
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward a stabilized lattice framework and surface structure of layered lithium-rich cathode materials with Ti modification†
Sihui
Wang
ab,
Yixiao
Li
a,
Jue
Wu
a,
Bizhu
Zheng
a,
Matthew J.
McDonald
a and
Yong
Yang
*ac
aState Key Laboratory of Physical Chemistry of Solid Surfaces, and Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: yyang@xmu.edu.cn; Fax: +86-592-2185753; Tel: +86-592-2185753
bTianjin Institute of Power Sources, Tianjin 300384, P. R. China
cSchool of Energy Research, Xiamen University, Xiamen 361005, P. R. China
First published on 12th March 2015
Abstract
Layered lithium-rich oxides have several serious shortcomings such as fast voltage fading and poor cyclic stability of energy density which greatly hinder their practical applications. Fabrication of a stable framework of layered lithium-rich oxides during charging–discharging is crucial for addressing the above problems. In this work, we show that Ti modification is a promising way to realize this target with bifunctional roles. For example, it is able to substitute Mn in the lattice framework and form a stable surface layer. It therefore leads to an improved retention of energy density of the Ti-modified Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0.04, 0.08, and 0.15) materials during cycling. The evolution of dQ/dV curves show that the layered/spinel phase transformation is suppressed owing to the introduction of strong Ti–O bonds in the framework. In addition, SEM, TEM, and EIS results confirm that a more uniform and stable interface layer is formed on Ti-modified Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0.04, 0.08, and 0.15) materials compared with the Ti-free counterpart. The stable interface layer on the lithium-rich oxides is also beneficial for further reducing side reactions, resulting in stable interface layer resistance. Therefore, the improved cycling performance of the material is due to both contribution of the more stable framework and enhanced electrode/electrolyte interface by Ti modification.
1. Introduction
Lithium-rich manganese-based materials xLi2MnO3·(1 − x) LiMO2 or Li[M1−xLix]O2 (M = Ni, Co, Mn, etc.) are attractive cathode materials for lithium ion batteries because of their characteristics of high capacity, high thermal stability, low cost and environmental friendliness. Typically, lithium-rich materials can deliver a high capacity of up to 250 mA h g−1 (even surpassing 300 mA h g−1 at high temperatures),1–5 which is nearly twice the capacity of commercial LiCoO2 materials. This high capacity makes lithium-rich materials promising candidates for meeting the high energy density demands of electric vehicles and other modern battery applications. However, lithium-rich materials are still far from practical applications due to their serious shortcomings such as substantial initial irreversibility, poor rate capability and significant capacity/voltage fading during cycling.For lithium-rich cathode materials, the initial charge process involves Li+ ions being first extracted from a layered LiMO2 component below ∼4.5 V, then being extracted from a monoclinic Li2MnO3 component above ∼4.5 V which is accompanied by oxygen loss.6 The extracted Li+ ions cannot be fully reinserted into the lattice, resulting in a large irreversible capacity loss and a low coulombic efficiency. Furthermore, during electrochemical cycling the capacity gradually decreases due to the conversion of a layered structure into a spinel structure,7 deterioration of the electrode/electrolyte interface,8 Jahn–Teller distortion,9,10 transition metal dissolution,10 and so on. The layered/spinel phase transformation also lowers the potential of the Mn4+/Mn3+ reduction reaction, makes the discharge profile shift to a lower voltage region (the phenomenon of voltage fading), and reduces the energy density of the lithium-rich cathode material.
Surface modification, element substitution and the use of electrolyte additives are all effective methods for improving the electrochemical performance of cathode materials for lithium ion batteries. Coating with Al2O3,11–13 TiO2,14 AlF3,8,15 C,16 MnOx,17 conductive polyaniline,18etc. has been found to reduce the initial irreversible capacity loss and improve the initial coulombic efficiency. Surface modification can also reduce the side reactions of the electrode/electrolyte interface, resulting in an improved cycling performance and rate capability. LiBOB, 3-hexylthiophene and LiDFOB have been investigated as electrolyte additives for lithium-rich cathode materials,19,20 and cells with such additives show better cycling performance. However, surface modification and using electrolyte additives have little positive or even negative effects on the working voltage fading during cycling, as voltage fading is the intrinsic characteristic of the layered/spinel phase transformation of lithium-rich materials.20 Therefore, in order to sustain the working voltage of a lithium-rich material, its structure must be stabilized during cycling. F substitution for some O atoms in Li[Li0.2Mn0.54Ni0.13Co0.13]O2 and Ru substitution for some Mn ions in Li2MnO3 have been proven to stabilize the voltage during cycling.21,22 And it appears that element substitution that improves structural stability is currently the most efficient method for suppressing voltage fading.
Ti has previously been utilized as a substitution element for lithium-rich cathode materials. In 2011, Deng et al. reported their investigation into the influence of Ti substitution on the oxygen loss and reversible capacity of lithium-rich cathodes.23 Their results show that Ti substitution can suppress oxygen loss ascribing to the stronger Ti–O bond and the lower M–O covalence compared with Mn–O and Ni–O bonds. However, the investigation of other electrochemical properties and working voltage evolution after Ti modification were excluded in Deng's work. Our previous work on Ti substitution in LiMn2O4 and LiNi0.5Mn1.5O4 materials has proven that Ti substitution can improve the structural stability of spinel materials.24–26 Therefore, we believe that Ti modification can not only substitute for partial Mn ions in the lithium-rich cathode material Li[Li0.2Mn0.54Ni0.13Co0.13]O2, but also modify the material's surface. In this work, the influence of Ti modification on the bulk/surface structure and electrochemical performance of Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) was systematically studied.
