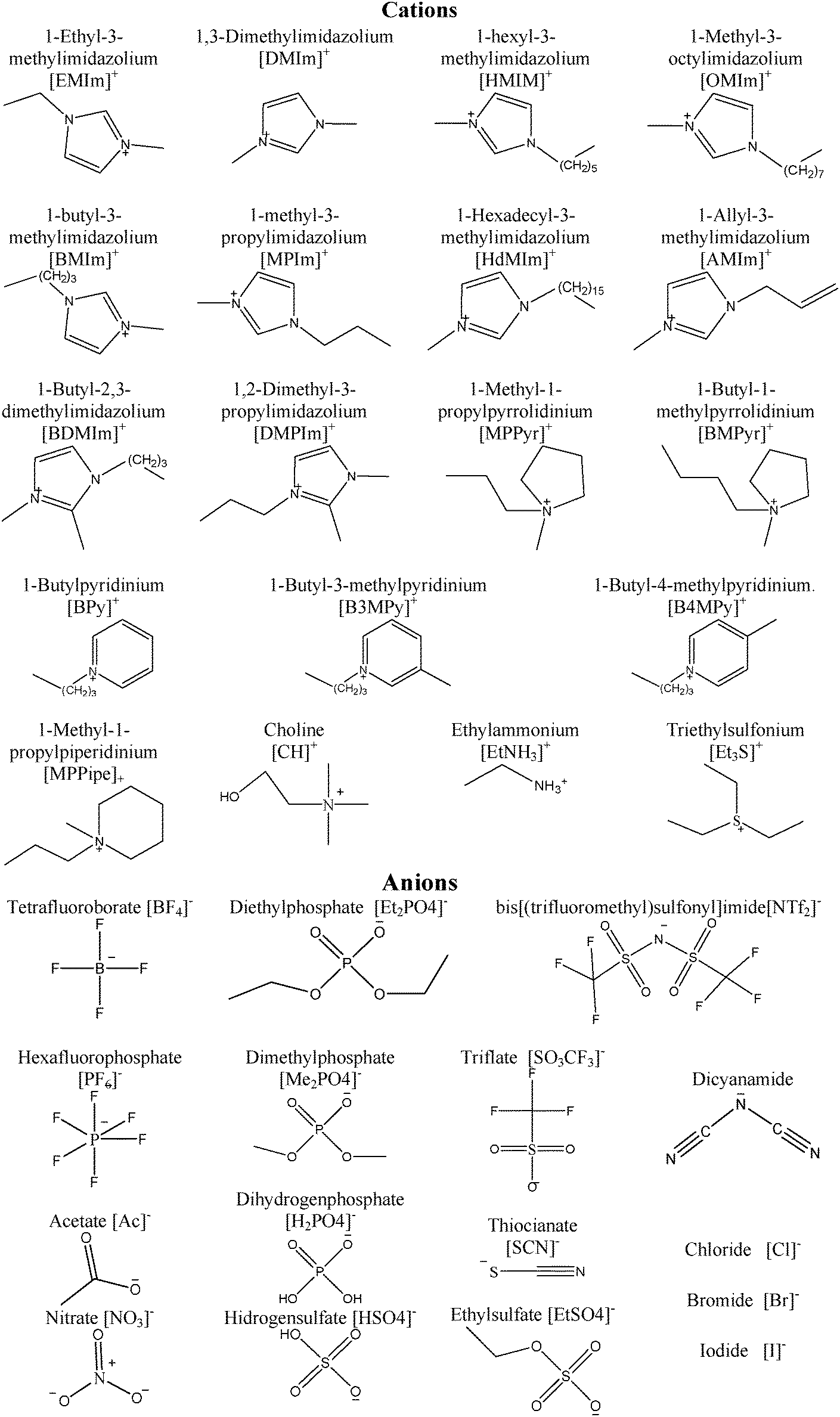
A density functional theory insight towards the rational design of ionic liquids for SO2 capture†
Received
6th January 2015
, Accepted 16th April 2015
First published on 20th April 2015
Abstract
A systematic density functional theory (DFT) analysis has been carried out to obtain information at the molecular level on the key parameters related to efficient SO2 capture by ionic liquids (ILs). A set of 55 ILs, for which high gas solubility is expected, has been selected. SO2 solubility of ILs was firstly predicted based on the COSMO-RS (Conductor-like Screening Model for Real Solvents) method, which provides a good prediction of gas solubility data in ILs without prior experimental knowledge of the compounds' features. Then, interactions between SO2 and ILs were deeply analyzed through DFT simulations. This work provides valuable information about required factors at the molecular level to provide high SO2 solubility in ILs, which is crucial for further implementation of these materials in the future. In our opinion, systematic research on ILs for SO2 capture increases our knowledge about those factors which could be controlled at the molecular level, providing an approach for the rational design of task-specific ILs.
1. Introduction
Air pollution is attracting increasing attention throughout the world. Among the main air pollutants, sulfur dioxide (SO2), which is mainly emitted through the combustion of fossil based fuels, is causing serious harm to the environment and human health.1,2 At the same time, SO2 is a useful source of many intermediates in chemical synthesis.3 As a matter of fact, there is general interest in the design and improvement of methods for SO2 capture. Although several methods have been developed for this purpose, all of them have several drawbacks. For instance, an effective method based on flue gas desulfurization (FGD) needs a large amount of water and subsequent treatment of the consequent waste, in order to prevent excessive amounts of calcium sulphate that lead to secondary pollution in the environment. Other methods, such as amine scrubbing, are affected by solvent loss and degradation due to the low volatility and stability of amine solutions.2,4,5
In recent years, ionic liquids (ILs) have demonstrated their effectiveness for acid-gas removal from flue gas such as SO22,3,5–9 and CO2.8–14 In addition, ILs contain unique properties, including good thermal and chemical stability, non-flammability and most distinctly they have almost null vapor pressure. All these features have been proved to be useful in chemical processes to replace volatile organic compounds. Nonetheless, the major advantage of ILs is the possibility to design task-specific solvents through the adequate cation–anion combinations, which requires a deep understanding of the structure–property relationship.9,15 There is a large collection of compounds (approximately about ∼106 when considering only “pure” ILs), and thus, system approaches on the ability of ILs for acid gas capture are useful in the selection of ILs for SO2 storage. Unfortunately, the larger number of ILs hinders systematic experimental studies on a huge number of ILs, due to the economical and temporal cost as well as limited experimental resources. Having mentioned the cost of experimental difficulties and cost hurdles associated with broad screening of ILs for acid-gas removal, density functional theory (DFT) simulations have proven their ability to provide valuable indications and guide to the experimentalists. As a matter of fact, DFT is a suitable tool for the analysis of the interactions between ILs and gas molecules at the nanoscopic level, which allow a deeper knowledge of the structure–property relationship. Most of the reported DFT studies only consider CO2.7,12,13,16,17 Though, some researches leading with SO2 capture have been reported.7,17
There are few recent studies that address utilization of ILs for gas capture at the molecular level, especially SO2 capture. Damas et al. have shown a systematic study of acid- and sour-gas mitigation alternatives (SO2, CO2 and H2S) by using ILs through DFT simulations, which mainly focuses on imidazolium cation based ILs.17 In this presented work, we broadened the study that was conducted by Damas et al. by including other cations such as piridinium or cholinium cations in combination with anions such as bis(trifluorosulfonyl)imide, triflate, or tetrafluoroborate as shown in Table 1 and Fig. 1.
Table 1 The selected family of ionic liquids studied in this work along with their estimated Henry's Law constants of SO2 (KH) at 303 K predicted using the COSMO-RS method
| No. |
Cation |
Anion |
Labelling |
K
H × 10−5/Pascal |
| 1 |
1-Ethyl-3-methylimidazolium |
Tetrafluoroborate |
[EMIm][BF4] |
3.69 |
| 2 |
1-Butyl-3-methylimidazolium |
Tetrafluoroborate |
[BMIm][BF4] |
3.55 |
| 3 |
1-Hexyl-3-methylimidazolium |
Tetrafluoroborate |
[HMIm][BF4] |
3.47 |
| 4 |
1-Methyl-3-octylimidazolium |
Tetrafluoroborate |
[OMIm][BF4] |
3.35 |
| 5 |
1-Butylpyridinium |
Tetrafluoroborate |
[BPy][BF4] |
3.72 |
| 6 |
1-Butyl-3-methylpyridinium |
Tetrafluoroborate |
[B3MPy][BF4] |
2.96 |
| 7 |
1-Butyl-4-methylpyridinium |
Tetrafluoroborate |
[B4MPy][BF4] |
2.96 |
| 8 |
1-Butyl-3-methylimidazolium |
Hexafluorophosphate |
[BMIm][PF6] |
2.76 |
| 9 |
1-Hexyl-3-methylimidazolium |
Hexafluorophosphate |
[HMIm][PF6] |
2.65 |
| 10 |
1-Methyl-3-octylimidazolium |
Hexafluorophosphate |
[OMIm][PF6] |
2.61 |
| 11 |
1-Butylpyridinium |
Hexafluorophosphate |
[BPy] [PF6] |
2.79 |
| 12 |
1-Butyl-3-methylpyridinium |
Hexafluorophosphate |
[B3MPy][PF6] |
2.47 |
| 13 |
1-Butyl-4-methylpyridinium |
Hexafluorophosphate |
[B4MPy][PF6] |
2.46 |
|
|
| 14 |
1-Ethyl-3-methylimidazolium |
Diethylphosphate |
[EMIm][Et2PO4] |
4.73 |
| 15 |
1,3-Dimethylimidazolium |
Dimethylphosphate |
[DMIm][Me2PO4] |
4.07 |
| 16 |
Choline |
Dihydrogenphosphate |
[CH][H2PO4] |
6.29 |
| 17 |
1-Ethyl-3-methylimidazolium |
Ethylsulfate |
[EMIm][EtSO4] |
4.63 |
| 18 |
1-Ethyl-3-methylimidazolium |
Hidrogensulfate |
[EMIm][HSO4] |
6.45 |
| 19 |
1-Ethyl-3-methylimidazolium |
Acetate |
[EMIm][Ac] |
3.85 |
| 20 |
Ethylammonium |
Nitrate |
[EtNH3][NO3] |
6.37 |
|
|
| 21 |
Triethylsulfonium |
Bis[(trifluoromethyl)sulfonyl]imide |
[Et3S][NTf2] |
3.52 |
| 22 |
1-Ethyl-3-methylimidazolium |
Bis[(trifluoromethyl)sulfonyl]imide |
[EMIm][NTf2] |
3.63 |
| 23 |
1-Methyl-3-propylimidazolium |
Bis[(trifluoromethyl)sulfonyl]imide |
[MPIm][NTf2] |
3.54 |
| 24 |
1,2-Dimethyl-3-propylimidazolium |
Bis[(trifluoromethyl)sulfonyl]imide |
[DMPIm][NTf2] |
3.19 |
| 25 |
1-Butyl-3-methylimidazolium |
Bis[(trifluoromethyl)sulfonyl]imide |
[BMIm][NTf2] |
3.50 |
| 26 |
1-Butyl-2,3-dimethylimidazolium |
Bis[(trifluoromethyl)sulfonyl]imide |
[BDMIm][NTf2] |
3.18 |
| 27 |
1-Hexyl-3-methylimidazolium |
Bis[(trifluoromethyl)sulfonyl]imide |
[HMIm][NTf2] |
3.50 |
| 28 |
1-Hexadecyl-3-methylimidazolium |
Bis[(trifluoromethyl)sulfonyl]imide |
[HdMIm][NTf2] |
3.69 |
| 29 |
1-Allyl-3-methylimidazolium |
Bis[(trifluoromethyl)sulfonyl]imide |
[AMIm][NTf2] |
3.56 |
| 30 |
1-Methyl-1-propylpyrrolidinium |
Bis[(trifluoromethyl)sulfonyl]imide |
[MPPyr][NTf2] |
3.39 |
| 31 |
1-Butyl-1-methylpyrrolidinium |
Bis[(trifluoromethyl)sulfonyl]imide |
[BMPyr][NTf2] |
3.36 |
| 32 |
1-Methyl-1-propylpiperidinium |
Bis[(trifluoromethyl)sulfonyl]imide |
[MPPipe][NTf2] |
3.33 |
| 33 |
1-Butylpyridinium |
Bis[(trifluoromethyl)sulfonyl]imide |
[BPy][NTf2] |
3.49 |
| 34 |
1-Butyl-3-methylpyridinium |
Bis[(trifluoromethyl)sulfonyl]imide |
[B3MPy][NTf2] |
3.29 |
| 35 |
1-Butyl-4-methylpyridinium |
Bis[(trifluoromethyl)sulfonyl]imide |
[B4MPy][NTf2] |
3.29 |
|
|
| 36 |
1-Ethyl-3-methylimidazolium |
Triflate |
[EMIM][SO3CF3] |
4.41 |
| 37 |
1-Butyl-3-methylimidazolium |
Triflate |
[BMIM][SO3CF3] |
4.20 |
| 38 |
1-Hexyl-3-methylimidazolium |
Triflate |
[HMIM][SO3CF3] |
4.17 |
| 39 |
1-Methyl-3-octhylimidazolium |
Triflate |
[OMIM][SO3CF3] |
4.11 |
| 40 |
1-Butyl-1-methylpyrrolidinium |
Triflate |
[BMPyr][SO3CF3] |
3.57 |
|
|
| 41 |
1-Ethyl-3-methylimidazolium |
Thiocianate |
[EMIM][SCN] |
4.17 |
| 42 |
1-Ethyl-3-methylimidazolium |
Dicyanamide |
[EMIM][DCA] |
4.30 |
| 43 |
1-Butyl-3-methylimidazolium |
Dicyanamide |
[BMIM][DCA] |
4.22 |
| 44 |
1-Butyl-1-methylpyrrolidinium |
Dicyanamide |
[BMPyr][DCA] |
2.88 |
|
|
| 45 |
1-Ethyl-3-methylimidazolium |
Chloride |
[EMIM][Cl] |
2.01 |
| 46 |
1-Butyl-3-methylimidazolium |
Chloride |
[BMIM][Cl] |
3.42 |
| 47 |
1-Allyl-3-methylimidazolium |
Chloride |
[AMIM][Cl] |
2.87 |
| 48 |
1-Ethyl-3-methylimidazolium |
Bromide |
[EMIM][Br] |
2.24 |
| 49 |
1-Butyl-3-methylimidazolium |
Bromide |
[BMIM][Br] |
3.53 |
| 50 |
1,3-Dimethylimidazolium |
Iodide |
[DMIm][I] |
3.39 |
| 51 |
1-Ethyl-3-methylimidazolium |
Iodide |
[EMIm][I] |
3.03 |
| 52 |
1-Methyl-3-propylimidazolium |
Iodide |
[MPIm][I] |
3.70 |
| 53 |
1-Butyl-3-methylimidazolium |
Iodide |
[BMIm][I] |
4.07 |
| 54 |
1-Hexyl-3-methylimidazolium |
Iodide |
[HMIm][I] |
4.50 |
| 55 |
1-Allyl-3-methylimidazolium |
Iodide |
[AMIm][I] |
4.07 |
 |
| | Fig. 1 Chemical structure for the ions involved in the selected family of ionic liquids. | |
In our opinion, the analysis of those ILs with high efficiency for SO2 capture through DFT tools should be a good starting point to shed some light on the main molecular factors related with efficient SO2 capture. Unfortunately, experimental studies dealing with SO2 capture by ILs are still scarce and reduced to a small number of selected ionic liquids. A key parameter in the selection of an ionic liquid for SO2 capture is gas solubility. It is well known that gas solubility in ILs can be predicted based on the COSMO-RS (Conductor-like Screening Model for Real Solvents) method.18 The COSMO-RS predicts thermodynamics properties of solvents on the basis of uni-molecular quantum chemical calculations for the individual molecules, which provides a good prediction of gas solubility data in ILs without prior experimental knowledge on the compound's properties.14 Thus, COSMO-RS is able to carry out fast screening on a huge number of ionic liquids, reducing the number of candidates for experimental studies, which also reduces try-and-error attempts and economical and temporal cost. Consequently, the COSMO-RS method was firstly used to carry out a quick screening on a big matrix of ILs. Then, an in-depth study of those ILs, which are expected to provide high SO2 solubility according to the COSMO-RS method, from a molecular point of view was done using DFT tools. The combination of first screening to select efficient ILs for SO2 capture using COSMO-RS analysis along with DFT analysis on the most adequate ILs have allowed us to obtain information about structure vs. property relations that control SO2 solubility in ILs, which is crucial for the rational design of task-specific ILs for SO2 absorption.
