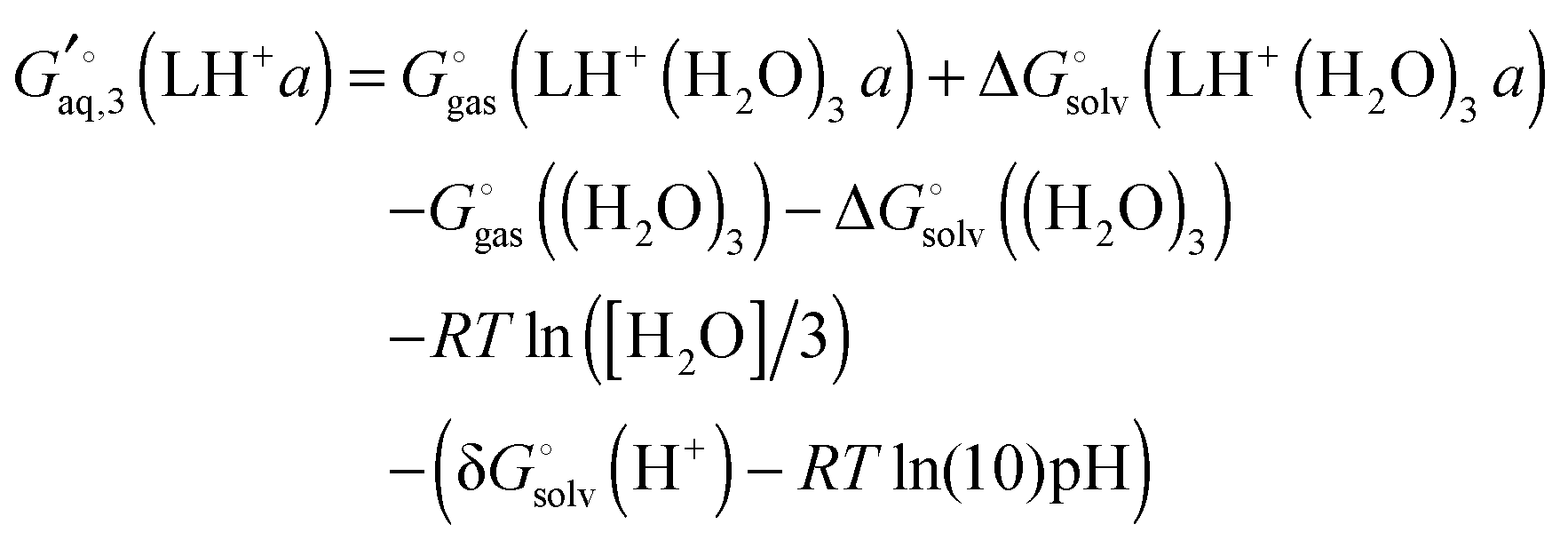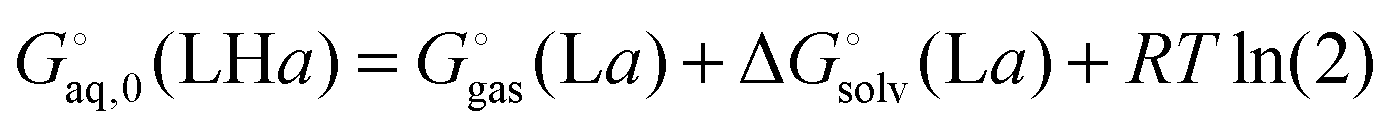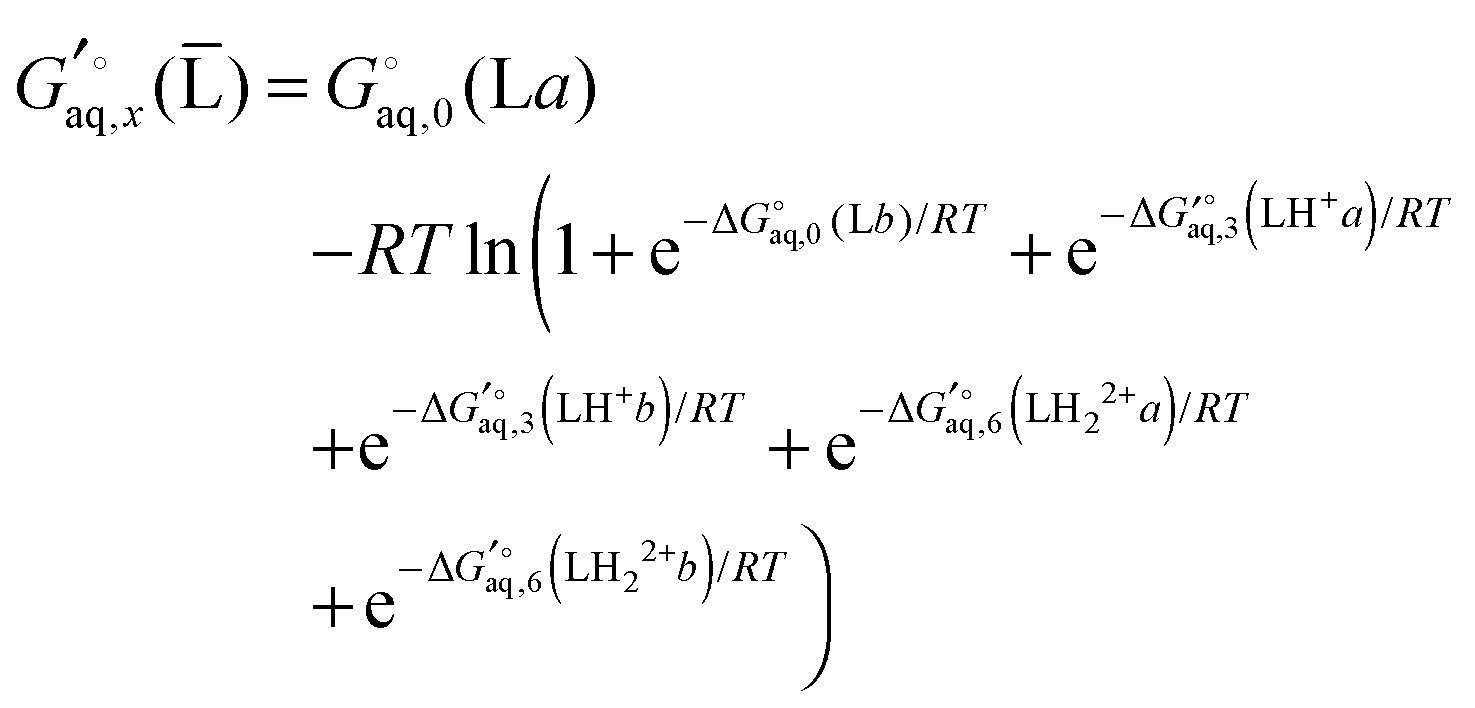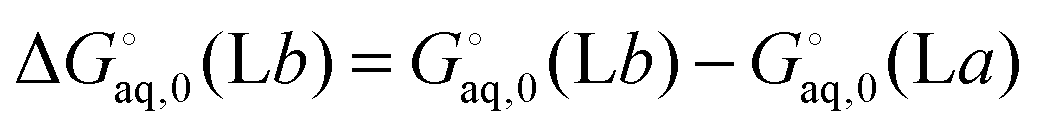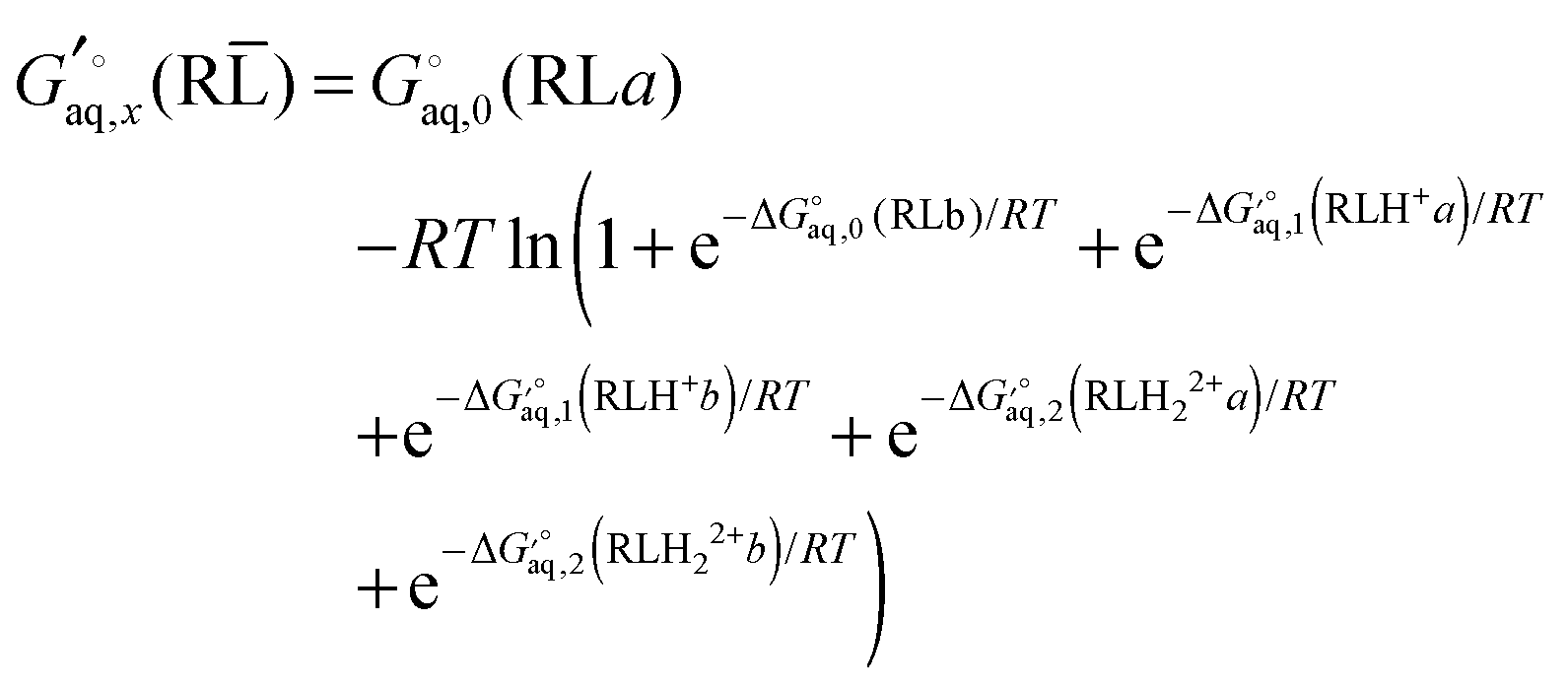DOI:
10.1039/C5CP00628G
(Perspective)
Phys. Chem. Chem. Phys., 2015,
17, 12441-12451
Predicting accurate absolute binding energies in aqueous solution: thermodynamic considerations for electronic structure methods
Received
31st January 2015
, Accepted 7th April 2015
First published on 9th April 2015
Abstract
Recent predictions of absolute binding free energies of host–guest complexes in aqueous solution using electronic structure theory have been encouraging for some systems, while other systems remain problematic. In this paper I summarize some of the many factors that could easily contribute 1–3 kcal mol−1 errors at 298 K: three-body dispersion effects, molecular symmetry, anharmonicity, spurious imaginary frequencies, insufficient conformational sampling, wrong or changing ionization states, errors in the solvation free energy of ions, and explicit solvent (and ion) effects that are not well-represented by continuum models. While I focus on binding free energies in aqueous solution the approach also applies (with minor adjustments) to any free energy difference such as conformational or reaction free energy differences or activation free energies in any solvent.

Jan H. Jensen
| Jan H. Jensen obtained his PhD in theoretical chemistry in 1995 from Iowa State University working with Mark Gordon, where he continued as a postdoc until he joined the faculty at the University of Iowa in 1997. In 2006 he moved to the University of Copenhagen, where he is now professor of bio-computational chemistry. In addition to his research interests in quantum biochemistry, he is interested in blended learning (receiving the University of Copenhagen teacher of the year award in 2013), open access publishing and open science. He is an active blogger (on Molecular Modeling Basics and Proteins and Wave Functions), active on Twitter and Google+ and initiated the overlay journal Computational Chemistry Highlights and the aggregator Computational Chemistry Daily. He welcomes further discussion on this Perspective on PubPeer. |
Introduction
The prediction of accurate absolute binding energies in aqueous solution is one of the holy grails of computational chemistry, mainly because of the potential use in rational drug design. “Accurate” is typically taken to be 1 kcal mol−1, which roughly corresponds to predicting the binding constant within an order of magnitude at room temperature and it is understood that the method must be generally applicable. The recent blind prediction challenge SAMPL4 has shown that this goal has yet to be met even for host–guest complexes that are significantly smaller than proteins.1 Interestingly, the entry that arguably performed best for one of the hosts (curcurbit[7]uril or CB7) was, for the first time, based on the rigid rotor-harmonic oscillator (RRHO) approximation and electronic structure theory and involved no direct parameterization against experimental binding free energies.2 This method reproduced 14 experimental CB7–guest binding free energies with a mean absolute deviation of 2.02 ± 0.46 kcal mol−1 suggesting that, perhaps, the holy grail is within reach. However, the mean absolute error was significantly larger for another host–guest system indicating that there remains some work to be done.
In this paper I summarize why electronic structure/RRHO-based approaches are starting to yield accurate binding free energies. I also discuss many of the possible sources of error when computing aqueous binding free energies with electronic structure theory and how to correct for them.