2. Experimental
Materials preparation
A coprecipitation–calcination method was used to prepare the lithium-rich cathode material Li1.2Mn0.54Ni0.13Co0.13O2. NaOH solution was added into a mixed solution of MnSO4·H2O, NiSO4·6H2O, and CoSO4·7H2O, and NH3·H2O was used to control the pH between 11 and 12 during the coprecipitation process. The as-prepared coprecipitation precursor was washed with deionized water to remove any unreacted compounds, and the sample was then dried at 120 °C to completely remove moisture. After drying, the precursor was mixed with Li2CO3 (5% excess) by ball milling. The mixture was pressed into pellets and precalcined at 480 °C in air for 6 h, followed by grinding and pressing into new pellets. Finally, the new pellets were calcined at 900 °C in air for 12 h and then quenched to room temperature to obtain the final material. To prepare the Ti-substituted lithium-rich cathode material Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0.04, 0.08, and 0.15), Ti(OC4H9)4 was first dissolved in ethanol where CH3COOH was used as the stabilizing agent. The Ti(OC4H9)4 solution was added into the mixed solution of MnSO4·H2O, NiSO4·6H2O, and CoSO4·7H2O, with the samples then undergoing coprecipitation and calcination as described above.Morphology and structure investigation
X-ray diffraction (XRD) patterns were recorded on a Panalytical X-pert diffractometer (PANalytical, Netherlands) at a scanning rate of 8 °C min−1 between 10° and 90°. The actual chemical compositions of the as-prepared materials were determined using inductively coupled plasma/atomic emission spectrometry (ICP-AES) (IRIS Intrepid II XSP, Thermo Electron). The morphology of the as-prepared materials and the cycled electrodes was characterized by scanning electron microscopy (SEM) on a S-4800 (HITACHI, Japan) microscope. Energy-dispersive X-ray spectroscopy (EDS) was used to identify the elements of the decomposition product on the surface of the cycled electrode. Transmission electron microscopy (TEM), high resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) analysis were carried out on a Tecnai F20 (Philips-FEI, Netherlands) apparatus. The valencies of transition metal ions were determined by X-ray photoelectron spectroscopy (XPS) using a Quantum 2000 ESCA spectrometer (Physical Electronics, USA).Electrochemical analysis
Electrochemical performance of the as-prepared materials was studied using CR2025 coin-type cells. 80 wt% active material, 10 wt% acetylene black, and 10 wt% poly-vinylidene fluoride binder (PVDF) in N-methyl-2-pyrrolidone (NMP) solvent were ground into slurry by ball milling, and then uniformly coated onto Al current collectors. These cathodes were dried at 120 °C in air to completely remove any water present. The cells consisting of the as-prepared electrode as a cathode, lithium metal as an anode, and Celgard 2300 as a separator were assembled in an argon-filled glove box. 1 M LiPF6/ethylene carbonate (EC) + dimethyl carbonate (DMC) (volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was used as an electrolyte. The cells were galvanostatically charged/discharged at different current densities over 2–4.7 V (vs. Li/Li+) at room temperature on Land CT 2001A battery testers. After cycling the batteries were disassembled in the glove box, the cathodes were thoroughly washed with dimethyl carbonate (DMC) solvent and dried, and then the materials were scraped off the electrodes if needed. Electrochemical impedance spectroscopy (EIS) was performed on a VersaSTAT MC Multichannel Potentiostat/Galvanostat (Princeton, America) analyzer at the charged state of 4.3 V. The frequency range was 100000–0.01 Hz or 100000–0.001 Hz with perturbation amplitude of ±10 mV.
:1) was used as an electrolyte. The cells were galvanostatically charged/discharged at different current densities over 2–4.7 V (vs. Li/Li+) at room temperature on Land CT 2001A battery testers. After cycling the batteries were disassembled in the glove box, the cathodes were thoroughly washed with dimethyl carbonate (DMC) solvent and dried, and then the materials were scraped off the electrodes if needed. Electrochemical impedance spectroscopy (EIS) was performed on a VersaSTAT MC Multichannel Potentiostat/Galvanostat (Princeton, America) analyzer at the charged state of 4.3 V. The frequency range was 100000–0.01 Hz or 100000–0.001 Hz with perturbation amplitude of ±10 mV.
3. Results and discussion
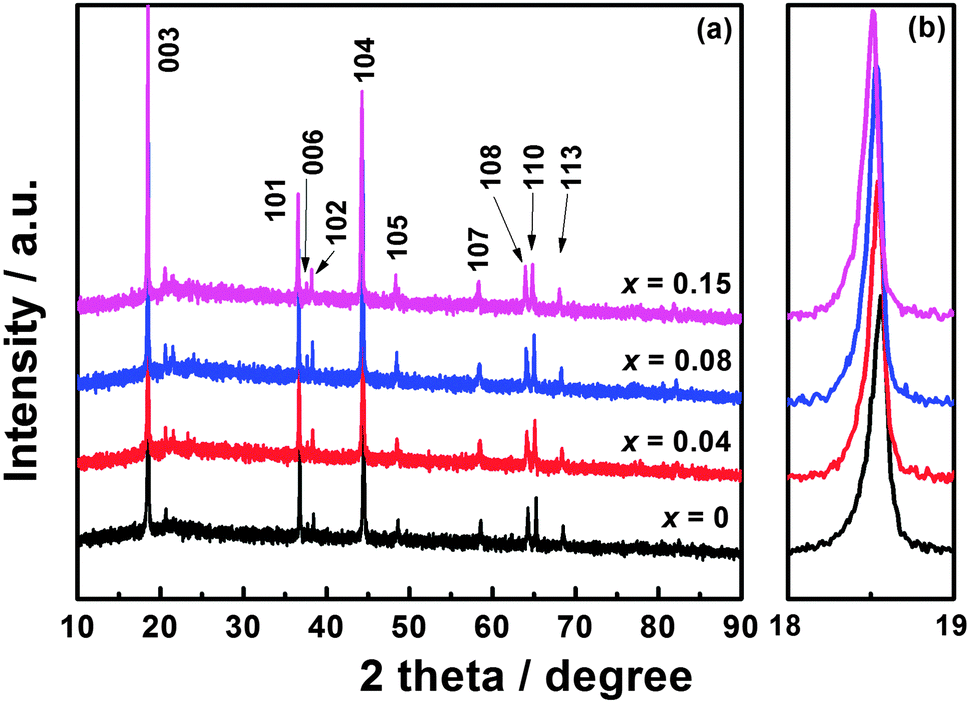
XRD patterns of Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) are shown in Fig. 1(a). For these materials, the strong diffraction peaks can be indexed to the hexagonal α-NaFeO2 structure with the space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The clear splitting of the (006)/(102) and (108)/(110) peaks demonstrates the well-defined layered structure.27 The weak diffraction peaks located between 20° and 25° are the characteristic peaks of monoclinic Li2MnO3 (space group C2/m), reflecting the superlattice ordering of Li and Mn in the transition metal layer.6 No impurity phase can be observed in any of these materials. As shown in Fig. 1(b), the diffraction peaks shift to a lower angle with increasing Ti content, which is caused by the larger Ti4+ ions substituting for partial Mn4+ ions.
