
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioinspired nanoreactors for the biomineralisation of metallic-based nanoparticles for nanomedicine
Jennifer
Bain
and
Sarah S
Staniland
*
Department of Chemistry, University of Sheffield, Brook Hill, Sheffield S3 7HF, UK. E-mail: s.s.staniland@sheffield.ac.uk
First published on 2nd April 2015
Abstract
This review explores the synthesis of inorganic metallic-based nanoparticles (MBNPs) (metals, alloys, metal oxides) using biological and biologically inspired nanoreactors for precipitation/crystallisation. Such nanoparticles exhibit a range of nanoscale properties such as surface plasmon resonance (nobel metals e.g. Au), fluorescence (semiconductor quantum dots e.g. CdSe) and nanomagnetism (magnetic alloys e.g. CoPt and iron oxides e.g. magnetite), which are currently the subject of intensive research for their applicability in diagnostic and therapeutic nanomedicine. For such applications, MBNPs are required to be biocompatible, of a precise size and shape for a consistent signal or output and be easily modified with biomolecules for applications. Ideally the MBNPs would be obtained via an environmentally-friendly synthetic route. A biological or biologically inspired nanoreactor synthesis of MBNPs is shown to address these issues. Biological nanoreactors for crystallizing MBNPs within cells (magnetosomes), protein cages (ferritin) and virus capsids (cowpea chlorotic mottle, cowpea mosaic and tobacco mosaic viruses), are discussed along with how these have been modified for applications and for the next generation of new materials. Biomimetic liposome, polymersome and even designed self-assembled proteinosome nanoreactors are also reviewed for MBNP crystallisation and further modification for applications. With the advent of synthetic biology, the research and understanding in this field is growing, with the goal of realising nanoreactor synthesis of MBNPs for biomedical applications within our grasp in the near future.
Introduction
Applications of metallic-based nanoparticles (MBNPs)
The field of metallic-based (metals, alloys and metal oxides) nanoparticles (MBNPs) is becoming ever more prominent due to the emergence of numerous applications across a wide variety of disciplines and fields. Perhaps the most prominent application, outlined in many recent nanoparticle and nanoreactor publications, is in the field of biomedicine. MBNPs display a range of useful nanoproperty characteristics; namely nanomagnetism for magnetic nanoparticles (MNPs), fluorescence for semiconductor quantum dots (QDs) and increased photon scattering and absorbance due to surface plasmon resonance (SPR) for nanoparticles (NPs) of nobel metals. These characteristics are extremely applicable in the field of biomedicine. For example the incorporation of magnetism into a NP designed for drug delivery allows this to be magnetically directed to the site, making it a targeted therapy, which would then require lower dosage and reduce toxicity to the rest of the body. Furthermore, such magnetic character can open up the possibility of further application as a diagnostic agent. MNPs are excellent diagnostic tools in magnetic resonance imaging (MRI), as the interaction of their local field with the MRI magnetic field can significantly enhance contrast in vivo. This is achieved by the MNP having a shorter T2 (spin–spin) relaxation time relative to the surrounding tissue, hence improving contrast between MNP tagged and untagged cells.1,2 The effect on relaxation time can be controlled by control of the MNP size, which in turn also controls magnetic properties, as more magnetic material will have increased T2 relaxation rates and thus provide greater contrast.2 Similarly, magnetic particle imaging (MPI) maps the position in vivo of magnetic tracer particles.3 Using this technique it is possible to image these tracer particles with an acquisition time of <0.1 s.3 Although as yet un-optimised, MPI is expected to be a more cost effective alternative to current techniques such as MRI.3 MNPs can also be utilised for magnetic hyperthermia therapy which relies on an alternating magnetic field, which switches the individual spins (for larger single-domain MNPs) or physically flips the whole particle (for smaller superparamagnetic MNPs). This internal or external magnetic switching can induce heating which is transferred to the surrounding area, damaging or killing the diseased tissue.4–6 There are thus two different mechanisms for heating that are highly dependent on the magnetic nanoparticle size, but within each regime the heating is dependent on the magnetic saturation and coercivity of the MNPs as well as the alternating field frequency. In the case of superparamagnetic NP which can physically rotate in the field, the interaction of the MNP's with the surrounding tissue can induce heating as the result of friction. Surface plasmon resonance (SPR) is exhibited by nobel metal NPs and can have similar potential in medical diagnostics and therapeutics. Although most commonly observed in gold or silver NPs any metal or alloy which has a large negative real dielectric constant can be tuned to display an SPR. The SPR effect is induced by irradiation with light. This irradiation causes conduction electrons driven by the electric field to oscillate at a specific resonant frequency which is dependent on the particle composition, size and structure. Similarly to MNPs, NPs with SPR characteristics can be used in heat dissipation, due to the conversion of photon energy to phonon (lattice vibrational) energy when exposed to light at a specific wavelength. The heat created by this conversion can then dissipate into surrounding tissues in vivo in a process analogous to magnetic hyperthermia treatment. Furthermore by controlling the nanoparticle structure and shape (i.e. rod, shell, particle) this plasmonic effect can be tuned to be in the visible or near infrared region, making a valuable biosensor for binding kinetics or to enhance contrast in techniques such as optical coherence tomography, used in non-invasive biopsies.7,8 Size tuneable photoluminesence is also achievable with quantum dots (QDs). QDs are semiconductor NPs whose excitations are in three spatial dimensions. This unique characteristic offers properties which again have the potential to vastly improve biosensing and imaging in medicine. QDs have an extremely broad absorption and narrow emission band which is tuneable with QD size. This means they offer superior fluorescence, lifetime and resistance to bleaching when compared to more conventional fluorescent biomarkers such as green fluorescent protein, and have proven to be successfully funtionalised, by conjugation to biological moieties. QDs are perceived to be inherently toxic due to their composition, which generally includes heavy metals such as cadmium. However work is ongoing to move away from these issues by investigating less toxic materials for QDs and using new synthesis methods discussed in this review to lessen toxic effects so their bioimaging potential can be realised.9,10 All of these applications particularly those in vivo demand strict control with respect to the particle properties and thus the composition, size and shape, but in many cases this can be hard to achieve using conventional synthetic routes, often involving extreme reaction conditions and toxic reagents. Much of the work to develop precise MBNPs is being carried out with the ultimate goal of realisation in medicine applications.6,11 Nanocomponents hold potential for improvement of drug delivery techniques,12–14 hyperthermic treatment of tumours,5,6 creation of diagnostic toolkits; e.g. MRI contrast agents6,15,16 and simultaneous diagnostic and therapeutic nanomaterials; termed theranostics.15,17–19 This drive for use of MBNPs in biomedical applications dictates a requirement for an extremely high level of consistency and reproducibility during MBNP synthesis with respect to the NP properties discussed above, not only from particle to particle but also from batch to batch. The need for tight constraint is a consequence of utilising nanoscale physical properties for in vivo application. Ideally, employing a synthetic route that operates at the nanoscale will offer higher levels of nanoscale precision over the process.Synthesis of metallic-based nanoparticles (MBNPs)
The mineralisation of MBNPs occurs when insoluble NPs are precipitated out of solution from soluble precursors. This is achieved when metal salts are oxidised (in the case of metal oxides) or reduced (in the case on metals, alloys and semiconductors). Controlling this process can be challenging due to factors such as multiple intermediate phases, unwanted oxidation, side reactions and by-products that can all contaminate the final product. Furthermore, crystallisation (nucleation and crystal growth) is governed by a complex interplay of both thermodynamic and kinetic factors that render the process and thus the resulting particle size and morphology of the population, particularly sensitive to reaction conditions. The best means of controlling the particle uniformity with respect to size and shape is to ensure simultaneous nucleation, via increased temperature and/or initiate an instant burst of nucleation by injection of a precursor into the hot reaction solution (hot injection). Such methods have been used to synthesis a range of monodispersed QDs20 and magnetic nanoparticles (MNPs).21 However, such synthetic routes, involve high temperature, toxic precursors and organic solvents, they are economically unfavourable, labour intensively and environmentally costly, particularly in a climate striving for greener chemical synthetic routes.22 To demonstrate the variation of NP products with different synthetic methods, we will consider the mineralisation of the iron oxide magnetite. Magnetite is an inverse spinel crystal lattice that contains iron in both ferrous and ferric oxidation states. This balance of oxidation states can easily be tipped to favour a range of competing iron oxides produced by subtly changing the reaction conditions such as temperature, precursor oxidation states, ratio, anions present, atmosphere etc. This can affect changes in the particle properties particularly its magnetism, but also the size and shape of the resulting crystal which may be critical to certain applications. There are a number magnetite precipitation routes, each having their advantages and disadvantages as well as different end crystal morphology.23 For example room temperature co-precipitation methods are green, but produce small ill-defined particles over a very large size distribution and are liable to contain contaminant iron oxides. Further to this, a precipitation method of heating a ferrous iron precursor produces larger more uniformed octahedral particles but the size distribution is still relatively broad. Contrarily, there are numerous high temperature methods which produce particles of defined size and shape but involve harsher chemical and physical conditions.21,23–25 Similar challenges can also be seen in the synthesis of the other MBNPs, such as QDs10 and gadolinium MRI contrast agents.26Conducting a chemical reaction within a confined space or compartment (termed nanoreactors) can alleviate such issues, especially if we consider biological nanoreactors.
