
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The impact of steam on the electronic structure of the selective propane oxidation catalyst MoVTeNb oxide (orthorhombic M1 phase)†
Christian
Heine
a,
Michael
Hävecker
ab,
Annette
Trunschke
a,
Robert
Schlögl
a and
Maik
Eichelbaum
*ac
aDepartment of Inorganic Chemistry, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: me@fhi-berlin.mpg.de; Fax: +49 (0)30 84134405; Tel: +49 (0)30 84134566
bSolar Energy Research, Helmholtz-Zentrum Berlin/BESSY II, Albert-Einstein-Straße 15, 12489 Berlin, Germany
cBasCat, UniCat BASF JointLab, TU Berlin, Marchstraße 6, 10587 Berlin, Germany
First published on 25th February 2015
Abstract
The selective propane oxidation catalyst MoVTeNb oxide M1 was investigated by microwave conductivity, synchrotron X-ray photoelectron, soft X-ray absorption and resonant photoelectron spectroscopy under reaction conditions to identify the influence of steam on the electronic bulk and surface properties. Steam significantly increases both the conversion of propane and the selectivity to the target product acrylic acid. The increased catalytic performance comes along with a decreased conductivity, a modification of the surface chemical and electronic structure with an enrichment of covalently bonded V5+ species to the extent of Mo6+, a decreased work function and hence polarity of the surface and a modified valence band structure. The higher degree of covalency in metal oxide bonds affects the mobility of the free charge carriers, and hence explains the decrease of the conductivity with steam. Furthermore we could prove that a subsurface space charge region depleted in electrons and thus an upward bending of the electronic band structure are induced by the reaction mixture, which is however not dependent on the steam content.
1 Introduction
Acrylic acid with an annual global production of 4.7 million tonnes (2012) is an important intermediate in the chemical industry for the production of polyacrylates and methacrylate esters for diverse applications, e.g. for superabsorbers. It is currently mostly manufactured with a yield of up to 80% by a two step heterogeneous catalytic process starting with the oxidation of propylene to acrolein followed by its oxidation to acrylic acid.1 Propylene is produced by steam cracking of the naphtha distillation fraction of crude oil. The endothermic steam cracking process is highly energy-consuming due to the high temperatures needed (up to 850 °C) and the necessary steam generation. The upcoming scarcity of crude oil resources and the corresponding expected price increase are serious factors that would reduce the cost effectiveness of the current acrylic acid manufacture process. A competitive alternative could be the direct oxidation of propane from natural gas. The so-far most promising catalyst for direct acrylic acid production from propane with a current acrylic acid yield of about 50% is the orthorhombic MoVTeNb oxide M1 phase (ICSD No. 5509; in our publication simply abbreviated as “M1”).2–5 M1 is a multimetal mixed oxide that can be synthesized with high phase purity by powder X-ray diffractometry (XRD). It should be noted that XRD phase purity does not exclude inhomogeneities such as cation disorder or dynamic overlayers terminating the crystal surface, which can be different from the bulk structure.6,7 The (ideal) crystal structure of M1 provides hexagonal and heptagonal channels perpendicular to the (001) basal plane partially occupied by tellurium. The (001) basal plane is formed by corner-linked MO6 octahedra (M = molybdenum or vanadium) building a six and seven membered ring structure. MO7 (M = mainly niobium) pentagonal bipyramids connect the ring structures by sharing edges with the octahedra. Along the c-direction the (001) basal planes are linked by oxygen atoms.In the oxidation of propane with oxygen on M1, water (i.e. steam) plays a peculiar role in the selective oxidation reaction. In dry feed containing only propane and oxygen, propylene formed by oxidative dehydrogenation (ODH) is the main product (besides CO and CO2), while the selectivity to acrylic acid is rather low.8 However, the selectivity to acrylic acid is substantially increased up to 60% if steam is added to the reaction feed.8,9 It was proposed that water increases the desorption rate of acrylic acid on the catalyst surface preventing its over-oxidation (e.g. by transforming surface carboxylates into acrylic acid), but it also changes the nature of the active sites.8 In general, recent studies indicate that the bulk phase of M1 provides the framework of the active surface which is formed only under reaction conditions.7,9,10 While during the catalytic reaction the bulk structure of M1 is unaffected,8 the surface structure changes depending on the composition of the gas phase. Near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) exhibited that the average oxidation states of vanadium and molybdenum are substantially increased at the surface.7,10 Molybdenum is only found in the formal oxidation state of 6+ on the surface, while in the bulk a ratio of molybdenum(V) and (VI) is needed to form the crystal structure.4 Vanadium is a redox active element in M1 with a gas phase-dependent oxidation state, as known from a careful analysis of the vanadium 2p3/2 core level. The addition of water to the propane feed, for triggering the production of acrylic acid, induces an increase in the V5+ concentration7,10 which is suggested to be essential for the alkane activation step.10,11 The V4+ concentration is unaffected by the water content of the propane feed7,8,10 supporting the assumption of the unique role of V5+ in the acrylic acid formation. Moreover, the tellurium content increases while the molybdenum content decreases with steam.7,10 That is why it has been suggested that tellurium is also part of the active site.9
In catalysis chemical modifications on the surface of the catalyst are in general interpreted in terms of a localized molecular model. However, regarding the crystalline, n-type semiconducting MoVTeNb oxide M112 the collective solid state properties such as the valence and conduction band structures, work function and electrical conductivity also have to be taken into account. It was proven recently by microwave conductivity, which is a contactless technique allowing the investigation of the electrical properties of catalysts in a fixed-bed reactor without electrode contact resistance limitations,13 and NAP-XPS studies that M1 acts as a semiconducting gas sensor by variation of the reaction conditions.12 The conductivity, work function and valence band onset are adjusted reversibly to the applied chemical potential of the gas phase. Consequently, an active charge transfer between the bulk and the gas phase (or adsorbates) was proven indicating the relevance of the semiconducting properties of the bulk phase in controlling the catalytically active surface structure.
In our contribution we want to identify how steam modifies the active surface layer catalyzing the selective oxidation of propane to acrylic acid, and if it also changes the collective electronic properties of the M1 phase. The latter was investigated by microwave conductivity measurements under dry and wet feed conditions at ambient pressure. The results are compared and interpreted with surface sensitive synchrotron-based in situ NAP-XPS and near-edge X-ray absorption fine structure (NEXAFS) investigations at 0.25 mbar under dry and wet propane oxidation conditions to identify structure–function relationships between the local electronic surface structure of M1, and its collective electronic and catalytic properties. Core level spectra of vanadium 2p3/2, molybdenum 3d, tellurium 3d and oxygen 1s, valence band spectra and the work function were analyzed. Moreover, the contribution of vanadium to the electronic structure of the valence band and gap states was investigated in detail by resonant photoelectron spectroscopy (resPES) at the vanadium 2p threshold (vanadium L2,3-edge).
2 Experimental
2.1 Sample
The MoVTeNb oxide M1 phase (internal ID.: 10790) was prepared according to a precipitation procedure described previously.12 Relative metal contents of 65 at% Mo, 6 at% Te, 19 at% V and 10 at% Nb were determined by X-ray fluorescence analysis. The surface area of the sample is 9.2 m2 g−1 as measured by Brunauer–Emmett–Teller (BET) nitrogen adsorption. The Rietveld analysis of the X-ray powder diffraction (XRD) pattern proved the phase purity of the used M1 powder. It has also been proven by in situ XRD that the bulk phase is stable under all applied reaction conditions.82.2 Microwave conductivity measurements
The electrical conductivity of M1 under reaction conditions was measured by the microwave cavity perturbation technique. In the depolarization regime, the semiconducting sample located in a microwave cavity causes a perturbation of the resonant electromagnetic field decreasing its quality factor Q and resonance frequency ω0.14 The dielectric function (permittivity) of the sample (ε = ε1 + iε2) can be computed by using the quality factor/resonance frequency of the perturbed (Qs/ωs) and unperturbed cavities (Q0/ω0).15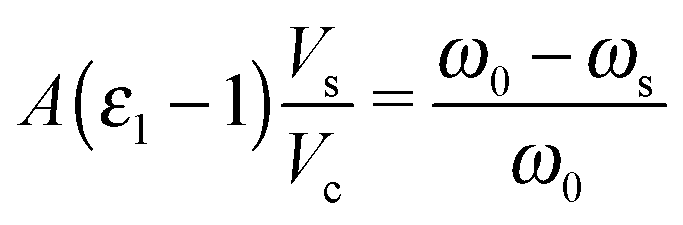 | (1) |
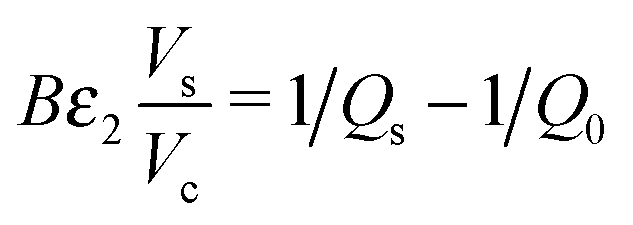 | (2) |
| σ = ε0ωsε2 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 333 N mL (g h)−1 (contact time: 0.27 s g N mL−1) with a catalyst mass of 90 mg and a bed length of 10.6 mm in a quartz tube reactor with a 3 mm inner diameter. A particle sieve fraction between 100 and 200 μm was used for the experiment. The measurement was performed under wet and dry feed conditions containing 3 vol% propane, 6 vol% oxygen and 5 vol% steam for the wet feed with residual N2 gas at 350 °C.
