
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Band gap narrowing in nitrogen-doped La2Ti2O7 predicted by density-functional theory calculations†
Junying
Zhang
*a,
Wenqiang
Dang
a,
Zhimin
Ao
b,
Scott K.
Cushing
c and
Nianqiang
Wu
*c
aDepartment of Physics, Beihang University, Beijing 100191, China. E-mail: zjy@buaa.edu.cn; Fax: +86-10-82317931; Tel: +86-10-82315351
bCenter for Clean Energy Technology, School of Chemistry and Forensic Science, University of Technology Sydney, P.O. Box 123, Broadway, Sydney, NSW 2007, Australia
cDepartment of Mechanical and Aerospace Engineering, West Virginia University, Morgantown, WV 26506-6106, USA. E-mail: nick.wu@mail.wvu.edu; Tel: +1-304-293-3326
First published on 25th February 2015
Abstract
In order to reveal the origin of enhanced photocatalytic activity of N-doped La2Ti2O7 in both the visible light and ultraviolet light regions, its electronic structure has been studied using spin-polarized conventional density functional theory (DFT) and the Heyd–Scuseria–Ernzerhof (HSE06) hybrid approach. The results show that the deep localized states are formed in the forbidden band when nitrogen solely substitutes for oxygen. Introducing the interstitial Ti atom into the N-doped La2Ti2O7 photocatalyst still causes the formation of a localized energy state. Two nitrogen substitutions co-exist stably with one oxygen vacancy, creating a continuum energy band just above the valence band maximum. The formation of a continuum band instead of mid-gap states can extend the light absorption to the visible light region without increasing the charge recombination, explaining the enhanced visible light performance without deteriorating the ultraviolet light photocatalytic activity.
1. Introduction
Since the discovery of photocatalysis by Fujishima and Honda,1 semiconductor-based materials have found a wide range of applications in removing organic pollutants from water and air, splitting water into hydrogen and oxygen, as well as reducing CO2.2–4 Of the numerous photocatalysts, layer-structured semiconductors have received extensive attention because the reduction and oxidation reactions, e.g. H2 and O2 evolution sites, are separated out.5–7 Perovskite-type photocatalysts consist of the network of corner-sharing octahedra that is beneficial to the mobility of charge carriers, leading to high photocatalytic activity.8–10 Perovskite-type La2Ti2O7 with a layered structure possesses the merits of both perovskite and layered structures, which exhibits high photocatalytic activity toward organic decomposition, water splitting and CO2 reduction. For example, layered perovskite La2Ti2O7(110) loaded with nickel showed a quantum yield as high as 12% in water splitting under ultraviolet (UV) irradiation, which exhibited much better photocatalytic activity than Pt-loaded TiO2.5 In the absence of a co-catalyst, La2Ti2O7 nanosheets demonstrated similar activity to Degussa P25 in photocatalytic hydrogen evolution from the water–ethanol solution.11 It has also been reported that 98% of Cr(VI) ions in water was removed by La2Ti2O7 after 3 hours of UV irradiation.12La2Ti2O7 has a wide band gap (3.0–4.0 eV), which depends on the synthesis method and the resulting morphology.5,11,13–19 Hence it shows photocatalytic activity only under UV light irradiation. Rh-doped La2Ti2O7 was active for H2 evolution from an aqueous methanol solution under visible light irradiation owing to the photo-excitation of localized energy levels resulting from the Rh3+doping.14 Similarly, Cr and/or Fe doping in La2Ti2O7 induced intensive light absorption in the visible light region and photocatalytic hydrogen production from the water–methanol solution due to the formation of a partially filled 3d band in the band gap.17,20 The recent experimental results demonstrated that the nitrogen-doped La2Ti2O7 nanosheets not only showed significant visible light photocatalytic activity but also exhibited enhanced ultraviolet light photocatalytic activity.13,21 Nitrogen doping in La2Ti2O7 narrowed the band gap rather than creating the localized mid-gap states, which was different from N-doped anatase TiO2. These results could not be explained by the first-principles calculations, which showed that nitrogen substituting for oxygen caused the formation of the localized energy state in the band gap of La2Ti2O7.22,23 Since the energy band gap is a critical factor governing the photocatalytic activity, and doping is the most common method for altering the energy band gap,2 it is essential to understand the effect of doping on the electronic band structure of a semiconductor.
