achieved for the HCN–Sr complex (and vice versa for HCN–Ca) by vibrational excitation,30 and in general, surface-located species migrate inside the droplet upon ionization.31–34
Here we demonstrate that photoexcitation of solvated Cr2 molecules results in dissociation to one atom in a solvated state (a7S3) and a second atom in a surface bound state (a5S2). Both fragments are sufficiently cooled to prevent ejection from the droplet. A resonant three photon ionization scheme is applied to the surface atom to verify its location and, at the same time, trigger solvation and recombination with its original partner. The stable interior location of the other fragment is proven with a two laser experiment, where both lasers are scanned individually.
2 Experimental
Chromium clusters (Cr2–Cr4) are formed inside helium nanodroplets by the pickup of single Cr atoms. The setup follows a Helium Nanodroplet Isolation (HENDI) apparatus, described in detail in ref. 35. In brief, HeN are formed by the supersonic expansion of high purity 4He gas through a cooled nozzle (diameter d = 5 μm, stagnation pressure p0 = 50 bar). Droplet sizes follow a log-normal distribution with a distribution maximum of![[N with combining circumflex]](https://www.rsc.org/images/entities/i_char_004e_0302.gif) = 6300 He atoms (mean droplet size
= 6300 He atoms (mean droplet size ![[N with combining macron]](https://www.rsc.org/images/entities/i_char_004e_0304.gif) = 16
= 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) for a nozzle temperature of 14 K. The collimated HeN beam is crossed at right angles by an effusive beam of Cr atoms from an electron bombardment Cr oven36 beneath the HeN beam. This setup ensures that no free atoms reach the detection region. To characterize the pick-up conditions, the number of Cr1,2,…+ ions formed by electron bombardment ionization is monitored as function of the oven heating power. A quadrupole mass spectrometer (QMS, Balzers QMG 422) is attached at the end of the measurement chamber to facilitate mass selective ion detection. This allows an assignment of the photoion signal to a specific Crn cluster size, by comparing the photoion yield to the electron bombardment Cr1,2,…+ yield as a function of heating power.
000) for a nozzle temperature of 14 K. The collimated HeN beam is crossed at right angles by an effusive beam of Cr atoms from an electron bombardment Cr oven36 beneath the HeN beam. This setup ensures that no free atoms reach the detection region. To characterize the pick-up conditions, the number of Cr1,2,…+ ions formed by electron bombardment ionization is monitored as function of the oven heating power. A quadrupole mass spectrometer (QMS, Balzers QMG 422) is attached at the end of the measurement chamber to facilitate mass selective ion detection. This allows an assignment of the photoion signal to a specific Crn cluster size, by comparing the photoion yield to the electron bombardment Cr1,2,…+ yield as a function of heating power.
Excitation spectra of the Crn–HeN (n = 2–4) complexes are recorded with resonance-enhanced multiphoton ionization (REMPI) spectroscopy.21 Laser pulses are obtained from excimer (XeCl, Radiant Dyes RD-EXC-200, 308 nm, ∼20 ns pulse duration) pumped dye lasers. For one-color REMPI, a pulsed dye laser beam (Lamda Physik, FL3002, dyes: Coumarin 2, Coumarin 102) intersects the doped HeN beam at right angles in the extraction region of the QMS. The mass window of the QMS is set to integrate the signal of the Crn+ isotopes and Crn+–He around the central cluster mass to increase the signal. To further increase the signal quality, two-color REMPI is used in addition.23 Therefore, a fraction of the 308 nm pump laser light is overlapped with the dye laser.
A pump–probe scheme is applied to observe the formation of ground state atoms after dissociating Cr2. Two dye laser beams (Radiant Dyes, DL-midi, dye: Coumarin 2; Lamda Physik, FL3002, dye: RDC360 neu) intersect the doped HeN beam, where the first one dissociates the molecules, while the second ionizes the produced ground state Cr atoms state selectively with a known REMPI scheme.21 In addition to the signal from dissociated Cr2, single Cr atom doped HeN cause a background signal, which is subtracted by a difference detection scheme (dissociation laser on minus off). The fluence of the dissociation laser is kept low in order to prevent ionization of the dissociation products (in contrast to the experiment described above).
In Fig. 1 the Cr2 potential energy curves37,38 and Cr atom energy levels39 that are of relevance for the presented experiments are shown, together with an excitation–ionization scheme indicated with arrows. The first step in this scheme is excitation of ground state Cr2 (4σ23dσ23dπ43dδ440) inside HeNvia the A1Σu+ ← X1Σg+ transition. The excited A1Σu+ state correlates to one ground state (a7S3, 3d54s) and one excited (z7P°, 3d54p) Cr atom, and, as will be shown below, dissociates via internal conversion to form one a7S3 and one a5S2 (3d54s) atom.
 | ||
| Fig. 1 Cr2 and Cr2+ potential energy curves37,38 (left) and Cr atom energy levels39 (right). A one-color ionization scheme is indicated by a combination of solid arrows (laser excitation) and dashed arrows (relaxation). Upon Cr2 excitation to the A1Σu+ state inside HeN, the molecule undergoes relaxation to predissociating states resulting in a7S3 Cr and a5S2 Cr atom states. While the first remains solvated inside the droplet, the latter migrates to the surface where it is ionized by a resonant three photon scheme. This triggers solvation and recombination to Cr2+ (top of the figure). The ionization path to probe the ground state atom with a second dye laser is indicated by dash-dotted arrows.21 A suggested ionization path that does not include Cr2 dissociation is indicated by gray arrows. | ||
3 Results
One-color REMPI excitation spectra of Cr2via the A1Σu+ ← X1Σg+ transition for Cr+ and Cr2+ detection are shown in Fig. 2(a). The broad band, stretching from 21500 to 22750 cm−1, is attributed to the Cr2 A1Σu+ ← X1Σg+ transition and the three pronounced peaks can be assigned to Cr atom  transitions (indicated by triangles39). Because the A1Σu+ ← X1Σg+ molecular band overlaps with the atomic transition energies, these peaks indicate the photoinduced dissociation with one of the fragments being in the a5S2 state, as will be discussed in detail below. The whole excitation–ionization process from a5S2 is indicated with solid, upwards pointing arrows in Fig. 1. Cr2 is excited to a predissociating state that leads to the formation of a ground state a7S3 and a metastable state a5S2 atom.41 The latter is excited via the
transitions (indicated by triangles39). Because the A1Σu+ ← X1Σg+ molecular band overlaps with the atomic transition energies, these peaks indicate the photoinduced dissociation with one of the fragments being in the a5S2 state, as will be discussed in detail below. The whole excitation–ionization process from a5S2 is indicated with solid, upwards pointing arrows in Fig. 1. Cr2 is excited to a predissociating state that leads to the formation of a ground state a7S3 and a metastable state a5S2 atom.41 The latter is excited via the  transition and ionized with either two (one-color REMPI) or one (two-color REMPI) photon. This ionization path selectively addresses a5S2 metastable Cr atoms and will be called a5S REMPI from now on. For further insight into the dynamics after dissociation, we present detailed scans across the
transition and ionized with either two (one-color REMPI) or one (two-color REMPI) photon. This ionization path selectively addresses a5S2 metastable Cr atoms and will be called a5S REMPI from now on. For further insight into the dynamics after dissociation, we present detailed scans across the  transition for the detection of Cr+ to Cr4+ in Fig. 3. Although one-color and two-color REMPI give equal results, the latter provides a better signal to noise ratio and was thus chosen. One 308 nm photon is used for ionization of excited
transition for the detection of Cr+ to Cr4+ in Fig. 3. Although one-color and two-color REMPI give equal results, the latter provides a better signal to noise ratio and was thus chosen. One 308 nm photon is used for ionization of excited 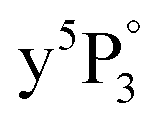 state atoms instead of two dye laser photons (cf.Fig. 1). The spectral signature is similar for detection of Cr1,2,3,4+, where the appearance of Cr3,4+ is connected to larger Crn (n = 3, 4,…) clusters, as will be discussed in Section 4.3.