333 N mL (g h)−1 (contact time: 0.27 s g N mL−1) with a catalyst mass of 90 mg and a bed length of 10.6 mm in a quartz tube reactor with a 3 mm inner diameter. A particle sieve fraction between 100 and 200 μm was used for the experiment. The measurement was performed under wet and dry feed conditions containing 3 vol% propane, 6 vol% oxygen and 5 vol% steam for the wet feed with residual N2 gas at 350 °C.
2.3 Near-ambient pressure X-ray photoelectron and X-ray absorption spectroscopy
X-ray photoelectron spectroscopy (XPS), resonant photoelectron spectroscopy (resPES) and near edge X-ray absorption fine structure (NEXAFS) studies were performed at the ISISS (Innovative Station for In Situ Spectroscopy) beamline at the synchrotron facility BESSY II in Berlin, Germany. Monochromatic light was used to record high resolution in situ XP and NEXAFS spectra. A detailed description of the differentially pumped hemispheric electron analyzer and the in situ vacuum chamber was reported before.19,20 For the measurements the sample powder was pressed into a self-supporting pellet of 8 mm diameter (1 ton pressing pressure). The experiment was performed at 25 Pa and 400 °C (heating rate 5 K min−1). The reaction feed contained 1 N ml min−1 propane, 2 N ml min−1 oxygen and 3 N ml min−1 helium for the dry feed conditions and 1 N ml min−1 propane, 2 N ml min−1 oxygen and 3 N ml min−1 steam for the wet feed conditions. The experiment was started in the wet feed, followed by dry and finally changed again to wet feed conditions.NEXAFS experiments were performed in the total electron yield (TEY) mode and in the Auger–Meitner electron yield (AMEY) mode. The photon energy was varied from 505 to 560 eV by continuously moving the monochromator at a constant velocity of 217 meV s−1 to record the vanadium L-edges and the oxygen K-edge. The exit slit setting was 111 μm. The energy positions of every raw data points were read back from the monochromator control. The generated electrons are accelerated to a Faraday cup by the electric field (applied voltage) and the photocurrent is measured. The AMEY data were acquired using an electron spectrometer setting the kinetic energy at 385 eV (pass energy 20 eV) to suppress contributions of the gas phase to the AMEY, in particular from molecular oxygen. Details about the methodology to suppress gas phase contributions in the NEXAFS signal were reported before.21
Core level spectra of O1s, V2p, Mo3d and Te3d were performed at a pass energy of 20 eV with constant kinetic electron energies of 150 and 650 eV, respectively, corresponding to inelastic mean free paths (IMFPs) of 0.6 nm (surface sensitive) and 1.6 nm (“bulk” sensitive). The IMFP was calculated for MoO3 using the model of Tanuma et al.22 Normalized core level intensities were evaluated after subtraction of a Shirley type background (BG) taking into account the photon energy dependence of the atomic subshell photoionization cross sections, using CASA data analysis software (Neil Farley, http://www.casaxps.com). Valence spectra and secondary electron cutoffs of the photoelectron spectra were recorded at a photon energy of 100 eV. A voltage of −18 V was applied between the sample and the photoelectron detector to measure the cutoff of the photoelectron spectrum. The electron escape vector was perpendicular to the surface of the pellet to avoid a distortion of the electron trajectories by the electric field.23 In the case of the resPES measurements at the vanadium 2p threshold, the valence band spectra were recorded at characteristic points of the vanadium L2,3-edge. The monochromator was operated in a higher diffraction order suppression mode (c = 1.4). Further details about the measurement are discussed in the results section.
3 Results and discussion
3.1 Microwave conductivity measurements
Microwave conductivity measurements of M1 in a fixed-bed reactor under propane oxidation conditions were performed at 350 °C and with a contact time of 0.27 s g N ml−1 to achieve a low propane conversion of <5% (differential conditions) for better comparison with the low pressure in situ XPS results (propane conversion of less than 1%) and to avoid concentration (and conductivity) gradients across the sample. Fig. 1a shows the microwave conductivity of M1 obtained under dry and wet feed conditions, while the simultaneously measured catalytic performance is plotted in Fig. 1b. In the wet feed containing 3% propane, 6% oxygen and 5% steam the selectivity to acrylic acid is significantly increased from 10% to 30%, as compared to that under the conditions without steam. The propane conversion increases also from 3% to 4%. In contrast, the selectivities to propylene (from 80% to 60%) and to the undesired total oxidation products CO and CO2 (from ca. 7% to 4%) decrease with steam. These reversible and stable trends and even the absolute values are basically in agreement with the catalytic data reported recently for similar conversions in a comprehensive reactivity study of M1 tested in a broad parameter field.8 Based on these results the effect of water was interpreted in terms of the suppression of a decomposition of acrylic acid to CO and CO2, explaining the increased selectivity to acrylic acid and the decreased selectivity to COx. However, the increased conversion has to be explained by an additional effect of water on the nature of the active sites with increased turn-over frequencies and decreased activation energies. The investigation of this latter effect is the focus of the present study.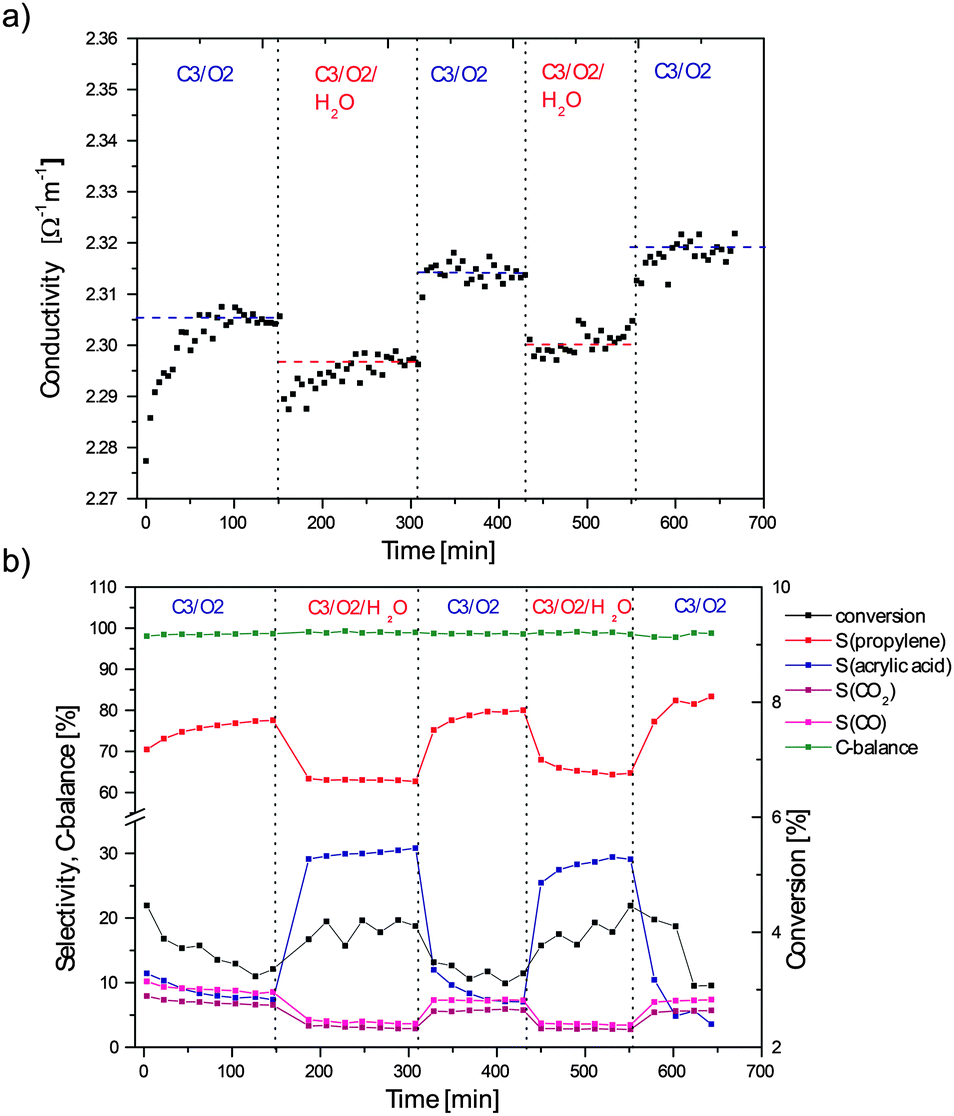 | ||
| Fig. 1 (a) Microwave conductivity of M1 under dry and wet propane feed conditions at 350 °C and (b) simultaneously measured catalytic data. | ||
Upon reaching the final temperature of 350 °C (time point “0” in Fig. 1a) the conductivity increased until it stabilized at a value of about 2.305 Ω−1 m−1 after 1.5 hours. This value is in the range of the conductivity measured for pure silicon at 350 °C, which is much smaller than that for doped silicon samples,24 but orders of magnitude higher than determined for the selective n-butane oxidation catalyst vanadyl pyrophosphate (VPO).25 The wet feed conditions were applied after the conductivity value remained constant for 1 hour. The feed change induced an immediate decrease followed by a slow increase in conductivity to a constant value still lower than the orignal conductivity in the dry feed. After applying the dry gas mixture for the second time the conductivity increased instantaneously to a constant value slightly above the value measured under the first dry conditions. The repeated change to wet and then again dry feed gave qualitatively the same results as obtained before with a generally lower conductivity in the presence of steam.
