
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeprotonation of a dinuclear copper complex of 3,5-diamino-1,2,4-triazole for high oxygen reduction activity†
Masaru
Kato
ab,
Ken'ichi
Kimijima‡
c,
Mari
Shibata
c,
Hideo
Notsu
c,
Kazuya
Ogino
c,
Kiyoshi
Inokuma
c,
Narumi
Ohta§
c,
Hiromitsu
Uehara
d,
Yohei
Uemura¶
d,
Nobuhisa
Oyaizu
b,
Tadashi
Ohba
d,
Satoru
Takakusagi
d,
Kiyotaka
Asakura
*d and
Ichizo
Yagi
*abc
aFaculty of Environmental Earth Science, Hokkaido University, N10W5, Kita-ku, Sapporo 060-0810, Japan. E-mail: iyagi@ees.hokudai.ac.jp
bGraduate School of Environmental Science, Hokkaido University, N10W5, Kita-ku, Sapporo 060-0810, Japan
cFuel Cell Cutting-Edge Center - Technology Research Association (FC-Cubic, TRA), 2-3-26 Aomi, Koto-ku, Tokyo 135-0064, Japan
dCatalysis Research Center, Hokkaido University, N21W10, Kita-ku, Sapporo 001-0021, Japan
First published on 3rd March 2015
Abstract
A dinuclear copper(II) complex of 3,5-diamino-1,2,4-triazole is one of the highly active copper-based catalysts for the oxygen reduction reaction (ORR) in basic solutions. Our in situ X-ray absorption near edge structure measurements revealed that deprotonation of the triazole ligand might cause coordination geometrical changes, resulting in the enhancement of the ORR activity.
Polymer electrolyte fuel cells (PEFCs) generate electricity from oxygen and hydrogen, where only water is produced without the formation of any toxic or greenhouse gases. PEFCs are sustainable power sources for mobile phones, laptop computers and vehicles, and have great potential to shift away from the fossil-fuel economy we have today toward a carbon-emission-free economy, where hydrogen will be used as an energy source. To develop practical PEFCs we need to overcome many technical obstacles, and one well-known bottleneck for the widespread applications of PEFCs is the development of low-cost catalysts for the oxygen reduction reaction (ORR) at the cathode. The best-known electrocatalyst for ORR is the carbon-supported platinum metal.1–4 Despite extensive efforts in searching for efficient Pt-based catalysts, platinum is scarce and the Pt-based cathodes still require a large overpotential of >200 mV for ORR. It is highly desirable to find low-cost electrocatalysts that can fulfill demands in PEFCs.
In nature multicopper oxidases (MCOs) such as laccases are able to catalyze the ORR with great efficiency.5 MCOs reduce oxygen to water at their multi-nuclear copper active site with almost no overpotential (ca. 20 mV).6–10 It is most unlikely that the enzymes would be used as biocathodes in practical PEFCs, since the enzymes are easily denatured and their footprints are large (ca. 160 nm3 for laccase),11 resulting in low stability and power outputs, respectively. The active site structures of MCOs, however, have inspired us to design artificial copper-based ORR catalysts, and many bio-inspired electrocatalysts have been extensively studied.12–15
A bio-inspired dinuclear copper(II) complex of 3,5-diamino-1,2,4-triazole (Cu-Hdatrz, Scheme 1)16 supported on carbon black shows high ORR activities in basic solutions.17 Although many studies on the Cu-Hdatrz catalyst have been done, for example, its tolerance to poisoning ions18 and its ORR activity in a lipid layer,19 its detailed catalytic mechanism has not been understood yet. The investigation of the chemical nature of Cu-Hdatrz under reaction conditions will give us insights into such high ORR activities in basic solutions, and will facilitate further development of bio-inspired ORR catalysts.
 | ||
| Scheme 1 Molecular structure of the dinuclear copper(II) complex of 3,5-diamino-1,2,4-triazole (Cu-Hdatrz). | ||
Here we report electrochemical and in situ X-ray absorption fine structure (XAFS) studies of the carbon-supported Cu-Hdatrz under neutral and basic conditions. In situ XAFS is a powerful technique used to probe the electronic structure or coordination geometry of metal-based electrocatalysts under catalytic conditions.20–22 In this work we studied X-ray absorption near-edge structure (XANES) spectra of Cu-Hdatrz at various bias potentials under neutral and basic conditions (pH 7, 10 and 13) to understand relationships between deprotonation of the active species and the ORR activity.