state atoms instead of two dye laser photons (cf.Fig. 1). The spectral signature is similar for detection of Cr1,2,3,4+, where the appearance of Cr3,4+ is connected to larger Crn (n = 3, 4,…) clusters, as will be discussed in Section 4.3.
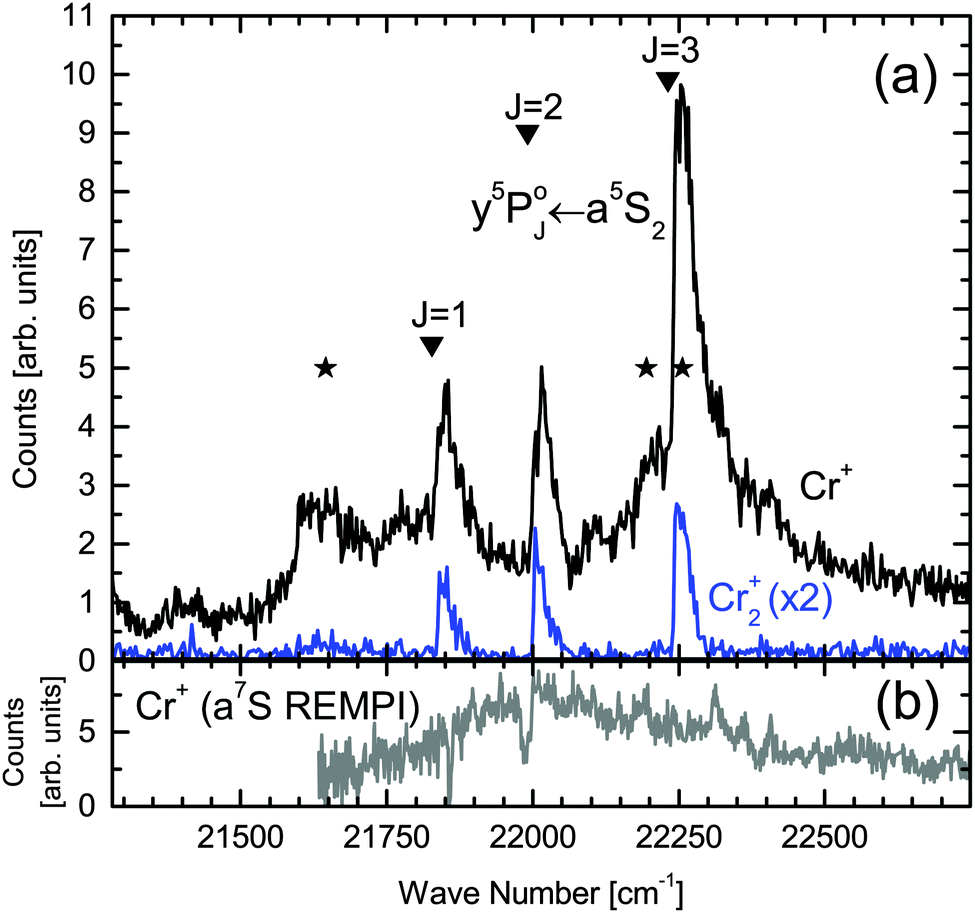 | ||
Fig. 2 (a) One-color REMPI spectra in the range of the Cr2 A1Σu+ ← X1Σg+ transition. Photoions are detected at the mass of Cr+ (black line) and Cr2+ (blue line, multiplied by 2). The bare Cr atom  transition energies are indicated with triangles.39 Asterisks mark the wavelengths at which the photoion yield is compared to the Cr1,2,…+ ion yield obtained with electron impact ionization in dependence on the pick-up oven temperature. (b) Excitation spectrum in the range of the Cr2 A1Σu+ ← X1Σg+ transition, recorded with a pump–probe laser scheme. The dissociating laser is scanned across the Cr2 band, while the second laser ionizes the produced ground state atoms with a7S REMPI (includes the transition energies are indicated with triangles.39 Asterisks mark the wavelengths at which the photoion yield is compared to the Cr1,2,…+ ion yield obtained with electron impact ionization in dependence on the pick-up oven temperature. (b) Excitation spectrum in the range of the Cr2 A1Σu+ ← X1Σg+ transition, recorded with a pump–probe laser scheme. The dissociating laser is scanned across the Cr2 band, while the second laser ionizes the produced ground state atoms with a7S REMPI (includes the  transition). Narrow dips and peaks are artifacts due to experimental instabilities. transition). Narrow dips and peaks are artifacts due to experimental instabilities. | ||
 | ||
Fig. 3 High resolution REMPI scan of the  transition within the Cr2 A1Σu+ ← X1Σg+ band (Fig. 2) for detection of Cr1,2,3,4+ ions (two-color ionization scheme with the XeCl laser at 308 nm as second wavelength). The bare Cr atom transition within the Cr2 A1Σu+ ← X1Σg+ band (Fig. 2) for detection of Cr1,2,3,4+ ions (two-color ionization scheme with the XeCl laser at 308 nm as second wavelength). The bare Cr atom  transition energy is indicated with a vertical line.39 transition energy is indicated with a vertical line.39 | ||
While the production of the metastable a5S2 atom as one of the dissociation products is verified with a5S REMPI (see Fig. 2(a) and 3), the verification of the other dissociation product – the a7S3 ground state atom – requires a second laser. It ionizes the a7S3 atom with a well known Cr one-color REMPI scheme21 that includes the  ground state transition (Fig. 1, dash-dotted arrows) and is called a7S REMPI from now on. The two-laser scheme opens the possibility to scan both laser wavelengths individually. First, the dissociation process is studied (Fig. 2(b)) by scanning the dissociation laser across the Cr2 excitation band and photoionizing the produced ground state atoms with a7S REMPI (
ground state transition (Fig. 1, dash-dotted arrows) and is called a7S REMPI from now on. The two-laser scheme opens the possibility to scan both laser wavelengths individually. First, the dissociation process is studied (Fig. 2(b)) by scanning the dissociation laser across the Cr2 excitation band and photoionizing the produced ground state atoms with a7S REMPI (![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 28204 cm−1). Note that a certain fraction of the HeN are originally doped with single a7S3 Cr atoms according to the pick-up statistics. Hence, for all two dye laser experiments a differential measurement is applied to subtract these atoms from the Cr2 dissociation signal.
