
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nature's lessons in design: nanomachines to scaffold, remodel and shape membrane compartments
Paul A.
Beales
*a,
Barbara
Ciani
*bc and
Alexa J.
Cleasby
bc
aSchool of Chemistry and Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds LS2 9JT, UK. E-mail: p.a.beales@leeds.ac.uk
bCentre for Membrane Interaction and Dynamics, Department of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: b.ciani@sheffield.ac.uk
cCentre for Chemical Biology, Department of Chemistry, University of Sheffield, Sheffield S3 7HF, UK
First published on 16th March 2015
Abstract
Compartmentalisation of cellular processes is fundamental to regulation of metabolism in Eukaryotic organisms and is primarily provided by membrane-bound organelles. These organelles are dynamic structures whose membrane barriers are continually shaped, remodelled and scaffolded by a rich variety of highly sophisticated protein complexes. Towards the goal of bottom-up assembly of compartmentalised protocells in synthetic biology, we believe it will be important to harness and reconstitute the membrane shaping and sculpting characteristics of natural cells. We review different in vitro membrane models and how biophysical investigations of minimal systems combined with appropriate theoretical modelling have been used to gain new insights into the intricate mechanisms of these membrane nanomachines, paying particular attention to proteins involved in membrane fusion, fission and cytoskeletal scaffolding processes. We argue that minimal machineries need to be developed and optimised for employment in artificial protocell systems rather than the complex environs of a living organism. Thus, well-characterised minimal components might be predictably combined into functional, compartmentalised protocellular materials that can be engineered for wide-ranging applications.
Introduction
Scientists have long looked to nature to provide solutions to some of our greatest problems, from therapeutics and medical materials, to the environment and energy production. As the oft-quoted physicist Richard Feynman said ‘nature's imagination is so much greater than man's, she's never going to let us relax.’This statement couldn't be more evident than in the stunningly complex and vastly interlinked systems that make up each and every living cell. Cells are some of the most incredibly adept devices we know and understanding them is the first step towards manipulating and harnessing cells as machines and applying them in novel ways.
One of the cell's most ingenious skills is also one of its most simple: compartmentalisation. Cell biologists have been intrigued for decades by the organisational abilities of cells and their clever methods of compartmentalisation. The ability to keeps things separate sounds simple, and yet, behind the scenes lies hundreds of interacting proteins and precise pathways that orchestrate and permit a multitude of mini-reactions to occur independently, repeatedly and simultaneously in the space of one cell. Eduard Buchner commented in his 1907 Nobel Lecture that ‘We are seeing cells more and more clearly as chemical factories, where the various products are manufactured in separate workshops, the enzymes acting as the overseers.’
This same complexity is one reason designing experiments to exclusively probe one aspect of a cell's function can be near impossible in vivo, and yield results difficult to interpret. Cell biology relies heavily on the use of model systems, which have considerably furthered the current understanding of how cells work, but lack the finer mechanistic detail.
Synthetic biology† or reconstitution studies, addresses this issue by offering a powerful way to study proteins in a stripped down system that can be tightly controlled. Liu & Fletcher1 outline the benefits succinctly; in vitro experiments can refine and confirm molecular processes outside of a complicating cellular environment, whilst providing insight into how these biological functions could be engineered towards new functions. It is this idea that lies at the heart of synthetic biology; creating an interface between biology and engineering and the understanding to turn cell biology into a physical and engineering discipline.
It's important to appreciate there is so much more to synthetic biology than uncovering the secrets of how cells work. In vitro reconstitution experiments are slowly revealing a whole toolkit of proteins with enormous potential – the full scope of which is yet to be realised and with only man's imagination as the limit to the possibilities. Occurring throughout cells, dynamic membrane remodelling is gained by the precise interplay of proteins and lipids that are able to generate, sense and stabilise membrane curvature.
Here, we introduce a handful of these proteins and the experimental setups that enabled the study of the mechanisms through which they shape membranes, together with examples of how this knowledge has been used to produce minimal systems of membrane remodelling tools. The scope of this review is to present advances in recent in vitro studies of membrane deformation mechanisms by natural systems as an opportunity for developing new instruments for the generation of ad hoc compartments leading to the engineering of functional protocells. First we give an overview of different model membrane systems and how they are best applied in studying these protein–membrane interactions and construction of protocellular systems. We then review recent mechanistic insights into the function of proteins involved in the sculpting and scaffolding of membranes gained from biophysical studies on reconstituted minimal systems. This leads us into a brief overview of the theory of membrane elasticity and the important additional insights of combining appropriate theoretical models with biophysical experiments in understanding biological mechanism. These biophysical insights form the basis of a discussion of how these lessons from nature can be best applied to the design of protocellular materials where we argue the merits of designing minimal machineries optimised for in vitro function and give recent examples of such reengineered or biomimetic nanomachines. We close with an outlook for the future development of nanomachines for dynamic control of compartmentalisation in engineered protocells, proposing how the field might proceed from biophysical insight of natural machineries to biological components that can be engineered into predictable protocellular machines.
Setting boundaries: in vitro model membrane systems
A range of model membrane systems exist that are useful for the study of reconstituted proteins and protein complexes. These can be categorised into supported membranes, which are membranes that have been formed on a supporting substrate, and unsupported membranes, which are unconstrained membranes that are free-floating in solution. While, as we will see, unsupported membrane systems have the greatest capacity for encapsulation of chemical processes, supported membrane systems allow the application of powerful surface-analytical techniques to the study of protein–membrane interactions.Substrate-supported membranes
Supported membranes can be formed upon a variety of solid substrates, with commonly used substrate materials including mica, gold, quartz and glass.2 These solid supports rigidly confine the membrane to their surface and thereby suppress the membrane's freedom to bend in response to interactions with proteins and other membrane-active matter. The attractive interactions that are required to exist between the membrane and its support in order for it to remain confined to its surface may also have other perturbations on the properties of these membranes that do not exist in unsupported systems. For example, changes in membrane dynamical properties are well known: lateral diffusion coefficients of lipids in surface supported membranes can be significantly slower than in unsupported model membranes.3 Furthermore, the morphology and dynamics of lipid domains in phase-separated membranes can exhibit significant differences between supported and unsupported membrane models.4–8 Despite these possible disadvantages, surface-supported systems are still widely used to study lipid membranes. The primary reason for this is the range of surface analytical techniques that allow membranes to be studied with greater resolution and sensitivity than is often achievable with unsupported model membranes. These techniques include atomic force microscopy (AFM) for high resolution topological imaging,4,5,7,9 AFM and surface force apparatus for measuring the mechanical forces and interaction forces of membranes,10 quartz crystal microbalance,11,12 surface plasmon resonance13 and total internal reflection fluorescence microscopy (TIRF-M)14 for membrane binding interactions, membrane electrochemistry (e.g. supported membranes on gold electrodes)15 and time-of-flight secondary ion mass spectrometry imaging for studying the lateral distribution of chemical composition within a membrane.16A variety of methods exist for creating substrate-supported model membranes. The most commonly used techniques are the vesicle fusion method, where small or large unilamellar vesicles, which are dispersed in an aqueous solution above the substrate, fuse with the supporting surface to form a single lipid bilayer, and the Langmuir–Blodgett (LB) method, where the substrate is passed twice through a lipid monolayer formed at an air–water interface by dipping the cleaned solid-surface into and out of the aqueous phase.17 Several methods have also been developed to create asymmetric supported lipid bilayers where each lipid monolayer has a different lipid composition.18–20 This mimics the lipid asymmetry known to occur in natural membranes and allows characterisation of how such asymmetry modulates the physical properties and behaviour of the membrane.
Due to solid membrane substrates not generally being relevant as natural biological interfaces and their hindrance for incorporation of transmembrane proteins, softer supports have been developed. These include supported membranes on polymer cushions or hydrogels and membranes extended from the surface by “molecular stilts”: functionalised lipids such as oligoethylene glycols with a thiol functionality at one end for covalent surface attachment and a sterol at the other to insert into the hydrophobic core of the membrane.21–25 Furthermore, techniques have been developed to create regions of unsupported membrane within a supported lipid membrane by forming the bilayer atop a microfabricated substrate that contains small holes. The supported membrane does not enter these holes but rather spans the gap, providing regions of membrane that do not directly interact with the substrate.26–28 Finally it should be noted that supported membranes have been formed on the surface of colloidal particles; this allows the dispersion of supported membranes into bulk solution and introduces curvature to the membranes comparable to those found in biological cells and organelles.29,30
Unsupported membranes
Unsupported membranes allow dispersion of lipid bilayers into the aqueous phase without the need for a supporting substrate. Several such model systems exist; we will briefly introduce lipid nanodiscs, droplet interface bilayers and small, large and giant unilamellar vesicles. We will give most emphasis to giant unilamellar vesicles (GUVs) as these are cell-sized model membranes capable of encapsulating chemical processes. Furthermore, GUVs are the most common model membrane used in the literature for reconstitution of functional protein complexes.(a) Lipid nanodiscs are lipid bilayer micelles whose edge tension is stabilised by α-helical scaffold proteins.31,32 Depending on the scaffold protein that is employed, these disc-like membrane fragments vary in diameter between approximately 10–18 nm with a narrow size distribution.33 These model membranes have primarily been employed as a more biologically relevant alternative to detergent solubilisation of integral membrane proteins for structural and functional assays.34–36 Most relevant to the study of membrane remodelling protein complexes is the use of lipid nanodiscs for single molecule fluorescence spectroscopy studies.37 Due to the small surface area of membrane available on a lipid nanodisc, experimental protocols can be designed such that only a single protein or protein complex will interact with a nanodisc. This scenario is much harder to achieve using model membrane systems based upon lipid vesicles, which have a much larger surface area per particle for interactions to take place and the number of interacting proteins per lipidic particle would follow a Poisson distribution.
(b) Droplet Interface Bilayers (DIBs) are formed using water-in-oil emulsions.38 Water droplets dispersed in an oil containing dissolved lipids recruit a lipid monolayer to the oil–water interface. When two of these droplets are brought into contact, oil is excluded from the interfacial region between the droplets such that a lipid bilayer membrane forms between the two aqueous compartments. Electrophysiological measurements have been used to study the ion permeability of these membranes in the presence of integral and peripheral membrane proteins.39–41 Experimental geometries have also been developed that allow simultaneous electrophysiological and fluorescence microscopy studies.42 As we will see, DIBs are also a starting point for some of the more advanced methods for fabrication of GUV model membranes. Notably, DIBs and vesicles, which will be discussed below, have the capacity to compartmentalise chemical entities and processes within an encapsulated volume and therefore are prime for a stepwise increase in complexity of reconstituted biological systems towards the bottom-up development of artificial cells and tissues.
(c) Small or Large Unilamellar Vesicles (SUVs or LUVs) are nanoscale or sub-micrometer unsupported membrane shells with an encapsulated aqueous compartment segregated from the bulk water phase. SUVs (<100 nm diameter) are usually formed by high energy sonication of multilamellar vesicles (MLVs; formed by the dispersion of dried lipid films into an aqueous buffer by vortexing).43 LUVs (100–1000 nm diameter) can be formed by several different methods; most commonly they are formed by multiple extrusions of MLVs through polycarbonate filters with monodisperse pore sizes of between 100–400 nm. Alternatively they can be formed by reverse phase evaporation or detergent extraction methods. Aqueous vesicle dispersions are often studied by spectroscopic or calorimetry techniques that report the statistical ensemble averaged behaviour of a large population of vesicles; example techniques include isothermal titration calorimetry, fluorescence spectroscopy, dynamic light scattering, electrophoretic light scattering and electron spin resonance spectroscopy. More recently techniques have been developed that allow single vesicle analysis of sub-micrometer vesicles, e.g. by anchoring these vesicles to a surface and using spatially-resolved surface analytical techniques.44,45 One of the advantages of SUV and LUV systems is the ability to create membranes of high curvature, thus allowing systematic study of curvature-dependent effects that influence protein–membrane interactions.46–48
(d) Giant Unilamellar Vesicles (GUVs) are vesicles that are >1 μm in diameter (most typically in the range of 10–30 μm) and so are observable by optical microscopy techniques. This allows experimental observations to be conducted at the single vesicle level. Beyond the average behaviour of a sample, single vesicle analysis can reveal the full distribution of vesicle behaviours, transient intermediate states and is sensitive to rare events. Due to their large size (similar to that of biological cells), GUV membranes have low curvature and are usually considered to be effectively flat for the purposes of interactions with nanoscale proteins and protein complexes. However methods have been developed to study curvature effects in GUVs by extruding cylindrical lipid tubules from the GUV surface that are a few tens of nanometers in diameter: the cylindrical curvature of these structures implies high curvature for only one of the two principal curvatures of the membrane, the second principal curvature being equal to zero.49–51 For high spherical membrane curvature (both principal curvatures are high), then SUVs or LUVs are more appropriate model systems.