General approach
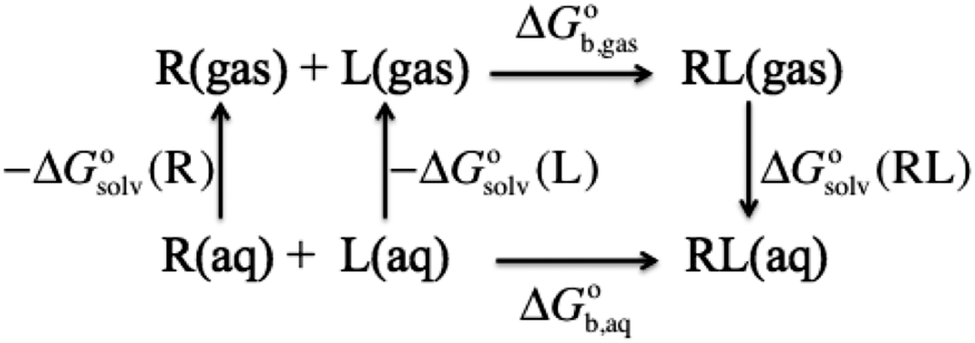
The general approach for predicting the standard free energy of binding 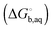 of a receptor (R or host) and ligand (L or guest) molecule in aqueous (aq) solution
of a receptor (R or host) and ligand (L or guest) molecule in aqueous (aq) solution| | | R(aq) + L(aq) ⇌ RL(aq) | (R1) |
using electronic structure theories is through a thermodynamic cycle (Fig. 1)| | 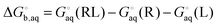 | (1) |
where| | 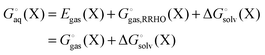 | (2) |
Egas(X),  , and
, and 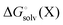 is the electronic energy, rigid rotor-harmonic oscillator (RRHO), and solvation free energy, respectively, of molecule X. Note that
is the electronic energy, rigid rotor-harmonic oscillator (RRHO), and solvation free energy, respectively, of molecule X. Note that  contains the zero point energy. The standard state (denoted by “°”) throughout this paper is 1 M, unless otherwise noted. The solvation free energy is typically computed using a continuum solvation model as described in detail below.
contains the zero point energy. The standard state (denoted by “°”) throughout this paper is 1 M, unless otherwise noted. The solvation free energy is typically computed using a continuum solvation model as described in detail below.
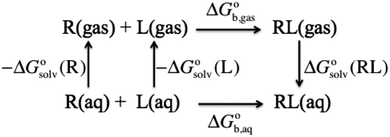 |
| | Fig. 1 Thermodynamic cycle for computing the binding free energy in aqueous solution for a ligand (L) binding to a receptor (R) to form a complex (RL). | |
The electronic energy
One of the reasons electronic structure-based approaches are starting to yield accurate binding free energies is the use of dispersion corrections3 in the evaluation of the electronic energy and the structure (as well as the vibrational frequencies as discussed below). Grimme4 has shown that dispersion typically makes a very big (>10 kcal mol−1) contribution to binding free energies of host–guest complexes. Dispersion corrections are therefore a must if DFT is used to compute the electronic binding energy. Furthermore, Grimme has shown that three-body dispersion makes a non-negligible (2–3 kcal mol−1) contribution to the electronic binding energy. For convergent methods this effect is only included in rather expensive methods that involve triple-excitations such as MP4 and CCSD(T).
Interestingly, it has been found that dispersion corrected, and short-range corrected, semiempirical methods such as DFTB or PM6, yield binding energies with accuracies similar to conventional DFT calculations with large basis sets. For example, Muddana and Gilson5 used PM6-DH+ to compute reasonably accurate relative binding energies for CB7–ligand complexes. On the other hand, Yilmazer and Korth6 found significant deviations for PM6-DH+ and similar methods when applied to larger protein–ligand models. Whether these minimal basis set-based methods are sufficiently flexible to handle large many-body polarization effects involving many charged groups remains to be determined. In any case, Grimme and co-workers have computed Egas(X) at the PW6B95-D3(BJ)/def2-QZVP//TPSS27-D3(BJ)/def2-TZVP level of theory with good results.2
Molecular thermodynamics
The translational, rotational and vibrational thermodynamic contribution to the binding free energy is very large (>10 kcal mol−1) and must be included for accurate results. Some years ago there was a bit of confusion in the literature about whether the RRHO approach was appropriate for condensed phase systems, but Zhou and Gilson7 have clarified this beautifully. The accuracy of the dispersion and hydrogen bond-corrected semi-empirical methods mentioned above has now made it feasible to compute the vibrational frequencies for typical host–guest complexes and this is another reason why electronic structure-based approaches are starting to yield accurate binding free energies. (They appear to be a qualitative step forward in accuracy compared to standard force fields in this regard.) For example, Grimme has computed  with PM6-D3H4 and HF-3c2 with good results.
with PM6-D3H4 and HF-3c2 with good results.
The standard state
Most electronic structure codes compute the RRHO energy corrections for an ideal gas, where the standard state is a pressure of 1 bar. As I'll discuss further below the solvation free energies are computed for a 1 M standard state so the gas phase free energy must be corrected accordingly| |  | (3) |
where V is the volume of an ideal gas a temperature T and R is the ideal gas constant. At 298 K this correction increases the free energy by 1.90 kcal mol−1.
It is tempting to argue that since the volume change in solution is negligible one should use the Helmholtz free energy  instead of the Gibbs free energy. However, as I discuss below, the solvation free energy corrects for the change in volume on going from the gas phase to solution, so the Gibbs free energy change should be used throughout.
instead of the Gibbs free energy. However, as I discuss below, the solvation free energy corrects for the change in volume on going from the gas phase to solution, so the Gibbs free energy change should be used throughout.
The vibrational enthalpy for NDDO based semiempirical methods
NDDO based semiempirical methods such as PM6 are parameterized against experimental standard enthalpies of formation 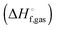 . However, in the case of intermolecular interactions such as hydrogen binding the parameterization was done by fitting
. However, in the case of intermolecular interactions such as hydrogen binding the parameterization was done by fitting 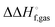 to ΔEgas values computed using electronic structure theory (Stewart 2007).8 The same is true for dispersion and hydrogen bond corrected PM6 methods. Thus, if a PM6 based method is used to compute the interaction energy the RRHO enthalpy corrections should still be included, i.e.
to ΔEgas values computed using electronic structure theory (Stewart 2007).8 The same is true for dispersion and hydrogen bond corrected PM6 methods. Thus, if a PM6 based method is used to compute the interaction energy the RRHO enthalpy corrections should still be included, i.e.| | 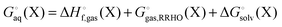 | (4) |
Molecular symmetry
Many host molecules and some guest molecules are symmetric and this affects the rigid-rotor rotational entropy (SRR) through the symmetry number (σ), which is a function of the molecular point group.| | 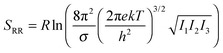 | (5) |
Here h and k are Planck's and Boltzmann's constant, respectively and Ix is the moment of inertia for principal axis x. In practice it can be very difficult to build large molecules with the correct point group and most studies use C1 symmetry. In this case the effect of symmetry must be added manually to the free energy| | 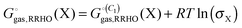 | (6) |
As an example, CB7 has D7h symmetry and a corresponding σ value of 14, in which case the correction contributes 1.56 kcal mol−1 to the free energy at 298 K.
Anharmonicity and low frequency modes
Host–guest complexes can exhibit very low frequency vibrations on the order of 50 cm−1 or less, which tend to dominate the vibrational entropy contribution.4 Many researchers have questioned whether the harmonic approximation is valid for such low frequency modes and this is an open research question. The main problem is that it is very difficult to compute the vibrational entropy exactly. Most methods for computing anharmonic effects are developed to obtain the 1 or 2 lowest energy states, but for very low frequency modes 10–20 states are likely significantly populated at room temperature and therefore contribute to the entropy.
In the absence of theoretical benchmarks, comparison to experiment can prove constructive. Kjærgaard and co-workers9,10 have recently measured standard binding free energies for small gas phase compounds and compared them to CCSD(T)/aug-cc-pV(T+d) calculations. For example, in the case of acetronitrile–HCl the measured binding free energy at 295 K is between 1.2 and 1.9 kcal mol−1, while the predicted value is 1.9 kcal mol−1 using the harmonic approximation.10 Since the errors in ΔE and the rigid-rotor approximation presumably are quite low, this suggest an error in the vibrational free energy of at most 0.7 kcal mol−1, despite the fact that the lowest vibrational frequency is only about 30 cm−1. Furthermore, the error can be reduced by 0.4 kcal mol−1 by scaling the harmonic frequencies by anharmonic scaling factors suggested by Shields and co-workers.11,12 Similar results were found for dimethylsulfide–HCl.9 So there are some indications that the harmonic approximation yields free energy corrections that are reasonable and possibly can be improved upon by relatively minor corrections.
On the other hand in a recent study Piccini and Sauer13 show that anharmonic effects need to be included to obtain agreement with the experimental binding free energy of methane to H-CHA zeolite. Specifically, they compute the vibrational binding free energy by computing the 1-dimensional potential energy surface for each low frequency mode and compute the vibrational energy levels and corresponding partition function numerically (as opposed to using the anharmonic fundamental frequency together with the harmonic oscillator partition function). This decreases the binding free energy by 2.5 kcal mol−1 compared to the standard harmonic oscillator treatment.
Grimme4 has taken a different approach by arguing that low-frequency modes resemble free rotations and using the corresponding entropy term for low frequency modes. This changes the RRHO free energy correction by 0.5–4 kcal mol−1, depending on the system.
Low frequencies are especially susceptible to numerical error and it is not unusual to see 1 or 2 imaginary frequencies of low magnitude in a vibrational analysis of a host–guest complex. Since imaginary frequencies are excluded from the vibrational free energy this effectively removes 1 or 2 low frequency contributions to the vibrational free energy. For example, a 30 cm−1 frequency contributes about 1.7 kcal mol−1 to the free energy at 298 K.
Imaginary frequencies resulting from a flat PES and numerical errors can often be removed by making the convergence criteria for the geometry optimization and electronic energy minimization more stringent and making the grid size finer in the case of DFT calculations. If the Hessian is computed using finite difference it is important to use central-differencing. If all else fails, it is probably better to pretend that the imaginary frequency is real and add the corresponding vibrational free energy contribution. However, this needs to be systematically tested.
Conformations
One of the main problems in computing accurate binding free energies is to identify the structures of the host, guest and (especially) the host–guest complex with the lowest free energy. Because both the RRHO and solvation energy contributions contribute greatly to the binding free energy change, simply finding the structure with the lowest electronic energy and computing the free energy only for that conformation is unlikely to result in the global free energy minimum.
For a molecule (X) with Nconf conformations the standard free energy is
| | 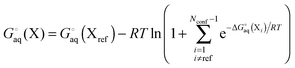 | (7) |
where
| | 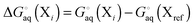 | (8) |
and where X
ref is some arbitrarily chosen reference geometry – for example the global minimum. With that choice for X
ref, conformations with free energies higher than 1.36 kcal mol
−1 contribute less than 0.1 to the sum at 298 K. So a significant number of very low free energy structures are needed to make even a 0.5 kcal mol
−1 contribution to the free energy. Conformations related by symmetry should not be included here as their effects are accounted for in the rotational entropy (see above). Note that if the binding measurements are done for racemic mixtures then all stereoisomers must be included in the sum.
Molecular charge and pH
Virtually all binding measurements in aqueous solution are performed in a buffer with a constant pH and many ligands and/or receptors contain one or more ionizable groups. The charge (q) of an ionizable (acid/base) group in aqueous solution depends on its pKa and the pH:| | 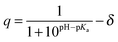 | (9) |
where δ is 1 for an acid and 0 for a base. This is an average charge for all the molecules in solution and will not be an integer. This section describes how to handle charges that different significantly from an integer value and/or change as a result of binding. The pKa can be computed using electronic structure theory or empirically using software such as Marvin.14 However, if the pKa value is perturbed by the binding the situation may be complicated further. Here I illustrate this point for a simple example where the ligand has a basic group that is neutral when deprotonated and the receptor is non-ionizable.| | | R(aq) + L(H+)(aq) ⇌ RL(H+)(aq) | (R2) |
The apparent equilibrium constant is then (throughout this paper I assume ideal solutions where the activity is equal to the concentration)| | 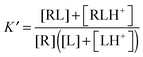 | (10) |
and the corresponding binding free energy is| | 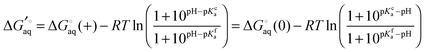 | (11) |
where 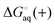 and
and 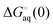 is the binding free energy computed using the charged (protonated) and neutral form of the ligand and pKca and pKfa are the pKa values the ligand bound to the receptor and the free ligand, respectively.
is the binding free energy computed using the charged (protonated) and neutral form of the ligand and pKca and pKfa are the pKa values the ligand bound to the receptor and the free ligand, respectively.
For example, Koner et al.15,16 have shown that binding of benzimidazole and derivatives to CB7 can increase the pKa of the ligand by as much as 4 pH units (from pKfa = 4.6 to pKca = 8.6) which results in a 3.3 kcal mol−1 pH-dependent correction to the binding free energy at pH 7. Put another way, using pKfa to determine the protonation state of the bound ligand would result in an 3.3 kcal mol−1 error in the binding free energy.
For many ligands of interest the pKfa can be estimated fairly accurately in a matter of second using programs such as Marvin. The effect of binding on pKfa can often be estimated by chemical intuition since hydrogen bonds to charged acid and basic groups tend to, respectively, lower or raise the pKa even further. For example, if an amine with pKfa = 9 binds to the receptor via hydrogen bonding, then pKca is likely higher than 9 and 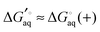 is a good approximation. However, if pKfa is close to 7 then pKca should be computed. Also, it is possible for charged ligands to change to their neutral state if they bind to hydrophobic or similarly charged receptors.
is a good approximation. However, if pKfa is close to 7 then pKca should be computed. Also, it is possible for charged ligands to change to their neutral state if they bind to hydrophobic or similarly charged receptors.
If pKfa is known with some degree of confidence (e.g. from experiment or Marvin) then pKca can be estimated by
| | 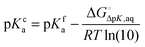 | (12) |
where
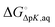
is the free energy change for this reaction
17| | | RLH+(aq) + L(aq) ⇌ RL(aq) + LH+(aq) | (R3) |
However, if one suspects that empirical p
Ka predictors such as Marvin give inaccurate results for p
Kfa then this value can be computed using quantum chemistry. Ho and Coote
18 have written a very useful summary of different approaches to such predictions. The accuracy for phenol and carboxyl p
Ka values are as low at 1 pH units (unfortunately they did not give a value for amines). However, if the p
Ka value is close to the pH of interest a 1 pH unit-error can lead to prediction of the wrong protonation and result in errors in the binding free energy on the order of 1–3 kcal mol
−1.