In a previous study probing the microwave conductivity of M1 as a function of dry alkane oxidation feeds (with different alkanes, ethane, propane, n-butane and different alkane/oxygen ratios), the conductivity decreased while going from reducing to oxidizing conditions indicating the n-type semiconductivity of M1 with electrons as majority charge carriers.12 In this study the work function, the valence band and core levels of M1 were additionally investigated by NAP-XPS. As a result, the work function of M1 increased from reducing to oxidizing gas atmospheres. The analysis of the peak area and shape of the core level spectra exhibited no significant changes of the surface elemental composition, but a slight change in the vanadium oxidation state. Furthermore, the core and valence levels shifted to lower energies in oxidizing and to higher energies in reducing feeds. This can be explained by a typical semiconductor response of a gas sensor, where the Fermi level of the solid phase is pinned to the surface state energy modified by the adsorbates.26 As a consequence, the conduction and valence bands and all core levels are bent towards the surface, as indicated by the consistent shift to lower binding energies, because charge carriers are transferred between the surface (adsorbates) and the bulk phase, while a subsurface space charge region and an oppositely charged surface layer are formed. The electrical conductivity σ(x) depends on the spatially varying energy interval between the conduction band onset EC and the Fermi energy EF in the space charge region26 after
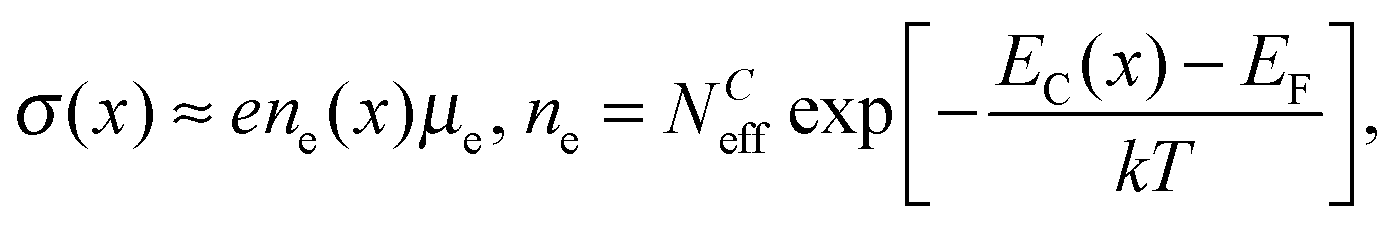 | (4) |
3.2 Core level spectra
We analyzed oxygen 1s, vanadium 2p, molybdenum 3d and tellurium 3d core level spectra of M1 under dry and wet feed conditions by NAP-XPS. Two questions will be addressed by this study: (1) can we prove a bending of bands under selective propane oxidation conditions and hence the existence of a space charge region extending from the surface into the bulk by measuring the binding energy of photoelectrons in different depths of the catalyst? (2) If band bending is observed, does it depend on steam, which would allow a correlation between the selectivity to acrylic acid, showing a strong dependence on steam content, and the semiconducting response of the catalyst? The core levels were recorded in a surface sensitive (photoelectron kinetic energy: 150 eV; inelastic mean free path (IMFP): 0.6 nm, meaning that 63% of the photoelectrons were released within this depth) and “bulk” sensitive (650 eV, IMFP: 1.6 nm) mode to study the extension of the space charge region from the surface into the bulk and to discriminate between upward (i.e. formation of a depletion layer) and downward band bending (accumulation layer). A steam-dependent band bending can be proven if the core level binding energies are systematically shifted between dry and wet reaction feeds. In Fig. 2a the surface-sensitively recorded O1s and V2p spectra are shown. It is obvious that the maximum of the O1s peak appears at the same binding energy position independent of the applied feed conditions. Moreover, difference spectra between measurements in dry and wet feed gave no hint for a specific contribution to the O1s spectrum that is directly caused by the addition of steam to the gas phase (Fig. S1, ESI†).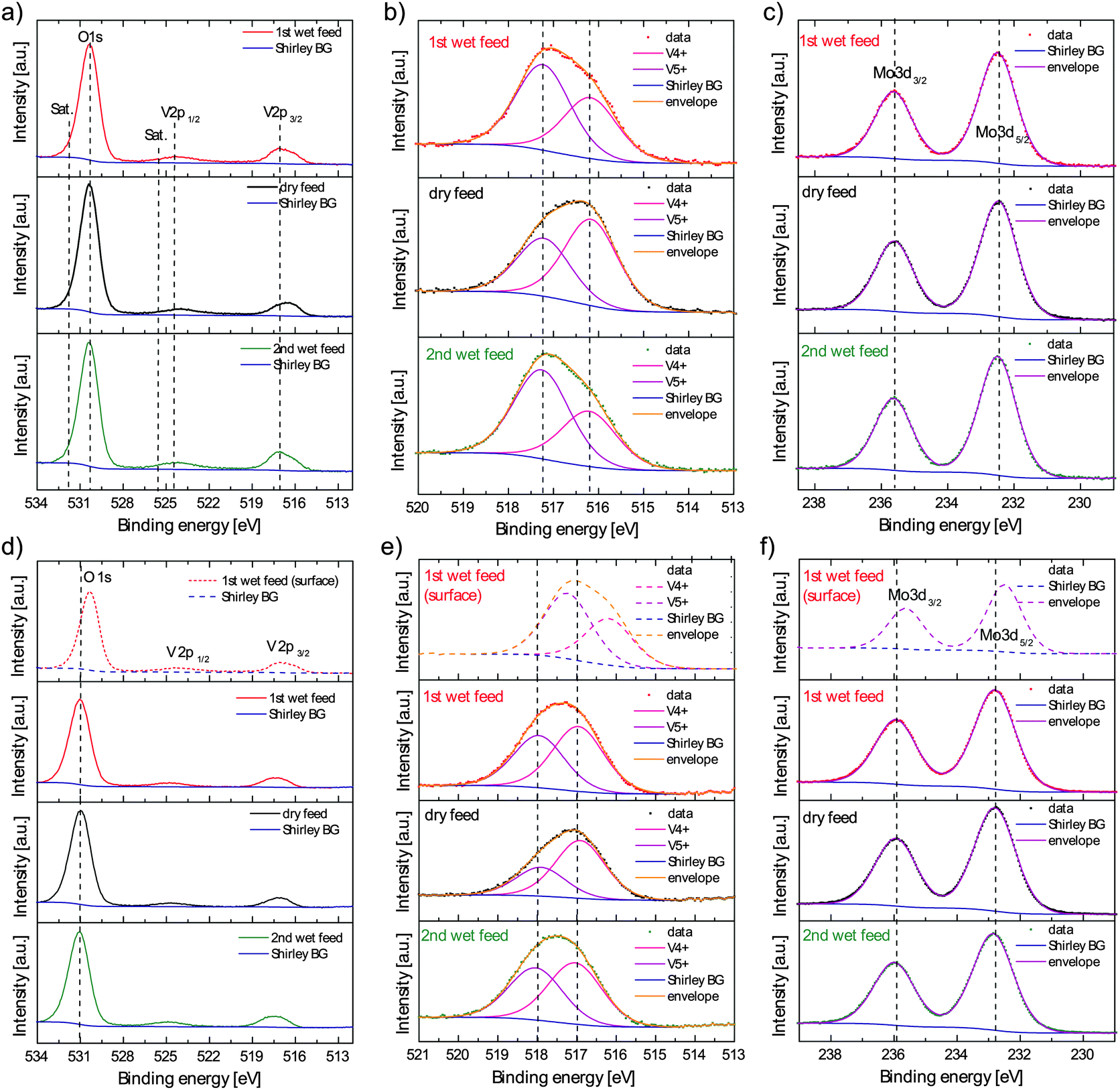 | ||
| Fig. 2 Surface sensitively recorded O1s and V2p (a), V2p3/2 (b), and Mo3d (c), and bulk sensitively recorded O1s and V2p (d), V2p3/2 (e) and Mo3d (f) core level spectra (always compared to a surface spectrum in the first row) in dry and wet propane oxidation feeds. | ||
Since the satellite of the V4+ contribution to the V2p3/2 spectrum overlaps with the V2p1/2 peak (Fig. 2a), only the undistorted V2p3/2 core level was used to analyze chemical shifts. The V2p3/2 spectra are shown in detail in Fig. 2b. The spectra were fitted with two Gaussian–Lorentz peaks corresponding to V4+ and V5+ components.7,8,10 Though a clear increase of the V5+ contribution is observed with steam, the absolute binding energies are for both oxidation state components independent of the feed.