m. The clear splitting of the (006)/(102) and (108)/(110) peaks demonstrates the well-defined layered structure.27 The weak diffraction peaks located between 20° and 25° are the characteristic peaks of monoclinic Li2MnO3 (space group C2/m), reflecting the superlattice ordering of Li and Mn in the transition metal layer.6 No impurity phase can be observed in any of these materials. As shown in Fig. 1(b), the diffraction peaks shift to a lower angle with increasing Ti content, which is caused by the larger Ti4+ ions substituting for partial Mn4+ ions.
 | ||
| Fig. 1 XRD patterns of Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15). | ||
The calculated lattice parameters and I003/I104 values are listed in Table 1. It has been reported that the peak intensity ratio of I003/I104 can be regarded as an indicator of cation mixing in the layered structure, and the cation mixing is small when this value is larger than 1.2.10,27,28 The Ti-modified materials have larger I003/I104 values compared with Li1.2Mn0.54Ni0.13Co0.13O2, suggesting that Ti modification is beneficial for reducing cation mixing and improving the electrochemical performance. However, the conductivity can be reduced after Ti substitution, leading to deteriorated performance. Variations in lattice parameters and volumes of Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) are exhibited in Fig. S1, ESI.† It can be seen that a, c, and V increase nearly linearly with increasing Ti content, indicating that Ti can be successfully incorporated into the lattice. Table S1, ESI† shows the chemical composition results of Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) obtained by ICP-AES analysis. The actual compositions of the as-prepared materials are in good agreement with their nominal compositions.
| a (Å) | c (Å) | V (Å3) | I 003/I104 | |
|---|---|---|---|---|
| x = 0 | 2.85572 | 14.24314 | 100.59 | 1.85 |
| x = 0.04 | 2.86191 | 14.26839 | 101.21 | 2.27 |
| x = 0.08 | 2.86563 | 14.27033 | 101.49 | 2.87 |
| x = 0.15 | 2.87256 | 14.29418 | 102.15 | 2.34 |
All the Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) materials exhibit very similar morphologies with the particle size of around 200–500 nm (Fig. S2, ESI†), indicating that Ti substitution does not affect grain growth in high temperature calcination. HRTEM images of Li1.2Mn0.54Ni0.13Co0.13O2 and Li1.2Mn0.39Ti0.15Ni0.13Co0.13O2 are shown in Fig. 2. The clear lattice fringes confirm the good crystallinity of the as-prepared lithium-rich materials. The calculated distances between two lattice fringes are 0.427 nm for Li1.2Mn0.54Ni0.13Co0.13O2, corresponding to the (020) planes of the monoclinic Li2MnO3, and 0.475 nm for Li1.2Mn0.39Ti0.15Ni0.13Co0.13O2, corresponding to the (001) planes of the monoclinic Li2MnO3 and/or the (003) planes of the rhombohedral LiMO2 phase. The insets in Fig. 2 show SAED patterns of the corresponding materials. The brighter points labelled with pink circles are reflections of the rhombohedral phase, and the dimmer points surrounded by blue ovals can be indexed to the monoclinic phase. The appearance of these two sets of reflections demonstrates the integration of the monoclinic phase with the rhombohedral phase. Ti modification does not change the structure of the lithium-rich cathode material. The valencies of transition metal ions were determined by XPS analysis (Fig. S3, ESI†), with transition metal ions in the states of Mn4+, Co3+ and Ni2+ in all these materials. This indicates that the equivalent Ti4+ substitution for partial Mn4+ ions does not affect the valencies of other ions.