If there are several (Nionz) ionizable groups then eqn (11) generalizes to
| | 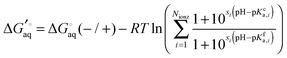 | (13) |
where
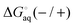
is the binding free energy when all acids and bases are deprotonated and protonated, respectively, the sum runs over all ionizable groups and
si is 1 and −1 if
i is a base or acid, respectively.
However, this assumes that the ionizable groups titrate independently of one another, i.e. that the pKa value of one group is independent of the protonation states of all other ionizable groups. If that is not the case then it is difficult to give a general expression for the pH-dependent free energy correction in terms of pKa values (though it can be derived for a specific case). Next I present an alternative approach, but note that in practice because one can obtain more accurate relative pKa values (using eqn (12)) or similar18 than absolute pKa values it may be worth the extra effort to derive the pH-dependent free energy correction in terms of pKa values.
Legendre transformed free energies
Instead a general expression can be written in terms of Legendre transformed free energies as suggested by Alberty19,20 and modified here to electronic structure calculations:21| | 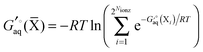 | (14) |
where 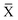 denotes an average over several protonation states of X, 2Nionz is the number of possible protonation states given Nionz sites and
denotes an average over several protonation states of X, 2Nionz is the number of possible protonation states given Nionz sites and| | 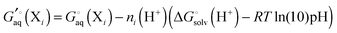 | (15) |
where ni(H+) is the number of ionizable protons in protonation state i, and 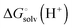 is the solvation free energy of the proton. So in the case of ligand L considered above, ni(H+) is 0 and 1 for L and LH+, respectively.
is the solvation free energy of the proton. So in the case of ligand L considered above, ni(H+) is 0 and 1 for L and LH+, respectively.
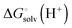 is usually taken from the literature where estimates vary between −264 and −266 kcal mol−1,22 which can add to the uncertainty in the predicted binding free energy change. There are at least two ways of reducing the error. One way is to maximize error cancelation by computing
is usually taken from the literature where estimates vary between −264 and −266 kcal mol−1,22 which can add to the uncertainty in the predicted binding free energy change. There are at least two ways of reducing the error. One way is to maximize error cancelation by computing 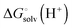 (using explicit solvent molecules as discussed below) using the same level of theory method use to compute
(using explicit solvent molecules as discussed below) using the same level of theory method use to compute 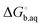 . The other way is to choose the value of
. The other way is to choose the value of 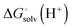 used as reference for the experimental solvation free energies of ions that are used to parameterize the continuum solvation model you use (Table 1). The first way is best if explicit solvent molecules are used to compute the solvation free energies of ions in the binding study and otherwise the second method is best.
used as reference for the experimental solvation free energies of ions that are used to parameterize the continuum solvation model you use (Table 1). The first way is best if explicit solvent molecules are used to compute the solvation free energies of ions in the binding study and otherwise the second method is best.
Table 1 Common continuum solvation models used with electronic structure theory, the level of theory used for parameterization and the solvation energy of the proton used as a reference for the experimental solvation energies of ions used in the parameterization. Adapted from Ho27
| Method |
Level of theory used for parameterization |
Solvation energy of proton used as reference for ions |
|
Ref. 28 IEF and CPCM give virtually identical results for water.
Ref. 29 UAHF spheres have been used with CPCM with good results.
Diffuse functions are used only for anions.
This parameterization has not been published and the information is taken from the Gaussian09 manual. The method has been benchmarked for CPCM by Takano and Houk.30
Ref. 31.
The parameterization was performed by minimizing the error for all six methods simultaneously and any of the six methods can be used with the same parameter set.
Ref. 32.
Ref. 33.
|
| IEFPCM-MSTa |
HF/6-31+G(d) |
−264.0 kcal mol−1 |
| DPCM-UAHFb |
HF/6-31(+)G(d)c |
−261.4 kcal mol−1 |
| PCM-UAKSd |
PBE1PBE/6-31G(d) |
Unknown |
| IEFPCM-SMDe,f |
M05-2X98/MIDI!6D |
−265.9 kcal mol−1 |
| M05-2X/6-31G* |
| M05-2X/6-31+G** |
| M05-2X/cc-pVTZ |
| B3LYP/6-31G* |
| HF/6-31G* |
| COSMO-RSg |
BP/TZVP |
Not specifically parameterized for ions |
| SM8h |
Independent of level of theory |
−265.9 kcal mol−1 |
Using Legendre transformed free energies, eqn (1) can be rewritten as
| | 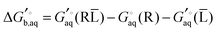 | (16) |
Since the electronic energy contribution to the standard free energy can be very large in magnitude this form is more easily evaluated
| | 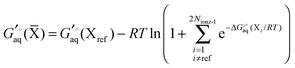 | (17) |
where
| | 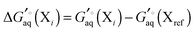 | (18) |
and where X
ref is some arbitrarily chosen reference protonation state, for example that for which
ni(H
+) = 0. The sum can be combined with that over different conformations [
eqn (7)] as discussed below.
Other ions and ionic strength
If the ligand and/or hosts contain ionizable groups then the binding measurements were likely performed in a buffer, with a certain ionic strength, to regulate pH. It is possible to include this effect in continuum solvation models such as the PCM method.23 However, given the relatively low (10–100 mM) concentrations usually used in the experiments this will only have a noticeable (>0.5 kcal mol−1) effect on the energetics involving multiply charged ions. As discussed below, the error in the computed solvation energy for such ions is already large and it is not clear whether it is worth including non-specific ionic strength effects in the computations. At high ion concentrations, it is possible that these ions bind at certain sites in the ligand, receptor, or ligand–receptor complex with sufficient probability that they must be included in the thermodynamics. If so the exact same equations and considerations outlined above for H+ also apply to, e.g. Cl− and pCl− (computed from the specified buffer concentration) is used instead of pH.
Solvation thermodynamics
Background
Most continuum models (CMs) of solvation compute the solvation free energy as the difference between the free energy in solution 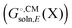 and the gas phase electronic energy (Egas(X))
and the gas phase electronic energy (Egas(X))| | 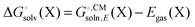 | (19) |
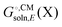 typically contains energy terms describing the electrostatic interaction of the molecule and the continuum as well as the van der Waals interactions with the solvent and free energy required to create the molecular cavity in the solvent (cavitation). The electrostatic interaction with the solvent alters the molecular wavefunction and is computed self-consistently. Usually the gas phase structure of X is used for the computation of
typically contains energy terms describing the electrostatic interaction of the molecule and the continuum as well as the van der Waals interactions with the solvent and free energy required to create the molecular cavity in the solvent (cavitation). The electrostatic interaction with the solvent alters the molecular wavefunction and is computed self-consistently. Usually the gas phase structure of X is used for the computation of 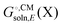 , though for COSMO-RS the structure is optimized in solution. There is typically no explicit RRHO contribution for
, though for COSMO-RS the structure is optimized in solution. There is typically no explicit RRHO contribution for 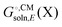 so the computational cost is comparable to that for Egas(X).
so the computational cost is comparable to that for Egas(X).
Some software packages automatically compute 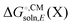 and Egas(X) in one run, while other packages only compute
and Egas(X) in one run, while other packages only compute 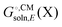 . Also, some programs just compute the electrostatic component of
. Also, some programs just compute the electrostatic component of 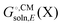 by default. However, the van der Waals and, especially, the cavitation component can make sizable contributions to the binding free energy and must be included for accurate results. It is worth noting that any hydrophobic contribution to binding will derive primarily from the change in cavitation energy.24
by default. However, the van der Waals and, especially, the cavitation component can make sizable contributions to the binding free energy and must be included for accurate results. It is worth noting that any hydrophobic contribution to binding will derive primarily from the change in cavitation energy.24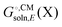 contains parameters (e.g. atomic radii) that are adjusted to reproduce experimentally measured solvation free energies
contains parameters (e.g. atomic radii) that are adjusted to reproduce experimentally measured solvation free energies
| | 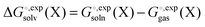 | (20) |
The standard state for both
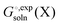
and
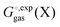
is generally chosen to be 1 M.
25,26 The latter is the reason a 1 M reference state also must be used when computing

.
Notice that the volume on going from the gas phase to solution is included in the solvation free energy
| | 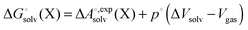 | (21) |
where Δ
Vsolv is the volume change in solution due to addition of the solute X to the neat solvent. For an ideal gas (
p°
Vgas =
RT) it follows that
| | 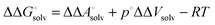 | (22) |
and
| | 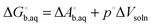 | (23) |
because the −
RT term is cancelled by a corresponding term in the translational enthalpy contribution to
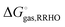
. Δ
Vsoln = ΔΔ
Vsolv is the change in the volume of the solution on upon binding.
Atomic radii
The solvation energy is computed using a set of atomic radii that define the solute–solvent boundary surface. These radii are usually obtained by fitting to experimentally measured solvation energies. Accurate solvation energies should not be expected from methods that use iso-electron density surfaces or van der Waals radii without additional empirical fitting. When using fitted radii one should use the same level of theory for the solute as was used in the parameterization (Table 1).
Ions
For neutral molecules solvation free energies can be measured with an accuracy of roughly 0.2 kcal mol−1 and reproduced theoretically to within roughly 0.5–1.0 kcal mol−1, depending on the method. However, the solvation energies of ions cannot be directly measured and must be indirectly inferred relative to a standard (usually the solvation energy of the proton). The experimentally obtained solvation energies are typically accurate to within 3 kcal mol−1 and can be reproduced computationally with roughly the same accuracy.22 The solvation energy of ions are therefore an especially likely source of error in binding free energies – especially if the ionic regions of the molecules become significantly desolvated due to binding.
Gas phase vs. solution optimization
The fitting of the radii described above is usually done using gas phase optimized structures only, i.e. any change in structure and corresponding rotational and vibrational effects are “included” in the radii via the parameterization. However, for ionic species gas phase optimization can lead to significantly distorted structures or even proton transfer and in these cases solution phase optimizations and, hence, vibrational frequency calculations, tend to be used. However, numerical noise in the continuum models can make it necessary to increase (i.e. make less stringent) the geometry convergence criteria and can lead to more imaginary frequencies than in the gas phase. One option is to compute the vibrational contribution to 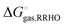 using gas phase optimized structures as Sure et al. have done.2
using gas phase optimized structures as Sure et al. have done.2
When using solution phase geometries the gas phase single point energies needed to evaluate 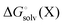 represent added computational expense one option is to use solution phase free energies to evaluate the binding free energies
represent added computational expense one option is to use solution phase free energies to evaluate the binding free energies
| | 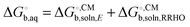 | (24) |
One problem with this approach is that
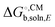
, unlike Δ
Egas, is not systematically improveable due to the empirical parameterization. For a more thorough discussion of this issue see Ho
et al.,
34 Ribeiro
et al.,
35 and Ho.
27
Cavities
The atomic radii and corresponding cavity generation algorithms are parameterized for small molecules. For more complex molecules such as receptors this can lead to continuum solvation of regions of molecules, e.g. deep in the binding pocket, that are not accessible to the molecular solvent. Furthermore, any solvent molecule inside such pocket is likely to be quite “un-bulk-like” and not well-represented by the bulk solvent or fixed by the underlying parametrization. However, how big an error this may introduce to the binding free energy is not really known, but certain models for the cavitation energy have been shown to give unrealistically large contributions to the binding free energy.36,37
Explicit water molecules
Adding explicit solvent molecules to the receptor and/or ligand can potentially lead to more accurate results. For example, including explicit water molecules around ionic sites reduces the strong dependence of the solvation energy on the corresponding atomic radii. Also, “un-bulk-like” water molecules now are treated more naturally and the risk of solvating non-solvent-accessible regions is reduced somewhat. However, adding explicit solvent molecules increases the computational cost by increasing the CPU time needed to compute energies, perform conformational searches, and compute vibrational frequencies.
There are several approaches to include the effect of explicit solvent molecules in the binding free energy. Bryantsev et al.38 suggest computing the solvation energy by
| | 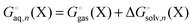 | (25) |
where
| |  | (26) |
(note that
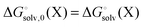
) and
| | 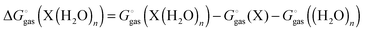 | (27) |
and
| | 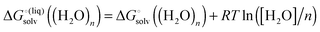 | (28) |
with “°(liq)” referring to a standard state of 55.34 M (the concentration of liquid water at 298 K), respectively. The term
RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
ln([H
2O]/
n) is the free energy required to change the standard state of (H
2O)
n from 1 M to 55.34/
n M.
Bryantsev et al. have shown that using this water cluster approach leads to a smooth convergence of the solvation free energy with respect to the cluster size n. The optimum choice of n is the one where an additional water molecule changes the solvation energy by less than a certain amount defined by the user. One can thereby compute the optimum number of water molecules for the receptor (n), ligand (m) and receptor–ligand complex (l) and then compute the change in solvation free energy as
| | 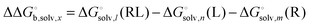 | (29) |
and computing Δ
Egas and
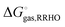
as before. One can show that this corresponds to the free energy change for this reaction
| | | R(H2O)m(aq) + L(H2O)n(aq) + (H2O)l(liq) ⇌ RL(H2O)l(aq) + (H2O)n(liq) + (H2O)m(liq) | (R4) |
In principle, the free energy is zero for
| | | (H2O)l(liq) ⇌ (H2O)n(liq) + (H2O)m(liq) + sgn(d)H2O|d|(liq) | (R5) |
where
d =
l −
m −
n and sgn(
d) returns the sign of
d. So the free energy change for
reaction (4) can also be computed as the free energy change for
| | | R(H2O)m(aq) + L(H2O)n(aq) ⇌ RL(H2O)l(aq) + sgn(d)H2O|d|(liq) | (R6) |
However, this is only approximately true in practice due to errors in the computed gas phase and solvation free energies. Furthermore,
reaction (6) does not really lead to any significant reduction in CPU time because the water cluster free energies only have to be computed once. However, if
reaction (6) is used then one must add an additional term correcting for the indistinguishability of water molecules
| | 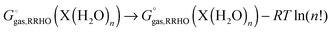 | (30) |
and similarly for the water clusters. Using
reaction (4) leads to a cancellation of this term and also maximizes error cancellation in the other energy terms. Similar considerations apply when using individual water molecules to the balance the reaction instead of water clusters
| | | R(H2O)m(aq) + L(H2O)n(aq) ⇌ RL(H2O)l(aq) + d(H2O)|d|(liq) | (R7) |
One of the main reasons
reaction (4) maximizes error cancellation is that the number and type of hydrogen bonds involving water molecules are very similar on each side of the equilibrium. This can also be achieved when using
reaction (6) or (7) by ensuring that
l =
m +
n, in which case the error cancellation may be comparable and will depend on the nature of the ligand, host, and water arrangement. However,
eqn (30) must still be used when using
reaction (6) or (7) in this way.