Herein, we investigate the influence of N-doping on the crystal and the electronic structures of La2Ti2O7 using first-principles calculations. We assume that nitrogen substitutes for oxygen when it is doped in La2Ti2O7 based on the experimental evidence.13 We check whether deep trap states will be formed in the forbidden band when nitrogen solely replaces oxygen. We also examine whether a localized energy state will still exist when the nitrogen substitutions co-exist with interstitial Ti atoms. Furthermore, we explore how the oxygen vacancies are used to stabilize the dopants, leading to an enhancement of visible light absorption without reducing the lifetime of the charge carrier. In particular, we investigate what is the case if one oxygen vacancy (VO) stably exists with two nitrogen substitutions (NS).
2. Calculation methods
The calculations have been performed using the Vienna ab initio simulation program (VASP).24 Adopting the Perdew–Burke–Ernzerhof (PBE) prescription, the generalized gradient approximation (GGA) was used to represent the electronic exchange–correlation energy during the geometry relaxation.25 The hybrid-functional Heyd–Scuseria–Ernzerhof (HSE06) method26 was also employed to calculate the electronic structure. The projector-augmented-wave (PAW) potentials were used to describe the electron–ion interactions.27 The valence configurations including valence and semicore electrons were 2s22p4 for oxygen, 2s22p3 for nitrogen, 3p63d34s1 for titanium and 5s25p65d16s2 for lanthanum. The cut-off energy was 450 eV and the Monkhorst–Pack k-point mesh was 6 × 8 × 4 for a 44-atom supercell. Structural relaxations were performed until the self-consistent total energy difference reached 10−5 eV and the residual forces on atoms fell below 0.02 eV Å−1.3. Results and discussion
The monoclinic La2Ti2O7 has a layered perovskite structure with P21 space group. The calculated lattice parameters using GGA are, respectively, a = 7.795 Å, b = 5.604 Å and c = 13.264 Å (Fig. 1(a)), agreeing well with the experimental values of a = 7.812 Å, b = 5.5440 Å, and c = 13.010 Å.28 For the supercell containing 44 atoms, there were 14 lattice sites of the oxygen atom for nitrogen to enter, giving rise to a doping concentration of 3.6 at% similar to the experimental value.13 We calculated the total energies for all the 14 configurations, and the relaxed geometry with the minimum total energy is shown in Fig. 1(b). It can be seen that doping N into La2Ti2O7 had little influence on the crystal structure. | ||
| Fig. 1 Supercells showing the optimized crystal structures of pristine La2Ti2O7 (a), N-doped La2Ti2O7 (b), La2Ti2O7 with the Ns + Tii complex (c) and La2Ti2O7 with complex defects (2NS + VO) (d). The grey, red, light-blue and blue spheres denote the Ti, O, La, and N atoms, respectively. | ||
Fig. 2 shows the density of states (DOS) of pristine La2Ti2O7 resulting from the calculations of the conventional DFT and the HSE06 hybrid function. It can be seen that the components of the DOS curves obtained with the two methods were very similar. The valence band maximum was composed of O 2p in majority and Ti 3p, Ti 3d and La 5p, La 5d in minority. The conduction band minimum was mainly constructed by Ti 3d with minor O 2p and a few La 5d states. Since all the elements contribute to the valence band maximum (VBM) and the conduction band minimum (CBM), the subtle stoichiometric change, e.g. by doping other elements, forming the vacancies or interstitial atoms, will definitely affect the electronic structure of the system.