= 28204 cm−1). Note that a certain fraction of the HeN are originally doped with single a7S3 Cr atoms according to the pick-up statistics. Hence, for all two dye laser experiments a differential measurement is applied to subtract these atoms from the Cr2 dissociation signal.
To gain information about the location of the a7S3 Cr atom, the dissociating laser wavelength is kept constant at = 21978 cm−1 and the a7S laser is scanned across the atomic 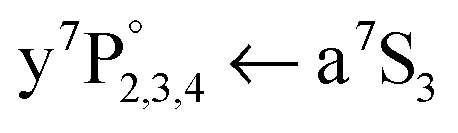 ground state transition. It was shown for single Cr atoms solvated inside HeN, that the
ground state transition. It was shown for single Cr atoms solvated inside HeN, that the 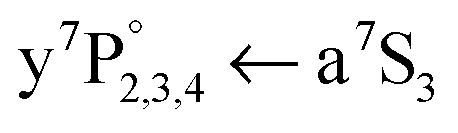 ground state transition is shifted and broadened by about 400 to 600 cm−1 due to the Pauli repulsion of the surrounding helium.21–23 As indicated in Fig. 1, after excitation to
ground state transition is shifted and broadened by about 400 to 600 cm−1 due to the Pauli repulsion of the surrounding helium.21–23 As indicated in Fig. 1, after excitation to 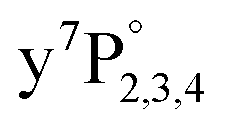 the atoms are ejected from the droplet while relaxing to energetically lower states (e.g.,
the atoms are ejected from the droplet while relaxing to energetically lower states (e.g.,  ).21–23 This mechanism enables a highly efficient ionization of bare atoms with one further laser photon, also through Cr autoionizing states that lie close to the ionization potential (g5D, e3D). The spectrum with the fixed dissociating laser and the a7S REMPI laser scanning across the ground state transition is shown in Fig. 4(a). Due to a difference scheme, the spectrum can be allocated to Cr atoms from dissociated Cr2, detected at the Cr+ mass. For comparison, the spectrum obtained with a7S REMPI of single Cr atoms in HeN (not via dissociation of Cr2) is plotted in Fig. 4(b) (from ref. 21). In both spectra, the broad structure is attributed to the in-droplet
).21–23 This mechanism enables a highly efficient ionization of bare atoms with one further laser photon, also through Cr autoionizing states that lie close to the ionization potential (g5D, e3D). The spectrum with the fixed dissociating laser and the a7S REMPI laser scanning across the ground state transition is shown in Fig. 4(a). Due to a difference scheme, the spectrum can be allocated to Cr atoms from dissociated Cr2, detected at the Cr+ mass. For comparison, the spectrum obtained with a7S REMPI of single Cr atoms in HeN (not via dissociation of Cr2) is plotted in Fig. 4(b) (from ref. 21). In both spectra, the broad structure is attributed to the in-droplet 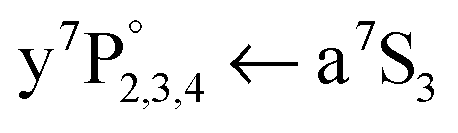 ground state excitation, which is very similar regarding shape and position. The narrow peaks (indicated with triangles) are allocated to the bare atom
ground state excitation, which is very similar regarding shape and position. The narrow peaks (indicated with triangles) are allocated to the bare atom  autoionizing transitions. The experimental conditions, i.e. droplet size, for the spectrum in Fig. 4(a) are optimized for Cr2 formation resulting in a lower signal to noise ratio and reduced autoionizing peaks.23 A very similar spectrum to Fig. 4(a) was obtained when delaying the a7S REMPI probe laser pulse by 20 ns with respect to the dissociating pump laser pulse, so that there is no temporal overlap between the pulses.
autoionizing transitions. The experimental conditions, i.e. droplet size, for the spectrum in Fig. 4(a) are optimized for Cr2 formation resulting in a lower signal to noise ratio and reduced autoionizing peaks.23 A very similar spectrum to Fig. 4(a) was obtained when delaying the a7S REMPI probe laser pulse by 20 ns with respect to the dissociating pump laser pulse, so that there is no temporal overlap between the pulses.