When using many explicit water molecules the error in the continuum solvation energies can be reduced by ensuring that the continuum solvation energy of a single water molecule matches the experimental value of −6.32 kcal mol−1 at 298.15 K as close as possible.
Enthalpy and entropy contributions to the binding free energy
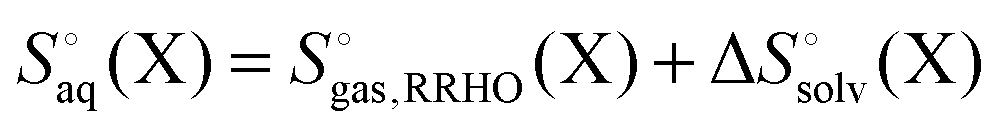
It is often instructive to decompose the binding free energy into enthalpy and entropy contributions. The standard enthalpy and entropy of molecule X in aqueous solution is| | 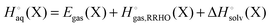 | (31) |
and| | 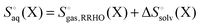 | (32) |
where the standard state eqn (3) and symmetry correction [eqn (6)] is applied to the entropy term. Thus, in order to compute these quantities one must compute the enthalpy and entropy of solvation, which can be done by the COSMO-RS32 and SM8T39 solvation methods. Chamberlin et al.39 have noted that most of the temperature dependence of the aqueous solvation free energy comes from the non-polar term so simply including the effect of temperature on the dielectric constant is unlikely to give accurate results. Plata and Singleton40 have recently shown that 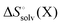 can make an appreciable contribution to the energy change for reaction energies.
can make an appreciable contribution to the energy change for reaction energies.
For a molecule (X) with Nconf conformations the standard enthalpy and entropy is
| | 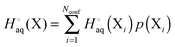 | (33) |
and
| | 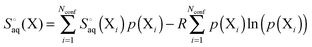 | (34) |
where
| |  | (35) |
and
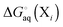
is computed relative to the conformation with the lowest free energy.
The Legendre transformed entropy and enthalpy is
| | 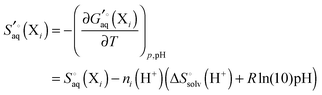 | (36) |
and
| | 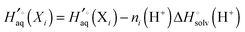 | (37) |
When comparing computed enthalpy and entropy changes to experimental measurements on systems with ionizable groups note that the observed values will depend on the buffer used
if protonation states change upon binding (see
e.g.ref. 41). Unless the experimental study has corrected for this effect by repeating the measurements in different buffers, this effect can contribute to the difference between the computed and experimental values.
A concrete example
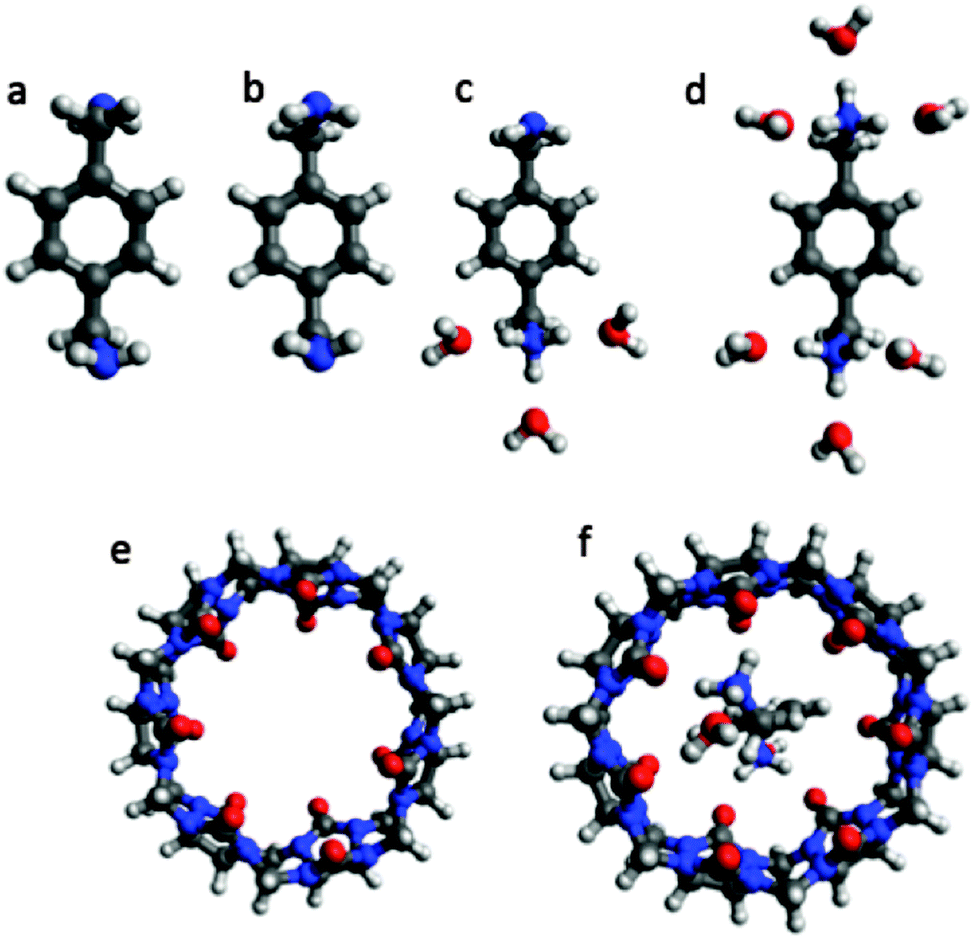
In this section I apply the key equations discussed above to a specific example: p-xylylenediamine (L, Fig. 2) binding to CB7 (R) for which a binding free energy of −9.9 ± 0.1 kcal mol−1 has been measured at pH 7.4 and 298 K.1 The conformations and other details such as the number of water molecules are just selected and constructed for illustration purposes only using the Avogadro program42 and the MMFF force field and should not be considered accurate.
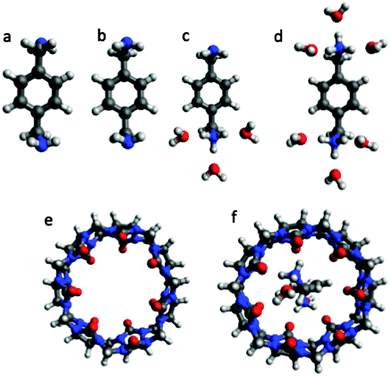 |
| | Fig. 2 Representative conformations of ligand L (p-xylylenediamine), receptor R (CB7), and a receptor–ligand complex RL used to illustrate the use of the equations presented in this paper. (a) La, (b) Lb, (c) LH+b, (d) LH22+b, (e) R, and (f) RLH22+a. The coordinates for the structures are available here: http://dx.doi.org/10.6084/m9.figshare.1290639. | |
CB7 has one conformation with D7h symmetry and no ionizable groups. It is assumed that the solvation energy can be computed accurately without explicit water molecules. Thus, the free energy is aqueous solution is
| | 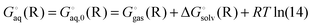 | (38) |
where
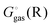
is computed in
C1 symmetry and 14 is the symmetry number (
σ) corresponding to the
D7h point group.
Ligand L has two basic groups and is assumed to have two conformations a and b for each protonation state. The pKa values for the basic groups are 9.2 and 9.8 according to Marvin, so both groups are likely 100% protonated at pH 7. However, for illustration purposes I will include all three protonation states in the computation of the free energy. Furthermore, I will assume that each charged amine group is microsolvated by three explicit water molecules.
The free energy of conformer a of the doubly protonated state (LH22+) is thus
| | 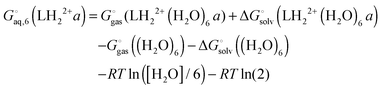 | (39) |
where the gas phase energy is computed in
C1 symmetry and 2 is the symmetry number of the
C2 point group. The lowest energy structure of (H
2O)
6 suggested by Bransyev
et al. can be used for compute
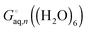
, or the effect of additional conformations can be included using
eqn (7). Finally, the Legendre transformed free energy [
eqn (15)] at pH 7 is computed by
| | 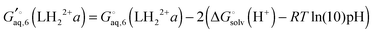 | (40) |
The corresponding free energy of conformer
b,
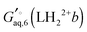
, which has
C2v symmetry and for which
σ is also 2, is computed in the same way. Notice that each conformation in principle can have different numbers of water associated with them. Similarly, the free energies of the singly protonated and neutral ligand (with
C1 and
C2 symmetry) is computed by
| | 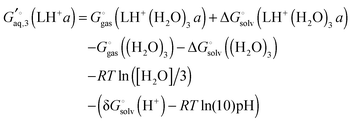 | (41) |
and
| | 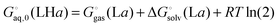 | (42) |
(here for conformer
a and similarly for conformer
b). Finally, the free energy of L averaged over conformations and protonation states is
| | 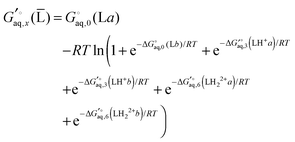 | (43) |
where
| | 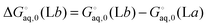 | (44) |
and similarly for the remaining terms in the sum. Notice that for each conformation there are three protonation states rather than (2
2) because the two singly protonated structures are equivalent.
For the host–guest complex I have assumed that each conformation can bind CB7 in only one way and that two explicit water molecules per protonated group is lost upon binding, so that
| | 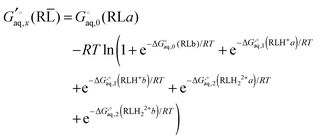 | (45) |
Note that the effect of the 28 equivalent binding modes to other oxygen atoms for
e.g. LH
22+a (
Fig. 2f) is accounted for by the symmetry factors. Finally, the binding free energy is computed using
eqn (16).
Protein–ligand binding
In order for the electronic structure approach to be used in drug design corresponding calculation have to be carried out on proteins, which are significantly larger than the hosts that have been used to benchmark the approach so far. QM/MM is of course the obvious choice for computing the geometries and gas phase energies, although linear scaling all QM methods such as the FMO43 method is also possible. Furthermore, continuum methods such as PCM have been adapted for large systems and interfaced to both QM/MM44 and the FMO method.45 Of course as the system size increases conformational sampling will become a bigger practical issue.
The main issue is the computation of vibrational frequencies for the protein and protein–ligand complex. The fast semi-empirical methods currently used for computing the vibrational frequencies (dispersion and hydrogen bond-corrected PM6 and DFTB as well as HF-3c) must be interfaced with QM/MM codes and/or be implemented in a linear scaling approach that allow for frequency calculations. Dispersion-corrected PM6 and DFTB are already implemented in AMBER, a FMO implementation of DFTB has recently been added to GAMESS46 and a similar HF-3c/FMO implementation is forthcoming from my lab.
Most QM/MM studies of enzyme catalysis constrain the geometry of a significant portion of the system to avoid spurious structural fluctuation far away from the active site contributing to the barrier. This may well be necessary for binding free energy calculations as well, in which case the effect of the constraints on the vibrational frequencies must be accounted for.47 Alternatively, only the Hessian of the un-constrained region can be computed.48
So while there is some code-adjustment to be done it may well be that the promising developments in electronic structure-based prediction of aqueous binding free energies may also be brought to bear on drug design within the next few years.
Summary and outlook
Recent predictions of absolute binding free energies of host–guest complexes in aqueous solution using electronic structure theory have been encouraging for some systems. It is interesting to consider the underlying innovations that have lead to the recent increase in accuracy in predicted binding free energies. Advances in computer hardware and coupled cluster algorithms made it possible to construct benchmark sets of accurate electronic binding energies for a diverse set of molecules. These benchmarks sets were then used to develop the dispersion corrections needed for accurate DFT-based electronic binding energies and the short-range (hydrogen bond) corrections to the semi-empirical methods needed to compute accurate vibrational frequencies for the RRHO free energy corrections. In fact methods like HF-3c,49 while containing empirical corrections, was developed without reference to any experimental data. Another interesting observation is that the dispersion and RRHO free energy contributions to the binding free energy have roughly the same magnitude, but opposite signs. So including just one of the corrections is likely to significantly increase the error relative to experiment and lead to the wrong conclusions regarding their importance.
While there have been reasonably accurate predictions for some host–guest systems, other systems remain problematic. In this paper I summarize some of the many factors that could easily contribute 1–3 kcal mol−1 at 298 K: three-body dispersion effects, molecular symmetry, anharmonicity, spurious imaginary frequencies, insufficient conformational sampling, wrong or changing ionization states, errors in the solvation free energy of ions, and explicit solvent (and ion) effects that are not well-represented by continuum models.
While I focus on binding free energies in aqueous solution it is worth noting that the approach also applies to any free energy difference in solution, such as conformational and reaction free energy differences or activation free energies. Furthermore, the equations apply to solvents other than water as long as the concentration of liquid water, the solvation free energy of the proton changed, and the parameterization of the continuum solvation model is changed to match the solvent of interest. Furthermore, while the recent successes with electronic structure-based approaches have been for host–guest complexes they can be extended to protein–ligand complexes with a few methodological improvements (mainly related to the computation of vibrational frequencies). Thus, it may well be that the promising developments in electronic structure-based prediction of aqueous binding free energies may also be brought to bear on drug design within the next few years.
Acknowledgements
I thank the following people for very illuminating discussions and/or comments on the manuscript: Vyacheslav Bryantsev, Chris Cramer, Mike Gilson, Stefan Grimme, Fahmi Himo, Junming Ho, Adrian Jinich, Andreas Klamt, Pedro Silva, Dan Singleton and Casper Steinmann.
References
- H. S. Muddana, A. T. Fenley, D. L. Mobley and M. K. Gilson, The SAMPL4 host–guest blind prediction challenge: an overview, J. Comput.-Aided Mol. Des., 2014, 1–13 CAS.