 | ||
| Fig. 2 DOS of pristine La2Ti2O7 calculated using the conventional DFT (a) and the HSE06 method (b). The dash line annotates the Fermi level. | ||
The indirect band gap of pristine La2Ti2O7 calculated using the conventional DFT was 2.9 eV (Fig. 3(a)), underestimating the experimental value due to the well-known limitations in the conventional DFT.29,30 When the HSE06 hybrid method was used, the calculated band gap was 4.4 eV (Fig. 3(b)), which was slightly larger than the typical experimental value obtained with the solid-state reaction (3.8 eV)14 and the molten salt synthesis method (3.9 eV).15
 | ||
| Fig. 3 Band structure of pristine La2Ti2O7 calculated using the conventional DFT (a) and the HSE06 method (b). The dash line annotates the Fermi level. | ||
Fig. 4 and Fig. S1 (ESI†) reveal the DOS and the band structure of N-doped La2Ti2O7. The zero energy point was set to the Fermi level of pristine La2Ti2O7. The systems with defects were corrected by aligning the average electrostatic potential (Vav) of La, Ti and O atoms located far from the defects and the Vav of the same elements in the pristine La2Ti2O7.31 When N substituted for O to form the NS defect, the localized deep energy states appeared in the middle of the forbidden band, which came from the mixture of N 2p, O 2p, Ti 3d and La 5d orbitals. Furthermore, Fig. 4(c) shows that the Ti d orbitals were hybridized with the N p orbitals at the VBM. As a result, mixing of N, O, Ti and La orbitals caused the add-on of shallow states above the VBM. Band gap narrowing of 0.30 eV and 0.41 eV occurred in comparison with the pristine material, respectively, for the value calculated using the conventional DFT and HSE06 methods. The NS defect shifted the direct transition at the G point to a similar energy as the indirect transition near the B and D points, comparing Fig. 3 with 4, allowing a larger increase in absorption near the band edge. Since N requires one more electron than an O atom from the La2Ti2O7 lattice, the unequal up and down-spin electrons result in the asymmetric DOS curve. These results agreed with the previous results calculated using the HSE06 method.22,23 In TiO2, first-principles calculations also showed that NS produced the deep localized states in the band gap,32–34 similar to the results of this work. The deep localized energy band was the un-occupied state. It can induce the limited visible light photocatalytic activity but it generally has an adverse effect on the UV light photocatalytic activity since the mid-gap states can act as the charge traps.35
 | ||
| Fig. 4 DOS (a), band structure (b) and partial electron density of the VBM at a 0.01 eV Å−3 isosurface level (c) of La2Ti2O7 with the NS defect calculated using the HSE06 method. The dash line annotates the Fermi level. | ||
Recently, the DFT plus the onsite Coulomb interaction (U) calculations show that a NS is strongly bound with a titanium atom at the interstitial site (Tii), giving rise to a defect-impurity band thermally connected with the host VBM and eliminating the localized NS-related states.31 The suggested model mechanism gives a reasonable explanation for the photocatalytic oxidation reactions as well as the red-shift of the light absorption edge observed in N-doped TiO2. In this La2Ti2O7 system, we also investigated the electronic structure when NS and Tii were bound together to form a complex defect. One Ti atom is inserted into the largest interstitial site in the 44-atom supercell so as to bring minimum lattice distortion to the system, as shown in Fig. 1(c). The point-defect formation energy was calculated using the formula:
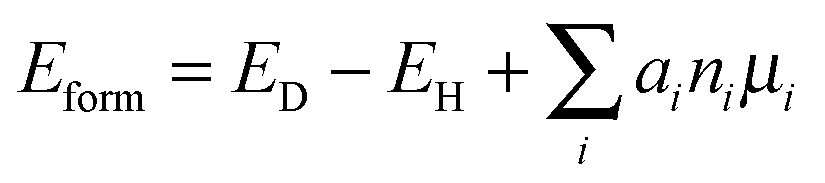 | (1) |
The formation energy of NS + Tii is plotted in Fig. S2 (ESI†). It can be seen that under O-poor and Ti-rich conditions, the formation energy is negative, which indicates that the N dopant and the interstitial Ti atoms are prone to co-exist. The formation energy comparison in Fig. S3 (ESI†) also indicates that formation of an interstitial Ti atom is energetically favourable when one oxygen atom is substituted by nitrogen atom under O-poor conditions.