While optical microscopy is often used to study GUVs, GUV experimental analysis can go well beyond the recording of qualitative pictures, as summarised in Table 1. Dense (e.g. sugar) solutions are often encapsulated within GUVs such that the encapsulated phase is more dense than the surrounding solution, causing the vesicles to sediment to the bottom of the sample dish, making them easier to find and observe.52 Methods have also been developed that allow reconstitution of integral membrane proteins into GUV model membranes.53
| Measurement | Technique | Brief description and comments | Key and example references |
|---|---|---|---|
| Membrane morphology | Optical microscopy | GUV morphology changes directly observed by optical microscopy (phase contrast, epifluorescence, fluorescence confocal) can give qualitative clues to the nature of material interactions with the membrane. | 52, 166 |
| Membrane fluidity | Fluorescence recovery after photobleaching (FRAP) | A region of the membrane is bleached of fluorescence with a high energy laser of a confocal microscope. The rate of fluorescence recovery by dyes from outside of the region of interest can be quantified into a lateral diffusion constant of lipids. This methods assumes homogeneous diffusion across a larger area of membrane (i.e. the photobleached region) | 164, 170, 171 |
| Fluorescence correlation spectroscopy (FCS) | The autocorrelation function of fluorescence fluctuations of a pixel on the GUV membrane is used to quantify the diffusion constant of fluorophore-labelled lipids or membrane-bound proteins. This is a more localised measurement than FRAP (above) and can be confined to the nanoscale using stimulated emission depletion microscopy FCS (STED-FCS172,173). | 172–180 | |
| Mechanical moduli | Micropipette aspiration | GUVs are aspirated into a micropipette; stress-strain plots are used to quantify κb, KA and the lysis stress and strain. κb is difficult to measure as this requires a lot of data points in the low tension regime. Membrane contour analysis (below) can be better for measuring κb. | 123–125, 181, 182 |
| Membrane contour analysis | The radial localisation of the GUV's membrane is determined with sub-pixel resolution. Fitting the power spectrum of membrane fluctuations allows the bending modulus to be quantified. | 183–188 | |
| Membrane permeability | Dye translocation kinetics | The rate of influx of water soluble fluorescent probes into GUVs can be used to quantify the membrane's permeability and the fractional permeable area of membrane. | 189–191 |
| Patch-clamp electrophysiology | Electrophysiological patch-clamp recordings have been achieved in GUVs to quantify ion permeability of membranes. | 192 | |
| Membrane phase behaviour | Fluorescent lipid partitioning | Imaging the partitioning of dyes between coexisting lipid phases in phase separated membranes allow the study of membrane phase behaviour and the construction of phase diagrams of lipid mixtures. | 193 |
| Lipid packing and hydration | Laurdan fluorescence | The generalised polarisation function of Laurdan obtained from ratiometric imaging of two environment-sensitive emission maxima reveals information about changes in the local structure and hydration of the membrane. | 165, 194, 195 |
| Membrane binding | Fluorescence localisation imaging | Imaging of fluorescent membrane-active compounds (e.g. proteins) can be used to semi-quantitatively investigate their binding at GUV membranes, including relative binding to different coexisting membrane phases or curvatures. | 49, 52, 99 |
| Flow cytometry | The colocalisation of fluorescent proteins or particles with GUVs in flow cytometry experiments can be used to quantify the kinetics and extent of adsorption. | 196 | |
Methods for preparation of GUVs are summarised in Fig. 1. The most popular method for GUV production is the electroformation method.54,55 Lipid films are dried from chloroform stock solutions onto Pt wire or ITO-coated glass electrodes. After rehydration with an aqueous solution (often a sucrose solution), an a.c. electric field protocol is applied to the electrodes for 1–2 h. The initial sine wave a.c. frequency usually ranges between 10–20 Hz and many protocols gradually reduce this frequency in a stepwise fashion during the electroformation procedure. The amplitude of the a.c. field that is used usually varies between 1.5–5 V, with the lower end of this range more appropriate when charged lipids form part of the lipid mixture. While electroformation usually produces a high yield of high quality GUVs, disadvantages of this method are that high salt or high (∼>20 mol%) anionic lipid content significantly suppress GUV yield or prevent their formation altogether. However it should be noted that this technique works very well for phosphocholine-rich membranes, the most predominant lipid type in Eukaryotic membranes. Physiological salt buffer can be added to the extravesicular medium after electroformation is complete. Protocols using a higher frequency a.c. field have been reported that facilitate GUV formation in physiological salt buffers.56 Alternatively, the gentle hydration method can be used to form GUVs, but this method gives a lower yield of high quality GUVs.57 Recently formation of GUVs assisted by a polyvinyl alcohol gel support has been reported that permits a wide range of lipid compositions and buffer conditions to be used.58
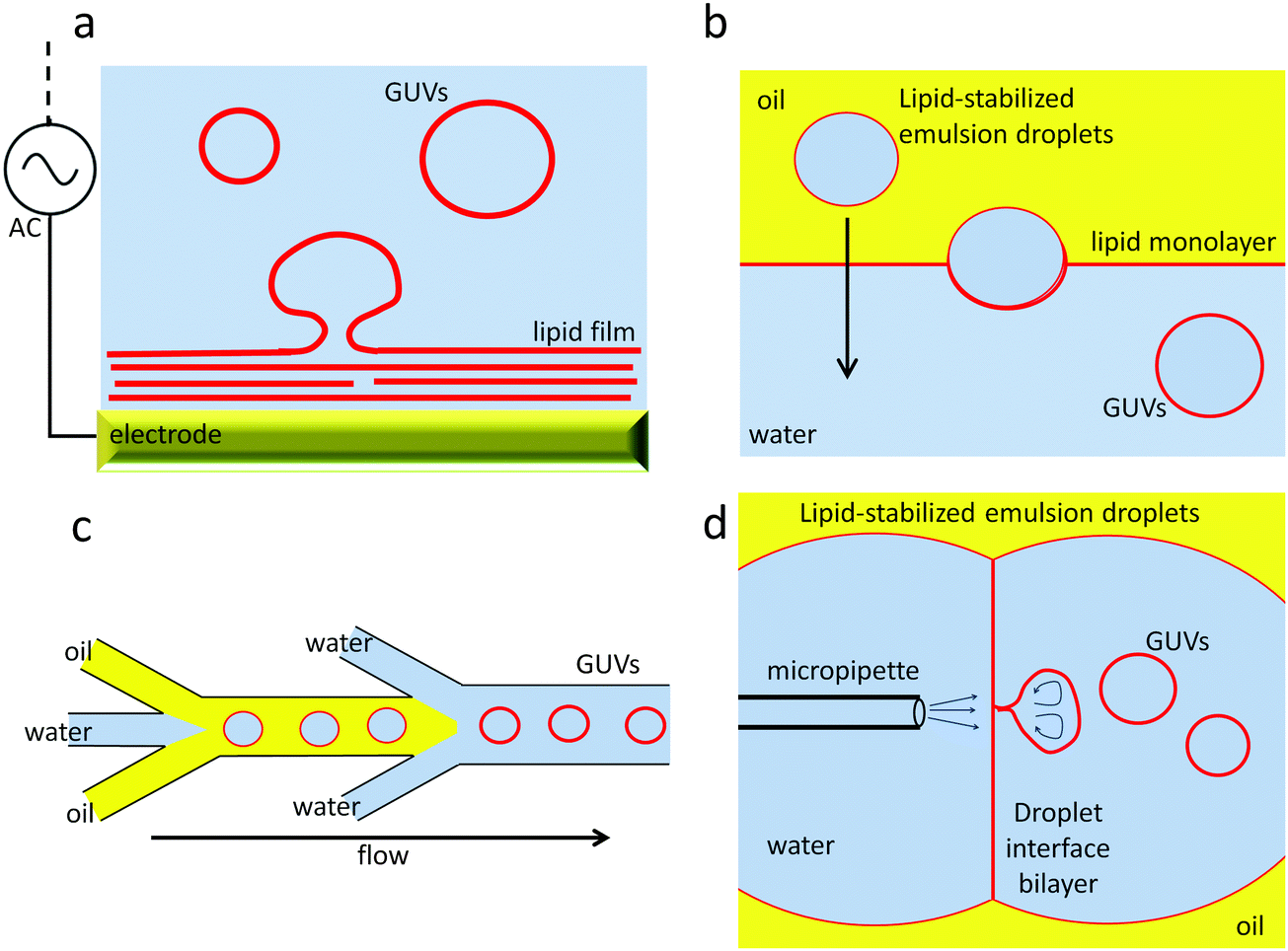 | ||
| Fig. 1 Overview of some of the GUV formation methods. (a) electroformation; (b) emulsion-transfer method; (c) microfluidic double emulsions; (d) microfluidic jetting. | ||
A further disadvantage of these GUV preparation methods is the challenge of encapsulation of different chemical entities inside GUVs compared to those in the extra-vesicular aqueous solution. Upon forming the GUVs in the presence of compounds required for encapsulation, similar concentrations will be present inside and outside the vesicles. Separation of unencapsulated compounds from the sample is challenging, with tradition approaches used for LUVs, such as size exclusion chromatography or dialysis, proving to be inefficient. More recent developments in GUV fabrication methods use emulsion-based assembly protocols and allow more efficient specific encapsulation of compounds. Emulsion phase transfer methods are based on the transfer of lipid stabilised water-in-oil droplets through an oil–water interface, where it picks up a second lipid monolayer, resulting in formation of a completed GUV in water with encapsulated contents.59 This method can also be adapted to produce asymmetric membranes.60 Microfluidic lab-on-a-chip methods are being developed for GUV fabrication, in particular using flow-focussing droplet microfluidics geometries.61,62 Recently droplet microfluidics and emulsion phase transfer strategies have been combined into a single on-chip GUV fabrication protocol.63 Microfluidic jetting has also been reported using DIBs as a starting point for GUV formation; jets of fluid cause GUVs to break away from the DIB.64 This method can be used for high encapsulation efficiency and formation of asymmetric membranes.65 DIBs in conjunction with the emulsion phase transfer method have been used to create multicompartment GUVs.66,67
Structure translates into function for membrane-bound compartments
The cell cytoplasm is crowded with molecules that each belong to different pathways. Membrane organelles organise cell content and streamline cellular functions. These membrane-bound compartments allow small molecules to permeate by diffusion whereas larger cargoes are encapsulated through remodelling of their surfaces in a process called endocytosis (Fig. 2). Phospholipid membranes provide organelles with unique morphologies that define their functions. Intracellular membrane organelles have highly complex shapes with a common characteristic: their large membrane area in comparison to the volume they enclose. Endoplasmic reticulum and Golgi apparatus membranes adopt typical tubulated structures, acting as storage and processing units for vesicular cargoes. Membrane tubules are also a feature of early endosomes, organelles that capture vesicles originated from endocytosed cargo and ‘mature’ into multivesicular bodies while moving along microtubules tracks (Fig. 3). While early endosomes adopt irregular structures, MVBs consist of vesicles inside larger vesicles that are conduits for compartmentalisation, sorting and transport of material to selected destinations68 (Fig. 3). There is a biological reason for this variety in organelle shape: larger surface areas facilitate cargo exchange between the intraluminal space of the organelle and the cytosol. Furthermore, larger surfaces allow for efficient separation of cargo at the membrane. This structure–function relationship existing in intracellular membrane compartments would ideally be translated into synthetic cellular systems to optimise their functional capabilities and efficiencies. So what is shaping membrane-bound compartments in functional biological cells? | ||
