
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A multi-reference study of the byproduct formation for a ring-closed dithienylethene photoswitch†
Jochen René
Matis
,
Jan Boyke
Schönborn
and
Peter
Saalfrank
*
Institut für Chemie, Universität Potsdam, Karl-Liebknecht-Straße 24, D-Potsdam, Germany. E-mail: peter.saalfrank@uni-potsdam.de
First published on 6th May 2015
Abstract
Photodriven molecular switches are sometimes hindered in their performance by forming byproducts which act as dead ends in sequences of switching cycles, leading to rapid fatigue effects. Understanding the reaction pathways to unwanted byproducts is a prerequisite for preventing them. This article presents a study of the photochemical reaction pathways for byproduct formation in the photochromic switch 1,2-bis-(3-thienyl)-ethene. Specifically, using single- and multi-reference methods the post-deexcitation reaction towards the byproduct in the electronic ground state S0 when starting from the S1–S0 conical intersection (CoIn), is considered in detail. We find an unusual low-energy pathway, which offers the possibility for the formation of a dyotropic byproduct. Several high-energy pathways can be excluded with high probability.
1 Introduction
Molecular photoswitches are compounds which are capable of a reversible change in their molecular structure between at least two structures upon external photostimulation. The isomers must exhibit different absorption spectra to facilitate the selective isomerization of one or the other form.1 For a plethora of molecules that fit this minimal requirement, highly desired secondary properties are thermal stability of the switched states, fatigue resistance, rapid response, and high sensitivity. Depending on their specific properties, photoswitches show potential as components for example in optical memory media or optically switchable, electronic devices.Diarylethene derivatives are quite auspicious systems for these applications.2 Dithienylethenes are a very well studied sub-class of these, as the low energy difference between the photo-active open-ring and closed-ring conformation (see Fig. 1) is quite favorable.3–5 In the system under study in this article for example the open-ring structure is only roughly 17 kJ mol−1 more stable on UMP2/6-31G(d) level than the closed structure. As a consequence, open-ring and closed-ring structures are separated by comparatively large barriers,6 guaranteeing high thermal stability of all photoactive isomers. At the same time the energy difference of the Franck–Condon points on the S1 state is very large and thus facilitates the selective excitation of the isomers. During the optical switching process the barrier is surpassed either from the open form by photocyclization upon irradiation with ultra-violet (UV) light, or from the closed form by cycloreversion (ring-opening) upon irradiation with visible (Vis) light as indicated in Fig. 1.
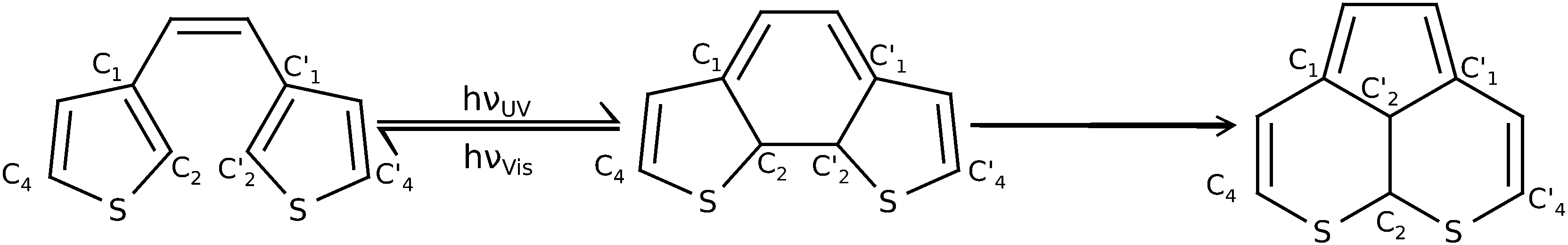 | ||
| Fig. 1 Photoswitching reaction between the closed-ring (middle) and the open-ring structure (left) of 1,2-bis-(3-thiophenyl)-ethene. The cyclization requires UV light, the cycloreversion requires visible light. The molecule on the right side is a photochemical byproduct detrimental to the use as a photoswitch. The labeled atoms (C1, C2 and S) are centers participating in the bonds that are broken and formed in the byproduct formation process. | ||
Despite the excellent properties of diarylethenes the formation of a byproduct remains a problem. The byproduct, also shown in Fig. 1, acts as a dead end in photoswitching and therefore leads to fatigue of the switch. The formation and the identity of this kind of byproduct was shown experimentally in ref. 7–9. For this reason a major concern of recent and ongoing research is the understanding and suppression of byproduct formation via dyotropic rearrangement, with the goal to increase the fatigue resistance of diarylethenes.10,11 The first mechanistic ideas were based upon theoretical studies by Celani et al.12 who suggest for a simpler system, cyclohexadiene (closed)/hexatriene (open), a two-step mechanism. Accordingly, one first photoexcites the closed-ring structure and then the system is funneled towards the S1/S0 conical intersection (CoIn). From there a path leading via an initial C2–C2′ bond elongation to a significant C1–C2′ bond length decrease can be followed, creating a triangular CoIn which is well-known from related molecules.13,14 Since a C1–C2′ bond needs to be formed in the byproduct of diarylethenes (cf.Fig. 1), this supports the idea of the closed-ring structure being the most probable starting point for byproduct formation.