 | ||
| Fig. 2 HRTEM images of (a) Li1.2Mn0.54Ni0.13Co0.13O2 and (b) Li1.2Mn0.39Ti0.15Ni0.13Co0.13O2. Insets show SAED patterns of the corresponding materials. | ||
The initial charge–discharge profiles of Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) at a current density of 40 mA g−1 over 2–4.7 V are presented in Fig. 3. The profiles are quite similar for all these materials, which show a gradually increasing voltage before 4.5 V and a long plateau at about 4.5 V during charge, and a sloping discharge profile. During the charge process, Ni2+ and Co3+ are oxidized to Ni4+ and Co4+ ions below 4.5 V, and Li and oxygen are removed at the 4.5 V plateau during the activation of the Li2MnO3 component. However, the extracted Li+ ions cannot be fully reinserted back into the lattice, leading to the observed large initial irreversible capacity loss. Li1.2Mn0.54Ni0.13Co0.13O2 delivers charge and discharge capacities of 312 and 268 mA h g−1, respectively, with a coulombic efficiency of 85.9%. In Fig. 3, the initial charge capacities of the Ti-modified materials are similar to that of Li1.2Mn0.54Ni0.13Co0.13O2, indicating that the strong Ti–O bond of these materials can also be broken and oxygen is extracted as occurs in Li1.2Mn0.54Ni0.13Co0.13O2. This is quite different from Deng's results that Ti-substituted materials show a shorter 4.5 V plateau compared with Li1.2Mn0.54Ni0.13Co0.13O2. The reason for this difference is still under investigation. However, since Ti substitution leads to a smaller amount of the activation product MnO2, the discharge capacities of Ti-modified materials are reduced because of the lower amount of Mn4+ ions available to be reduced to Mn3+ ions. The discharge capacities of Li1.2Mn0.50Ti0.04Ni0.13Co0.13O2, Li1.2Mn0.46Ti0.08Ni0.13Co0.13O2, and Li1.2Mn0.39Ti0.15Ni0.13Co0.13O2 are 257, 245 and 230 mA h g−1, respectively, with the reduced coulombic efficiencies of 82.4%, 78.5% and 72.8%.
 | ||
| Fig. 3 Initial charge–discharge profiles of Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) at the current density of 40 mA g−1 over 2–4.7 V. | ||
To investigate the cycling performance, the cathodes were cycled at the current density of 40 and 200 mA g−1 over 2–4.7 V. In Fig. 4(a), the capacities at the current density of 40 mA g−1 gradually decrease during cycling, which is ascribed to the deteriorated structure and electrode/electrolyte interface, dissolution of the metal ions, Jahn–Teller distortion, etc. Ti modification leads to a lower initial discharge capacity; however, it improves the cycling stability of lithium-rich materials. After 100 cycles, the electrodes with Ti content of x = 0, 0.04, 0.08 and 0.15 deliver similar discharge capacities of 202, 203, 204 and 203 mA h g−1, and the capacity retention ratio improves from 75.4% to 79.0%, 83.3% and 88.3%, respectively. By contrast, when cycled at 200 mA g−1, the discharge capacity more sharply decreases due to the large polarization, and all the electrodes show faster capacity fading compared with those cycled at 40 mA g−1. However, Ti-modified materials still exhibit more stable cycling performance compared with the Ti-free sample. For example, Li1.2Mn0.54−xTixNi0.13Co0.13O2 with Ti content of x = 0.04, 0.08 and 0.15 can deliver discharge capacities of 136, 156 and 150 mA h g−1 after 150 cycles, with associated capacity retention ratios of 61.2%, 75.6% and 81.3%, respectively, which are much better than the capacity of 125 mA h g−1 and the capacity retention ratio of 37.1% for Li1.2Mn0.54Ni0.13Co0.13O2. The bonding energy of the Ti–O bond is higher than that of the Mn–O bond; therefore, Ti modification can improve the structural stability of lithium-rich materials, which improves the cycling performance as observed.
 | ||
| Fig. 4 Cycling performance of Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) at current densities of (a) 40 mA g−1 and (b) 200 mA g−1 over 2–4.7 V. | ||
Fig. 5(a)–(d) show the evolution of charge–discharge profiles during cycling for Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) at the current density of 200 mA g−1. In Fig. 5(a), Li1.2Mn0.54Ni0.13Co0.13O2 exhibits a gradually reduced capacity, accompanied by the shifting of discharge profiles to lower voltages. The voltage fading is attributed to the layered/spinel phase transformation.29,30 For the Ti-modified materials shown in Fig. 5(b)–(d), the capacity–voltage fading is mitigated due to the improved framework stability coming from the introduction of more stable Ti–O bonds into the lattice. The corresponding dQ/dV curves are shown in Fig. 5(a′)–(d′). For all these materials, the oxidation peak at about 4.0 V during the initial charge process is attributed to the oxidation of Ni2+ and Co3+ to Ni4+ and Co4+ ions in the layered component, respectively. After that, the Li2MnO3 component is activated at about 4.5 V, with Li and oxygen being extracted simultaneously and forming the MnO2 component. As this activation process is irreversible, no corresponding reduction peak is observed in the discharge process. Co4+/3+ and Ni4+/3+/2+ reduction processes take place at about 3.8 V, and the activation product MnO2 is reduced at around 3.4 V during the initial discharge process. Compared with the Ti-modified materials, Li1.2Mn0.54Ni0.13Co0.13O2 shows a more pronounced reduction peak at around 3.4 V. Ti modification reduces the amount of MnO2 created during the initial activation, therefore, less Mn4+ is reduced to Mn3+, which in turn leads to a lower capacity below 3.5 V. The oxidation peak at about 4.0 V and the reduction peak at about 3.8 V gradually lose their intensity during further cycling, and the reduction peak of Mn4+/Mn3+ at about 3.4 V shifts to a lower potential which can be ascribed to the layered/spinel phase transformation. In Fig. 5(a′), the 3.4 V reduction peak shifts to about 2.71 V after 150 cycles, with the peak intensity being greatly reduced. This phenomenon demonstrates that the newly formed spinel phase gradually loses its capacity, which is in agreement with the observed fast capacity loss of the Li1.2Mn0.54Ni0.13Co0.13O2 material. The shifting of the 3.4 V reduction peak is suppressed by Ti modification, and the peak positions after 150 cycles are at about 2.83 V, 2.92 V and 2.94 V for materials with Ti content of x = 0.04, 0.08 and 0.15, respectively. In addition, the peak intensities below 3.0 V are steady or even slightly increased for the Ti-modified materials, suggesting that the capacity fading of the spinel phase is effectively suppressed, resulting in a more stable cycling performance.