| Fig. 2 Key cellular events that require membrane fission and fusion. Membrane fission is typical of events such as endocytosis. It is necessary for the generation of transport vesicles, which fuse with sorting stations, namely the endosomes and the multivesicular bodies. Membrane fusion events are typical of exocytosis processes, such viral particles budding and the release of exosomal vesicles. | ||
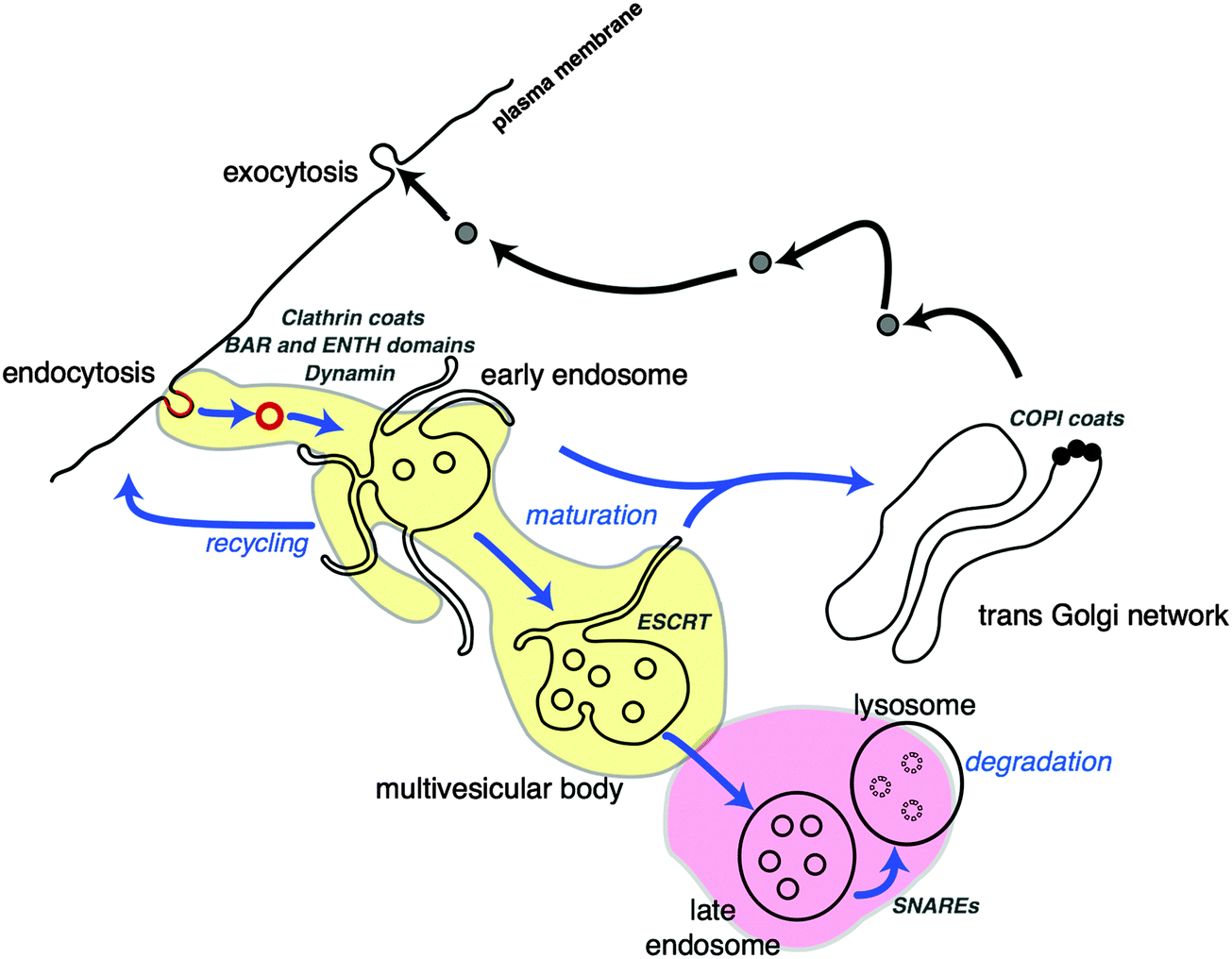 | ||
| Fig. 3 Cellular fission and fusion events in trafficking of cargo via membrane compartments. Endocytosed cargo is enveloped in clathrin coated vesicles (red), with the aid of fission proteins (BAR and ENTH domains). Clathrin-coated vesicles are incorporated into early endosomal structures. The tubules of endosome membranes are shaped by molecular motors (kinesin, dynein), while moving along microtubule (blue arrows) towards the centre of the cell. Some vesicular cargo is also recycled to the cell surface. During movement, the endosomes ‘mature’ into multivesicular bodies (MVB), changing their shape and protein composition (yellow area). MVBs are generated by accumulation of cargo destined for degradation by the lysosome (pink area) via membrane budding and scission events performed by the ESCRT complexes. The trans-Golgi network (TGN) is another sorting station for vesicular cargo that utilises COPI protein coats for membrane scaffolding. This figure is a derivative of Fig. 1 from ref. 68, used under CC BY 3.0 licence. | ||
Tools for the job: in vitro reconstitution of molecular systems for sculpting membranes
The complex shape of intracellular organelles is moulded by physical forces acting on phospholipid membrane bilayers. The mechanical tools exerting localised forces that induce membranes' deformation are organised assemblies of proteins that recognise, anchor and remodel vesicular structures. There are two main types of membrane remodelling events: fusion and fission. Here we will cover some of the most recent work aimed at understanding how structures of individual and assembled proteins drive these processes.Membrane fusion
Membrane fusion occurs in a wide variety of cellular activities, especially those that involve endocytosis or exocytosis of material.69 It has only been in the last two decades that the molecular machinery and mechanisms of membrane fusion have been uncovered. Molecules, or ‘cargo’, are constantly being moved around cellular compartments and delivered to precise intracellular destinations. This traffic and transport of cargoes in, out and throughout the cell occurs without compromising the structure of any cellular organelle thereby leaking their contents; this is achieved via fission and fusion of membrane vesicles. Transport vesicles filled with cargo bud from a donor organelle, and are then transported to an acceptor organelle, where they dock to the membrane and fuse, thereby releasing their cargo (Fig. 3). Here we look at some of the main structural and mechanistic aspects of the main players in membrane fusion and targeting: SNARE complexes.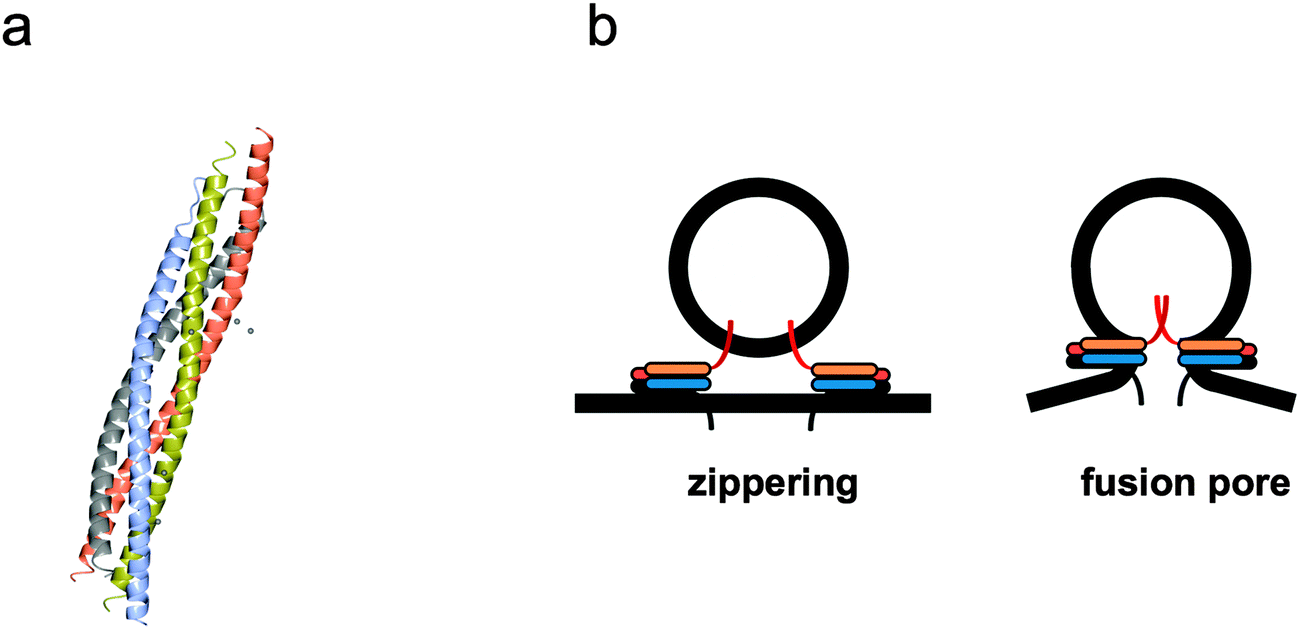 | ||
| Fig. 4 Mechanism of membrane fusion and fission by SNARE complexes. (a) Tetrameric coiled coil structure of an assembled neuronal SNARE complex involved in synaptic exocytosis (pdb code 1SFC). (b) Complementary v- and t-SNARE coiled coils sitting on donor vesicle and target membranes ‘zipper’ to bring the bilayers closer and initiate ‘fusion pore’ formation. The individual α-helical coiled coil chains in (a) and the respective cartoons representations in (b) are identically colour coded. | ||
The first observation of content mixing was seen using an in vitro fluorescence-based assay using liposomes.72 This work suggested SNARE proteins form the minimum machinery required for fusion. SNARE complexes are specific, as v-SNAREs do not interact with other v-SNARE containing liposomes or protein free vesicles. The interaction occurs via a unique hairpin-like structure formed by the SNARE complex, named SNAREpin, and is similar to structures formed by some viral fusion proteins.73 The Weber paper has been cited ~2000 times since its publication in 1998 and received much attention, not only due to the cell biology questions it answered, but also the prospect of studying membrane fusion in a fully reconstituted system.
Assembly of a stable SNARE complex occurs spontaneously in solution when using free SNAREs and is energetically favourable. In cells this is controlled by a series of regulatory proteins, but can formation of the SNARE complex and fusion be controlled in synthetic cell systems? SNARE complexes can assemble before fusion and these unfused intermediates accumulate at lower temperatures, therefore something other than high concentrations is required to induce fusion. It was noticed that prefusion ‘priming’ steps occur more slowly in a cell free system, possibly as the proteins need to topically rearrange themselves into a circular array to give rise to the ‘fusion pore’ (Fig. 4b) that initiates fusion.74,75 Some viral proteins behave in a similar fashion, forming pre-fusion intermediates at low temperatures that only fuse once the temperature has increased or been triggered by a low pH (such as within a late endosome).76
Lipid bilayer nanodiscs were used recently to demonstrate that one SNARE only is required for maximum rates of membrane fusion, but release of membrane contents on physiological timescales requires zippering of multiple SNAREs into SNAREpin structures, suggesting that SNARE complexes prevent the fusion pore from resealing during cargo release.77 SNARE-driven fusion in liposomes occurs at rates comparable to physiological fusion following removal of an inhibitory N-terminal domain in t-SNAREs, without affecting the rate of the v- t-SNARE complex formation itself. Full length v-/t-SNARE reconstituted liposomes in the presence of Ca2+ also enables membrane fusion at near physiological rate, confirming the role of Ca2+ as a regulatory factor in fusion.78
The size of the lipid vesicles and the lipid composition itself may also affect the efficiency of fusion. For instance, recently we have learned that for physiological concentrations of SNARE proteins to perform efficient membrane fusion there is a specific requirement for small neutral lipids that do not form bilayer structures (i.e., phosphatidylethanolamine). In the absence of these types of lipids, vesicles can still dock and form trans-SNARE complexes but cannot remodel membranes for fusion.79
It might be expected that in a synthetic system with minimal machinery that fusion would occur more slowly than in vivo due to the absence of other components that could facilitate fusion, for example SNAP and NSF. NSF, stimulated by SNAPs, use ATP hydrolysis to dissociate the stable SNARE complex and seem to allow further rounds of fusion to occur. The possibility remains that they also act to regulate fusion, potentially through the inhibitory N-terminal domain of t-SNAREs. It is generally agreed that fusion occurs through a fusion pore, more recently termed a ‘porosome’, whereby several SNARE complexes form at the site where fusion is to take place and arrange themselves into a ring structure giving the membrane a dimpled appearance.80 The detailed structure of porosomes has been revealed using reconstituted fusion pores in large unilamellar lipid vesicles (LUVs) and examined by transmission electron microscopy (TEM). This work revealed a basket-shaped morphology with a SNARE complex ring at the base of the fusion pore and also that the porosome was still functional.81
Measurement of content mixing of large biomolecules within vesicles can be elegantly performed by incorporation of fluorescently labelled DNA hairpins inside vesicles. Using FRET to quantify real fusion events, a decrease in FRET signal would occur when a labelled DNA strand encounters a complementary strand from another vesicle.82 This approach discounted instances of vesicle aggregation or vesicle rupture, which can obscure actual membrane fusion, and allows a highly reliable method for detecting fusion. Adopting this method showed that SNARE complex alone is the minimal machinery required for fusion pore formation and expansion (large enough to allow cargo transfer up to 11 kDa).
An interesting characteristic of SNARE proteins is their preference for phase-separated lipid bilayers, or lipid ‘rafts’, as it was suggested from in vivo work showing SNAREs' preference for cholesterol-rich regions. Lipid rafts represent liquid-ordered regions of the membrane, rich in saturated lipids and cholesterol, which act as platforms for an array of interactions concerned with protein intracellular trafficking. These micro-regions are more ordered and tightly packed than the surrounding bilayer, but are largely inaccessible to direct observation in vivo. However, it was found that SNAREs in fact have a higher affinity for liquid-disordered regions of membrane83 as demonstrated by monitoring the localisation of fluorescently-labeled SNAREs reconstituted in phase separated GUVs.