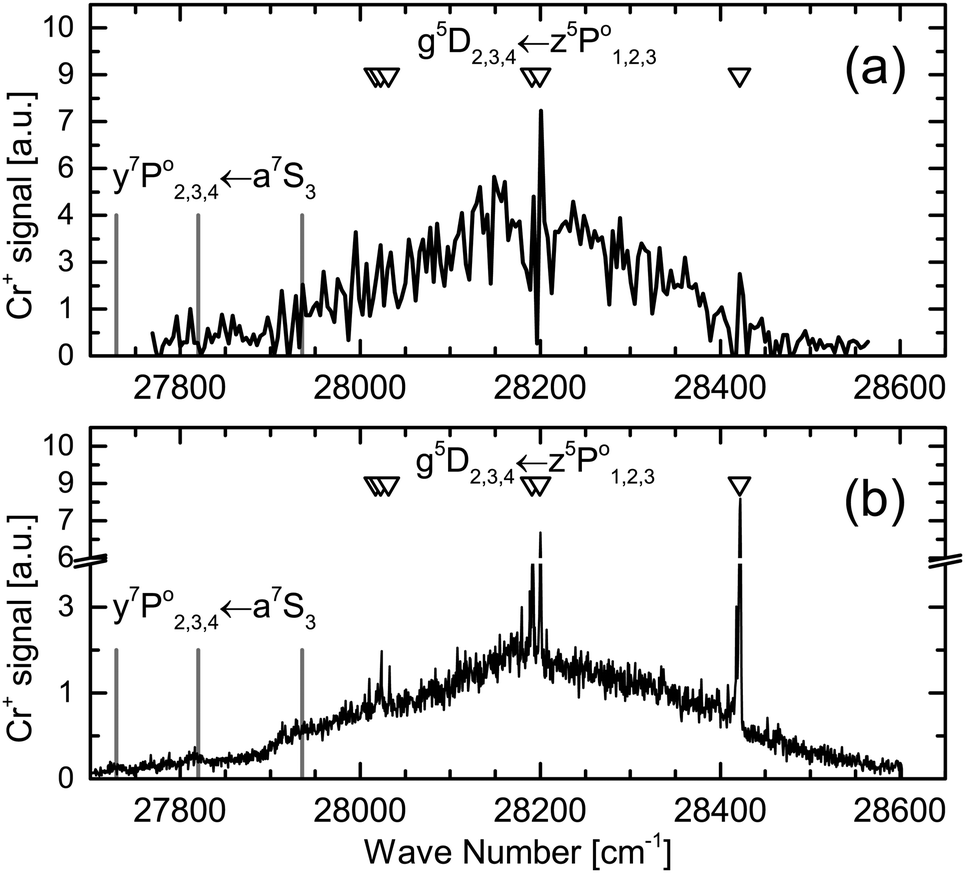 | ||
Fig. 4 a7S3 REMPI spectra of ground state Cr atoms in HeN. (a) Spectrum recorded in a two laser experiment, where the Cr2 dissociation laser is fixed ( = 21978 cm−1) and the second laser (a7S3 REMPI) is scanned. A difference scheme (dissociation laser on minus off) was applied. (b) a7S3 REMPI spectrum for single Cr-atom doped HeN.21 The bare Cr atom ground state transitions 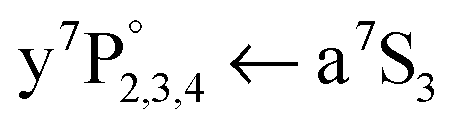 are indicated as vertical lines and transitions to autoionizing states with triangles. are indicated as vertical lines and transitions to autoionizing states with triangles. | ||
4 Discussion
4.1 Predissociation
Photoinduced dissociation in HeN has been observed for various species.13,25,28,42 Predissociation of Cr2 upon excitation to A1Σu+ has been observed in gas phase37,41,43 and in matrix.40 It was concluded, that the A1Σu+ state is crossed by at least one perturbing state and one predissociating state, where possible assignments were discussed by Andersson.37 Riley et al.41 propose the nonradiative transition from the A1Σu+ to another bound state. The latter is rapidly dissociated by a state correlating to the separate atom limit states Cr (a7S3) and Cr (a5S2, a5D).41 From the observation of the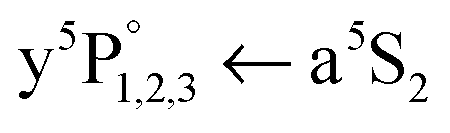 and
and  atomic transitions, we clearly identify the population of the a5S2 and a7S3 atom states and assign the following dissociative process:
atomic transitions, we clearly identify the population of the a5S2 and a7S3 atom states and assign the following dissociative process: | (1) |
4.2 Separation to a surface located a5S2 and a solvated a7S3 atom
The fate after dissociation is dictated by the interaction of the Cr atoms in the respective state (a7S3, a5S2) with He atoms.44,45 In principle, the atoms can remain solvated inside the HeN, stay bound on the surface, or detach from the HeN with a certain probability to take a few He atoms along. Since the droplet acts as a heat bath, kinetic energy can be dissipated to prevent ejection.8,9 An excess energy of about 0.4 eV (3200 cm−1), given by the difference of excitation energy and the dissociation limit (cf.Fig. 1), can be compensated by the evaporation of 640 He atoms. So the location of the dopants after dissociation is governed by the electronic states of the atoms. Experimentally, the location of the two fragments is obtained from the lineshapes of the a7S REMPI (via ) and the a5S REMPI (via
) and the a5S REMPI (via ) spectra. As observed for single Cr atoms in earlier experiments21–23 and as predicted by theoretical studies,44,45 the ground state (a7S3) atom remains solvated inside the droplet. The metastable state (a5S2) atom, in contrast, favors a surface location, which is suggested by DFT calculations (ref. 45) and is supported by the experimental observations, as follows.
) spectra. As observed for single Cr atoms in earlier experiments21–23 and as predicted by theoretical studies,44,45 the ground state (a7S3) atom remains solvated inside the droplet. The metastable state (a5S2) atom, in contrast, favors a surface location, which is suggested by DFT calculations (ref. 45) and is supported by the experimental observations, as follows.
We start with the a5S2 atom, because the in-droplet A1Σu+ ← X1Σg+ molecule band overlaps in energy with the atom  transition, leading to excited Cr* (y5P°) atoms which are photoionized by absorption of two photons (Fig. 1), so that Cr+ ions are detected upon detachment from the droplet. In the Cr+ signal in Fig. 2(a), all three peaks of the
transition, leading to excited Cr* (y5P°) atoms which are photoionized by absorption of two photons (Fig. 1), so that Cr+ ions are detected upon detachment from the droplet. In the Cr+ signal in Fig. 2(a), all three peaks of the 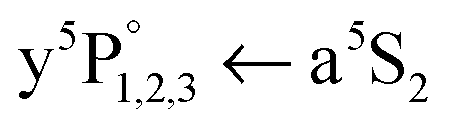 transition are observable. Considering the detailed Cr+ scan (Fig. 3) of the most intense
transition are observable. Considering the detailed Cr+ scan (Fig. 3) of the most intense  peak, the onset of the main feature is shifted approximately 5 cm−1 to the blue relative to the bare atom transition energy. A maximum is reached at 22240 cm−1 followed by an almost steady signal decrease, giving a total peak width of ∼50 cm−1. These spectral characteristics are comparable to transitions of heavier alkali metal atoms that reside in a surface dimple,27,35,46 which is a first indication for the surface location of the a5S2 atom (others will follow). At the bare atom transition energy (indicated with a vertical line in the detailed scan in Fig. 3), the absence of sharp atomic lines proves that hardly any atoms are ejected from the droplet upon dissociation, encouraging the interpretation in terms of the effective kinetic energy dissipation upon predissociation.
peak, the onset of the main feature is shifted approximately 5 cm−1 to the blue relative to the bare atom transition energy. A maximum is reached at 22240 cm−1 followed by an almost steady signal decrease, giving a total peak width of ∼50 cm−1. These spectral characteristics are comparable to transitions of heavier alkali metal atoms that reside in a surface dimple,27,35,46 which is a first indication for the surface location of the a5S2 atom (others will follow). At the bare atom transition energy (indicated with a vertical line in the detailed scan in Fig. 3), the absence of sharp atomic lines proves that hardly any atoms are ejected from the droplet upon dissociation, encouraging the interpretation in terms of the effective kinetic energy dissipation upon predissociation.