When NS and Tii co-existed, the electronic structures were very similar to the model where N solely doped with La2Ti2O7, i.e., localized mid-gap states formed as shown in Fig. 5 and Fig. S4 (ESI†). According to the hybrid results, the two localized energy bands, composed of mainly Ti 3d and the least O 2p states, covered a large range below the CBM, unlike the system with only the NS defect where one deep energy band lies in the middle of the band gap. Furthermore, the mixed N 2p and O 2p energy band (denoted L1) was located at 0.4 eV above the VBM, rather than being close to the VBM as in the N mono-doped system. The hybridization among the N p and Ti d orbitals can be clearly observed using the partial electron densities in Fig. 5(c) and (d). The localized energy states were occupied, thus the Fermi level was substantially increased to just below the CBM. Again, the deep localized mid-gap energy states (denoted L2) lead to low charge mobility and can act as the charge-carrier traps, which are unfavourable to the photocatalytic activity.35 The DOS and the band structure obtained using the conventional DFT also indicated the formation of the localized energy state at 0.3 eV above the VBM (Fig. S4, ESI†).
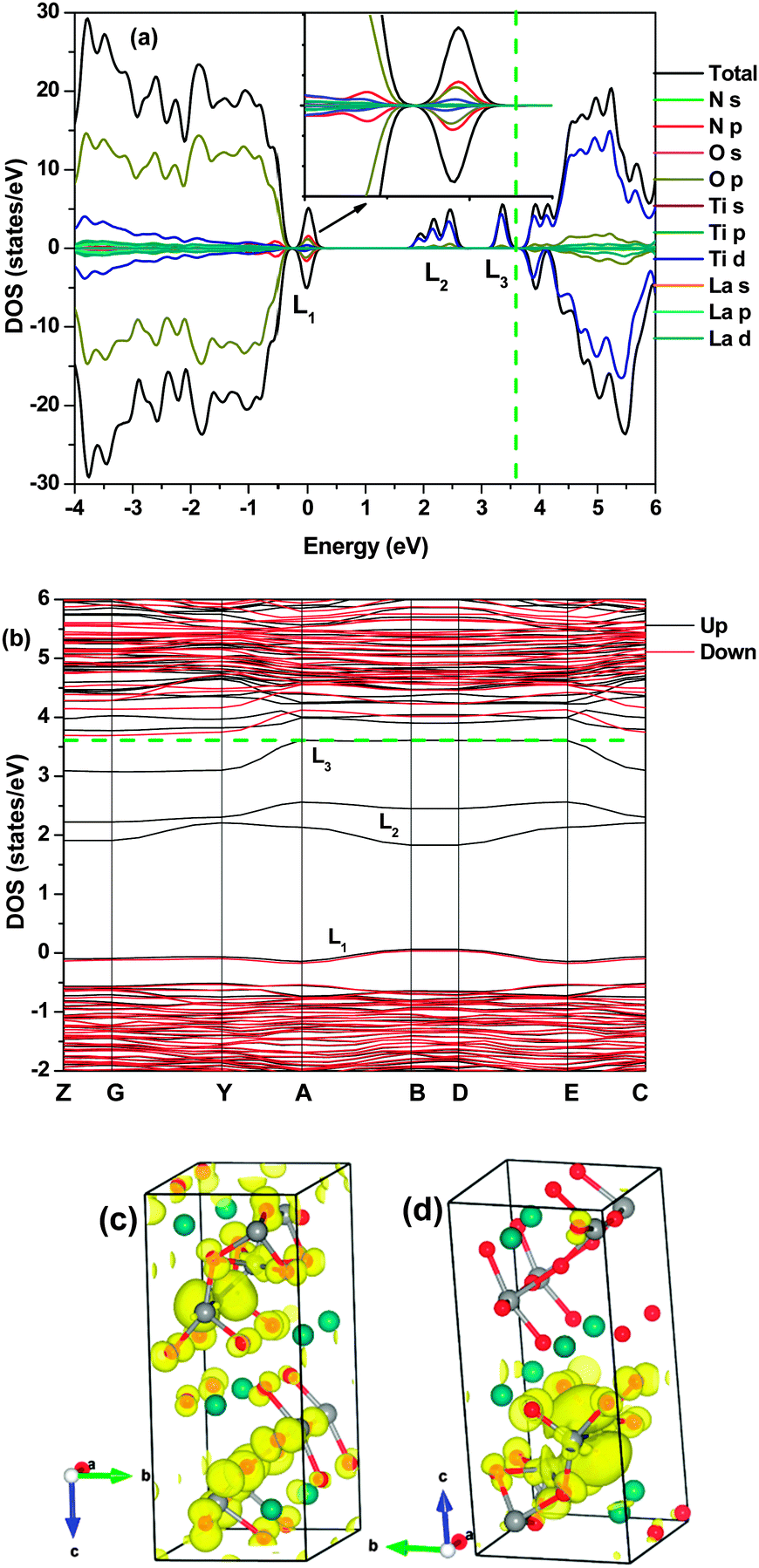 | ||
| Fig. 5 DOS (a), band structure (b), partial electron densities of the VBM (c) and the localized energy level (L1) at point D (d) at a 0.01 eV Å−3 isosurface level of La2Ti2O7 with the NS and the Tii defects in a supercell calculated using the HSE06 method. The dash line annotates the Fermi level. | ||