Intriguingly, enveloped viruses also use membrane fusion machineries to enter a cell. These machineries are functionally similar to, but mechanistically distinct from the SNAREs. In analogy to SNAREs, coiled-coil structures are pivotal in the fusion mechanism of many viral proteins, and the formation of a low energy membrane structure via the fusion pore. Typical examples are gp41, a protein that adopts a functional six helix bundle allowing HIV-1 fusion with the cell membrane,84 and influenza hemagglutinin, which undergo pH-driven conformational change to form a fusion-competent coiled coil trimer.85 It has been common to study the membrane interaction of viral fusion peptide fragments in reconstituted systems to demonstrate the mechanistic diversity between different viral fusion machineries.86,87 However, an example of functional reconstitution of a full fusion protein has been reported with the assembly of the full GPC envelope glycoprotein of Hemorrhagic-Fever Arenavirus into proteoliposomes. In this study, pH-dependent fusion was demonstrated using fluorescence lipid-mixing and content-mixing assays.88
Membrane bending and fission
Membrane fission shares some similarities with fusion, such as the formation of a pore-like structure, and the ability to overcome the energetically unfavourable process of membrane bending. During fission, vesicle budding occurs either away from or into the original cellular compartment. Frolov et al. have recently presented an excellent review on the geometry of fission.89 Membrane fission is a multistep process that requires the action by key proteins that initiate the process through membrane deformation. Well-studied examples of proteins that remodel the membrane to induce or aid fission are BAR and ENTH domains, and the ESCRT machinery. Protein assembly on the inner or the outer part of membrane compartments dictate the topology of fission. There are few characterised mechanisms that promote membrane deformation prior to fission (Fig. 5). Some protein modules mediate fission by shaping the exterior of a nascent budded vesicle like clathrin, and COPI coats, and oligomerised proteins containing ENTH and BAR domains. A different approach is instead used by ESCRT complexes, which assemble on what will become the interior surface of the lipid bilayer delimiting a vesicle. For this reason, ESCRT complexes can also participate in processes such as cell division, performing the last step of fission of the plasma membrane.90 | ||
| Fig. 5 Membrane remodelling proteins use various methods in order to generate curvature. During protein insertion, hydrophobic protein domains asymmetrically inserted in the lipid bilayer generate membrane curvature; this is a mechanism used by some BAR proteins. In protein crowding, it is assumed that the energy for membrane deformation is generated by collisions between proteins in close proximity on the surface. This mechanism of membrane deformation is possibly used by BAR and ENTH proteins in addition to insertion into bilayers. Protein shape defines a mechanism where membrane bending is generated by curved shaped proteins with lipid binding domains, such as clathrin and COPI coats. Regions of negative and positive curvature are indicated (blue). Last, in protein scaffolding, membrane curvature is generated by assembling an organised protein scaffold or ‘lattice’ structure. Membrane deformation can occur where components of the scaffold are anchored to the membrane through protein–lipid interactions or as shown by adaptor proteins (green); this method is used by ESCRT proteins. | ||
 | ||
| Fig. 6 Mechanism of membrane fusion and fission by BAR domains (a) structure of the N-BAR domain of hBRAP1/B1N2 showing the curved shape of the dimer (pdb code 4I1Q). (b) BAR domain sensing membrane curvature through its structure and shaping further bending via oligomerisation. The colour coding of protein chains represented by the crystallographic structures is identical to colour coding used for the protein subunits in the cartoon representations. | ||
During BAR domain interactions with the membrane, lipid clustering can occur.92 The idea of proteins preferentially binding to liquid ordered phases, namely ‘lipid rafts’, was previously explored in experiments conducted with SNAREs. Phosphoinositide-clustering is an effect observed for mammalian I-BAR domains96 but is also displayed by BAR and F-BAR domains, which effectively ‘freeze’ the dynamics of phosphoinositides and, in turn, whole regions of the membrane rich in these acidic lipids. The link between sensing curvature and inducing bending in membranes was an active role proposed for protein density.97 It is possible to create an environment where membrane tension, curvature and protein density can be systematically controlled, using GUVs as a membrane model system. Optical tweezers can be used to create nanotubes with different shape and sizes by pulling of membranes from GUVs. Protein density at the GUV surface and tubules can be measured by fluorescence, whereas the pulling force and tube radius gives a measure of curvature and tension. The radius of the tubules is directly proportional to membrane tension in the GUV. When the interaction of amphiphysin (an N-BAR protein enriched at clathrin coated necks during endocytosis) with membranes is analysed vs. degree of curvature and tension, simultaneous roles of BAR proteins as curvature sensors and inducers are revealed.50 For instance, low densities of amphiphysin sense but do not significantly induce curvature. At high densities, amphiphysin strongly deforms membranes via the formation of a scaffold (Fig. 6b). Similar behaviour was also observed previously for dynamin, a GTPase involved in membrane fission and for endophilin, an endocytic protein enriched at neural synapses.98,99
Some BAR domain containing proteins can be regulated by autoinhibition such as in the case of pacsin. Pacsin is a F-BAR protein found primarily in neurons that can be studied in vitro using liposomes enriched with Folch lipids (a type of lipid found primarily in brain tissue for signalling). The interaction between pacsin and dynamin (an F-BAR containing protein) demonstrated how different BAR domain-containing proteins can efficiently regulate each other. It has been shown that pacsin exists in an autoinhibited state in vitro and in vivo.100 Full length pacsin binds with less affinity to the membrane than its F-BAR domain alone and also prefers less curved surfaces. In comparison, endophilin, an N-BAR protein does not show any significant difference in membrane deformation activity between full-length endophilin and its N-BAR domain alone. This evidence suggests that N-BAR proteins may induce curvature by a different mechanism to F-BAR proteins, possibly such as protein insertion and that relieving F-BAR proteins autoinhibition is a mechanism to regulate membrane deformation.
In contrast to BAR domains, ENTH domain structures are not naturally curved but utilise an N-terminal amphipathic helix to ‘wedge’ into membranes promoting deformation and even tubulation at high concentrations. Epsin NH2-terminal homology (ENTH) domain is a membrane-binding motif first identified in the endocytotic protein, epsin. The ENTH domain can bind to certain acidic lipids, particularly phospholipids, including PI(4,5)P2 and PI(1,4,5)P3. ENTH-domain proteins are involved in clathrin-mediated membrane budding, whereby a clathrin protein coat forms around budding vesicles. In particular, ENTH proteins can recruit clathrin adaptors to the site of membrane budding enabling regulation and organisation of clathrin on membranes. In vitro, isolated ENTH domains promote fission of vesicles when added to lipid bilayers but cannot act in concert like BAR domains and facilitate membrane fission generated by dynamin.101
The ESCRT machinery consists of four distinct complexes generally acting in sequential mechanism. ESCRT-0 and ESCRT-I perform specific cargo recognition and concentration at sites of MVB intraluminal vesicle formation, whereas ESCRT-II interacts with and anchors to acidic lipids in the endosomal membrane allowing ESCRT-III to form an organised structure on the membrane surface at the site of fission and initiate budding102 (Fig. 7). ESCRT-III is the core membrane scission machinery that performs membrane fission. It creates filamentous circular arrays from polymerisation of its subunits, delineating spirals narrowing in size and ultimately generating variable size necks for nascent vesicles.103
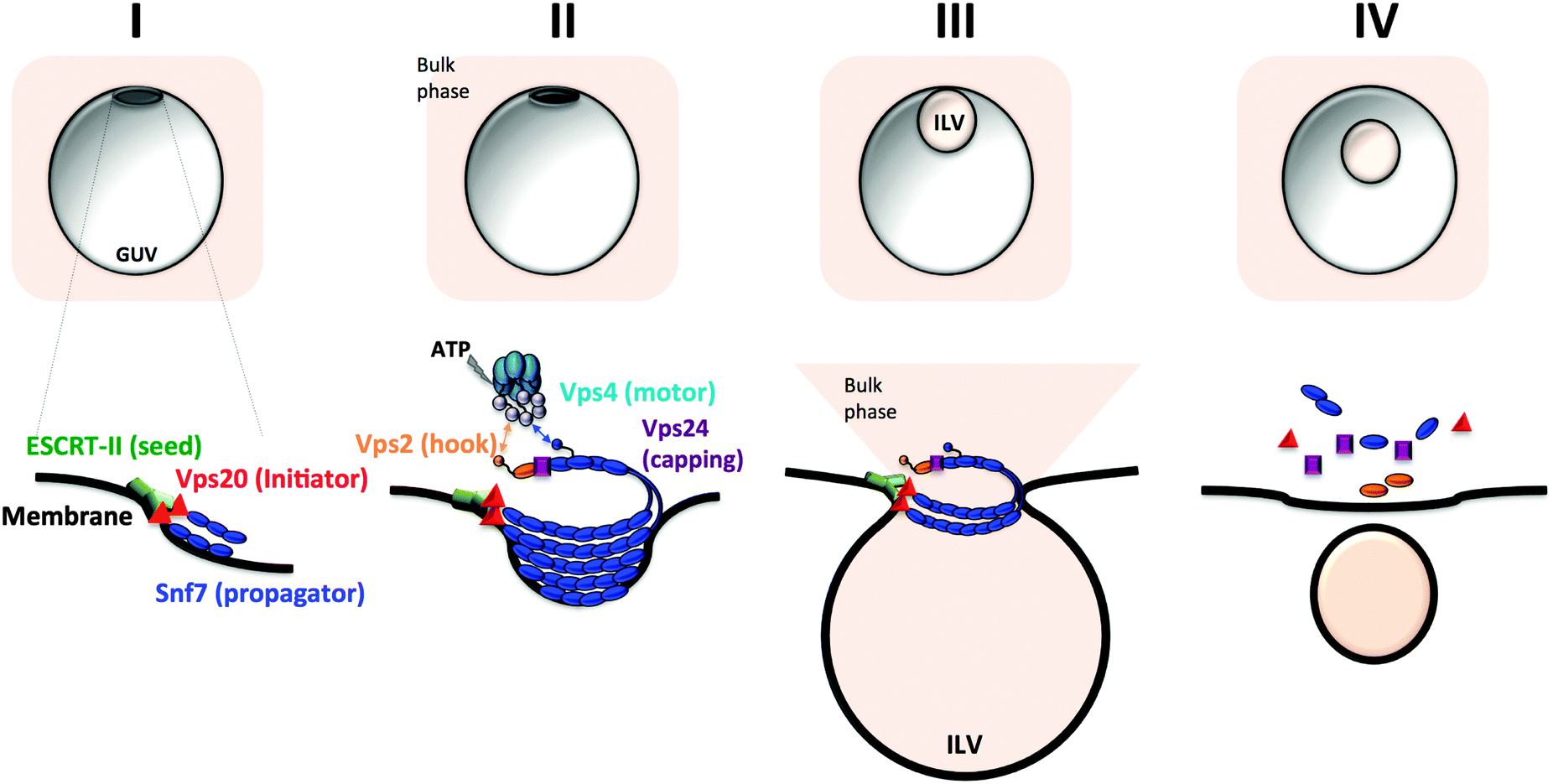 | ||
| Fig. 7 Generation of intraluminal vesicular compartments by the core ESCRT membrane scission machinery. (I) Initial membrane bending by ESCRT-II (green cylinders) and seeding of ESCRT-III assembly. (II) The subunit Vps20 (red triangle) initiates Snf7 polymerisation and ESCRT-III propagation into a spiral filament (blue beaded assembly). The three-dimensional spiral topology of ESCRT-III induces further membrane invagination and defines the size of the vesicle neck. A capping subunit (purple cube, Vps24) stops filament elongation. (III) Neck constriction and scission of the intraluminal budded vesicle occurs via Vps4-mediated ESCRT-III polymer disassembly. The ATPase Vps4 binds the ESCRT-III filament via the anchor subunit (Vps2, golden bead) (IV) Full ESCRT-III disassembly in preparation for a second round of intraluminal vesicle formation (ILV). | ||
Much is known about the structure of the filament array of ESCRTs and how they sequester cargo into intraluminal vesicles, however it is only recently that reconstitution of MVB formation has been successfully reproduced. Elegant work using GUVs demonstrated a coordinated assembly of ESCRT complexes on phospholipid capable of inducing a region of negative curvature that initiates budding and stabilises the neck of a nascent vesicle. Ultimately, narrowing of the vesicle neck by gradual disassembly of the complex drives intraluminal vesicle fission. In analogy to BAR domains, ESCRT-III proteins exist in an autoinhibited state, which has to be released for initiating complex assembly. The enzyme VPS4 provides the mechano-chemical force necessary to activate ESCRT-III for assembly on the endosomal membrane, disassembly of the complex and ultimately recycling ESCRT subunits, returning them in the monomeric inhibited state. This assembly-disassembly cycle allows for further rounds of fission to occur thereby generating continuous cargo encapsulation.104–106
So far, we can only predict via computational modelling how ESCRT-III polymers generate inward budding in membranes. In the ‘dome’ model, ESCRT-III polymers assembled into a three-dimensional spiral define the necks of a nascent vesicle and provide the physical force to push the membrane.107 In the ‘coatless’ model, ESCRT complexes localise only at the neck region of nascent membrane buds and bend the membrane aided by inducing lateral lipid phase separation.108–110 Intriguingly, the high redundancy of ESCRT-III in Nature suggests that varying the content of different ESCRT-III subunits may give origins to different polymeric structures,111 possibly generating vesicles of different sizes.