We consider it important to mention that the Cr 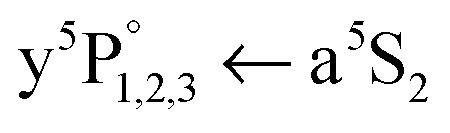 excitation observed here on the HeN surface is of completely different nature than the z5P° ← a5S2 excitation of bare Cr atoms and small Cr–Hen (n = 1, 2,…) exciplexes observed in our previous studies.23 This becomes obvious by comparing the corresponding line shapes. In the two-color REMPI scheme in ref. 23, 308 nm excitation and droplet mediated relaxation results in bare a5S2 Cr atoms and Cr–Hen exciplexes, both being ejected from the HeN. The corresponding excitation spectrum is composed of a sharp atom line accompanied by a wing to the blue (due to Cr–Hen). Here, in contrast, we find hardly any signal at the bare atom line position (Fig. 3) and observe the onset of the peak 5 cm−1 to the blue. Although the XeCl laser was present for recording the spectra in Fig. 3, a bare atom transition peak is hardly detectable. Systematic studies showed an increased bare atom signal for higher XeCl laser fluence, which starts causing the ejection process.
excitation observed here on the HeN surface is of completely different nature than the z5P° ← a5S2 excitation of bare Cr atoms and small Cr–Hen (n = 1, 2,…) exciplexes observed in our previous studies.23 This becomes obvious by comparing the corresponding line shapes. In the two-color REMPI scheme in ref. 23, 308 nm excitation and droplet mediated relaxation results in bare a5S2 Cr atoms and Cr–Hen exciplexes, both being ejected from the HeN. The corresponding excitation spectrum is composed of a sharp atom line accompanied by a wing to the blue (due to Cr–Hen). Here, in contrast, we find hardly any signal at the bare atom line position (Fig. 3) and observe the onset of the peak 5 cm−1 to the blue. Although the XeCl laser was present for recording the spectra in Fig. 3, a bare atom transition peak is hardly detectable. Systematic studies showed an increased bare atom signal for higher XeCl laser fluence, which starts causing the ejection process.
We now turn to the location and environment of the other dissociation fragment – the a7S ground state atom – based on the two dye laser experiment. The first laser excites the dissociating A1Σu+ ← X1Σg+ Cr2 transition and the second laser the a7S REMPI. Fig. 4(a) shows the a7S REMPI spectrum, recorded from the dissociation fragment. The fact that a7S atoms are detected by the differential scheme proves the proposed predissociation mechanism. The spectrum is compared to the reference spectrum obtained from single atom doped HeN (Fig. 4(b)).21 The spectral similarity of the broad structure, which represents the in-droplet broadened  transition, indicates a well defined separation after dissociation of the ground state atom from the a5S2 atom on the surface. If both dissociation fragments resided inside the HeN, one would expect a spectral line shift like it was observed for Mg atoms
transition, indicates a well defined separation after dissociation of the ground state atom from the a5S2 atom on the surface. If both dissociation fragments resided inside the HeN, one would expect a spectral line shift like it was observed for Mg atoms  .47 Hence, the spectral similarity of a7S3 atoms from dissociated Cr2 and original a7S3 atoms is a further indication of the interior location of the ground state fragment and the surface location of the a5S2 atom fragment.
.47 Hence, the spectral similarity of a7S3 atoms from dissociated Cr2 and original a7S3 atoms is a further indication of the interior location of the ground state fragment and the surface location of the a5S2 atom fragment.
To show the stable position of both fragments after dissociation, the probe laser was time-delayed (Δτ ∼ 20 ns) to avoid a temporal overlap between the dissociation pump laser and the ionizing probe laser pulse. The pump–probe delay has no significant influence on the spectrum. Hence, no indication for a recombination or spatial approach between both neutral fragments within the delay time is found. We thus conclude that the Cr a5S2 atom is stable on the HeN surface, where it is available for further excitation.
It is important to point out that despite the attractive interaction between the center-located ground state and the surface-located metastable state atoms, molecule formation does not occur. This indicates an energy barrier between the two atoms as a result of the confining potential energy curve of the solvated atom and the attractive Cr2 (a7S3–a5S2) potential energy curve, in a similar way as it was recently calculated for the Rb–Xe van der Waals system in a He500 droplet.27
4.3 Cr2,3,4+ from Cr2 and small Crn cluster
The characteristic atomic transition peaks are also present in the REMPI spectra employing Cr2+ detection (see Fig. 2(a) and 3), with exactly the same position and shape as for Cr+ detection. This is very surprising, because the Cr atom
transition peaks are also present in the REMPI spectra employing Cr2+ detection (see Fig. 2(a) and 3), with exactly the same position and shape as for Cr+ detection. This is very surprising, because the Cr atom  transition obviously has to be a step in the ionization process leading from Cr2 to Cr2+. The observation becomes plausible when we build on the results from before, namely that upon Cr2 dissociation the a5S2 atom is residing on the surface while the a7S3 atom remains solvated inside the droplet. After resonant ionization of the surface atom with a5S REMPI (see Fig. 1), the Cr+ ion has a fair chance to be pulled inside the HeN32,34,45 where it recombines with its original partner to form Cr2+. Forming the Cr2+ a2Σg+ ground state (Cr (7S) + Cr+ (6S), binding energy = 1.30 eV38), releases approximately the same amount of energy to the droplet as the initial Cr2 formation, evaporating ∼2100 He atoms. A certain fraction of the droplets is now completely evaporated by this energy input which produces detectable, bare Cr2+ ions. Remaining Cr2+ doped droplets that survived are not detected within the QMS mass window.
transition obviously has to be a step in the ionization process leading from Cr2 to Cr2+. The observation becomes plausible when we build on the results from before, namely that upon Cr2 dissociation the a5S2 atom is residing on the surface while the a7S3 atom remains solvated inside the droplet. After resonant ionization of the surface atom with a5S REMPI (see Fig. 1), the Cr+ ion has a fair chance to be pulled inside the HeN32,34,45 where it recombines with its original partner to form Cr2+. Forming the Cr2+ a2Σg+ ground state (Cr (7S) + Cr+ (6S), binding energy = 1.30 eV38), releases approximately the same amount of energy to the droplet as the initial Cr2 formation, evaporating ∼2100 He atoms. A certain fraction of the droplets is now completely evaporated by this energy input which produces detectable, bare Cr2+ ions. Remaining Cr2+ doped droplets that survived are not detected within the QMS mass window.