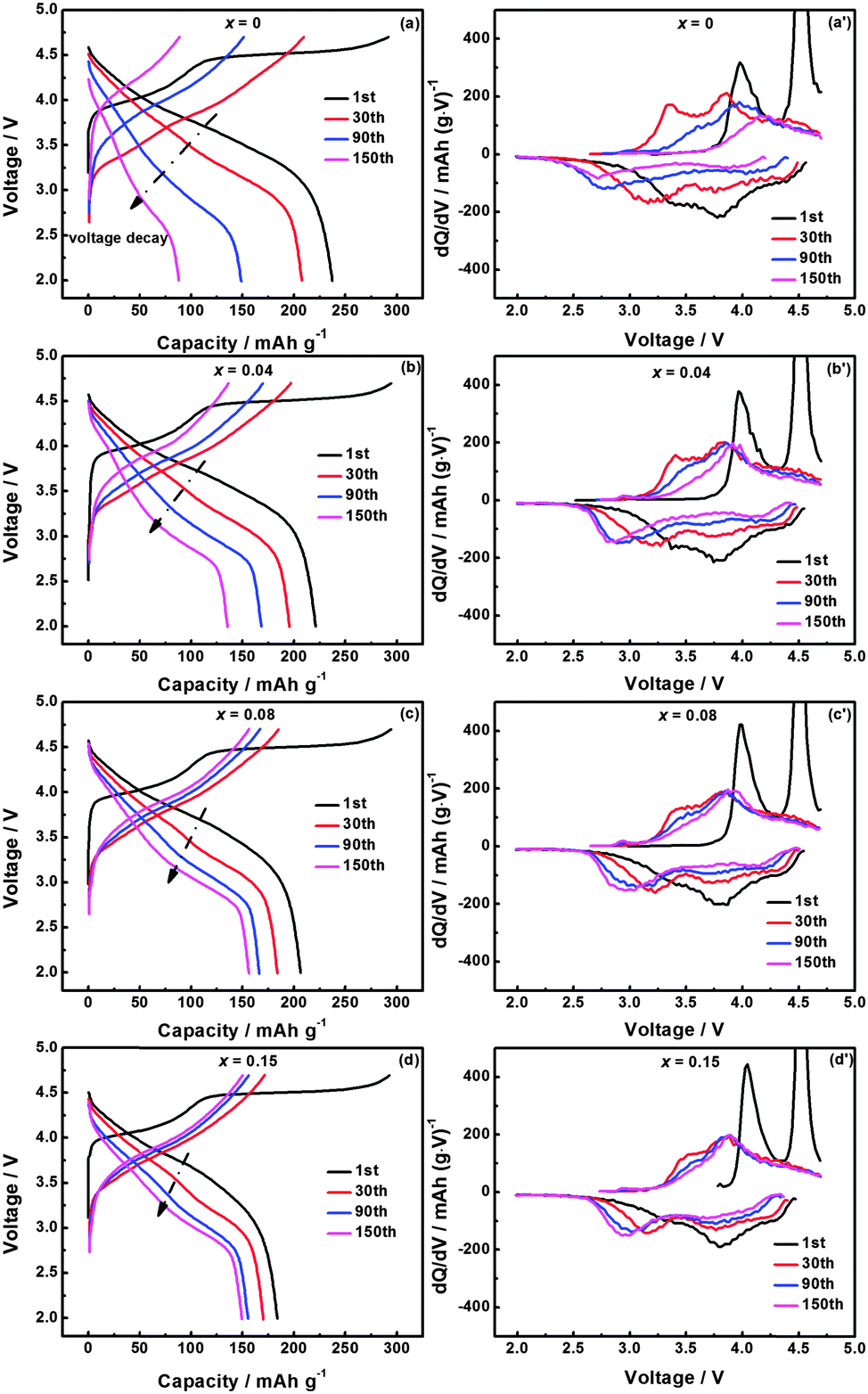 | ||
| Fig. 5 (a–d) Evolution of charge–discharge profiles and (a′)–(d′) dQ/dV curves of the corresponding cycle numbers for Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15). | ||
To get a more intuitive idea of the evolution of the voltage, mid-point voltages of these materials are plotted in Fig. 6. It is clear that the mid-point voltage of lithium-rich materials decreases as the cycling goes on. All these materials show similar mid-point voltages at the initial cycle, however, Li1.2Mn0.54Ni0.13Co0.13O2 shows faster voltage fading compared with the Ti-modified materials, illustrating again the benefits of Ti modification in stabilizing the structure and suppressing the voltage fading of the lithium-rich cathode material. As far as we know, this is the first report on sustaining the voltage by Ti substitution for lithium-rich cathode materials. In addition, the difference in mid-point voltages between materials with Ti content of x = 0.08 and 0.15 is very small, suggesting that there is no need to introduce a large amount of Ti into the lattice for sustaining the voltage. Fig. S4, ESI† shows the energy density of these materials during cycling. The Li1.2Mn0.54Ni0.13Co0.13O2 material exhibits a fast energy density decrease because of its unstable capacity and working voltage. Given that Ti modification improves the cycling performance and mitigates the voltage fading, a more stable energy density is the natural result. The discharge capacities and energy densities of Ti-modified materials are smaller than Li1.2Mn0.54Ni0.13Co0.13O2 during the initial few cycles, however, after 60–80 cycles the situation reverses and the energy densities of Ti-modified materials are higher than that of Li1.2Mn0.54Ni0.13Co0.13O2.
 | ||
| Fig. 6 Mid-point voltage of Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) materials cycled at a current density of 200 mA g−1. | ||
The 1st discharge profiles of different materials galvanostatically charged/discharged at current densities of 40, 200, 400 and 1000 mA g−1 are shown in Fig. 7(a)–(d). The discharge capacity decreases as the current density increases due to the large polarization occurring at high current densities. For all these materials, the variation in discharge capacities at different current densities mainly comes from the contributions below 3.5 V, indicating that the reaction below 3.5 V governs the rate performance of lithium-rich cathode materials. Fig. 7(e) and (f) show that the rate performance is negatively affected by Ti modification, and only the material with a Ti content of x = 0.04 exhibits similar capacity retention compared with Ti-free Li1.2Mn0.54Ni0.13Co0.13O2.