In order to overcome the flaw of byproduct formation, multiple modifications of the basic dithienylethene molecular body have experimentally been realized. Irie et al. suggested that methyl substituents at C4 and C4′ suppress rearrangement.6 Other substituents, preferably with π-conjugation, in these positions can also be beneficial.15–17 Irie et al. also suggested the methylation of the C2 and C2′ positions (see Fig. 1) which yields a significant increase of stability.6 Nevertheless this modification is often left out in theoretical models.18–20 Finally, another successful modification is the variation of the ethene-like bridging unit, replacing it by groups like maleic anhydride,21 perfluoro-cycloalkenes22 or cyclopentenes,15,17 or replacing the hydrogens with cyano groups.23 Especially cyclopentenes and their fluorinated counterparts are intensively synthesized and studied nowadays as this structural moiety supports the planarity of the structure and thus prevents the already rather slow unintended process of thermal cycloreversion. In addition the lone electron pairs of the flourine atoms extend the π-conjugation of the system even further.22
On the basis of time-dependent density functional theory, Hania et al.15 identified the S4 state, also labeled 2B according to C2 symmetry nomenclature, as the final state of the initial photoirradiation of the closed-ring isomer of a (substituted) diarylethene. Experimentally they found that the molecule relaxes on a sub-picosecond timescale to the almost degenerate S3 (3A) state. For dithienylethene, relaxation towards the S2 (1B) state was discussed and found theoretically by Guillaumont et al.,18 as well as a further relaxation pathway towards the S1 (2A) and, finally, S0 (1A) state. The S0 state is reached through a S0/S1 CoIn. From there, only the possibilities leading to the open-ring and closed-ring structures were discussed in these early studies, while the byproduct formation was not considered.
At this point Patel et al.24 proposed a greater variety of ground state (S0) reaction pathways, in analogy to the hexatriene/cyclohexadiene study of Celani et al., for various functionalised dithienylethenes. They computed transition barriers between the closed-ring form, a so-called bicyclohexane (BCH, right structure in Fig. 2), and the byproduct. In particular, BCH serves as a possible intermediate for byproduct formation in a thermal mechanism. All calculations were done on the (single-reference) unrestricted hybrid density functional level of theory. On the UB3LYP/6-31G(d) level for example, the thermal pathway leading from the closed form to BCH and from there to the byproduct, was accompanied by barriers of about 50 kcal mol−1 (more than 2 eV, for closed to BCH) and 20 kcal mol−1 (ca. 1 eV, for BCH to byproduct), respectively. In particular the first barrier is very high, probably too high for a thermal process. Also, photochemical byproduct formation was discussed briefly. Here it was postulated that after passage through the S1/S0 CoIn, a methylcyclopentene diradical (MCPD, left structure in Fig. 2) might form as a possible intermediate on the way to the byproduct. However, this path could not be verified computationally.
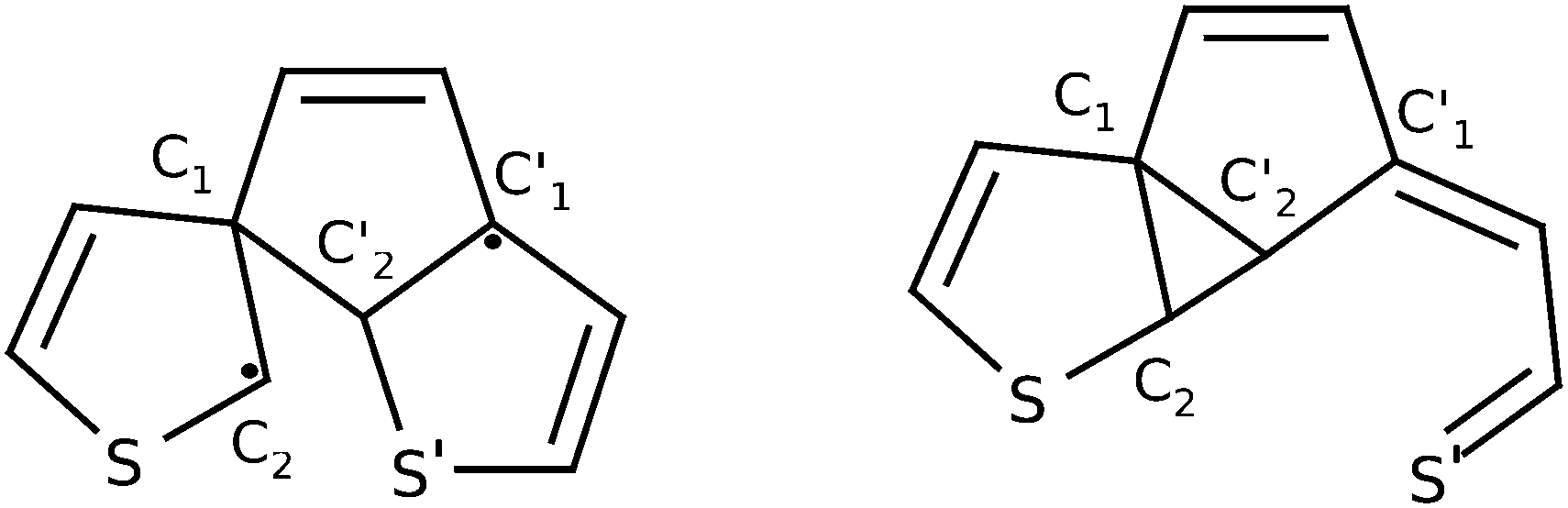 | ||
| Fig. 2 The methylcyclopentene diradical (MCPD, left) and the bicyclohexane form (BCH, right) of 1,2-bis-(3-thiophenyl)-ethene. | ||
While Patel et al.24 focused on (thermal) ground state processes, very recently Mendive-Tapia et al.20 studied the reaction pathways for various diarylethenes toward the byproduct focusing on the excited states, using multi-reference methods. They proposed that the key for photochemical formation of the byproduct is the non-adiabatic decay from the excited state to the ground state at a geometry between closed-ring and that of the byproduct. As the closed-ring structure faces barriers on the excited state surface in the direction of cycloreversion as well as for C2–S (C2′–S′) bond breaking, both reactions (formation of byproduct and re-formation of the closed form) seem possible. Detailed investigations of the fate of the molecule after returning to the ground state were not provided in ref. 20, however.