A carbon black-supported CuII-Hdatrz was synthesized from CuSO4·5H2O, Hdatrz and Ketjenblack ECP300 (KB), based on the method in the literature with slight modifications (see ESI†).17 KB is a carbon black material with a relatively high BET surface area (813 m2 g−1), compared to Vulcan (247 m2 g−1), which was used as a carbon support in the original catalyst.17 Carbon supports with high surface areas, in principle, allow us to load well-dispersed catalysts, which are suitable for fluorescence XANES measurements with high data quality.
Cyclic voltammograms (CVs) of the copper(II) complex of Hdatrz supported on KB (Cu/KB) were recorded under Ar to understand the redox behavior of Cu/KB under non-catalytic conditions (Fig. 1). At pH 7 an oxidation wave at +0.73 V vs. RHE and a reduction wave at +0.39 V vs. RHE were observed, and these waves were assigned as: the oxidation wave to CuII and the reduction wave to CuI, respectively. The peak separation between these peaks was approximately 0.33 V, which was much greater than 0 V for electrochemical active species strongly adsorbing on electrode surfaces, indicating that the electrochemical redox behavior of Cu-Hdatrz was irreversible. We also recorded CVs of Cu/KB at pH 4, pH 10 and pH 13, and obtained similar results for the peak separation. Thus, the redox reaction of CuII-Hdatrz may involve structural changes such as geometrical changes around the metal center.
 | ||
| Fig. 1 CVs of Cu/KB in Britton–Robinson buffered aqueous solutions (0.04 M) containing 0.1 M NaClO4 at pH 4 (solid line in black), 7 (dashed line in red), 10 (dotted line in green) and 13 (alternate long and short dashed line in blue). All CVs shown here were recorded at a sweep rate of 100 mV s−1 under Ar. | ||
The ORR activity of Cu/KB was studied using rotating ring disc electrodes (RRDEs) in buffered aqueous solutions at pH 4, 7, 10 and 13. Linear sweep voltammograms of Cu/KB were recorded at 1600 rpm under oxygen (Fig. 2). An onset potential of Cu/KB for ORR was observed at +0.77 V vs. RHE at pH 7 and the diffusion-limited current reached ca. −1.0 mA. A quite similar ORR activity was observed for Cu-Hdatrz on Vulcan under the same conditions (Fig. S1, ESI†), suggesting that there was no obvious effect of the carbon supports (KB and Vulcan) on the catalytic activity. The ring current of Cu/KB is less than 5 μA at pH 7, indicating almost no hydrogen peroxide generation during ORR. Thus, Cu/KB is able to selectively reduce molecular oxygen to water.
 | ||
| Fig. 2 (A) Ring and (B) disc current of linear sweep voltammograms of Cu/KB using a RRDE (disc: glassy carbon with 0.196 cm2; ring: Pt) at 1600 rpm at pH 4 (black), pH 7 (red), pH 10 (green) and pH 13 (blue) under oxygen. The voltammograms were recorded at a sweep rate of 20 mV s−1 scanning in the positive direction. Britton–Robinson buffered aqueous solution (0.04 M) containing 0.1 M NaClO4 was used as the electrolyte solution. A constant bias potential of 1.2 V vs. RHE was applied to the Pt ring electrode. (C) A plot of onset potential for ORR vs. pH. The plot gave a linear relationship with Eonset = 0.026 × pH + 0.57. | ||
The ORR activity of Cu/KB depends on pH, and Cu/KB is more active in basic solutions than in the neutral solution: the onset potentials for ORR were shifted in the positive direction with increasing pH, and the plot of the onset potential against pH gave a linear relationship with a slope of 26 mV per pH (Fig. 2C), which is equivalent to −33 mV per pH using a pH-independent reference electrode. This value is approximately half the value of −60 mV per pH, implying that the rate-limiting step involves the transfer of two electrons and one proton. The same results were reported for CuII-Hdatrz supported on a carbon black of Vulcan, where the reduction of two copper centers and the concomitant protonation of a bridging OH− or O2− ligand occur.17 These results suggest that carbon supports may not influence the ORR mechanism of Cu-Hdatrz in the pH range from 4 to 13.