Both DFT calculations and experiments have shown that nitrogen doping leads to formation of oxygen vacancies in bulk TiO2.36,37 The complex defects (NS + VO) alter the band gap, and also cause the formation of a mid-gap localized energy state.35,38,39 The 3d states of Ti3+ below the conduction band, which are associated with oxygen vacancies, have been found to act as the electron–hole recombination sites, leading to the reduction of photocatalytic activity.35 Herein we also found that the formation energy was substantially reduced after VO was introduced into the N-doped La2Ti2O7 lattice (Fig. S3, ESI†), which indicated that oxygen vacancies were easily formed during the introduction of N into the lattice. But the influence of complex defects on the electronic structures was greatly different for La2Ti2O7 and TiO2. In order to achieve the charge neutrality, the model was built to contain two NS and one VO (Fig. 1(d)). As shown in Fig. 6 and Fig. S5 (ESI†), the localized energy state completely disappeared when two NS co-existed with one VO. The mixed N 2p, O 2p and Ti 3d orbitals formed a continual energy band on the top of the valence band (obtained using the HSE06 method) or thermally connected with the VBM (obtained using the conventional DFT), which accounted for the parallel red shift of the whole optical absorption edge of the N-doped sample.13 The band gap was narrowed by 0.85 eV and 1.15 eV, which were determined by the conventional DFT and HSE06 approaches, respectively. For the HSE06 calculation result, the VBM shifted upwards by 1.07 eV while the CBM shifted downwards by 0.14 eV in comparison with pristine La2Ti2O7. The VBM and CBM variation trends obtained using the theoretical calculation were in good agreement with the experimental results reported for the N-doped La2Ti2O7 nanosheets.13 As well, the added bands created a broad direct transition at a similar energy as the indirect band gap, improving the light absorption. This was in agreement with the added band edge as seen experimentally with N doping in La2Ti2O7, wherein the absorption can be described by the superimposed direct and indirect band gaps.13,21