To verify Cr2 doped HeN as the parent species of the Cr2+ ions, the Cr2+ ion yield was recorded while increasing the Cr oven temperature, and hence, the probability distribution for the formation of different Crn cluster sizes.36 The Cr2+ ion signal shows a clear dependence on the pick-up oven temperature. For reference, the number of Cr1,2,3,…+ ions obtained with electron impact ionization of the doped HeN, is also recorded in dependence on the pick-up oven temperature. Keeping possible fragmentation in mind, the comparison allows an assignment of the Cr2+ signal (Fig. 2(a), blue line, 22257 cm−1) to Cr2 doped HeN.
signal (Fig. 2(a), blue line, 22257 cm−1) to Cr2 doped HeN.
We mention that in addition to the Cr + Cr+ → Cr2+ recombination, another recombination path is possible, although unlikely. According to DFT calculations,45 Cr* y5P° atoms have a stable position on the surface and inside the HeN, with a small energy barrier in between. Hence, the Cr* (y5P°) atom might move back inside the droplet to form an excited Cr2 molecule with the Cr (a7S3) ground state atom. As above, evaporation of a certain fraction of the droplets and subsequent two photon ionization would lead to Cr2+ detection. Irrespective of the recombination path, the recombination process is restricted to the volume of the droplet. The Cr2+ detection after a5S REMPI proves the location of both dissociation fragments to be on the droplet.
Now we consider HeN doped with Cr3,4 clusters. Fig. 3 shows that at the Cr atom 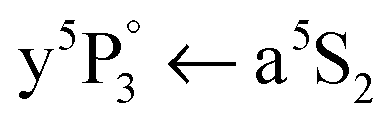 transition also Cr3+ and Cr4+ ion clusters are detected. This means, HeN doped with Cr3 and Cr4 (probably also Cr5,…) clusters can undergo a similar excitation process as Cr2, including the characteristic a5S REMPI of a surface located atom and subsequent recombination with the solvated fragments. Because of the strong Cr2 bonding nature, Cr3 is composed of a dimer plus a loosely bonded atom48,49 and Cr4 of two dimers with strong intradimer but weak interdimer bonding.50 This dimerization effect controls the cluster growth up to Cr11, yielding similarities between the ground state photoabsorption spectra of dimers and small chromium clusters.50–52 The detection of Cr3+ and Cr4+ therefore suggests that the Cr2 dissociation process and surface migration of one atom is not disturbed by the presence of a further Cr atom or dimer in the droplet.
transition also Cr3+ and Cr4+ ion clusters are detected. This means, HeN doped with Cr3 and Cr4 (probably also Cr5,…) clusters can undergo a similar excitation process as Cr2, including the characteristic a5S REMPI of a surface located atom and subsequent recombination with the solvated fragments. Because of the strong Cr2 bonding nature, Cr3 is composed of a dimer plus a loosely bonded atom48,49 and Cr4 of two dimers with strong intradimer but weak interdimer bonding.50 This dimerization effect controls the cluster growth up to Cr11, yielding similarities between the ground state photoabsorption spectra of dimers and small chromium clusters.50–52 The detection of Cr3+ and Cr4+ therefore suggests that the Cr2 dissociation process and surface migration of one atom is not disturbed by the presence of a further Cr atom or dimer in the droplet.
4.4 Cr2 excitation spectrum
Now we turn to the Cr2 A1Σu+ ← X1Σg+ excitation band, in particular to the broad band stretching from 21500 to 22750 cm−1 that is present in the Cr+ signal of the one-color REMPI scheme, shown as black curve in Fig. 2(a). We will now show that the signal of the broad band can be assigned to Cr2 molecules and arises from direct multiphoton ionization and subsequent dissociation to Cr and Cr+, an ionization path that does not include Cr2 predissociation. Outside the spectral regions of the 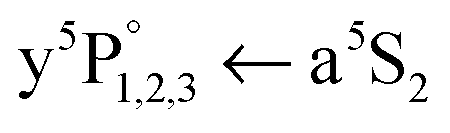 transitions, i.e., without resonance enhancement, the ionization probability of both Cr atoms resulting from dissociation is estimated to be lower than a resonant ionization via vibronic levels of the Cr2 molecule before dissociation (see Fig. 1). Hence, we account resonant multiphoton ionization of Cr2 in combination with efficient dissociation to Cr and Cr+ to be responsible for the observed signal.
transitions, i.e., without resonance enhancement, the ionization probability of both Cr atoms resulting from dissociation is estimated to be lower than a resonant ionization via vibronic levels of the Cr2 molecule before dissociation (see Fig. 1). Hence, we account resonant multiphoton ionization of Cr2 in combination with efficient dissociation to Cr and Cr+ to be responsible for the observed signal.
It is instructive to compare the band obtained with one-color REMPI (Fig. 2(a)) to Fig. 2(b), which shows the Cr+ signal of the two-laser a7S REMPI scheme where the dissociating laser is scanned. It is important to recall the following two points: (1) the a7S REMPI is obtained from resonant ionization of the center located ground state atoms that are produced via Cr2 predissociation. (2) The a7S REMPI is exclusively sensitive to Cr2 molecules and not to larger Crn clusters, as discussed in Section 4.2. Therefore, Fig. 2(b) represents the Cr2 excitation spectrum, which shows good overall agreement with the one-color REMPI band in Fig. 2(a). The extra features in the one-color REMPI spectrum in Fig. 2(a) with respect to the a7S REMPI spectrum in Fig. 2(b) indicate the presence of resonances in this ionization path. This interpretation is supported by monitoring the one-color REMPI Cr+ yield at the spectral positions marked with asterisks in Fig. 2(a) while increasing the evaporation oven temperature and comparing this signal to electron bombardment ion yield, as above. It is found that at all three spectral positions the Cr+ ion signal can be assigned predominantly to Cr2 and not to larger Crn cluster.