2. Theoretical methodology
2.1. COSMO-RS method
Four different approaches can be performed to describe ionic liquids according to the COSMO-RS method. According to Palomar et al., these approximations are labelled [C + A]GAS, [C + A]COSMO, [CA]GAS and [CA]COSMO.14 The [C + A] model uses isolated ions to simulate ionic liquids systems, while ion-paired structures are used in [CA]. System optimizations can be carried out in gas-phase using quantum chemistry methods (GAS subscript), or the continuum solvation COSMO model (COSMO subscript). The [C + A]GAS approach (i.e. independent ionic structures optimized in the gas phase) predicts gas solubility data in slightly better agreement with the experiments. Palomar et al. also concluded that all COSMOS-RS approaches provide similar good capability to predict Henry's law constants for ionic liquid. Nonetheless, the [C + A]GAS model allows us to perform analysis with a reduced computational time, since only optimized ion structures in the gas phase are needed, which is especially useful for screening purposes.
The [C + A]GAS model was employed in this work, which is based on two main steps: (i) quantum chemical optimization for the molecular involved species and (ii) COSMO-RS statistical calculations. Firstly, the isolated ions and SO2 were optimized at the B3LYP/6-311+G(d,p) level using Gaussian 09 (Revision D.01) package,19 which was also instructed to provide the COSMO files. For these structures, COSMO files were calculated at the BVP86/TZVP/DGA1 theoretical level and used as input in the COSMOthermX program18 to estimate Henry's law constants. The COSMO-RS model parameterization used for all calculations was BPTZVPC21-0111.
In this work, Henry's law constants (KH) for SO2 were selected as a measure of absorbing capability. Henry's constants are directly calculated by COSMOthermX code. The details of theory of COSMO-RS can be found in the original work of Klamt et al.18 Briefly, Henry's law constants can be defined as the ratio between the liquid phase concentration of SO2 and its partial vapour pressure in the gas phase:
where
Pi and
xi are the partial vapour pressure of a compound i (SO
2 in our study) in the gas phase and its molar fraction in the liquid.
γ∞i is the activity coefficient of the compound at infinite dilution, and
PSi is the saturated pure compound vapor pressure of the gas. Those parameters are directly provided by the COSMOthermX code.
2.2. DFT simulations
Systems composed by one isolated molecule (i.e. isolated ions and SO2) up to the system composed by both ions and SO2 were optimized. Optimized minima were checked through their vibrational frequencies. For those simulations wherein two or more molecules are present, different starting points were employed in order to study different relative dispositions, focusing our attention on the disposition of minimal energy. All these calculations were carried out using a B3LYP-D2 functional. B3LYP20 has been selected since it has been proven to show appreciable performance over a previously studied wide range of systems,21 while dispersion corrections (D2) are adequate since we dealt with systems with dispersive interactions such as hydrogen bonds.22 In addition, other works dealing with the performance of dispersion corrected functionals to study ionic liquid concluded that dispersion correction could significantly decrease mean absolute deviations for binding energies up to 10.0 kJ mol−1 or lower in comparison with the MP2 method.23 All atomic elements, except iodine, were described with the standard Pople basis set 6-311+G(d,p). For iodine, a small core Stuttgart–Dresden–Bonn effective core potential was used (SDB-cc-pVTZ).24 Interaction energies (BE) related with SO2 capture were computed as the energy difference between the complex and the sum of the energy of each component. For example, BE for IL⋯SO2 was calculated as:| | | BE = EIL–SO2 − (Ecat + Eani + ESO2) | (2) |
Binding energies were also estimated by considering the IL as a whole (BE′), i.e., the binding energy due to the interaction between the IL and the gas molecule:| | | BE′ = EIL–SO2 − (EIL + ESO2) | (3) |
where EIL–SO2, Ecat, Eani, EIL and ESO2 stand for the energies of IL⋯SO2, cation, anion, IL and SO2, respectively. For those systems composed of two or more molecules, computed energies were corrected according to the counterpoise method to avoid basis set supper position error (BSSE).25
It has been shown that there is a specific charge transfer interaction between SO2 and the ions.26 There are different methods to calculate charge distributions, such as the Mulliken method,27 whose basis set dependence is well known.28 ChelpG scheme29 has demonstrated its suitability for ILs.12,30 Thus, atomic charges were also computed according to both ChelpG and Mulliken schemes. Intermolecular interactions where analyzed in the framework of Bader's theory (Atoms in Molecules, AIM).31 In this context, intermolecular interactions are characterized through critical points (CP). Although four kind of critical points were obtained, we focused on bond critical points (BCP), which raises the criteria for considering the presence of intermolecular interactions.31 AIM analysis was carried out with the MultiWFN code.32 All the above-mentioned calculations were carried out with Gaussian 09 (Revision D.01) package.19
3. Results and discussion
3.1. COSMO-RS analysis: selection of the optimal IL family
As said, the first step in our study was the selection of an optimal family of ILs with high SO2 solubility. The SO2 absorption capacities were evaluated in terms of Henry's law constants (KH) predicted according to the COSMO-RS method. COSMO-RS is a predictive method for thermodynamic equilibrium of fluids, which uses a statistical thermodynamic approach based on the results of uni-molecular quantum chemical calculations. The efficiency of COSMO-RS to predict the solubility behaviour of different solutes in ILs was evaluated by comparing both the experimental and computed (according to the COSMO-RS method) Henry's constants.14,18,33,34 Although some publications have reported that COSMO-RS systematically overestimates the Henry's constants, it provides a reasonable linear fit between the calculated and experimental values.14,34
In this work, COSMO-RS approach has been used to perform a fast screening on SO2 solubility in ILs. Although several properties, such as σ-surfaces, screening charge density, σ-profiles, and histograms of screening charge can be computed with COSMO-RS, we have focused on Henry's law constants for SO2 as a measure of absorbing capability. For this, KH (at 303 K) was estimated for a matrix of ≃7600 ILs formed through a combination of cations based on imidazoluim, piperidinium, choline, ammonium cations paired with anions such as halogens, phosphates, tetrafluoroborate, dicyanamide or bis(trifluorosulfonyl)imide (see Table S1, ESI†). In addition to low Henry's law constants, only those ILs with an adequate viscosity profile for industrial applications as suitable ILs for SO2 capture were considered. Thus, a set of 55 ILs (see Table 1 and Fig. 1) was selected for a deeper DFT analysis. Table 1 and Fig. 2 gather the computed Henry's law constants of the selected ionic liquids. All selected ILs yield KH within the range of 2.5 × 105 to 6.5 × 105 Pascal at 303 K. These values are smaller (which means higher solubility) than those reported by González-Miquel et al. (of around 30 × 105 Pascal–60 × 105 Pascal) for CO2 absorption.35 Then, high efficiency for SO2 capture can be expected for selected ILs. Note that most of the selected ILs are based on cations such as imidazolium, pyridinium or piperazinium and anions such as [BF4]−, [PF6]−, [NTf2]− triflate or halides. Then, the combination of these anions would be adequate to design ILs for SO2 capture with high efficiencies.
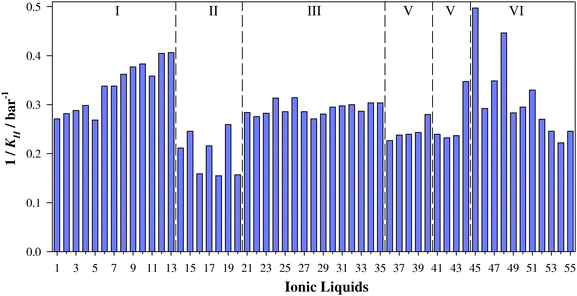 |
| | Fig. 2 Inverse of Henry's Law constants of SO2 in ILs (1/KH) at 303 K predicted using the COSMO-RS method. | |
3.2. DFT analysis
As a first approximation, SO2 capture at the molecule level could be related with the strength of the interactions between the ions and the SO2 molecule. In this work, the interaction strength has been mainly analyzed based on binding energies (BE). Prior to analysis of SO2 capture by selected ILs, ion⋯SO2 and ionic pairs were also briefly assessed. Such information could be useful to rationalize the behavior of IL⋯SO2 systems.
3.2.1. Ion⋯SO2 systems.
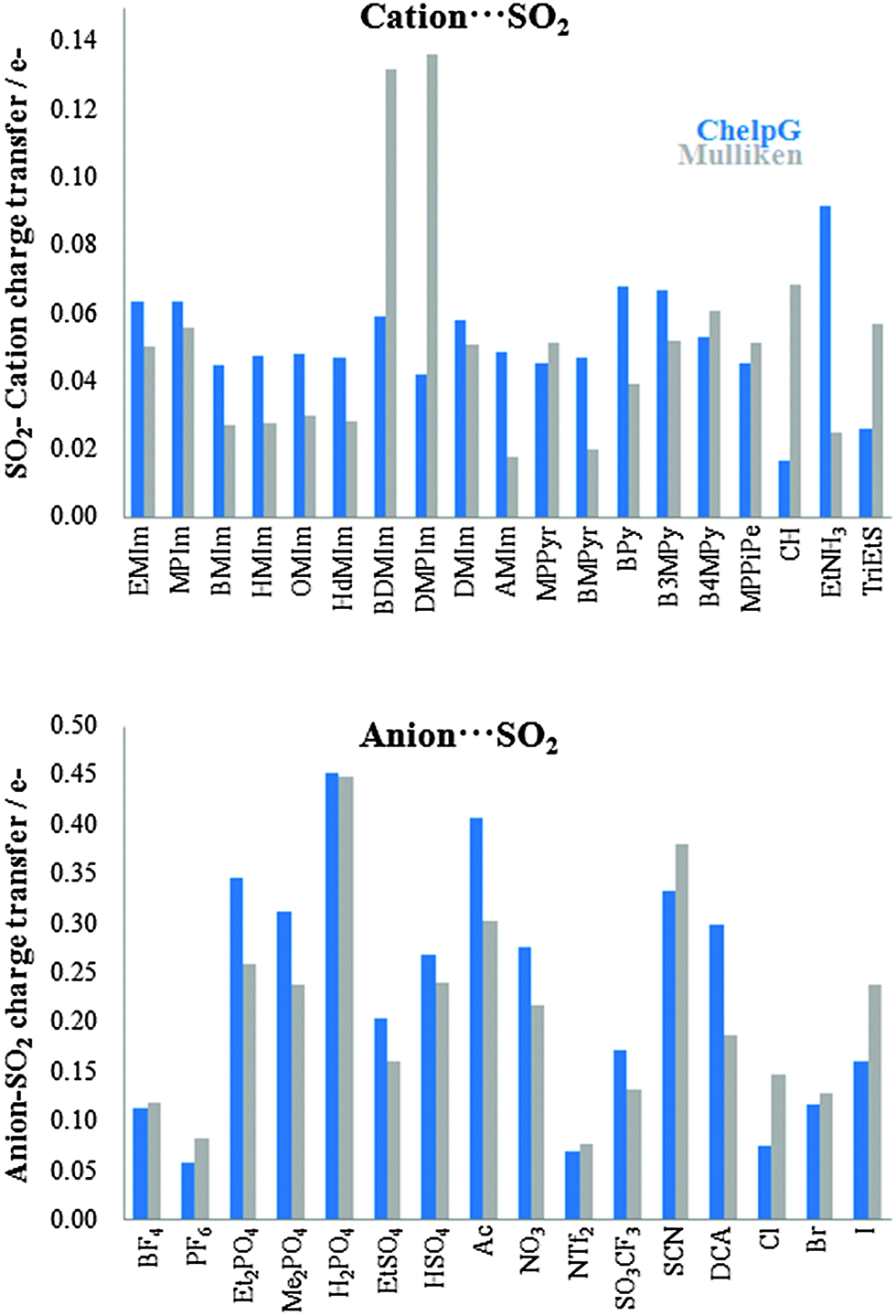
Fig. 3 shows computed binding energies (|BE|) for anion⋯SO2 interactions. In general, the selected cations provide similar |BE|, whose values lie between 31.70 kJ mol−1 ([BMPyr]+) and 42.92 kJ mol−1 ([CH]+), except [EtNH3]+ which yields the largest cation ⋯SO2 values, (|BE| = 58.60 kJ mol−1). In concordance with Damas's work, the binding energy for the imizadolium family decreases upon alkyl side chain elongation. In fact, from [EMIM]+ to [HMIM]+, BE varies by only 1.83 kJ mol−1. However, larger alkyl side chains such as [OMIM]+ and [HdMIM]+ lead to a slight increase in BE upon chain elongation. Anion⋯SO2 binding energies are, in general, larger than cation⋯SO2 binding energies, with values varying between 41.91 kJ mol−1 ([NTf2]−) and 123.37 kJ mol−1 ([H2PO4]−). Some ions can be classified according to their chemical structure (such as those based on phosphate or sulfate anions). Thus, |BE| of those ones based on dialkyl phosphate slightly decreases (≃4.00 kJ mol−1) upon alkyl chain elongation. The alkyl chain absence in [H2PO4]− leads to |BE| values of 29.12 kJ mol−1 which is greater than that of [Et2PO4]−. Similar patterns are noted for sulfate-based ions, wherein the presence of an ethyl chain leads to a diminution of 16.48 kJ mol−1. As concerns as halides, |BE| = 52.18 kJ mol−1 (in average). In order to compare BE values with experimental data, IL 22 ([EMIM][NTf2]) has been selected as its CO2 capture performance has been demonstrated experimentally.11 According to eqn (2), CO2 capture by IL 22 yields |BE|′ = 36.58 kJ mol−1. This energy could be considered as a low limit, from which higher |BE| would be adequate to provide high SO2 affinities.