 | ||
| Fig. 7 Rate capabilities of Li1.2Mn0.54−xTixNi0.13Co0.13O2 with the Ti content of (a) x = 0, (b) x = 0.04, (c) x = 0.08, and (d) x = 0.15. A comparison of discharge capacity and normalized discharge capacity for different materials is shown in (e) and (f), respectively. | ||
The structure of Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) after 150 cycles at a current density of 200 mA g−1 was examined by XRD technique. As shown in Fig. 8(a), all the materials still exhibit a layered structure, while the superlattice diffraction peaks located at 20–25° disappear, suggesting the substantial activation of the Li2MnO3 component. Though dQ/dV curves confirm the layered/spinel phase transformation, diffraction peaks of the spinel phase cannot be detected due to its low content or amorphous characteristic. As shown in Fig. 8(b) and (c), for Li1.2Mn0.54Ni0.13Co0.13O2 and Li1.2Mn0.39Ti0.15Ni0.13Co0.13O2 materials the (108) diffraction peak shifts to a lower angle, and the (110) diffraction peak shifts to a higher angle after cycling, resulting in an increased separation between them. The behaviour of (108) and (110) peaks indicates the c-axis expansion and the presence of ions with a smaller ionic radius in the transition metal planes.31 As ions with a high valence such as Ni3+ (0.56 Å) or Ni4+ (0.48 Å) are smaller than those with low valence such as Ni2+ (0.69 Å), the amount of transition metal ions that can be oxidized/reduced to provide capacity is reduced during cycling. This is consistent with the decreasing capacity and the reduced oxidation/reduction peak intensity at about 4.0 V/3.8 V in dQ/dV curves during cycling.
 | ||
| Fig. 8 XRD patterns of (a) Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) electrodes after 150 cycles at a current density of 200 mA g−1. (b) and (c) A comparison of XRD patterns between the pristine material (black line) and the cycled electrode (red line) for Li1.2Mn0.54Ni0.13Co0.13O2 and Li1.2Mn0.39 Ti0.15Ni0.13Co0.13O2, respectively. | ||
SEM images of Li1.2Mn0.54−xTixNi0.13Co0.13O2 electrodes tested at a current density of 40 mA g−1 for 100 cycles are shown in Fig. 9. In Fig. 9(a), there are many areas of “dark” compounds covering the surface of the Li1.2Mn0.54Ni0.13Co0.13O2 electrode. The amount and thickness of the “dark” compounds decrease with the increase of Ti content (Fig. 9(b)–(d)), and no “dark” compounds are observed for the Li1.2Mn0.39Ti0.15Ni0.13Co0.13O2 electrode. This phenomenon is also seen in images of electrodes that were cycled at 200 mA g−1 (Fig. S5, ESI†). From Fig. 9(a′′)–(d′′), we can see that the surfaces of cathode materials are rougher than their corresponding pristine powders, indicating that surface films are formed during cycling. The elements of the “dark” surface compounds were determined by the EDS technique, with the EDS mappings shown in Fig. 10 confirming a large amount of F and P in the “dark” compounds. It is reasonable to deduce that these “dark” compounds may originate from the decomposition of the electrolyte during cycling, which typically forms insoluble LiF, LixPFyOz, etc., precipitating on the surface of the electrodes. TEM images of Li1.2Mn0.54−xTixNi0.13Co0.13O2 materials after 150 cycles at the current density of 200 mA g−1 are shown in Fig. 11. The interface film of Li1.2Mn0.54Ni0.13Co0.13O2 in Fig. 11(a) is nonuniform, and its thickness is quite different even for one nanoparticle. However, more uniform surface films are formed on the surface of Ti-modified materials, and the thicknesses of surface films are 8.1, 6.1 and 5.7 nm for materials with the Ti content of x = 0.04, 0.08 and 0.15, respectively. The uniform surface films can suppress the side reactions on the electrode/electrolyte interface, reduce the impedance of the electrode and improve the cycling performance.