Our article complements the research done in ref. 24 and 20, by thoroughly exploring a thus far not further characterized plateau region on the ground state potential energy surface near the S0/S1 CoIn. The accessibility and topology of this region may play a decisive role for the photochemical formation of the byproduct. The point where our findings fit into the current state of research is indicated schematically in Fig. 3. Our paper aims to study in detail the photoinduced byproduct formation in diarylethenes, in particular its ground state part, i.e., after the system has passed the S1/S0 CoIn, or “photochemical funnel”. Using single- and multi-reference methods, a structure similar to the mentioned MCPD molecule was found, which can play the role of a first intermediate on S0 towards (by)-products, after the S1/S0 CoIn has been reached from higher states. Further, several other local minima and transition states were identified between this first intermediate and the final products. More and less likely pathways are revealed, including those along a BCH-like structure. All of this is done for 1,2-bis-(3-thienyl)-ethene, whose moderate size allows us to use accurate quantum chemical methods and which serves as a natural starting point to study fatigue of an “unprotected” diarylethene. Thereby, our work will fill in the more complex ground state connection between the CoIn seam the byproduct and cyclic isomer, for the system under study and the method used, and refine what is called a thermal pathway in ref. 24. This can be considered as a starting point which we intend to expand on more complex molecules in future studies to understand in detail the reasons for the increased stability induced by more complex substitution patterns.
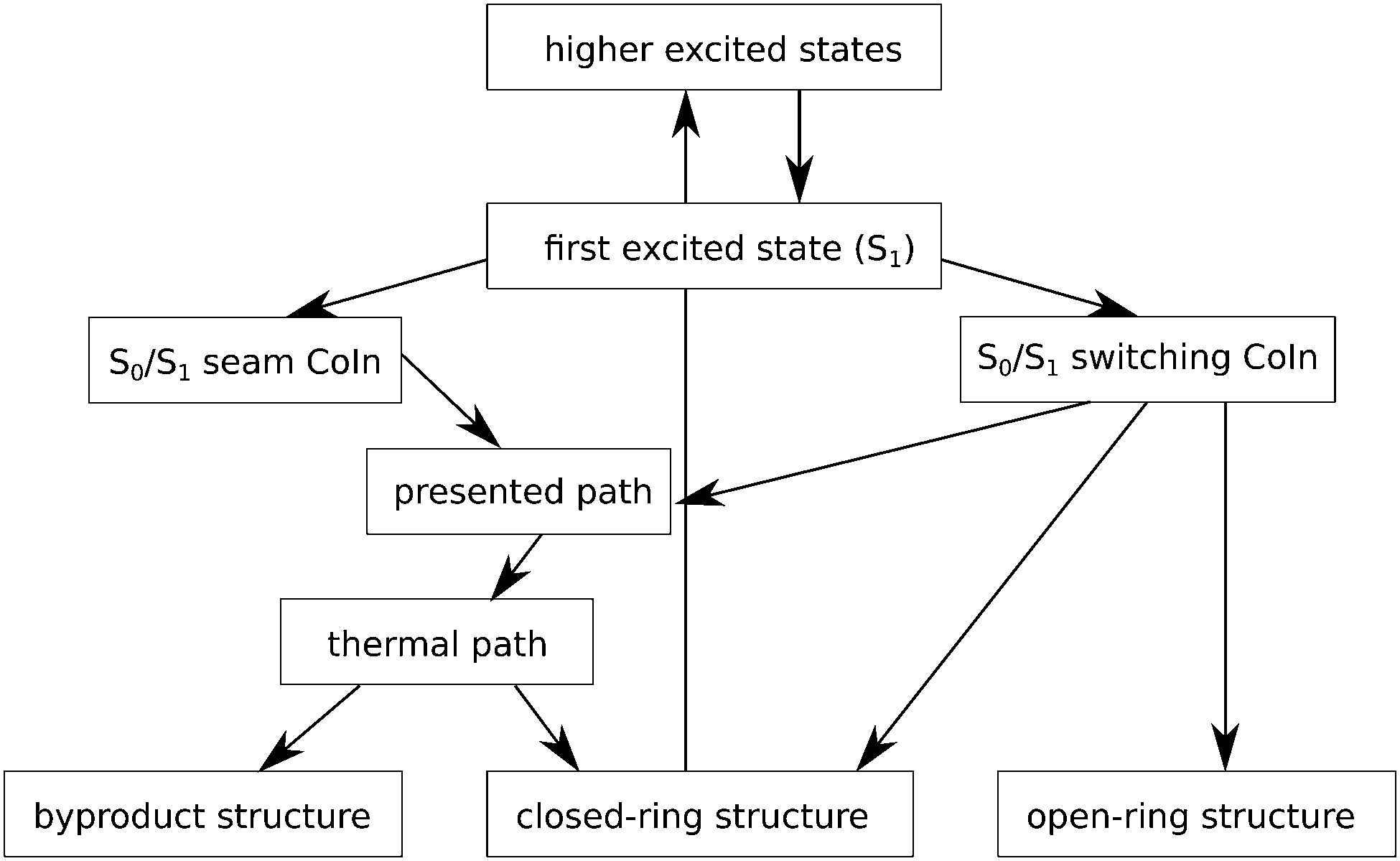 | ||
| Fig. 3 Photochemical reaction pathways for 1,2-bis-(3-thiophenyl)-ethene. After an excitation into S4 state (here subsumed under higher excited states) the molecule in funneled down to the S1 state. There occurs a barrier-free transition to the switching conical intersection which offers open- and closed-ring molecules, as well as the path established in this article (highlighted in grey) that lead to the cyclic molecule and the byproduct. This path contains a region similar to the thermal path presented in ref. 24 in the very end. Alternatively a barrier can be overcome to reach a part of the intersection seam (denoted as S0/S1 seam CoIn) which immediately leads into the ground state region presented in this article where the closed-ring molecule and the byproduct are possible products. The two different deexcitation regions on S1 are discussed in ref. 20. | ||
The article is organized as follows: Section 2 will focus on the computational methods. In Section 3 our investigation of the reaction pathway itself will be laid out in detail with emphasis on the reaction steps leading to the byproduct formation. Section 4 will summarize our work and its implications for the understanding of the dithienylethenes, and suggest future investigations.