The Mo3d doublet is depicted in Fig. 2c. Independent of the applied feed conditions this core level can be fitted with just one component7,8,10 corresponding to Mo6+.27 Again, the maximum of this spectrum is not shifted between dry and wet propane oxidation feeds, which is in agreement with the other core level spectra, but in apparent contradiction to the previous assumption correlating the conductivity change with a gas phase dependent modification of the band bending, as indicated in Fig. 4.
In Fig. 2d, e and f the bulk sensitively recorded O1s, V2p3/2 and Mo3d core level spectra are shown and compared by the surface sensitive investigations. The maxima of the bulk sensitively measured core level spectra of O1s, V2p, Mo3d and also Te3d (Fig. S2, ESI†) are shifted by about 0.6 eV to higher binding energies. This trend shows clearly that the core levels measured in a deeper region of the catalyst are systematically shifted to higher binding energies, meaning that the difference between the respective core and the Fermi level (at 0 eV binding energy per definition) increases with increasing probing depth. This effect can have two possible reasons. First, the core level shifts could be caused by charging of the surface by the X-ray beam.28 The photon intensity increases with increasing photon energy at the ISISS beam line. Thus a possible charging of the catalyst surface is more pronounced at higher photon energies. However, charging affects the peak shape of the core levels inducing asymmetries. Since the Mo3d spectra shown in Fig. 2c and f, which can be fitted with one component, are highly symmetric, charging can be excluded as explanation. Another possible reason for the binding energy shift is an upward bending of the valence and conduction bands at the M1 catalyst surface. In the case of the n-type semiconducting M1 phase the upward band bending is induced by the formation of an electron-depleted space charge region (depletion layer). The surface of M1 is indeed more oxidized than the bulk as was proven previously.7,8,10,12 This oxidized surface provides electron acceptor states which act as traps for mobile electrons in the bulk and obviously cause a positively charged depletion layer extending into the bulk. It should also be mentioned that in XPS a surface photovoltage can arise due to the excitation of electrons by X-rays to higher states (final state effect).29 The induced electron and hole charge carriers are separated due to the band bending, i.e. electrons are accelerated into the bulk and holes to the surface, the electric field within the space charge region is compensated, and the upward bending of the bands is decreased. The same circumstance can also be present in our study – even though the applied high temperature and the usage of a polycrystalline sample at a usually high electron–hole recombination rate should make it rather unlikely – but does not influence the conclusion, since it would only decrease the initial binding energy shifts.
Consequently, under propane oxidation conditions a depletion layer is formed proving the semiconducting response of the catalyst with charge transfer between the surface and the bulk under working conditions. However, this depletion layer or upward band bending is not influenced further by steam. This indicates that the absolute number of charge carriers in the catalyst is not affected by the presence of water in the gas phase.
3.3 Valence band spectra and secondary electron cutoff (work function measurements)
Since steam does not influence the band bending, its influence on the valence band and the work function was elucidated. Changes in the valence band structure can denote modified bonding motifs of the surface metal oxide moieties, while a change in the work function would directly indicate a modification of the dipole moments and hence polarity of the surface. Valence band spectra of M1 in wet and dry feeds are shown in Fig. 3. The absolute intensities of the valence band measured in the wet feed were increased by a factor of two in comparison to the dry feed. Interestingly, this effect was also observed in ultra-high vacuum valence band studies of transition metal oxides investigating the effect of chemisorbed water,30,31 though the reason remained unclear.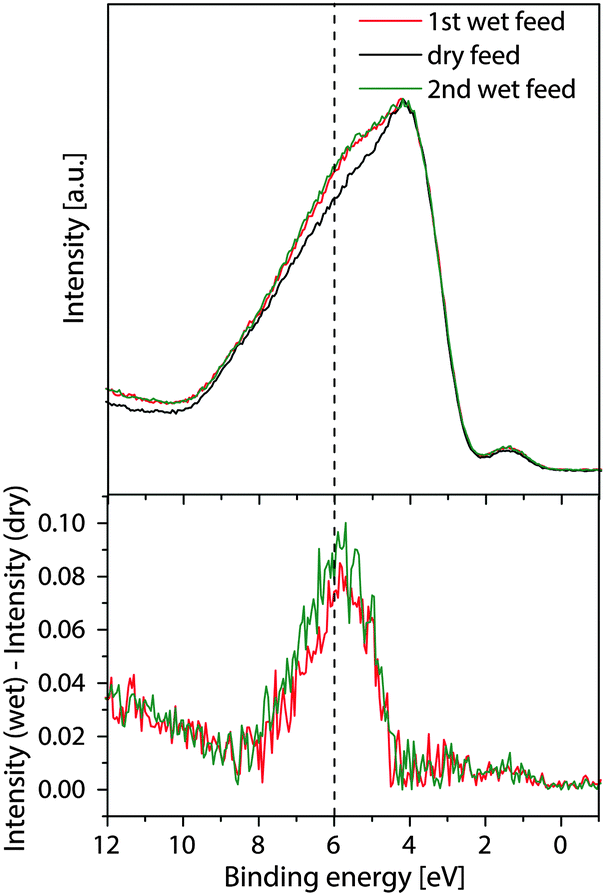 | ||
| Fig. 3 Valence band spectra (normalized to their maximum) and difference spectra between wet and dry feeds recorded at 100 eV photon energy. | ||
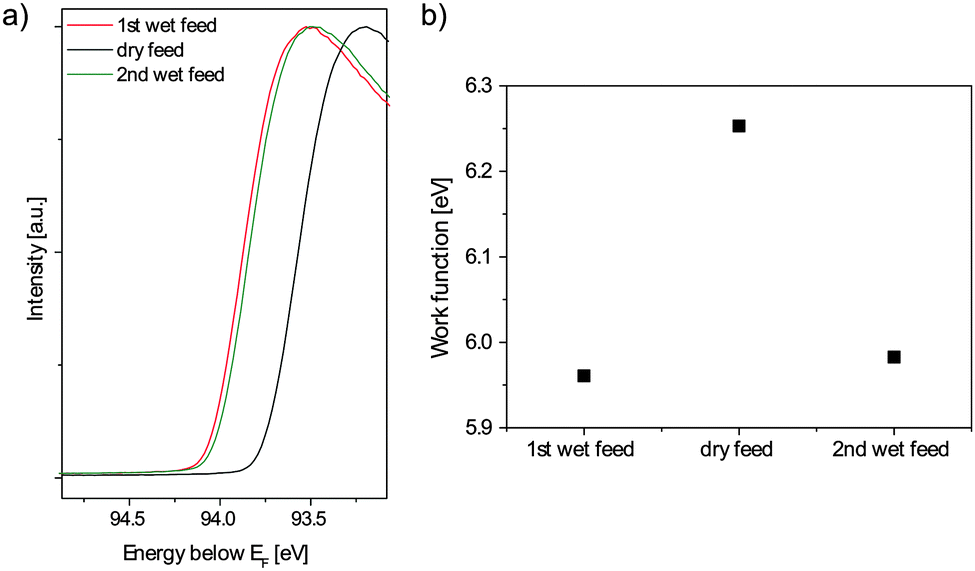 | ||
| Fig. 4 (a) Secondary electron cutoff and (b) calculated work function of M1 in dry and wet propane oxidation feeds. | ||
For a better comparison all valence band spectra depicted in Fig. 3 were normalized to their maximum. As a result, no binding energy shifts of the valence band edge dependent on steam are observed. This is in agreement with the non-shifted core level energies, hence definitely excluding the effect of water on the subsurface depletion layer and band bending. Besides, a clear difference between the relative contributions of the different components to the valence band is observed upon the addition of steam (bottom of Fig. 3). The relative spectral intensity between 4 and 8 eV is clearly enhanced in the wet feed. An increased intensity with steam is also measured above the binding energy of 10 eV. The feature at 1.5 eV is basically not affected relative to the maximum of the valence band. The assignment of different contributions to the valence band will be discussed in the resonant photoelectron spectroscopy section.