 | ||
| Fig. 9 SEM images of Li1.2Mn0.54−xTixNi0.13Co0.13O2 electrodes after 100 cycles at a current density of 40 mA g−1. (a)–(a′′): x = 0, (b)–(b′′): x = 0.04, (c)–(c′′): x = 0.08, and (d)–(d′′): x = 0.15. The magnifications are 300×, 8000× and 30000×. | ||
 | ||
| Fig. 10 (a) SEM image of the “dark” surface compounds on a cycled Li1.2Mn0.54Ni0.13Co0.13O2 electrode and the corresponding EDS mappings of (b) F, (c) P, (d) O, (e) C, (f) Mn, (g) Co, and (h) Ni. | ||
 | ||
| Fig. 11 TEM images of Li1.2Mn0.54−xTixNi0.13Co0.13O2 materials after 150 cycles at a current density of 200 mA g−1. (a) x = 0, (b) x = 0.04, (c) x = 0.08, and (d) x = 0.15. | ||
EIS measurements were carried out to further analyze the effect of Ti substitution. Nyquist plots shown in Fig. 12 have three regions. The semicircle in the high-frequency region is attributed to the surface film resistance (Rsf), the semicircle in the intermediate-frequency is assigned to the charge transfer resistance (Rct), and the tail in the low-frequency region represents the Warburg impedance, which is related to Li+ diffusion in the bulk of the materials.32,33 The fitting data for Rsf and Rct are given in Table S2, ESI.†Fig. 12(a)–(c) demonstrate that Rct is the dominant resistance for lithium-rich cathode materials during cycling. The Ti-substituted materials show larger Rct values compared with Li1.2Mn0.54Ni0.13Co0.13O2, which is caused by the low electronic conductivity after Ti substitution. During cycling, the conversion of a layered structure to the spinel phase, the dissolution of transition metal ions, the rearrangement of the lattice, etc., lead to the increase of Rct values for these materials. Since the layered/spinel phase transformation is suppressed and the framework becomes more stable due to Ti modification, Ti-modified materials show a sluggish growth of Rct. However, the effect of the low electronic conductivity is so strong that Li1.2Mn0.39Ti0.15Ni0.13Co0.13O2 exhibits the largest Rct value throughout the cycling process, despite this material also having the most stable structure. Fig. 12(a′)–(c′) show the magnifications of Nyquist plots in the high frequency regions. Fig. 12(a′) shows that the differences in Rsf values of the 2nd cycle for these materials are very small. The Rsf value gradually increases during cycling for Li1.2Mn0.54Ni0.13Co0.13O2, while it remains stable at a low value for the Ti-modified materials. The increased Rsf of the Ti-free electrode suggests great changes in the surface structure or composition, possibly forming a thick surface film or compounds inhibiting Li+ ion transmission. This coincides with the SEM and TEM results in Fig. 9–11. The stable surface film and the lattice structure are beneficial for suppressing side reactions at the electrode/electrolyte interface, reducing the amount of Li+ ions forming the new surface film, and improving the charge transfer and Li+ ion diffusion. Therefore, the cycling stability of lithium-rich cathode materials is enhanced by Ti modification.
 | ||
| Fig. 12 Nyquist plots of Li/Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) during cycling at a current density of 200 mA g−1. The frequency ranges were 100000–0.01 Hz for the 2nd cycle, and 100000–0.001 Hz for the 100th and 150th cycles. | ||
Diffusion coefficients of Li+ for Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) during the 3rd discharge process were calculated using the GITT method. In Fig. 13, the diffusion coefficients of Li+ can be divided into three regions. Between 4.5 and 3.6 V, the Li+ ions are inserted into the layered MO2 structure, yielding similar diffusion coefficients for all the four materials. The plateau between 3.6 V and 3.25 V may be attributed to Li+ ions being inserted into the MnO2 component formed by the activation of Li2MnO3, and the diffusion coefficients in this region decrease with increasing Ti content. The concentration polarization is quite large at the end of the discharge process, therefore, the diffusion coefficients decrease faster in the 3.25–3.0 V region compared with those in the other two regions. This phenomenon is similar to that described in Yu's investigation of 0.5Li2MnO3·0.5LiMn0.42Ni0.42Co0.16O2 material.34 It is clear that Ti substitution mainly influences the diffusion coefficients when Li+ ions are inserted into MnO2. The more the Ti ions substitute in the lattice, the larger the resistance of Li+ ion diffusion is. Furthermore, the low diffusion coefficients are directly related to the poor rate performance of the Ti-modified lithium-rich cathode materials.
 | ||
| Fig. 13 Diffusion coefficients of Li+ for Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, and 0.15) during the 3rd discharge process. | ||
4. Conclusion
The cycling performance of Li1.2Mn0.54−xTixNi0.13Co0.13O2 (x = 0, 0.04, 0.08, 0.15) has been significantly improved by Ti modification, which is attributed to the bifunctional role of Ti in enhancing the framework and electrode/electrolyte interface stability. Mid-point voltage fading is mitigated for Ti-modified materials which is ascribed to the suppression of the layered/spinel phase transformation. The stable capacity and working voltage result in more stable retention of energy density after Ti modification.On the other hand, Ti modification decreases the amount of electrolyte decomposition product containing F and P on the surface of lithium-rich cathode electrodes. In addition, a more uniform interface layer is formed on the surface of Ti-modified materials, leading to a stabilized interfacial resistance. The resistance of Li1.2Mn0.54Ni0.13Co0.13O2 rapidly increases during cycling because of the layered/spinel phase transformation and an unstable interface layer, which results in the rapid decline of the discharge capacity. Ti modification is also proven to influence the diffusion of Li+ ions in the activation product MnO2 between 3.6 V and 3.25 V during the discharge process, which deteriorates the rate performance.
The ability of Ti to suppress voltage fading is particularly important, and the lessons learned here could be applied to the design of new lithium ion battery electrodes that can meet the urgent demands of high-energy density.
Acknowledgements
This work was financially supported by the National Basic Research Program of China (973 program, Grant No. 2011CB935903), National Natural Science Foundation of China (Grant No. 21321062) and National Foundation for Fostering Talents in Basic Science (Grant No. J1310024).Notes and references
- X. Jin, Q. Xu, X. Yuan, L. Zhou and Y. Xia, Electrochim. Acta, 2013, 114, 605–610 CrossRef CAS PubMed.
- Y. Jiang, Z. Yang, W. Luo, X. Hu and Y. Huang, Phys. Chem. Chem. Phys., 2013, 15, 2954–2960 RSC.
- J. Wang, B. Qiu, H. Cao, Y. Xia and Z. Liu, J. Power Sources, 2012, 218, 128–133 CrossRef CAS PubMed.
- X. Zhang, C. Yu, X. Huang, J. Zheng, X. Guan, D. Luo and L. Li, Electrochim. Acta, 2012, 81, 233–238 CrossRef CAS PubMed.
- C. S. Johnson, N. Li, C. Lefief and M. M. Thackeray, Electrochem. Commun., 2007, 9, 787–795 CrossRef CAS PubMed.
- M. M. Thackeray, S.-H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC.
- M. Gu, I. Belharouak, J. Zheng, H. Wu, J. Xiao, A. Genc, K. Amine, S. Thevuthasan, D. R. Baer, J.-G. Zhang, N. D. Browning, J. Liu and C. Wang, ACS Nano, 2013, 7, 760–767 CrossRef CAS PubMed.