The predissociation process, which is observed by the formation of ground state a7S and metastable state a5S atoms, has been reported for the A1Σu+ ← X1Σg+ excitation in literature.37,40,41,43 We note that the Cr2 might additionally be formed in higher multiplicities as it was observed for Ag2 in HeN.53 However, currently we cannot identify other multiplicities. For the strongly bound Cr2 singlet ground state (binding energy = 1.42 eV (11450 cm−1)38), the majority of droplets are expected to survive the formation for a size distribution maximum of = 6300 and an energy of 5 cm−1 dissipated per evaporated He atom. In gas phase, the A1Σu+ ← X1Σg+ Cr2 transition is observed at an energy of 21751 cm−1 (0–0 band, 2.7 eV).41,43,54 Cr2 excitation spectra in other solid rare gas matrices yield usually shifted and broadened spectral features, but cannot provide a consistent picture about the magnitude of the shift.40,51,52,55,56
5 Conclusions
In conclusion, we have investigated the electronic A1Σu+ ← X1Σg+ excitation of Cr2 molecules located inside HeN with resonant multiphoton ionization spectroscopy, applying one- and two-laser schemes. The ionization spectra show, in addition to the droplet broadened molecular excitation structure, three pronounced peaks which we allocate to the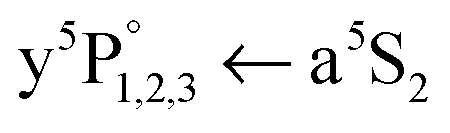 Cr atomic transitions. We conclude that the Cr2 molecule dissociates upon excitation41 into a solvated, ground state (a7S3) atom and a surface-located, metastable (a5S2) atom. The latter is ionized by resonance-enhanced multiphoton ionization. The conclusion is based on (a) the
Cr atomic transitions. We conclude that the Cr2 molecule dissociates upon excitation41 into a solvated, ground state (a7S3) atom and a surface-located, metastable (a5S2) atom. The latter is ionized by resonance-enhanced multiphoton ionization. The conclusion is based on (a) the  transition line shape (50 cm−1 width and a 5 cm−1 blueshift of the onset with respect to the bare atom lines), (b) a two laser experiment with REMPI detection of the produced a7S3 atoms, and (c) theoretical simulations of the a5S2 atom location.45 Surprisingly, we detect these three
transition line shape (50 cm−1 width and a 5 cm−1 blueshift of the onset with respect to the bare atom lines), (b) a two laser experiment with REMPI detection of the produced a7S3 atoms, and (c) theoretical simulations of the a5S2 atom location.45 Surprisingly, we detect these three  atom peaks not only at the Cr+ ion mass, but also at Cr2,3,4+ masses. Cr2+ detection demonstrates that upon photoionization, a fraction of the surface atoms migrates back inside the droplet to recombine with their ground state counterpart. The Cr3,4+ detection indicates that this pairwise dissociation and surface migration process is not disturbed by the presence of additional Cr atoms, a fact which we attribute to the strong dimerization effect in small Cr clusters.50
atom peaks not only at the Cr+ ion mass, but also at Cr2,3,4+ masses. Cr2+ detection demonstrates that upon photoionization, a fraction of the surface atoms migrates back inside the droplet to recombine with their ground state counterpart. The Cr3,4+ detection indicates that this pairwise dissociation and surface migration process is not disturbed by the presence of additional Cr atoms, a fact which we attribute to the strong dimerization effect in small Cr clusters.50
The elucidated mechanisms open a possibility for photoinduced chemistry mediated by HeN. Chemical reactions in HeN doped with both surface located (e.g., alkali-metal or alkaline earth metal atoms) and solvated species could be triggered by photoexcitation of the latter to a surface located state. The bond formation can then be followed, e.g., with time-resolved femtosecond spectroscopy.
Acknowledgements
The authors thank Friedrich Lindebner for experimental assistance as well as Martin Ratschek and Johann V. Pototschnig for computational support and Florian Lackner for careful reading. The research was supported by the Austrian Science Fund (FWF) under grant number P22962-N20 and by the European Commission and the Styrian Government within the ERDF program.References
- C. Callegari and W. E. Ernst, Helium Droplets as Nanocryostats for Molecular Spectroscopy – from the Vacuum Ultraviolet to the Microwave Regime, Handbook of High-Resolution Spectroscopy, John Wiley & Sons, Chinchester, 2011 Search PubMed.
- J. M. Merritt, J. Kupper and R. E. Miller, Phys. Chem. Chem. Phys., 2005, 7, 67–78 RSC.
- J. M. Merritt, S. Rudić and R. E. Miller, J. Chem. Phys., 2006, 124, 084301 CrossRef CAS PubMed.
- S. Rudić, J. M. Merritt and R. E. Miller, J. Chem. Phys., 2006, 124, 104305 CrossRef PubMed.
- J. Küpper and J. M. Merritt, Int. Rev. Phys. Chem., 2007, 26, 249–287 CrossRef.
- A. M. Morrison, J. Agarwal, H. F. Schaefer and G. E. Douberly, J. Phys. Chem. A, 2012, 116, 5299–5304 CrossRef CAS PubMed.
- K. von Haeften, A. Metzelthin, S. Rudolph, V. Staemmler and M. Havenith, Phys. Rev. Lett., 2005, 95, 215301 CrossRef CAS.
- D. Stolyarov, E. Polyakova and C. Wittig, J. Phys. Chem. A, 2004, 108, 9841–9846 CrossRef CAS.
- A. Braun and M. Drabbels, J. Chem. Phys., 2007, 127, 114305 CrossRef PubMed.
- K. Nauta and R. E. Miller, Science, 1999, 283, 1895–1897 CrossRef CAS.
- K. Nauta and R. E. Miller, Science, 2000, 287, 293–295 CrossRef CAS.
- G. E. Douberly and R. E. Miller, J. Phys. Chem. A, 2007, 111, 7292–7302 CrossRef CAS PubMed.
- G. E. Douberly, J. M. Merritt and R. E. Miller, Phys. Chem. Chem. Phys., 2005, 7, 463–468 RSC.
- T. Liang, S. D. Flynn, A. M. Morrison and G. E. Douberly, J. Phys. Chem. A, 2011, 115, 7437–7447 CrossRef CAS PubMed.
- F. Ancilotto, P. B. Lerner and M. W. Cole, J. Low Temp. Phys., 1995, 101, 1123–1146 CrossRef CAS.
- F. Federmann, K. Hoffmann, N. Quaas and J. D. Close, Phys. Rev. Lett., 1999, 83, 2548–2551 CrossRef CAS.
- N. B. Brauer, S. Smolarek, E. Loginov, D. Mateo, A. Hernando, M. Pi, M. Barranco, W. J. Buma and M. Drabbels, Phys. Rev. Lett., 2013, 111, 153002 CrossRef.