Compartmentalisation in nature
Broadly speaking there are two branches to the field of synthetic biology: top-down and bottom-up. Top-down is an extension of biotechnology into cellular metabolic engineering, which has sought to explain biological observations from a systems perspective in order to design and engineer new processes within a living organism. This methodology has distinct advantages: the most compelling being that adaption of a living organism intrinsically offers the benefits of a living system such as growth and regeneration. Furthermore, the products are intrinsically biocompatible (an essential property for biomedical application), while the research also serves to further our understanding of the mechanisms of these systems and processes in vivo. However, there are limitations when one considers the extent to which a process can deviate from the natural. i.e. the process must be within the boundaries of what is possible within a living cell.
By contrast, the bottom-up methodology has its roots in the fields of bionanoscience and protocells. It focuses on the individual building block components and using these and mimics thereof to design and build completely new artificial biological systems from first principles. Much of the work in the field is based around the design of entirely synthetic components such as vesicle compartments and incorporation of processes usually only observed in cells into these structures, summarised in the reviews by Vriezema and Dzieciol.28,30 The advantages to this methodology are that it allows for complete control over each element in the system and the creation of novel materials, far removed from natural products, which not only aids our understanding of biological processes ex vivo but also opens up opportunities for broader application of such systems. Here the key challenges are in biocompatibility of the products and scale-up of the processes.
For both disciplines, the engineering of complex systems is based on the principle of reuse and mimicry of components, tools and methodologies in a systematic way to achieve desired functionality of new (unnatural) systems. The flagship example of synthetic biology that defines what has been learnt from the top-down principle and subsequently uses the bottom up methodology to build a synthetic cell or protocell, which has been extensively explored in multiple other discussions.29,30,32,33 The field has been reinforced by pioneering research by those such as Venter et al., responsible for the synthetic bacterial cell; which has proved hugely advocatory for synthetic biology in the public domain.34
While much of the work in the field of synthetic biology is outside the scope of this review, parallels can be drawn between the emergence of synthetic biology and the biomimetic routes to mineralisation within biological and bioinspired nanoreactors discussed throughout this review.
(1) Heterogeneous nucleation is aided by providing a surface, in this case the membrane of the nanoreactor, for metal ions to nucleate on. This means that the concentration of metal ions does not need to be at supersaturation levels for nucleation to commence. Furthermore the surface of the nanoreactor can be designed around the location of nucleation sites to promote specific crystal growth.36–38 Once crystal growth is underway, specifically designed active sites or proteins in the membrane can interact with the growing crystal to block growth on specific crystal faces, thus controlling the particle morphology.37
(2) The overall nanoreactor structure provides a perfect template for NP morphology. The nucleation and propagation steps of crystallization occurs within the confinement of the nanoreactor boundaries, leading to precise sized and morphologically monodisperse populations of particles.39 Furthermore and less obvious, biotemplating can be achieved on the exterior of the structure to form precise mineralised shell MBNPs, which will also be briefly reviewed.
(3) The compartment provides control over the form and availability of precursors and thus affects the reaction and resulting NPs produced. This is a particularly sensitive reaction parameter, with outcomes changing significantly even between different forms of compartmentalisation.28 Studies of crystal growth in confined volumes40 and substrates41 confirm this hypothesis. The crystallisation of calcium carbonate both in picolitre droplets and in an angular wedge showed that for a finite concentration, slower crystallisation rates and shorter precipitation time resulted in higher quality particles. These studies also show that removal of the confined environment can reduce mineral stability. This sentiment is echoed by intravesicular biomineralisation study which notes that cells can kinetically stabilise internal growth of minerals.42
(4) Taking a biomimetic approach to the crystallisation of MBNPs inherently requires greener environmental conditions, with reactions occurring under ambient conditions in aqueous medium. Bulk synthetic reactions performed under these conditions lead to poor particle composition, uniformity and quality, however formation within a biological nanoreactor results in high quality monodispersed NPs for the reasons outlined above, removing the need for harsh solvent or high temperatures.
(5) The biomineralisation of MBNPs in a nanoreactor leads to biocompatible materials that are ready for biomedical applications, without an express need for further processing. For example, precipitation of magnetite within the vesicle would make it responsive in a magnetic field, or compartmentalisation of QD synthesis removes toxicity and functionalises a vesicle for fluorescence detection. Additionally, if novel therapeutics and targets were required, the biocompatible nanoreactors can easily be further functionalized. Furthermore, as well as templating, the nanoreactors could be further functionalised by encapsulating different NPs for multimodal use.43
Therefore many groups, are working towards the development and improvement of nanoreactor design and biomineralisation reactions for in situ precipitation of MBNPs. This can be achieved by a number of different approaches. One uses a more top-down “biokleptic” approach by adapting nanoreactors found in nature, such as protein cages44,45 and viruses.46 Another uses cellular vesicles as bio-inspiration for the design and synthesis of artificial vesicle nanoreactors from the bottom-up. Vesicles can self-assemble from almost any amphiphilic material, by numerous routes47–50 and in certain cases assembly is possible from a non-amphiphilic starting material51 allowing for a wide variety of properties to be incorporated. Most commonly, vesicles are formed from lipid molecules (liposomes) or specifically designed amphiphilic polymers (polymersomes). Such building blocks can also be controlled to self-assemble into structures such as micelles or nanotubes. Vesicles are suitable nanoreactors for a wide variety of purposes, such as incorporation and functionality of membrane proteins, encapsulation of therapeutics for in vivo drug delivery and as analytical tools in a range of fields, which have been widely discussed in numerous other comprehensive reviews.17,48,49,52
Other reviews have outlined both the properties and potential application of both naturally occurring and synthetically engineered nanoreactors.28,53–55 The concept covered in this review is to use biological nanoreactors (cells, protein cages, viruses) or seek inspiration from nature to design biomimetic nanoreactors (liposomes, polymersomes) which can compartmentalise the biomineralisation of MBNPs that result in more precise particle sizes and morphology under milder reaction conditions. Such biocompatible vesicle material can then be further functionalised for a wide variety of biomedical application.
The review
Biomineralisation in natural organisms
Compartmentalised biomineralisation reactions occurs in numerous organisms ranging from humans to algae.56 Biomineralisation is the crystallisation or precipitation of an inorganic material within or around a cell or organism.57 There are two distinct types of biomineralisation, the first is “biologically induced mineralisation” in which the precipitation of a mineral is a by-product of a biochemical process. In these cases there is often little to no control over the mineral formed. The second type is “biologically controlled mineralisation” whereby the organism deliberately forms the mineral to fulfil a function (i.e. protective (shell), structural (skeleton), attack and defence (claws and teeth)). In this case the minerals are intricately designed with respect to requirements such as size, shape, toughness etc. Transmembrane metal ion transport proteins or morphological controlling proteins are commonly utilised to control crystallisation of the resulting particles with superior properties when compared to equivalent synthesised particles.These mechanisms can be seen in the precipitation of a wide variety of minerals the vast expanse of which cannot be discussed thoroughly as part of this review. However some key examples include the biomineralisation of calcite, which highlights the controlled, complex and often beautiful architectures that can be achieved by nature. Such architectures can be seen in multiple species of mollusc and coral which biomineralise their hard outer shell; vital for protection. The coccolith is also an excellent example of this process; formed in intracellular vesicles and subsequently exorcised, these structures comprise individual biomineralised CaCO3 plates which then assemble into spheres (Fig. 1i).57–59 Coccoliths have provided a model system for the study of biomineralisation mechanisms and are indicative of the high levels of complexity that can be achieved if one adopts a bioinspired method. This sentiment is reflected in the lesser studied biomineralisation of silicates in diatom algae, again extensive and complex architectures are formed by the polymerisation of biogenic silica, similarly used as armour for the organism's survival.60 Understanding the mechanism by which these organisms biomineralise can inform new routes to biomimetic and bioinspired synthesis of novel and improved materials.61–63 Biomineralisation can vary in size from nanometres to metres (when one considers biomineralisation of bones in large animals). However, in the context of this review compartments for biomineralisation tend to fall in the range of 8 nm up to 2 μm diameters, indicating nanoreactors can be utilised for the precipitation of monodisperse minerals over a range of sizes.28,35,64,65