In situ XANES spectra of Cu/KB were recorded at pH 7, 10 and 13 under catalytic (under O2) and non-catalytic conditions (under N2) to gain insights into active species during ORR. The potential dependent XANES spectra of Cu/KB are shown in Fig. 3. The spectra show isosbestic points, indicating that CuII-Hdatrz was electrochemically converted to one reduced species, and a characteristic peak appeared at ~8981 eV in the edge region upon electrochemical reduction. This peak is assigned to the 1s → 4pπ transition of the CuI complex.22–24 Thus, CuII-Hdatrz was electrochemically reduced and converted to the CuI species.
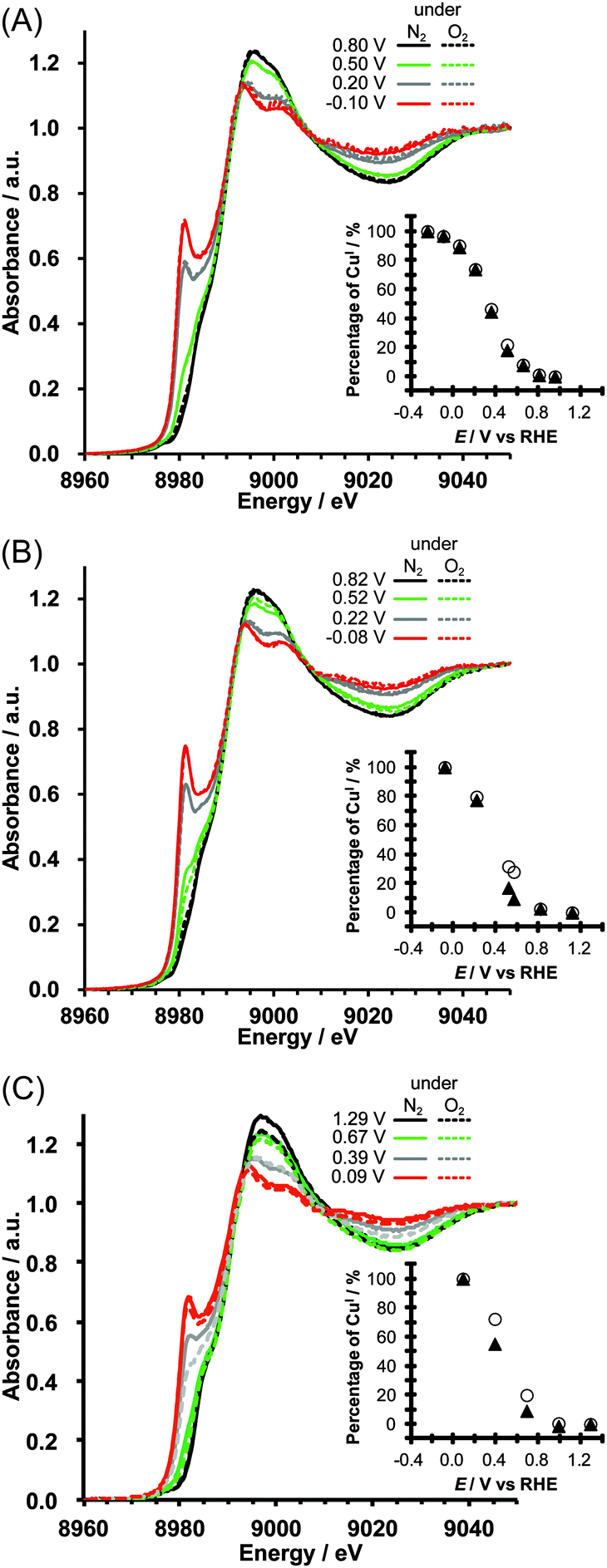 | ||
| Fig. 3 In situ fluorescence XANES spectra of Cu/KB (A) at pH 7, (B) at pH 10 and (C) pH 13. The solid line and dashed lines indicate the XANES spectra that were recorded under nitrogen and oxygen, respectively. Insets: plots of percentages of CuIvs. applied potential (open circle: under nitrogen; filled triangle: under oxygen). The percentages were calculated based on the results of the linear combination analysis of the XANES spectra of Cu/KB and fully reduced Cu/KB. | ||
Linear combination fitting analysis was performed for the XANES spectra of Cu/KB, and percentages of CuI species were calculated, assuming that all CuII-Hdatrz molecules were reduced at −0.25 V vs. RHE at pH 7, at −0.08 V vs. RHE at pH 10 and at +0.09 V vs. RHE at pH 13 (insets in Fig. 3). Although at pH 7 there was no difference between the amounts of CuI under N2 and O2 (Fig. 3A), more amounts of CuI were observed under N2 than those under oxygen at pH 10 (Fig. 3B) and pH 13 (Fig. 3C). Thus, it is clear that the electrochemical behavior of Cu-Hdatrz under basic conditions is different from that under the neutral conditions.