- R. Sure, J. Antony and S. Grimme, Blind Prediction of Binding Affinities for Charged Supramolecular Host–Guest Systems: Achievements and Shortcomings of DFT-D3, J. Phys. Chem. B, 2014, 118(12), 3431–3440 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- S. Grimme, Supramolecular binding thermodynamics by dispersion-corrected density functional theory, Chem. – Eur. J., 2012, 18(32), 9955–9964 CrossRef CAS PubMed.
- H. S. Muddana and M. K. Gilson, Calculation of host–guest binding affinities using a quantum-mechanical energy model, J. Chem. Theory Comput., 2012, 8(6), 2023–2033 CrossRef CAS PubMed.
- N. D. Yilmazer and M. Korth, Comparison of molecular mechanics, semi-empirical quantum mechanical, and density functional theory methods for scoring protein–ligand interactions, J. Phys. Chem. B, 2013, 117(27), 8075–8084 CrossRef CAS PubMed.
- H. X. Zhou and M. K. Gilson, Theory of free energy and entropy in noncovalent binding, Chem. Rev., 2009, 109(9), 4092–4107 CrossRef CAS PubMed.
- J. J. P. Stewart, Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements, J. Mol. Model., 2007, 13(12), 1173–1213 CrossRef CAS PubMed.
- N. Bork, L. Du and H. G. Kjaergaard, Identification and Characterization of the HCl–DMS Gas Phase Molecular Complex via Infrared Spectroscopy and Electronic Structure Calculations, J. Phys. Chem. A, 2014a, 118(8), 1384–1389 Search PubMed.
- N. Bork, L. Du, H. Reiman, T. Kurtén and H. G. Kjaergaard, Benchmarking Ab Initio Binding Energies of Hydrogen-Bonded Molecular Clusters Based on FTIR Spectroscopy, J. Phys. Chem. A, 2014b, 118(28), 5316–5322 Search PubMed.
- B. Temelso, K. A. Archer and G. C. Shields, Benchmark structures and binding energies of small water clusters with anharmonicity corrections, J. Phys. Chem. A, 2011, 115(43), 12034–12046 CrossRef CAS PubMed.
- B. Temelso and G. C. Shields, The role of anharmonicity in hydrogen-bonded systems: the case of water clusters, J. Chem. Theory Comput., 2011, 7(9), 2804–2817 CrossRef CAS.
- G. Piccini and J. Sauer, Effect of Anharmonicity on Adsorption Thermodynamics, J. Chem. Theory Comput., 2014, 10(6), 2479–2487 CrossRef CAS.
- Marvin, http://www.chemaxon.com/marvin/sketch/index.php, 2014.
- A. L. Koner, I. Ghosh, N. I. Saleh and W. M. Nau, Supramolecular encapsulation of benzimidazole-derived drugs by cucurbit[7] uril, Can. J. Chem., 2011, 89(2), 139–147 CrossRef CAS PubMed.
- M. O. Kim, P. G. Blachly, J. W. Kaus and J. A. McCammon, Protocols Utilizing Constant pH Molecular Dynamics to Compute pH-Dependent Binding Free Energies, J. Phys. Chem. B, 2015, 119, 861–872 CrossRef CAS PubMed.
- H. Li, A. D. Robertson and J. H. Jensen, The determinants of carboxyl pKa values in turkey ovomucoid third domain, Proteins: Struct., Funct., Bioinf., 2004, 55(3), 689–704 CrossRef CAS PubMed.
- J. Ho and M. L. Coote, A universal approach for continuum solvent pKa calculations: are we there yet?, Theor. Chem. Acc., 2010, 125(1–2), 3–21 CrossRef CAS PubMed.
-
R. A. Alberty, Thermodynamics of biochemical reactions, John Wiley & Sons., 2005 Search PubMed.
- R. A. Alberty, A. Cornish-Bowden, R. N. Goldberg, G. G. Hammes, K. Tipton and H. V. Westerhoff, Recommendations for terminology and databases for biochemical thermodynamics, Biophys. Chem., 2011, 155(2), 89–103 CrossRef CAS PubMed.
- A. Jinich, D. Rappoport, I. Dunn, B. Sanchez-Lengeling, R. Olivares-Amaya, E. Noor and A. Aspuru-Guzik, Quantum Chemical Approach to Estimating the Thermodynamics of Metabolic Reactions, Sci. Rep., 2014, 4, 7022 CrossRef CAS PubMed.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, Aqueous solvation free energies of ions and ion-water clusters based on an accurate value for the absolute aqueous solvation free energy of the proton, J. Phys. Chem. B, 2006, 110(32), 16066–16081 CrossRef CAS PubMed.
- M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Ab initio study of ionic solutions by a polarizable continuum dielectric model, Chem. Phys. Lett., 1998, 286(3), 253–260 CrossRef CAS.
- C. J. Cramer and D. G. Truhlar, General parameterized SCF model for free energies of solvation in aqueous solution, J. Am. Chem. Soc., 1991, 113(22), 8305–8311 CrossRef CAS.
- A. Ben-Naim, Standard thermodynamics of transfer, Uses and misuses, J. Phys. Chem., 1978, 82(7), 792–803 CrossRef CAS.
- A. Ben-Naim and Y. Marcus, Solvation thermodynamics of nonionic solutes, J. Chem. Phys., 1984, 81(4), 2016–2027 CrossRef CAS PubMed.
- J. Ho, Are thermodynamic cycles necessary for continuum solvent calculation of pKas and reduction potentials?, Phys. Chem. Chem. Phys., 2015, 17(4), 2859–2868 RSC.
- C. Curutchet, A. Bidon-Chanal, I. Soteras, M. Orozco and F. J. Luque, MST continuum study of the hydration free energies of monovalent ionic species, J. Phys. Chem. B, 2005, 109(8), 3565–3574 CrossRef CAS PubMed.
- V. Barone, M. Cossi and J. Tomasi, A new definition of cavities for the computation of solvation free energies by the polarizable continuum model, J. Chem. Phys., 1997, 107(8), 3210–3221 CrossRef CAS PubMed.
- Y. Takano and K. N. Houk, Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules, J. Chem. Theory Comput., 2005, 1(1), 70–77 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- F. Eckert and A. Klamt, Fast solvent screening via quantum chemistry: COSMO-RS approach, AIChE J., 2002, 48(2), 369–385 CrossRef CAS PubMed.
- A. V. Marenich, R. M. Olson, C. P. Kelly, C. J. Cramer and D. G. Truhlar, Self-consistent reaction field model for aqueous and nonaqueous solutions based on accurate polarized partial charges, J. Chem. Theory Comput., 2007, 3(6), 2011–2033 CrossRef CAS.
- J. Ho, A. Klamt and M. L. Coote, Comment on the correct use of continuum solvent models, J. Phys. Chem. A, 2010, 114(51), 13442–13444 CrossRef CAS PubMed.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, Use of solution-phase vibrational frequencies in continuum models for the free energy of solvation, J. Phys. Chem. B, 2011, 115(49), 14556–14562 CrossRef CAS PubMed.
- S. Genheden, J. Kongsted, P. Söderhjelm and U. Ryde, Nonpolar Solvation Free Energies of Protein– Ligand Complexes, J. Chem. Theory Comput., 2010, 6(11), 3558–3568 CrossRef CAS.
- S. Genheden and U. Ryde, Improving the Efficiency of Protein–Ligand Binding Free-Energy Calculations by System Truncation, J. Chem. Theory Comput., 2012, 8(4), 1449–1458 CrossRef CAS.
- V. S. Bryantsev, M. S. Diallo and W. A. Goddard III, Calculation of solvation free energies of charged solutes using mixed cluster/continuum models, J. Phys. Chem. B, 2008, 112(32), 9709–9719 CrossRef CAS PubMed.
- A. C. Chamberlin, C. J. Cramer and D. G. Truhlar, Extension of a temperature-dependent aqueous solvation model to compounds containing nitrogen, fluorine, chlorine, bromine, and sulfur, J. Phys. Chem. B, 2008, 112(10), 3024–3039 CrossRef CAS PubMed.
- R. E. Plata and D. A. Singleton, A Case Study of the Mechanism of Alcohol-Mediated Morita Baylis-Hillman Reactions. The Importance of Experimental Observations, J. Am. Chem. Soc., 2015, 137, 3811–3826 CrossRef CAS PubMed.
- F. Dullweber, M. T. Stubbs, Đ. Musil, J. Stürzebecher and G. Klebe, Factorising ligand affinity: a combined thermodynamic and crystallographic study of trypsin and thrombin inhibition†, J. Mol. Biol., 2001, 313(3), 593–614 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: An advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 17 CAS.
- D. G. Fedorov, T. Nagata and K. Kitaura, Exploring chemistry with the fragment molecular orbital method, Phys. Chem. Chem. Phys., 2012, 14(21), 7562–7577 RSC.
- H. Li, C. S. Pomelli and J. H. Jensen, Continuum solvation of large molecules described by QM/MM: a semi-iterative implementation of the PCM/EFP interface, Theor. Chem. Acc., 2003, 109(2), 71–84 CrossRef CAS.
- D. G. Fedorov, K. Kitaura, H. Li, J. H. Jensen and M. S. Gordon, The polarizable continuum model (PCM) interfaced with the fragment molecular orbital method (FMO), J. Comput. Chem., 2006, 27(8), 976–985 CrossRef CAS PubMed.
- Y. Nishimoto, D. G. Fedorov and S. Irle, Density-Functional Tight-Binding Combined with the Fragment Molecular Orbital Method, J. Chem. Theory Comput., 2014, 10(11), 4801–4812 CrossRef CAS.
- A. Ghysels, D. Van Neck, V. Van Speybroeck, T. Verstraelen and M. Waroquier, Vibrational modes in partially optimized molecular systems, J. Chem. Phys., 2007, 126(22), 224102 CrossRef CAS PubMed.
- H. Li and J. H. Jensen, Partial Hessian vibrational analysis: the localization of the molecular vibrational energy and entropy, Theor. Chem. Acc., 2002, 107(4), 211–219 CrossRef CAS PubMed.
- R. Sure and S. Grimme, Corrected small basis set Hartree–Fock method for large systems, J. Comput. Chem., 2013, 34(19), 1672–1685 CrossRef CAS PubMed.
|
| This journal is © the Owner Societies 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 
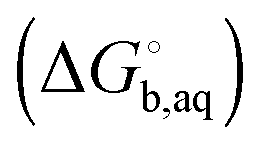 of a receptor (R or host) and ligand (L or guest) molecule in aqueous (aq) solution
of a receptor (R or host) and ligand (L or guest) molecule in aqueous (aq) solution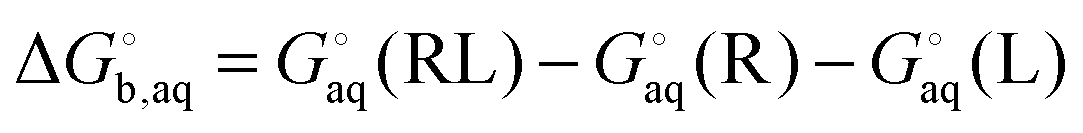
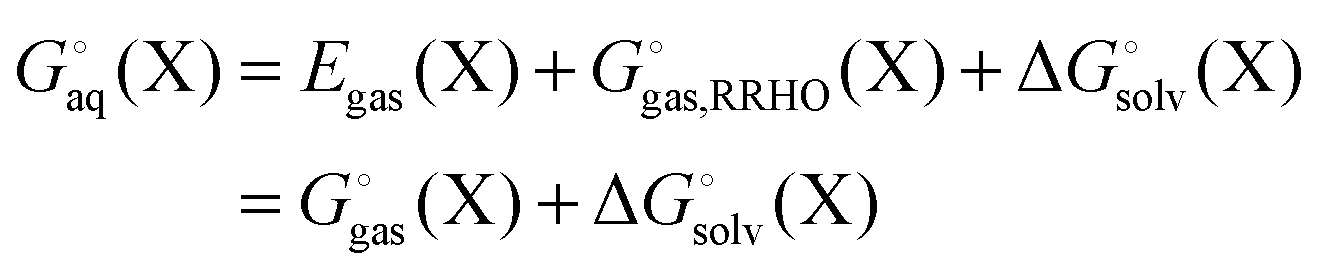
 , and
, and 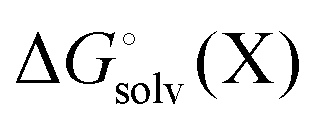 is the electronic energy, rigid rotor-harmonic oscillator (RRHO), and solvation free energy, respectively, of molecule X. Note that
is the electronic energy, rigid rotor-harmonic oscillator (RRHO), and solvation free energy, respectively, of molecule X. Note that  contains the zero point energy. The standard state (denoted by “°”) throughout this paper is 1 M, unless otherwise noted. The solvation free energy is typically computed using a continuum solvation model as described in detail below.
contains the zero point energy. The standard state (denoted by “°”) throughout this paper is 1 M, unless otherwise noted. The solvation free energy is typically computed using a continuum solvation model as described in detail below.
 with PM6-D3H4 and HF-3c2 with good results.
with PM6-D3H4 and HF-3c2 with good results.