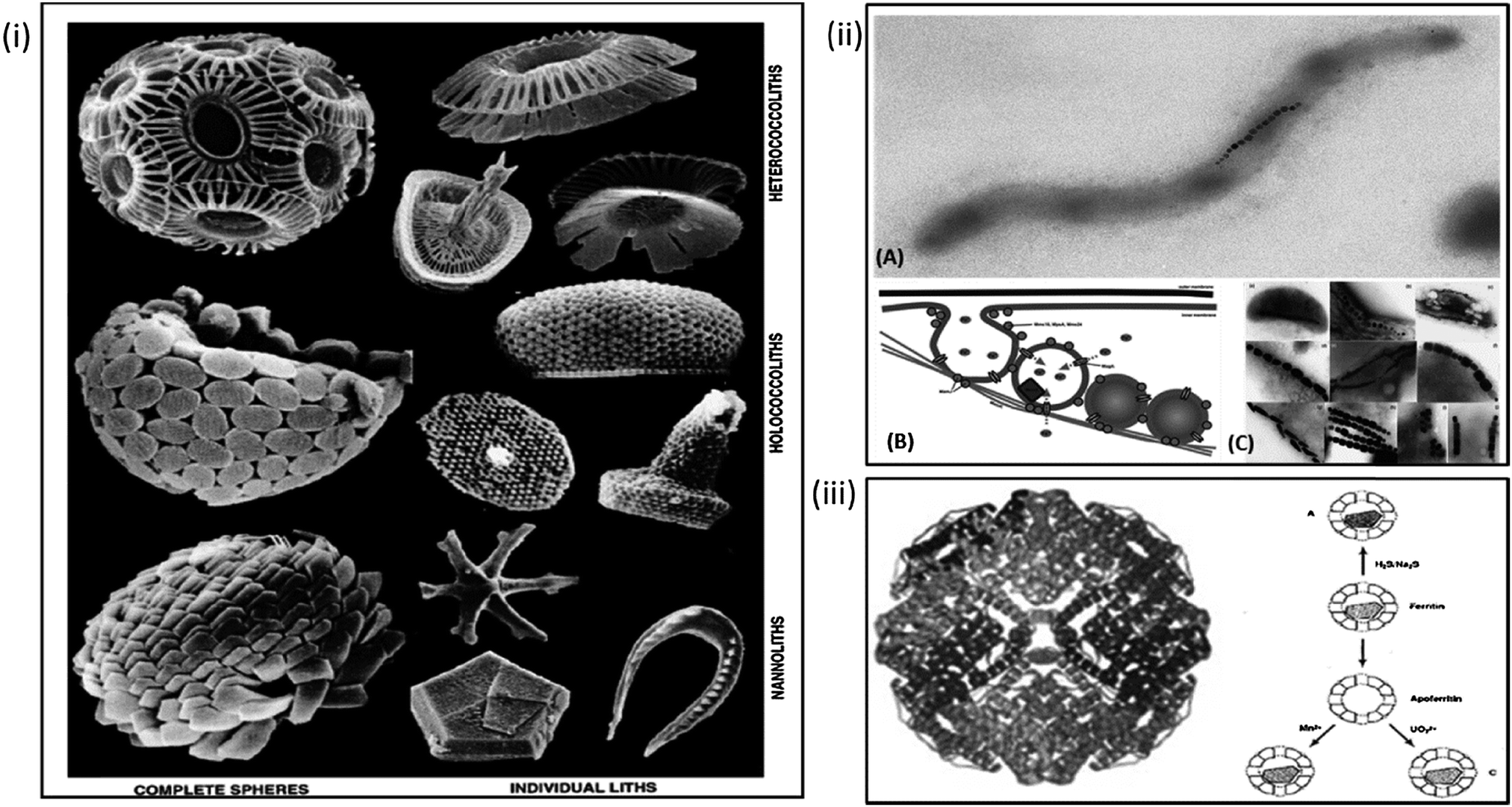 | ||
| Fig. 1 Natural biomineralisation within organisms. Clockwise from left: (i) architectures observed in nature as the result of calcium carbonate biomineralisation in coccoliths. (Adapted with permission from Young et al.59); (ii) formation and biomineralisation of the magnetosome in magnetic bacteria. (A) Example of magnetospirillum magnetotacticum MS-1. (B) Schematic of formation of the magnetosome vesicle, recruitment of iron and morphological control exerted by bacterial proteins in (reproduced with permission from Arakaki et al.72). (C) The various particle morphologies observed in different species of magnetic bacteria (reproduced with permission from Schuler, FEMS Microbiology rev. 2008). (iii) Structure of iron storage protein ferritin and possible biomineralisation pathways of ferritin and apoferritin as suggested by Mann et al. (Reproduced with permission from Ueno et al.63 and Mann et al.97). | ||
Crystallisation of magnetite is perhaps one of the most common forms of metal oxide biomineralised materials found in nature. examples of this process are wide ranging, with biomagnetite being found everywhere from chiton teeth to the NPs within the brains of insects, birds and humans.56,66 Arguably the most intricate and well-studied example of magnetite biomineralisation is the formation of the magnetosome in magnetic bacteria (MTB).67
Sufficient ferrous concentrations are only reached in MTB due to the compartmentalisation of the reaction within vesicles formed by invagination of the cytoplasmic membrane to form a liposome.69,81 A hypothesis confirmed by Stolz,82 with the discovery that magnetosomes are only present in cases where a sufficiently high ferrous ion sources are available.71 Lipidiomic and electron microscopy analyses show that the bacterial vesicles contain a suite of proteins specific to the magnetosome.69 Further studies such as gene knockout mutagenesis83,84 genomic studies70 and proteomic studies85–87 are trying to understand the role of these proteins. Numerous reviews detail what is already known about the formation of the magnetosomes and their potential for application.27,70,72,75,88,89 However, briefly the mechanism is comprised of a number of steps. Initially the magnetosome nanoreactor recruits iron through transmembrane iron-transporter proteins, which also function as antiporters, as iron is recruited into the magnetosome core protons are pumped out, raising the pH of the internal magnetosome environment. The resulting pH increase within a supersaturated iron environment leads to the precipitation of a single magnetite nanocrystal within the magnetosome core. The formation of which is controlled with respect to size and shape by specific biomineralisation proteins (Fig. 1ii). If we can fully understand the formation of the magnetosome from first principles; from the formation of the vesicle, to the role of each protein involved and how they work in concert to form the resulting MNPs, we can use this knowledge to attempt the bottom-up creation of an artificial magnetosome and thus enable superior biomimetic biomineralisation within nanoreactors.
Until we can create a fully biomimetic magnetosome, work is ongoing to understand the extent to which bacterial magnetosomes can be modified and functionalised.88 Magnetosomes properties have proven to be superior in several applications when compared with synthetic analogues88 and there is a concerted effort to functionalise magnetosomes for a range of extensive applications,90–94 from targeted therapeutics, to DNA extraction, immunoassays and bio-sensing of toxic substances. The extensive list of possibilities outlined by Lang et al.88 and Matsunaga et al.90–94 demonstrates the potential of magnetosomes ex vivo in nanomedicine.
Despite their potential the anaerobic and slow growing nature of MTB makes cultures difficult to work with and yields are commonly low, making industrial scale-up economically impractical. Bacterial magnetosomes directly for application may not prove to be industrially scalable, but it is envisaged that we could harvest their potential by biomimetically recreating the system.
Biokleptic nanoreactors
Biokleptic is a term that means to use a component directly from nature (as opposed to biomimetic, meaning mimicking a natural component). Nature produces a wide range of nano and microscale compartment structures that have the potential to act as nanoreactors. The advantage of using a compartment/vesicle directly extracted from nature is to utilise the natural advantages of their self-assembly properties and capacity to be genetically modified. As such, in this section we will concentrate on the simplest self-assembly protein cages as nanoreactors for MBNP crystallisation, namely examples of cage-proteins, virus and capsids and finally the decoration of such structures with biomineralisation proteins to directly control crystallisation.Manipulation of the native ferritin protein structure has also been achieved. For example Shin et al. has shown how the internal diameter of the ferritin protein cage can be altered by control of the internal chemical composition. In a study involving the precipitation of Au–Ag alloy NPs within the core of apoferritin it was possible to increase and decrease NP diameter by alteration of ratio of Au to Ag.104 The group were able to gradually increase the alloy NP diameter by 0.7 nm; from 5.6 to 6.3 nm by slow addition of Ag. Mutation of the inner binding sites by the introduction or deletion of residues in the proteins core can dictate both the species and coordination of crystals biomineralised. This has been demonstrated in the case of Pd complex manipulation changing from a dinuclear to trinuclear complex by changing the Pd coordinating residue from a histidine to an alanine though mutagenesis.62
Douglas et al.'s work on the virus protein cages: cowpea chlorotic mottle virus (CCMV) and cowpea mosaic virus (CMPV) are excellent examples of how the inherent properties of such structures can be capitalized upon. Exploitation of CCMV's intrinsic pH dependent gates and positively charged inner membrane; arising from the amino acid subunits which make up the viruses protein cage have thus far created a model environment for the nucleation and precipitation of both paratungstate and decavanadate, whilst the uncharged exterior of the cage remains unaffected ensuring precipitation is localised to the cage's interior.44,105 The viral capsid undergoes reversible swelling above pH 6.5 which increases the capsid size by 10% and in turn removes the viruses intrinsic RNA, an effect of the capsids loosely coupled structure.44,106 (Fig. 2i) This process opens up to 60 pores in the virus structure promoting diffusion in and out of the capsid core and leaving the cage free for NP synthesis, particularly where conditions above physiological pH are required. The fact that this process is reversible means that the reaction can be very carefully controlled by simply lowering the pH back below 6.5, closing the pores with the resulting particles being constrained within the monodisperse capsids. (Fig. 2i) Many other groups have also looked into the CCMV's viability as an in vitro nanoreactor due to the viral capsids unique properties, and have yielded reactions such as creation of an environment for single enzyme studies by disassembling the protein cage (at <pH 5) for the purposes of enzyme incorporation again utilising the effects of pH.46 Also explored is the CCMV's binding capability to Gd3+ for MRI application. The cage has proven to be an ideal biotemplate due to the large number of possible binding sites available. This increases the payload that can be delivered for efficient application.107 CCMV functionality has also been investigated as a potential mimic for iron storage protein ferritin, again using the high number of inherent cage binding sites for accumulation and oxidative hydrolysis of ferrous ions.108 Similar work has also been carried out by the Evans lab on CPMV exploring its potential for mineralisation of gold, silica and multiple iron mineral NPs particularly exploring their viability for biomedical applications.109–112 Such a switchable process in which a nanoreactor self-assembles around the reactant, whereby the encapsulation can be pH controlled and nucleation can be electrostatically induced would be extremely difficult to replicate synthetically.