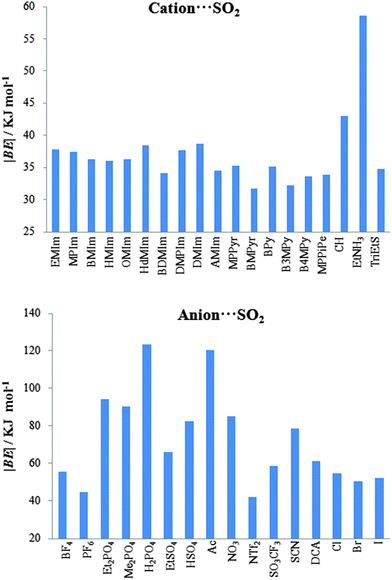 |
| | Fig. 3 Computed binding energies (in absolute value, |BE|) of cation⋯SO2 (up) and anion⋯SO2 (bottom) systems. | |
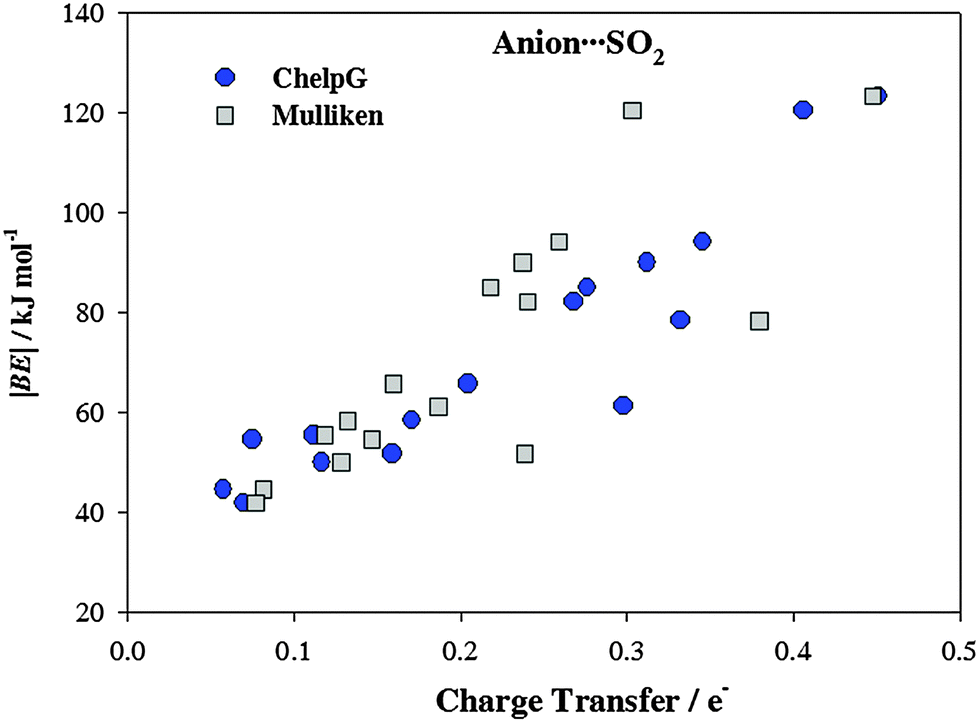
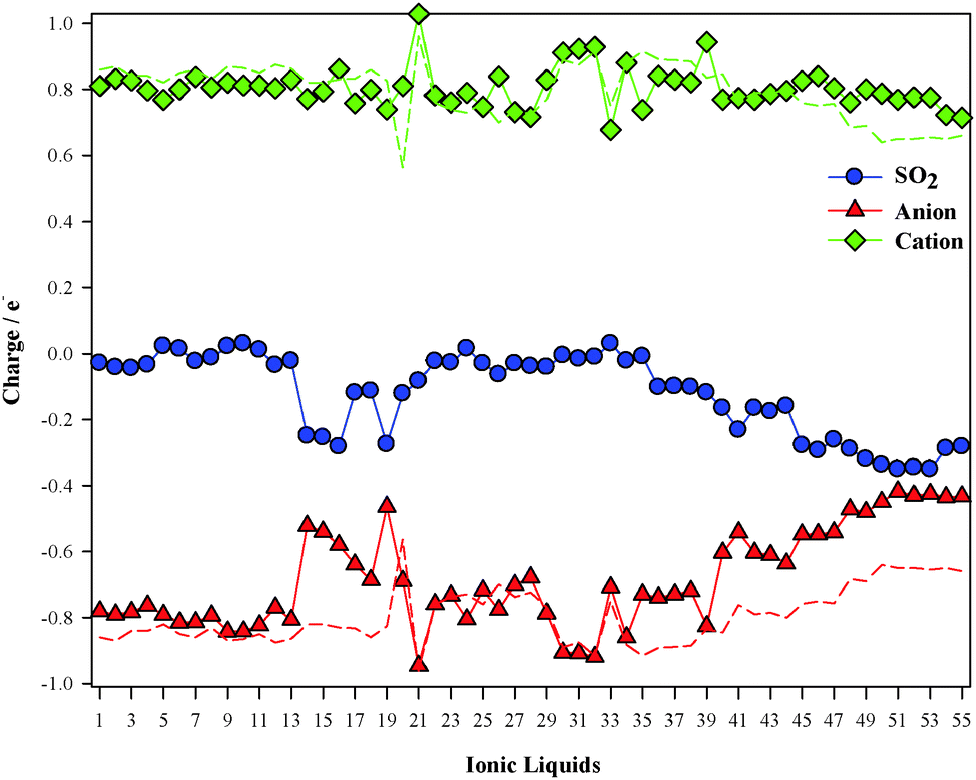
It has been proven that there is a charge transfer interaction between SO2 and the anion motif of ILs. This charge transfer interaction is proportional to the anion basicity and plays an important role on the gas adsorption capacity.26Fig. 4 collects the charge transfers between the cation/anion and SO2 molecule. For cation (anion)⋯SO2 systems, the total charge over the SO2 molecule takes positive (negatives) values, which means that charge is transferred from the SO2 up to the cation (from the anion up to the SO2). Broadly, charge populations according to the Mulliken scheme are smaller than those computed using the ChelpG model. According to ChelpG (Mulliken) atomic charges, charge transfer between cations and the SO2 molecule is, on average, 0.05 (0.05) electrons. Thus, van der Waals interactions are one of the main contribution to the |BE| for cation–SO2 systems, which is in concordance with lower |BE| values than anion–SO2 systems. Now, the total charge over SO2 molecule is 0.23(0.21) electrons for anion–SO2 systems. These higher values are in concordance with greater anion appetency to interact with the SO2 molecule due to a charge transfer interactions. Fig. 5 shows the relationship between binding energies and charge transfer of anion ⋯SO2 systems (a similar pattern has not been found for cation⋯SO2 systems), which follows a linear behavior for most anions.
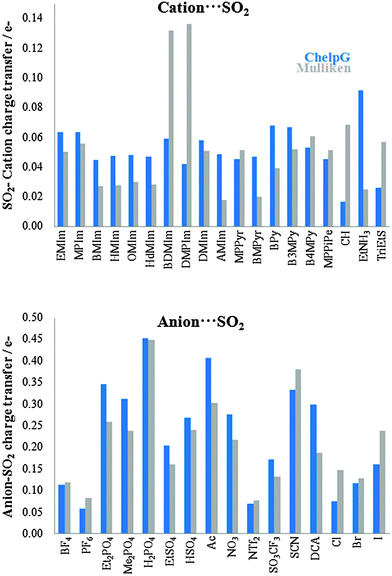 |
| | Fig. 4 Charge transfer of cation⋯SO2 (up) and anion⋯SO2 (bottom) systems. | |
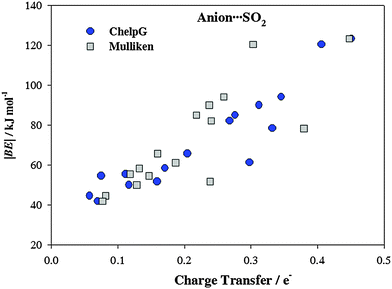 |
| | Fig. 5 Binding energies (|BE|) vs. charge transfer of anion⋯SO2 systems. | |
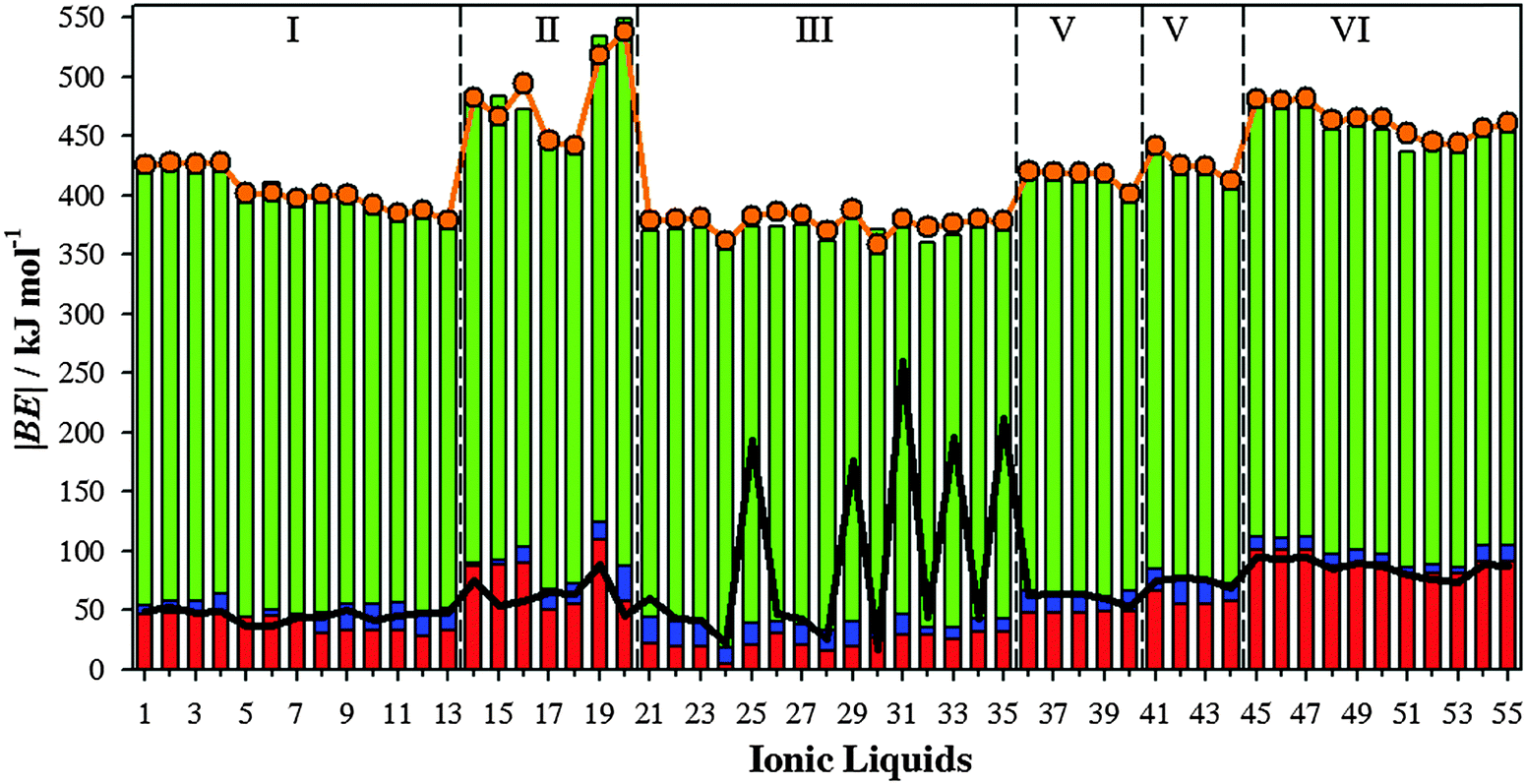
3.2.2. Ionic liquids.
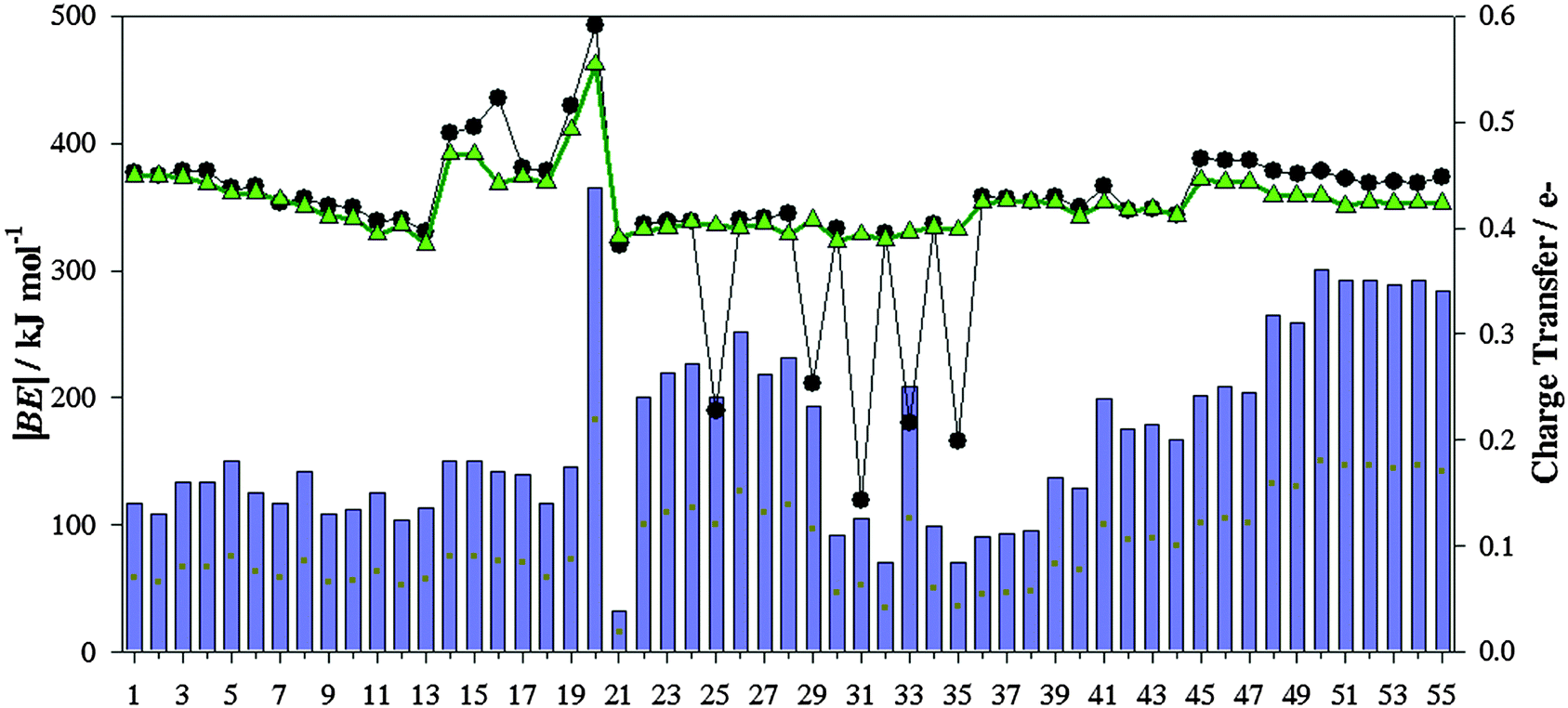
Fig. 6 gathers computed |BE| of the isolated ionic pair and the charge transfer (CT) between ions according to the ChelpG scheme. Most ILs yield |BE| between 318.99 kJ mol−1 (IL 21) and 492.70 kJ mol−1 (IL 20), while ILs 25, 29, 31, 33 and 35 provide the smallest values, around 173.09 kJ mol−1. As known, the columbic attraction between opposite charges is the main force between both ions forming the ionic liquid. Even though, other intermolecular forces can also be present. Both the charge transfer and BE follow similar patterns (Fig. 6), i.e., the columbic interaction between both positive and negative charges is one of the main contributions to the binding energy. ILs with the smallest |BE|, i.e. IL 25, 29, 31, 33 and 35, are those wherein high charge transfer does not provide high binding energies, which points out that other interactions (such as hydrogen bonds) also represent an important contribution (intermolecular interactions between ions are below described for some ILs). ILs based on halide anions (45–55) show increasing CT with the halide electronegativity. Those effects are stronger from chloride to bromide halides. CTs and binding energies depend on both the cation and anion nature as well. For instance, those ILs based on imidazol cations and [NTF2]− anions (except ILs 25 and 29) yield similar |BE| (≃339.0 kJ mol−1).