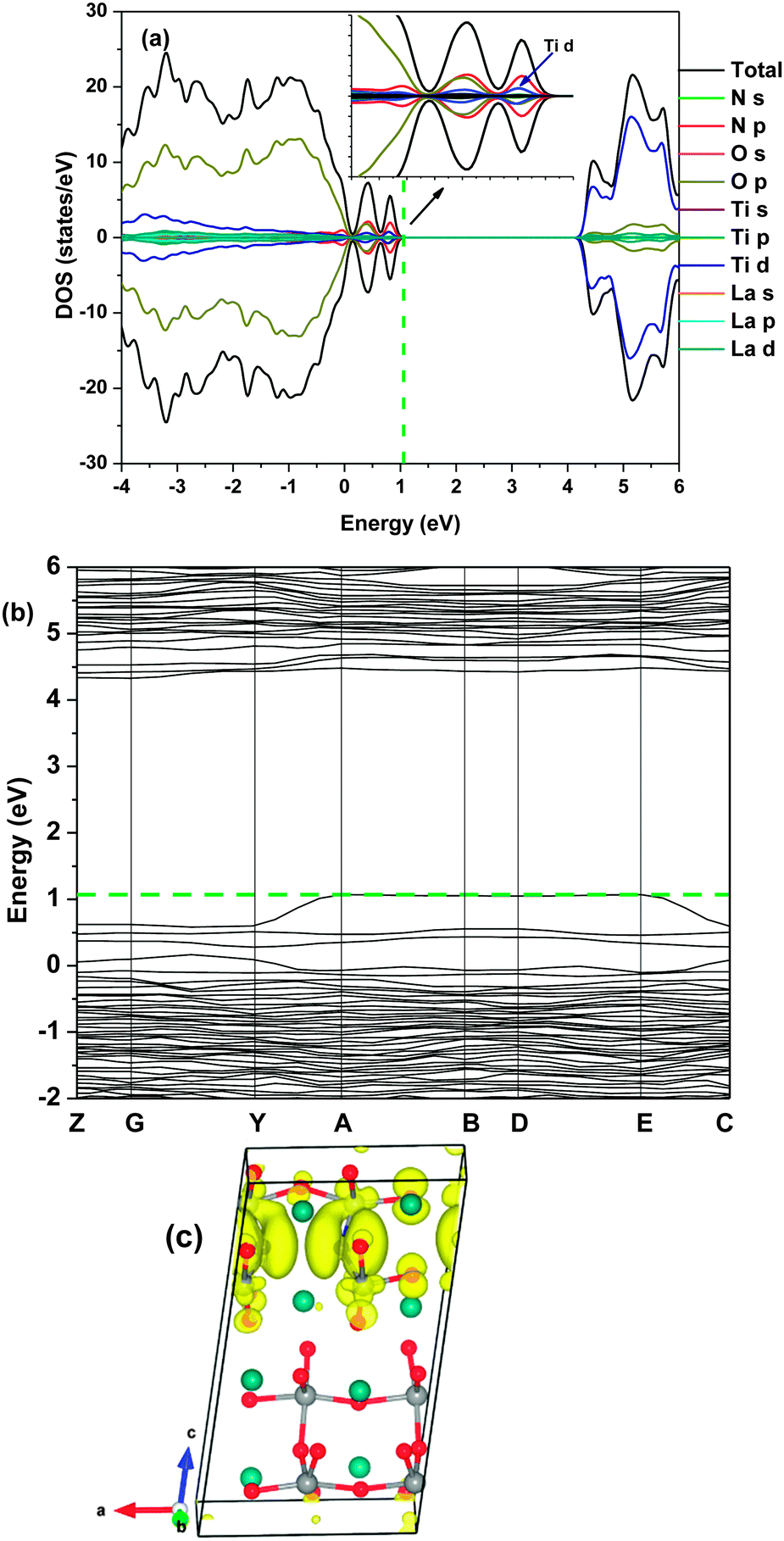 | ||
| Fig. 6 DOS (a), the band structure (b) and partial electron densities of the VBM at a 0.01 eV Å−3 isosurface level (c) of La2Ti2O7 with two NS and one VO defects in a supercell calculated using the HSE06 method. The dash line annotates the Fermi level. | ||
In TiO2, the N dopant concentration has an effect on the electronic structure of N-doped TiO2.31 If a high concentration of N (e.g. 6.25 at%) was employed in the calculation, the N 2p state was mixed with O 2p, leading to band gap narrowing rather than localized deep energy state formation.40 If a low concentration of N (e.g. 1.56 at%) was used in the calculation, a deep localized energy state formed in the forbidden band.31,34,35 In order to study the influence of the nitrogen concentration on the electronic structure, we also investigated the DOS and the band structure of La2Ti2O7 with 7.1 at% of N concentration, corresponding to the system containing two NS and one VO in a supercell in Fig. 1(d). By comparing Fig. 4 and 7, it can be confirmed that doping N alone into La2Ti2O7 led to the formation of a localized deep energy state in the forbidden gap, independent of the concentration change. For the two systems containing either two NS (Fig. 7 and Fig. S6, ESI†) or two NS plus one VO (Fig. 6 and Fig. S5, ESI†), the electronic structures were very similar, except that the localized deep energy state disappeared completely in the latter case. This has further confirmed that the localized energy state can be removed and the band gap be narrowed simultaneously only when NS and VO co-exist in La2Ti2O7.