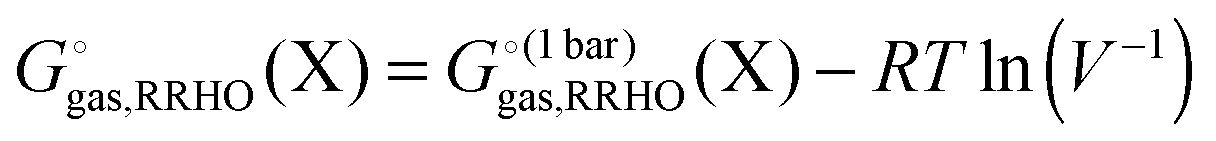
 instead of the Gibbs free energy. However, as I discuss below, the solvation free energy corrects for the change in volume on going from the gas phase to solution, so the Gibbs free energy change should be used throughout.
instead of the Gibbs free energy. However, as I discuss below, the solvation free energy corrects for the change in volume on going from the gas phase to solution, so the Gibbs free energy change should be used throughout.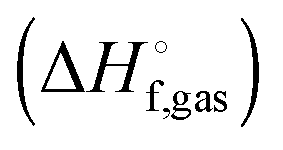 . However, in the case of intermolecular interactions such as hydrogen binding the parameterization was done by fitting
. However, in the case of intermolecular interactions such as hydrogen binding the parameterization was done by fitting 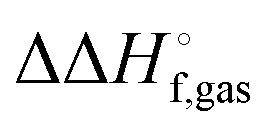 to ΔEgas values computed using electronic structure theory (Stewart 2007).8 The same is true for dispersion and hydrogen bond corrected PM6 methods. Thus, if a PM6 based method is used to compute the interaction energy the RRHO enthalpy corrections should still be included, i.e.
to ΔEgas values computed using electronic structure theory (Stewart 2007).8 The same is true for dispersion and hydrogen bond corrected PM6 methods. Thus, if a PM6 based method is used to compute the interaction energy the RRHO enthalpy corrections should still be included, i.e.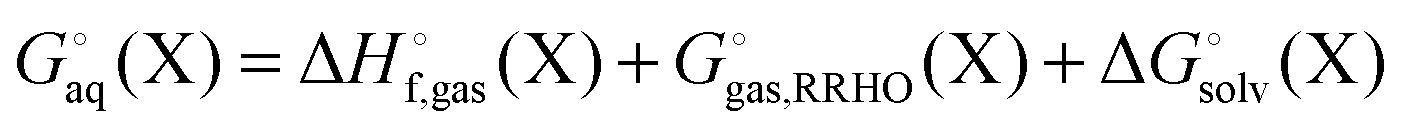
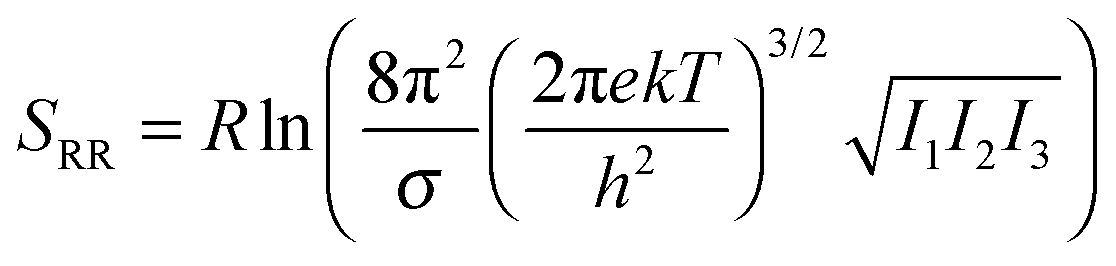
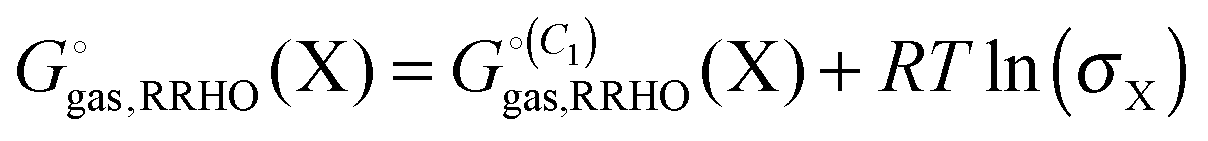
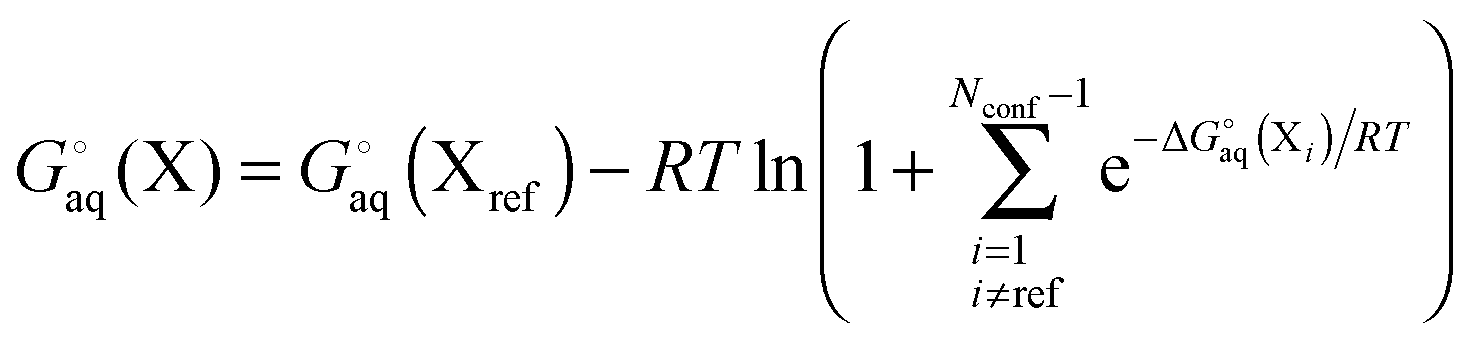
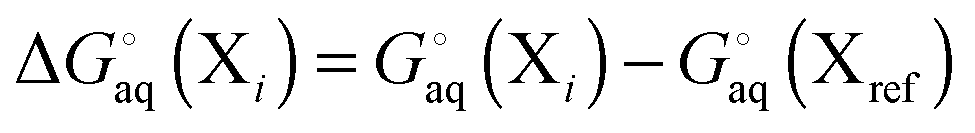
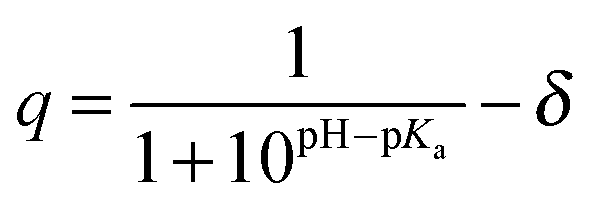
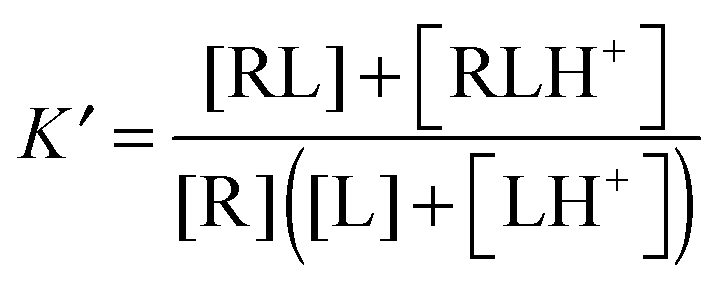
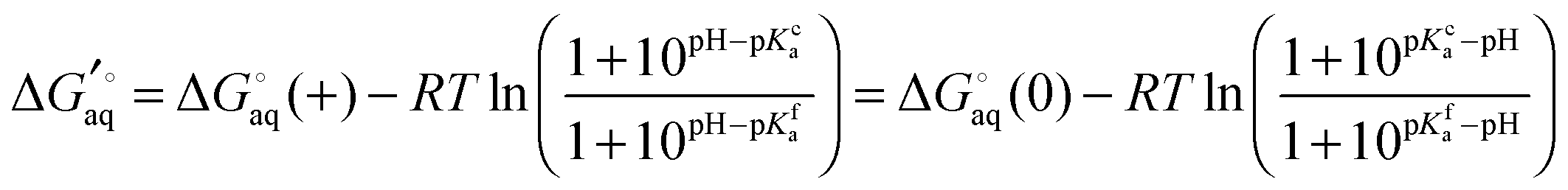
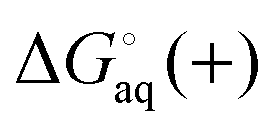 and
and 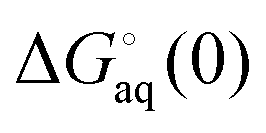 is the binding free energy computed using the charged (protonated) and neutral form of the ligand and pKca and pKfa are the pKa values the ligand bound to the receptor and the free ligand, respectively.
is the binding free energy computed using the charged (protonated) and neutral form of the ligand and pKca and pKfa are the pKa values the ligand bound to the receptor and the free ligand, respectively.
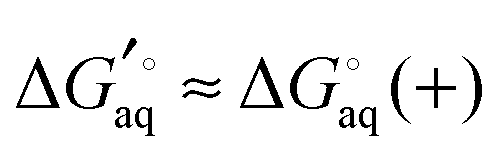 is a good approximation. However, if pKfa is close to 7 then pKca should be computed. Also, it is possible for charged ligands to change to their neutral state if they bind to hydrophobic or similarly charged receptors.
is a good approximation. However, if pKfa is close to 7 then pKca should be computed. Also, it is possible for charged ligands to change to their neutral state if they bind to hydrophobic or similarly charged receptors.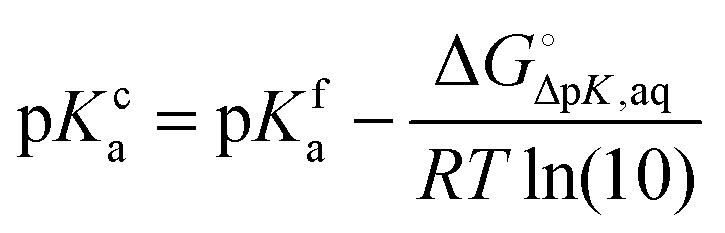
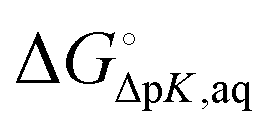 is the free energy change for this reaction17
is the free energy change for this reaction17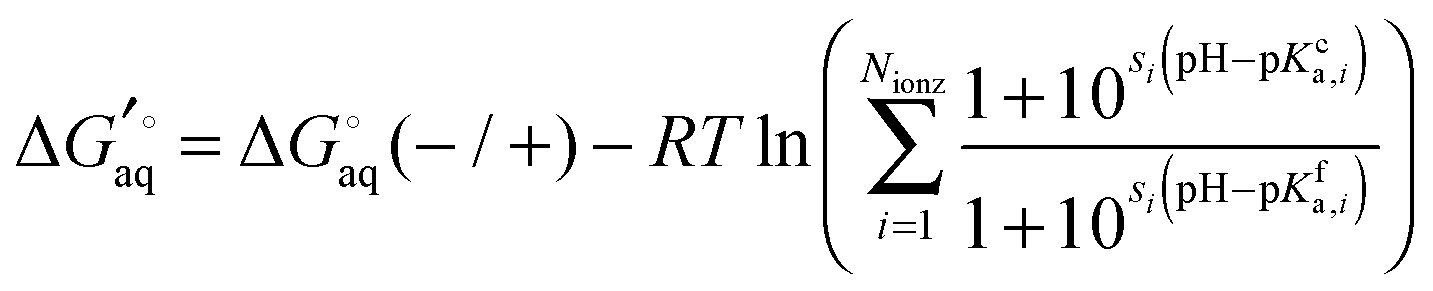
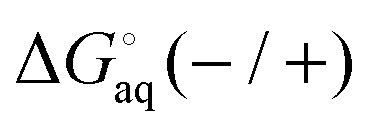 is the binding free energy when all acids and bases are deprotonated and protonated, respectively, the sum runs over all ionizable groups and si is 1 and −1 if i is a base or acid, respectively.
is the binding free energy when all acids and bases are deprotonated and protonated, respectively, the sum runs over all ionizable groups and si is 1 and −1 if i is a base or acid, respectively.
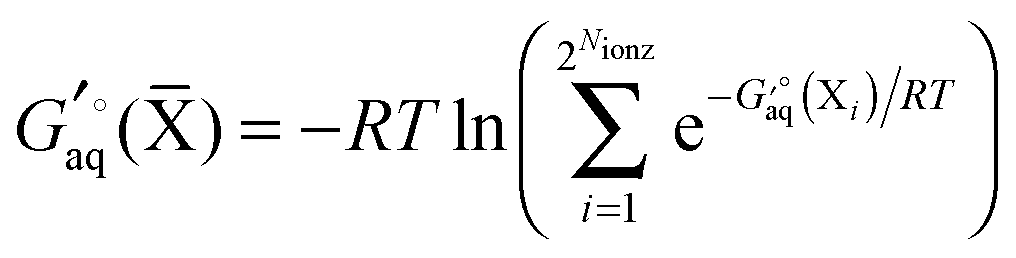
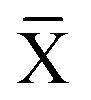 denotes an average over several protonation states of X, 2Nionz is the number of possible protonation states given Nionz sites and
denotes an average over several protonation states of X, 2Nionz is the number of possible protonation states given Nionz sites and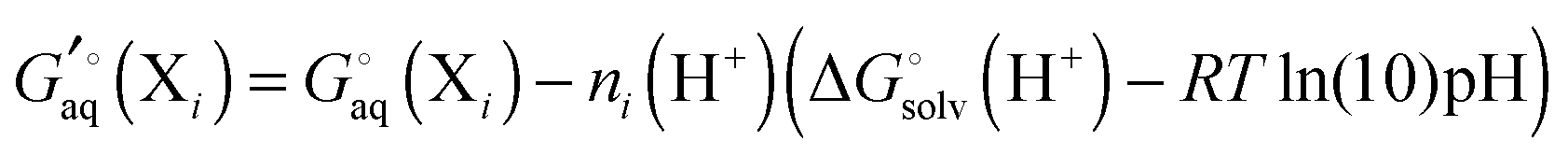
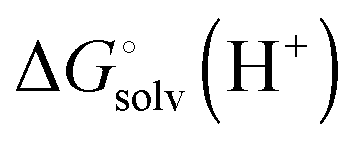 is the solvation free energy of the proton. So in the case of ligand L considered above, ni(H+) is 0 and 1 for L and LH+, respectively.
is the solvation free energy of the proton. So in the case of ligand L considered above, ni(H+) is 0 and 1 for L and LH+, respectively.