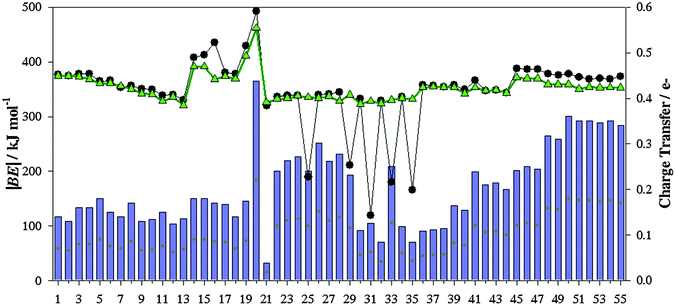 |
| | Fig. 6 Computed binding energies (in absolute value, |BE|) of ionic pairs (black line), along charge transfer computed according to the ChelpG scheme (blue bar). Binding energies of ionic pairs using their geometries in the presence of SO2 are also collected (green line). | |
3.2.3. SO2 capture by ionic liquids.
Binding energies of IL⋯SO2 systems have been used as a measurement of the interaction strength between selected ILs and SO2 molecule. Fig. 7 collects |BE| (according eqn (2)) of IL⋯SO2 systems, which has been decomposed as a sum of the ionic pair, cation⋯SO2 and anion⋯SO2 contributions. Thus, using the optimized IL⋯SO2 geometries, contributions from cation–anion, cation⋯SO2 and anion⋯SO2 have been also calculated. BE energies were also estimated taking into account the ILs as a whole (eqn (2), |BE′|). All these quantities are also provided in Fig. 7. The largest contribution to the binding energy comes from the interaction between both ions. For an easier comparison, this contribution has been also represented in Fig. 6. For most ILs, the SO2 molecule only induces a scarce weakening on the interaction between ions (lower |BE|). However, ILs with the lowest |BE| in the absence of SO2 (ILs 25, 29, 31, 33 and 35, see Fig. 6) are those wherein the SO2 molecule steers to a strengthening on the interaction between ions. This is due to the phenomena that the new arrangement between ions of SO2 improves the interaction between both ions and their interactions with the gas molecule, which is described in detail below.
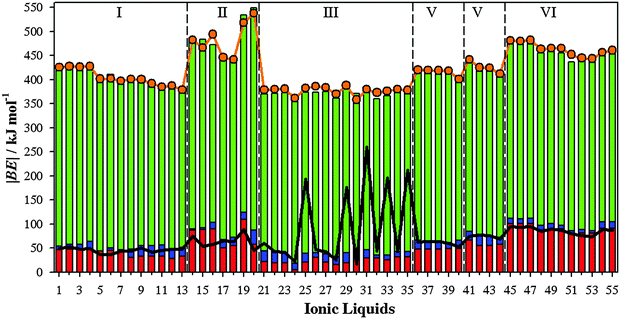 |
| | Fig. 7 Computed binding energies (in absolute value, |BE|) of ILs⋯SO2 (orange line), along anion⋯SO2 contributions (red bar), cation⋯SO2 (blue bar) and cation⋯anion (green bar). Computed binding energies of ILs⋯SO2 considering the IL as a whole are also collected (black line). | |
Regarding ion⋯SO2 contributions, cation⋯SO2 one is, in general, much lower than anion⋯SO2 contributions. Even if, the behavior of both ion⋯SO2 contributions and its relationship with |BE|′ depends on the analyzed IL. For instance, anion⋯SO2 contributions present similar values to |BE′| for ILs 1–6 (based on tetrafluoroborate anion) and 45–55 (based on halides), i.e., anion⋯SO2 interactions stand for the main contribution to the total binding energies of these IL–SO2 systems. Hence, for those ILs based on [BF4]− (1–6) and halide (45–55) anions, the SO2 adsorption process is mainly governed by the anion. For ILs based on triflate, thiocianathe or dicyanamide (ILs 36–44), the sum of both ion⋯SO2 contribution yields similar values of |BE′|. In consequence, the SO2 capture using ILs 36–44 would be guided by both ions. Based on average values, binding energies of cation/anion–SO2 systems (Fig. 3) yield values ≃37.09 kJ mol−1/72.37 kJ mol−1. However, cation/anion⋯SO2 contributions to the binding energy (Fig. 7) are of around 13.94 kJ mol−1/52.03 kJ mol−1. For both ions, interaction energies reduce ≃21.9 kJ mol−1 due to the presence of the paired ion. Ions became less negative, since they transfer charge up to both the cation and SO2 molecule. However, both ions strongly interact between them, hindering cation/anion⋯SO2 interactions. Afresh, this general trend depends on the selected family. For example, for [BF4]−/[Cl]−/[Br]−/[I]− anion |BE| = 55.45 kJ mol−1/54.64 kJ mol−1/50.07 kJ mol−1/51.83 kJ mol−1, while anion–SO2 contributions to the total |BE′| for ILs 1–6 (which are those based on tetrafluoroborate anion) are ≃69.28 kJ mol−1, and ≃90.07 kJ mol−1 for those ILs based on halides (45–55). As seen above, anion⋯SO2 interactions are mainly ruled by the anion⋯SO2. Both factors point out that the CT between both ions would increase anion basicity, as well as its interaction strength with the SO2 molecule. Bearing in mind a value of around 36.0 kJ mol−1 (estimated for CO2 capture by [EMIM][Tf2N] IL) as a low limit, almost ILs yield larger values, |BE′| ≃ 45.0 kJ mol−1. According to these raised values, an efficient SO2 capture can be expected. Once more, ILs 25, 29, 31, 33 and 35 do not follow the general trend, since their binding energies despising ionic contribution (|BE′|) are much larger than the sum of both ion⋯SO2 contributions.
Total charges over both ions and the SO2 molecule are displayed in Fig. 8. The gas usually gets a negative charge, i.e., there is a charge transfer for the anion up to the SO2 molecule. Charge populations over both ions for ILs in the absence of SO2 are also included in Fig. 8. According to the ChelpG scheme, cationic charges slight vary due to the SO2 molecule, while anionic charges suffer drastic lessening due to the charge transfer up to the SO2 molecule.
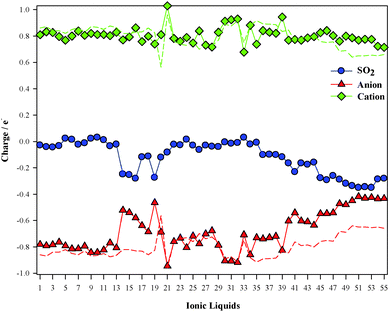 |
| | Fig. 8 Computed charge over SO2 (blue), cation (green) and anion (red) according to the ChelpG scheme. Dotted lines correspond to ion charge for isolated ILs. | |
In short, anion⋯SO2 interactions play an important role in SO2 capture by ILs. When both ions are considered, anion⋯SO2 strengths will be affected by cation–anion interactions. We have defined the binding energies of IL–SO2 systems (BE, according eqn (2)) as a function of the BE of ion–SO2 systems (Section 3.2.1. and Fig. 3) and ionic pairs (Section 3.2.2. and Fig. 6):
| | | BEIL–SO2 = (aBECAT–SO2)x + (bBEANI–SO2)y + (cBEIL)z | (4) |
where BE
IL–SO2, BE
CAT–SO2, BE
ANI–SO2, BE
IL are the binding energies of the IL–SO
2, cation–SO
2, anion–SO
2 and anion–cation systems, respectively, while
a,
b,
c,
x,
y and
z are adjustable parameters.
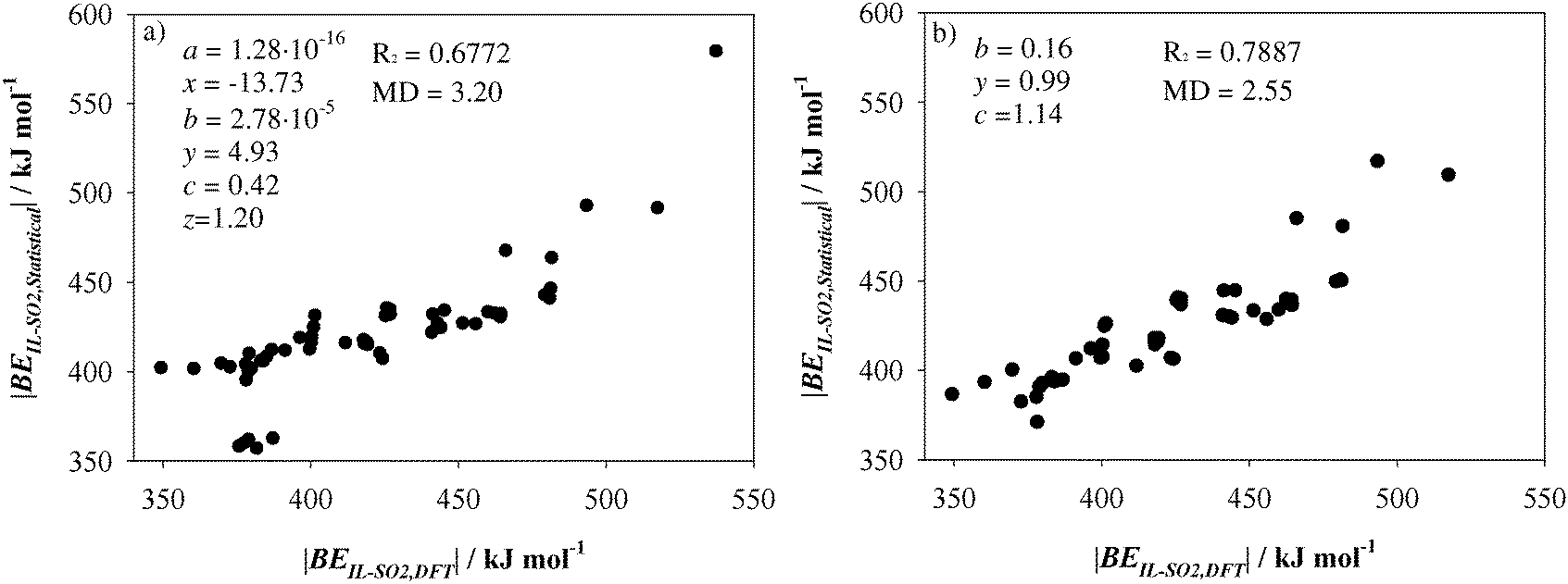
Fig. 9a plots the results of a statistical analysis after expressing BE
IL–SO2 according to
eqn (4).
Fig. 9a gathers the data collected for the whole set of ILs. Most of them yield a linear behavior between BE
IL–SO2 estimated from the IL–SO
2 optimized systems (BE
IL–SO2,DFT) and those ones after the fit of
eqn (4) (BE
IL–SO2,Statistical). Hence, the total binding energy of IL–SO
2 systems, which takes into account both anion–cation and anion–SO
2 interactions, could be directly obtained through the optimization of ion⋯SO
2 systems and ILs. The fit yields
R2 = 0.6772 and medium deviation (MD) = 3.20 kJ mol
−1, which could be considered an acceptable value despite the variety in the chemical structure of the selected ionic liquid. The largest errors correspond to ILs 25, 29, 31, 33 and 35 (|BE
IL–SO2,Statistical| ≃ 360.0 kJ mol). As seen above, those ILs suffered and strengthening of the interaction between ions due to the presence of the SO
2 molecule occurred. According to
eqn (4), no important differences on binding energies for ILs are expected upon SO
2 presence. On the other hand, IL 20 (|BE
IL–SO2,Statistical| = 578.34 kJ mol) is based on [EtNH
3]
+ cation. [EtNH
3]
+–SO
2 provided the highest binding energy among all studied cations. According with
a parameter (
a = 1.28 × 10
−16), contribution from BE
CAT–SO2 is close to zero. Hence, the above expression is only applicable to those ILs wherein the anion plays the main role on SO
2 capture and for those ILs which do not suffer important geometrical arrangements in the presence of the gas molecule. Note that the
z parameter is close to one. As a result, we defined
eqn (4) based only on BE
ANI–SO2 and BE
IL. BE
IL–SO2,Statistical as follows:
| | | BEIL–SO2,Statistical = b(BEANI–SO2)y + cBEIL | (5) |
The fit was repeated despising ILs 20, 25, 29, 31, 33 and 35. As seen in Fig. 9b, there is a notable improvement in the fit performance with R2 = 0.7887 and MD = 2.55 kJ mol−1. It could be concluded that SO2 capture by ILs is mainly governed by ILs, while interactions between ions are also an important parameters. Since BE between ions is much higher than anion–SO2 ones, BEIL grants the most important contribution to BEIL–SO2,Statistical. Then, for those ILs with similar BEIL, the efficiency in SO2 capture will be ruled by the anion. In addition, eqn (5) allows estimating BEIL–SO2 only through the optimization of anion–SO2 and cation–anion systems, which can be considered a useful insight into the rational design of ILs for SO2 capture.
3.2.4. Representative ionic liquids for SO2 capture.
Up to now, properties for IL⋯SO2 interactions have been analyzed for the whole family of selected ILs based on binding energies. As seen, the SO2 absorption capacity is often governed by anion⋯SO2 interactions, although cations have also an important role. Even though, cation–SO2 contributions to the total |BE′| are always lower than cation–SO2 binding energies, while this general trend was not found for anion⋯SO2 contributions. For instance, anion⋯SO2 contributions to the total |BE′| for ILs (1–4, which are based on imidazolium cations paired with tetrafluoroborate anion) are higher than binding energies for anion–[BF4]− systems, while the opposite trend was noted for ILs 22–29 (also based on imidazol derived cations, but paired with the [NTf2]− anion). In addition, a statistical analysis has shown that BE of IL–SO2 systems mainly depends on anion–SO2 and cation–anion interactions. The diversity in the nature of both ions forming the family of studied ILs hinders the search of structure–property relationships. Therefore the IL family has been divided into six sets (labelled as I–VII, see Fig. 2 and 7), wherein ILs within the same sets have similar features regarding the chemical structure of their ions. For each one, the most representative ILs have been selected, whose intermolecular interactions where analyzed within the context of the AIM theory to obtain some information on the SO2 capture mechanism at the nanoscopic level.