2 Computational approach
To characterize special points of interest on the ground state (S0) PES of dithienylethene such as CoIns, transition states, and (local) minima, methods of increasing sophistication were adopted. First, ground state minima were calculated with Unrestricted Møller–Plesset perturbation theory of second order (UMP2) with the 6-31G25 and 6-31G(d)26 basis sets, using the GAUSSIAN 09 package.27 Based upon these results the first ten electronic states were calculated with the state-averaged CASSCF approach,28 including ten active electrons in ten active orbitals, abbreviated as SA-10-CASSCF(10,10), with the MOLPRO 2012 package.29,30 The active space was chosen such that it includes as many (π,π*) orbitals as possible. For the open-ring isomer this yielded ten (π,π*) orbitals while for the closed-ring species the active space was composed of ten (π,π*) orbitals supplemented by two (σ,σ*) orbitals. The active σ orbitals were chosen in a way that they form the C2–C2′ bond which is built during cyclization. Additional calculations with larger active spaces and more active electrons yielded no additional information and no significant change in relative energies. This was done to get a rough overview of low-lying electronic states and orbital contributions. The number of states was, in later simulations, reduced to the number of states actually necessary for the specific calculation. Thus at the S1/S0 CoIn two states were considered and averaged, while for the ground state reaction path we only calculated the ground state making all ground state calculations, both CASSCF and CASPT2, single state calculations.The S1/S0 CoIn was located on the SA-2-CASSCF(10,10)/6-31G(d) level of theory. Minimum energy path (MEP) calculations were executed by constrained quadratic steepest descent (QSD)31 on CASSCF(10,10)/6-31G and CASSCF(10,10)/6-31G(d) levels.
The local minima and transition states were pre-optimized on the UMP2/6-31G(d) level. For the localization of the transition states the Quasi Synchronous Transit method32 with three input geometries (QST3) was used. They were verified by intrinsic reaction coordinate (IRC) calculations.33 Several structures along the path exhibit a significant multiconfigurational character (see ESI† for details), thus the resulting geometries were refined by single-state (SS) CASSCF calculations on the CASSCF(10,10)/6-31G(d) level.
For all CASSCF-optimized structures the energies were finally refined by single-point energy calculations with the computationally demanding CASPT2(10,10)/6-31G(d) method, i.e., multi-reference perturbation theory to the second order on a CASSCF(10,10) reference wave function, as implemented in MOLPRO 2012.34 We finally stress that all structures below exhibit singlet electronic wavefunctions.
3 Results
3.1 Photochemical preparation of an initial, reactive intermediate
In what follows, we shall characterize stationary points on the S0 PES, leading from the S1/S0 CoIn all the way to the byproduct, and also (back) to the closed-ring form of the molecule. Of course, there is also a (direct) pathway to the open form, however, the latter is of no concern in this work. As outlined above in the photochemical process we assume, also in line with previous theory, that first a photoexcitation of the closed-ring molecule took place, followed by excited-state relaxation towards the S1/S0 CoIn from where the molecule will react further in the ground state. In order to estimate photoreactivity (e.g., towards the byproduct), it will be interesting to identify barrier heights for transitions between local minima on S0, and compare them to the energy of the S1/S0 CoIn, which is a valid starting point for analysis. Local minima act in addition as possible intermediate traps if vibrational relaxation is very efficient. Thus, stationary points on S0 (minima and transition states) are of great interest also for photochemistry. It should be said, however, that in this stationary, non-dynamical picture we can not reliably make any statements about product distributions. Also, in a dynamical process obviously the molecule will reach the ground state not only precisely at the CoIn, but rather in an extended region around it. However, in the following we map a large portion of the ground state potential energy landscape, so it is likely that molecules that cross over to S0 somewhat displaced from the S1/S0 CoIn will still end up on the reaction surface we propose.The S1/S0 CoIn, optimized on the SA-2-CASSCF(10,10)/6-31G(d) level of theory, is shown in Fig. 4(a). Detailed geometries are given in the ESI.† Both “wanted” directions of the photoreaction, ring-opening and cyclization, are strongly coupled to the C2–C2′ distance.20,24 Interestingly for the S1/S0 CoIn, we find a C1–C2′ distance which is even shorter (2.046 Å) than the C2–C2′ distance (2.156 Å). Thus, the formation of a five-membered ring with a completed C1–C2′ bond, a necessary prerequisite for the byproduct, becomes a strong alternative to (re-)formation of a six-ring product (or the open form), especially when the high hyperthermal energies of the molecule after electronic relaxation are considered (see below). This idea lead to the prediction of the MCPD structure already in ref. 24, as a possible intermediate with a complete C1–C2′ bond.
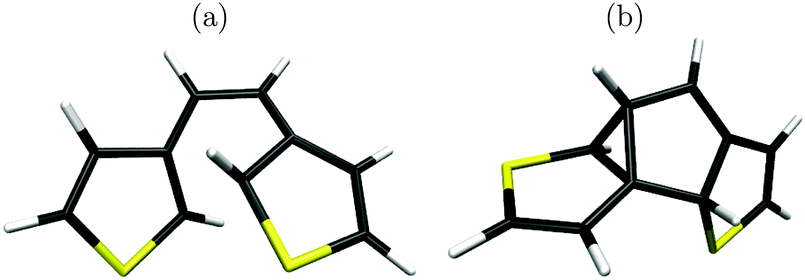 | ||
| Fig. 4 (a) The S1/S0 CoIn of dithienylethene, optimized on the SA-2-CASSCF(10,10)/6-31G(d) level of theory. (b) First intermediate, denoted as 1 below, optimized with CASSCF(10,10)/6-31G(d). | ||
We carried out extensive MEP searches in order to see if such a structure exists. Starting from the S1/S0 CoIn a CASSCF(10,10)/6-31G calculation, later reoptimized with CASSCF(10,10)/6-31G(d) gave a stable species with the structure depicted in Fig. 4(b). It contains indeed, an intact C1–C2′ bond with a distance of 1.513 Å, however, it is not the biradical as postulated in ref. 24 and shown in Fig. 2, left. Rather, a three-membered ring is formed by pairing (compared to Fig. 2) two unpaired electrons – see also Fig. 5 below. We shall see shortly that this molecule is a low-energy species. Therefore we have evidence for the accessibility of a C5-ring intermediate close to the CoIn, similar (but not identical to) MCPD. In the following, we shall denote this intermediate also as species 1.