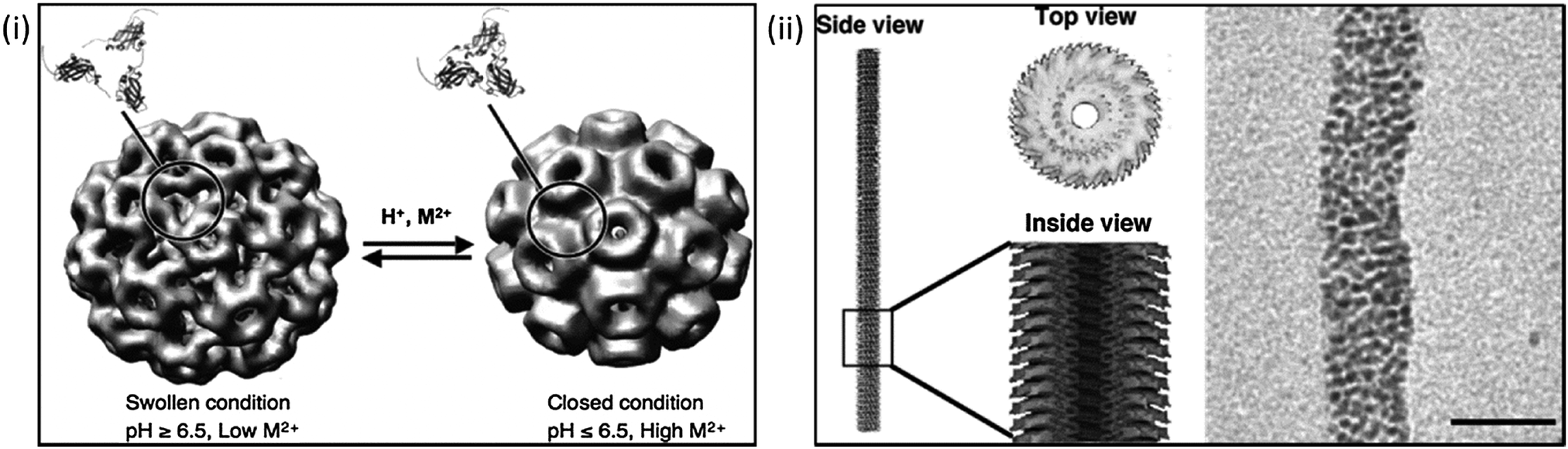 | ||
| Fig. 2 Using biological templates for biomineralisation. Left: (i) manipulation of cowpea chlorotic mottle virus (CCMV) using pH conditions to aid mineralisation reactions. (Reproduced with permission from Uchida et al.39); right: (ii) cryo-electron micrograph and reconstruction of tobacco mosaic virus, and its use as template for the mineralisation of Pt nanoparticles. Scale bars 20 nm (Reproduced with permission from Uchida et al.39 and Górzny et al.116). | ||
Protein cage templated biomineralisation is not limited to precipitation within the structures core. Biomineralisation on the exterior of CPMV has also been explored by modification of surface charge for the precipitation of both cobalt and iron oxide NPs of extremely narrow size distribution.109 Another similar structure widely studied as a potential biotemplate is the tobacco mosaic virus (TMV). Its rod-like structure is composed of helical RNA and 2130 coat proteins, the exterior of the virus can be functionalised for the binding of metal ions making it an ideal template for metal biomineralisation. The virus has been used for the pH controlled precipitation of Ag NPs and QDs along the internal cavity of the virus.113 The central channel of the virus has also been exploited for the biomineralisation of both FePt3 and CoPt nanowires.114 Like CCMV, the templating capability of TMV has been shown to be ideal in countless studies in which a narrow worm-like or a nano-tube of specific diameter architecture is required. Górzny et al. have shown that it is possible to use the virus structure as a template for the synthesis of Pt NPs, this increases the surface area and overall stability of the Pt nanostructures when compared to more conventional particles115,116 (Fig. 2ii). Pejoux et al. have taken a similar approach to biotemplating of Ag2S mineralisation on the outer shell of an enzyme protein cage.117 The group exploited the properties of the shell by tuning the catalytic activity to induce crystallisation, tailoring enzymatic activity to both initiate and inhibit S2− production as the reaction requires for semiconductor growth in the presence of the optimum concentration. This approach has been applied to other systems suggesting it can be utilised in the synthesis of numerous other semiconductors117 in which consistently sized semiconductor NPs are required with a hollow core available for further functionality.
The diatom controls the biomineralisation of silica in a similar way using biomineralisation proteins and peptides. Again these can be added to an in vitro precipitation to improve silica formation. The isolation of a polycationic peptide called a silaffin has been shown to exert control over the precipitation of silica nanosphere networks. A lysine–lysine section within the peptide structure has proven to be responsible for the formation of the network.120 These effects are also observed with the polyanionic proteins extracted from mollusc abalone shell whose in vivo function is crystal phase switching from aragonite to calcite during the formation of nucleating protein sheets. Purification and in vitro use of these proteins can initiate the same changes in crystal phase observed within the proteins natural environment.121
Many more studies of this kind are ongoing, and some work is now focusing on the use of ferritin as a template for the study of these and other proteins, such as Mms6.122 Whilst this is by no means an exhaustive list of bioadditives, it demonstrates the versatility that can be achieved when we “borrow” biomineralisation protein from nature to use as additives in chemical reactions. The next step is to combine these additives into MBNP precipitation nanoreactors to simulate the proteins natural functional environment, which is already underway, with ferritin for example. Research of this type is invaluable in both confirming the natural function of these proteins; which can often be hard to identify within a biological system and to improve and simplify nanoreactor synthetic methods of mimicking biomineralisation.
Biomimetic systems
Natural biomineralisation and biokleptic templates of protein cages such as viral capsids have their limitations; constrained by only utilising the components that nature has to offer. Such nanoreactors may not be robust enough to reaction conditions outside ambient condition parameters. Taking a bottom-up approach i.e. using synthetic vesicles, means that should any new specification arise outside the biological parameters, it could be accommodated by the synthetic host, in which chemical and physical adaptions are possible. For example, if a larger compartment for mineralisation is required this can easily be optimised in vesicles with techniques such as extrusion.123 This flexibility is much less achievable with biological components such as protein cages whose evolution has been optimised by nature for their specific natural role and not the new role imposed upon them. Thus a change such as altering the nanoreactor size is unlikely to be as easily engineered.Other issues encountered when using naturally occurring biotemplates are those of solubility and an intolerance to the conditions required for synthetic routes to precipitation, such as high temperatures and wide ranging pH changes, for example those used in the partial oxidation of magnetite.23,35 It is in situations such as these that research must turn to synthetic alternatives for compartmentalisation reactions and design of biomimetic nanoreactors. The benefit of creating a nanoreactor from a synthetic analogue is the ability to engineer into a scaffold the properties required for a specific mineralisation reaction124,125 which is not possible in biological systems. A key problem with crystallisation of many types of MBNPs is the harsh chemical conditions required for their uniformed synthesis. Compartmentalising within a nanoreactor massively reduces the requirement for such conditions or in some cases completely eradicates the need for organic solvents and harmful chemicals. This is particularly evident in the work by both Genc et al.126 and Pejoux et al.117 where in both cases compartmentalisation was utilised for the precipitation of gold and sulphide NPs respectively. In both examples organic solvents and harsh conditions are normally required, but by carrying out the reaction in the confines of a nanoreactor such as a lipid environment126 or an enzyme nanoreactor117 allows for the compartmentalisation to enable a move toward greener chemistry synthetic routes.