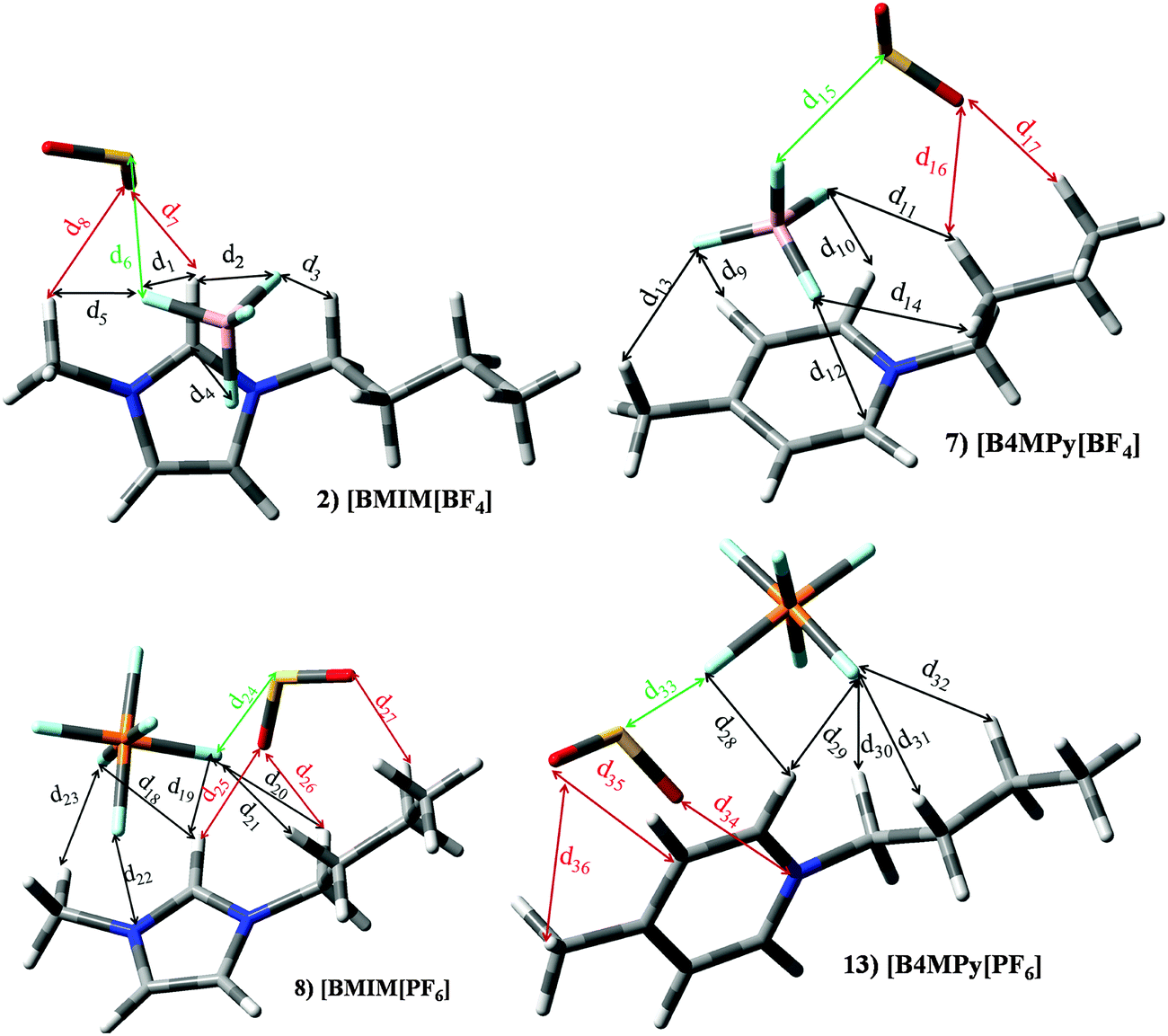
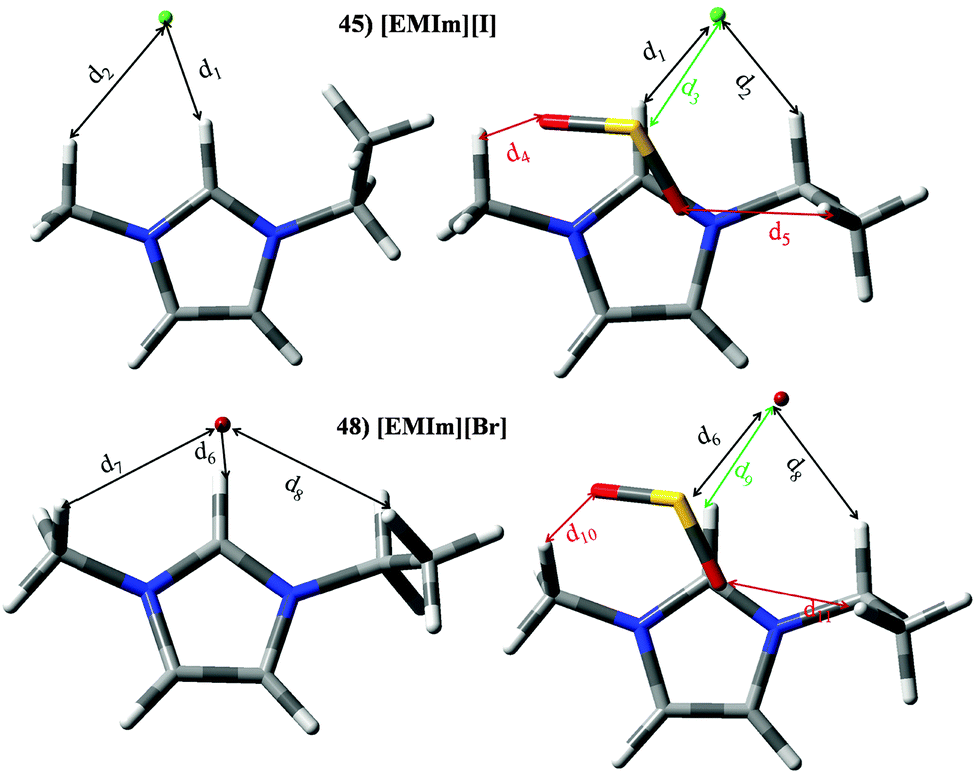
Set I (ILs 1–13) includes ILs based on imidazolium ([Im]+) or pyridinium ([Py]+) cations paired with [BF4]− or [PF6]− anions. ILs based on imidazolium and [BF4]− (ILs 1–4) yields similar KH ≃ 3.6 × 105 Pascal and |BE′| ≃ 49.65 kJ mol−1. [Im][PF6] based ILs (8–10) render smaller KH (≃2.7 × 105 Pascal); however this improvement in KH is not observed on |BE′| (≃45.19 kJ mol−1). For pyridinium based ILs (5–7 and 11–13) The replacement of [Im]+ by [Py]+ does not lead to important changes on KH and |BE′|. Though, the alkyl chain length in the cation, as well as the presence of [BF4]− or [PF6]− anions have an effect of thermophysical properties such as viscosity or density.30,36 ILs included in set I would provide similar SO2 capture efficiency (based on KH and |BE′| values). As a matter of fact, several papers highlight the effect on macroscopical properties as a function of the selected ions elsewhere.30,36,37 In order to discuss the effects on different ions at the molecular level, besides previously described parameters, the interaction mechanisms of [BMIm][BF4] (IL 2), [BMIm][PF6] (IL 8), [B4MPy][BF4] (IL 7) and [B4MPy][PF6] (IL 13) have been deeply analyzed as representative compounds of this set. Intermolecular interactions were localized and featured through the AIM theory (we have focused on electronic density values, ρ, for the main intermolecular interactions). Fig. 10 plots their optimized structures in the presence of the SO2 molecule (optimized geometries for isolated ILs are not represented since the presence of SO2 does not carry out important changes on the relative disposition between ions), whereas bond length and AIM features of intermolecular interactions are reported in Table 2. In the absence of the SO2 molecule, several anion–cation interactions are established. The main interactions are formed between F and H in position 2 of the imidazolium/pyridinium ring, whose d (intermolecular distance) and ρ are ≃2.240 and 0.0140 a.u., respectively. In this sense, it is well known that the main interaction in imidazilium based ILs is carried out through the H atom in position 2.17 The presence of the SO2 molecule leads to an intermolecular distance elongation and electronic density decrease, in concordance with lower |BE| of ILs using their geometries in the presence of SO2. As seen below, this effect is also noted for almost all ILs under study. Anion–SO2 interactions are mainly characterized by a BCP between F and S, labeled as d6, d15, d24 and d33 for ILs 2, 7, 8 and 13, respectively, whose ρ are 0.0273 a.u., 0.0189 a.u., 0.0135 a.u., 0.0092 a.u., respectively. Similar patterns are noted for anion–SO2 contribution to the total binging energies (Fig. 7). Cation–SO2 interactions take place through O (SO2) and H (cation). These H are mainly located on the alkyl side chain. For ILs based on [B3MPy]+/[B4MPy]+ based ILs (6/7 and 12/13), the presence of methyl chain in position 3/4 brings slight improvement on KH and BE with respect to [BPy]+. This methyl group in position 3/4 allows an additional intermolecular interaction (e.g., d13 for IL 7) with SO2, which is absent for [BPy]+.
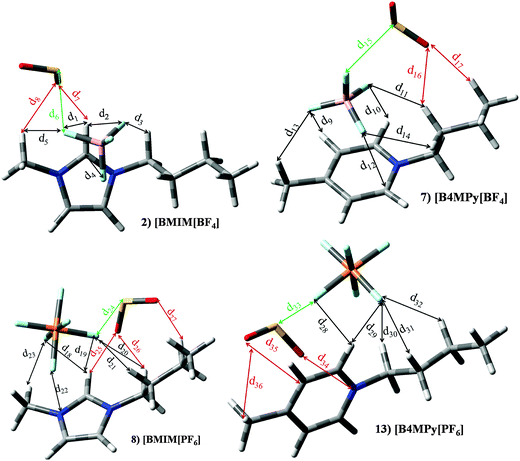 |
| | Fig. 10 Optimized geometries of [BMIm][BF4] (2), [B4MPy][BF4] (7), [BMIm][PF6] (8) and [B4MPy][PF6] (13) in the presence of the SO2 molecule. Main intermolecular interactions are also displayed. Atom colour code: C (gray), oxygen (red) sulphur (yellow), hydrogen (white), nitrogen (blue), boron (pink), phosphorous (orange) and fluorine (light blue). See Table 2 for a more detailed description on intermolecular interactions. | |
Table 2 Intermolecular distances (d) along the electronic density values (ρ) of [BMIm][BF4] (2), [B4MPy][BF4] (7), [BMIm][PF6] (8) and [B4MPy][PF6] (13) ionic liquids. See Fig. 10 for labeling
| |
IL |
IL⋯SO2 |
|
d/Å |
ρ/a.u. |
d/Å |
ρ/a.u. |
|
For isolated IL, this interaction take places between F and H in position 2.
For isolated IL, this interaction takes places between F and C in position 2.