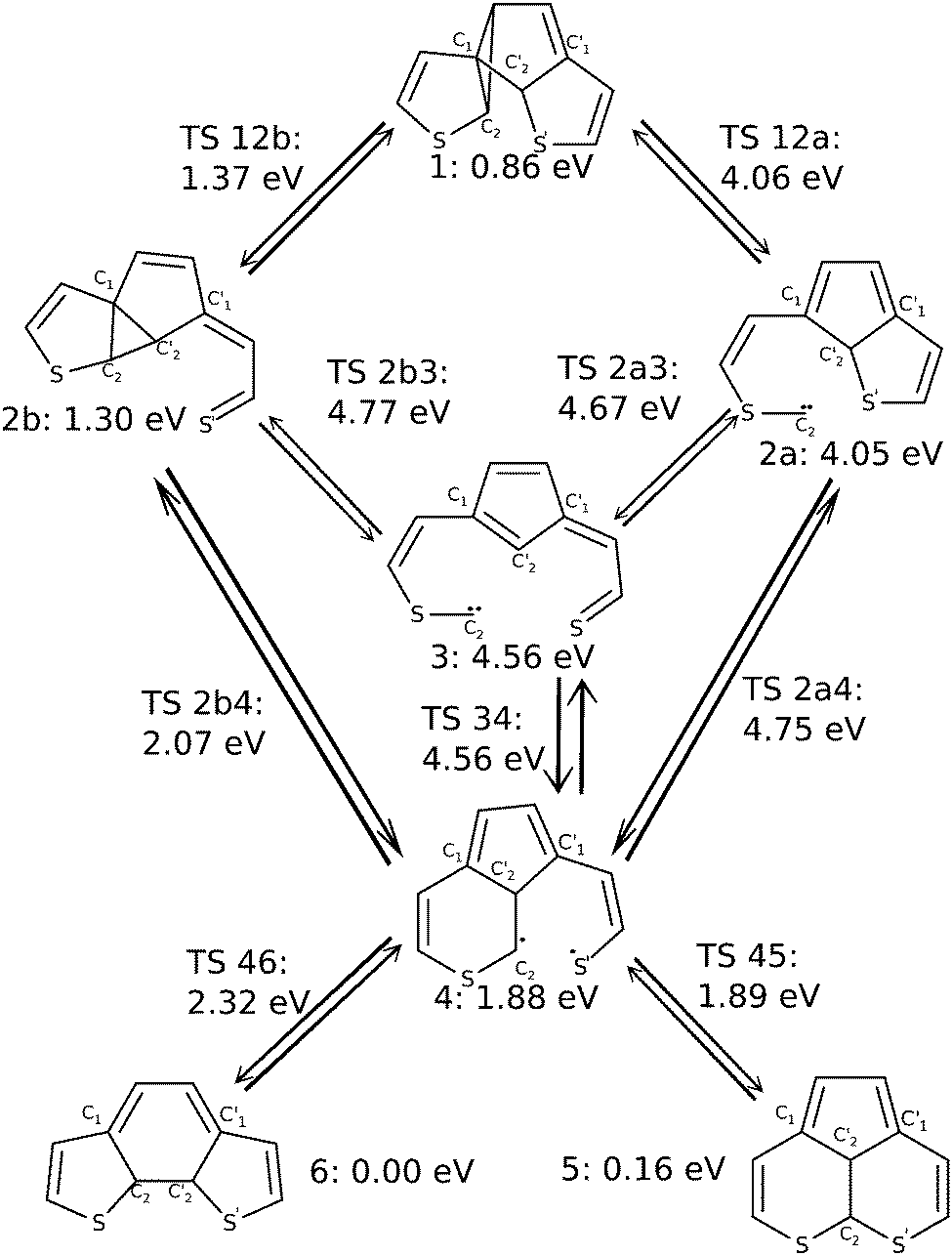 | ||
| Fig. 5 Sketch of stationary points (minima and transition states) on the ground state PES of dithienylethene, as investigated in this work. Minima are named by single numbers, n, and transition states connecting two minima n and m are denoted as TS nm. CASPT2(10,10)/6-31G(d) energies, relative to the (most stable) closed-ring structure, 6, are also shown. | ||
3.2 Stationary points on the ground state potential energy surface
We now consider possible reaction pathways, starting from structure 1. Fig. 5 summarizes these possibilities by showing stable compounds (indicated by single numbers, 1, 2a, 2b, 3, 4, 5, and 6) and transition states connecting different species (indicated as TS nm). Also, CASPT2(10,10)/6-31G(d) energies, all relative to the closed-ring structure 6 are indicated.Comparing the local minimum structure 1 with the byproduct 5 shows that the former consists of three five-membered rings and the byproduct of one five-membered ring and two six-membered rings which have a C–C bond in common (cf.Fig. 5). Obviously, there is no way to yield the byproduct without breaking and reforming C–C bonds. By basic reasoning the pathway that requires the least number of bonds to be simultaneously broken, can be expected to be the most likely. Nevertheless, other possible pathways were investigated too.
In order to achieve the relevant atomic rearrangements, the C1–C2 bond of one 5-ring of 1 and the C2–S bond of the other 5-ring have to be cleaved. This opens up two possible branches in the reaction path, depending on which bond is cleaved first (a concerted path could not be found in our study). The first path, shown as the right branch in Fig. 5, begins with breaking the C1–C2 bond leading to structure 2a, which is a reactive intermediate with an almost planar structure, and, based on investigation of the molecular orbitals and the electron density, a lone pair at C2. Cleaving the C2′–S′ bond then yields compound 3 in Fig. 5, which is also a minimum structure, formally still with a lone pair at C2, based on an inspection of molecular orbitals and electron density as above. From structure 3, a biradical 4 can be formed by making a C2–C2′ bond. Note that there is also a direct path from 2a to 4, without the detour via3. Along this latter route the carbon lone pair of C2 attacks C2′ forming the C2–C2′ bond, and cleaving the C2′–S′ bond in the sense of an SN2 reaction. Finally, the two unpaired electrons of compound 4 (which way ever formed), can pair in a new bond and yield the expected byproduct 5 according to Fig. 5. Alternatively, from structure 4, we can react back to the closed-ring structure 6. This requires shortening of the C1–C2 distance, elongating the C1–C2′ bond, and reforming the C2′–S′ bond.