Membrane transport across the liposome bilayer can be initiated by careful control of both the inner and outer pHs. Osmotic effects can be used to aid transmembrane diffusion, by increase or decrease of the outer vesicular pH it is possible to force diffusion across the membrane.133 Ongoing studies are working toward better quantifying the effect of pH on membrane transport.134,135 It has also been demonstrated that pH changes can result in full structural changes.136 The presence of a lipid bilayer (wall to the nanoreactors) can often aid compartmentalised reactions, by adsorption of ions on to the membrane surface which then facilitates the consequent nucleation. This nucleation and subsequent crystal growth process is limited to the size of the internal vesicle diameter, ensuring the production of NPs of monodisperse size; this effect is observed in the growth of CdS, ZnCdS and HgCdS nanocrystal QDs within the core of liposome.137,138 The authors were successful in controlling synthesis to 1 particle per vesicle using membrane mediated crystal growth137 a ratio which could not have been achieved with post synthesis incorporation into vesicles, for biomedical application to lessen QD toxicity issues.
Liposomes offer the perfect vehicle for a biomimetic approach to synthetic biomineralisation. An excellent example is that of biomimicing the biomineralisation of a magnetosome within MTB.42 Vesicles are an ideal environment to create this supersaturated environment, evident in the work of Mann and Hannington42,139 and their work toward the creation of a biomimetic magnetosome. Compartmentalisation of a reaction forces a supersaturated environment and the consequent nucleation processes involved in crystallisation of the magnetite MNPs. This approach has also been applied to the biomimetic biomineralisation of calcite, aragonite, hydroxyapatite and silica140 demonstrating flexibility and diversity with regards to the reactions which can be incorporated into a single biomimetic system. Ongoing research has expanded this field further such as, the design of the “magnetonion” a multilamellar vesicle, resembling an onion-like structure in which MNPs sit within the layers of the “onion” by Faure et al.141 and Sangregorio et al.142 both of these systems build on Mann and Hannington's biomimetic system, but none have thus far achieved a comprehensive mimetic of a magnetosome functional for bioapplication. Much of the work in our own lab builds on Mann's initial biomimetic research,143 with ours focusing on trying to further understand the formation and biomineralisation processes observed in the magnetosome. To this effect, in our lab we are currently developing an artificial magnetosome nanoreactor.
Other groups such as Chakrabarti et al., have also explored taking a biomimetic approach to the creation of a nanoreactor, by the addition of a divalent specific ion channel. The study has shown how biological components can be incorporated into synthetic vesicles to mimic the ion transport processes observed in nature. This was proven by the loading both Fe2+ and Ba2+ into preformed liposomes via liposomal incorporation of Ca2+ ionophore A23187.144 Studies such as this and others134 are critical to the development of synthetic biology for the creation of novel biomimetic biomineralisation nanoreactors.
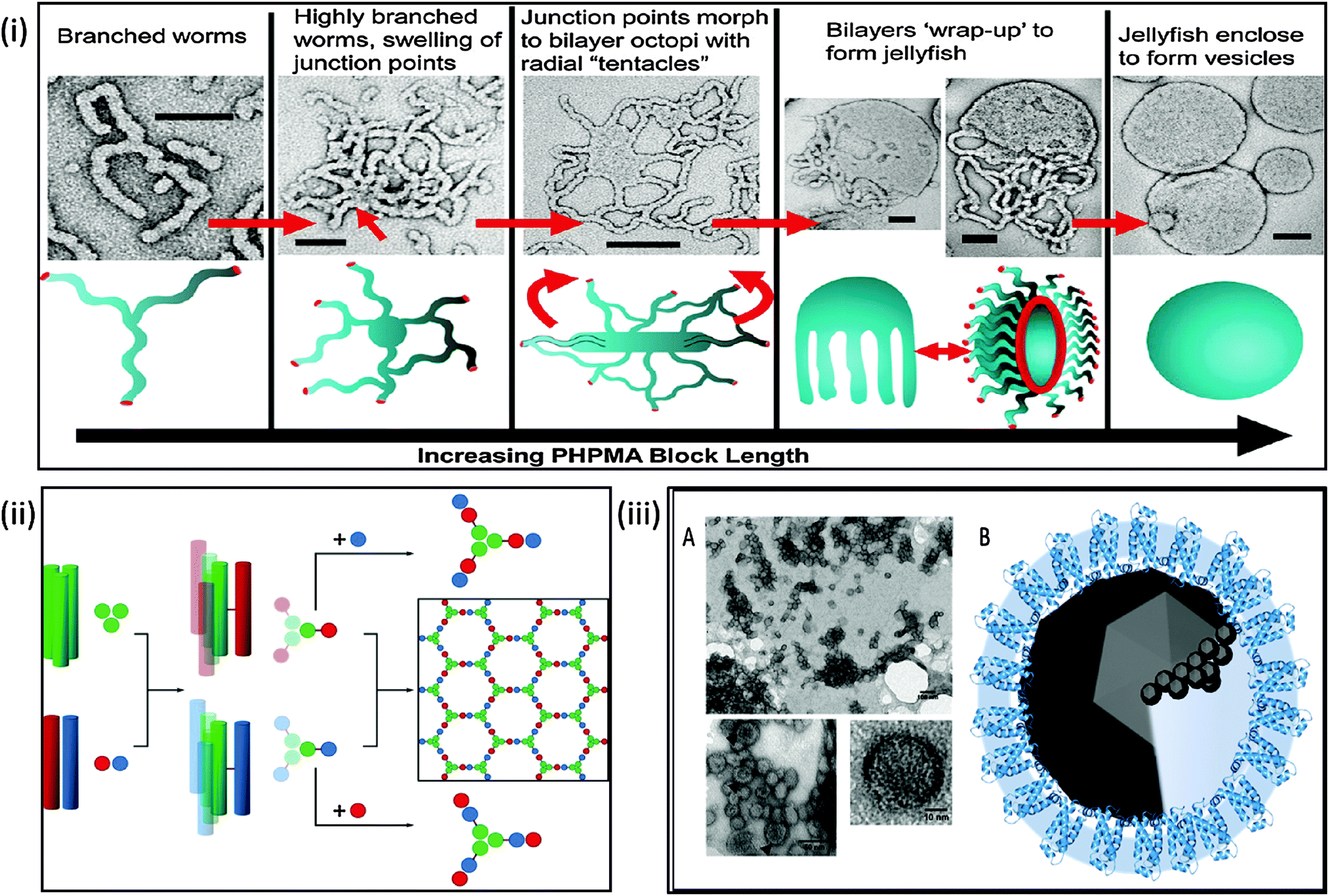 | ||
| Fig. 3 Biomimetic mineralisation systems. Top: (i) schematic and electron microscopy interpretation of architectures achievable during polymersome synthesis demonstrating the potential for control of the formation of a polymeric nanoreactor; (reprinted with permission from A. Blanazs, et al., J. Am. Chem. Soc., 2011 Copyright (2011) American Chemical Society). Bottom left: (ii) schematic of peptide SAGE cage synthesis which takes a bottom-up approach to nanoreactor design (reproduced with permission from Fletcher et al.159); bottom right: (iii) (A) transmission electron micrograph of self-assembled MmsF proteinosomes. (B) Schematic of potential proteinosome biomineralisation pathways (adapted with permission from Rawlings et al.36). | ||
Polymersomes as nanoreactors is a relatively new field, with increasing success. Meier et al. have successfully incorporated biological components into polymer membranes,156 this work has been further advanced by numerous other groups by incorporation of both protein channels and enzymes.157 Much of this work is outlined in the review by Meier et al.158 which demonstrates a framework of polymersome nanoreactors for the study of ion transporters, membrane proteins and as biomineralisation scaffolds. Therefore this suggests that block copolymers vesicles are now competing with liposomes in their use as vehicles for both the study of biological moieties and as successful nanoreactors.
Conclusions and future outlook
The use of a nanoreactor to biomineralise MBNPs has advanced many areas of research and the development of future applications. Studies of biological components in vitro have already advanced understanding of biomineralisation processes in nature.36,37,119There is continuing exploration of biology's effectiveness in internal compartmentalisation of chemical reactions, and how this can result in perfectly biomineralised MBNPs. This knowledge is invaluable in understanding the multiple physiological processes and the function of numerous biological components involved in both the transport and morphological control observed for biomineralisation in natural nanoreactors, which serves to aid fundamental understanding of the processes and advance the development of novel nanomaterials.
Advancement in this field has found that synthesis of MBNPs within a nanoreactor mimicking biomineralisation offers a range of superior properties and functionality. The benefits for nanomedicinal materials are extensive, with the scope of applicability of MBNPs ever widening. Such benefits include: a higher degree of precision and reproducibility with respect to size and morphology across the entire population of particles; the fact this can be “switched” by stimuli such as pH change; the intrinsic inclusion of a biocompatible coating, which can be readily biofunctionalised for further application. MBNPs can also often prove toxic in vivo. Compartmentalisation of such reactions removes the need for post formation processing and coating to remove toxicity. Additionally, this superior control over MBNP formation is achieved under ambient environmentally-friendly reaction conditions. Research now allows us to finely tune both the structure and properties of a nanoreactor. From the mutation of single amino acids to change the outcome of mineralisation reaction, to completely altering the architecture and size of a nanoreactor in the case of PGMA–PHMPA polymersomes. It can be seen that the further we move towards fully synthetic nanoreactors, the levels of complexity observe in naturally occurring nanoreactors are removed, and we able to then fully tailor the properties to our requirements.