|
| 2 – [BMIm][BF4] |
|
d
1
|
2.233 |
0.0143 |
2.788 |
0.0076 |
|
d
2
|
2.106 |
0.0178 |
2.457 |
0.0121 |
|
d
3
|
2.502 |
0.0082 |
2.502 |
0.0091 |
|
d
4
|
2.873 |
0.0101 |
2.892 |
0.0102 |
|
d
5
|
2.222 |
0.0130 |
2.447 |
0.0100 |
|
d
6
|
2.531 |
0.0273 |
|
d
7
|
2.518 |
0.0087 |
|
d
8
|
2.118 |
0.0170 |
|
|
| 7 – [B4MPy][BF4] |
|
d
9
|
2.298 |
0.0129 |
2.910 |
0.0097 |
|
d
10
|
2.119 |
0.0173 |
2.549 |
0.0148 |
|
d
11
|
2.671 |
0.0117 |
2.484 |
0.0081 |
|
d
12
|
2.322 |
0.0097 |
2.870 |
0.0105 |
|
d
13
|
2.479 |
0.0077 |
|
d
14
|
2.289 |
0.0120 |
2.706 |
0.0188 |
|
d
15
|
2.716 |
0.0089 |
|
d
16
|
2.496 |
0.0082 |
|
d
17
|
2.429 |
0.0097 |
|
|
| 8 – [BMIm][PF6] |
|
d
18
|
2.406 |
0.0113 |
2.421 |
0.0116 |
|
d
19
|
2.320 |
0.0138 |
2.607 |
0.0101 |
|
d
20
|
2.413 |
0.0107 |
2.701 |
0.0067 |
|
d
21
|
2.479 |
0.0100 |
2.315 |
0.0118 |
|
d
22
|
2.698 |
0.0142 |
2.803 |
0.0094 |
|
d
23
|
2.488 |
0.0097 |
2.648 |
0.0206 |
|
d
24
|
2.652 |
0.0135 |
|
d
25
|
2.209 |
0.0084 |
|
d
26
|
2.535 |
0.0071 |
|
d
27
|
2.548 |
0.073 |
|
|
| 13 – [B4MPy][PF6] |
|
d
28
|
2.328 |
0.0116 |
2.363 |
0.0128 |
|
d
29
|
2.124 |
0.0167 |
2.078 |
0.0181 |
|
d
30
|
2.313 |
0.0113 |
2.481 |
0.0145 |
|
d
31
|
2.588 |
0.0114 |
2.446 |
0.0095 |
|
d
32
|
2.698 |
0.0057 |
|
d
33
|
2.648 |
0.0092 |
|
d
34
|
2.955 |
0.0065 |
|
d
35
|
3.218 |
0.0059 |
|
d
36
|
2.718 |
0.0058 |
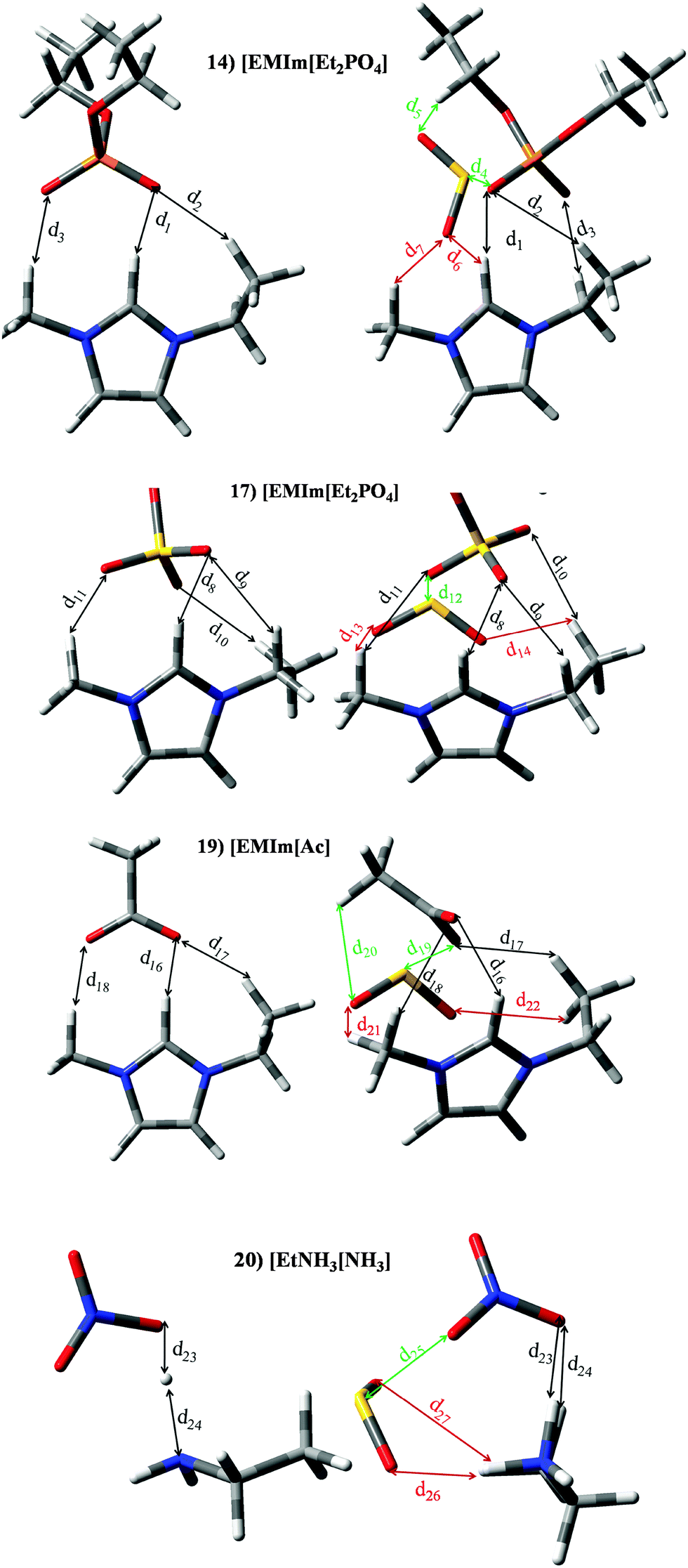
ILs based on phosphate, sulfate, acetate or nitrate anions are located in set II. Most of them are also based on [EMIm]+ cation. Interaction energies of anion⋯SO2 systems (see Fig. 3); [Et2PO4]− (99.24 kJ mol−1), [EtSO4]− (65.75 kJ mol−1), [Ac]− (120.47 kJ mol−1) and [NO3]− (85.06 kJ mol−1), are larger than those estimated for [EMIm]+ (33.77 kJ mol−1) and [EtNH3]+ (58.60 kJ mol−1) cations. Analogous behaviour is noted for both ion⋯SO2 contribution to the binding energy, i.e., anion⋯SO2 > cation⋯SO2. Nevertheless, the sum of both contributions is higher than |BE′| (see Fig. 7). In concordance with |BE| computed for cation/anion⋯SO2, anion⋯SO2 contribution to the total BE is larger. Within this set we have focused on [EMIm][Et2PO4] (14), [EMIm][EtSO4] (17) and [EMIm][Ac] (19) ILs. A detailed analysis of [CH][H2PO4] at the molecular level and their application for SO2 capture will be studied in a separate work in the future. The structures of [EMIm][Et2PO4] (14), [EMIm][EtSO4] (17) and [EMIm][Ac] (19) in the presence and absence of SO2 are reported in Fig. 11. [EtNH3][NO3] (20) IL has been also selected to obtain some insight up to the behaviour of this IL. In the absence of the SO2 molecule, the main interaction between imidazolim cation and the corresponding anion is carried out by a hydrogen bond between the O atom (anion) and H in position 2 of the imidazolium ring (labelled d1, d8 and d16 for ILs 14, 17 and 19 respectively). Again, the presence of the SO2 molecule brings a diminution of the interaction between both ions. As seen in Fig. 7 for IL 17, contribution from anion⋯SO2 interaction to the |BE| is larger than cation⋯SO2 interaction, which agrees with larger ρ values for d12 regarding to interaction between cation and SO2, i.e., d13 and d14 (similar behaviour can be drawn for ILs 14 and IL19). The SO2 molecule interacts with the anion through an intermolecular bond between the S and one oxygen atom located in the anion. [EtNH3][NO3] (20) presents the highest charge transfer and |BE| between ions in the absence of SO2 (see Fig. 6). As seen in Fig. 11, there is a proton transfer between ions. In fact, the distance between [NO3]− and H (d23) is 1.045 Å, while the distance between N and H (d24) is 1.595 Å. ChelpG charges have shown that such O has an atomic charge of −0.58 (larger than the −0.46 e− over the other O), while the positive charge over this H is 0.39 (charge over remaining H linked to N is of around 0.26 e−). This effect is not observed in the presence of the SO2 molecule. The adsorption of SO2 by IL 20 is carried out by a strong interaction between SO2 and the anion (d25), while there are dual interactions between SO2 and the cation (d26 and d27, being the latter the weakness). Once more, a larger electronic density for d35 (respect to d26) agrees with the greater contribution from SO2⋯interaction to |BE′|.
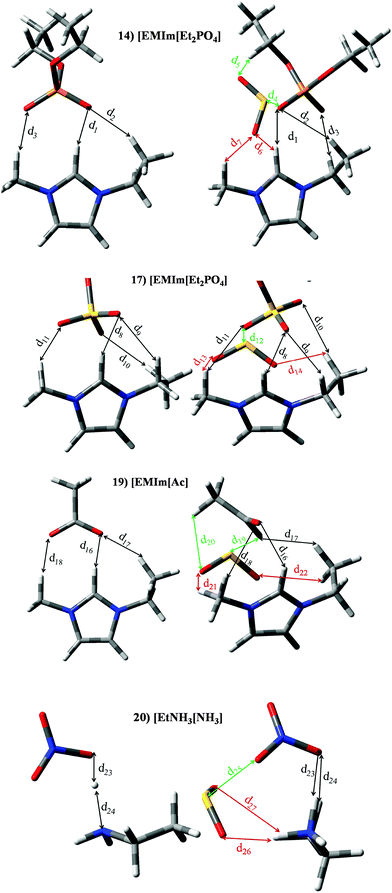 |
| | Fig. 11 Optimized geometries of [EMIm][Et2PO4] (14), [EMIm][EtSO4] (17), [EMIm][Ac] (19) and [EtNH3][NO3] (20). Main intermolecular interactions are also displayed. Atom colour code: C (gray), oxygen (red) sulphur (yellow), hydrogen (white), nitrogen (blue)and phosphorous (orange). See Table 3 for a more detailed description on intermolecular interactions. | |
Table 3 Intermolecular distances (d) along the electronic density values (ρ) of [EMIm][Et2PO4] (14), [EMIm][EtSO4] (17), [EMIm][Ac] (19) and [EtNH3][NO3] (20). See Fig. 11 for labeling
| |
IL |
IL⋯SO2 |
|
d/Å |
ρ/a.u. |
d/Å |
ρ/a.u. |
| 14 – [EMIm][Et2PO4] |
|
d
1
|
1.756 |
0.0299 |
1.793 |
0.0369 |
|
d
2
|
2.304 |
0.0120 |
2.780 |
0.0194 |
|
d
3
|
1.962 |
0.0253 |
2.119 |
0.0064 |
|
d
4
|
2.238 |
0.0575 |
|
d
5
|
2.510 |
0.0088 |
|
d
6
|
2.578 |
0.0087 |
|
d
7
|
2.242 |
0.0144 |
|
|
| 17 – [EMIm][EtSO4] |
|
d
8
|
2.055 |
0.0231 |
2.052 |
0.0225 |
|
d
9
|
2.510 |
0.0095 |
2.500 |
0.0096 |
|
d
10
|
2.417 |
0.0103 |
2.513 |
0.0087 |
|
d
11
|
2.184 |
0.0159 |
2.555 |
0.0082 |
|
d
12
|
2.243 |
0.0396 |
|
d
13
|
2.419 |
0.0109 |
|
d
14
|
2.444 |
0.0081 |
|
d
15
|
2.576 |
0.0059 |
|
|
| 19 – [EMIm][Ac] |
|
d
16
|
1.654 |
0.0278 |
2.110 |
0.0205 |
|
d
17
|
2.352 |
0.0109 |
2.491 |
0.0097 |
|
d
18
|
2.002 |
0.0240 |
2.427 |
0.0104 |
|
d
19
|
2.189 |
0.0658 |
|
d
20
|
2.667 |
0.0077 |
|
d
21
|
3.665 |
0.0128 |
|
d
22
|
2.583 |
0.0082 |
|
|
| 20 – [EMIm][NO3] |
|
d
23
|
1.596 |
0.0072 |
1.639 |
0.0564 |
|
d
24
|
1.045 |
2.625 |
0.0078 |
|
d
25
|
2.350 |
0.0454 |
|
d
26
|
1.820 |
0.0328 |
|
d
27
|
2.620 |
0.0074 |
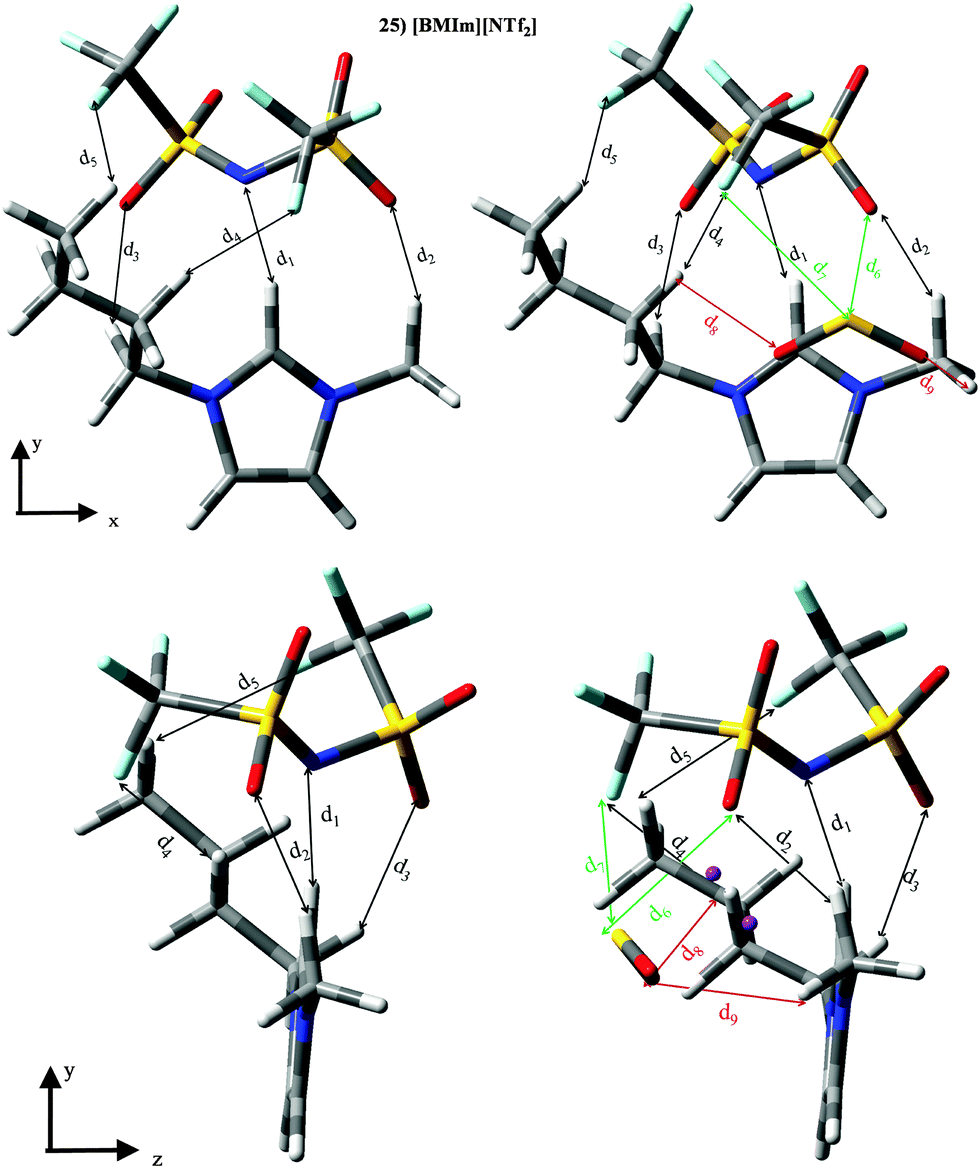
ILs based on the [NTf2]− anion (set III) are the largest group, whose KH ≃ 3.4 × 105 Pascal and |BE′| ≃ 38.45 kJ mol−1 (despising ILs 25, 29, 31, 33 and 35). For ILs 25, 29, 31, 33 and 35, |BE′| is much larger than sum of both ion⋯SO2 contributions. Furthermore, ILs 25, 29, 31, 33 and 35 are the only ones whose interactions between ions are strengthened in the presence of the SO2 molecule (see Fig. 6). The SO2 brings a rearrangement between ions which improves their mutual interaction and also their interactions with SO2. To obtain information about this fact, we have focused on IL [BMIm][NTf2]. Optimized geometries as well as the main results from intermolecular interaction analysis are collected in Fig. 12 and Table 4. [BMIm][NTf2] ILs yields five intermolecular interactions (d1–d5) between both ions, wherein the one between the N (anion) and the H (cation) is position 2 is the strongest one. Although the same interactions between both ions are found in the presence of the SO2 molecule, all of them suffer an elongation/decrease on intermolecular distances/electronic density values. SO2 molecule is able to for two bonds with the anion, i.e., d6 (S⋯O) and d7 (S⋯F), being the latter much weaker than S⋯O interaction. Further, two O⋯H bonds (d8 and d9) are noted between SO2 and cation molecules. Although SO2 causes a weakening of the interaction between ions (based on electronic density values), it also allows the formation of a cage, with their corresponding cage critical points (CCP). Concretely, two cage critical points (represented as purple points along the yz view) are found, whose electronic density is 0.0027 a.u. and 0.0018 a.u. The presence of both CCP points out to a charge delocalization process between different motifs. Results described for this IL could be extrapolated to ILs 29, 31, 33 and 35, i.e., larger |BE′| values and stronger interaction between ions are due to the charge delocalization process. This charge delocalization brings an increase on inter ionic interaction (with respect to isolated IL), and |BE′| is higher than the sum of both ion⋯SO2 contributions. Although, CCPs are also found for other ILs, they own much lower electronic density values.