The secondary electron cutoff of the photoelectron spectrum of M1 in wet and dry feeds recorded with an excitation energy of 100 eV is shown in Fig. 4a. The cutoff shifts reversibly to higher binding energies upon the addition of water to the feed. With the known energy position of the cutoff Ecutoff, measured at half maximum of the leading edge, and the photon energy Eℏω, the work function Φ can be calculated:23
| Φ = Eℏω − Ecutoff | (5) |
Work function changes can originate from a modification of the band bending and/or of the surface dipolar structure, i.e. by a change in the surface electron affinity.26 Since the effect of steam on band bending can be excluded based on the valence and core level spectra, the work function change results obviously from modified surface dipoles. As reported previously, steam significantly changes the surface composition of M1.7,8,10 While the relative metal abundance of vanadium(V) (from 5 to 15%) and tellurium (from 16 to 25%) increases, a decrease in molybdenum from 55 to 35% is observed. It is therefore comprehensible that this drastic compositional change might impact the dipole momenta on the surface and hence changes also the work function of the catalyst. However, a decrease of the work function upon the adsorption of water was also observed in ultraviolet photoelectron spectroscopy studies of the binary oxides V2O330 and α-Fe2O3.31 Hence also the formation of, e.g., OH groups that can neutralize polar surfaces, and/or probably trigger a surface reconstruction due to the reversible formation of metal hydroxides (e.g. of molybdenum), could give rise to the observed steam-induced reduction of the work function.
Summing up, steam does not change the charge carrier density of conduction electrons at the surface and subsurface of M1 since the band bending is not influenced. However, it modifies reversibly the valence band structure and surface polarity of the catalyst. In situ NEXAFS and resonant photoelectron spectroscopy were therefore performed to investigate these findings in more detail.
3.4 Near-edge X-ray absorption fine structure (NEXAFS) spectroscopy
The influence of steam on the electronic structure of M1 was further investigated by near-ambient pressure NEXAFS experiments. In Fig. 5a the vanadium L3-edge spectrum of M1 recorded in the total electron yield (TEY) mode is shown under dry and wet propane oxidation conditions at 400 °C. For a better comparison, the linear-background corrected spectra were normalized to their maximum. Six prominent features can be identified. The intensities of features I (514.9 eV) and II (515.5 eV) decrease under wet feed conditions, while features III (516.0 eV) and IV (516.4 eV) remain unaffected. Features V (dry: 517.4 eV, wet: 517.6 eV) and VI (dry: 517.9 eV, wet: 518.2 eV) are reversibly shifted by 0.2–0.3 eV to higher photon energies in the steam containing feed. Moreover, the relative intensity of feature V is decreased in the wet feed and can then only be identified as a shoulder on the low energy side of the maximum.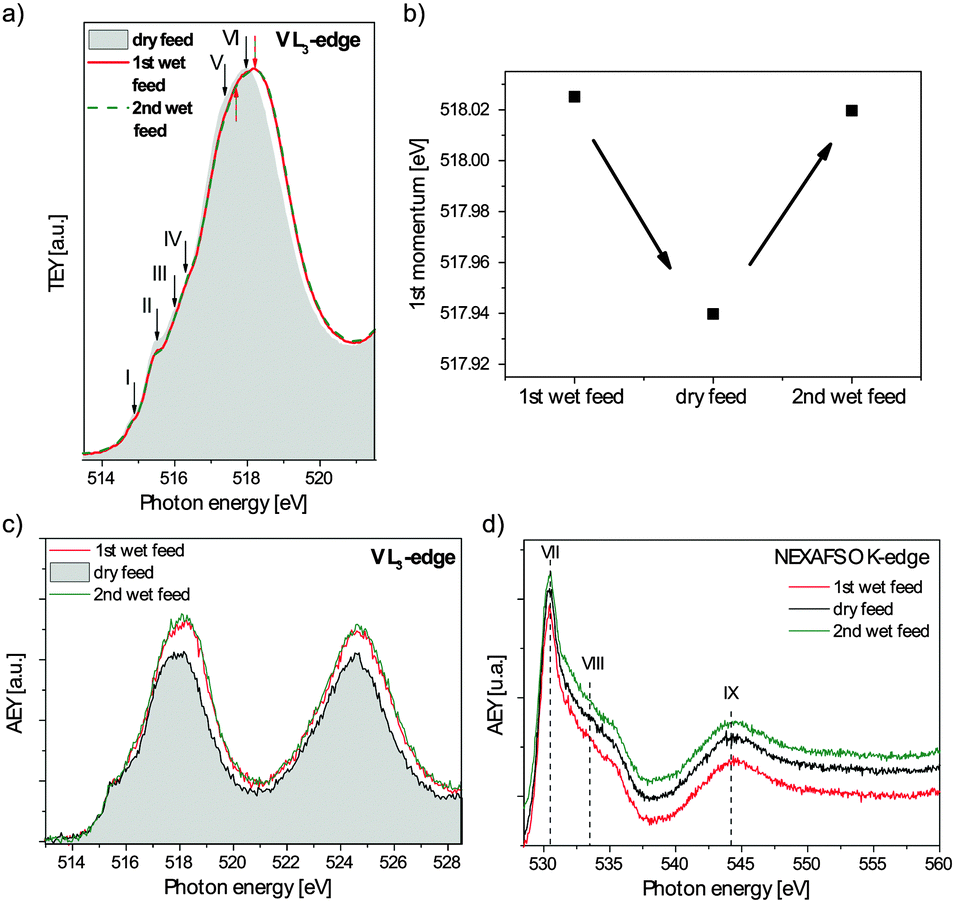 | ||
| Fig. 5 (a) Vanadium L3-edge (TEY mode), (b) 1st momentum of the vanadium L3-edge, (c) vanadium L2,3-edges (AMEY mode), and (d) oxygen K-edge in both wet and dry feeds (stacked plot with a constant off-set). | ||
It could be observed that energetic shifts in the spectral weight of the V L3-edge spectrum correlate with the average vanadium oxidation state of the material.36 This relationship can be understood in terms of the effective nuclear potential which affects the position of the electronic states relative to the vacuum level and the relative position of the corresponding core level.37 In the case of vanadium in the oxidation state 5+ the effective nuclear potential is larger than that for vanadium 4+. Thus, the vanadium(V) L3-edge is shifted to higher photon energies in comparison to the spectrum of a vanadium(IV) or (III) compound. For a more quantitative discussion of such changes, the 1st momentum of the spectral distribution at the V L3-edge can be calculated in order to precisely determine the spectral weight as a function of the gas composition.36 The 1st momentum M is calculated as
| M = 1/A∫ℏωTEYdℏω, | (6) |
The bulk crystal phase M1 is composed of vanadium atoms with formal oxidation states of 4+ and 5+ in an octahedral coordination sphere.2,4 According to density functional theory/restricted open shell configuration interaction single (DFT/ROCIS) calculations of V4+ and V5+ in distorted octahedral oxygen ligand fields, the low photon energy onset of the vanadium L3-edge consists of transitions into 3dxy,xz (in V5+ also 3dyz) orbitals,38,39 which could correspond to features I and II in the experimental spectrum (Fig. 5a). These orbitals are located between (3dxy) or above and below (3dxz, 3dyz) the V–O–V bridging bonds in the basal plane of the [VO]6 coordination polyhedra. Thus the decreased intensity of features I and II under wet feed conditions could be assigned to a structural modification of the 3dxy, 3dxz, and 3dyz orbitals, but a clear assignment to concrete bonding motifs in M1 is difficult. While according to the calculation the main peak of a V4+ L3-edge contains transitions into 3dyz, 3dxy, 3dx2−y2 and 3dz2 orbitals, it consists for V5+ species mainly of transitions into 3dx2−y2, while the contribution of 3dz2 is mainly found at the high energy onset. Hence a predominant shift upon a change in the oxidation state of vanadium from 4+ to 5+ can be observed for the transition into 3dz2. Thus a large shift of feature V to higher energies can be likely assigned to the 3dz2 transition and hence to an increase of the vanadium(V) concentration on the surface. Since an increase in energy for the transition into the 3dz2 orbital is correlated with the shortening of the vanadyl bond in a distorted octahedral oxygen ligand field,38,39 steam obviously affects basically the V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond motif on the surface of M1.