Supporting membrane structure and dynamics
Changes in cell shape control fundamental processes such as cell division and cell motility. Building cells from scratch or creating transport systems mimicking cellular structures requires reconstitution of an organised cytoskeleton interacting with a membrane. Here we will review some of the reconstitution studies of cytoskeletal components for the purpose of investigating the mechanisms of membrane shaping.Keeping in shape and keeping active: the role of cytoskeleton in membrane deformation
The cytoskeleton provides membrane structures with shape control through mechanical modulation of networks of filamentous protein polymers. The major players in cellular shape change are the actin cytoskeleton that dynamically changes membrane morphology, and tubulin, which following polymerisation, supplies the tracks onto which membrane vesicles transport cargo across a larger organelle. Actin filamentous networks provide the necessary membrane tension for shaping membranes into protrusions and invaginations or simply to provide a framework for stability. Furthermore microtubules and actin filaments display directionality of polymerisation, a property conferred by their coupling to molecular motors.Experimental approaches employing supported lipid bilayers and liposomes have helped to shed light on the mechanism of membrane shaping through actin polymerisation. An increasing number of in vitro studies have demonstrated how to successfully reconstitute actin networks inside and outside liposomes112 or on lipid bilayers.113 A reconstituted actin cytoskeleton can be coupled to a vesicle's interior to provide mechanical shape and stability to the membrane compartment. For instance, spectrin, a cytoskeletal protein component of erythroid cells, can be reconstituted as a shell at the outer membrane of GUVs by addition of ATP, providing a rigid framework for vesicle shape.114 Co-encapsulation of myosin motors with actin filaments induces contraction of the meshwork, ultimately reproducing membrane contractility.115In vitro reconstitution studies of actin shells anchored outside or inside liposomes have revealed how the strength of anchoring to membranes controls cortical actin remodelling,116 providing a simple model for polarity of actin shells and mechanistic basis for a cell's movement. When actin layers are assembled and anchored outside liposomes, the action of molecular motors generates cortical tension, which results in opposite effects on the membranes. High tension causes cortical actin to retract to one side of the liposome, whereas low tension coupled with strong anchoring between actin and membranes, results in vesicles shrinkage. In a model where an actin shell is assembled and anchored inside liposome vesicles, actomyosin filaments retract away from the membrane if the strength of anchoring is weak, whereas they are pulled towards the membrane when the attachment of the cortex is strong.
The cytoskeleton is also crucial for facilitating membrane scission in cell division events. For instance, a reconstituted minimal complex of a bacterial homologue of tubulin (FtsZ), and a single accessory protein (FtsA) has been shown to be competent for cell division when incorporated into liposome vesicles.117 The beautiful simplicity of the bacterial division machinery demonstrates that the main requirement for membrane severing is an organised assembly of proteins, without the need for excess membrane.
However, cytoskeletal cortices do not only play an active role in moulding a cell's plasma membrane but they define shape changes of intracellular membrane compartments. Membrane tubulation is an event initiated either by proteins that can scaffold positive membrane curvature (such as COPI or BAR proteins), or generated by the pulling forces of molecular motors attached to localised regions of the organelle's membrane. Growth and retraction of membrane tubules from larger organelles is another mean of cargo transport in the membrane trafficking system of a cell, in addition to vesicles. This dynamic motion of tubular structures is due to the presence of molecular motors that can walk along the fast growing tip of cytoskeletal microtubules (plus end motors, e.g. kinesins) or in the opposite direction (minus end motors, e.g. dyneins). Reconstitution of membrane tubule ‘pulling’ processes has been reproduced using both cell extracts and minimal components.118 An alternative means to generate tubulation is to ‘push’ from the inside of a vesicle. Encapsulated microtubule components can create long protrusions when polymerisation is induced.119
Curvaceous models: membrane elasticity theory
Theoretical modeling combined with in vitro model systems is a powerful tool to characterise and quantify the physical properties of the protein components of this membrane remodelling toolbox. When the proteins or protein complexes reviewed above interact with membranes, they generate changes in the shape or structure of the membrane interface. Theoretical models to understand these processes usually use the Helfrich Hamiltonian.120 This is a phenomenological model for membrane elasticity proposed by Wolfgang Helfrich in 1973.121 The membrane is treated using a continuum model where the lipid bilayer is considered to be a fluid elastic sheet, i.e., the molecular scale details of the membrane structure are averaged out into bulk mechanical properties of the bilayer that determine its behaviour. This model also assumes that the membrane is a thin sheet such that the thickness of the membrane is very small compared to the lateral extent of the membrane surface. The Helfrich Hamiltonian (felastic) describes the elastic contribution (per unit area) to the free energy of the membrane,| felastic = fbend + fstretch | (1) |
The stretching term fstretch = τ, where τ is the surface tension of the membrane. This term is often neglected as the energy cost of stretching a lipid membrane is much greater than that of bending the membrane. Therefore, in many cases the stretching of the membrane is not considered to be significant. Membrane stretching can also be described by an elastic area expansion modulus (KA), where the change in free energy per unit area upon stretching the membrane Δfstretch = (KA/2)[ΔA/A0]2, where ΔA is the change in membrane area and A0 is the initial membrane area.
Typical values for KA for lipid membranes have been found to be in the range 0.1–0.2 J m−2, or 25–50kBT nm−2.122 It should be noted that lipid membranes are not particularly stable under stretching deformations; they rupture upon fractional area increases (ΔA/A0) between 2–5%.123–125
The bending contribution to the free energy of a membrane is therefore the most significant. This can be described by two mechanical moduli:
 | (2) |
It has been shown that values for the Gaussian modulus κG are constrained by the bending modulus κb such that −1 ≤ κG/κb ≤ 0.7κG is challenging to determine directly from experiments, but the few experimental studies of κG in amphiphile systems have found that κG ≈ −0.8κb.126,127 Conveniently, due to the Gauss–Bonnet theorem, the Gaussian curvature term in eqn 2 only contributes to the change in free energy of the system if the membrane undergoes a topological transition (e.g. fusion, fission, poration). The Gaussian elasticity term can therefore be ignored for bending of a membrane of fixed topology, i.e. when only considering the membrane bending capabilities of these proteins, prior to scission, merger or poration events that may occur later in the membrane remodelling process.
Eqn 2 can also often be further simplified due to the fact that the lipid composition of the membrane is symmetric across the membrane for almost all experimental model membrane systems. When topological changes in the membrane do occur, it is often useful to treat the elastic energies of the two lipid monolayers of the membrane separately with a finite spontaneous curvature, C0. However for bending at fixed topology, the elastic energy of the whole lipid bilayer is considered together and due to the symmetry of the lipid composition in each monolayer, C0 = 0. Therefore the total free energy of model membranes for bending at fixed topology can be simplified to
Felastic = ∫felastic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) dA = 2κb∫H2dA dA = 2κb∫H2dA | (3) |
An example of the application of theoretical models is the modelling of the interaction of F-BAR domains with lipid membranes.134 Combining simulations and membrane elasticity theory predicts an initial binding energy of an F-BAR dimer to the membrane of 4kBT, which reduces to 2.3kBT at equilibrium during membrane tubulation. Therefore this shows that single F-BAR dimers at low density on a membrane would not individually be able to generate enough energy to bend a membrane with a bending modulus of 20kBT. Multiple F-BAR dimers would be required to act in concert at high density on a membrane in order to manipulate its curvature, in agreement with experimental observations reviewed above.
Ultimately, it should be noted that although the Helfrich model considers the membrane to be a continuous sheet with no molecular details. However, the membrane mechanical moduli in this model can be directly related to the intermolecular interactions between lipids by considering the lateral stress profile in the membrane.135,136 This can allow consideration of protein-induced changes in the mechanical moduli of membranes due to the ways in which proteins modulate the interactions between lipids, for example by direct insertion of part of the protein complex into the membrane.137
Finally, it is worth highlighting that computational studies have also proved an extremely practical tool to provide suggestive insights into the molecular-scale mechanisms of membrane remodelling processes, and interactions between membranes and protein complexes. While the focus of this perspective is the in vitro reconstitution of membrane remodelling machineries, there is a wealth of literature in which computer simulation approaches have provided new mechanistic ideas impossible to gather by traditional experimental work. In addition to the work on the F-BAR domain discussed above, important contributions to the possible molecular mechanisms underlying membrane remodelling include simulations of the membrane interactions of N-BAR domains138,139 and SNARE proteins.140,141
Rationalisation of the toolbox for membrane remodelling
If natural protein components are to be used as tools to generate new functional synthetic cell-like materials, they will need to be easy to handle and compatible with other components when incorporated into more complex systems. It would be highly challenging to engineer synthetic cells from component parts that were not well characterised and simplified machineries. The highly elaborate nature of many of these natural protein complexes is designed for optimal function in complex in vivo cellular environments. This complexity is unlikely to be required when protein complexes are used as parts within minimal in vitro systems. There are two possible approaches to developing simplified minimal components for an in vitro membrane remodelling toolbox: (i) to use protein engineering to redesign the proteins as minimal systems without additional, redundant functionality; (ii) to take inspiration from the function of these proteins to design novel, synthetic components that perform the required in vitro tasks. Little work has been done to establish such minimal machineries but examples of success in this approach will be given below.Looking to the future: is the design of minimal systems for moulding membranes possible?