Escosura et al.35 writes that whilst the creation of a fully functional biomimetic nanocontainer may be just out of reach of today's research, nobody can doubt that great understanding and numerous beneficial bioinspired nanoreactors with applications in multiple fields have been and will be discovered along the way. With this sentiment in mind the pathway to the future biomimetic production of monodisperse MBNP has shed real light on the nano and microscale encapsulated biomineralisation process. Although the fully functional biomimetic nanocontainer is out of reach of today's research, tomorrow is getting ever closer and we believe we are on the verge of this goal. In order to achieve this for nanoreactor synthesis of MBNP there is vast opportunities in exploiting a combination of biological materials and designed artificial self-assembly materials to design and engineer nanoreactors of ideal specification (robust to chemical environment, biocompatible, mono-dispersed, switchable etc.) and incorporate other functionality such as further biomedical therapeutics on exterior and biomineralisation controlling species on the interior. Ultimately, the design and mixing of biology and biomimicry has led to nanoreactors that are perfectly poised to move from the bench to the bedside.
Acknowledgements
We would like to thank the EPSRC (EP/I032355/2) for funding the authors. We would also like to thank Dr Andrea Rawlings, Mr Scott Bird and Miss Lori Somner (University of Sheffield, Department of Chemistry) and Dr Jonathan Bramble (IBIOS, University of Nottingham) for their support and useful discussions.References
- Q. A. Pankhurst, J. Connolly, S. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- Y.-w. Jun, Y.-M. Huh, J.-s. Choi, J.-H. Lee, H.-T. Song, S. Kim, S. Yoon, K.-S. Kim, J.-S. Shin, J.-S. Suh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 5732–5733 CrossRef CAS PubMed.
- Y. Du, P. Lai, C. Leung and P. Pong, Int. J. Mol. Sci., 2013, 14, 18682–18710 CrossRef CAS PubMed.
- E. Alphandéry, F. Guyot and I. Chebbi, Int. J. Pharm., 2012, 434, 444–452 CrossRef PubMed.
- C. S. S. R. Kumar and F. Mohammad, Adv. Drug Delivery Rev., 2011, 789–808 CrossRef CAS PubMed.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- S. Zeng, K.-T. Yong, I. Roy, X.-Q. Dinh, X. Yu and F. Luan, Plasmonics, 2011, 6, 491–506 CrossRef CAS.
- S. Ghaderi, B. Ramesh and A. M. Seifalian, J. Drug Targeting, 2011, 19, 475–486 CrossRef CAS PubMed.
- H. Ron, Environ. Health Perspect., 2006, 114, 165–172 CrossRef.
- Q. Pankhurst, N. Thanh, S. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef.
- J. Dobson, Drug Dev. Res., 2006, 67, 55–60 CrossRef CAS PubMed.
- J. Dobson, Nanomedicine, 2006, 1, 31–37 CrossRef CAS PubMed.
- D. C. Drummond, M. Zignani and J. Leroux, Prog. Lipid Res., 2000, 39, 409–460 CrossRef CAS.
- S. M. Janib, A. S. Moses and J. A. MacKay, Adv. Drug Delivery Rev., 2010, 62, 1052–1063 CrossRef CAS PubMed.
- A. Singh, F. Dilnawaz, S. Mewar, U. Sharma, N. R. Jagannathan and S. K. Sahoo, ACS Appl. Mater. Interfaces, 2011, 3, 842–856 CAS.
- W. T. Al-Jamal and K. Kostarelos, Acc. Chem. Res., 2011, 44, 1094–1104 CrossRef CAS PubMed.
- P. B. Santhosh and N. P. Ulrih, Cancer Lett., 2013, 8–17 CrossRef CAS PubMed.
- S. Svenson, Mol. Pharmaceutics, 2013, 848–856 CrossRef CAS PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- C.-H. Ho, C.-P. Tsai, C.-C. Chung, C.-Y. Tsai, F.-R. Chen, H.-J. Lin and C.-H. Lai, Chem. Mater., 2011, 23, 1753–1760 CrossRef CAS.
- P. Raveendran, J. Fu and S. L. Wallen, J. Am. Chem. Soc., 2003, 125, 13940–13941 CrossRef CAS PubMed.
- A. E. Regazzoni, G. A. Urrutia, M. A. Blesa and A. J. G. Maroto, J. Inorg. Nucl. Chem., 1981, 43, 1489–1493 CrossRef CAS.
- S. Asuha, B. Suyala, X. Siqintana and S. Zhao, J. Alloys Compd., 2011, 509, 2870–2873 CrossRef CAS PubMed.
- Y. K. Jeong, D. K. Shin, H. J. Lee, K. S. Oh, J. H. Lee and D. H. Riu, in Science of Engineering Ceramics III, ed. T. Ohji, T. Sekino and K. Niihara, 2006, vol. 317–318, pp. 203–206 Search PubMed.
- H. Steen and V. Schwenger, Pediatr. Nephrol., 2007, 22, 1239–1242 CrossRef PubMed.
- S. E. Greene and A. Komeili, Curr. Opin. Cell Biol., 2012, 24, 490–495 CrossRef CAS PubMed.
- D. M. Vriezema, M. Comellas Aragonès, J. A. Elemans, J. J. Cornelissen, A. E. Rowan and R. J. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS PubMed.
- S. Mann, Acc. Chem. Res., 2012, 45, 2131–2141 CrossRef CAS PubMed.
- A. J. Dzieciol and S. Mann, Chem. Soc. Rev., 2012, 41, 79–85 RSC.
- S. Mann, Biomimetic materials chemistry, John Wiley & Sons, 1995 Search PubMed.
- R. Roodbeen and J. van Hest, BioEssays, 2009, 31, 1299–1308 CrossRef CAS PubMed.
- V. Noireaux, R. Bar-Ziv, J. Godefroy, H. Salman and A. Libchaber, Phys. Biol., 2005, 2, P1 CrossRef CAS PubMed.
- D. G. Gibson, J. I. Glass, C. Lartigue, V. N. Noskov, R.-Y. Chuang, M. A. Algire, G. A. Benders, M. G. Montague, L. Ma, M. M. Moodie, C. Merryman, S. Vashee, R. Krishnakumar, N. Assad-Garcia, C. Andrews-Pfannkoch, E. A. Denisova, L. Young, Z.-Q. Qi, T. H. Segall-Shapiro, C. H. Calvey, P. P. Parmar, C. A. Hutchison, H. O. Smith and J. C. Venter, Science, 2010, 329, 52–56 CrossRef CAS PubMed.
- A. de la Escosura, R. J. Nolte and J. J. Cornelissen, J. Mater. Chem., 2009, 19, 2274–2278 RSC.
- A. E. Rawlings, J. P. Bramble, R. Walker, J. Bain, J. M. Galloway and S. S. Staniland, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16094–16099 CrossRef CAS PubMed.
- Y. Amemiya, A. Arakaki, S. S. Staniland, T. Tanaka and T. Matsunaga, Biomaterials, 2007, 28, 5381–5389 CrossRef CAS PubMed.
- M. Tanaka, E. Mazuyama, A. Arakaki and T. Matsunaga, J. Biol. Chem., 2011, 286, 6386–6392 CrossRef CAS PubMed.
- M. Uchida, M. T. Klem, M. Allen, P. Suci, M. Flenniken, E. Gillitzer, Z. Varpness, L. O. Liepold, M. Young and T. Douglas, Adv. Mater., 2007, 19, 1025–1042 CrossRef CAS PubMed.
- C. J. Stephens, Y.-Y. Kim, S. D. Evans, F. C. Meldrum and H. K. Christenson, J. Am. Chem. Soc., 2011, 133, 5210–5213 CrossRef CAS PubMed.
- C. J. Stephens, S. F. Ladden, F. C. Meldrum and H. K. Christenson, Adv. Funct. Mater., 2010, 20, 2108–2115 CrossRef CAS PubMed.
- S. Mann, J. P. Hannington and R. J. P. Williams, Nature, 1986, 324, 565–567 CrossRef CAS.
- Y. Mai and A. Eisenberg, Acc. Chem. Res., 2012, 1657–1666 CrossRef CAS PubMed.
- T. Douglas and M. Young, Nature, 1998, 393, 152–155 CrossRef CAS PubMed.
- M. Allen, D. Willits, J. Mosolf, M. Young and T. Douglas, Adv. Mater., 2002, 14, 1562–1565 CrossRef CAS.
- M. Comellas-Aragones, H. Engelkamp, V. I. Claessen, N. A. J. M. Sommerdijk, A. E. Rowan, P. C. M. Christianen, J. C. Maan, B. J. M. Verduin, J. J. L. M. Cornelissen and R. J. M. Nolte, Nat. Nanotechnol., 2007, 2, 635–639 CrossRef CAS PubMed.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS.
- M. L. Immordino, F. Dosio and L. Cattel, Int. J. Nanomed., 2006, 1, 297–315 CrossRef CAS PubMed.
- A. Jesorka and O. Orwar, Annual Review of Analytical Chemistry, 2008, vol. 1, pp. 801–832 Search PubMed.
- B. Maherani, E. Arab-Tehrany, M. R. Mozafari, C. Gaiani and M. Linder, Curr. Nanosci., 2011, 7, 436–452 CrossRef CAS.