- J. M. Zheng, Z. R. Zhang, X. B. Wu, Z. X. Dong, Z. Zhu and Y. Yang, J. Electrochem. Soc., 2008, 155, A775–A782 CrossRef CAS PubMed.
- D. Luo, G. Li, X. Guan, C. Yu, J. Zheng, X. Zhang and L. Li, J. Mater. Chem. A, 2013, 1, 1220–1227 CAS.
- S. J. Shi, J. P. Tu, Y. D. Zhang, Y. J. Zhang, C. D. Gu and X. L. Wang, Electrochim. Acta, 2013, 109, 828–834 CrossRef CAS PubMed.
- W. He, J. Qian, Y. Cao, X. Ai and H. Yang, RSC Adv., 2012, 2, 3423–3429 RSC.
- Y. Wu and A. Manthiram, Electrochem. Solid-State Lett., 2006, 9, A221–A224 CrossRef CAS PubMed.
- Y. Wu and A. Manthiram, Solid State Ionics, 2009, 180, 50–56 CrossRef CAS PubMed.
- J. M. Zheng, J. Li, Z. R. Zhang, X. J. Guo and Y. Yang, Solid State Ionics, 2008, 179, 1794–1799 CrossRef CAS PubMed.
- Y.-K. Sun, M.-J. Lee, C. S. Yoon, J. Hassoun, K. Amine and B. Scrosati, Adv. Mater., 2012, 24, 1192–1196 CrossRef CAS PubMed.
- J.-H. Ju, S.-W. Cho, S.-G. Hwang, S.-R. Yun, Y. Lee, H. M. Jeong, M.-J. Hwang, K. M. Kim and K.-S. Ryu, Electrochim. Acta, 2011, 56, 8791–8796 CrossRef CAS PubMed.
- J. Wang, X. He, R. Kloepsch, S. Wang, B. Hoffmann, S. Jeong, Y. Yang and J. Li, Energy Technol., 2014, 2, 188–193 CrossRef CAS.
- D.-h. Cho, H. Yashiro, Y.-K. Sun and S.-T. Myung, J. Electrochem. Soc., 2014, 161, A142–A148 CrossRef CAS PubMed.
- Y. Zhu, Y. Li, M. Bettge and D. P. Abraham, Electrochim. Acta, 2013, 110, 191–199 CrossRef CAS PubMed.
- I. Bloom, L. Trahey, A. Abouimrane, I. Belharouak, X. Zhang, Q. Wu, W. Lu, D. P. Abraham, M. Bettge, J. W. Elam, X. Meng, A. K. Burrell, C. Ban, R. Tenent, J. Nanda and N. Dudney, J. Power Sources, 2014, 249, 509–514 CrossRef CAS PubMed.
- J. Zheng, X. Wu and Y. Yang, Electrochim. Acta, 2013, 105, 200–208 CrossRef CAS PubMed.
- M. Sathiya, K. Ramesha, G. Rousse, D. Foix, D. Gonbeau, A. S. Prakash, M. L. Doublet, K. Hemalatha and J. M. Tarascon, Chem. Mater., 2013, 25, 1121–1131 CrossRef CAS.
- Z. Q. Deng and A. Manthiram, J. Phys. Chem. C, 2011, 115, 7097–7103 CAS.
- S. Wang, J. Yang, X. Wu, Y. Li, Z. Gong, W. Wen, M. Lin, J. Yang and Y. Yang, J. Power Sources, 2014, 245, 570–578 CrossRef CAS PubMed.
- G. He, Y. Li, J. Li and Y. Yang, Electrochem. Solid-State Lett., 2010, 13, A19–A21 CrossRef CAS PubMed.
- M. Lin, S. H. Wang, Z. L. Gong, X. K. Huang and Y. Yang, J. Electrochem. Soc., 2013, 160, A3036–A3040 CrossRef CAS PubMed.
- L. Zhang, W. Borong, L. Ning and W. Feng, Electrochim. Acta, 2014, 118, 67–74 CrossRef CAS PubMed.
- X. Wei, S. Zhang, Z. Du, P. Yang, J. Wang and Y. Ren, Electrochim. Acta, 2013, 107, 549–554 CrossRef CAS PubMed.
- J. R. Croy, D. Kim, M. Balasubramanian, K. Gallagher, S.-H. Kang and M. M. Thackeray, J. Electrochem. Soc., 2012, 159, A781–A790 CrossRef CAS PubMed.
- D. Mohanty, A. S. Sefat, J. Li, R. A. Meisner, A. J. Rondinone, E. A. Payzant, D. P. Abraham, D. L. Wood Iii and C. Daniel, Phys. Chem. Chem. Phys., 2013, 15, 19496–19509 RSC.
- Y. Li, M. Bettge, B. Polzin, Y. Zhu, M. Balasubramanian and D. P. Abraham, J. Electrochem. Soc., 2013, 160, A3006–A3019 CrossRef CAS PubMed.
- Q. Y. Wang, J. Liu, A. V. Murugan and A. Manthiram, J. Mater. Chem., 2009, 19, 4965–4972 RSC.
- X. Yang, X. Wang, Q. Wei, H. Shu, L. Liu, S. Yang, B. Hu, Y. Song, G. Zou, L. Hu and L. Yi, J. Mater. Chem., 2012, 22, 19666–19672 RSC.
- H. Yu, Y. Wang, D. Asakura, E. Hosono, T. Zhang and H. Zhou, RSC Adv., 2012, 2, 8797–8807 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Variations of lattice parameters and volume, ICP data, SEM images, XPS results, energy density evolution and impedance fitting results. See DOI: 10.1039/c5cp00853k |
| This journal is © the Owner Societies 2015 |