Considering the amounts of CuI calculated (Fig. 3) and two simplified electron transfer steps (Scheme 2), we concluded that the second electron transfer might be faster in basic solutions than in the neutral solution. The first step is the electron transfer from the electrode or KB to the CuII-Hdatrz complex and the second step is the electron transfer from the CuI species to molecular oxygen (the ORR step). The XANES spectra at pH 7 under N2 and O2 exhibited the same relative amounts of CuI at each potential, indicating that CuII-Hdatrz can be immediately reduced to the CuI species even in the presence of oxygen. At pH 10 and pH 13, less relative amounts of CuI were observed under O2 than those under N2, suggesting that the ORR was faster than the reduction of CuII-Hdatrz by the electrode or KB at pH 10. The second electron transfer (the ORR step) may be faster in basic solutions than in the neutral solution, which is consistent with the high ORR activity observed in the basic solution (Fig. 2).
 | ||
| Scheme 2 Simplified electron transfer steps in the Cu-Hdatrz/KB system: (i) electron transfer from the electrode/KB to CuII-Hdatrz and (ii) electron transfer from CuI-Hdatrz to O2 (the ORR step). | ||
Since XANES spectra are sensitive to coordination geometrical changes around the element measured, the XANES spectra of Cu/KB recorded at pH 7, pH 10 and pH 13 were overlaid to compare their shapes (Fig. 4). Although almost similar shapes of the spectra were observed at pH 7 and pH 10 in the same oxidation state, the XANES spectra at pH 13 were different from those at pH 7 and pH 10, indicating that the coordination geometries of the CuII and CuI centers were the same in the pH range from 7 to 10, but these might be different at pH 13. Furthermore, spectral differences were observed between the XANES spectra under nitrogen and oxygen at pH 13 (Fig. S2, ESI†). Since the pKa value of Hdatrz was reported to be 12.12,25 it is most likely that deprotonation occurred in the Hdatrz ligands at pH 13. Thus, deprotonation of Cu-Hdatrz in basic solutions may affect the electronic state of the Hdatrz ligands and induce coordination geometrical changes, resulting in a highly active ORR catalyst in basic solutions.
 | ||
| Fig. 4 In situ XANES spectra of Cu/KB in the oxidation state of CuII (A) under N2 and (B) under O2, and in the oxidation state of CuI (C) under N2 and (D) under O2. The red, green and blue traces indicate the spectra recorded at pH 7, pH 10 and pH 13, respectively. Bias potentials applied are as follows: +0.95 V vs. RHE at pH 7, +1.11 V vs. RHE at pH 10, +1.29 V vs. RHE at pH 13 for CuII; −0.25 V vs. RHE at pH 7, −0.08 V vs. RHE at pH 10 and +0.09 V vs. RHE at pH 13 for CuI. | ||
In summary, the electrochemical ORR activity of Cu/KB was studied in neutral and basic aqueous solutions using the in situ electrochemical XANES techniques. Our electrochemical studies revealed that the faster ORR kinetics of Cu/KB resulted in the higher ORR catalytic activity under basic conditions, compared to that under the neutral conditions. The coordination geometry at pH 13 was different from those at pH 7 and pH 10, and this difference might be caused by deprotonation of the Hdatrz ligand. The structural information of the deprotonated CuI species or O2-bound intermediates in basic solutions may be a key to understand the high ORR activity of Cu/KB or to design highly active Cu-based ORR catalysts.