O bond motif on the surface of M1.
Previous XPS measurements of M1 indicated that not only the average oxidation state, but also the total vanadium content on the surface increases in steam-containing feeds.8 However, the aforementioned TEY NEXAFS mode cannot be used for a comparison of absolute signal intensities in different feeds due to secondary electrons created by electron impact ionisation of gas phase molecules (“environmental electrons”) that contribute strongly to the TEY signal and are hence gas phase dependent.40 In Fig. 5c vanadium L2,3-edge spectra recorded in the Auger–Meitner electron yield (AMEY) mode, normalized to the synchrotron ring current and linear-background corrected to the intensity value at 512 eV, are shown under wet and dry feed conditions in order to compare the absolute resonance intensities. In the AMEY mode Auger–Meitner electrons with a high kinetic energy are detected, which are less strongly scattered in the gas phase. The analyzer was set to a kinetic energy of 385 eV to suppress contributions of the gas phase (mainly O2) to the signal.21 In the wet feed the intensity of the vanadium L2,3-edge increases reversibly in comparison to the dry feed conditions. Assuming that the electron scattering and cascading processes in the M1 solid phase are hardly affected by the chemical modification of the surface due to the wet feed conditions, the AMEY intensity of the vanadium L2,3-edge should be proportional to the number of absorption centers. Even if the gas phase still disturbs the absolute measurement, since in the wet feed helium is replaced by steam which should induce an increase in photon absorption and inelastic photoelectron scattering, the AMEY signal would then have to decrease with steam. Since the addition of water leads to an increased signal instead, the vanadium content is obviously indeed enriched at the surface, hence supporting the XPS results.
In Fig. 5d a stacked plot of the oxygen K-edge measured also in the AMEY mode is shown. In all spectra three dominant features can be identified. Feature VII is located at 531 eV, a shoulder (feature VIII) can be found between 532 and 536 eV and a further feature IX is identified at about 545 eV. No change in the shape of the spectra due to the addition of water to the feed can be observed. The oxygen K-edge of M1 is comparable with the oxygen K-edges of MoO341 and V2O5.42 MoO3 also has three features which nearly coincide, with respect to their positions and shape, with the identified features in M1. Features VII and VIII can also be observed in V2O5, in contrast to feature IX which differs with respect to its position and shape. The main metal component in M1 is molybdenum. Its coordination to oxygen is similar to that observed in MoO3. In both oxides molybdenum is coordinated by six oxygen atoms in a distorted octahedral ligand field. In this context features VII (π* resonance) and VIII (σ* resonance) can be assigned to transitions into orbitals with the antibonding Mo4d–O2p character indicating covalent bonds between molybdenum and oxygen atoms,41 while feature IX is attributed to transitions into antibonding orbitals with Mo5s–O2p character.41 In a pure ionic oxide, the oxygen 2p orbitals are completely filled and no resonance occurs. Thus the oxygen K-edge dominated by contributions from molybdenum–oxygen bonds indicates that molybdenum to a large extent is covalently bonded in M1.
3.5 Resonant photoelectron spectroscopy (resPES) at the vanadium L2,3-edge
In order to investigate the electronic structure of the valence band in detail, resPES is the method of choice. In resPES contributions of a particular element to the valence band can be enhanced by selective excitation,43 if the normal photoelectron channel interferes with a second, element-specific excitation channel. In the case of vanadium excited at the V2p threshold both processes can be described in the following way:42| 2p63dn + ℏω → 2p63dn−1 + e− | (7) |
| 2p63dn + ℏω → [2p53dn+1]* → 2p63dn−1 + e− | (8) |
| |A + B|2 = |A|2 + |B|2 + cross term | (9) |
The valence band of M1 was recorded at photon energies corresponding to the vanadium L2,3-absorption edge (Fig. 6a). To check whether the enhancement is due to a resPES process or just an overlap (incoherent enhancement) of the valence band with the Auger–Meitner decay channel, the valence band recorded at different photon energies has beeen plotted versus the kinetic energy of the photoelectrons. The O KLL Auger–Meitner emission spectra, which could in principal overlap with the valence band, are shown in Fig. 6b. In the case of a non-resonant overlap, the apparent enhancement appears at constant kinetic energies. This is however not observed in our experiment (Fig. 6c). The valence band spectra, normalized to the ring current, plotted versus the binding energy are shown in Fig. 6d. Since the onsets of the valence band spectra coincide, no background correction of the spectra was necessary. In all spectra the off-resonance valence band spectrum recorded at 512 eV is additionally plotted (dashed red line) to highlight the resonance enhancement. It can be clearly observed that features at binding energies of 1.5 eV and in the range of 6–10 eV are enhanced, if they are excited at photon energies corresponding to the maxima of the vanadium L2,3-edge. A plot of the valence band intensities at 1.5 and 6 eV versus the applied photon energy is shown in Fig. 6e. As a result, the excitation energy dependence of the enhancement of the valence band spectrum follows exactly the vanadium L3- and L2-edge spectra with maxima at 517.9 and 524.5 eV, respectively.
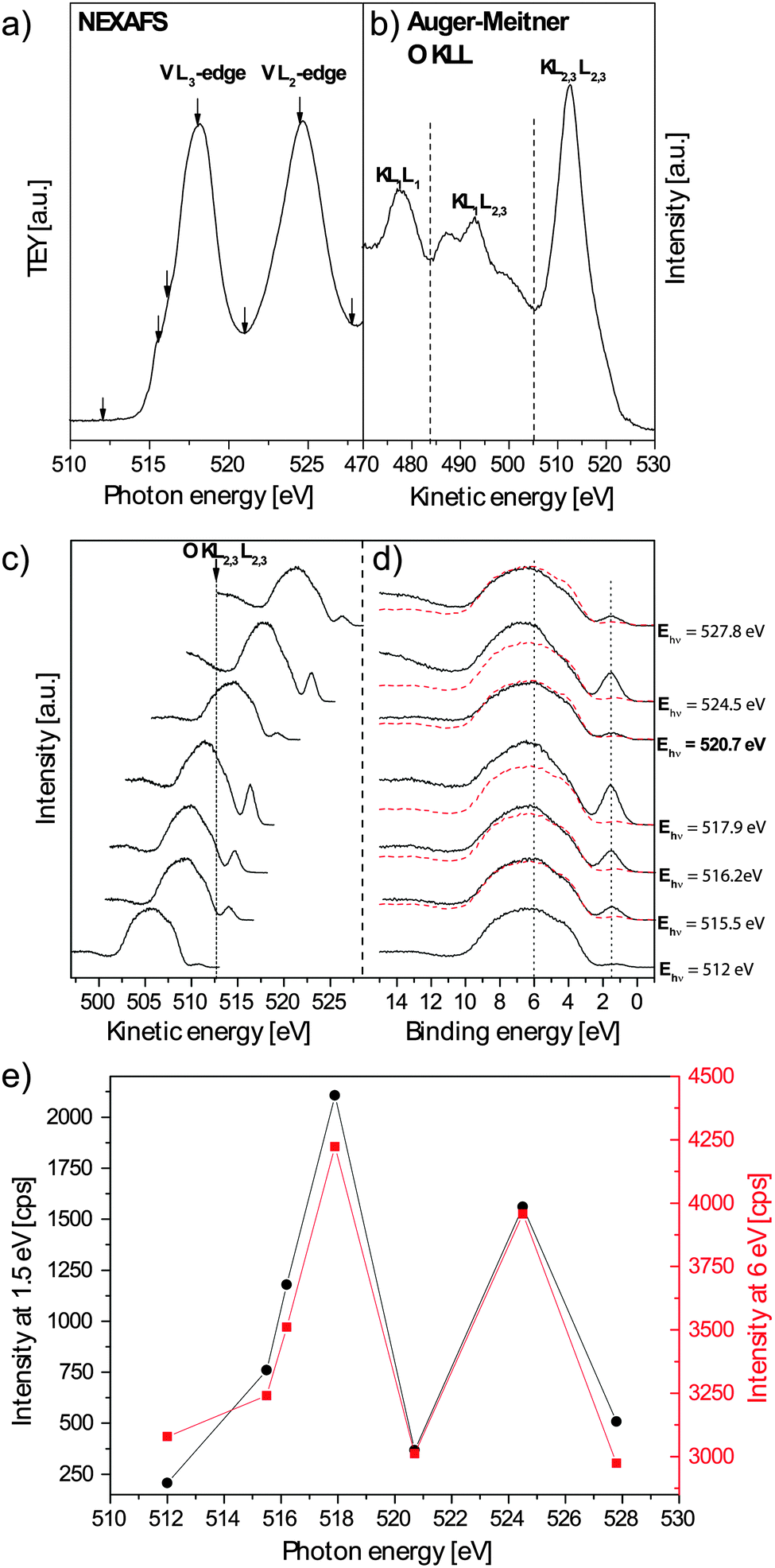 | ||
| Fig. 6 (a) Vanadium L2,3-edge with marked (arrows) photon energies at which the resPES spectra were recorded. (b) Oxygen KLL Auger–Meitner spectrum, (c) plot of resPES valence band spectra versus kinetic energy, (d) resPES valence band spectra versus binding energy, (e) intensity of the valence band at 1.5 and 6 eV binding energies versus excitation photon energy. | ||
Zimmerman et al. performed a resPES study of V2O3 and V2O5.42 In their investigation they identified a resonant enhancement for V2O3 at 1 eV binding energy which they attributed to an occupied vanadium 3d state with the oxygen 2p admixture. Additionally, they found an enhancement of above 6 eV, which was also observed for V2O5. In an ionic bonding model of binary vanadium oxides the valence band is composed of occupied O2p states, while the conduction band is formed by unoccupied V3d states. In contrast, in a covalent model allowing a hybridization of V3d with O2p states, the valence band has O2p and V3d character. Thus even V2O5, without 3d valence electrons in an ionic model, contains partially occupied V3d (or mixed V3d/O2p) states.42,44,45 Zimmermann et al. attributed the enhancement above 6 eV to the admixture of V3d states to the O2p valence band suggesting a strong covalent V–O bonding character in V2O5.42 Their conclusion is supported by DFT calculations42,44 and was also seen in resPES measurements at the V3p threshold.46,47