- D. Mateo, A. Hernando, M. Barranco, E. Loginov, M. Drabbels and M. Pi, Phys. Chem. Chem. Phys., 2013, 15, 18388–18400 RSC.
- F. Lindebner, A. Kautsch, M. Koch and W. E. Ernst, Int. J. Mass Spectrom., 2014, 365–366, 255–259 CrossRef CAS PubMed.
- F. Cargnoni and M. Mella, J. Phys. Chem. A, 2011, 115, 7141–7152 CrossRef CAS PubMed.
- A. Kautsch, M. Hasewend, M. Koch and W. E. Ernst, Phys. Rev. A: At., Mol., Opt. Phys., 2012, 86, 033428 CrossRef.
- A. Kautsch, M. Koch and W. E. Ernst, J. Phys. Chem. A, 2013, 117, 9621–9625 CrossRef CAS PubMed.
- M. Koch, A. Kautsch, F. Lackner and W. E. Ernst, J. Phys. Chem. A, 2014, 118, 8373–8379 CrossRef CAS PubMed.
- A. Braun and M. Drabbels, Phys. Rev. Lett., 2004, 93, 253401 CrossRef.
- A. Braun and M. Drabbels, J. Chem. Phys., 2007, 127, 114303 CrossRef PubMed.
- E. Polyakova, D. Stolyarov and C. Wittig, J. Chem. Phys., 2006, 124, 214308 CrossRef CAS PubMed.
- J. Poms, A. W. Hauser and W. E. Ernst, Phys. Chem. Chem. Phys., 2012, 14, 15158–15165 RSC.
- A. Masson, M. Briant, A. Hernando, N. Halberstadt, J.-M. Mestdagh and M.-A. Gaveau, J. Chem. Phys., 2012, 137, 184310 CrossRef CAS PubMed.
- E. Lugovoj, J. P. Toennies and A. Vilesov, J. Chem. Phys., 2000, 112, 8217–8220 CrossRef CAS PubMed.
- G. E. Douberly, P. L. Stiles, R. E. Miller, R. Schmied and K. K. Lehmann, J. Phys. Chem. A, 2010, 114, 3391–3402 CrossRef CAS PubMed.
- L. An der Lan, P. Bartl, C. Leidlmair, R. Jochum, S. Denifl, O. Echt and P. Scheier, Chem. – Eur. J., 2012, 18, 4411–4418 CrossRef CAS PubMed.
- M. Theisen, F. Lackner and W. E. Ernst, Phys. Chem. Chem. Phys., 2010, 12, 14861–14863 RSC.
- M. Theisen, F. Lackner, G. Krois and W. E. Ernst, J. Phys. Chem. Lett., 2011, 2, 2778–2782 CrossRef CAS.
- X. Zhang and M. Drabbels, J. Chem. Phys., 2012, 137, 051102 CrossRef PubMed.
- M. Koch, J. Lanzersdorfer, C. Callegari, J. S. Muenter and W. E. Ernst, J. Phys. Chem. A, 2009, 113, 13347–13356 CrossRef CAS PubMed.
- M. Ratschek, M. Koch and W. E. Ernst, J. Chem. Phys., 2012, 136, 104201 CrossRef PubMed.
- K. Andersson, Chem. Phys. Lett., 1995, 237, 212–221 CrossRef CAS.
- C.-X. Su, D. A. Hales and P. Armentrout, Chem. Phys. Lett., 1993, 201, 199–204 CrossRef CAS.
- A. Kramida, Y. u. Ralchenko, J. Reader and NIST ASD Team, NIST Atomic Spectra Database (ver. 5.0), National Institute of Standards and Technology, Gaithersburg, MD, 2012, online. Available: http://physics.nist.gov/asd, 2013, may 16 Search PubMed.
- M. J. Pellin and D. M. Gruen, J. Chem. Phys., 1983, 79, 5887–5893 CrossRef CAS PubMed.
- S. J. Riley, E. K. Parks, L. G. Pobo and S. Wexler, J. Chem. Phys., 1983, 79, 2577–2582 CrossRef CAS PubMed.
- J. Higgins, C. Callegari, J. Reho, F. Stienkemeier, W. E. Ernst, K. K. Lehmann, M. Gutowski and G. Scoles, Science, 1996, 273, 629–631 CAS.
- V. Bondybey and J. English, Chem. Phys. Lett., 1983, 94, 443–447 CrossRef CAS.
- J. V. Pototschnig, M. Ratschek, A. W. Hauser and W. E. Ernst, Phys. Chem. Chem. Phys., 2014, 16, 9469–9478 RSC.
- M. Ratschek, J. V. Pototschnig, A. W. Hauser and W. E. Ernst, J. Phys. Chem. A, 2014, 118, 6622–6631 CrossRef CAS PubMed.
- G. Auböck, J. Nagl, C. Callegari and W. E. Ernst, Phys. Rev. Lett., 2008, 101, 035301 CrossRef.
- A. Przystawik, S. Göde, T. Döppner, J. Tiggesbäumker and K.-H. Meiwes-Broer, Phys. Rev. A: At., Mol., Opt. Phys., 2008, 78, 021202 CrossRef.
- H. Cheng and L.-S. Wang, Phys. Rev. Lett., 1996, 77, 51–54 CrossRef CAS.
- L.-S. Wang, H. Wu and H. Cheng, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 12884–12887 CrossRef CAS.
- J. I. Martínez and J. A. Alonso, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 205409 CrossRef.
- L. Fang, B. Davis, H. Lu and J. R. Lombardi, J. Phys. Chem. A, 2001, 105, 9375–9378 CrossRef CAS.
- J. Derouault and M. Dalibart, Z. Phys. D: At., Mol. Clusters, 1991, 19, 211–214 CrossRef CAS.
- A. Przystawik, P. Radcliffe, S. Göde, K. H. Meiwes-Broer and J. Tiggesbäumker, J. Phys. B: At., Mol. Opt. Phys., 2006, 39, S1183–S1189 CrossRef CAS.
- D. L. Michalopoulos, M. E. Geusic, S. G. Hansen, D. E. Powers and R. E. Smalley, J. Phys. Chem., 1982, 86, 3914–3916 CrossRef CAS.
- E. P. Kündig, M. Moskovits and G. A. Ozin, Nature, 1975, 254, 503–504 CrossRef.
- M. Vala, R. Pyzalski, J. Shakhsemampour, M. Eyring, J. Pyka, T. Tipton and J. C. Rivoal, J. Chem. Phys., 1987, 86, 5951–5957 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2015 |