- S. K. P. Velu, M. Yan, K.-P. Tseng, K.-T. Wong, D. M. Bassani and P. Terech, Macromolecules, 2013, 46, 1591–1598 CrossRef CAS.
- D. E. Discher, V. Ortiz, G. Srinivas, M. L. Klein, Y. Kim, D. Christian, S. Cai, P. Photos and F. Ahmed, Prog. Polym. Sci., 2007, 32, 838–857 CrossRef CAS PubMed.
- Y. Lu, L. Dong, L.-C. Zhang, Y.-D. Su and S.-H. Yu, Nano Today, 2012, 7, 297–315 CrossRef CAS PubMed.
- M. Bonini, D. Berti and P. Baglioni, Curr. Opin. Colloid Interface Sci., 2013, 18, 459–467 CrossRef CAS PubMed.
- K. T. Kim, S. A. Meeuwissen, R. J. Nolte and J. C. van Hest, Nanoscale, 2010, 2, 844–858 RSC.
- J. L. Kirschvink and J. W. Hagadorn, The Biomineralisation of Nano-and Micro-Structures, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2000, pp. 139–150 Search PubMed.
- H. Skinner and A. Jahren, Treatise Geochem., 2003, 8, 117–184 Search PubMed.
- P. Westbroek, J. Young and K. Linschooten, J. Protozool., 1989, 36, 368–373 CrossRef PubMed.
- J. R. Young, S. A. Davis, P. R. Bown and S. Mann, J. Struct. Biol., 1999, 126, 195–215 CrossRef CAS PubMed.
- M. Hildebrand, Chem. Rev., 2008, 108, 4855 CrossRef CAS PubMed.
- C. M. Niemeyer and C. A. Mirkin, Nanobiotechnology: concepts, applications and perspectives, John Wiley & Sons, 2006 Search PubMed.
- S. Abe, J. Niemeyer, M. Abe, Y. Takezawa, T. Ueno, T. Hikage, G. Erker and Y. Watanabe, J. Am. Chem. Soc., 2008, 130, 10512–10514 CrossRef CAS PubMed.
- T. Ueno, M. Suzuki, T. Goto, T. Matsumoto, K. Nagayama and Y. Watanabe, Angew. Chem., 2004, 116, 2581–2584 CrossRef PubMed.
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896–910 CrossRef CAS PubMed.
- T. S. Koblenz, J. Wassenaar and J. N. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC.
- J. L. Kirschvink, A. Kobayashi-Kirschvink and B. J. Woodford, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7683–7687 CrossRef CAS.
- R. Blakemore, Science, 1975, 190, 377 CrossRef CAS.
- D. Faivre and D. Schüler, Chem. Rev., 2008, 108, 4875–4898 CrossRef CAS PubMed.
- Y. A. Gorby, T. J. Beveridge and R. P. Blakemore, J. Bacteriol., 1988, 170, 834 CAS.
- C. Jogler and D. Schüler, Annu. Rev. Microbiol., 2009, 63, 501–521 CrossRef CAS PubMed.
- D. H. Nies, Mol. Microbiol., 2011, 82, 792–796 CrossRef CAS PubMed.
- A. Arakaki, H. Nakazawa, M. Nemoto, T. Mori and T. Matsunaga, J. R. Soc., Interface, 2008, 5, 977–999 CrossRef CAS PubMed.
- R. B. Frankel, R. P. Blakemore and R. S. Wolfe, Science, 1979, 203, 1355 CAS.
- R. B. Frankel and R. P. Blakemore, Bioelectromagnetics, 1989, 10, 223–237 CrossRef CAS PubMed.
- D. A. Bazylinski and R. B. Frankel, Nat. Rev. Microbiol., 2004, 2, 217–230 CrossRef CAS PubMed.
- M. Smith, P. Sheehan, L. Perry, K. O'Connor, L. Csonka, B. Applegate and L. Whitman, Biophys. J., 2006, 91, 1098–1107 CrossRef CAS PubMed.
- W. F. Guerin and R. P. Blakemore, Appl. Environ. Microbiol., 1992, 58, 1102–1109 CAS.
- R. P. Blakemore, Annu. Rev. Microbiol., 1982, 36, 217–238 CrossRef CAS PubMed.
- R. Frankel, Chin. J. Oceanol. Limnol., 2009, 27, 1–2 CrossRef PubMed.
- H. Nudelman and R. Zarivach, Front. Microbiol., 2014, 5, 9 Search PubMed.
- A. Komeili, Z. Li, D. K. Newman and G. J. Jensen, Science, 2006, 311, 242–245 CrossRef CAS PubMed.
- J. F. Stolz, S.-B. R. Chang and J. L. Kirschvink, Nature, 1986, 321, 849–851 CrossRef.
- D. Murat, V. Falahati, L. Bertinetti, R. Csencsits, A. Körnig, K. Downing, D. Faivre and A. Komeili, Mol. Microbiol., 2012, 684–699 CrossRef CAS PubMed.
- R. Uebe, K. Junge, V. Henn, G. Poxleitner, E. Katzmann, J. M. Plitzko, R. Zarivach, T. Kasama, G. Wanner and M. Pósfai, Mol. Microbiol., 2011, 818–835 CrossRef CAS PubMed.
- K. Grünberg, E.-C. Müller, A. Otto, R. Reszka, D. Linder, M. Kube, R. Reinhardt and D. Schüler, Appl. Environ. Microbiol., 2004, 70, 1040–1050 CrossRef.
- T. Matsunaga, M. Nemoto, A. Arakaki and M. Tanaka, Proteomics, 2009, 9, 3341–3352 CrossRef CAS PubMed.
- M. Tanaka, Y. Okamura, A. Arakaki, T. Tanaka, H. Takeyama and T. Matsunaga, Proteomics, 2006, 6, 5234–5247 CrossRef CAS PubMed.
- C. Lang, D. Schüler and D. Faivre, Macromol. Biosci., 2007, 7, 144–151 CrossRef CAS PubMed.
- T. Prozorov, D. A. Bazylinski, S. K. Mallapragada and R. Prozorov, Mater. Sci. Eng., R, 2013, 74, 133–172 CrossRef PubMed.
- T. Tanaka, K. Maruyama, K. Yoda, E. Nemoto, Y. Udagawa, H. Nakayama, H. Takeyama and T. Matsunaga, Biosens. Bioelectron., 2003, 19, 325–330 CrossRef CAS.
- T. Yoshino, F. Kato, H. Takeyama, M. Nakai, Y. Yakabe and T. Matsunaga, Anal. Chim. Acta, 2005, 532, 105–111 CrossRef CAS PubMed.
- T. Tanaka and T. Matsunaga, Anal. Chem., 2000, 72, 3518–3522 CrossRef CAS.
- K. Sode, S. Kudo, T. Sakaguchi, N. Nakamura and T. Matsunaga, Biotechnol. Tech., 1993, 7, 688–694 CrossRef CAS.
- B. Yoza, A. Arakaki and T. Matsunaga, J. Biotechnol., 2003, 101, 219–228 CrossRef CAS.
- F. C. Meldrum, T. Douglas, S. Levi, P. Arosio and S. Mann, J. Inorg. Biochem., 1995, 58, 59–68 CrossRef CAS.
- K. K. W. Wong and S. Mann, Adv. Mater., 1996, 8, 928–932 CrossRef CAS PubMed.
- S. Mann and F. C. Meldrum, Adv. Mater., 1991, 3, 316–318 CrossRef CAS PubMed.
- B. Warne, O. I. Kasyutich, E. L. Mayes, J. A. Wiggins and K. K. Wong, IEEE Trans. Magn., 2000, 36, 3009–3011 CrossRef CAS.
- F. C. Meldrum, B. R. Heywood and S. Mann, Science, 1992, 257, 522–523 CAS.
- K. Zeth, S. Offermann, L.-O. Essen and D. Oesterhelt, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13780–13785 CrossRef CAS PubMed.
- W. Zhong, D. Alexeev, I. Harvey, M. Guo, D. J. Hunter, H. Zhu, D. J. Campopiano and P. J. Sadler, Angew. Chem., Int. Ed., 2004, 43, 5914–5918 CrossRef CAS PubMed.
- J. Schemberg, K. Schneider, U. Demmer, E. Warkentin, A. Müller and U. Ermler, Angew. Chem., 2007, 119, 2460–2465 CrossRef PubMed.
- D. Alexeev, H. Zhu, M. Guo, W. Zhong, D. J. Hunter, W. Yang, D. J. Campopiano and P. J. Sadler, Nat. Struct. Mol. Biol., 2003, 10, 297–302 CAS.
- Y. Shin, A. Dohnalkova and Y. Lin, J. Phys. Chem. C, 2010, 114, 5985–5989 CAS.
- J. E. Johnson and J. A. Speir, J. Mol. Biol., 1997, 269, 665–675 CrossRef CAS PubMed.
- J. A. Speir, S. Munshi, G. Wang, T. S. Baker and J. E. Johnson, Structure, 1995, 3, 63–78 CrossRef CAS.