In M1 the situation is more complicated because besides vanadium, it also contains molybdenum, tellurium and niobium atoms. However, vanadium is coordinated by oxygen in a distorted octahedron as in V2O5 or V2O3. Thus the conclusion drawn for the resPES studies of V2O3 and V2O5 can be used to interpret the resonance spectra of M1 at the vanadium 2p threshold. Consequently, the enhancement at 1.5 eV binding energy in M1 can be assigned to a partially occupied V3d state. The enhancement between the binding energies 6 and 10 eV can be interpreted in terms of hybridization of V3d with O2p states similar to the results obtained for V2O3 and V2O5. Thus vanadium shows also in M1 a strong covalent bonding character. These results are in agreement with the interpretation of the oxygen K-edge spectra with σ* and π* resonance components (Fig. 5d), which is mainly attributed to the hybridization between molybdenum 4d and oxygen 2p orbitals and hence points to a strong covalent molybdenum–oxygen bonding character.
As discussed before, steam induced a significant relative increase of the signal intensity at binding energies in the 6–10 eV range of the valence band (Fig. 3). This can now be interpreted as an enhancement of the vanadium 3d state admixture to the valence band. This observation agrees with the results obtained from NEXAFS AMEY mode measurements at the L2,3-edge (Fig. 5c) and from NAP-XPS results,7,10 where an increased vanadium abundance on the surface was observed upon the addition of water to the feed. In the studies on the adsorption of water on metal oxides, peaks in the valence band spectrum between 6 and 12 eV were assigned to undissociated, strongly chemisorbed, and dissociated water.48,49 For example, for ceria on platinum, signals at 10.8 and 7.6 eV were assigned to the 3s and 1p molecular orbitals of hydroxyl groups.49 Consequently, the increased emission above 10 eV observed for the M1 phase might also arise from the formation of OH groups on the surface of the catalyst upon dissociative adsorption of water. However, in V2O5 a very broad satellite of the valence band, which is also seen in other transition metal oxides occurs between the valence band and the oxygen 2s level above 10 eV.47,50 If a feature is enhanced in the valence band, the corresponding satellite is also enhanced because the main line(s) (valence band) and the satellite belong to the same ground state(s). Thus V3d states could also contribute to the increased intensity above 10 eV upon steam addition.
Since the peak at 1.5 eV binding energy can now be assigned to a (partially) filled vanadium 3d state, steam obviosly does not influence its occupation (Fig. 3). Interestingly, it is known from NAP-XPS studies that only the V5+ concentration increases upon steam addition, while the V4+ concentration remains constant.7,8,10 The constant and not steam-influenced intensity at 1.5 eV representing basically vanadium with 3d electrons such as V4+ is therefore in good agreement with these findings, while the enhancement of the valence band at 6–10 eV might be due to surface-enriched, covalently bonded V5+ species.
A changed valence (and likely conduction band) structure also means that the dispersion relation of the energy band and therefore the effective mass of the charge carriers is modified. According to the Drude–Lorentz model used to describe charge carrier dynamics in semiconductors,51 a modified effective mass m* changes the plasmon frequency ωp = nee2/(ε0m*) (with ne being the charge carrier density of conduction electrons, e the elementary charge and ε0 the vacuum permittivity) and therefore the mobility of the charge carriers μe = e2τ/m* (with τ being the relaxation time). This affects finally the conductivity as shown by eqn (4). Thus the modification of the effective mass and hence electron mobility would explain the decreased conductivity in a steam containing propane oxidation feed (cf.Fig. 1a). And furthermore, a higher covalency will also decrease the dipole moment in a metal oxide and consequently its work function, in agreement with the experimental observations.
4 Conclusions
The selective propane oxidation catalyst MoVTeNbOx (orthorhombic M1 phase) was investigated by microwave conductivity, synchrotron X-ray photoelectron, soft X-ray absorption and resonant photoelectron spectroscopy under reaction conditions to identify the influence of steam on its bulk and surface electronic properties. The investigation of steam-induced structural modifications might help us to understand the observed steam-induced increase of the selectivity to the target product acrylic acid. We found that steam decreases the electrical conductivity, increases the concentration of V5+ and Te6+ and decreases the relative amount of Mo6+ on the surface, decreases the work function and modifies the valence band structure. Furthermore, we could observe that the catalyst forms a subsurface space charge region depleted in electrons (upward band bending) under reaction conditions. This proves that the catalyst acts like a semiconducting gas sensor (chemiresistor), where the (bulk) conductivity can be used to sensor electronic changes on the surface. However, since the addition of steam to the reaction feed does not affect the extent of band bending, it obviously does not influence the number of charge carriers in the space charge region by an additional bulk surface charge transfer. This could mean that bulk charge carriers do not participate in the formation of acrylic acid. The presence of band bending and hence the formation of a high surface potential barrier, which electrons have to overcome on their way from the bulk to the surface, could be however necessary to terminate the bulk surface charge transfer, limit the formation of active oxygen species on the surface and to achieve finally the high selectivity to acrylic acid, while suppressing its total oxidation to CO2. The surface potential barrier and the limited bulk surface charge transfer would also explain the extraordinary stability of the MoVTeNbOx M1 bulk phase under reaction conditions.However, steam modifies strongly the electronic and chemical structure of the surface. V L3-edge NEXAFS experiments point to a particular modification of the vanadyl bond motif (VO) on the surface and changes in the valence band spectra suggest an enrichment of covalently bonded V5+ surface species. The increased covalent bonding character in the multi-metal oxide and the decreased work function indicating a decreased surface polarity might induce a decrease of the charge carrier mobility. This effect would hence explain the observed decreased conductivity in the steam-containing reaction feed. These results point to the conclusion that steam is basically needed to form the active surface layer that can selectively oxidize propane to acrylic acid.
Acknowledgements
We thank Dr Maricruz Sanchez-Sanchez for the synthesis of the MoVTeNb oxide M1 sample. The HZB staff is acknowledged for their continual support of the high-pressure electron spectroscopy activities of the FHI at BESSY II.References
- F. Cavani, Catal. Today, 2010, 157, 8–15 CrossRef CAS PubMed.
- P. DeSanto Jr, D. J. Buttrey, R. Grasselli, W. Pyrz, C. Lugmair, A. F. Volpe Jr, T. Vogt and B. Toby, Top. Catal., 2006, 38, 31–40 CrossRef PubMed.