Minimal protein-based systems acting upon membranes are surely useful to investigate mechanisms of membrane remodelling but they also provide us with a framework for extracting basic design rules for function, which can then be employed for creating reduced systems. To date, several protein complexes have been simplified or even de novo designed for membrane fusion, fission and vesicle transport. The SNARE system for membrane fusion is certainly the most amenable to reproduction. SNAREs function on a simple principle: bringing membrane bilayers into close physical proximity by means of formation of an extremely stable protein complex. Bioinspired systems based on this principle have been created by de novo designing a minimal SNARE pair from complementary coiled coil peptides. The reduced SNARE system for membrane fusion is based on a simple heterodimeric coiled coil pair and has been subsequently refined by including moieties for membrane anchoring.142,143 Intriguingly, most protein complexes involved in membrane remodelling are held together by coiled coil interactions. For instance, the structural features of BAR domains are arrays of anti-parallel α-helical coiled-coils adopting curved shapes and using a positively charged surface to interact with lipids. The extensive knowledge we have acquired in the area of coiled coil design,144,145 including the computational-guided de novo design of protein assembly curvature,146 make the engineering of functional membrane sculpting systems more achievable. Coiled coil modules have already been extensively used as cytoskeleton biomimicry147 and recently, have been designed to self-assemble into organised cages resembling the structure of clathrin coats.148,149 It is not too far fetched to think that soon we could use these systems to design de novo membrane remodelling tools from the bottom up.Engineering of a minimal machinery that performs membrane fission in a controllable manner is however challenging. Any curvature-inducing protein system can eventually promote membrane fission at high densities. However, the creation of new vesicles in a controlled manner requires a multistep mechanism including factors to induce initial membrane bending (Clathrin and COPI coats and even actin filaments), entities to stabilise and promote further budding (BAR and ENTH domain-containing proteins) and regulatory factors that promote membrane fission mechanically-driven by nucleotide hydrolysis (dynamin). Since the development of the PURE system for in vitro protein expression, assembling a minimal machinery to recreate cell division using model membranes has moved a step closer. In situ expression of membrane deforming proteins using the PURE system has been shown to produce extensive membrane deformation in liposomes preparations upon the activity of expressed N-BAR domains.150
Initiation of membrane scission in a model vesicle, for mimicking a dividing cell, is a process that must begin from the lumen of the vesicle and requires proteins that can generate narrow necks and induce negative curvature, such as I-BAR proteins or ESCRT complexes. In situ expression of minimal ESCRT machineries inside GUVs or liposomes could be the first step towards recreating membrane-bound compartments competent for cell division. The drawback of using complexes such as ESCRTs is the high redundancy of the functional assembly, making reconstitution of such a system for large scale production (e.g. for biotechnology applications) challenging. Monocellular ancient organisms such as archea bacteria, for instance, utilise ESCRT complexes with a reduced number of components in comparison to yeast or humans, given their simpler metabolism. These ESCRTs modules can be regarded as a source or an inspiration for assembly of ‘divisomes’, or remodelling of membranes with appropriate lipid composition.151
Interesting developments in the design of minimal vesicular transport systems include coupling of liposomes to molecular motors capable of walking on molecular tracks. Liposomes coated with bundled actin have been used to build simple transport systems based on myosin motors.152,153 Complementary DNA strands are an efficient manner to anchor membrane vesicles to a kinesin molecule gliding on microtubules.154 In such system, loading, transport and unloading of fluorescent cargo-loaded liposomes can be reproduced using a flow-cell – tuning of the strength of binding between the DNA strands allows for vesicle unloading. Alternatively, coupling of cargo vesicles can be achieved via streptavidin–biotin linkage and quantum dots coupled to the trailing ends of actomyosin filaments via capping proteins.155
Inspired by the function of natural proteins, several synthetic alternatives have been shown to mimic properties of these proteins at lipid biomembranes. DNA nanotechnology is proving to be one useful tool in this approach.156 DNA–lipid conjugates have been successfully applied to mimic SNARE fusion proteins, membrane adhesion receptors, tetherins and transmembrane pores.157–160,161 Supramolecular chemistry has also provided synthetic organic macromolecules that can, for example, trigger membrane adhesion or poration.162,163 Synthetic nanoparticles may also prove useful mimics of membrane remodeling proteins in minimal systems. While much of the existing work on nanoparticle–membrane interactions was not motivated by biomimicry of natural membrane-active proteins, nanoparticles have been shown to be able to bend and scaffold the membranes of GUVs.164–167
Outlook: towards engineering functional protocells with minimal machineries
We have reviewed examples of the successful in vitro reconstitution of membrane remodeling protein complexes and shown how these minimal systems have provided new insight into their in vivo biological properties and functions and their regulation by other proteins. However towards the higher goal of using biological components to build synthetic cell-like materials, much progress needs to be made. Relatively few examples exist of successful engineering of minimal membrane remodeling machineries more suited to optimal function within model membrane systems. Further efforts are required to generate a toolbox of these specialised in vitro components. To the best of our knowledge, no work has been reported that successfully combines two or more of these minimal membrane machineries into a single system; compatibility of these components may prove challenging in order to mitigate mutual interference of membrane interactions and function. Furthermore, it would be of interest to demonstrate an increase in the complexity of these reconstituted minimal systems through coupling membrane remodelling processes with other functional biomolecular modules, such as mechanosensitive proteins and channels.168,169In order to develop advanced minimal cells by bottom-up assembly techniques that incorporate scaffolding, shaping and membrane vesicle trafficking capabilities, we suggest that a process of development described in Fig. 8 should be followed. Firstly, the components need to be characterised using biophysical investigative approaches. These components then need to be stripped back to minimal functional entities to provide maximum simplicity in their design and function. This process will require multiple iterations between protein complex redesign and biophysical characterisation. Thirdly, two or more independent membrane-active components need to be successfully reconstituted into model systems to demonstrate mutual compatibility and minimise unwanted interference between individual components. This will require the simplified, minimal components to behave in a predictable manner in how they interface with other components of the synthetic biology toolbox when combined together in systems of increasing complexity. Finally, incorporating metabolic processes within the compartments of synthetic cells can further increase complexity and functionality.
 | ||
| Fig. 8 The iterative process in the design of a minimal toolbox for membrane remodelling. | ||
Nature shows us how control of dynamic chemical compartmentalisation is required to regulate a large number of parallel processes within composite cellular systems; an important feature that synthetic biologists will wish to harness and exploit. The pathway to realising functional cell-like materials has become much clearer over the past 5–10 years with many significant advances in the understanding, design and application of in vitro model membrane–protein interaction systems. Now the next steps need to be taken in establishing minimal standardised parts and tools to generate compartmentalisation, and show that they can be successfully incorporated into organised functional multicomponent systems of increasing complexity.
Acknowledgements
We would like to thank the EPSRC Directed Assembly Network ‘Beyond the Molecule’ for facilitating and funding the initial collaboration between the authors. PAB would like to thank the E.U. Marie Curie career integration grant BioNanoMuTT (PCIG09-GA-2011-293643). The electron microscopy image for the graphical abstract was kindly gifted by Prof Philip Woodman (School of Life Science, University of Manchester). We also would like to apologise to the numerous authors whose work is not cited for reasons of space.References
- A. P. Liu and D. A. Fletcher, Nat. Rev. Mol. Cell Biol., 2009, 10, 644–650 CrossRef CAS PubMed.
- E. T. Castellana and P. S. Cremer, Surf. Sci. Rep., 2006, 61, 429–444 CrossRef CAS PubMed.
- M. Przybylo, J. Sykora, J. Humpolickova, A. Benda, A. Zan and M. Hof, Langmuir, 2006, 22, 9096–9099 CrossRef CAS PubMed.
- U. Bernchou, H. Midtiby, J. H. Ipsen and A. C. Simonsen, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2849–2858 CrossRef CAS PubMed.
- U. Bernchou, J. H. Ipsen and A. C. Simonsen, J. Phys. Chem. B, 2009, 113, 7170–7177 CrossRef CAS PubMed.
- D. Chen and M. M. Santore, Langmuir, 2014, 30, 9484–9493 CrossRef CAS PubMed.
- E. I. Goksu, J. M. Vanegas, C. D. Blanchette, W.-C. Lin and M. L. Longo, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 254–266 CrossRef CAS PubMed.
- V. D. Gordon, P. A. Beales, G. C. Shearman, Z. Zhao, J. M. Seddon, W. C. K. Poon and S. U. Egelhaaf, in Advances in Planar Lipid Bilayers and Liposomes, ed. A. Iglic and C. V. Kulkarni, 2014, vol. 20, pp. 137–154 Search PubMed.
- S. D. Connell, G. Heath, P. D. Olmsted and A. Kisil, Faraday Discuss., 2013, 161, 91–111 RSC.
- C. Das, K. H. Sheikh, P. D. Olmsted and S. D. Connell, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 82, 041920 CrossRef.
- G. E. Rydell, A. B. Dahlin, F. Hook and G. Larson, Glycobiology, 2009, 19, 1176–1184 CrossRef CAS PubMed.
- N.-J. Cho, C. W. Frank, B. Kasemo and F. Hook, Nat. Protoc., 2010, 5, 1096–1106 CrossRef CAS PubMed.
- A. Dahlin, M. Zach, T. Rindzevicius, M. Kall, D. S. Sutherland and F. Hook, J. Am. Chem. Soc., 2005, 127, 5043–5048 CrossRef CAS PubMed.
- J. M. Moran-Mirabal, J. B. Edel, G. D. Meyer, D. Throckmorton, A. K. Singh and H. G. Craighead, Biophys. J., 2005, 89, 296–305 CrossRef CAS PubMed.
- L. J. C. Jeuken, S. D. Connell, P. J. F. Henderson, R. B. Gennis, S. D. Evans and R. J. Bushby, J. Am. Chem. Soc., 2006, 128, 1711–1716 CrossRef CAS PubMed.
- M. L. Kraft, P. K. Weber, M. L. Longo, I. D. Hutcheon and S. G. Boxer, Science, 2006, 313, 1948–1951 CrossRef CAS PubMed.
- P. S. Cremer and S. G. Boxer, J. Phys. Chem. B, 1999, 103, 2554–2559 CrossRef CAS.
- V. Kiessling, C. Wan and L. K. Tamm, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 64–71 CrossRef CAS PubMed.
- J. Yuan, C. Hao, M. Chen, P. Berini and S. Zou, Langmuir, 2015, 29, 221–227 CrossRef PubMed.
- W.-C. Lin, C. D. Blanchette, T. V. Ratto and M. L. Longo, Biophys. J., 2006, 90, 228–237 CrossRef CAS PubMed.
- M. Tanaka and E. Sackmann, Nature, 2005, 437, 656–663 CrossRef CAS PubMed.
- M. L. Wagner and L. K. Tamm, Biophys. J., 2000, 79, 1400–1414 CrossRef CAS.
- S. Majd and M. Mayer, Angew. Chem., Int. Ed., 2005, 44, 6697–6700 CrossRef CAS PubMed.
- A. S. Achalkumar, R. J. Bushby and S. D. Evans, Soft Matter, 2010, 6, 6036–6051 RSC.
- N. Boden, R. J. Bushby, S. Clarkson, S. D. Evans, P. F. Knowles and A. Marsh, Tetrahedron, 1997, 53, 10939–10952 CrossRef CAS.
- M. C. Peterman, J. M. Ziebarth, O. Braha, H. Bayley, H. A. Fishman and D. M. Bloom, Biomed. Microdevices, 2002, 4, 231–236 CrossRef CAS.
- A. Studer, X. Han, F. K. Winkler and L. X. Tiefenauer, Colloids Surf., B, 2009, 73, 325–331 CrossRef CAS PubMed.
- O. Worsfold, N. H. Voelcker and T. Nishiya, Langmuir, 2006, 22, 7078–7083 CrossRef CAS PubMed.
- M. Tanaka, M. Tutus, S. Kaufmann, F. F. Rossetti, E. Schneck and I. M. Weiss, J. Struct. Biol., 2009, 168, 137–142 CrossRef CAS PubMed.
- A.-L. Troutier and C. Ladaviere, Adv. Colloid Interface Sci., 2007, 133, 1–21 CrossRef CAS PubMed.
- A. Nath, W. M. Atkins and S. G. Sligar, Biochemistry, 2007, 46, 2059–2069 CrossRef CAS PubMed.
- I. G. Denisov, Y. V. Grinkova, A. A. Lazarides and S. G. Sligar, J. Am. Chem. Soc., 2004, 126, 3477–3487 CrossRef CAS PubMed.
- T. K. Ritchie, Y. V. Grinkova, T. H. Bayburt, I. G. Denisov, J. K. Zolnerciks, W. M. Atkins and S. G. Sligar, in Single Molecule Tools, Pt A: Fluorescence Based Approaches, ed. N. Duzgunes, Methods in Enzymology, 2009, vol. 464, pp. 11–31 Search PubMed.
- H. Katayama, J. Wang, F. Tama, L. Chollet, E. P. Gogol, R. J. Collier and M. T. Fisher, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3453–3457 CrossRef CAS PubMed.
- T. Raschle, S. Hiller, T.-Y. Yu, A. J. Rice, T. Walz and G. Wagner, J. Am. Chem. Soc., 2009, 131, 17777–17779 CrossRef CAS PubMed.
- T. H. Bayburt and S. G. Sligar, FEBS Lett., 2010, 584, 1721–1727 CrossRef CAS PubMed.
- A. Nath, A. J. Trexler, P. Koo, A. D. Miranker, W. M. Atkins and E. Rhoades, in Single Molecule Tools, Pt A: Fluorescence Based Approaches, ed. N. G. Walter, Methods in Enzymology, 2010, vol. 472, pp. 89–117 Search PubMed.
- H. Bayley, B. Cronin, A. Heron, M. A. Holden, W. L. Hwang, R. Syeda, J. Thompson and M. Wallace, Mol. BioSyst., 2008, 4, 1191–1208 RSC.
- G. Maglia, A. J. Heron, W. L. Hwang, M. A. Holden, E. Mikhailova, Q. Li, S. Cheley and H. Bayley, Nat. Nanotechnol., 2009, 4, 437–440 CrossRef CAS PubMed.
- R. Syeda, M. A. Holden, W. L. Hwang and H. Bayley, J. Am. Chem. Soc., 2008, 130, 15543–15548 CrossRef CAS PubMed.
- J. Xu, T. K. Vanderlick and P. A. Beales, PLoS One, 2013, 8, e69492 CAS.
- J. R. Thompson, B. Cronin, H. Bayley and M. I. Wallace, Biophys. J., 2011, 101, 2679–2683 CrossRef CAS PubMed.
- F. Szoka and D. Papahadjopoulos, Annu. Rev. Biophys. Bioeng., 1980, 9, 467–508 CrossRef CAS PubMed.
- J. J. Benkoski and F. Hook, J. Phys. Chem. B, 2005, 109, 9773–9779 CrossRef CAS PubMed.
- Y.-H. M. Chan, P. Lenz and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18913–18918 CrossRef CAS PubMed.
- J. R. Wetterau and A. Jonas, J. Biol. Chem., 1982, 257, 961–966 Search PubMed.
- T. Wieprecht, M. Beyermann and J. Seelig, Biophys. Chem., 2002, 96, 191–201 CrossRef CAS.