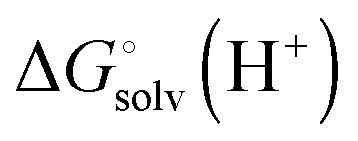 is usually taken from the literature where estimates vary between −264 and −266 kcal mol−1,22 which can add to the uncertainty in the predicted binding free energy change. There are at least two ways of reducing the error. One way is to maximize error cancelation by computing
is usually taken from the literature where estimates vary between −264 and −266 kcal mol−1,22 which can add to the uncertainty in the predicted binding free energy change. There are at least two ways of reducing the error. One way is to maximize error cancelation by computing 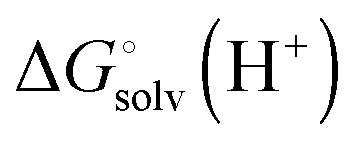 (using explicit solvent molecules as discussed below) using the same level of theory method use to compute
(using explicit solvent molecules as discussed below) using the same level of theory method use to compute 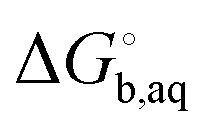 . The other way is to choose the value of
. The other way is to choose the value of 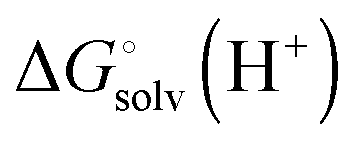 used as reference for the experimental solvation free energies of ions that are used to parameterize the continuum solvation model you use (Table 1). The first way is best if explicit solvent molecules are used to compute the solvation free energies of ions in the binding study and otherwise the second method is best.
used as reference for the experimental solvation free energies of ions that are used to parameterize the continuum solvation model you use (Table 1). The first way is best if explicit solvent molecules are used to compute the solvation free energies of ions in the binding study and otherwise the second method is best.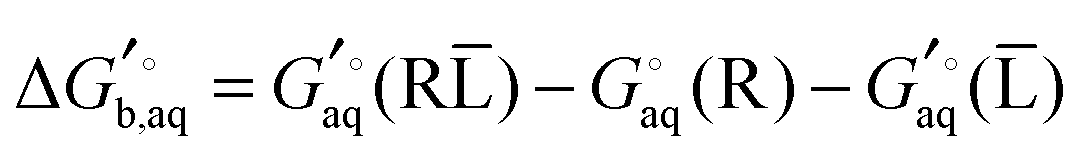
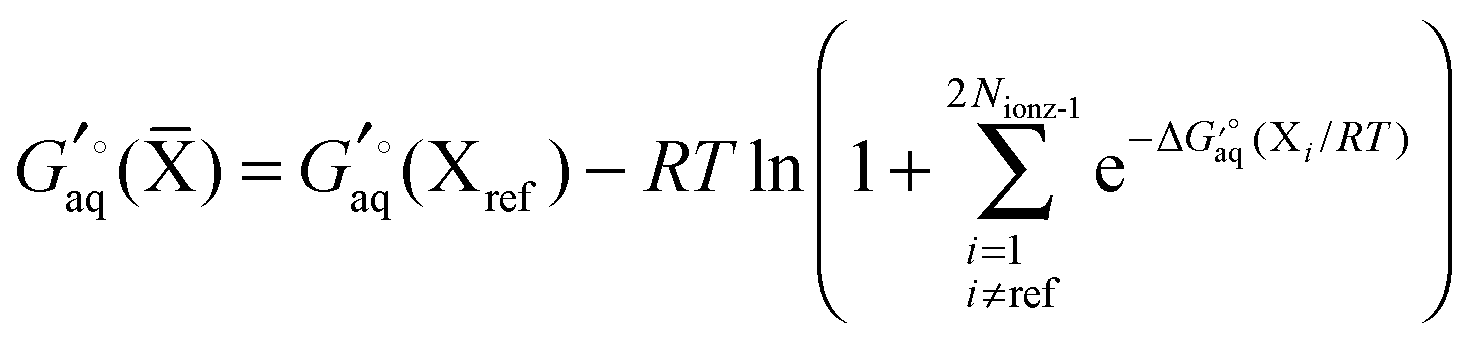
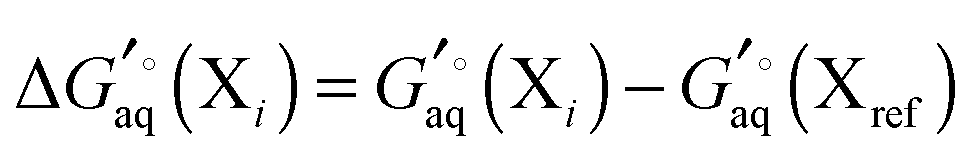
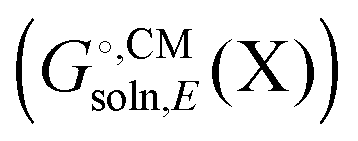 and the gas phase electronic energy (Egas(X))
and the gas phase electronic energy (Egas(X))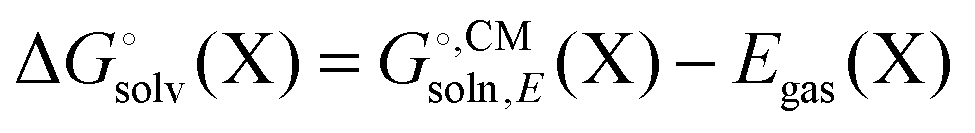
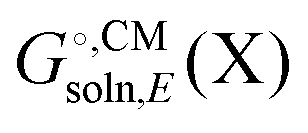 typically contains energy terms describing the electrostatic interaction of the molecule and the continuum as well as the van der Waals interactions with the solvent and free energy required to create the molecular cavity in the solvent (cavitation). The electrostatic interaction with the solvent alters the molecular wavefunction and is computed self-consistently. Usually the gas phase structure of X is used for the computation of
typically contains energy terms describing the electrostatic interaction of the molecule and the continuum as well as the van der Waals interactions with the solvent and free energy required to create the molecular cavity in the solvent (cavitation). The electrostatic interaction with the solvent alters the molecular wavefunction and is computed self-consistently. Usually the gas phase structure of X is used for the computation of 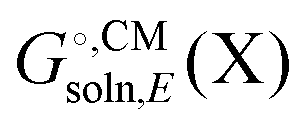 , though for COSMO-RS the structure is optimized in solution. There is typically no explicit RRHO contribution for
, though for COSMO-RS the structure is optimized in solution. There is typically no explicit RRHO contribution for 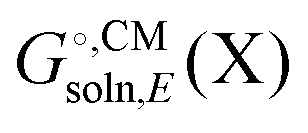 so the computational cost is comparable to that for Egas(X).
so the computational cost is comparable to that for Egas(X).
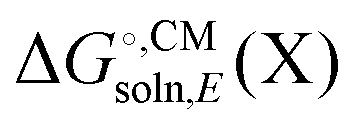 and Egas(X) in one run, while other packages only compute
and Egas(X) in one run, while other packages only compute 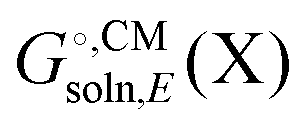 . Also, some programs just compute the electrostatic component of
. Also, some programs just compute the electrostatic component of 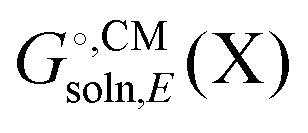 by default. However, the van der Waals and, especially, the cavitation component can make sizable contributions to the binding free energy and must be included for accurate results. It is worth noting that any hydrophobic contribution to binding will derive primarily from the change in cavitation energy.24
by default. However, the van der Waals and, especially, the cavitation component can make sizable contributions to the binding free energy and must be included for accurate results. It is worth noting that any hydrophobic contribution to binding will derive primarily from the change in cavitation energy.24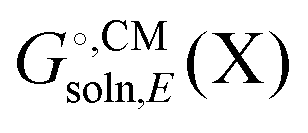 contains parameters (e.g. atomic radii) that are adjusted to reproduce experimentally measured solvation free energies
contains parameters (e.g. atomic radii) that are adjusted to reproduce experimentally measured solvation free energies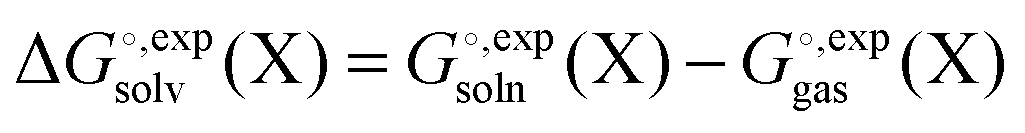
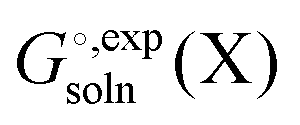 and
and 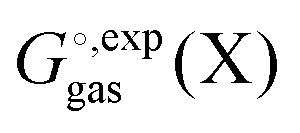 is generally chosen to be 1 M.25,26 The latter is the reason a 1 M reference state also must be used when computing
is generally chosen to be 1 M.25,26 The latter is the reason a 1 M reference state also must be used when computing  .
.
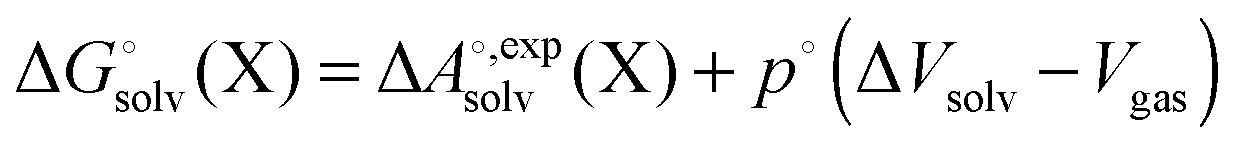
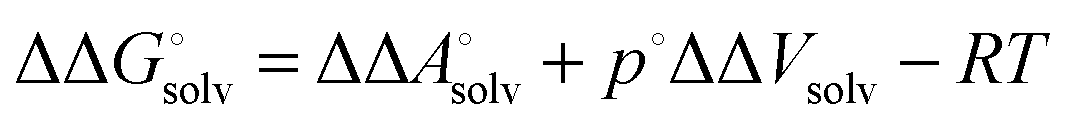
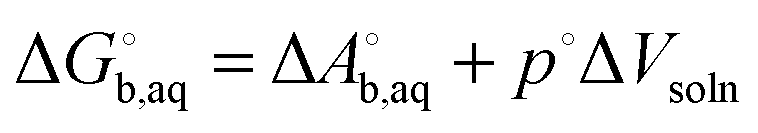
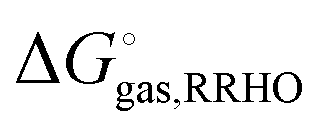 . ΔVsoln = ΔΔVsolv is the change in the volume of the solution on upon binding.
. ΔVsoln = ΔΔVsolv is the change in the volume of the solution on upon binding.
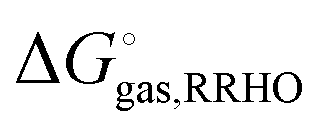 using gas phase optimized structures as Sure et al. have done.2
using gas phase optimized structures as Sure et al. have done.2
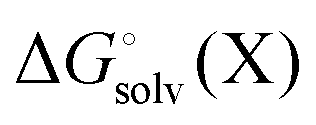 represent added computational expense one option is to use solution phase free energies to evaluate the binding free energies
represent added computational expense one option is to use solution phase free energies to evaluate the binding free energies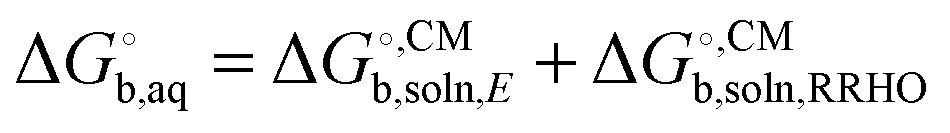
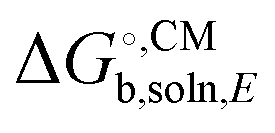 , unlike ΔEgas, is not systematically improveable due to the empirical parameterization. For a more thorough discussion of this issue see Ho et al.,34 Ribeiro et al.,35 and Ho.27
, unlike ΔEgas, is not systematically improveable due to the empirical parameterization. For a more thorough discussion of this issue see Ho et al.,34 Ribeiro et al.,35 and Ho.27
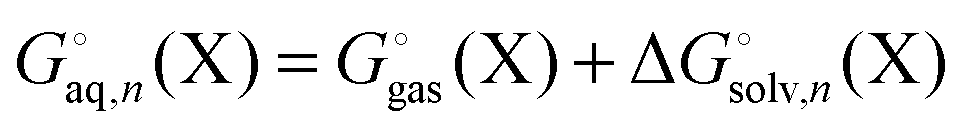
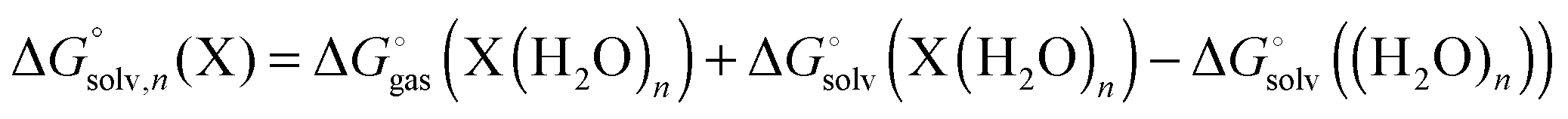
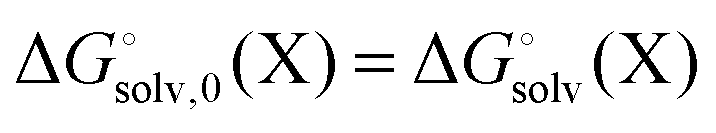 ) and
) and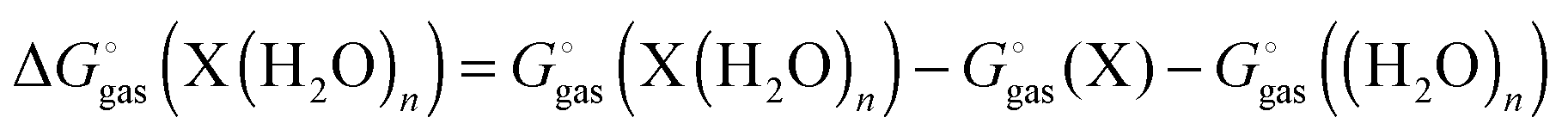
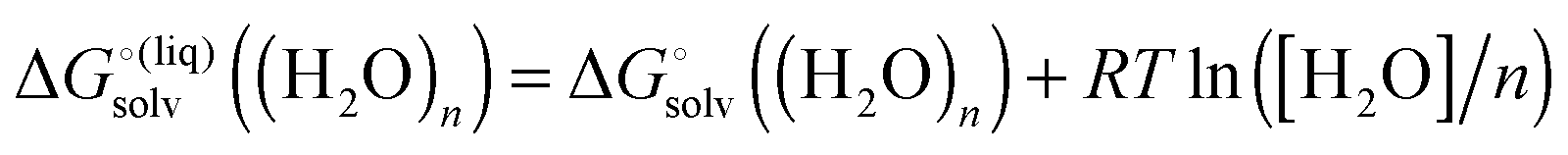
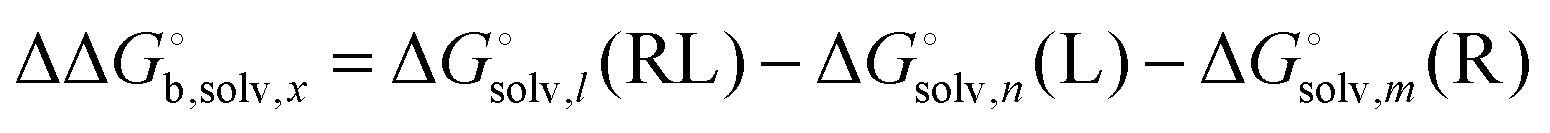
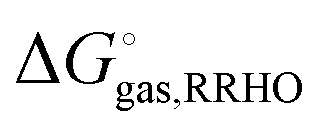 as before. One can show that this corresponds to the free energy change for this reaction
as before. One can show that this corresponds to the free energy change for this reaction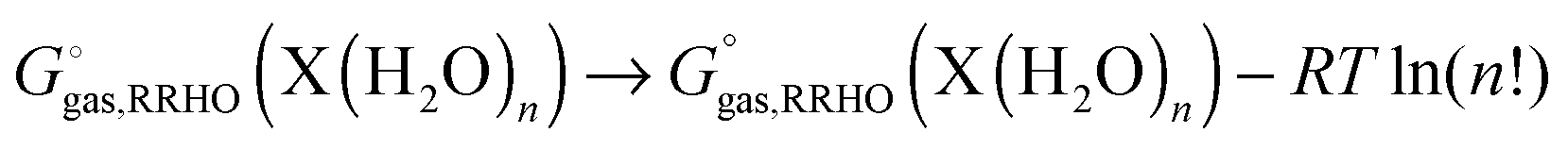
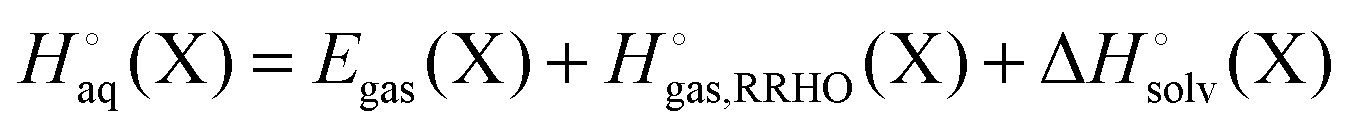
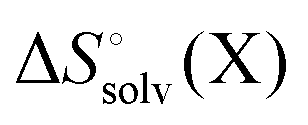 can make an appreciable contribution to the energy change for reaction energies.