As mentioned above a second major path leading from 1 to (by-)products, can be realized by initial cleavage of the C2′–S′ (rather than C1–C2) bond. This is shown in the left branch of Fig. 5, giving structure 2b. To form 2b, not only C2′–S′ must be broken but also C2 needs to break the distorted three-membered ring with the ethene carbon to form another three-membered ring structure with C1 and C2′. The opened and the central ring are still coplanar to each other. Note that intermediate 2b is the BCH structure shown in Fig. 2. After this first step the reaction pathway branches similarly to above. On the one hand it is possible to cleave the C1–C2 bond to yield structure 3 as before, with nothing changed but the order of the cleaved bonds. From there we can react further to 4 as before. Again, there is also a direct path from 2b to 4. Along this path the C1–C2 bond is elongated without actually being broken. While energetic aspects will be discussed in more detail in Section 3.3, we already state here that the pathway just mentioned (1 → 2b → 4 →…), involves the structures that are lowest in energy and is thus expected to be the most likely path leading to closed-ring and byproduct structures.
We should also like to emphasize a difference between the reaction paths found here (on MP2 single-reference and CASSCF multi-configuration levels of theory), and the one developed by Patel et al.24 (on single-reference U3BLYP and UM052x levels of theory). According to our study for the most simple dithienylethene, the BCH structure 2b is not a local minimum between closed-ring structure 6 and byproduct 5, as proposed (for a number of dithienylethenes) in ref. 24: on neither UMP2/6-31G(d) nor CASSCF(10,10)/6-31G(d) levels were we able to locate proper transition states TS 2b5 or TS 2b6 between 2b and 5 or 6, respectively. Instead we find another local minimum 4 in a flat “plateau region” of the PES (see below), which then is directly connected to the final products via dyotropic rearrangement (TS 46), or a nearly barrierless C2–S′ bond formation (viaTS 45).
3.3 A mechanism for byproduct formation
Now we discuss in more detail the byproduct formation pathway starting from the first intermediate 1 in Fig. 5, that is most likely to occur based on our MEP calculations and energetic arguments. As outlined above, we assume that the photoexcited molecule will relax to the S1/S0 CoIn, from where it proceeds towards product(s) and byproduct. The molecule will carry at least the potential energy corresponding to the S1/S0 CoIn geometry when entering S0, and at most the energy gained from optical excitation from S0 to the bright state S4, at the Franck–Condon point corresponding to the closed-ring geometry. Due to thermal relaxation, probably the lower bound is more realistic. Comparing the energy deposited in the molecule to transition barriers in the ground state, will allow us to identify the most probable reactive routes.The energy difference between the closed-ring structure (at its ground state geometry) and the Franck–Condon point for vertical excitation to the optically bright S4 state is 6.39 eV on the CASPT2(10,10)/6-31G(d)//CASSCF(10,10)/6-31G(d) level of theory. The potential energy at the S1/S0 CoIn is 3.50 eV on the CASPT2(10,10)/6-31G(d)//SA-CASSCF(10,10)/6-31G(d) level. Due to the high excitation level the vertical excitation energy to the S4 level is likely to be overestimated with our calculation setup, but note that this number is only used as an orientation and that our simulations of the ground and S1 state can be considered to be significantly more accurate. Nonetheless, the molecule can be expected to roughly contain an internal energy between 3.50 and 6.39 eV when it enters the ground state through the S1/S0 CoIn. These energies need to be compared to those of intermediates and transition states, which are (for all stationary points indicated in Fig. 5), listed in Table 1. In the table, we give energies relative to the most stable species, as obtained with three different methods of gradually increasing sophistication: UMP2/6-31G(d), CASSCF(10,10)/6-31G(d), and CASPT2(10,10)/6-31G(d)//CASSCF(10,10)/6-31G(d). The latter method is expected to be the most reliable, and was already reported in Fig. 5. It is also the method which should be compared to the energy window between 3.50 eV and 6.39 eV just mentioned. All further statements will refer to this level of theory. We note that the lowest-energy structure is the closed-ring species 6 for UMP2 and CASPT2(10,10), and 5, the byproduct, is the second-most stable species, about 0.2 eV higher in energy. CASSCF alone (without perturbative corrections), however, predicts 6 to be slightly less stable than 5.
| Stationary point | UMP2 | CASSCF | CASPT2 |
|---|---|---|---|
| 1 | 1.60 | 1.20 | 0.86 |
| TS 12a | 5.53 | 3.36 | 4.06 |
| TS 12b | 4.36 | 1.20 | 1.37 |
| 2a | 4.50 | 3.06 | 4.05 |
| TS 2a3 | 5.53 | 3.44 | 4.67 |
| TS 2a4 | 5.98 | 4.02 | 4.75 |
| 2b | 1.37 | 1.40 | 1.30 |
| TS 2b3 | 4.49 | 3.49 | 4.77 |
| TS 2b4 | 2.18 | 1.93 | 2.07 |
| 3 | 4.67 | 3.32 | 4.56 |
| TS 34 | 4.76 | 4.08 | 4.77 |
| 4 | 2.21 | 1.95 | 1.88 |
| TS 45 | 2.19 | 1.94 | 1.89 |
| 5 | 0.21 | 0.00 | 0.16 |
| TS 46 | 2.39 | 2.28 | 2.32 |
| 6 | 0.00 | 0.05 | 0.00 |
Based on the given energies our basic considerations already render the right hand reaction path marked as a in Fig. 5 as very unlikely: transition states TS 12a, TS 2a3 and TS 2a4 are all above 4 eV, the latter two (with 4.67 and 4.75 eV) well above. This has to do with the high energies of species 2a and 3, of 4.05 eV and 4.56 eV, respectively. The barriers along the a path are thus lower than the vertical S0 → S4 excitation energy, but higher than the energy of the S1/S0 CoIn. Since the latter is probably the more relevant measure for the available energy, these pathways are unlikely, even more so when compared to paths discussed in the following.