- A. Tonnesen, S. M. Christensen, V. Tkach and D. Stamou, Biophys. J., 2014, 106, 201–209 CrossRef CAS PubMed.
- A. Tian and T. Baumgart, Biophys. J., 2009, 96, 2676–2688 CrossRef CAS PubMed.
- B. Sorre, A. Callan-Jones, J.-B. Manneville, P. Nassoy, J.-F. Joanny, J. Prost, B. Goud and P. Bassereau, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5622–5626 CrossRef CAS PubMed.
- B. Sorre, A. Callan-Jones, J. Manzi, B. Goud, J. Prost, P. Bassereau and A. Roux, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 173–178 CrossRef CAS PubMed.
- P. A. Beales, C. L. Bergstrom, N. Geerts, J. T. Groves and T. K. Vanderlick, Langmuir, 2011, 27, 6107–6115 CrossRef CAS PubMed.
- P. Girard, J. Pécréaux, G. Lenoir, P. Falson, J.-L. Rigaud and P. Bassereau, Biophys. J., 2004, 87, 419–429 CrossRef CAS PubMed.
- M. I. Angelova and D. S. Dimitrov, Faraday Discuss. Chem. Soc., 1986, 81, 303–311 RSC.
- M. I. Angelova, S. Soleau, P. Meleard, J. F. Faucon and P. Bothorel, Preparation of giant vesicles by external AC electric-fields – kinetics and applications, 1992, vol. 89 Search PubMed.
- L. R. Montes, A. Alonso, F. M. Goñi and L. A. Bagatolli, Biophys. J., 2007, 93, 3548–3554 CrossRef CAS PubMed.
- S. Manley and V. D. Gordon, Current Protocols in Cell Biology, John Wiley & Sons, Inc., 2001 Search PubMed.
- A. Weinberger, F.-C. Tsai, G. H. Koenderink, T. F. Schmidt, R. Itri, W. Meier, T. Schmatko, A. Schröder and C. Marques, Biophys. J., 2013, 105, 154–164 CrossRef CAS PubMed.
- S. Pautot, B. J. Frisken and D. A. Weitz, Langmuir, 2003, 19, 2870–2879 CrossRef CAS.
- S. Pautot, B. J. Frisken and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10718–10721 CrossRef CAS PubMed.
- H. C. Shum, D. Lee, I. Yoon, T. Kodger and D. A. Weitz, Langmuir, 2008, 24, 7651–7653 CrossRef CAS PubMed.
- L. R. Arriaga, S. S. Datta, S.-H. Kim, E. Amstad, T. E. Kodger, F. Monroy and D. A. Weitz, Small, 2014, 10, 950–956 CrossRef CAS PubMed.
- K. Karamdad, R. V. Law, J. M. Seddon, N. J. Brooks and O. Ces, Lab Chip, 2015, 15, 557–562 RSC.
- J. C. Stachowiak, D. L. Richmond, T. H. Li, A. P. Liu, S. H. Parekh and D. A. Fletcher, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4697–4702 CrossRef CAS PubMed.
- D. L. Richmond, E. M. Schmid, S. Martens, J. C. Stachowiak, N. Liska and D. A. Fletcher, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9431–9436 CrossRef CAS PubMed.
- Y. Elani, A. Gee, R. V. Law and O. Ces, Chem. Sci., 2013, 4, 3332–3338 RSC.
- Y. Elani, R. V. Law and O. Ces, Nat. Commun., 2014, 5, 5305 CrossRef CAS PubMed.
- P. G. Woodman and C. E. Futter, Curr. Opin. Cell Biol., 2008, 20, 408–414 CrossRef CAS PubMed.
- S. Martens and H. T. McMahon, Nat. Rev. Mol. Cell Biol., 2008, 9, 543–556 CrossRef CAS PubMed.
- M. K. Bennett, Curr. Opin. Cell Biol., 1995, 7, 581–586 CrossRef CAS.
- T. Söllner, M. K. Bennett, S. W. Whiteheart, R. H. Scheller and J. E. Rothman, Cell, 1993, 75, 409–418 CrossRef.
- T. Weber, B. V. Zemelman, J. A. McNew, B. Westermann, M. Gmachl, F. Parlati, T. H. Söllner and J. E. Rothman, Cell, 1998, 92, 759–772 CrossRef CAS.
- T. Stegmann, J. M. Delfino, F. M. Richards and A. Helenius, J. Biol. Chem., 1991, 266, 18404–18410 CAS.
- J. R. Monck and J. M. Fernandez, Curr. Opin. Cell Biol., 1996, 8, 524–533 CrossRef CAS.
- S.-J. Cho, M. Kelly, K. T. Rognlien, J. A. Cho, J. K. H. Hörber and B. P. Jena, Biophys. J., 2002, 83, 2522–2527 CrossRef CAS.
- M. Tsurudome, R. Glück, R. Graf, R. Falchetto, U. Schaller and J. Brunner, J. Biol. Chem., 1992, 267, 20225–20232 CAS.
- L. Shi, Q.-T. Shen, A. Kiel, J. Wang, H.-W. Wang, T. J. Melia, J. E. Rothman and F. Pincet, Science, 2012, 335, 1355–1359 CrossRef CAS PubMed.
- F. Parlati, T. Weber, J. A. McNew, B. Westermann, T. H. Söllner and J. E. Rothman, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12565–12570 CrossRef CAS.
- M. Zick, C. Stroupe, A. Orr, D. Douville, W. T. Wickner and A. T. Brunger, eLife, 2014, 3, e01879 Search PubMed.
- W. J. Cho, A. Jeremic, K. T. Rognlien, M. G. Zhvania, I. Lazrishvili, B. Tamar and B. P. Jena, Cell Biol. Int., 2004, 28, 699–708 CrossRef CAS PubMed.
- B. P. Jena, S.-J. Cho, A. Jeremic, M. H. Stromer and R. Abu-Hamdah, Biophys. J., 2003, 84, 1337–1343 CrossRef CAS.
- J. Diao, Z. Su, Y. Ishitsuka, B. Lu, K. S. Lee, Y. Lai, Y.-K. Shin and T. Ha, Nat. Commun., 2010, 1, 1–6 CrossRef PubMed.
- K. Bacia, C. G. Schuette, N. Kahya, R. Jahn and P. Schwille, J. Biol. Chem., 2004, 279, 37951–37955 CrossRef CAS PubMed.
- G. B. Melikyan, R. M. Markosyan, H. Hemmati, M. K. Delmedico, D. M. Lambert and F. S. Cohen, J. Cell Biol., 2000, 151, 413–424 CrossRef CAS.
- S. C. Harrison, Nat. Struct. Mol. Biol., 2008, 15, 690–698 CAS.
- R. M. Epand, Biochim. Biophys. Acta, 2003, 1614, 116–121 CrossRef CAS.
- B. Apellaniz, N. Huarte, E. Largo and J. L. Nieva, Chem. Phys. Lipids, 2014, 181, 40–55 CrossRef CAS PubMed.
- C. J. Thomas, S. Shankar, H. E. Casquilho-Gray, J. York, S. R. Sprang and J. H. Nunberg, PLoS One, 2012, 7, e51114 CAS.
- V. A. Frolov, A. Escalada, S. A. Akimov and A. V. Shnyrova, Chem. Phys. Lipids, 2014, 185, 129–140 CrossRef PubMed.
- M. Agromayor and J. Martin-Serrano, Trends Cell Biol., 2013, 23, 433–441 CrossRef CAS PubMed.
- S. Suetsugu, S. Kurisu and T. Takenawa, Physiol. Rev., 2014, 94, 1219–1248 CrossRef CAS PubMed.
- H. Zhao, A. Michelot, E. V. Koskela, V. Tkach, D. Stamou, D. G. Drubin and P. Lappalainen, Cell Rep., 2013, 4, 1213–1223 CrossRef CAS PubMed.
- J. C. Stachowiak, E. M. Schmid, C. J. Ryan, H. S. Ann, D. Y. Sasaki, M. B. Sherman, P. L. Geissler, D. A. Fletcher and C. C. Hayden, Nat. Cell Biol., 2012, 14, 944–949 CrossRef CAS PubMed.
- M. M. Kozlov, F. Campelo, N. Liska, L. V. Chernomordik, S. J. Marrink and H. T. McMahon, Curr. Opin. Cell Biol., 2014, 29, 53–60 CrossRef CAS PubMed.
- M. Simunovic, C. Mim, T. C. Marlovits, G. Resch, V. M. Unger and G. A. Voth, Biophys. J., 2013, 105, 711–719 CrossRef CAS PubMed.
- J. Saarikangas, H. Zhao, A. Pykäläinen, P. Laurinmäki, P. K. Mattila, P. K. J. Kinnunen, S. J. Butcher and P. Lappalainen, Curr. Biol., 2009, 19, 95–107 CrossRef CAS PubMed.
- B. Sorre, A. Callan-Jones, J. Manzi, B. Goud, J. Prost, P. Bassereau and A. Roux, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 173–178 CrossRef CAS PubMed.
- C. Zhu, S. L. Das and T. Baumgart, Biophys. J., 2012, 102, 1837–1845 CrossRef CAS PubMed.
- A. Roux, G. Koster, M. Lenz, B. Sorre, J.-B. Manneville, P. Nassoy and P. Bassereau, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4141–4146 CrossRef CAS PubMed.
- S. L. Goh, Q. Wang, L. J. Byrnes and H. Sondermann, PLoS One, 2012, 7, e51628 CAS.
- S. Neumann and S. L. Schmid, J. Biol. Chem., 2013, 288, 25119–25128 CrossRef CAS PubMed.
- J. H. Hurley and P. I. Hanson, Nat. Rev. Mol. Cell Biol., 2010, 11, 556–566 CrossRef CAS PubMed.
- S. Lata, G. Schoehn, J. Solomons, R. Pires, H. G. Göttlinger and W. Weissenhorn, Biochem. Soc. Trans., 2009, 37, 156–160 CrossRef CAS PubMed.
- M. A. Y. Adell, G. F. Vogel, M. Pakdel, M. Muller, H. Lindner, M. W. Hess and D. Teis, Nat. Cell Biol., 2014, 205, 400 Search PubMed.
- T. Wollert, C. Wunder, J. Lippincott-Schwartz and J. H. Hurley, Nature, 2009, 457, 172–177 CrossRef PubMed.
- T. Wollert and J. H. Hurley, Nature, 2010, 464, 864–869 CrossRef CAS PubMed.
- G. Fabrikant, S. Lata, J. D. Riches, J. A. G. Briggs, W. Weissenhorn and M. M. Kozlov, PLoS Comput. Biol., 2009, 5, e1000575 Search PubMed.
- B. Różycki, E. Boura, J. H. Hurley and G. Hummer, PLoS Comput. Biol., 2012, 8, e1002736 Search PubMed.
- M. Mercker and A. Marciniak-Czochra, Biophys. J., 2015, 108, 833–843 CrossRef CAS PubMed.
- E. Boura, V. Ivanov, L.-A. Carlson, K. Mizuuchi and J. H. Hurley, J. Biol. Chem., 2012, 287, 28144–28151 CrossRef CAS PubMed.
- A. G. Cashikar, S. Shim, R. Roth, M. R. Maldazys, J. E. Heuser, P. I. Hanson and W. Sundquist, eLife, 2014, 3, e02184 Search PubMed.
- C. Campillo, P. Sens, D. Köster, L.-L. Pontani, D. Lévy, P. Bassereau, P. Nassoy and C. Sykes, Biophys. J., 2013, 104, 1248–1256 CrossRef CAS PubMed.
- S. K. Vogel, F. Heinemann, G. Chwastek and P. Schwille, Cytoskeleton, 2013, 70, 706–717 CrossRef CAS PubMed.
- I. López-Montero, R. Rodríguez-García and F. Monroy, J. Phys. Chem. Lett., 2012, 3, 1583–1588 CrossRef.
- F.-C. Tsai, B. Stuhrmann and G. H. Koenderink, Langmuir, 2011, 27, 10061–10071 CrossRef CAS PubMed.
- K. Carvalho, F.-C. Tsai, F. C. Tsai, E. Lees, R. Voituriez, G. H. Koenderink and C. Sykes, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16456–16461 CrossRef CAS PubMed.
- M. Osawa and H. P. Erickson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11000–11004 CrossRef CAS PubMed.
- C. Leduc, O. Campàs, J.-F. Joanny, J. Prost and P. Bassereau, Biochim. Biophys. Acta, 2010, 1798, 1418–1426 CrossRef CAS PubMed.
- P. M. Shaklee, S. Semrau, M. Malkus, S. Kubick, M. Dogterom and T. Schmidt, ChemBioChem, 2010, 11, 175–179 CrossRef CAS PubMed.
- M. M. Kozlov, S. L. Leikin and V. S. Markin, J. Chem. Soc., Faraday Trans. 2, 1989, 85, 277–292 RSC.