can make an appreciable contribution to the energy change for reaction energies.
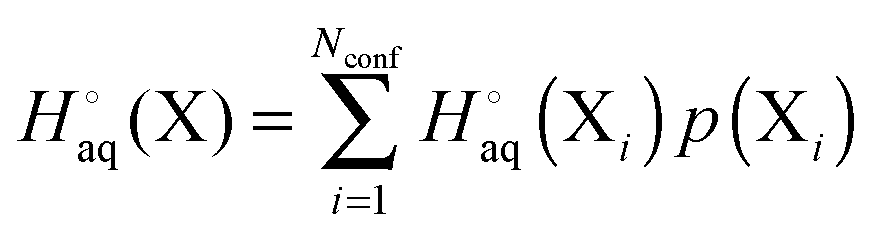
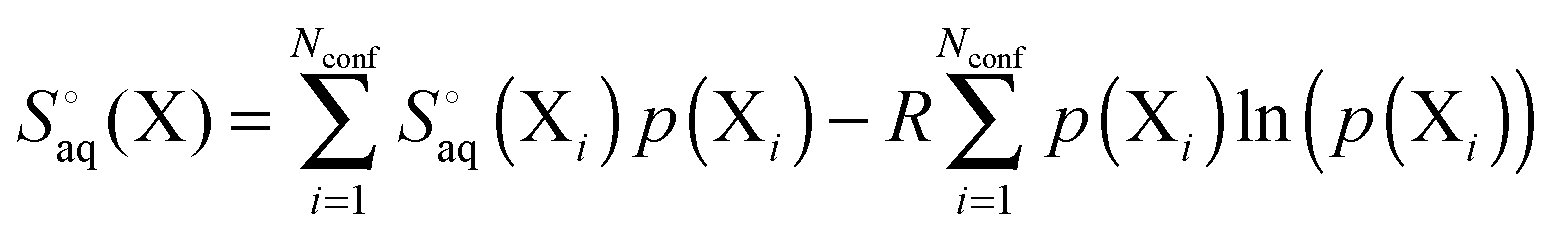
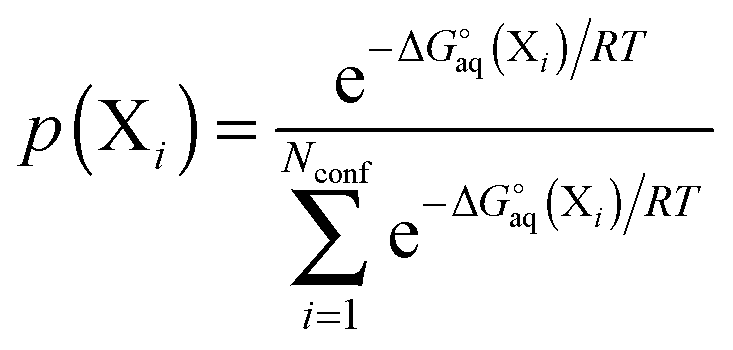
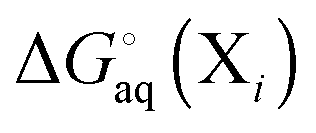 is computed relative to the conformation with the lowest free energy.
is computed relative to the conformation with the lowest free energy.
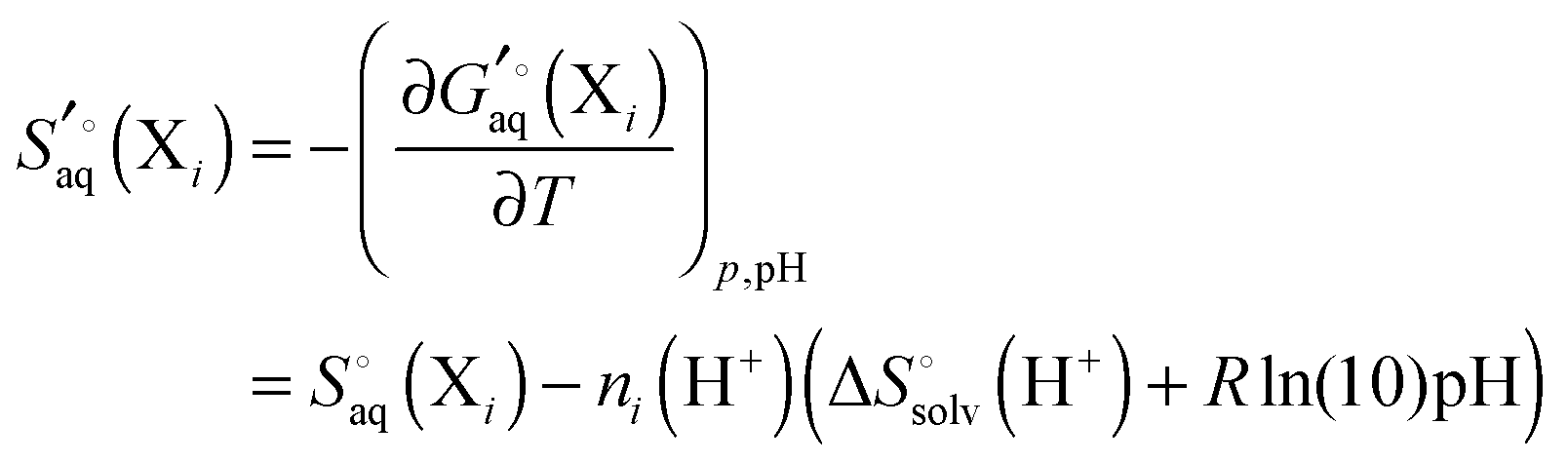
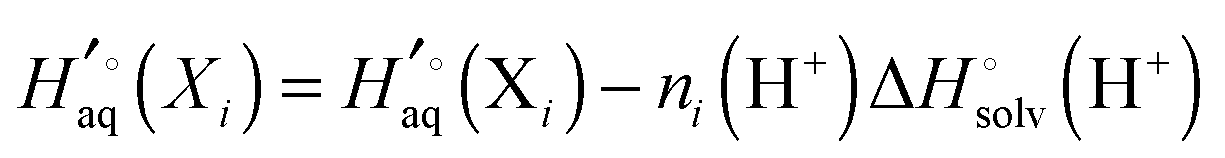
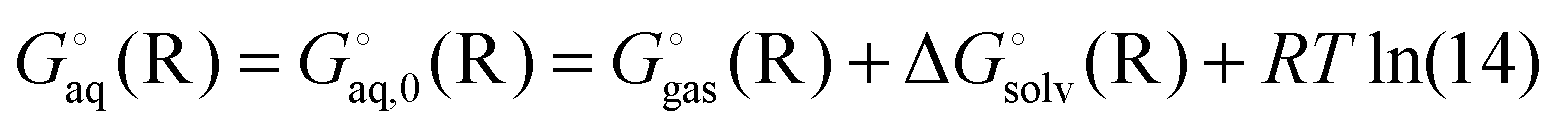
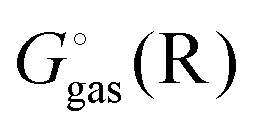 is computed in C1 symmetry and 14 is the symmetry number (σ) corresponding to the D7h point group.
is computed in C1 symmetry and 14 is the symmetry number (σ) corresponding to the D7h point group.
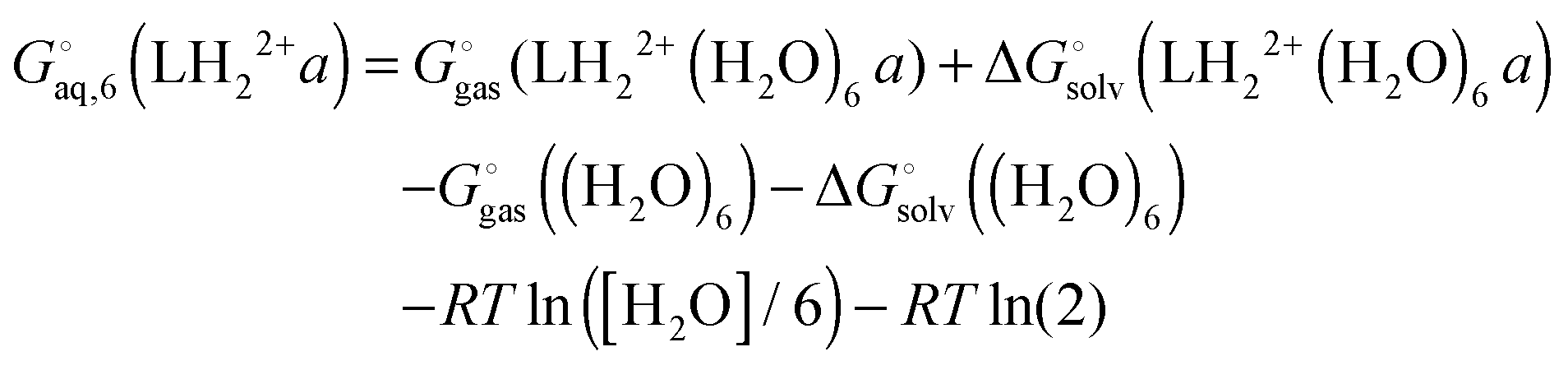
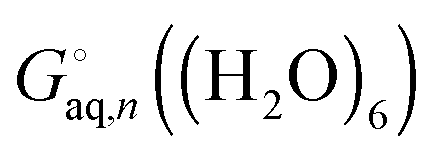 , or the effect of additional conformations can be included using eqn (7). Finally, the Legendre transformed free energy [eqn (15)] at pH 7 is computed by
, or the effect of additional conformations can be included using eqn (7). Finally, the Legendre transformed free energy [eqn (15)] at pH 7 is computed by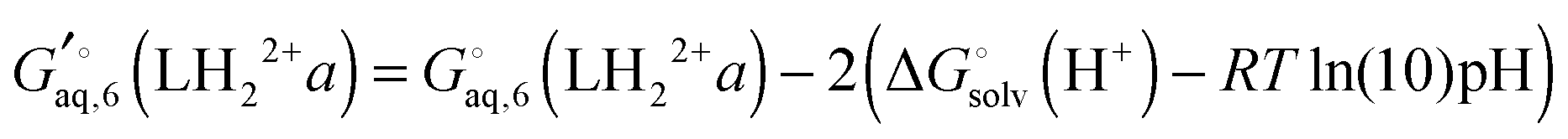
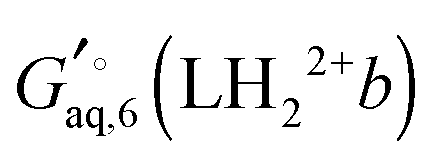 , which has C2v symmetry and for which σ is also 2, is computed in the same way. Notice that each conformation in principle can have different numbers of water associated with them. Similarly, the free energies of the singly protonated and neutral ligand (with C1 and C2 symmetry) is computed by
, which has C2v symmetry and for which σ is also 2, is computed in the same way. Notice that each conformation in principle can have different numbers of water associated with them. Similarly, the free energies of the singly protonated and neutral ligand (with C1 and C2 symmetry) is computed by