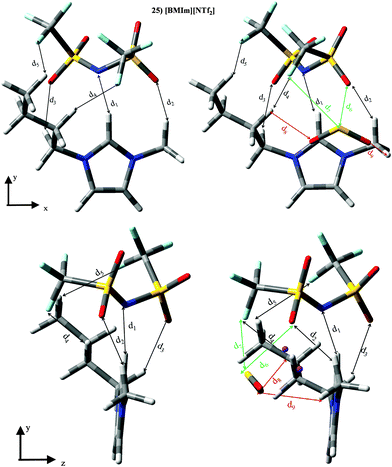 |
| | Fig. 12 Optimized geometries of [BMIm][NTf2] (25), [BMPyr][NTf2] (31) and [B4MPy][NTf2] (35). Main intermolecular interactions are also displayed. Atom colour code: C (gray), oxygen (red) sulphur (yellow), hydrogen (white), nitrogen (blue) and phosphorous (orange). See Table 4 for a more detailed description on intermolecular interactions. | |
Table 4 Intermolecular distances (d) along the electronic density values (ρ) of [BMIm][NTf2] (25). See Fig. 12 for labeling
| |
IL |
IL⋯SO2 |
|
d/Å |
ρ/a.u. |
d/Å |
ρ/a.u. |
| 25 – [BMIm][NTf2] |
|
d
1
|
1.952 |
0.0303 |
2.020 |
0.0266 |
|
d
2
|
2.186 |
0.0150 |
2.419 |
0.0104 |
|
d
3
|
2.290 |
0.0130 |
2.352 |
0.0115 |
|
d
4
|
2.564 |
0.0063 |
2.594 |
0.0058 |
|
d
5
|
2.553 |
0.0024 |
2.623 |
0.0058 |
|
d
6
|
2.683 |
0.0218 |
|
d
7
|
3.208 |
0.0062 |
|
d
8
|
2.739 |
0.0068 |
|
d
9
|
2.479 |
0.0099 |
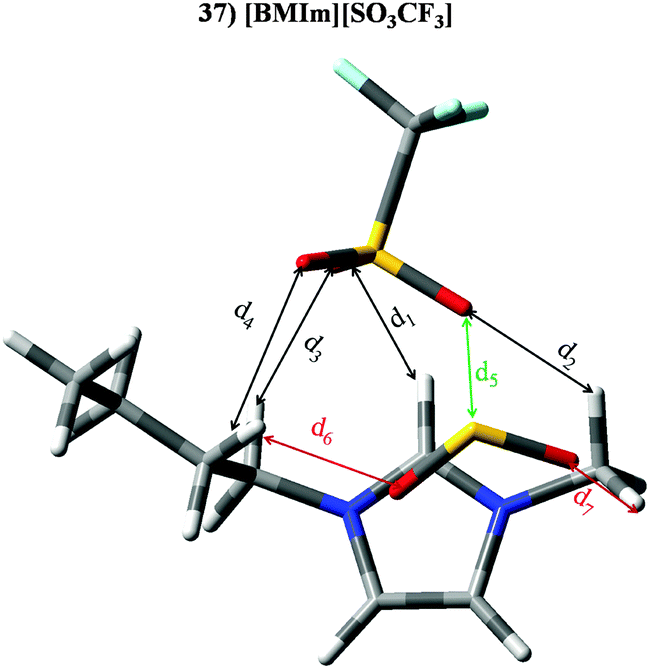
ILs based on the triflate anion ([SO3CF3]−) are within set IV (IL 36–40). Those ones also based on imidazolium cations (36–39) provide KH ≃ 4.22 × 105 Pascal and |BE′| ≃ 60.30 kJ mol−1, which is due to the sum of both ion⋯SO2 contributions. [BMPyr][SO3CF3] (IL40) yields KH = 3.57 × 105 Pascal and |BE′| = 51.97 kJ mol−1, mainly due to the anion⋯SO2 contribution. Larger anion⋯SO2 contributions (Fig. 7) to the binding energy mimic the previously reported compound for ion⋯SO2 binding energies (Fig. 3). Fig. 13 and Table 5 gather optimized geometries and intermolecular parameters for [BMIm][SO3CF3] (37). Results obtained for this IL could be extrapolated for the whole set IV. The main interaction between both ions takes places through O corresponding to the anion and H in position 2 located in the cation (d1), whose ρ and distances are more affected by the SO2 molecule, which causes its weakening. However, the remaining interactions are slightly affected by the gas molecule. Thus, binding energy for IL 37 is very similar to contribution from inter ionic interaction to the BE estimated for the IL 37⋯SO2 system (see Fig. 6). The adsorption of SO2 by IL 37 is mainly carried out through O (anion)⋯S(SO2) interaction (labelled as d5). Even if, SO2 molecule also owns two intermolecular O⋯H bonds with alkyl H atoms located in the cation (d6 and d7). In concordance with ion⋯ contributions to |BE′|, anion⋯SO2 interaction (based on its larger electronic density value) is greater than cation⋯SO2 interaction.
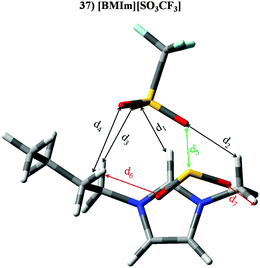 |
| | Fig. 13 Optimized geometries of [BMIm][SO3CF3] (37) in the presence of SO2 (similar results are obtained for isolated IL). Main intermolecular interactions are also displayed. Atom colour code: C (gray), oxygen (red) sulphur (yellow), hydrogen (white), nitrogen (blue) and fluorine (light blue). See Table 5 for a more detailed description on intermolecular interactions. | |
Table 5 Intermolecular distances (d) along the electronic density values (ρ) of [BMIm][SO3CF3] (37). See Fig. 13 for labeling
| |
IL |
IL⋯SO2 |
|
d/Å |
ρ/a.u. |
d/Å |
ρ/a.u. |
| 37 – [BMIm][SO3CF3] |
|
d
1
|
2.025 |
0.0239 |
2.184 |
0.0070 |
|
d
2
|
2.280 |
0.0130 |
2.626 |
0.0109 |
|
d
3
|
2.520 |
0.0081 |
2.439 |
0.0089 |
|
d
4
|
2.594 |
0.0088 |
2.544 |
0.0351 |
|
d
5
|
2.476 |
0.0096 |
|
d
6
|
2.464 |
0.0107 |
|
d
7
|
2.451 |
0.0070 |
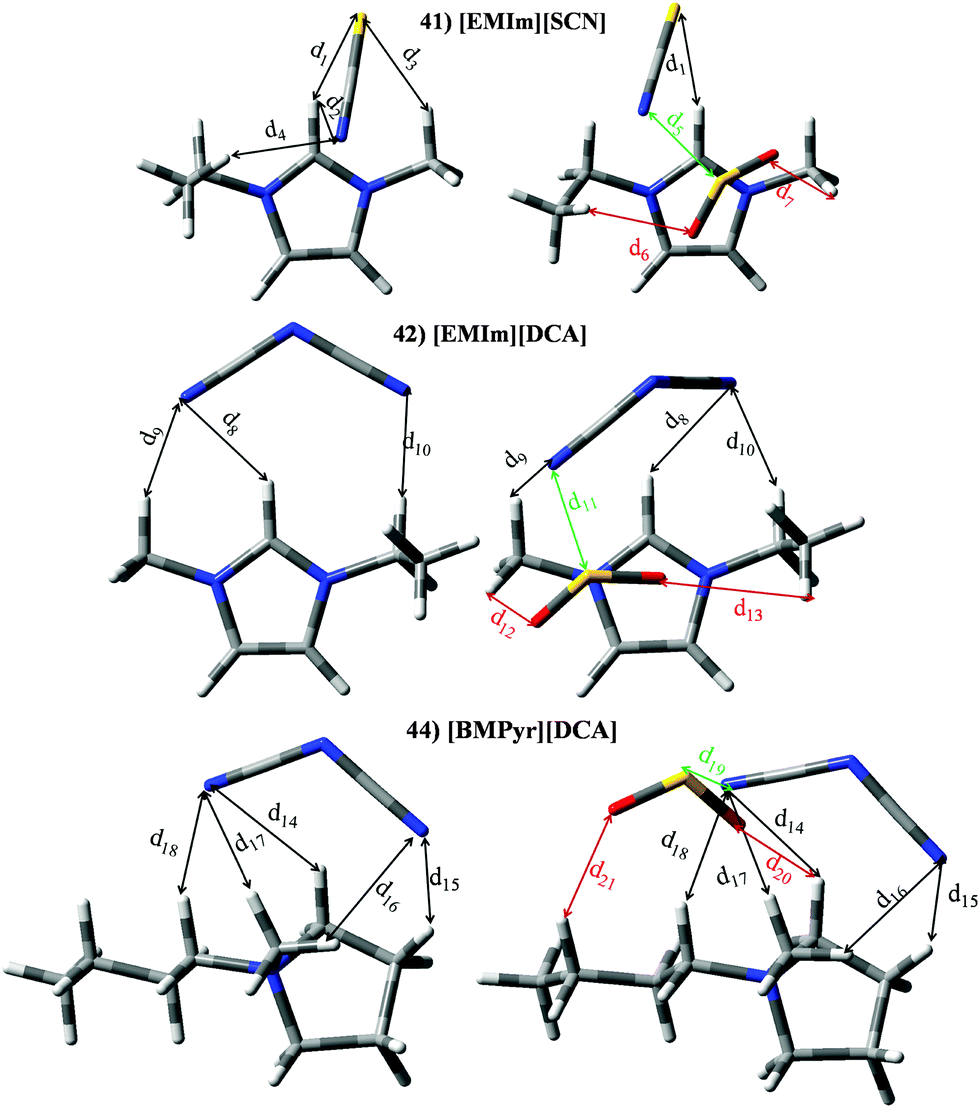
Set V (IL 41 44) comprises those ILs whose anions have at least one CN group, i.e., thiocianate ([SCN]−) and dicyanamide ([DCA]−). ILs based on the imidazolium cation (41–43) supply KH ≃ 4.23 × 105 Pascal and |BE′| ≃ 76.21 kJ mol−1, while [BMPyr][DCA] (IL 44) yields KH = 2.88 × 105 Pascal and |BE′| = 68.20 kJ mol−1. Isolated ions provided |BE| = 78.40 kJ mol−1 and 61.29 kJ mol−1, while |BE| for imidazolium and [BMPyr] + are ≃ 36.99 kJ mol−1 and 31.70 kJ mol−1, respectively. Anew, the trend perceived for the interaction between anions (cation) and SO2 in the absence of the cation (anion) is also found for both ion⋯SO2 contributions to the binding energies. [EMIM][SCN] (41) brings a |BE′| similar to anion⋯SO2 contribution, while |BE′| for [DCA]− based ILs comes from both ion⋯SO2 contributions. Fig. 14 and Table 6 reports optimized geometries for [EMIm][SCN] (41), [EMIm][DCA] (42) and [BMPyr][DCA] (44). As expected (in the absence of SO2), both ions interact with the [EMIM]+ cation through its H in position 2. S and N terminal atoms from [SCN]− anion are able to interact with these H atoms (d1 and d2). Further, the S atom also provides an intermolecular interaction with methyl hydrogen (d3). Although, [DCA]− owns two CN groups, only one of them interacts with H in position 2 (d8), even though two interactions with alkyl H atoms are also found (d9 and d10). Regarding IL 44, only one N group interacts with the main position provided by the cation (d14), although other intermolecular H bonds (with lower ρ) are also present (d15–d18). According to electronic density values, ionic interactions are stronger for [SCN]− anion, which agrees with its higher |BE| vales (see Fig. 6). ILs 41, 42 and 44 show similarities regarding to the interactions with the gas molecule. Thus, the main interaction is carried out between one terminal N (anion) and the central S atom (d5, d11 or d19 for IL 41, 42 or 44, respectively). Although electronic density for d5 (0.387 a.u.) is smaller than electronic density for d11 and d19 (≃0.423 a.u.), larger charge transfer from the [SCN]− anion up to the gas (see Fig. 8) agrees with greater [SCN]−⋯SO2 contribution in IL41–SO2 system (Fig. 7). The SO2 molecule also interacts (through both hydrogen atoms) with the cation (d6 and d7, d12 and d13 or d20 and d21 for IL 41, 42 or 44, respectively). Instead the selected IL, the sum of the electronic density for both intermolecular bonds is ≃0.0190 a.u. Thus, cation⋯SO2 contribution to the BE is similar for all ILs within set V.
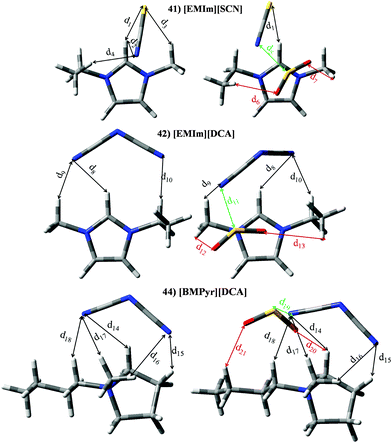 |
| | Fig. 14 Optimized geometries of [EMIM][SCN] (41), [EMIM][DCA] (42) and [BMPyr][DCA] (44). Main intermolecular interactions are also displayed. Atom colour code: C (gray), oxygen (red) sulphur (yellow), hydrogen (white), nitrogen (blue) and fluorine (light blue). See Table 6 for a more detailed description on intermolecular interactions. | |
Table 6 Intermolecular distances (d) along the electronic density values (ρ) of [EMIm][SCN] (41), [EMIm][DCA] (42) and [BMPyr][DCA] (44). See Fig. 14 for labeling
| |
IL |
IL⋯SO2 |
|
d/Å |
ρ/a.u. |
d/Å |
ρ/a.u. |
| 41 – [EMIm][SCN] |
|
d
1
|
2.567 |
0.0152 |
2.572 |
0.0141 |
|
d
2
|
2.907 |
0.0142 |
|
d
3
|
2.841 |
0.0092 |
|
d
4
|
2.532 |
0.0093 |
|
d
5
|
2.369 |
0.0387 |
|
d
6
|
2.506 |
0.0089 |
|
d
7
|
2.513 |
0.0107 |
|
|
| 42 – [EMIm][DCA] |
|
d
8
|
2.348 |
0.0137 |
2.480 |
0.0115 |
|
d
9
|
2.180 |
0.0177 |
2.395 |
0.0051 |
|
d
10
|
2.161 |
0.0181 |
2.535 |
0.0048 |
|
d
11
|
2.431 |
0.0430 |
|
d
12
|
2.376 |
0.0118 |
|
d
13
|
2.630 |
0.0074 |
|
|
| 44 – [BMPyr][DCA] |
|
d
14
|
2.750 |
0.0084 |
2.745 |
0.0088 |
|
d
15
|
2.462 |
0.0106 |
2.406 |
0.0097 |
|
d
16
|
2.689 |
0.0075 |
2.534 |
0.0117 |
|
d
17
|
2.550 |
0.0104 |
2.585 |
0.0103 |
|
d
18
|
2.294 |
0.0143 |
2.463 |
0.0105 |
|
d
19
|
2.464 |
0.0105 |
|
d
20
|
2.617 |
0.0103 |
|
d
21
|
2.448 |
0.0088 |
Set VI is devoted to those ILs based on imidazolium cations and halides (IL 44–55). Keeping constant the halide, [EMIM]+ cation always provides the lowest KH values, while for the same cation KH increases from chloride to bromide. Similar trends are noted for |BE′| (see Fig. 7), i.e., high KH is related with low |BE′|. The elected halides in this work gave |BE| values (≃52.18 kJ mol−1) lower than other anions; even though this |BE| is larger than those obtained for imidazolium cations (≃35.99 kJ mol−1). For ILs 45–55, cation⋯SO2 and anion⋯SO2 contributions take values of around 8.00 kJ mol−1 and 88 kJ mol−1. Halide effects on the SO2 adsorption mechanism have been analyzed for ILs based on the [EMIM]+ cation as a function of the anion. Optimized structure for [EMIM][Cl]and [EMIM][Br] (optimized structures for [EMIM][[I] is not displayed since similar results to [EMIM][Br] are obtained) are shown in Fig. 15, while the main structural parameter of intermolecular interactions along their electronic density values are collected in Table 7. As expected, the main interaction between both ions is a hydrogen bond between the halide and the H atom located in position 2 (d1 or d6 for IL 45 or 48/51, respectively), which is weakened in the presence of SO2 molecule. For IL⋯SO2 systems, S⋯X (X = Cl, Br or I) is the main interaction (labelled as d3 or d9 for IL 45 or 48/51, respectively), while two O⋯H intermolecular bonds are also found between SO2 and the cation. As seen in Table 6, electronic density for d6 is much greater than those of cation⋯SO2 interactions in concordance with its larger contribution from [Cl]−⋯SO2 interaction. The same behaviour is also noted for ILs 48 and 51.