- M. Allen, J. W. Bulte, L. Liepold, G. Basu, H. A. Zywicke, J. A. Frank, M. Young and T. Douglas, Magn. Reson. Med., 2005, 54, 807–812 CrossRef CAS PubMed.
- T. Douglas, E. Strable, D. Willits, A. Aitouchen, M. Libera and M. Young, Adv. Mater., 2002, 14, 415–418 CrossRef CAS.
- A. A. Aljabali, J. E. Barclay, O. Cespedes, A. Rashid, S. S. Staniland, G. P. Lomonossoff and D. J. Evans, Adv. Funct. Mater., 2011, 21, 4137–4142 CrossRef CAS PubMed.
- A. A. Aljabali, S. Shukla, G. P. Lomonossoff, N. F. Steinmetz and D. J. Evans, Mol. Pharmaceutics, 2012, 10, 3–10 CrossRef PubMed.
- A. M. Wen, S. Shukla, P. Saxena, A. A. Aljabali, I. Yildiz, S. Dey, J. E. Mealy, A. C. Yang, D. J. Evans and G. P. Lomonossoff, Biomacromolecules, 2012, 13, 3990–4001 CrossRef CAS PubMed.
- N. F. Steinmetz, S. N. Shah, J. E. Barclay, G. Rallapalli, G. P. Lomonossoff and D. Evans, Small, 2009, 5, 813–816 CrossRef CAS PubMed.
- E. Dujardin, C. Peet, G. Stubbs, J. N. Culver and S. Mann, Nano Lett., 2003, 3, 413–417 CrossRef CAS.
- R. Tsukamoto, M. Muraoka, M. Seki, H. Tabata and I. Yamashita, Chem. Mater., 2007, 19, 2389–2391 CrossRef CAS.
- M. Ł. Górzny, A. Walton, M. Wnk, P. Stockley and S. Evans, Nanotechnology, 2008, 19, 165704 CrossRef PubMed.
- M. Ł. Górzny, A. S. Walton and S. D. Evans, Adv. Funct. Mater., 2010, 20, 1295–1300 CrossRef PubMed.
- C. Pejoux, R. de la Rica and H. Matsui, Small, 2010, 6, 999–1002 CrossRef CAS PubMed.
- A. Arakaki, J. Webb and T. Matsunaga, J. Biol. Chem., 2003, 278, 8745–8750 CrossRef CAS PubMed.
- J. M. Galloway, A. Arakaki, F. Masuda, T. Tanaka, T. Matsunaga and S. S. Staniland, J. Mater. Chem., 2011, 21, 15244–15254 RSC.
- N. Kröger, R. Deutzmann and M. Sumper, Science, 1999, 286, 1129–1132 CrossRef.
- A. M. Belcher, X. H. Wu, R. J. Christensen, P. K. Hansma, G. D. Stucky and D. E. Morse, Nature, 1996, 381, 56–58 CrossRef CAS.
- M. Neeman, R. Zarivach, M. Radoul, B. Cohen, M. Vandsburger and L. Lewin, US 13/856,788, 2013 Search PubMed.
- L. D. Mayer, M. J. Hope and P. R. Cullis, Biochim. Biophys. Acta, Biomembr., 1986, 858, 161–168 CrossRef CAS.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- R. Genc, G. Clergeaud, M. Ortiz and C. O'Sullivan, Biomater. Sci., 2014, 1128–1134 RSC.
- Y.-C. Tan, K. Hettiarachchi, M. Siu, Y.-R. Pan and A. P. Lee, J. Am. Chem. Soc., 2006, 128, 5656–5658 CrossRef CAS PubMed.
- P. A. Monnard, J. Membr. Biol., 2003, 191, 87–97 CrossRef CAS PubMed.
- C. C. Tester, C.-H. Wu, S. Weigand and D. Joester, Faraday Discuss., 2012, 159, 345–356 RSC.
- P. Walde, K. Cosentino, H. Engel and P. Stano, ChemBioChem, 2010, 11, 848–865 CrossRef CAS PubMed.
- S. Pautot, B. J. Frisken and D. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10718 CrossRef CAS PubMed.
- S. Pautot, B. J. Frisken and D. A. Weitz, Langmuir, 2003, 19, 2870–2879 CrossRef CAS.
- S. Mann, M. J. Kime, R. G. Ratcliffe and R. J. P. Williams, J. Chem. Soc., Dalton Trans., 1983, 771–774 RSC.
- M. Megens, C. E. Korman, C. M. Ajo-Franklin and D. A. Horsley, Biochim. Biophys. Acta, Biomembr., 2014, 2420–2424 CrossRef CAS PubMed.
- E. Mamasheva, C. O'Donnell, A. Bandekar and S. Sofou, Mol. Pharmaceutics, 2011, 8, 2224–2232 CrossRef CAS PubMed.
- C.-Y. Leung, L. C. Palmer, B. F. Qiao, S. Kewalramani, R. Sknepnek, C. J. Newcomb, M. A. Greenfield, G. Vernizzi, S. I. Stupp and M. J. Bedzyk, ACS Nano, 2012, 6, 10901–10909 CrossRef CAS PubMed.
- M. T. Kennedy, B. A. Korgel, H. G. Monbouquette and J. A. Zasadzinski, Chem. Mater., 1998, 10, 2116–2119 CrossRef CAS.
- B. A. Korgel and H. G. Monbouquette, Langmuir, 2000, 16, 3588–3594 CrossRef CAS.
- S. Mann and J. P. Hannington, J. Colloid Interface Sci., 1988, 122, 326–335 CrossRef CAS.
- S. Mann and R. J. P. Williams, J. Chem. Soc., Dalton Trans., 1983, 311–316 RSC.
- C. Faure, M.-E. Meyre, S. Trépout, O. Lambert and E. Lebraud, J. Phys. Chem. B, 2009, 113, 8552–8559 CrossRef CAS PubMed.
- C. Sangregorio, J. K. Wiemann, C. J. O'Connor and Z. Rosenzweig, J. Appl. Phys., 1999, 85, 5699–5701 CrossRef CAS PubMed.
- S. Mann, J. P. Hannington and R. J. P. Williams, Nature, 1986, 324, 565–567 CrossRef CAS.
- A. C. Chakrabarti, J. A. Veiro, N. S. Wong, J. J. Wheeler and P. R. Cullis, Biochim. Biophys. Acta, Biomembr., 1992, 1108, 233–239 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 267–277 Search PubMed.
- C. W. Evans, M. Fitzgerald, T. D. Clemons, M. J. House, B. S. Padman, J. A. Shaw, M. Saunders, A. R. Harvey, B. Zdyrko, I. Luzinov, G. A. Silva, S. A. Dunlop and K. S. Iyer, ACS Nano, 2011, 5, 8640–8648 CrossRef PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- F. Zhang, J. A. Smolen, S. Zhang, R. Li, P. N. Shah, S. Cho, H. Wang, J. E. Raymond, C. L. Cannon and K. L. Wooley, Nanoscale, 2015, 7, 2265–2270 RSC.
- J.-F. Lutz, Nat. Chem., 2010, 2, 84–85 CrossRef CAS PubMed.
- Q. Zhang, E. E. Remsen and K. L. Wooley, J. Am. Chem. Soc., 2000, 122, 3642–3651 CrossRef CAS.
- P. B. Zetterlund, Polym. Chem., 2011, 2, 534–549 RSC.
- R. P. Brinkhuis, F. P. Rutjes and J. C. van Hest, Polym. Chem., 2011, 2, 1449–1462 RSC.
- M. Massignani, H. Lomas and G. Battaglia, Polymersomes: A Synthetic Biological Approach to Encapsulation and Delivery, 2010, pp. 1–40 Search PubMed.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS PubMed.
- W. Meier, C. Nardin and M. Winterhalter, Angew. Chem., Int. Ed., 2000, 39, 4599–4602 CrossRef CAS.
- O. Onaca, M. Nallani, S. Ihle, A. Schenk and U. Schwaneberg, Biotechnol. J., 2006, 1, 795–805 CrossRef CAS PubMed.
- V. Malinova, S. Belegrinou, D. de Bruyn Ouboter and W. P. Meier, Organic Electronics, Springer, 2010, pp. 213–258 Search PubMed.
- J. M. Fletcher, R. L. Harniman, F. R. H. Barnes, A. L. Boyle, A. Collins, J. Mantell, T. H. Sharp, M. Antognozzi, P. J. Booth, N. Linden, M. J. Miles, R. B. Sessions, P. Verkade and D. N. Woolfson, Science, 2013, 340, 595–599 CrossRef CAS PubMed.
- H. Gradišar, S. Božič, T. Doles, D. Vengust, I. Hafner-Bratkovič, A. Mertelj, B. Webb, A. Šali, S. Klavžar and R. Jerala, Nat. Chem. Biol., 2013, 9, 362–366 CrossRef PubMed.
- Y.-T. Lai, E. Reading, G. L. Hura, K.-L. Tsai, A. Laganowsky, F. J. Asturias, J. A. Tainer, C. V. Robinson and T. O. Yeates, Nat. Chem., 2014, 1065–1071 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2015 |