 | ||
| Fig. 7 DOS (a) and the band structure (b) of La2Ti2O7 with two NS defects in a supercell calculated using the HSE06 method. The dash line annotates the Fermi level. | ||
In comparison with the systems containing NS and NS + Tii, the Ti 4d orbitals contributed more to the VBM in La2Ti2O7 containing both NS and VO, as clearly demonstrated by the DOS shown in the inset of Fig. 6(a). This indicated that hybridization of orbitals between non-metals and metals was enhanced when NS and VO existed simultaneously, which can also be verified by the partial electron density of the VBM of La2Ti2O7 containing different defects. In comparison with other configurations, strong orbital hybridization between N and Ti occurred in La2Ti2O7 containing both NS and VO, as shown in Fig. 6(c). The extended nature of the valence band edge caused by the hybridized N 2p and Ti 3d states is beneficial to the migration of photo-induced holes, which increases the oxidation capability of the photocatalysts.31,41 As a result, the N doped La2Ti2O7 nanosheets perform well in photocatalytic decomposition of methyl orange.13 N doping is an effective method to realize the visible light photocatalytic activity. In short, if N doping into La2Ti2O7 occurs in a reducing atmosphere, the presence of VO will reduce the band gap and eliminate the localized energy state, leading to enhancement in the photocatalytic activity in both the ultraviolet and visible light regions.
4. Conclusions
The conventional DFT and HSE06 hybrid methods were employed to investigate the influence of the N substitutions co-existing with the oxygen vacancy and the interstitial Ti atom on the electronic structures. Doping N alone into La2Ti2O7 caused the formation of the deep localized energy state, independent of the doping concentration. When interstitial Ti co-existed with NS, the localized energy state was still present. However, when NS co-existed with VO, the localized energy state was completely removed and the band gap can be substantially narrowed. Furthermore, the added DOS altered the VBM, which facilitated the migration of photo-induced holes and the oxidation power of the photocatalyst, introduced optical transitions at lower energy to increase light absorption. As a result, the NS plus VO model in La2Ti2O7 can accurately interpret the fact that N doping into La2Ti2O7 can not only lead to strong visible light photocatalytic activity but also enhance the performance under UV irradiation. This work reveals that tuning the non-neutral point defects with the oxygen vacancy is an effective strategy for alleviating the problem associated with doping to increase light absorption, which have implications in engineering the band gap of semiconductor photocatalysts.Acknowledgements
This work was supported by PhD Program Foundation of the Ministry of Education of China (Grant No. 20121102110027), the National Science Foundation of China (Grant No. 51472013 and 91222110), the high-performance computing platform of Network Information Centre in Beihang University, and the National Computational Infrastructure (NCI) through the merit allocation scheme, and used NCI resources and facilities in Canberra, Australia.Notes and references
- K. Honda and A. Fujishima, Nature, 1972, 238, 37 CrossRef.
- H. Tong, S. X. Ouyang, Y. P. Bi, N. Umezawa, M. Oshikiri and J. H. Ye, Adv. Mater., 2012, 24, 229 CrossRef CAS PubMed.
- J. Y. Li and N. Q. Wu, Catal. Sci. Technol., 2015, 5, 1360 CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- H. G. Kim, D. W. Hwang, J. Kim, Y. G. Kim and J. S. Lee, Chem. Commun., 1999, 1077 RSC.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- Y. Miseki, H. Kato and A. Kudo, Energy Environ. Sci., 2009, 2, 306 CAS.
- R. Abe, M. Higashi, K. Sayama, Y. Abe and H. Sugihara, J. Phys. Chem. B, 2006, 110, 2219 CrossRef CAS PubMed.
- T. Takata, Y. Furumi, K. Shinohara, A. Tanaka, M. Hara, J. N. Kondo and K. Domen, Chem. Mater., 1997, 9, 1063 CrossRef CAS.
- Y. F. Huang, Y. L. Wei, S. H. Cheng, L. Q. Fan, Y. B. Li, J. M. Lin and J. H. Wu, Sol. Energy Mater. Sol. Cells, 2010, 94, 761 CrossRef CAS PubMed.
- K. W. Li, Y. Wang, H. Wang, M. K. Zhu and H. Yan, Nanotechnology, 2006, 17, 4863 CrossRef CAS.