For the same reason the reaction path labeled b that contains structure 3 as an intermediate is also unlikely. This leaves the sub-branch of path b in Fig. 5, which leads via the local minima 1, 2b and 4 to possible (by-)products 5 and 6, as the most probable one. Along this path all energies for intermediates as well as transition states, are well below 3.50 eV, the S1/S0 CoIn energy. In particular the lowest-energy path towards the byproduct, 1 → 2b → 4 → 5 is promising and will be discussed in further detail below. This path with computed geometries for intermediate minima and connecting transition states is also depicted in Fig. 6 for further illustration.
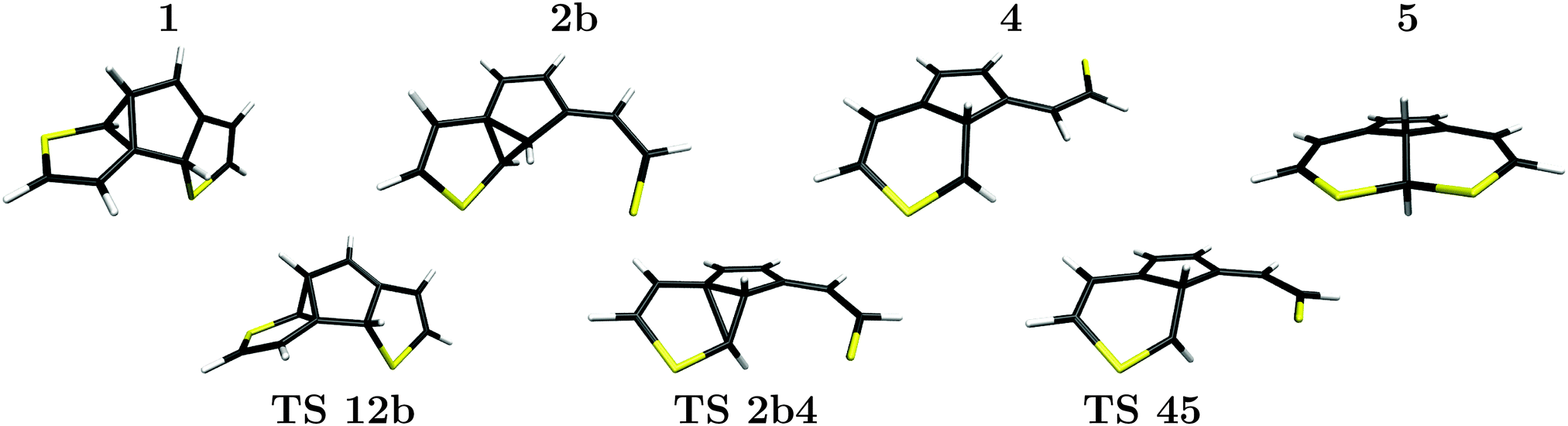 | ||
| Fig. 6 Spatial structures of the local minima (1, 2b, 4, 5, upper line) leading to the byproduct along the lowest-energy path, and corresponding transition states (TS 12b, TS 2b4, TS 45, lower line), optimized on the CASSCF(10,10)/6-31G(d) level of theory. | ||
Accordingly, the pathway presented here starts from structure 1 which contains no spiro-like units but consists of two five-membered rings connected by a three-membered ring including one of the ethene carbon atoms. This structure is a low-energy species, just 0.86 eV above the closed-ring structure 6, and 0.70 eV above the byproduct 5. During the transition between the structures 1 and 2b, the C2 carbon atom moves below the central ring from a C0 (ethene carbon)–C1–C2 ring structure to a C1–C2′–C2 ring and the C2′–S bond breaks. The 2b structure (see Fig. 6) closely resembles BCH as found by Patel et al.24 (see also Fig. 2). The central ring and the “loose” sulfur arm resulting in the cleaved C2–S bond are almost coplanar, merely the sulfur atom is pointing out of the plane. The next transition state, TS 2b4 shows the process of ring deformation where the five-membered and three-membered ring merges into a single six-membered ring structure, while the rest of the system becomes more planar. At the end of the transition the local minimum 4 is reached. In its relaxed structure, the sulfur atom of the opened ring points away from the rest of the structure, as shown in Fig. 6. Structures 2b and 4 have energies of 1.30 eV and 1.88 eV, hence starting from 2b the reaction path enters a rather flat energy landscape with only small barriers in between. Our IRC calculations, though, all lead to the mentioned minima which makes us confident that we found all the relevant ones.
From structure 4 two also rather shallow transition states, TS 45 (1.89 eV) and TS 46 (2.32 eV) allow for the formation of the byproduct and the closed-ring isomer, respectively. The higher barrier of 2.32 eV (0.43 eV when starting from 4) towards the more stable closed-ring structure 6 suggests that this is the product of thermodynamic control. The lower barrier of 1.89 eV (0.01 eV when starting from 4) leading to the energetically less preferred byproduct 5, on the other hand, can be considered as the product of kinetic reaction control. Since the system has excess energy from the photo-relaxation process and also due to the shallow barriers, the differentiation between these processes based on static properties alone should be taken with care, however, and none of the pathways can be ruled out.
We also note a feature that could favor the formation of the byproduct which is the orientation of the sulfur arm in structures 2b, TS 2b4, 4 and TS 45. It seems that structures along the way towards the byproduct 5, are all connected by a similar reaction coordinate in the region of the transition states. In a dynamical process with (photochemical) excess energy, one can therefore imagine that the local minimum 4 can be bypassed and the transition from 2b to 5 could proceed in one go via both transition states. To tread the full reaction path including the intermediate minimum 4, the sulfur arm of the formerly cleaved C2–S bond has to revert away from and back to the rest of the molecule. The central atom and the conjugated bonds of both ring structures are almost coplanar in both, the TS 45 transition and the final byproduct 5, as all heavy atoms apart from C2 and C2′ are part of the aromatic system.