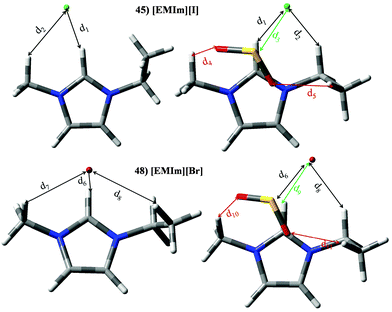 |
| | Fig. 15 Optimized geometries of [EMIm][Cl] (45) and [EMIM][Br] (48). Similar geometries are obtained for [EMIM][I] (51). Main intermolecular interactions are also displayed. Atom colour code: C (gray), oxygen (red) sulphur (yellow), hydrogen (white), nitrogen (blue), chloride (green) and bromide (garnet). See Table 7 for a more detailed description on intermolecular interactions. | |
Table 7 Intermolecular distances (d) along the electronic density values (ρ) of [EMIm][Cl] (45), [EMIm][Br] (48) and [EMIM][I] (51). See Fig. 15 for labeling
| |
IL |
IL⋯SO2 |
|
d/Å |
ρ/a.u. |
d/Å |
ρ/a.u. |
| 45 – [EMIm][Cl] |
|
d
1
|
1.982 |
0.0264 |
2.515 |
0.0149 |
|
d
2
|
2.750 |
0.0096 |
2.681 |
0.0111 |
|
d
3
|
2.589 |
0.0474 |
|
d
4
|
2.668 |
0.0110 |
|
d
5
|
2.367 |
0.0115 |
|
|
| 48 – [EMIm][Br] |
|
d
6
|
2.798 |
0.0242 |
2.581 |
0.0157 |
|
d
7
|
2.865 |
0.0101 |
|
d
8
|
2.861 |
0.0095 |
2.754 |
0.0117 |
|
d
9
|
2.709 |
0.0455 |
|
d
10
|
2.112 |
0.0189 |
|
d
11
|
2.321 |
0.0125 |
|
|
| 51 – [EMIm][I] |
|
d
6
|
2.987 |
0.0218 |
2.753 |
0.0149 |
|
d
7
|
3.041 |
0.0088 |
|
d
8
|
3.075 |
0.0099 |
2.963 |
0.0095 |
|
d
9
|
2.775 |
0.0455 |
|
d
10
|
2.121 |
0.0182 |
|
d
11
|
2.383 |
0.0134 |
4. Conclusions
This contribution reports a density functional theory (DFT) on several ILs, for which high SO2 solubility is expected. This work is divided into three parts: (i) we selected a set of ILs which should provide high efficiency for SO2 capture. For this, a screening of a large number of ILs via the COSMO-RS method was done; (ii) binding energies between SO2 and ILs were analyzed intensely through DFT simulations for a set of 55 ILs, which provided high efficiency in SO2 capture according to the COSMO-RS method; (iii) intermolecular interaction for some representative ILs were deeply studied through the AIM theory aimed at obtaining some information on the SO2 adsorption mechanism at the molecular level. The results evidenced the ability of the selected cations and anions to interact with the SO2 molecule, which is stronger for anion⋯SO2 interactions. Thus, anion⋯SO2 interactions are ruled by a strong charge transfer from the anion to SO2 molecule. For the ILs⋯SO2 system, the total binding energy (BE) has been decomposed in the contributions from the interactions between ions, anion⋯SO2 and cation⋯SO2. The interaction between both ions always provided the largest contribution to the total binding energy. Then, the binding energy related with SO2 capture by ILs was also calculated considering the ILs as a whole (BE′). A value of around 36.58 kJ mol−1 (for CO2 capture by [EMIM][NTf2] IL, which was taken as a pivotal reference for comparison purposes) as a low limit; all ILs yield larger binding energies. Most of them provide values of around 45.0 kJ mol−1. Therefore, all of them would provide high SO2 capture efficiency. Through the comparison between ion⋯SO2 contributions and BE′, we could obtain some information on what ions mainly govern the SO2 capture within the ILs. In most cases, SO2 capture would be mainly ruled out by the anion or by both ions. Even if the SO2 capture mechanism at the molecule level depends on each ILs, some common features as found for related ions. Even though, a statistical analysis of binding energies of IL–SO2 systems as a function of ion–SO2 and cation–anion ones brings to light that SO2 adsorption by ILs at the molecular level is mainly ruled by anion–SO2 interaction and cation–anion as well. Thus, qualitative trends on SO2 capture by ILs can be obtained only based on the study of anion–SO2 and isolated ILs systems. Systematic research on ILs for SO2 capture allow increase of our knowledge about those factors which could be controlled at the molecular level, allowing an approach up to the rational design of task-specific ILs for future applied studies.
Acknowledgements
Gregorio García acknowledges the funding by Junta de Castilla y León (Spain), cofunded by European Social Fund, for a postdoctoral contract. Santiago Aparicio acknowledges the funding by Ministerio de Economía y Competitividad (Spain, project CTQ2013-40476-R) and Junta de Castilla y León (Spain, project BU324U14). Mert Atilhan acknowledges support of an NPRP grant (No: 6-330-2-140) from the Qatar National Research Fund. The statements made herein are solely the responsibility of the authors.
References
- S. J. Smith, J. van Aardenne, Z. Klimont, R. J. Andres, A. Volke and S. Delgado Arias, Atmos. Chem. Phys., 2011, 11, 1101–1116 CAS.
- G. Cui, C. Wang, J. Zheng, Y. Guo, X. Luo and H. Li, Chem. Commun., 2012, 48, 2633–2635 RSC.
- J. Huang, A. Riisager, P. Wasserscheid and R. Fehrmann, Chem. Commun., 2006, 4027–4029 RSC.
- Z. Z. Yang, L. N. He, Y. N. Zhao and B. Yu, Environ. Sci. Technol., 2013, 47, 1598–1605 CAS.
- D. Yang, M. Hou, H. Ning, J. Ma, X. Kang, J. Zhang and B. Han, Reversible Capture of SO2 through Functionalized Ionic Liquids, ChemSusChem, 2013, 6(7), 1191–1195 CrossRef CAS PubMed.
- S. Tian, Y. Hou, W. Wu, S. Ren and C. Zhang, RSC Adv., 2013, 3, 3572–3577 RSC; X. L. Yuan, S. J. Zhang and X. M. Lu, J. Chem. Eng. Data, 2007, 52, 596–599 CrossRef CAS; S. Ren, Y. Hou, W. Wu, Q. Liu, Y. Xiao and X. Chen, J. Phys. Chem. B, 2010, 114, 2175–2179 CrossRef PubMed; G. Cui, J. Zheng, X. Luo, W. Lin, F. Ding, H. Li and C. Wang, Angew. Chem., Int. Ed., 2013, 52, 10620–10624 CrossRef PubMed.
- G. Yu and X. Chen, J. Phys. Chem. B, 2011, 115, 3466–3477 CrossRef CAS PubMed.
- S. Ren, Y. Hou, S. Tian, X. Chen and W. Wu, J. Phys. Chem. B, 2013, 117, 482–2486 Search PubMed.
- Z. Lei, C. Dai and B. Chen, Chem. Rev., 2013, 114, 1289–1326 CrossRef PubMed.
- S. Aparicio and M. Atilhan, Energy Fuels, 2010, 24, 4989–5001 CrossRef CAS; S. Aparicio and M. Atilhan, J. Phys. Chem. B, 2012, 116, 9171–9185 CrossRef PubMed; F. Karadas, M. Atilhan and S. Aparicio, Energy Fuels, 2010, 24, 5817–5828 CrossRef; D. H. Zaitsau, A. V. Yermalayeu, S. P. Verevkin, J. E. Bara and A. D. Stanton, Ind. Eng. Chem. Res., 2013, 52, 16615–16621 CrossRef; C. Wang, X. Luo, H. Luo, D. E. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 4918–4922 CrossRef PubMed; X. Zhang, H. Dong, Z. Zhao, S. Zhang and Y. Huang, Carbon capture with ionic liquids: overview and progress, Energy Environ. Sci., 2012, 5, 6668–6681 Search PubMed.
- F. Karadas, B. Köz, J. Jacquemin, E. Deniz, D. Rooney, J. Thompson, C. T. Yavuz, M. Khraisheh, S. Aparicio and M. Atilhan, Fluid Phase Equilib., 2013, 351, 74–86 CrossRef CAS PubMed.
- V. Sanz, R. Alcalde, M. Atilhan and S. Aparicio, J. Mol. Model., 2014, 20, 1–14 CrossRef CAS PubMed.
- S. Aparicio, M. Atilhan, M. Khraisheh, R. Alcalde and J. Fernández, J. Phys. Chem. B, 2011, 115, 12487–12498 CrossRef CAS PubMed.
- J. Palomar, M. González-Miquel, A. Polo and F. Rodríguez, Ind. Eng. Chem. Res., 2011, 50, 3452–3463 CrossRef CAS.
-
J. Earle-Martyn and R. Seddon-Kenneth, Green Solvents for the Future, in Clean Solvents, American Chemical Society, 2002 RSC; J. S. Wilkes, Green Chem., 2002, 4, 73–80 RSC; R. D. Rogers and K. R. Seddon, Science, 2003, 792–793 CrossRef CAS PubMed.
- O. Hollóczki, Z. Kelemen, L. Könczöl, D. Szieberth, L. Nyulászi, A. Stark and B. Kirchner, ChemPhysChem, 2013, 14, 315–320 CrossRef CAS PubMed; K. M. Gupta and J. Jiang, J. Phys. Chem. C, 2014, 118, 3110–3118 Search PubMed; P. Gu, R. Lü, S. Wang, Y. Lu and D. Liu, Comput. Theor. Chem., 2013, 1020, 22–31 CrossRef PubMed; F. Yan, M. Lartey, K. Damodaran, E. Albenze, R. L. Thompson, J. Kim, M. Haranczyk, H. B. Nulwala, D. R. Luebke and B. Smit, Phys. Chem. Chem. Phys., 2013, 15, 3264–3272 RSC; C. Wu, T. P. Senftle and W. F. Schneider, Phys. Chem. Chem. Phys., 2012, 14, 13163–13170 RSC; B. Gurkan, B. F. Goodrich, E. M. Mindrup, L. E. Ficke, M. Massel, S. Seo, T. P. Senftle, H. Wu, M. F. Glaser, J. K. Shah, E. J. Maginn, J. F. Brennecke and W. F. Schneider, J. Phys. Chem. Lett., 2010, 1, 3494–3499 CrossRef.
- G. B. Damas, A. B. A. Dias and L. T. Costa, J. Phys. Chem. B, 2014, 118, 9046–9064 CrossRef CAS PubMed.
-
A. Klamt, COSMO-RS: From Quantum Chemistry to Fluid Phase Thermodynamcis and Drug Design, Elsevier, Amsterdam, 2005 CrossRef CAS PubMed; F. Eckert and A. Klamt, AIChE J., 2002, 48, 369–385 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef PubMed; A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2012, 112, 289–320 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- T. Schwabe and S. Grimme, Phys. Chem. Chem. Phys., 2007, 9, 3397–3406 RSC; S. Zahn and B. Kirchner, J. Phys. Chem. A, 2008, 112, 8430–8435 CrossRef CAS PubMed; S. Grimme, W. Hujo and B. Kirchner, Phys. Chem. Chem. Phys., 2012, 14, 4875–4883 RSC; E. I. Izgorodina, U. L. Bernard and D. R. MacFarlane, J. Phys. Chem. A, 2009, 113, 7064–7072 CrossRef PubMed; S. Zahn, F. Uhlig, J. Thar, C. Spickermann and B. Kirchner, Angew. Chem., Int. Ed., 2008, 47, 3639–3641 CrossRef PubMed.
- J. Marten, W. Seichter, E. Weber and U. Bohme, CrystEngComm, 2008, 10, 541–547 RSC.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024 CrossRef CAS PubMed; S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef PubMed.
- R. A. Ando, L. J. A. Siqueira, F. C. Bazito, R. M. Torresi and P. S. Santos, J. Phys. Chem. B, 2007, 111, 8717–8719 CrossRef CAS PubMed; L. J. A. Siqueira, R. A. Ando, F. C. C. Bazito, R. M. Torresi, P. S. Santos and M. C. C. Ribeiro, J. Phys. Chem. B, 2008, 112, 6430–6435 CrossRef PubMed.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833 CrossRef CAS PubMed.
- J. J. Philips, M. A. Hudspeth, Jr, P. M. Browne and J. E. Peralta, Chem. Phys. Lett., 2010, 495, 146–150 CrossRef CAS PubMed.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS PubMed.
- I. Bandrés, R. Alcalde, C. Lafuente, M. Atilhan and S. Aparicio, J. Phys. Chem. B, 2011, 115, 12499–12513 CrossRef PubMed.
-
R. F. W. Bader, Atoms in Molecules: a Quantum Theory, Oxford, 1990 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. Palomar, V. R. Ferro, J. S. Torrecilla and F. Rodríguez, Ind. Eng. Chem. Res., 2007, 46, 6041–6048 CrossRef CAS; J. Palomar, J. S. Torrecilla, V. R. Ferro and F. Rodríguez, Ind. Eng. Chem. Res., 2009, 48, 2257–2265 CrossRef; A. Klamt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 699–709 CrossRef PubMed; M. A. Diedenhofen and A. Klamt, Fluid Phase Equilib., 2010, 294, 31–38 CrossRef PubMed; X. Zhang, Z. Liu and W. Wang, AIChE J., 2008, 54, 2717–2728 CrossRef PubMed; M. B. Miller, D. L. Chen, H. B. Xie, D. R. Luebke, J. Karl Johnson and R. M. Enick, Fluid Phase Equilib., 2009, 287, 26–32 CrossRef PubMed; R. Franke, B. Hannebauer and S. A. Jung, Chem. Eng. Technol., 2010, 33, 251–257 CrossRef PubMed; N. A. Manan, C. Hardacre, J. Jacquemin, D. W. Rooney and T. G. Youngs, J. Chem. Eng. Data, 2009, 54, 2005–2022 CrossRef.
- M. González-Miquel, J. Palomar and S. Omar, Ind. Eng. Chem. Res., 2011, 50, 5739–5748 CrossRef.
- M. González-Miquel, J. Bedia, J. Palomar and F. Rodríguez, J. Chem. Eng. Data, 2014, 59, 212–217 CrossRef.
- M. Atilhan, J. Jacquemin, D. Rooney, M. Khraisheh and S. Aparicio, Ind. Eng. Chem. Res., 2013, 52, 16774–16785 CrossRef CAS.
- G. García, M. Atilhan and S. Aparicio, Chem. Phys. Lett., 2014, 610, 267–272 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp00076a |
|
| This journal is © the Owner Societies 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a