- Y. Moro-oka, W. Ueda and K.-H. Lee, J. Mol. Catal. A: Chem., 2003, 199, 139–148 CrossRef CAS.
- H. Murayama, D. Vitry, W. Ueda, G. Fuchs, M. Anne and J. Dubois, Appl. Catal., A, 2007, 318, 137–142 CrossRef CAS PubMed.
- D. Vitry, J.-L. Dubois and W. Ueda, J. Mol. Catal. A: Chem., 2004, 220, 67–76 CrossRef CAS PubMed.
- W. D. Pyrz, D. A. Blom, N. R. Shiju, V. V. Guliants, T. Vogt and D. J. Buttrey, Catal. Today, 2009, 142, 320–328 CrossRef CAS PubMed.
- A. Celaya Sanfiz, T. W. Hansen, D. Teschner, P. Schnörch, F. Girgsdies, A. Trunschke, R. Schlögl, M. Hong Looi and S. B. Abd Hamid, J. Phys. Chem. C, 2010, 114, 1912–1921 Search PubMed.
- R. Naumann d’Alnoncourt, L.-I. Csepei, M. Hävecker, F. Girgsdies, M. E. Schuster, R. Schlögl and A. Trunschke, J. Catal., 2014, 311, 369–385 CrossRef PubMed.
- R. Schlögl, Top. Catal., 2011, 54, 627–638 CrossRef PubMed.
- M. Hävecker, S. Wrabetz, J. Kröhnert, L. I. Csepei, R. N. d’Alnoncourt, Y. V. Kolen’ko, F. Girgsdies, R. Schlögl and A. Trunschke, J. Catal., 2012, 285, 48–60 CrossRef PubMed.
- X. Rozanska, R. Fortrie and J. Sauer, J. Phys. Chem. C, 2007, 111, 6041–6050 CAS.
- C. Heine, M. Hävecker, M. Sanchez-Sanchez, A. Trunschke, R. Schlögl and M. Eichelbaum, J. Phys. Chem. C, 2013, 117, 26988–26997 CAS.
- M. Eichelbaum, R. Stöer, A. Karpov, C.-K. Dobner, F. Rosowski, A. Trunschke and R. Schlögl, Phys. Chem. Chem. Phys., 2012, 14, 1302–1312 RSC.
- J. C. Slater, Rev. Mod. Phys., 1946, 18, 441–512 CrossRef.
- L.-F. Chen, C. K. Ong, C. P. Neo, V. V. Varadan and V. K. Varadan, Microwave Electronics: Measurement and Materials Characterization, Wiley, 2004 Search PubMed.
- H. Looyenga, Physica, 1965, 31, 401–406 CrossRef CAS.
- D. C. Dube, J. Phys. D: Appl. Phys., 1970, 3, 1648–1652 CAS.
- C. Heine, M. Hävecker, F. Girgsdies, A. Trunschke, R. Schlögl and M. Eichelbaum, Appl. Phys. A: Mater. Sci. Process., 2013, 112, 289–296 CrossRef CAS PubMed.
- H. Bluhm, M. Hävecker, A. Knop-Gericke, M. Kiskinova, R. Schlögl and M. Salmeron, MRS Bull., 2007, 32, 1022–1030 CrossRef CAS.
- E. M. Vass, M. Hävecker, S. Zafeiratos, D. Teschner, A. Knop-Gericke and R. Schlögl, J. Phys.: Condens. Matter, 2008, 20, 184016 CrossRef.
- M. Hävecker, M. Cavalleri, R. Herbert, R. Follath, A. Knop-Gericke, C. Hess, K. Hermann and R. Schlögl, Phys. Status Solidi B, 2009, 246, 1459–1469 CrossRef.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1988, 11, 577–589 CrossRef CAS.
- M. G. Helander, M. T. Greiner, Z. B. Wang and Z. H. Lu, Appl. Surf. Sci., 2010, 256, 2602–2605 CrossRef CAS PubMed.
- G. L. Pearson and J. Bardeen, Phys. Rev., 1949, 75, 865–883 CrossRef CAS.
- C. Heine, M. Havecker, E. Stotz, F. Rosowski, A. Knop-Gericke, A. Trunschke, M. Eichelbaum and R. Schlogl, J. Phys. Chem. C, 2014, 118, 20405–20412 CAS.
- H. Lüth, Electronic Structure of Surfaces, Solid Surfaces, Interfaces and Thin Films, Springer Verlag, 2001, pp. 265–328 Search PubMed.
- R. Tokarz-Sobieraj, K. Hermann, M. Witko, A. Blume, G. Mestl and R. Schlögl, Surf. Sci., 2001, 489, 107–125 CrossRef CAS.
- B. J. Tielsch and J. E. Fulghum, Surf. Interface Anal., 1996, 24, 28–33 CrossRef.
- D. K. Schroder, Meas. Sci. Technol., 2001, 12, R16 CrossRef CAS.
- R. L. Kurtz and V. E. Henrich, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 6699–6706 CrossRef CAS.
- R. L. Kurtz and V. E. Henrich, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 3413–3421 CrossRef CAS.
- W. Li and D. Y. Li, J. Chem. Phys., 2005, 122, 064708 CrossRef CAS PubMed.
- V. E. Heinrich and P. A. Cox, The Surface Science of Metal Oxides, Univ. Pr., Cambridge, 1996 Search PubMed.
- J. Meyer, K. Zilberberg, T. Riedl and A. Kahn, J. Appl. Phys., 2011, 110, 033710 CrossRef PubMed.
- M. T. Greiner, L. Chai, M. G. Helander, W. M. Tang and Z. H. Lu, Adv. Funct. Mater., 2012, 22, 4557–4568 CrossRef CAS.
- M. Hävecker, A. Knop-Gericke, R. W. Mayer, M. Fait, H. Bluhm and R. Schlögl, J. Electron Spectrosc. Relat. Phenom., 2002, 125, 79–87 CrossRef.
- J. Chen, C. Kirn, B. Frähberger, B. Devries and M. Touvelle, Surf. Sci., 1994, 321, 145–155 CrossRef CAS.
- D. Manganas, M. Roemelt, T. Weyhermüller, R. Blume, M. Hävecker, A. Knop-Gericke, S. DeBeer, R. Schlögl and F. Neese, Phys. Chem. Chem. Phys., 2014, 16, 264–276 RSC.
- D. Manganas, M. Roemelt, M. Hävecker, A. Trunschke, A. Knop-Gericke, R. Schlögl and F. Neese, Phys. Chem. Chem. Phys., 2013, 15, 7260–7276 RSC.
- N. J. Shevchik and D. A. Fischer, Rev. Sci. Instrum., 1979, 50, 577–581 CrossRef CAS PubMed.
- M. Cavalleri, K. Hermann, S. Guimond, Y. Romanyshyn, H. Kuhlenbeck and H.-J. Freund, Catal. Today, 2007, 124, 21–27 CrossRef CAS PubMed.
- R. Zimmermann, R. Claessen, F. Reinert, P. Steiner and S. Hufner, J. Phys.: Condens. Matter, 1998, 10, 5697–5716 CrossRef CAS.
- N. Martensson, O. Karis and A. Nilsson, J. Electron Spectrosc. Relat. Phenom., 1999, 100, 379–393 CrossRef CAS.
- V. Eyert and K.-H. Höck, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 12727–12737 CrossRef CAS.
- G. A. Sawatzky and D. Post, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20, 1546–1555 CrossRef CAS.
- K. E. Smith and V. E. Henrich, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 9571–9580 CrossRef CAS.
- S. Shin, Y. Tezuka, T. Ishii and Y. Ueda, Solid State Commun., 1993, 87, 1051–1054 CrossRef CAS.
- W. R. Flavell, A. G. Thomas, J. Hollingworth, S. Warren, S. C. Grice, P. M. Dunwoody, C. E. J. Mitchell, P. G. D. Marr, D. Teehan, S. Downes, E. A. Seddon, S. Downes, E. A. Seddon, V. R. Dhanak, K. Asai, Y. Koboyashi and N. Yamada, Faraday Discuss., 1999, 114, 407–420 RSC.
- U. Berner, K. Schierbaum, G. Jones, P. Wincott, S. Haq and G. Thornton, Surf. Sci., 2000, 467, 201–213 CrossRef CAS.
- K. W. Goodman and V. E. Henrich, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 10450–10456 CrossRef CAS.
- R. Ulbricht, E. Hendry, J. Shan, T. F. Heinz and M. Bonn, Rev. Mod. Phys., 2011, 83, 543–586 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: O1s and Te3d X-ray photoelectron spectra of MoVTeNb oxide. See DOI: 10.1039/c5cp00289c |
| This journal is © the Owner Societies 2015 |