- W. Helfrich, Z. Naturforsch., C: Biochem., Biophys., Biol., Virol., 1973, 28, 693–703 CAS.
- D. Marsh, Chem. Phys. Lipids, 2006, 144, 146–159 CrossRef CAS PubMed.
- M. L. Longo and H. V. Ly, in Methods in Molecular Biology, ed. A. M. Dopico, 2007, vol. 400, pp. 421–437 Search PubMed.
- W. Rawicz, K. C. Olbrich, T. McIntosh, D. Needham and E. Evans, Biophys. J., 2000, 79, 328–339 CrossRef CAS.
- H. V. Ly and M. L. Longo, Biophys. J., 2004, 87, 1013–1033 CrossRef CAS PubMed.
- R. H. Templer, B. J. Khoo and J. M. Seddon, Langmuir, 1998, 14, 7427–7434 CrossRef CAS.
- D. P. Siegel and M. M. Kozlov, Biophys. J., 2004, 87, 366–374 CrossRef CAS PubMed.
- T. Baumgart, B. R. Capraro, C. Zhu and S. L. Das, Annu. Rev. Phys. Chem., 2011, 62, 483–506 CrossRef CAS PubMed.
- J. C. M. Holthuis and A. K. Menon, Nature, 2014, 510, 48–57 CrossRef CAS PubMed.
- A. S. Wang, A. Kundu, B. Fong, J. Fitzgerald, B. Larijani and D. Poccia, Biol. Bull., 2013, 224, 218–226 CAS.
- G. K. Voeltz and W. A. Prinz, Nat. Rev. Mol. Cell Biol., 2007, 8, 258–264 CrossRef CAS PubMed.
- N. Ramakrishnan, P. B. S. Kumar and R. Radhakrishnan, Phys. Rep., 2014, 543, 1–60 CrossRef CAS PubMed.
- G. S. Ayton and G. A. Voth, Curr. Opin. Struct. Biol., 2009, 19, 138–144 CrossRef CAS PubMed.
- H. Yu and K. Schulten, PLoS Comput. Biol., 2013, 9, e1002892 CAS.
- D. Marsh, Biochim. Biophys. Acta, Rev. Biomembr., 1996, 1286, 183–223 CrossRef CAS.
- D. Marsh, Biophys. J., 2007, 93, 3884–3899 CrossRef CAS PubMed.
- D. Marsh, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 1545–1575 CrossRef CAS PubMed.
- H. Cui, C. Mim, F. X. Vázquez, E. Lyman, V. M. Unger and G. A. Voth, Biophys. J., 2013, 104, 404–411 CrossRef CAS PubMed.
- E. Lyman, H. Cui and G. A. Voth, Phys. Chem. Chem. Phys., 2011, 13, 10430–10436 RSC.
- M. Lindau, B. A. Hall, A. Chetwynd, O. Beckstein and M. S. P. Sansom, Biophys. J., 2012, 103, 959–969 CrossRef CAS PubMed.
- M.-P. Durrieu, P. J. Bond, M. S. P. Sansom, R. Lavery and M. Baaden, ChemPhysChem, 2009, 10, 1548–1552 CrossRef CAS PubMed.
- H. Robson Marsden and A. Kros, Angew. Chem., Int. Ed., 2010, 49, 2988–3005 CrossRef CAS PubMed.
- M. Rabe, C. Schwieger, H. R. Zope, F. Versluis and A. Kros, Langmuir, 2014, 30, 7724–7735 CrossRef CAS PubMed.
- D. N. Woolfson, G. J. Bartlett, M. Bruning and A. R. Thomson, Curr. Opin. Struct. Biol., 2012, 22, 432–441 CrossRef CAS PubMed.
- B. Ciani, S. Bjelic, S. Honnappa, H. Jawhari, R. Jaussi, A. Payapilly, T. Jowitt, M. O. Steinmetz and R. A. Kammerer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19850–19855 CrossRef CAS PubMed.
- K. Park, B. W. Shen, F. Parmeggiani, P.-S. Huang, B. L. Stoddard and D. Baker, Nat. Struct. Mol. Biol., 2015, 22, 167–174 CAS.
- A. L. Boyle and D. N. Woolfson, Supramolecular Chemistry: From Molecules to Nanomaterials, 2012 Search PubMed.
- J. M. Fletcher, R. L. Harniman, F. R. H. Barnes, A. L. Boyle, A. Collins, J. Mantell, T. H. Sharp, M. Antognozzi, P. J. Booth, N. Linden, M. J. Miles, R. B. Sessions, P. Verkade and D. N. Woolfson, Science, 2013, 340, 595–599 CrossRef CAS PubMed.
- H. Gradišar, S. Božič, T. Doles, D. Vengust, I. Hafner-Bratkovič, A. Mertelj, B. Webb, A. Šali, S. Klavžar and R. Jerala, Nat. Chem. Biol., 2013, 9, 362–366 CrossRef PubMed.
- Z. Nourian, A. Scott and C. Danelon, Syst. Synth. Biol., 2014, 8, 237–247 CrossRef PubMed.
- T. Härtel and P. Schwille, Front. Microbiol., 2014, 5, 257 Search PubMed.
- S. Iwabuchi, T. Takahashi and K. Hatori, Biochem. Biophys. Res. Commun., 2012, 422, 164–168 CrossRef CAS PubMed.
- H. Takatsuki, K. M. Rice, S. Asano, B. S. Day, M. Hino, K. Oiwa, R. Ishikawa, Y. Hiratsuka, T. Q. Uyeda and K. Kohama, Small, 2010, 6, 452–457 CrossRef CAS PubMed.
- S. Hiyama, Y. Moritani, R. Gojo, S. Takeuchi and K. Sutoh, Lab Chip, 2010, 10, 2741 RSC.
- M. Persson, M. Gullberg, C. Tolf, A. M. Lindberg, A. Månsson and A. Kocer, PLoS One, 2013, 8, e55931 CAS.
- P. A. Beales and T. K. Vanderlick, Adv. Colloid Interface Sci., 2014, 207, 290–305 CrossRef CAS PubMed.
- L. Simonsson, M. E. Kurczy, R. Trouillon, F. Hook and A.-S. Cans, Sci. Rep., 2012, 2, 824 Search PubMed.
- P. A. Beales and T. K. Vanderlick, Biophys. J., 2009, 96, 1554–1565 CrossRef CAS PubMed.
- J. R. Burns, E. Stulz and S. Howorka, Nano Lett., 2013, 13, 2351–2356 CrossRef CAS PubMed.
- D. Serien, C. Grimm, J. Liebscher and A. Herrmann, New J. Chem., 2014, 38, 5181–5185 RSC.
- M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932–936 CrossRef CAS PubMed.
- C. P. Wilson, C. Boglio, L. Ma, S. L. Cockroft and S. J. Webb, Chem. – Eur. J., 2011, 17, 3465–3473 CrossRef CAS PubMed.
- R. J. Mart, K. P. Liem, X. Wang and S. J. Webb, J. Am. Chem. Soc., 2006, 128, 14462–14463 CrossRef CAS PubMed.
- S. Zhang, A. Nelson and P. A. Beales, Langmuir, 2012, 28, 12831–12837 CrossRef CAS PubMed.
- A. H. Churchman, R. Wallace, S. J. Milne, A. P. Brown, R. Brydson and P. A. Beales, Chem. Commun., 2013, 49, 4172–4174 RSC.
- Y. Yu and S. Granick, J. Am. Chem. Soc., 2009, 131, 14158–14159 CrossRef CAS PubMed.
- B. Wang, L. Zhang, S. C. Bae and S. Granick, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18171–18175 CrossRef CAS PubMed.
- E. S. Haswell, R. Phillips and D. C. Rees, Structure, 2011, 19, 1356–1369 CrossRef CAS PubMed.
- J. Árnadóttir and M. Chalfie, Annu. Rev. Biophys., 2010, 39, 111–137 CrossRef PubMed.
- D. Axelrod, D. E. Koppel, J. Schlessinger, E. Elson and W. W. Webb, Biophys. J., 1976, 16, 1055–1069 CrossRef CAS.
- D. M. Soumpasis, Biophys. J., 1983, 41, 95–97 CrossRef CAS.
- C. Eggeling, C. Ringemann, R. Medda, G. Schwarzmann, K. Sandhoff, S. Polyakova, V. N. Belov, B. Hein, C. von Middendorff, A. Schoenle and S. W. Hell, Nature, 2009, 457, 1159–1162 CrossRef CAS PubMed.
- A. Honigmann, V. Mueller, H. Ta, A. Schoenle, E. Sezgin, S. W. Hell and C. Eggeling, Nat. Commun., 2014, 5, 5412 CrossRef CAS PubMed.
- Q. Q. Ruan, M. A. Cheng, M. Levi, E. Gratton and W. W. Mantulin, Biophys. J., 2004, 87, 1260–1267 CrossRef CAS PubMed.
- E. Haustein and P. Schwille, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 151–169 CrossRef CAS PubMed.
- J. Korlach, P. Schwille, W. W. Webb and G. W. Feigenson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8461–8466 CrossRef CAS.
- P. Schwille, J. Korlach and W. W. Webb, Cytometry, 1999, 36, 176–182 CrossRef CAS.
- T. Weidemann, J. Muecksch and P. Schwille, Curr. Opin. Struct. Biol., 2014, 28, 69–76 CrossRef CAS PubMed.
- S. Ramadurai, A. Holt, V. Krasnikov, G. van den Bogaart, J. A. Killian and B. Poolman, J. Am. Chem. Soc., 2009, 131, 12650–12656 CrossRef CAS PubMed.
- S. Bleicken, M. Otsuki and A. J. Garcia-Saez, Curr. Protein Pept. Sci., 2011, 12, 691–698 CrossRef CAS.
- K. Olbrich, W. Rawicz, D. Needham and E. Evans, Biophys. J., 2000, 79, 321–327 CrossRef CAS.
- S. D. Shoemaker and T. K. Vanderlick, Biophys. J., 2003, 84, 998–1009 CrossRef CAS.
- Y. Z. Yoon, J. P. Hale, P. G. Petrov and P. Cicuta, J. Phys.: Condens. Matter, 2010, 22, 062101 CrossRef PubMed.
- T. Baumgart, S. Das, W. W. Webb and J. T. Jenkins, Biophys. J., 2005, 89, 1067–1080 CrossRef CAS PubMed.
- J. Henriksen, A. C. Rowat and J. H. Ipsen, Eur. Biophys. J. Biophys. Lett., 2004, 33, 732–741 CrossRef CAS PubMed.
- J. Pecreaux, H. G. Dobereiner, J. Prost, J. F. Joanny and P. Bassereau, Eur. Phys. J. E: Soft Matter Biol. Phys., 2004, 13, 277–290 CrossRef CAS PubMed.
- S. Semrau, T. Idema, L. Holtzer, T. Schmidt and C. Storm, Phys. Rev. Lett., 2008, 100, 088101 CrossRef.
- T. Betz and C. Sykes, Soft Matter, 2012, 8, 5317–5326 RSC.
- G. Fuertes, A. J. Garcia-Saez, S. Esteban-Martin, D. Gimenez, O. L. Sanchez-Munoz, P. Schwille and J. Salgado, Biophys. J., 2010, 99, 2917–2925 CrossRef CAS PubMed.
- P. Schoen, A. J. Garcia-Saez, P. Malovrh, K. Bacia, G. Anderluh and P. Schwille, Biophys. J., 2008, 95, 691–698 CrossRef CAS PubMed.
- C. L. Bergstrom, P. A. Beales, Y. Lv, T. K. Vanderlick and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6269–6274 CrossRef PubMed.
- S. Aimon, J. Manzi, D. Schmidt, J. A. Poveda Larrosa, P. Bassereau and G. E. S. Toombes, PLoS One, 2011, 6, e25529 CAS.
- T. Baumgart, G. Hunt, E. R. Farkas, W. W. Webb and G. W. Feigenson, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 2182–2194 CrossRef CAS PubMed.
- L. A. Bagatolli and E. Gratton, Biophys. J., 1999, 77, 2090–2101 CrossRef CAS.
- T. Parasassi, E. K. Krasnowska, L. Bagatolli and E. Gratton, J. Fluoresc., 1998, 8, 365–373 CrossRef CAS.
- M. Lamblet, B. Delord, L. Johannes, D. van Effenterre and P. Bassereau, Eur. Phys. J. E: Soft Matter Biol. Phys., 2008, 26, 205–216 CrossRef CAS PubMed.
Footnote |
| † Synthetic biology is defined as “(a) the design and construction of new biological parts, devices and systems and (b) the re-design of existing natural biological systems for useful purposes.” |
| This journal is © the Owner Societies 2015 |