4 Summary and conclusions
The ground state reaction pathway of 1,2-bis-(3-thienyl)-ethene leading to the photo-fatigue byproduct via dyotropic rearrangement has been studied. Three computational methods were used in order to verify the existence of local minima and transition states on single- and multi-configuration level with additional multi-referential perturbation theory calculations for energies. The stationary points found define a full reaction pathway which complements the thermal, ground state pathway investigated by Patel et al.,24 and the excited state photo pathway by Mendive-Tapia et al.,20 establishing it as a third possibility to form this detrimental photoproduct.Within this work, an MCPD-type starting structure whose existence has been claimed but not been proven so far was found by minimum energy path calculations starting for the S1/S0 CoIn. We were able to propose multiple pathways open to 1,2-bis-(3-thienyl)-ethene after photo-deexcitation and assessed their likelihood based on energetic arguments. Within our studies we were able to show that it is very well possible that the final “decision” of whether the ring-closed isomer or the unwanted byproduct is formed, is only made during the last stages along a prolonged ground state isomerization process. For that reason, the ground state processes cannot be left out of a discussion of the byproduct formation in 1,2-bis-(3-thienyl)-ethene.
Our study of the branching mechanism toward closed-ring and byproduct structure opens up the way to mechanistically driven substitution patterns that might help guiding future experimental and theoretical studies and can help to predict the effect of substitution patterns for this promising class of compounds. In fact, understanding and consequently being able to tailor fatigue resistant switches is a major driving force of our ongoing research.
Acknowledgements
We are thankful for fruitful discussions with Prof. S. Hecht (Humboldt University, Berlin). The authors gratefully acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG) via collaborative research center SFB 658 (project C2).References
- M. Irie, Chem. Rev., 2000, 100, 1685 CrossRef CAS PubMed.
- A. Aviram, J. Am. Chem. Soc., 1988, 110, 5687 CrossRef CAS.
- J. M. Tour, M. Kozaki and J. M. Seminario, J. Am. Chem. Soc., 1998, 120, 8486 CrossRef CAS.
- J. M. Seminario and J. M. Tour, in Molecular Electronics – Science and Technology, ed. A. Aviram and M. A. Ratner, New York Academy of Science, New York, 1998, vol. 69 Search PubMed.
- Y. Yokoyama, S. Uchida, Y. Yokoyama, Y. Sugawara and Y. Kurita, J. Am. Chem. Soc., 1996, 118, 3100 CrossRef CAS.
- M. Irie and M. Mohri, J. Org. Chem., 1988, 53, 803 CrossRef CAS.
- M. Irie, T. Lifka, K. Uchida, S. Kobatake and Y. Shindo, Chem. Commun., 1999, 747 RSC.
- K. Higashigushi, K. Matsuda, S. Kobatake, S. T. Yamada, T. Kawai and M. Irie, Bull. Chem. Soc. Jpn., 2000, 73, 2389 CrossRef.
- M. Herder, B. M. Schmidt, L. Grubert, M. Pätzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738 CrossRef CAS PubMed.
- Y.-C. Jeong, D. G. Park, E. Kim, K.-H. Ahn and S. I. Yang, Chem. Commun., 2006, 1881 RSC.
- R. Göstl, B. Kobin, L. Grubert, M. Pätzel and S. Hecht, Chem. – Eur. J., 2012, 18, 14282 CrossRef PubMed.
- P. Celani, S. Ottani, M. Olivucci, F. Bernardi and M. A. Robb, J. Am. Chem. Soc., 1994, 116, 10141 CrossRef CAS.
- F. Bernardi, M. Olivucci and M. A. Robb, Isr. J. Chem., 1993, 22, 265 CrossRef.
- Y. M. Wu and A. Kupperman, Chem. Phys. Lett., 1993, 201, 178 CrossRef CAS.
- P. R. Hania, R. Telesca, L. N. Lucas, A. Pugzlys, J. van Esch, B. L. Feringa, J. G. Snijders and K. Duppen, J. Phys. Chem., 2002, 106, 8498 CrossRef CAS.
- L. N. Lucas, J. van Esch, R. M. Kellogg and B. L. Feringa, Chem. Commun., 2002, 7136 Search PubMed.
- J. J. D. de Jong, L. N. Lucas, R. Hania, A. Pugzlys, R. M. Kellogg, B. L. Feringa, K. Duppen and J. H. van Esch, Eur. J. Org. Chem., 2003, 1887 CrossRef CAS.
- D. Guillaumont, T. Kobayashi, K. Kanda, H. Miyasaka, K. Uchida, S. Kobatake, K. Shibata, S. Nakamura and M. Irie, J. Phys. Chem., 2002, 106, 7222 CrossRef CAS.
- Y. Asano, A. Murakami, T. Kobayashi, A. Goldberg, D. Guillaumont, S. Yaboshita, M. Irie and S. Nakamura, J. Am. Chem. Soc., 2004, 126, 12112 CrossRef CAS PubMed.
- D. Mendive-Tapia, A. Perrier, M. J. Bearpark, M. A. Robb, B. Lasorne and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 18463 RSC.
- H.-G. Cho and B.-S. Cheong, Bull. Korean Chem. Soc., 1998, 19, 308 CAS.
- M. Hanazawa, R. Sumiya, Y. Horikawa and M. Irie, J. Chem. Soc., Chem. Commun., 1992, 206 RSC.
- T. Koshido, T. Kawai and K. Yoshino, J. Phys. Chem., 1995, 99, 6610 CrossRef.
- P. D. Patel, I. A. Mikhailov, K. D. Belfield and A. E. Masunov, Int. J. Quantum Chem., 2009, 109, 3711 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS.
- G. A. Petersson, A. Bennett, T. G. Tensfeld, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2913 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- H.-J. Werner and W. Meyer, J. Chem. Phys., 1980, 73, 2342 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, WIREs Comput. Mol. Sci., 2011, 2, 242 CrossRef.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. M. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2012.1, a package of ab initio programs, 2012 Search PubMed.
- J. Sun and K. Ruedenberg, J. Chem. Phys., 1993, 98, 9707 CrossRef CAS.
- C. Peng and H. B. Schlegel, Isr. J. Chem., 1993, 33, 449 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS.
- H.-J. Werner, Mol. Phys., 1996, 89, 645 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp00987a |
| This journal is © the Owner Societies 2015 |