- Q. L. Yang, S. Z. Kang, H. Chen, W. B. Bu and J. Mu, Desalination, 2011, 266, 149 CrossRef CAS PubMed.
- F. K. Meng, Z. L. Hong, J. Arndt, M. Li, M. J. Zhi, F. Yang and N. Q. Wu, Nano Res., 2012, 5, 213 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Y. Moriya, K. Maeda and K. Domen, Catal. Sci. Technol., 2013, 3, 2098 CAS.
- D. W. Hwang, H. G. Kim, J. S. Jang, S. W. Bae, S. M. Ji and J. S. Lee, Catal. Today, 2004, 93–95, 845 CrossRef CAS PubMed.
- D. Arney, B. Porter, B. Greve and P. A. Maggard, J. Photochem. Photobiol., A, 2008, 199, 230 CrossRef CAS PubMed.
- D. W. Hwang, H. G. Kim, J. S. Lee, J. Kim, W. Li and S. H. Oh, J. Phys. Chem. B, 2005, 109, 2093 CrossRef CAS PubMed.
- Z. M. Shao, S. Saitzek, P. Roussel, A. Ferri, Ol. Mentré and R. Desfeux, CrystEngComm, 2014, 16, 8517 RSC.
- A. Nashim, S. Martha and K. M. Parida, RSC Adv., 2014, 4, 14633 RSC.
- S. J. Hu, L. C. Jia, B. Chi, J. Pu and L. Jian, J. Power Sources, 2014, 266, 304 CrossRef CAS PubMed.
- F. K. Meng, J. T. Li, Z. L. Hong, M. J. Zhi, A. Sakla, C. C. Xiang and N. Q. Wu, Catal. Today, 2013, 199, 48 CrossRef CAS PubMed.
- P. Liu, J. Nisar, B. Pathak and R. Ahuja, Phys. Chem. Chem. Phys., 2013, 15, 17150 RSC.
- P. Liu, J. Nisar, B. S. Sa, B. Pathak and R. Ahuja, J. Phys. Chem. C, 2013, 117, 13845 CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- H. W. Schmalle, T. Williams and A. Reller, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 235 CrossRef.
- L.J. Sham and M. Schlüter, Phys. Rev. Lett., 1983, 51, 1888 CrossRef.
- L. J. Sham and M. Schlut, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 3883 CrossRef CAS.
- N. Umezawa and J. H. Ye, Phys. Chem. Chem. Phys., 2012, 14, 5924 RSC.
- C. D. Valentin, G. Pacchioni and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 085116 CrossRef.
- Z. Lin, A. Orlov, R. M. Lambert and M. C. Payne, J. Phys. Chem. B, 2005, 109, 20948 CrossRef CAS PubMed.
- J. B. Varley, A. Janotti and C. G. Van de Walle, Adv. Mater., 2011, 23, 2343 CrossRef CAS PubMed.
- J. Wang, D. N. Tafen, J. P. Lewis, Z. L. Hong, A. Manivannan, M. J. Zhi, M. Li and N. Q. Wu, J. Am. Chem. Soc., 2009, 131, 12290 CrossRef CAS PubMed.
- C. D. Valentin, G. Pacchioni, A. Selloni, S. Livraghi and E. Giamello, J. Phys. Chem. B, 2005, 109, 11414 CrossRef PubMed.
- M. Batzill, E. H. Morales and U. Diebold, Phys. Rev. Lett., 2006, 96, 026103 CrossRef.
- H. C. Wu, Y. S. Lin and S. W. Lin, Int. J. Photoenergy, 2013, 2013, 289328 Search PubMed.
- P. P. González-Borrero, H. S. Bernabé, N. G. C. Astrath, A. C. Bento, M. L. Baesso, M. V. Castro Meira, J. S. de Almeida and A. Ferreira da Silva, Appl. Phys. Lett., 2011, 99, 221909 CrossRef PubMed.
- K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef PubMed.
- H. Kawazoe, H. Yanagi, K. Ueda and H. Hosono, MRS Bull., 2000, 25, 28 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp00157a |
| This journal is © the Owner Societies 2015 |