Further in situ XAFS studies on the Cu-Hdatrz complex are under way to obtain more detailed structural information in basic solutions.
Acknowledgements
This work has been supported by NEDO and performed under the approval of the Photon Factory Program Advisory Committee (Proposal no. 2010G200 & 2013G173).Notes and references
- A. A. Gewirth and M. S. Thorum, Inorg. Chem., 2010, 49, 3557–3566 CrossRef CAS PubMed.
- S. Guo, S. Zhang and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526–8544 CrossRef CAS PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102–121 CrossRef CAS PubMed.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 CAS.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- C. F. Blanford, R. S. Heath and F. A. Armstrong, Chem. Commun., 2007, 1710–1712 RSC.
- N. Mano, V. Soukharev and A. Heller, J. Phys. Chem. B, 2006, 110, 11180–11187 CrossRef CAS PubMed.
- P. Olejnik, B. Palys, A. Kowalczyk and A. M. Nowicka, J. Phys. Chem. C, 2012, 116, 25911–25918 CAS.
- V. Climent, Y. Fu, S. Chumillas, B. Maestro, J.-F. Li, A. Kuzume, S. Keller and T. Wandlowski, J. Phys. Chem. C, 2014, 118, 15754–15765 CAS.
- M. S. Thorum, C. A. Anderson, J. J. Hatch, A. S. Campbell, N. M. Marshall, S. C. Zimmerman, Y. Lu and A. A. Gewirth, J. Phys. Chem. Lett., 2010, 1, 2251–2254 CrossRef CAS PubMed.
- K. Piontek, M. Antorini and T. Choinowski, J. Biol. Chem., 2002, 277, 37663–37669 CrossRef CAS PubMed.
- D. Das, Y.-M. Lee, K. Ohkubo, W. Nam, K. D. Karlin and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 4018–4026 CrossRef CAS PubMed.
- C. C. L. McCrory, X. Ottenwaelder, T. D. P. Stack and C. E. D. Chidsey, J. Phys. Chem. A, 2007, 111, 12641–12650 CrossRef CAS PubMed.
- M. A. Thorseth, C. S. Letko, T. B. Rauchfuss and A. A. Gewirth, Inorg. Chem., 2011, 50, 6158–6162 CrossRef CAS PubMed.
- M. A. Thorseth, C. E. Tornow, E. C. M. Tse and A. A. Gewirth, Coord. Chem. Rev., 2013, 257, 130–139 CrossRef CAS PubMed.
- E. Aznar, S. Ferrer, J. Borras, F. Lloret, M. Liu-Gonzalez, H. Rodriguez-Prieto and S. Garcia-Granda, Eur. J. Inorg. Chem., 2006, 5115–5125 CrossRef CAS.
- M. S. Thorum, J. Yadav and A. A. Gewirth, Angew. Chem., Int. Ed., 2009, 48, 165–167 CrossRef CAS PubMed.
- M. S. Thorum, J. M. Hankett and A. A. Gewirth, J. Phys. Chem. Lett., 2011, 2, 295–298 CrossRef CAS.
- C. J. Barile, E. C. M. Tse, Y. Li, T. B. Sobyra, S. C. Zimmerman, A. Hosseini and A. A. Gewirth, Nat. Mater., 2014, 13, 619–623 CrossRef CAS PubMed.
- K. A. Kuttiyiel, K. Sasaki, Y. Choi, D. Su, P. Liu and R. R. Adzic, Energy Environ. Sci., 2012, 5, 5297–5304 CAS.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- T. P. Kofman, Russ. J. Org. Chem., 2001, 37, 1158–1168 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and figures of our spectroelectrochemical cell, electrochemical ORR and in situ XANES spectra. See DOI: 10.1039/c4cp05595k |
| ‡ Present address: Institute of Materials Structure Science, High Energy Accelerator Research Organization, KEK, Oho 1-1, Tsukuba, 305-0801, Japan. |
| § Present address: Environment and Energy Materials Division, National Institute for Materials Science, 1-1 Namiki, Tsukuba, 305-0044, Japan. |
| ¶ Present address: Division of Electronic Structure, Department of Materials Molecular Science, Institute for Molecular Science, Myodaiji, Okazaki 444-8585, Japan. |
| This journal is © the Owner Societies 2015 |